

Abstract
Background
Schizophrenia often presents in adolescence, but current treatment guidelines are based largely on studies of adults with psychosis. Over the past decade, the number of studies on treatment of adolescent‐onset psychosis has increased. The current systematic review collates and critiques evidence obtained on the use of various atypical antipsychotic medications for adolescents with psychosis.
Objectives
To investigate the effects of atypical antipsychotic medications in adolescents with psychosis. We reviewed in separate analyses various comparisons of atypical antipsychotic medications with placebo or a typical antipsychotic medication or another atypical antipsychotic medication or the same atypical antipsychotic medication but at a lower dose.
Search methods
We searched the Cochrane Schizophrenia Group Register (October 2011), which is based on regular searches of BIOSIS, CENTRAL, CINAHL, EMBASE, MEDLINE and PsycINFO. We inspected references of all identified studies and contacted study authors and relevant pharmaceutical companies to ask for more information.
Selection criteria
We included all relevant randomised controlled trials (RCTs) that compared atypical antipsychotic medication with placebo or another pharmacological intervention or with psychosocial interventions, standard psychiatric treatment or no intervention in children and young people aged 13 to 18 years with a diagnosis of schizophrenia, schizoaffective disorder, acute and transient psychoses or unspecified psychosis. We included studies published in English and in other languages that were available in standardised databases.
Data collection and analysis
Review authors AK and SSD selected the studies, rated the quality of the studies and performed data extraction. For dichotomous data, we estimated risk ratios (RRs) with 95% confidence intervals (CIs) using a fixed‐effect model. When possible, for binary data presented in the 'Summary of findings' table, we calculated illustrative comparative risks. We summated continuous data using the mean difference (MD). Risk of bias was assessed for included studies.
Main results
We included 13 RCTs, with a total of 1112 participants. We found no data on service utilisation, economic outcomes, behaviour or cognitive response. Trials were classified into the following groups.
1. Atypical antipsychotics versus placebo Only two studies compared one atypical antipsychotic medication with placebo. In one study, the number of non‐responders treated with olanzapine was not different from the number treated with placebo (1 RCT, n = 107, RR 0.84, 95% CI 0.65 to 1.10); however, significantly more (57% vs 32%) people left the study early (1 RCT, n = 107, RR 0.56, 95% CI 0.36 to 0.87) from the placebo group compared with the olanzapine group. With regard to adverse effects, young people treated with aripiprazole had significantly lower serum cholesterol compared with those given placebo (1 RCT, n = 302, RR 3.77, 95% CI 1.88 to 7.58).
2. Atypical antipsychotics versus typical antipsychotics When the findings of all five trials comparing atypical antipsychotic medications with a typical antipsychotic medication were collated, no difference in the mean end point Brief Psychiatric Rating Scale (BPRS) score was noted between the two arms (5 RCTs, n = 236, MD ‐1.08, 95% CI ‐3.08 to 0.93). With regard to adverse effects, the mean end point serum prolactin concentration was much higher than the reference range for treatment with risperidone, olanzapine and molindone in one of the studies. However, fewer adolescents who were receiving atypical antipsychotic medications left the study because of adverse effects (3 RCTs, n = 187, RR 0.65, 95% CI 0.36 to 1.15) or for any reason (3 RCTs, n = 187, RR 0.62, 95% CI 0.39 to 0.97).
3. One atypical antipsychotic versus another atypical antipsychotic The mean end point BPRS score was not significantly different for people who received risperidone compared with those who received olanzapine; however, the above data were highly skewed. Overall no difference was noted in the number of people leaving the studies early because of any adverse effects between each study arm in the three studies comparing olanzapine and risperidone (3 RCTs, n = 130, RR 1.15, 95% CI 0.44 to 3.04). Specific adverse events were not reported uniformly across the six different studies included in this section of the review; therefore it was difficult to do a head‐to‐head comparison of adverse events for different atypical antipsychotic medications.
4. Lower‐dose atypical antipsychotic versus standard/higher‐dose atypical antipsychotic Three studies reported comparisons of lower doses of the atypical antipsychotic medication with standard/higher doses of the same medication. One study reported better symptom reduction with a standard dose of risperidone as compared with a low dose (1 RCT, n = 257, RR ‐8.00, 95% CI ‐13.75 to ‐2.25). In another study, no difference was reported in the number of participants not achieving remission between the group receiving 10 mg/d and those who received 30 mg/d of aripiprazole (1 RCT, n = 196, RR 0.84, 95% CI 0.48 to 1.48). Similarly in the other study, authors reported no statistically significant difference in clinical response between the two groups receiving lower‐dose (80 mg/d) and higher‐dose (160 mg/d) ziprasidone, as reflected by the mean end point BPRS score (1 RCT, n = 17, MD ‐4.40, 95% CI ‐19.20 to 10.40).
Authors' conclusions
No convincing evidence suggests that atypical antipsychotic medications are superior to typical medications for the treatment of adolescents with psychosis. However, atypical antipsychotic medications may be more acceptable to young people because fewer symptomatic adverse effects are seen in the short term. Little evidence is available to support the superiority of one atypical antipsychotic medication over another, but side effect profiles are different for different medications. Treatment with olanzapine, risperidone and clozapine is often associated with weight gain. Aripiprazole is not associated with increased prolactin or with dyslipidaemia. Adolescents may respond better to standard‐dose as opposed to lower‐dose risperidone, but for aripiprazole and ziprasidone, lower doses may be equally effective. Future trials should ensure uniform ways of reporting.
Keywords: Adolescent, Humans, Antipsychotic Agents, Antipsychotic Agents/adverse effects, Antipsychotic Agents/therapeutic use, Aripiprazole, Benzodiazepines, Benzodiazepines/adverse effects, Benzodiazepines/therapeutic use, Molindone, Molindone/adverse effects, Molindone/therapeutic use, Olanzapine, Piperazines, Piperazines/adverse effects, Piperazines/therapeutic use, Psychotic Disorders, Psychotic Disorders/drug therapy, Quinolones, Quinolones/adverse effects, Quinolones/therapeutic use, Randomized Controlled Trials as Topic, Risperidone, Risperidone/adverse effects, Risperidone/therapeutic use, Schizophrenia, Schizophrenia/drug therapy
Plain language summary
Atypical antipsychotic medications for adolescents with psychosis
Schizophrenia and other serious mental illnesses often begin in adolescence, and treatment of adolescents with psychosis usually involves use of antipsychotic drugs. Newer drugs (atypical antipsychotics) are more popular than older ones (typical antipsychotics). However, this determination is based on the generalisation of adult treatment to a younger age group, with evidence from studies on adults generally guiding the treatment of adolescents. Adolescents may respond differently to medication compared with adults. This review looks at evidence derived from trials in which the participants are adolescents receiving atypical or typical antipsychotics or a placebo (dummy treatment) and/or high or low doses of medication. A total of 13 trials consisting of 1112 people between 13 and 18 years of age are included. Most studies were short‐term trials (completed within 12 weeks). In the main, no convincing evidence shows that newer drugs (atypical antipsychotics) are better than older ones (typical antipsychotics) in terms of their ability to treat the symptoms of psychosis. However, newer drugs may be more acceptable for young people to take because they produce fewer side effects in the short term. Furthermore, very little evidence is available to support the superiority of one atypical antipsychotic over another atypical antipsychotic. The nature of side effects also differs markedly between medications. For example, treatment with olanzapine, risperidone and clozapine is associated with weight gain, but aripiprazole is not associated with weight gain. Some evidence indicates that adolescents respond better to standard‐dose as opposed to lower‐dose risperidone. However, for aripiprazole and ziprasidone, a lower dose and a standard dose may be equally effective. Longer, clearer and more detailed research trials that use systematic ways of reporting and comparing the side effects of different antipsychotic drugs are much needed. So too is a research focus on other important outcomes such as hospital admission, service use, costs, behaviour change and possible improvements in people’s thinking. Until such research is completed, very little evidence suggests that newer drugs (atypical antipsychotics) are better than older drugs (typical antipsychotics) for the treatment of adolescents with schizophrenia.
This plain language summary has been written by Benjamin Gray, Service User and Service User Expert, Rethink Mental Illness (Email: ben.gray@rethink.org).
Summary of findings
Summary of findings for the main comparison. Atypical antipsychotics compared with placebo (only short term).
| Atypical antipsychotics compared with placebo (only short term) | ||||||
| Patient or population: individuals with psychosis Settings: Intervention: atypical antipsychotics Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Atypical antipsychotics | |||||
| Weight gain | Study population | RR 3.56 (1.14 to 11.11) | 107 (1 study) | ⊕⊕⊕⊝ moderatea | ||
| 86 per 1000 | 305 per 1000 (98 to 952) | |||||
| Moderate | ||||||
| 86 per 1000 | 306 per 1000 (98 to 955) | |||||
| Weight gain ≥7% of baseline | Study population | RR 3.12 (1.34 to 7.27) | 106 (1 study) | ⊕⊕⊕⊕ high | ||
| 147 per 1000 | 459 per 1000 (197 to 1000) | |||||
| Moderate | ||||||
| 147 per 1000 | 459 per 1000 (197 to 1000) | |||||
| High prolactin at any time during treatment | Study population | RR 4.7 (2.25 to 9.82) | 107 (1 studies) | ⊕⊕⊕⊕ high | ||
| 171 per 1000 | 806 per 1000 (386 to 1000) | |||||
| Moderate | ||||||
| 171 per 1000 | 804 per 1000 (385 to 1000) | |||||
| Change in corrected QT | Mean change in corrected QT in the intervention groups was 6.3 lower (12.51 to 0.09 lower) | 107 (1 study) | ⊕⊕⊕⊕ high | |||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aWide confidence interval.
Summary of findings 2. Atypical compared with typical antipsychotics (only short term).
| Atypical compared with typical antipsychotics (only short term) | ||||||
| Patient or population: individuals with psychosis Settings: Intervention: atypical antipsychotics Comparison: typical antipsychotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Typical antipsychotics | Atypical antipsychotics | |||||
| Worse or no improvement | Study population | RR 3.3 (0.41 to 26.81) | 21 (1 study) | ⊕⊕⊝⊝ lowa,b | ||
| 91 per 1000 | 300 per 1000 (37 to 1000) | |||||
| Moderate | ||||||
| 91 per 1000 | 300 per 1000 (37 to 1000) | |||||
| Anticholinergic adverse effects | Study population | RR 0.2 (0.05 to 0.8) | 40 (1 study) | ⊕⊕⊕⊝ moderatec | ||
| 500 per 1000 | 100 per 1000 (25 to 400) | |||||
| Moderate | ||||||
| 500 per 1000 | 100 per 1000 (25 to 400) | |||||
| Drop in the absolute neutrophil count below 1500 per mm3 | Study population | RR 12 (0.75 to 192.86) | 21 (1 study) | ⊕⊝⊝⊝ very lowb,d | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Leaving the study early because of adverse effects | Study population | RR 3.3 (0.41 to 26.81) | 21 (1 study) | ⊕⊕⊕⊝ moderatea | ||
| 91 per 1000 | 300 per 1000 (37 to 1000) | |||||
| Moderate | ||||||
| 91 per 1000 | 300 per 1000 (37 to 1000) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aWide confidence interval. bThe high incidence of neutropenia in the clozapine group has been reported but not adequately discussed. cAlthough one of the studies said it was randomised and double blind, the authors did not provide a description. dVery wide confidence interval.
Summary of findings 3. Atypical vs atypical antipsychotics (only short term).
| Atypical vs atypical antipsychotics (only short term) | ||||||
| Patient or population: individuals with psychosis Settings: Intervention: Atypical antipsychotics Comparison: Atypical antipsychotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Atypical antipsychotics | Atypical antipsychotics | |||||
| No improvement (risperidone vs olanzapine) | Study population | RR 0.50 (0.24 to 1.07) | 111 (2 studies) | ⊕⊕⊕⊝ moderatea | ||
| 451 per 1000 | 468 per 1000 (291 to 654) | |||||
| Moderate | ||||||
| 413 per 1000 | 429 per 1000 (260 to 618) | |||||
| No improvement (clozapine vs olanzapine) | Study population | RR 0.5 (0.24 to 1.03) | 39 (1 study) | ⊕⊕⊕⊕ high | ||
| 667 per 1000 | 333 per 1000 (160 to 687) | |||||
| Moderate | ||||||
| 667 per 1000 | 333 per 1000 (160 to 687) | |||||
| Use of other antipsychotics | Study population | RR 0.5 (0.05 to 4.9) | 28 (1 study) | ⊕⊕⊕⊝ moderateb | ||
| 143 per 1000 | 71 per 1000 (7 to 700) | |||||
| Moderate | ||||||
| 143 per 1000 | 72 per 1000 (7 to 701) | |||||
| Drug‐induced diabetes (clozapine vs olanzapine) | Study population | RR 3 (0.13 to 67.91) | 28 (1 study) | ⊕⊕⊕⊝ moderateb | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Elevated prolactin (risperidone vs quetiapine) | Study population | RR 10 (1.53 to 65.41) | 22 (1 study) | ⊕⊕⊕⊝ moderatec | ||
| 91 per 1000 | 909 per 1000 (139 to 1000) | |||||
| Moderate | ||||||
| 91 per 1000 | 910 per 1000 (139 to 1000) | |||||
| Weight gain in kg (risperidone vs olanzapine) | The mean weight gain in kg (risperidone vs olanzapine) in the intervention groups was 2.5 lower (4.21 to 0.79 lower) | 76 (1 study) | ⊕⊕⊕⊕ high | |||
| Leaving the study early because of weight gain (risperidone vs olanzapine) | Study population | RR 0.37 (0.02 to 8.01) | 19 (1 study) | ⊕⊕⊝⊝ lowb,d | ||
| 100 per 1000 | 37 per 1000 (2 to 801) | |||||
| Moderate | ||||||
| 100 per 1000 | 37 per 1000 (2 to 801) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aRisk ratio reduction (RRR) or risk ratio increase (RRI) greater than 25%. bWide confidence interval. cOpen‐label study with blind midpoint and end point assessments. dThis was an open‐label study.
Summary of findings 4. Atypical (standard‐dose) vs atypical (low‐dose) antipsychotics (only short term).
| Atypical (standard‐dose) vs atypical (low‐dose) antipsychotics (only short term) | ||||||
| Patient or population: individuals with psychosis Settings: Intervention: atypical antipsychotics (standard dose) Comparison: atypical antipsychotics (low dose) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Atypical antipsychotics (low dose) | Atypical antipsychotics (standard dose) | |||||
| No response | Study population | RR 0.54 (0.38 to 0.75) | 255 (1 study) | ⊕⊕⊕⊕ high | ||
| 496 per 1000 | 268 per 1000 (189 to 372) | |||||
| Moderate | ||||||
| 496 per 1000 | 268 per 1000 (188 to 372) | |||||
| Symptomatic hyperprolactinaemia | See comment | See comment | Not estimable | 257 (1 study) | ⊕⊕⊕⊕ high | |
| Use of antiparkinsonian medications | Study population | RR 4.86 (1.91 to 12.38) | 257 (1 study) | ⊕⊕⊕⊕ high | ||
| 38 per 1000 | 184 per 1000 (72 to 469) | |||||
| Moderate | ||||||
| 38 per 1000 | 185 per 1000 (73 to 470) | |||||
| Weight gain (standard‐dose vs low‐dose risperidone) | Study population | RR 3.32 (1.47 to 7.49) | 257 (1 study) | ⊕⊕⊕⊕ high | ||
| 53 per 1000 | 176 per 1000 (78 to 397) | |||||
| Moderate | ||||||
| 53 per 1000 | 176 per 1000 (78 to 397) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence. High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Background
Description of the condition
Schizophrenia is a neurodevelopmental disorder (Owen 2011) that often starts during adolescence (Ballageer 2005). Late adolescence is a critical period in brain development, possibly making individuals of this age particularly vulnerable to onset of schizophrenia (Gogtay 2011; Rapoport 2011). The disorder is characterised by positive psychotic symptoms, negative symptoms and cognitive symptoms, along with other features that impact the socio‐occupational functioning of the young person (APA 1994). Although a diagnosis of psychosis is less stable in adolescentsthan in adults (Werry 1991), the subgroup of people with onset of schizophrenia between the ages of 13 to 18 falls into the category of 'Adolescent schizophrenia' (Hollis 2000a; Werry 1992). It must be noted that 'childhood‐onset' (APA 1994) and 'very early onset' schizophrenia, that is, schizophrenia that occurs in those younger than 13 years, are extremely rare and will not be considered directly here. Childhood‐onset schizophrenia has been reviewed separately by some of the authors of the current review (Kennedy 2007; Kennedy 2007a).
The criteria for diagnosing adolescent‐onset schizophrenia are similar to those used for adult‐onset schizophrenia, as per the International Classification of Diseases, Tenth Revision (ICD‐10) (WHO 1992) and The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM‐IV) (APA 1994). Young people with adolescent‐onset schizophrenia have more affective symptoms and increased behaviour problems (Werry 1991) as compared with those developing adult‐onset schizophrenia. Also, adolescent‐onset schizophrenia has a more severe and unremitting course with a poorer outcome (Hollis 2000b). Reticence amongst clinicians to make a formal diagnosis of schizophrenia is understandable in many cases, even if the likelihood of a full or prolonged remission after a first episode of psychosis is reduced (Hollis 2000b). Additional factors, such as a more distinct negative picture of symptoms, an insidious onset and poor premorbid functioning, may conspire to confuse or delay a diagnosis (and potentially the implementation of an appropriate treatment plan). In a clinical setting, psychosis in adolescents is often associated with use of cannabis and other illicit drugs (Schubart 2010; Zammit 2010) and may initially be labelled as episodic drug‐induced psychosis until the diagnosis is well established.
Description of the intervention
Evidence from studies on adults generally guides the treatment of schizophrenia and other psychotic disorders in adolescents. Amongst pharmacological treatment options, atypical antipsychotics are more popular than typical antipsychotic medications for treatment of adolescents with psychosis (Imran 2011). The current review will evaluate the evidence base for using atypical antipsychotic medications in adolescents with psychosis. The evidence base for typical antipsychotic medications on their own is being evaluated separately in another review (Datta 2011).
How the intervention might work
To date, all medications with proven antipsychotic activity block D2 receptors to some degree (Kumra 2008a). Atypical antipsychotics help patients clinically by occupying, albeit transiently, D2 receptors and then dissociating rapidly to allow normal dopamine neurotransmission. This has a slight impact on serum prolactin levels, helps in sparing cognition and obviates extrapyramidal adverse effects to some degree. One theory for this atypical nature is that the newer medications block 5‐HT2A receptors and at the same time block dopamine receptors and that, somehow, this serotonin‐dopamine balance confers atypicality (Kapur 2001; Kumra 2008a; Seeman 2002). Aripiprazole, which is a partial dopamine agonist, is also classified as an atypical antipsychotic medication. It must be noted that existing evidence for treatment of psychotic disorders in adolescents suggests fairly good and comparable efficacy of both typical and atypical antipsychotics (Clark 1998; Crossley 2010). Although improved short‐term tolerability of atypical antipsychotic medications is leading to increased use amongst adolescent patients (Imran 2011), it must be noted that the new atypical antipsychotics tend to have specific adverse effects of their own (Buchanan 2010).
Why it is important to do this review
Generalisation of adult‐based evidence to the younger age group is in part due to the importance wielded by antipsychotic drugs and widespread confidence in these products. However, response rates, tolerability and other outcome measures might differ significantly owing to significant differences between adolescents and adults with psychosis (e.g. prominence of negative symptoms, increased frequency of extrapyramidal adverse effects). It must be noted that clarification is needed as to whether there is an evidence‐based rationale for treating adolescents in the same way as adults because specific trials for this subgroup are few. The current trend of increased popularity of atypical antipsychotic medications over typical antipsychotic medications needs to be reviewed.
Objectives
To investigate the effects of atypical antipsychotic medications in adolescents with psychosis. We reviewed in separate analyses various comparisons of atypical antipsychotic medications with placebo or a typical antipsychotic medication or another atypical antipsychotic medication or the same atypical antipsychotic medication but at a lower dose.
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials. We included in a sensitivity analysis trials described as 'double blind' but for which randomisation was implied (see Sensitivity analysis). If their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion did result in statistically significant differences, we added the data from these lower‐quality studies to the results of the better trials but presented such data within a subcategory. We excluded quasi‐randomised studies, such as those allocating by alternate days of the week. In studies where the group of adolescents treated with atypical antipsychotic medications were given additional treatments, we included data only if the adjunct treatment was evenly distributed between groups and if only the participants receiving atypical antipsychotic medications were randomly assigned.
Types of participants
We considered for this review adolescents, aged 13 to 17 years, with schizophrenia or related disorders, including schizophreniform disorder, schizoaffective disorder and delusional disorder, by any means of diagnosis.
We were interested in making sure that information is as relevant as possible to the current care of people with psychosis/schizophrenia. If data were available, we intended to highlight the current clinical state (acute, early post‐acute, partial remission, remission) and stage (first episode, early illness, persistent) and whether identified studies focused primarily on people with particular problems (e.g. negative symptoms, treatment‐resistant illnesses).
Types of interventions
1. Atypical antipsychotic medications
Atypical antipsychotic medications include risperidone, olanzapine, quetiapine, ziprasidone, aripiprazole, amisulpiride, paliperidone, lurasidone and clozapine. We anticipated that most randomised trials on adolescents with psychosis would use antipsychotic medications within the British National Formulary therapeutic dose range, as found in a survey of clinical practice in the UK (Imran 2011). The mean effective chlorpromazine equivalent dose used in trials of antipsychotic medications is likely to be variable depending on the medication studied (Andreasen 2010). A review published a few years ago pointed out that a dose equivalent to 100 mg/d of chlorpromazine was equivalent to 2 mg/d of risperidone, 5 mg/d of olanzapine, 75 mg/d of quetiapine, 60 mg/d of ziprasidone and 7.5 mg/d of aripiprazole (Woods 2003). This has been refined further by Andreasen 2010, who reported that 100 mg/d of chlorpromazine was equivalent to 1.32 mg/d of risperidone, 4.75 mg/d of olanzapine, 142 mg/d of quetiapine, 50.5 mg/d of ziprasidone, 6.42 mg/d of aripiprazole and 108 mg/d of clozapine.
2. Control treatment
Control treatment included placebo or a typical antipsychotic medication or in some cases other atypical antipsychotic medications or the same atypical antipsychotic medication given at a lower dose.The different comparisons described above were analysed separately, as were studies using low‐dose antipsychotic medications. For the purpose of this review, we defined low‐dose antipsychotic medication as less than 150 mg of chlorpromazine equivalent per day, as this dose was lower than that given in the treatment arm of all trials measuring the effectiveness of antipsychotic medications (Andreasen 2010).
Types of outcome measures
We divided all outcomes into short term (less than six months), medium term (seven to 12 months) and long term (over one year).
Primary outcomes
1. Global state
1.1 Clinically significant response on global state, as defined by each of the studies
2. Clinical response
2.1 Clinically significant response on psychotic symptoms, as defined by each of the studies 2.2 Relapse
3. Global functioning
3.1 Clinically significant response on global functioning, as defined by each of the studies 3.2 Average score/change on global functioning, as defined by each of the studies
4. Adverse effects
Any reported adverse effects, as described by each of the studies
5. Service utilisation outcomes
Hospital admission, as reported by individual studies
Secondary outcomes
1. Global state
1.1 Average score/change on global state
2. Clinical response
2.1 Average score/change on psychotic symptoms 2.2 Clinical response on cognitive symptoms, as defined by each of the studies
3. Social functioning
3.1 Average score/change on social skills 3.2 Educational status/occupational status 3.3 Compliance with (a) drug treatment and (b) other non‐drug treatments
4. Adverse effects/events
4.1 Death, suicide or natural causes 4.2 Leaving the study early 4.3 Incidence of clinically significant depression/anxiety 4.4 Dependency 4.5 General adverse effects 4.5 Specific adverse effects 4.6 Average score on adverse effects
5. Service utilisation
5.1 Days in hospital
6. Economic outcomes
7. Quality of life/satisfaction with care for recipients of care or carers
7.1 Significant change in quality of life/satisfaction, as defined by each of the studies 7.2 General impression of carer/other 7.3 Average score/change on quality of life/satisfaction
Search methods for identification of studies
Electronic searches
Cochrane Schizophrenia Group Trials Register (October 2011)
We searched the register using the phrase:
[(*youth* OR *young* OR *pediatri* OR *paediatric* OR *teenag* OR *child* OR *adolesc* in title of REFERENCE) OR (*adoles* OR *child* OR *young adult* in participants of STUDY)]
This register is compiled by systematic searches of major databases, handsearches and searches of conference proceedings (see group module).
Searching other resources
1. Reference searching
We inspected references of all identified studies to look for further relevant studies.
2. Personal contact
We contacted the first author of each included study to ask for information regarding unpublished trials.
Data collection and analysis
Selection of studies
AK and SSD independently inspected citations from the searches and identified relevant abstracts. SDW independently re‐inspected a random 20% sample to ensure reliability. When disputes arose, we acquired the full report for more detailed scrutiny. We obtained full reports of abstracts meeting the review criteria, and SSD inspected them. Again, SDW re‐inspected a random 20% of reports to ensure reliable selection. When it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Data extraction and management
1. Extraction
Review author AK extracted data from all included studies. To ensure reliability, SSD independently extracted data from a random sample of these studies, representing 10% of the total. Again, we discussed any disagreement, documented decisions and, if necessary, contacted authors of studies for clarification. With remaining problems, SDW helped to clarify issues, and we documented these final decisions. We extracted data presented only in graphs and figures whenever possible, but data were included only if two study authors independently reached the same result. We attempted to contact study authors through an open‐ended request to ask for missing information or clarification whenever necessary. If studies were multi‐centre, when possible, we extracted separately data relevant to each component centre.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a. the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument had not been written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should be a self‐report or a report completed by an independent rater or relative (not the therapist). We realise that often this is not reported clearly. In 'Description of studies', we noted whether or not this was the case.
2.3 End point versus change data
Advantages are associated with both end point and change data. Change data can remove from the analysis a component of between‐person variability. On the other hand, calculation of change requires two assessments (baseline and end point), which can be difficult to perform in unstable and difficult to measure conditions such as schizophrenia. We decided to use primarily end point data and to use change data only in cases where the former were not available. End point and change data could be combined in the analysis, as we used mean differences (MDs) rather than standardised mean differences throughout (Higgins 2011, Chapter 9.4.5.2).
2.4 Skewed data
Continuous data on clinical and social outcomes often are not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion: (a) Standard deviations (SDs) and means are reported in the paper or are obtainable from the authors; (b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise, the mean is unlikely to be an appropriate measure of the centre of the distribution; Altman 1996); and (c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), which can include values from 30 to 210), the calculation described above was modified to take the scale starting point into account. In these cases, skew is present if 2 SD > (S – S min), where S is the mean score and S min is the minimum score. End point scores on scales often have a finite start point and end point, and these rules can be applied. When continuous data are presented on a scale that includes the possibility of negative values (such as change data), it is difficult to tell whether or not data are skewed. We entered skewed data from studies of fewer than 200 participants as other data within the data analyses section, rather than including them in a statistical analysis. The problem posed by skewed data is reduced when means are examined if the sample size is large and can be entered into syntheses.
2.5 Common measure
To facilitate comparison between trials, we intended to convert to a common metric (e.g. mean days per month) variables that could be reported in different metrics, such as days in hospital (mean days per year, per week or per month).
2.6 Conversion of continuous to binary
When possible, we attempted to convert outcome measures to dichotomous data by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' and 'not clinically improved'. It is generally assumed that a 50% reduction in a scale‐derived score such as the BPRS (Overall 1962) or the PANSS (Kay 1986) could be considered a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off as presented by the original study authors.
2.7 Direction of graphs
When possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for the atypical antipsychotic medication. When keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'not improved'), we reported data in such a way that the area to the left of the line indicates an unfavourable outcome. We noted this in the relevant graphs.
2.8 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and the GRADE profiler (GRADEPRO) to import data from Review Manager 5 (Review Manager) when creating 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence derived from each included study in the comparison, the magnitude of effect of the interventions examined and the sum of available data on all outcomes that we had rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
1. Global state
1.1 Clinically significant response on global state, as defined by each of the studies
2. Clinical response
2.1 Clinically significant response on psychotic symptoms, as defined by each of the studies
3. Global functioning
3.1 Clinically significant response on global functioning, as defined by each of the studies
4. Adverse effects
4.1 Extrapyramidal symptoms 4.2 Weight gain
5. Leaving the study early
Assessment of risk of bias in included studies
AK worked independently to assess risk of bias by using the criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of an article and included sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
If the raters disagreed, we assigned the final rating by consensus, with the involvement of another member of the review group. In cases where inadequate details of randomisation and other characteristics of trials are provided, we contacted study authors to request further information. We reported non‐concurrence in quality assessment, but if disputes arose as to which category a trial is to be allocated, again, we undertook resolution by discussion.
We noted the level of risk of bias in the text of the review and in the Table 1
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that the RR is more intuitive (Boissel 1999) than the odds ratio and that odds ratios tend to be interpreted as RRs by clinicians (Deeks 2000). For binary data presented in the 'Summary of findings' table, where possible, we calculated illustrative comparative risks as the number needed to treat for an additional harmful outcome (NNTH) statistic with its confidence intervals, which is intuitively attractive to clinicians but is problematic in terms of its accurate calculation in both meta‐analyses and interpretation (Hutton 2009) (see Differences between protocol and review).
2. Continuous data
For continuous outcomes, we estimated mean differences (MDs) between groups. We preferred to refrain from calculating effect size measures (standardised mean difference (SMD)). However, if scales of considerable similarity had been used, we would have presumed a small difference in measurement, and we would have calculated effect size and transformed the effect back to the units of one or more of the specific instruments used.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or by practice), but analysis with pooling of clustered data poses problems. First, authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
When clustering was not accounted for in primary studies, we had planned to present the data in a table to indicate the presence of a probable unit of analysis error. No such data were found in the search. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intraclass correlation coefficients for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). In cases where clustering had been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study, with adjustment for the clustering effect.
We sought statistical advice and have been advised that the binary data presented in a report should be divided by a 'design effect'. This is calculated by using the mean number of participants per cluster (m) and the intraclass correlation coefficient (ICC) [Design effect = 1 + (m – 1) * ICC] (Donner 2002). If the ICC is not reported, it can be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed by taking into account ICCs and relevant data as documented in the report, synthesis with other studies may be possible with the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carryover effect, which occurs if an effect (e.g. pharmacological, physiological, psychological) of a treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase, participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we used only data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
In cases where a study involved more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary, we simply added these and combined them within the two‐by‐two table. If data were continuous, we combined them in keeping with the formula provided in Section 7.7.3.8 ('Combining Groups') of the Cochrane Handbook for Systematic Reviews of Interventions. When the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We choose that, for any particular outcome, should more than 50% of data be unaccounted for, we will not reproduce these data or use them within analyses. If, however, more than 50% of data in one arm of a study were lost, but the total loss was less than 50%, we addressed this within the 'Summary of findings' tables by down‐rating quality. Finally, we also downgraded quality within the 'Summary of findings' tables should loss be 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). Those leaving the study early were assumed to have the same rates of negative outcome as those who completed the study, with the exception of the outcomes of death and adverse effects. For these outcomes, the rates of those who stayed in the study—in that particular arm of the trial—were also used as the rates of those who did not stay until completion.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 50% and completer‐only data were reported, we reproduced these.
3.2 Standard deviations
If standard deviations were not reported, we first tried to obtain the missing values from the authors. If the data were not available, in cases where measures of variance for continuous data were missing, but an exact standard error and confidence intervals were available for group means, and either a P value or a T value was available for differences in means, we were able to calculate standard deviations according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): When only the standard error (SE) is reported, standard deviations (SDs) are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Handbook (Higgins 2011) present detailed formulae for estimating SDs from P values, T or F values, confidence intervals, ranges or other statistics. If these formulae do not apply, we would calculate the SDs according to a validated imputation method that is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcomes, thus losing information. We nevertheless examined the validity of the imputations in a sensitivity analysis that excluded imputed values.
3.3 Last observation carried forward
We anticipated that in some studies, the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation used to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, in cases where LOCF data have been used in the trial, if less than 50% of the data were assumed, we reproduced these data and indicated that they are the products of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations that we had not predicted would arise. When such situations or participant groups arose, we fully discussed these.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods that we had not predicted would arise. When such methodological outliers arose, we fully discussed these.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
Heterogeneity between studies was investigated by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on both magnitude and direction of effects and strength of evidence for heterogeneity (e.g. P value from Chi2 test, confidence interval for I2). We interpreted an I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic as evidence of substantial levels of heterogeneity (see Section 9.5.2; Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (see Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in Section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We tried to locate protocols of included randomised trials. If the protocol was available, we compared outcomes in the protocol and in the published report. If the protocol was not available, we compared outcomes listed in the methods section of the trial report with actually reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes when 10 or fewer studies were analysed, or when all studies were of similar size. In other cases in which funnel plots were possible, we had access to statistical advice for their interpretation. However, as the authors of this review have been Cochrane reviewers for many years, we were able to interpret the data appropriately.
Data synthesis
We understand that no closed argument has been put forth for preference in the use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us, and the random‐effects model takes into account differences between studies even if no statistically significant heterogeneity is noted. However, a disadvantage of the random‐effects model is that it puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can inflate or deflate the effect size. We chose random‐effects models for all analyses. However, the reader can choose to inspect the data using the fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Dose and antipsychotic use
We anticipated subgroup analyses comparing higher doses of atypical antipsychotic medication with lower doses of the same antipsychotic medication, where available. We also analysed separately the data from studies on adolescents with treatment‐resistant schizophrenia, when available.
We anticipated subgroup analyses comparing one atypical antipsychotic medication with another antipsychotic medication, when available.
1.2 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of atypical antipsychotic medications for adolescents with schizophrenia in general. However, we could not obtain from the individual trials data on subgroups of young people in the same clinical state or stage and with similar problems.
2. Investigation of heterogeneity
If inconsistency was high, we reported this. First, we investigated whether data had been entered correctly. Second, if data were correct, we visually inspected the graph and successively removed studies outside of the company of the rest to see whether homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present the data. If this did not occur, we did not pool the data but discussed the issues. We knew of no supporting research for this 10% cut‐off, but we are investigating the use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity was obvious, we simply stated hypotheses regarding this for future reviews or versions of this review. We did not anticipate undertaking analyses related to these hypotheses.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way that implied randomisation. For primary outcomes, we intended to include these studies, and if no substantive difference was evident when the implied randomised studies were added to those with better descriptions of randomisation, we employed all data from these studies. We did not undertake sensitivity analysis, as none of the included studies had implied randomisation.
2. Assumptions for lost binary data
When assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of primary outcomes when we compared our assumption with completer data only. If a substantial difference was noted, we reported the results and discussed them but continued to employ our assumption.
When assumptions had to be made regarding missing data on SDs (see Dealing with missing data), we aimed to compare the findings of primary outcomes when we compared our assumption with completer data only. We intended to undertake a sensitivity analysis to test how prone results are to change when 'completer' data only are compared with imputed data using the above assumption. If a substantial difference was seen, we reported results and discussed them but continued to employ our assumption. We did not include any study for which we made assumptions about missing data on SDs.
3. Risk of bias
We analysed the effects of excluding trials judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available): allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of effect estimates, then we included data from these trials in the analysis.
4. Imputed values
We intended to undertake a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster randomised trials. We included no cluster randomised trials among the included studies.
If we noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with data from the other trials contributing to the outcome, but we presented them separately.
5. Fixed effect and random effects
We synthesised all data using a random‐effects model; however, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether the greater weights assigned to larger trials with greater event rates altered the significance of the results compared with the more evenly distributed weights in the random‐effects model. If susbtantial differences were noted, we presented these.
Results
Description of studies
Please see Characteristics of included studies; Characteristics of excluded studies; and Characteristics of ongoing studies.
Results of the search
The 2011 search identified 2771 references. Of these, 13 met our inclusion criteria and 43 had to be excluded (Figure 1).
1.
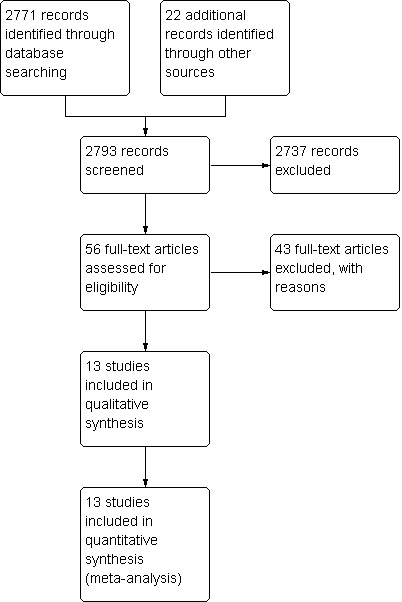
Review flow diagram.
Included studies
1. General
Thirteen studies (40 reports) met the inclusion criteria (Aranda 2007; DelBello 2008; Findling 2008; Haas 2009; Huo 2007; Jensen 2008; Kryzhanovskaya 2009; Kumra 1996; Kumra 2008; Sikich 2004; Sikich 2008; Swadi 2010; Xiong 2004). All were randomised and most were double blind. Swadi 2010, however, was a single‐blind study, and three trials were described as open label (Aranda 2007; DelBello 2008; Jensen 2008). For Huo 2007 and Xiong 2004, blinding was unclear. The quality of studies varied, but findings were comparable (Figure 2).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
2. Length of trials
Most included studies reported data on short‐term follow‐up (up to 12 weeks). Aranda 2007, however, reported data on medium‐term follow‐up (13 to 26 weeks). No trial reported on long‐term follow‐up (over 26 weeks).
3. Participants
A total of 1112 adolescent participants were involved in these trials (Figure 1). Findling 2008 had the highest number of participants—302. Haas 2009 had 257 participants, and Kryzhanovskaya 2009 and Sikich 2008 had 107 and 116 participants, respectively. The remaining trials were very small, with between 17 and 60 participants. Most included studies used Diagnostic and Statistical Manual (DSM) diagnostic criteria. Kumra 2008 had participants with treatment‐resistant schizophrenia. More boys than girls were enrolled in these studies (627 boys, 434 girls; Aranda 2007 did not specify the gender of participants). The age range across the studies was between 6 and 22 years of age. Two studies (Kumra 1996; Kumra 2008) were specifically focused on adolescents with treatment‐resistant schizophrenia.
4 Setting
Six studies were described as taking place in hospital or inpatient settings (Aranda 2007; Haas 2009; Kumra 1996; Kumra 2008;Swadi 2010; Xiong 2004). Six took place in both inpatient and outpatient settings (Findling 2008; Huo 2007; Jensen 2008; Kryzhanovskaya 2009; Sikich 2004; Sikich 2008). The setting was unclear with DelBello 2008.
5. Interventions
Antipsychotic drugs were administered in a wide range of doses. Daily dose ranges of typical antipsychotic drugs used as interventions were as follows: perphenazine 10 to 24 mg, haloperidol 5 to 27 mg, molindone mean 59.9 mg (SD 33.5), chlorpromazine 50 to 400 mg. The atypical antipsychotic drugs used were quetiapine 100 to 800 mg, olanzapine 2.5 to 30 mg, aripiprazole 10 to 30 mg, risperidone 0.15 to 6 mg, clozapine 25 to 700 mg, and ziprasidone 80 to 160 mg. Findling 2008 and Kryzhanovskaya 2009 used placebo as one of the comparators. Haas 2009 used different doses of risperidone in association with psychotherapy and psychoeducation.
6. Outcomes
Studies reported on global outcomes in several ways. Six trials used Clinical Global Impression (CGI; Guy 1976) scores to measure global clinical improvement in the short term (Findling 2008; Haas 2009; Kumra 2008; Kryzhanovskaya 2009; Sikich 2004; Sikich 2008). Findling 2008; Kumra 1996 and Kumra 2008 used the Children's Global Assessment Scale to assess global functioning (CGAS; Schaffer 1983).
Trials used several scales to measure mental state. Seven studies (DelBello 2008; Huo 2007; Kryzhanovskaya 2009; Kumra 1996; Sikich 2004; Sikich 2008; Xiong 2004) reported outcomes of mental state using the Brief Psychiatric Rating Scale (BPRS; Overall 1962). The Positive and Negative Symptom Scale (PANSS; Kay 1987) was used by Haas 2009 and Jensen 2008 to report outcomes. Kumra 1996 and Kumra 2008 also used the Scale for the Assessment of Negative Symptoms (SANS; Andreasen 1983) and the Scale for the Assessment of Positive Symptoms (SAPS; Andreasen 1984) to report mental state. Most studies reported usable data on adverse effects. Many studies used the Treatment Emergent Symptoms Scale (TESS; Guy 1976) and the Abnormal Involuntary Movements Scale (AIMS; Guy 1976) to report adverse effects. Seven studies (Findling 2008; Haas 2009; Jensen 2008; Kryzhanovskaya 2009; Kumra 2008; Sikich 2004; Sikich 2008) reported usable data on weight gain. Data regarding quality of life were reported by only one study (Findling 2008)
Aranda 2007; Findling 2008; Haas 2009; Jensen 2008; Kryzhanovskaya 2009; Kumra 1996; and Kumra 2008 reported the reasons for participants leaving the study early.
6.1 Outcome scales: details of the rating scales used to provide usable data
6.1.1 Global state
6.1.1.1 Clinical Global Impression Scale—CGI Scale (Guy 1976) This scale is used to assess illness severity and clinical improvement. The CGI is a seven‐point scoring system, with low scores denoting decreased severity and/or overall improvement. Sometimes studies report CGI‐I (CGI‐Improvement) and CGI‐S (CGI‐Severity) scores separately.
6.1.1.2 Childrens Global Assessment Scale—CGAS (Schaffer 1983) The CGAS is used to provide a global measure of functioning in children and adolescents. On a scale of 0 to 100, the assessment provides a single global rating. Higher scores indicate better functioning.
6.1.2 Mental state
6.1.2.1 Brief Psychiatric Rating Scale—BPRS (Overall 1962) The BPRS is a clinician‐ or researcher‐administered scale that is used to assess the severity of an abnormal mental state. The original scale consists of 16 items, but a revised 18‐item scale is commonly used. A 21‐item scale is also available specifically for use in children. Each item is scored on a seven‐point scale ranging from 'not present' to 'extremely severe', scoring from 0 to 6 or 1 to 7. Higher scores indicate more severe symptoms.
6.1.2.2 Positive and Negative Syndrome Scale—PANSS (Kay 1987) This schizophrenia scale includes 30 items. Each item is scored on a seven‐point scoring system ranging from absent to extreme. The PANSS has three subscales that are used to measure positive symptoms (PANSS‐P), negative symptoms (PANSS‐N) and general psychopathology. A higher score indicates greater severity.
6.1.2.3 Scale for the Assessment of Negative Symptoms—SANS (Andreasen 1983) The SANS scale assesses five symptom complexes to obtain clinical ratings of negative symptoms in patients with schizophrenia. They include affective blunting, alogia, avolition‐apathy, anhedonia‐asociality and disturbance of attention, with higher score meaning more severe symptoms.
6.1.2.4 Scale for the Assessment of Positive Symptoms—SAPS (Andreasen 1984) This scale has been designed to assess positive symptoms of schizophrenia. It serves as a complementary tool to the SANS. The positive symptoms it assesses are hallucinations, delusions, bizarre behaviour and positive formal thought disorder. In addition to using a clinical interview, the investigator draws on other sources of information such as direct observation and reports from the patient's family and nurses and from the patient.
6.1.2.5 Bunney‐Hamburg Psychosis Rating Scale—B‐HPRS (Bunney 1963) The B‐HPRS is a 15‐point scale that provides a clinical rating of severity of psychosis. The rating varies from no symptoms to incapacitating symptoms. Scores range from 1—no symptoms of psychosis, to 15—incapacitating symptoms of psychosis.
6.1.2.6 Overt Aggression Scale—OAS (Yudofsky 1986)
The OAS is used in children and adults to quantify aggression. It covers verbal aggression, aggression against self, aggression against others and aggression against objects.
6.1.2.7 Young Mania Rating Scale—YMRS (Young 1978)
The YMRS includes 11 items and is widely used to assess mental state in adolescents with bipolar disorder. It has good sensitivity and specificity. Clinical studies have demonstrated effectiveness of the parent version of the scale.
6.1.2.8 Hamilton Depression Rating Scale—HAM‐D (Hamilton 1960)
The HAM‐D is one of the most popular scales used in psychiatry to quantify severity of depression. It consists of 17 items, and most items score 0 to 4, although some of the items score 0 to 2.
6.1.2.9 Adult and Child Functional Assessment Scale (Hodges 1990)
The Adult and Child Functional Assessment Scale is used in one of the studies included in this review for assessment of mental state. A higher score on this scale indicates more severe problems.
6.1.3 Adverse effects scales
6.1.3.1 Treatment Emergent Symptoms Scale—TESS (Guy 1976) This checklist provides assessment of a variety of characteristics of different adverse events, including severity, relationship to the drug and temporal relation (timing after a drug dose, duration and pattern during the day). It also includes assessment of contributing factors and course and action taken to counteract the adverse effect. Symptoms can be listed a priori or can be recorded by the investigator as observed.
6.1.3.2 Simpson Angus Scale—SAS (Simpson 1970) The SAS is a 10‐item rating scale that has been used widely for assessment of neuroleptic medication–induced movement disorders in research settings. It consists of one item measuring gait (hypokinesia), six items measuring rigidity and three items measuring glabellar tap, tremor and salivation, respectively.
6.1.3.3 Abnormal Involuntary Movements Scale—AIMS (Guy 1976) This 12‐item scale is used to monitor antipsychotic‐induced movement disorders. It includes different items for oral and facial movements, movement of trunk and extremities and global judgements. Each item in the scale is scored from 0 to 4, with higher score indicating greater severity.
6.1.3.4 Barnes Akathisia Rating Scale—BARS (Barnes 1989) This scale comprises items rating the observable, restless movements that characterise akathisia, a subjective awareness of restlessness and any distress associated with the condition. Items are rated from 0—normal to 3—severe. In addition, an item for rating global severity (from 0—absent to 5—severe) is included. A low score indicates low levels of akathisia.
6.1.4 Quality of life
6.1.4.1 Paediatric Quality of Life Enjoyment and Satisfaction Questionnaire—PQ‐LES‐Q (Endicott 2006) The PQ‐LES‐Q is a 15‐item scale with a total score ranging from 14 to 70. The total score is calculated by adding scores for the first 14 items. Each item on the scale is scored by the young person from 1 to 5. Higher scores indicate greater enjoyment and satisfaction.
6.2 Redundant data
Conducting a trial requires enormous effort, which has been the case in the included studies for this review. Trialists rated and recorded huge quantities of data but failed to report findings adequately, rendering them unusable. For example, continuous data were reported on global mental state without variances, making them difficult to use.
6.3 Missing outcomes
We found no usable data for the outcomes of death, service utilisation (e.g. days in hospital), cognitive functioning, educational status, engagement with services, social skills and economic outcomes.
6.4 Primary outcomes
Several studies reported data on primary outcomes. Many other outcomes that we felt were of secondary importance were also reported. We do recognise that this information may be of primary interest to others.
Excluded studies
We excluded a total of 43 studies. Seven studies were not randomised (Antropov 1981; Jenner 2004; Liang 2003; McConville 2003; Newton 2005; Sela 2003; 谭友果 2002). Twenty‐one studies did not meet our criteria for the age range (Bertelsen 2005; Chen 2007; Davidson 2004; Gao 2007; Killackey 2006; Leblanc 2006; Leclerc 2006; Linszen 2006; Mathai 2004; McGlashan 2003; McGorry 2007; Power 2004; Stain 2006; van Nimwegen 2006; Wang 2007; Xiu 2004; Yang 2007; Yao 2003; Yi 2006; Zhang 2007; Zhou 2007). Four studies provided interventions that did not meet our inclusion criteria. Malik 1980 compared two typical antipsychotics, namely, loxapine versus trifluoperazine. Ueland 2004 used the comparison arms of cognitive remediation programme (CRP) and psychoeducational treatment programme (PTP) versus PTP. Loxapine versus haloperidol were the comparator arms for Versiani 1978. Wykes 2007 compared CRP with treatment as usual, and study authors did not describe what treatment as usual consisted of. Three studies did not report data for participants between 13 and 18 years of age (杨玲 2004; Amminger 2006; Berger 2007). Eight studies either reported insufficient data or reported them in a way that we found unusable (Buchsbaum 2007; Johnson 2004; Klier 2005; Lv 2004; Otsuka 2005; Schepp 1999; Tandon 2005; van Bruggen 2003). We attempted to contact authors of Tandon 2005 and van Bruggen 2003 but did not hear back from them.
Studies awaiting assessment
Currently, no studies are awaiting assessment.
Ongoing studies
Five studies are ongoing (Alaghband‐rad 2006; AstraZeneca 2004; AstraZeneca 2005; Bechdolf 2007; Pfizer 2005). Although study protocols were published a few years ago, we could not find full papers. Enquiries were made to the trialists to request more information, but no further information became available.
Risk of bias in included studies
Please also refer to the 'Risk of bias' table in the Characteristics of included studies.
Allocation
All 13 studies were described to be randomised. Four studies (Jensen 2008; Kumra 1996; Kumra 2008; Sikich 2004) provided descriptions of methods used to generate the sequence. Kumra 2008 and Sikich 2008 described adequate allocation concealment.
Blinding
Seven of 13 studies were described as double blind. Only three studies gave further explanation about blinding. Kryzhanovskaya 2009 mentioned a double‐blind phase followed by an open‐label phase. Kumra 1996 specifically said that raters, treating physicians and nurses were blind to interventions. In Sikich 2008, blinding was maintained even after one of the three arms had been discontinued. Four studies were described as single blind or open label (Aranda 2007; DelBello 2008; Jensen 2008; Swadi 2010). In the studies of Huo 2007 and Xiong 2004, the blinding was unclear.
Incomplete outcome data
Many of the included studies used intention‐to‐treat analysis to account for participants who left the study early. Two Chinese studies (Huo 2007; Xiong 2004) had no participants who left early, and investigators reported data for all participants. Aranda 2007 only partially addressed the issue by reporting data incompletely for participants who left the study early.
Selective reporting
Although most of the studies reported outcomes in sufficient detail, some studies were not so open. Aranda 2007 reported multiple outcome measures but only partially (e.g. reporting no SD for a continuous scale). Kumra 1996 highlighted positive findings but failed to discuss in adequate detail the high incidence of neutropenia in the clozapine group.
Other potential sources of bias
Most of the included studies were well‐designed randomised controlled trials. In DelBello 2008, participants were selected from ten different centres, and broad inclusion criteria were applied with a small number of participants (N = 17). Kumra 1996 was a well‐designed trial but it included few participants (N = 21). Sikich 2004 did not use correction techniques to take into account multiple analyses on a small sample (N = 51). The authors justified this by stating that these analyses were 'exploratory' rather than 'inferential'. The studies described as single blind or open label are prone to further bias because of lack of double blinding and prone to interviewer or assessor biases.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
1. Comparison 1: Atypical antipsychotics vs placebo (only short term)
Two studies compared atypical antipsychotic medications with placebo (Findling 2008; Kryzhanovskaya 2009).
1.1 Global state
Global state as measured on the CGI‐S was reported by Kryzhanovskaya 2009. No significant difference was noted between olanzapine and placebo (1 RCT, n = 107, RR 0.84, 95% CI 0.65 to 1.10) with regard to the number of non‐responders.
1.2 Mental State
The number of non‐responders in one study (Kryzhanovskaya 2009) was not significantly different between participants receiving olanzapine and those given placebo (1 RCT, n = 107, RR 0.84, 95% CI 0.65 to 1.10). In another study (Findling 2008), however, the number of non‐responders receiving aripiprazole 10 mg/d was greater than the number given placebo (1 RCT, n = 197, RR 0.72, 95% CI 0.56 to 0.94).
1.3 Adverse effects
Significantly more people (Kryzhanovskaya 2009) had weight gain > 7% of their baseline pretreatment weight in the group receiving olanzapine over placebo (1 RCT, n = 107, RR 3.56, 95% CI 1.14 to 11.11). The mean weight gain for the group of young people receiving olanzapine was 4.3 kg as compared with 0.1 kg (P < 0.001) for the placebo group. Significantly more young people treated with olanzapine (Kryzhanovskaya 2009) developed treatment‐emergent serum high prolactin concentration at any time during treatment (81.0% vs 16.7%, P = 0.008) as compared with the placebo group. The number of people with clinically significant high serum prolactin concentration at the end of the study was significantly higher for the olanzapine group (1 RCT, n = 107, RR 4.70, 95% CI 2.25 to 9.82).
In another study (Findling 2008), the authors reported no significant difference in weight gain > 5% between the group receiving aripiprazole and the group given placebo (1 RCT, n = 202, RR 4.41, 95% CI 0.98 to 19.91). Findling 2008 reported the total number of adolescents with clinically significant low prolactin for all three groups. Taken together, all adolescents treated with aripiprazole, that is, the number of people in the aripiprazole arms of the trial, had significantly lower serum prolactin concentration (1 RCT, n = 302, RR 3.77, 95% CI 1.88 to 7.58) as compared with the placebo group.
1.4 Leaving the study early
Significantly more (57% vs 32%) people left the study early (1 RCT, n = 107, RR 0.56, 95% CI 0.36 to 0.87) from the placebo group as compared with the olanzapine group (Kryzhanovskaya 2009). In the treatment arm, 10 of a total of 72 young people (14%) allocated to the olanzapine arm left the study because of lack of efficacy as compared with 18 of 35 young people (51%) allocated to the placebo arm, who left the study for the same reasons. In this trial, only 5 (7%) young people left the intervention arm (olanzapine) as the result of adverse effects. In the other study (Findling 2008), no difference was noted between the intervention arm and the placebo armwith regard to leaving the study early (1 RCT, n = 202, RR 1.76, 95% CI 0.86 to 3.63).
1.5 Quality of Life
The mean end point of quality of life score was not included in the analysis, as the data were highly skewed.
2. Comparison 2. Atypical vs typical antipsychotic medications (only short term)
Five studies compared atypical antipsychotic medications with typical antipsychotic medications (Huo 2007; Kumra 1996; Sikich 2004; Sikich 2008; Xiong 2004). Of these studies, Sikich 2004 and Sikich 2008 had three arms and used two atypical antipsychotic medications and one typical antipsychotic medication. We have provided separately the data for all comparisons.
2.1 Global state
In the study Kumra 1996, the mean end point CGAS score clearly favoured young people treated with clozapine (1 RCT, n = 21, RR 17.00, 95% CI 7.74 to 26.26) compared with haloperidol. However, the two groups did not differ in terms of the number of participants showing no improvement (1 RCT, n = 21, RR 3.30, 95% CI 0.41 to 26.81). The study by Sikich 2004 did not show significant improvement in the mean end point of CGI‐I scores for adolescents treated with risperidone as compared with haloperidol (1 RCT, n = 34, MD ‐0.60, 95% CI ‐1.45 to 0.25) or for those treated with olanzapine as compared with haloperidol (1 RCT, n = 31, MD ‐0.70, 95% CI ‐1.55 to 0.15). We could not calculate the mean end point CGI score for Sikich 2008, as the authors reported two different scores at two time points: CGI‐S score for baseline data and mean change in CGI‐I score following improvement. Huo 2007 and Xiong 2004 did not report global state.
2.2 Mental State
Mean end point BPRS score was reported by five studies included in the analysis (Huo 2007; Kumra 1996; Sikich 2004; Sikich 2008; Xiong 2004). No significant difference in the mean end point BPRS score was noted between atypical antipsychotic medications and typical antipsychotic medications (5 RCTs, n = 236, MD ‐1.08, 95% CI ‐3.08 to 0.93). For studies with three arms, of which two used atypical antipsychotic medications, we chose for the above analysis the group treated with atypical antipsychotic medication that included the bigger number of participants. For the above calculation, we included only one comparison per study, as otherwise a misleadingly higher number of total participants would have been included in the typical antipsychotic medication group (comparator group) as the result of duplication. When we looked at each of the studies individually, comparisons for the mean end point BPRS score between atypical and typical antipsychotic medications crossed the line of no effect for all studies in the forest plot. Mean end point total PANSS score calculated from the figures reported by Sikich 2008 showed significant improvement with olanzapine (1 RCT, n = 75, MD 27.00, 95% CI 15.27 to 38.73) and risperidone (1 RCT, n = 81, MD 32.90, 95% CI 19.70 to 46.10) as compared with molindone. Although Kumra 1996 reported mean end point SANS and SAPS scores, the data were highly skewed and have not been included in the current analysis.
2.3 Adverse effects
No significant difference between atypical and typical antipsychotic medications was reported by Huo 2007 and Xiong 2004 for extrapyramidal side effects such as tremors (2 RCTs, n = 100, RR 0.46, 95% CI 0.21 to 1.04) and restlessness (2 RCTs, n = 100, RR 0.71, 95% CI 0.24 to 2.10). Kumra 1996 reported that participants receiving clozapine were three times more likely to have drowsiness on treatment as compared with those given haloperidol (1 RCT, n = 21, RR 3.30, 95% CI 1.23 to 8.85, NNTH 2, 95% CI 2 to 17). Although not reaching statistical significance, 50% of the participants (5 of 10 participants) receiving clozapine in the study by Kumra 1996 had a drop in absolute neutrophil count to below 1500 per mm3. None of the participants in the haloperidol group experienced this adverse effect (1 RCT, n = 21, RR 12, 95% CI 0.75 to 192.86). For the same study, 2 of 10 participants taking clozapine had seizures. This is clinically significant, although the risk ratio for seizures while taking clozapine as compared with haloperidol was not statistically significant (1 RCT, n = 21, RR 5.45, 95% CI 0.29 to 101.55).
The mean end point body weight was not greater for adolescents treated with risperidone (1 RCT, n = 81, MD 0.60, 95% CI ‐8.31 to 9.51) or olanzapine (1 RCT, n = 75, MD 2.90, 95% CI ‐6.30 to 12.10) as compared with molindone (Sikich 2008). In this study, mean serum cholesterol concentration showed a statistically significant increase at the end of the treatment period (1 RCT, n = 75, MD 25.60, 95% CI 5.84 to 45.36) for adolescents treated with olanzapine as compared with those given molindone. The serum cholesterol concentration was not increased at the end of the study (Sikich 2008) for adolescents treated with risperidone (1 RCT, n = 75, MD ‐1.50, 95% CI ‐21.01 to 18.01). The mean end point serum prolactin concentration for all three groups (risperidone, olanzapine and molindone) in the study done by Sikich 2008 was much higher than the normal reference range, but no difference was reported for the mean end point serum prolactin concentration as compared with molindone for the the group of adolescents receiving atypical antipsychotic medications.
2.4 Leaving the study early
Although this did not reach statistical significance, 3 of the 10 young people treated with clozapine left the study by Kumra 1996 as the result of adverse effects, of which two were due to a drop in neutrophil count (1 RCT, n = 21, RR 3.30, 95% CI 0.41 to 26.81). When all studies that reported reasons for leaving the study early were taken together (Kumra 1996; Sikich 2004; Sikich 2008), fewer adolescents receiving atypical antipsychotic medications left the study because of adverse effects (3 RCTs, n = 187, RR 0.65, 95% CI 0.36 to 1.15) or for any reason (3 RCTs, n = 187, RR 0.62, 95% CI 0.39 to 0.97).
3. Comparison 3. Atypical vs atypical antipsychotic medication (only short term)
The number of studies comparing one atypical antipsychotic medication with another for adolescents with psychosis is increasing (Jensen 2008; Kumra 2008; Sikich 2004; Sikich 2008; Swadi 2010).
3.1 Global state
For two studies (Sikich 2004; Sikich 2008), the numbers of participants with no improvement in CGI score were similar for the groups receiving risperidone and olanzapine (2 RCTs, n = 111. RR 1.04, 95% CI 0.70 to 1.54). In another study (Swadi 2010), which compared quetiapine and risperidone, no significant difference was reported in the numbers of participants showing no improvement in CGI score (1 RCT, n = 22, RR 1.20, 95% CI 0.52 to 2.79). The mean end point CAGS score was not significantly different (1 RCT, n = 39, MD 4.10, 95% CI ‐6.71 to 14.91) for participants receiving clozapine and those taking olanzapine in a study by Kumra 2008. However, the mean end point CGI‐I score (Kumra 2008) was significantly better for the group of adolescents receiving clozapine as compared with those given olanzapine (1 RCT, n = 39, MD ‐1.07, 95% CI ‐1.9 to ‐0.22).
3.2 Mental State
The mean end point BPRS score was not different in two studies (Sikich 2004; Sikich 2008) that compared risperidone and olanzapine, which are not included in the analysis as the data were skewed. Even if one assumes normal distribution, the difference between the two treatment arms (olanzapine vs risperidone) is not significant when the mean end point BPRS scores are compared. Similarly, Jensen 2008 reported that similar numbers of participants in the groups receiving risperidone or quetiapine showed no response, as defined by less than 40% reduction in baseline PANSS score (1 RCT, n = 19, RR 0.48, 95% CI 0.17 to1.31). When we compared risperidone with quetiapine in the study by Jensen 2008, no difference between the groups was noted regarding the number of participants who did not improve (1 RCT, n = 29, RR 0.33, 95% CI 0.06 to 1.73). In the study by Swadi 2010, which compared risperidone with quetiapine, similar numbers of participants in both groups did not show response on the PANSS score at the end of the study (1 RCT, n = 22, RR 1.67, 95% CI 0.52 to 5.33). The study by Kumra 2008 reported a similar mean end point score on BPRS for participants receiving clozapine and olanzapine (1 RCT, n = 39, MD ‐2.9, 95% CI ‐10.13 to 4.33). However, categorical analysis of the data provided by Kumra 2008 on the number of people who did not respond (defined as less than 30% reduction in BPRS score) showed that results favoured clozapine over olanzapine (1 RCT, n = 39, RR 0.14, 95% CI 0.03 to 0.60).
3.3 Adverse effects
Not much difference was observed in some of the studies included in this review between medications used in the two arms of each trial (various atypical antipsychotics) regarding the mean end point body weight. Data reported by Sikich 2008 showed that the mean end point body weight was similar for adolescents treated with risperidone and those given olanzapine (1 RCT, n = 76, MD ‐2.30, 95% CI ‐9.97 to 5.37). However, the mean change in body weight showed that those treated with olanzapine had on average gained 6.1 + 3.6 kg by the end of treatment as compared with an average gain of 3.6 + 4 kg for those treated with risperidone. The mean change in body weight was statistically significant in this study.
Kumra 2008 reported no significant difference in the number of people who gained ≥ 7% of baseline body weight between groups of adolescents treated with olanzapine and clozapine (1 RCT, n = 39, RR 1.75, 95% CI 0.33 to 9.34). However it is important to remember that no difference between the two arms, does not mean that this is not a clinical issue. It may be that both arms included a high proportion of people who experienced weight gain. To give an example, Jensen 2008 reported that 8 of 9 adolescents taking risperidone and 6 of 10 adolescents treated with olanzapine gained more than 7% of their baseline body weight. Kumra 2008 mentioned that 66% of people started on clozapine (12 of 18) and 66% of adolescents started on olanzapine (14 of 21) reported increased appetite by the end of the study. In the study by Sikich 2008, participants treated with olanzapine had higher mean end point serum cholesterol concentration as compared with those taking risperidone (1 RCT, n = 76, MD ‐27.10, 95% CI ‐50.13 to ‐4.07). The serum cholesterol concentration for participants treated with olanzapine showed an average increase of 19.9 + 23.9 mg/dL at the conclusion of the study as compared with an average decrease of 10.2 + 26.7 mg/dL for those taking risperidoneof . These data are skewed and should be viewed with caution. As can be easily seen from the wide standard deviation, a few participants in both the risperidone and the olanzapine groups showed increased serum cholesterol concentration. The only study that reported drug‐induced diabetes was Kumra 2008. Only one of 18 adolescents treated with clozapine developed drug‐induced diabetes in this study. Although this finding is not statistically significant when compared with the control group, in which no participants developed diabetes, it needs to be viewed with caution given the short‐term nature of the study.
The serum prolactin concentration was increased much beyond the normal range by the end of the study (Sikich 2008) for both groups of adolescents treated with atypical antipsychotic medications. However, no significant difference was noted between those who received risperidone and those who took olanzapine (1 RCT, n = 76, MD ‐2.30, 95% CI ‐9.97 to 5.37). Swadi 2010 reported that a significantly greater number (10 of 11) of adolescents receiving risperidone as compared with quetiapine had raised serum prolactin concentration (1 RCT, n = 14, RR 4.44, 95% CI 0.60 to 32.77).
No difference in the number of participants reporting muscle stiffness or akathisia was noted between adolescents who received olanzapine and those who were given risperidone (1 RCT, n = 19, RR 2.22, 95% CI 0.53 to 9.37) or quetiapine and risperidone (1 RCT, n = 19, RR 4.44, 95% CI 0.60 to 32.77) in the study by Jensen 2008. In the study by Swadi 2010, no significant difference was reported between groups receiving risperidone versus quetiapine regarding their scores on the Barnes Akathisia Scale, the Simspson Angus Akathisia Scale and the Abnormal Involuntary Movement Scale. Kumra 2008, Sikich 2004 and Sikich 2008 did not provide details of extrapyramidal symptoms in their study, except when they had been a reason for withdrawal from the study.
3.4 Leaving the study early
In the study by Kumra 2008, 11 of a total of 39 participants recruited left the study early. Of these 11 participants, six treated with olanzapine and one treated with clozapine left the study because of non‐response, two left the clozapine arm of the trial because of weight gain and one left the olanzapine arm as a result of neutropenia.
No difference in the number of people leaving the trial early because of side effects was reported for those treated with risperidone or olanzapine (3 RCTs, n = 130, RR 1.21, 95% CI 0.51 to 2.87). Two of 10 adolescents who were treated with quetiapine left the study (Jensen 2008) because of non‐response. In total, one of 10 young people (Jensen 2008) from the risperidone group, four of 10 from the quetiapine group and four of 10 from the olanzapine group left the study. In total, only one young person from the olanzapine group left the study because of weight gain.
4. Comparison 4. Atypical (higher dose) vs atypical (lower dose) antipsychotic medications (only short term)
Three studies compared lower and higher doses of the same atypical antipsychotic medications (Findling 2008; DelBello 2008; Haas 2009). We divided the groups that had used less than or equal to 150 mg of chlorpromazine equivalent doses and groups that had used higher doses of the same medications. This cut‐off is based on the paper published by Andreasen 2010, in which the authors had calculated the standardised chlorpromazine equivalent doses of antipsychotic medications used in the CATIE study (Lieberman 2005). The chlorpromazine equivalent daily dose was attributed to ziprasidone in this paper (112 mg of ziprasidone was equated to 188 mg of chlorpromazine). Hence, we chose the cut‐off of 150 mg or more of chlorpromazine equivalent per day to define higher dose as compared with low dose, which is less than 150 mg of chlorpromazine equivalent.
4.1 Global state
Haas 2009 studied the comparative effects of a low dose versus a standard dose of risperidone. This multi‐centred trial reported that the standard dose of risperidone (1.5 mg to 6.0 mg) was found to be superior to the low dose of risperidone (0.15 mg to 0.6 mg) regarding the outcome of no response as assessed by the CGI‐I Scale (1 RCT, n = 255, RR 0.54, 95% CI 0.38 to 0.75). The authors reported that the higher dose of risperidone was more effective in reducing the mean end point CGI‐I score (1 RCT, n = 255, MD ‐0.60, 95% CI ‐0.93 to ‐0.27). Another study (Findling 2008) compared 30 mg of aripiprazole with 10 mg of aripiprazole. In this study (Findling 2008), young people who received aripiprazole 30 mg per day did better on the CGI Scale (1 RCT, n = 196, MD ‐0.20, 95% CI ‐0.48 to 0.08). DelBello 2008 reported findings of a small study that compared low‐dose (80 mg/d) versus high‐dose (160 mg/d) ziprasidone. Only some of the data (n = 17) reported by DelBello 2008 were used in this review; adolescents who had a primary diagnosis of bipolar disorder were excluded. The mean end point CGI score was not significantly different in the groups receiving different doses of ziprasidone (1 RCT, n = 17, MD 0.20, 95% CI ‐0.83 to 1.23). Taken together, participants treated with higher doses (greater than 150 mg chlorpromazine equivalent per day) had better mean end point CGI‐I scores (3 RCT, n = 468, MD ‐0.34, 95% CI ‐0.55 to ‐0.13) as compared with participants taking lower doses (less than 150 mg chlorpromazine equivalent per day).
4.2 Mental State
Haas 2009 reported that the mean end point PANSS score showed significantly greater symptom reduction with standard/higher doses of risperidone (1 RCT, n = 257, RR ‐8.00, 95% CI ‐13.75 to ‐2.25). However, it is important to put this in the perspective of relatively low mean baseline PANSS scores for the experimental group 96.4 (SD 15.39) and the comparator group 93.3 (SD 14.14). The difference in the mean change in PANSS score of ‐23.6 (SD 22.83) in the higher‐dose risperidone group and ‐12.5 (SD 20.32) in the low‐dose risperidone group, although statistically significant, is not clinically that important in that the total shift in PANSS score of 23.6 represents only 22% reduction from the baseline mean PANSS score of 96.4. In other words, no category shift in clinically significant symptoms can be seen. This is reflected in our analysis using categorical data (as reported by the study authors) on the number of people who achieved remission (defined as greater than 30% reduction in PANSS score), which shows that low versus high dose did not make a statistically significant difference (1 RCT, n = 113, RR 0.78, 95% CI 0.59 to 1.03). When findings were taken togather (Findling 2008; Haas 2009), standard dose versus low dose did not result in a difference in the mean end point PANSS score (2 RCTs, n = 451, MD ‐3.49, 95% CI ‐7.26 to 0.28).
Findling 2008 reported no statistically significant difference between groups receiving 10 mg/d and 30 mg/d of aripiprazole (1 RCT, n = 196, RR 0.84, 95% CI 0.48 to 1.48) for the outcome of not achieving remission at six weeks. In another study (DelBello 2008), authors reported no statistically significant difference between groups receiving a lower dose (80 mg/d) versus a higher dose (160 mg/d) of ziprasidone, as reflected by the mean end point BPRS score (1 RCT, n = 17, MD ‐4.40, 95% CI ‐19.20 to 10.40).
4.3 Adverse effects
It is not surprising that low‐dose risperidone (Haas 2009) was associated with significantly less‐frequent extrapyramidal symptoms (1 RCT, n = 254, RR 3.31, 95% CI 1.86 to 5.87). Data show that 23 of 125 young people receiving high‐dose risperidone and 8 of 132 young people given low‐dose risperidone reported dystonia. Although a significantly greater incidence of dystonia was seen in the higher‐dose group (1 RCT, n = 257, RR 3.04, 95% CI 1.41 to 6.53), the occurrence of dystonia in both groups is clinically significant. This study demonstrates that risperidone can cause dystonia even at low doses, but the risk is increased with increasing dose.
A lower incidence of drug‐induced Parkinsonism in the group taking aripiprazole 30 mg (1 RCT, n = 202, RR 2.03, 95% CI 1.17 to 3.52) was reported by Haas 2009, but extrapyramidal side effects did not differ between the two groups in the other study (Findling 2008).
Interesting findings regarding hyperprolactinaemia were reported in the study by Haas 2009. A significantly greater number of participants with serum prolactin above 100 ng/mL were described in the group that received a standard dose of risperidone (1 RCT, n = 257, RR 46.46, 95% CI 6.50 to 332.17). However, standard‐dose and low‐dose groups did not differ significantly regarding the number of participants who had symptomatic hyperprolactinaemia (1 RCT, n = 257, RR 3.70, 95% CI 0.78 to 17.45). In the trial reported by Findling 2008, serum prolactin was reduced by the end of treatment with aripiprazole. The mean change in serum prolactin was ‐11.93 (SD 23.29) ng/mL for the group treated with aripiprazole 10 mg/d and ‐15.14 (SD 26.87) ng/mL for the group treated with aripiprazole 30 mg/d. This difference between the two groups must be viewed with caution because the data are highly skewed, as expressed by the large standard deviation in the mean change in serum prolactin in both groups. The mean serum cholesterol concentration was reduced by ‐7.43 (SD 27.99) mg/dL and ‐5.01 (SD 23.28) mg/dL for participants treated with aripiprazole 10 mg and 30 mg, respectively. The difference in mean change between the two groups was not statistically significant, and the data were skewed. However, the fact that the cholesterol level did not increase for most of the young people is clinically important, given that dyslipidaemia is associated with many of the other atypical antipsychotic medications.
The study by DelBello 2008 included participants with psychosis and participants with bipolar disorder; for the purpose of this review, we included only the data on adolescents with schizophrenia and schizoaffective disorder. We did not include the adverse effects reported by DelBello 2008, as investigators did not provide a breakdown of adverse effects specifically for the subgroup of participants (schizophrenia and schizoaffective disorders) included in this review.
4.4 Leaving the study early
A total of 28% of participants in the group receiving a standard dose of risperidone left the study (Haas 2009) prematurely as compared with 38% in the control arm receiving low‐dose risperidone. The most common reason cited for discontinuation was inadequate response. In the group receiving aripiprazole 10 mg, 7.1% of participants left the study prematurely as compared with 4.1% in the group receiving aripiprazole 30 mg. DelBello 2008 reported that more participants (55%) who were receiving a higher dose of ziprasidone left the study early, but this was not statistically significant when compared with the lower‐dose group (1 RCT, n = 17, RR 0.59, 95% CI 0.26 to 1.36). In the study by Haas 2009, no difference was noted between the groups receiving low‐dose versus standard‐dose risperidone regarding treatment‐emergent self‐injury/aggression (1 RCT, n = 257, RR 0.35, 95% CI 0.04 to 3.34) or worsening of any psychiatric symptoms (1 RCT, n = 257, RR 1.32, 95% CI 0.36 to 4.80).
Discussion
Summary of main results
1. Search
The current review identified more studies published in the last 4 years than earlier in which atypical antipsychotic medications were used for treatment of adolescents with psychosis. Of the 13 studies included in this review, 8 were published during or after 2008. Research in the area of psychosis in adolescence seems to be active. However, given that 1 in 5 patients with schizophrenia develop the illness before turning 18, more research on early stages of the illness is needed. This would be in line with further investigation of the neurodevelopmental hypothesis of mental illnesses. Use of evidence‐based interventions early on for adolescents with psychosis provides ample scope to reduce the duration of untreated psychosis and hence influence the overall course of the illness.
2. Strengths and weaknesses
The review uses standardised search methodology and has included studies published in languages other than English. It is a comprehensive review on the topic that looks into the details of published evidence and focusses not only on comparisons of different medications but also on different doses of the same medication when reported by published trials. The main weakness of the review is its inability to collate data from various studies systematically in single forest plots, as many of the studies have used different outcome measures and are heterogeneous in reporting of side effects; thus their findings cannot be combined.
1. Atypical antipsychotic medications vs placebo (only short term)
The two studies reported comparative efficacy of atypical antipsychotic medications versus placebo.
1.1 Global state
Kryzhanovskaya 2009 did not show any difference in the measure of global state between olanzapine and placebo.
1.2 Mental state
The study by Kryzhanovskaya 2009 compared olanzapine (mean dose 11.0 + 4.0 mg/d) and placebo, whereas the other study (Findling 2008) compared aripiprazole 10 mg/d and 30 mg/d with placebo using three arms. The mean dose of olanzapine used by Kryzhanovskaya 2009 is lower than the usual clinical dose used for treatment of adolescents with psychosis and may explain the negative finding. In the study by Findling 2008, on the other hand, 46% of adolescents in the treatment arm (aripiprazole 10 mg) did not achieve remission as compared with 64% in the placebo arm. Although the above difference is statistically significant, the authors have defined remission as a score not exceeding 3 (mild symptoms) on items P1, P2, P3, N1, N4, N6, G5 and G9 of the PANSS. The mean end point total PANSS score, calculated from the data published in the paper, is not statistically significant between the treatment and placebo arms. Thus the results of the study should be viewed with caution.
1.3 Adverse effects
Kryzhanovskaya 2009 reported that more young people who were treated with placebo had exacerbation of schizophrenia, and this was statically significant. This is also reflected in the fact that significantly more people left the study early from the placebo group than from the olanzapine group because of lack of efficacy. This is understandable for a study that used placebo as the control arm. Increased weight gain and treatment‐emergent high prolactin levels were reported in the olanzapine arm (Kryzhanovskaya 2009) even for the lower mean dose of medication used. This implies that adolescents are sensitive to some of the side effects of olanzapine even at this lower dose, although as a group, they may not benefit in terms of therapeutic efficacy when compared with placebo. No significant weight gain was described with aripiprazole 30 mg as compared with placebo.
1.4 Quality of life
Only one study (Findling 2008) reported quality of life for participants treated with aripiprazole 30 mg compared with placebo. The data were skewed and could not be rationally interpreted. More studies should report quality of life.
2. Atypical vs Typical antipsychotic medications (only short term)
Five studies compared atypical antipsychotic medications with typical antipsychotic medications.
2.1 Global state
The mean end point CGAS score was significantly improved in the group of participants treated with haloperidol as compared with clozapine in the study by Kumra 1996. This could be a result of the fact that the study was short term, and functional improvement takes longer than clinical improvement, especially with clozapine. The studies by Sikich et al (Sikich 2004; Sikich 2008) did not individually report improvement in the global state with atypical antipsychotic medications.
2.3 Mental state
Second‐generation antipsychotic medications remain the drug of first choice for most UK clinicians who are treating adolescents with psychosis in inpatient settings, as was reported in a recent survey (Imran 2011). However, in the current review, the mean end point BPRS score was not statistically different in all five studies for the atypical antipsychotic group as compared with the group treated with typical antipsychotic medications. This is not in keeping with currently reported prescribing patterns (Imran 2011). On the Bunney‐Hamburg Rating Scale, participants treated with clozapine had a better outcome as reported by one of the included studies (Kumra 1996).
2.4 Adverse effects
Most adverse effects including extrapyramidal adverse effects, treatment‐emergent hyperprolactinaemia and anticholinergic adverse effects were similar for atypical and typical antipsychotic medications. Less weight gain was reported with some of the typical antipsychotic medications. However, because all side effects are not reported uniformly, it is difficult to make a head‐to‐head comparison between studies.
3. Atypical antipsychotic vs atypical antipsychotic medications (only short term)
Five studies compared two different antipsychotic medications for adolescents with psychosis.
3.1 Global state
Two studies by Sikich et al (Sikich 2004; Sikich 2008) reported no difference in the end point global state between risperidone and olanzapine. The end point global state also was not different for Kumra 2008, who compared clozapine and olanzapine.
3.2 Mental State
No difference in the outcome of mental state was noted in the studies that compared one atypical antipsychotic medication with another. However, most participants improved as compared with baseline score.
3.3 Adverse effects
All studies reported similar and comparable adverse effects (extrapyramidal adverse effects and weight gain) for most medications. Olanzapine, risperidone and clozapine were associated with increased body weight. For the studies included in our review, no difference was noted in the number of people putting on weight who were treated with olanzapine, risperidone or clozapine; however, we do appreciate that differences in the degree of weight gain have been described by other reviews that examined second‐generation antipsychotics, which could employ more robust analysis because they included non‐psychotic conditions, as well as psychosis, in adolescents and children (De Hert 2011). De Hert 2011 reported that ziprasidone was associated with the least weight gain, followed by aripiprazole, quetiapine, risperidone and olanzapine in ascending order. Olanzapine was associated with increased mean end point serum cholesterol concentration as compared with risperidone (Sikich 2008). More people had elevated serum prolactin when treated with risperidone as compared with quetiapine (Swadi 2010).
3.4 Leaving the study early
Similar numbers of people left the study early because of non‐response when treated with olanzapine as compared with risperidone (Jensen 2008; Sikich 2004; Sikich 2008).
4. Atypical (higher‐dose) vs atypical (lower‐dose) antipsychotic medications (only short term)
Three studies compared higher doses of an antipsychotic medication with lower doses of the same antipsychotic medication.
4.1 Global state
Some evidence shows that risperidone 1.5 to 6 mg is more likely to improve the global state when compared with the very low dose of 0.15 to 0.6 mg/d for adolescents with psychosis (Haas 2009). However, no difference has been noted between the final global state achieved by aripiprazole 30 mg/d and 10 mg/d (Findling 2008) and similarly between ziprasidone 160 mg/d and 80 mg/d (DelBello 2008).
4.2 Mental state
In most comparisons of mental state, the lower dose (< 150 mg chlorpromazine equivalent) was equally efficacious as the higher dose of the same antipsychotic medication (> 150 mg chlorpromazine equivalent). However, on the PANSS, the higher dose of risperidone fared better.
4.3 Adverse effects
Lower dose was associated with lesser side effects in general. However,the number of young people who had symptomatic hyperprolactinaemia was similar to the number who moved from being overweight to obese, even on lower doses of risperidone. In the study by Haas 2009, no difference was observed between groups receiving low‐dose versus standard‐dose risperidone with regard to treatment‐emergent self‐injury/aggression or worsening of any psychiatric symptoms.
Overall completeness and applicability of evidence
1. Completeness
No outcome in this review involves longer‐term follow‐up; this makes the review incomplete, as psychosis is often a long‐term condition. On the other hand, some of the studies have large and impressive sample sizes. Adverse effects were not reported by researchers in a uniform manner, making it difficult to compare findings across studies. No data on hospital and service utilisation outcomes, economic outcomes, behaviour or cognitive response were available; such data would have made the review more relevant to clinical practice in a milieu of patient‐centred care.
2. Applicability
One in five patients with schizophrenia experiences onset of illness during adolescence. Hence this review is relevant to the practice of psychiatry at this transitional period from adolescence to adulthood. Some trials are large and are representative of adolescents with psychosis in the community. The studies have been conducted in various social, cultural and political settings, making the findings of the review generalisable across the world. However, the participants seem to be adherent to treatment and do not have other co‐morbidities such as cannabis abuse. In the real world, non‐adherence and comorbid substance misuse are more of a rule than an exception.
Quality of the evidence
The atypical antipsychotic medications have been popular only in the last 15 years; therefore most of the trials were conducted recently as opposed to many other intervention trials. Hence, many of the studies included in this review are of high quality, were designed with the framework of the CONSORT guidelines in mind and were published in reputable journals.
Potential biases in the review process
We are not aware of any biases in the review process. We have made every effort to identify all relevant trials. We may have failed to identify small studies because of a degree of publishing bias, but we do not think it likely that we have failed to identify large relevant studies.
Agreements and disagreements with other studies or reviews
Atypical antipsychotic medications are the drug of first choice for most UK clinicians, according to a recent survey (Imran 2011). Similar data from other countries highlight the increased popularity of atypical antipsychotic medications. In Israel alone, prescription of atypical antipsychotic medications has increased by more than 50% over a ten‐year period (Gilat 2011). Although the evidence base on the topic is growing, a gap in knowledge on the use of atypical antipsychotic medications in children and adolescents has been identified (Almandil 2011; Caccia 2011). Our review agrees with another review published by Caccia 2011, who concluded that amongst different antipsychotic medications, the differences between children/adolescents and adults were greater with respect to type and severity of adverse effects than with respect to clinical efficacy of individual medications. In another review (Ardizzone 2010), the authors report that risperidone was associated with extrapyramidal adverse effects in children and adolescents. This notion is supported in our review by the findings of two recent Chinese RCTs comparing risperidone with typical antipsychotic medications. We report that the incidence of extrapyramidal symptoms with risperidone treatment is similar to that reported with typical antipsychotic medications. Our finding that aripiprazole is not associated with weight gain and hyperprolactinaemia has also been reported by Ardizzone 2010. However, many of the reviews published on atypical antipsychotic medications have included a wide range of studies/psychiatric conditions in children and adolescents; they have seldom focused specifically on adolescents with psychosis. We believe that treatment response and side effects may be linked not only by the medication but also by the primary psychiatric disorder. Hence we did not include studies that evaluated the use of antipsychotic medications in non‐psychotic conditions such as affective disorder, autism, etc. Consequently, our review includes a less heterogeneous participant population as compared with many other reviews on antipsychotic medication use in children and adolescents. The current review has tried to specifically address the issue of atypical antipsychotic medication use in adolescents with psychosis.
Authors' conclusions
Implications for practice.
1. For adolescents with psychosis
No convincing evidence supports the superiority of newer atypical antipsychotic medications over typical antipsychotic medications for the treatment of adolescents with psychosis. Patients fared very similarly on typical antipsychotic medications as compared with newer atypical antipsychotic medications with regard to clinical response, but they differed in some respects in terms of frequency and severity of adverse effects. Very little evidence supports the superiority of one atypical antipsychotic medication over another, but all atypical antipsychotic medications were associated with improvement at the end of six weeks when compared with baseline. One of the main considerations for choice of medication should be response or lack of response to past trials with a specific medication and acceptable adverse effects for the patient. Treatment with olanzapine, risperidone and clozapine was often associated with weight gain. On the other hand, aripiprazole was not associated with weight gain, increased prolactin (increased prolactin can lead to sexual side effects) or dyslipidaemia in most patients. Some evidence shows that adolescents respond better to standard doses as opposed to lower doses of medications (< 150 mg of chlorpromazine equivalent). For aripiprazole, a lower dose (10 mg) was as effective as a higher dose (30 mg) in the treatment of adolescents with psychosis.
2. For clinicians
No convincing evidence supports the superiority of newer atypical antipsychotic medications over typical antipsychotic medications for the treatment of adolescents with psychosis. However, adolescents are prone to side effects, even at low mean doses of olanzapine. Because very little evidence supports the superiority of one atypical antipsychotic (risperidone, olanzapine, clozapine, quetiapine) over another, the main considerations for choice of medication should be response or lack of response to past trials with a specific medication and adverse effects. Olanzapine was found to be consistently associated with weight gain across studies. Risperidone was found to be associated with extrapyramidal symptoms similar to those seen with typical antipsychotic medications. Aripiprazole was not associated with increased body weight, hyperprolactinaemia or dyslipidaemia in most patients. Some evidence supports the use of at least standard doses of antipsychotic medications as compared with lower doses (< 150 mg chlorpromazine equivalent), as the global response is better with standard dose than with doses less than 150 mg chlorpromazine equivalent. For aripiprazole, a lower dose (10 mg) and a higher dose (30 mg) were equally efficacious in the treatment of adolescents with psychosis.
3. For managers, policy makers and funders
Very little evidence supports the exclusive use of atypical antipsychotic medications over typical antipsychotic medications in adolescents with psychosis with regard to improvement in mental state. Many of the adverse effects are also comparable. Policy makers should not write off typical antipsychotic medications and should encourage a balanced approach in the use of different medications. Trainees and senior clinicians should be trained on the use of both atypical and typical antipsychotic medications as opposed to the current practice of almost exclusive use of atypical antipsychotic medications as the first line of treatment.
Implications for research.
The controlled studies report only short‐term outcome measures for adolescents treated with atypical antipsychotic medications. More studies are needed with flexible dosing of olanzapine and other atypical antipsychotic medications for adolescents with psychosis. Side effects noted in studies are not reported in a uniform way, making comparisons across studies difficult. For future studies, standardised reporting of side effects is advocated. The role of typical antipsychotic medications for treatment of adolescents with psychosis should be studied and reviewed separately in a systematic review.
Acknowledgements
The Cochrane Schizophrenia Group Editorial Base in Nottingham produces and maintains standard text for use in the Methods section of their reviews. We have used this text as the basis of what appears here and have adapted it as required.
The Trial Search Co‐ordinator of the Cochrane Schizophrenia Group, Samantha Roberts, carried out the search of the register.
We would like to acknowledge and thank Ben Gray and Philipp Rothe for their contributions and comments.
Ben wrote our plain language summary and Philipp acted as a peer reviewer.
Appendices
Appendix 1. Text presented in published protocol for 'dealing with missing data'
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we did not reproduce these data or use them within analyses. If, however, more than 50% of those in one arm of a study are lost, but the total loss is less than 50%, we marked such data with (*) to indicate that such a result may well be prone to bias.
2. Binary
In the case where attrition for a binary outcome is between 0 and 50%, and where these data are not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). We assumed that those who left the study early had the same rates of negative outcome as those who completed the study, with the exception of the outcomes of death and adverse effects. For these outcomes, we used the rates of those who stayed in the study—in that particular arm of the trial—as the rates for those who did not complete the study. We undertook a sensitivity analysis to test how prone the primary outcomes are to change when 'completer' data only are compared with the intention‐to‐treat analysis using the above assumptions.
Data and analyses
Comparison 1. Atypical antipsychotics vs placebo (only short term).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: No response (CGI‐S) | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.65, 1.10] |
| 2 Mental state: 1. No response | 2 | 304 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.63, 0.92] |
| 2.1 No response (BPRS‐C) | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.65, 1.10] |
| 2.2 Not achieving remission (PANSS) | 1 | 197 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.56, 0.94] |
| 3 Mental state: 2. Change in PANSS score (data skewed, high score = good) | Other data | No numeric data | ||
| 4 Adverse effects: 1. Different adverse effects (binary measures) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Cardiovascular—dizziness | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.92 [0.37, 23.30] |
| 4.2 Central nervous system—somnolence | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.26 [1.15, 59.61] |
| 4.3 Endocrine—clinically significant low prolactin | 1 | 302 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.77 [1.88, 7.58] |
| 4.4 Endocrine—treatment‐emergent high prolactin at any time during treatment | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.70 [2.25, 9.82] |
| 4.5 General deterioration—exacerbation of schizophrenia | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.09, 0.89] |
| 4.6 General deterioration—use of benzodiazepines | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.35, 0.92] |
| 4.7 Metabolic—increased appetite | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.94 [0.59, 6.45] |
| 4.8 Metabolic—weight gain | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.56 [1.14, 11.11] |
| 4.9 Metabolic—weight gain ≥ 5% of baseline | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.41 [0.98, 19.91] |
| 4.10 Metabolic—weight gain ≥ 7% of baseline | 1 | 106 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.12 [1.34, 7.27] |
| 4.11 Metabolic—treatment‐emergent high triglycerides at any time | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.38 [1.31, 4.30] |
| 5 Adverse effects: 2. Different adverse effects (continuous measures—mean changes) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Cardiovascular—corrected QT, QT/ms from baseline to end point (high score = poor | 1 | 107 | Mean Difference (IV, Fixed, 95% CI) | ‐6.3 [‐12.51, ‐0.09] |
| 5.2 Endocrine—prolactin, μg/L from baseline to end point (high score = poor) | 2 | 282 | Mean Difference (IV, Fixed, 95% CI) | 3.30 [‐1.72, 8.31] |
| 5.3 Hepatic—ALT, U/L from baseline to end point (high score = poor) | 1 | 104 | Mean Difference (IV, Fixed, 95% CI) | 26.6 [11.34, 41.86] |
| 5.4 Hepatic—total bilirubin, mg/dL from baseline to end point (high score = poor) | 1 | 104 | Mean Difference (IV, Fixed, 95% CI) | ‐3.70 [‐6.30, ‐1.10] |
| 5.5 Metabolic—weight, kg from baseline to end point (high score = poor) | 1 | 106 | Mean Difference (IV, Fixed, 95% CI) | 4.2 [2.99, 5.41] |
| 5.6 Metabolic—BMI, kg/m² from baseline to end point (high score = poor) | 1 | 106 | Mean Difference (IV, Fixed, 95% CI) | 1.5 [1.06, 1.94] |
| 5.7 Metabolic—triglycerides, mg/dL from baseline to end point (high score = poor) | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 37.2 [8.88, 65.52] |
| 5.8 Renal—uric acid, μmol/L from baseline to end point (high score = poor) | 1 | 104 | Mean Difference (IV, Fixed, 95% CI) | 38.40 [18.88, 57.92] |
| 6 Leaving study early: 1. Various reasons (olanzapine vs placebo) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 For any reason | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.36, 0.87] |
| 6.2 Because of adverse effects (elevated liver enzyme) | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.42 [0.31, 95.43] |
| 6.3 Because of lack of efficacy | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.14, 0.52] |
| 7 Leaving study early: 2. Any reason (aripiprazole vs placebo) | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.76 [0.86, 3.63] |
| 8 Quality of life: 1. Mean end point PQ‐LES‐Q score at 6 weeks (data skewed, high score = good) | Other data | No numeric data |
1.1. Analysis.
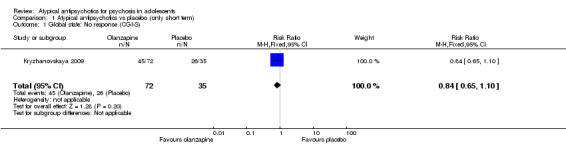
Comparison 1 Atypical antipsychotics vs placebo (only short term), Outcome 1 Global state: No response (CGI‐S).
1.2. Analysis.
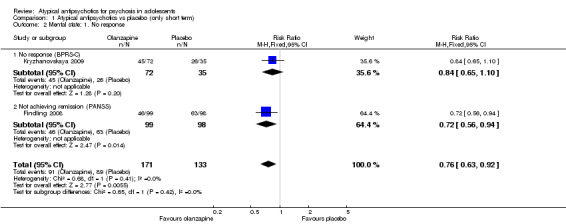
Comparison 1 Atypical antipsychotics vs placebo (only short term), Outcome 2 Mental state: 1. No response.
1.3. Analysis.
Comparison 1 Atypical antipsychotics vs placebo (only short term), Outcome 3 Mental state: 2. Change in PANSS score (data skewed, high score = good).
| Mental state: 2. Change in PANSS score (data skewed, high score = good) | ||||
|---|---|---|---|---|
| Study | Treatment | Mean | SD | N |
| Findling 2008 | Aripiprazole 10mg | 67 | 157 | 99 |
| Findling 2008 | Aripiprazole 30mg | 66.3 | 164.9 | 97 |
| Findling 2008 | Placebo | 73.8 | 152.99 | 98 |
1.4. Analysis.
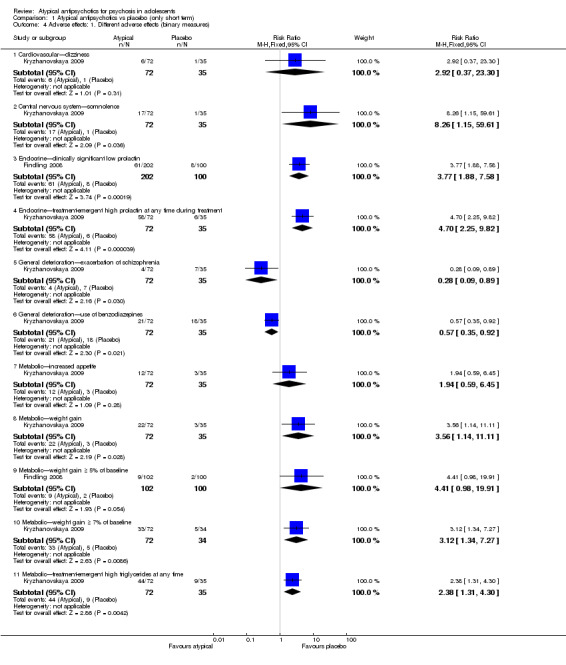
Comparison 1 Atypical antipsychotics vs placebo (only short term), Outcome 4 Adverse effects: 1. Different adverse effects (binary measures).
1.5. Analysis.
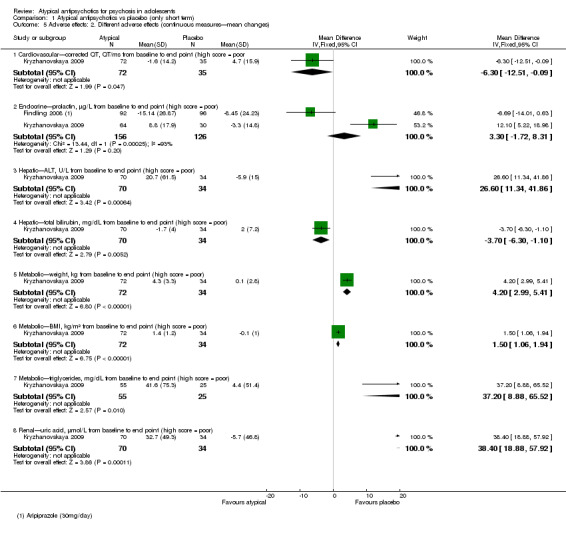
Comparison 1 Atypical antipsychotics vs placebo (only short term), Outcome 5 Adverse effects: 2. Different adverse effects (continuous measures—mean changes).
1.6. Analysis.
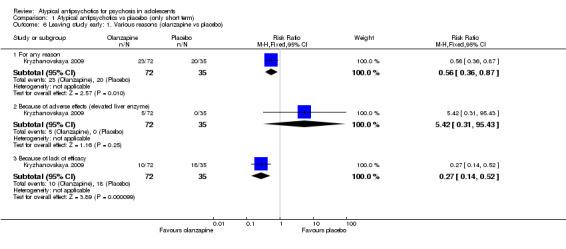
Comparison 1 Atypical antipsychotics vs placebo (only short term), Outcome 6 Leaving study early: 1. Various reasons (olanzapine vs placebo).
1.7. Analysis.
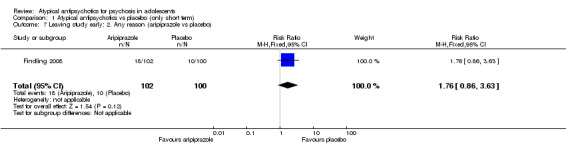
Comparison 1 Atypical antipsychotics vs placebo (only short term), Outcome 7 Leaving study early: 2. Any reason (aripiprazole vs placebo).
1.8. Analysis.
Comparison 1 Atypical antipsychotics vs placebo (only short term), Outcome 8 Quality of life: 1. Mean end point PQ‐LES‐Q score at 6 weeks (data skewed, high score = good).
| Quality of life: 1. Mean end point PQ‐LES‐Q score at 6 weeks (data skewed, high score = good) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Findling 2008 | Aripiprazole 30mg | 50.2 | 90 | 98 |
| Findling 2008 | Placebo | 48.8 | 94.4 | 98 |
Comparison 2. Atypical vs typical antipsychotics (only short term).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Worse or no improvement | 1 | 21 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.3 [0.41, 26.81] |
| 2 Global state: 2a. Mean end point score (CGAS, high score = good) | 1 | 21 | Mean Difference (IV, Fixed, 95% CI) | 17.0 [7.74, 26.26] |
| 3 Global state: 2b. Mean end point score (CGI‐I, high score = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4 Mental state: 1. No improvement (BPRS, high score = poor) | 2 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.38, 2.62] |
| 5 Mental state: 2a. Mean end point scores (various scales, high score = poor) | 5 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 B‐HPRS | 1 | 21 | Mean Difference (IV, Fixed, 95% CI) | ‐3.60 [‐6.64, ‐0.56] |
| 5.2 BPRS | 5 | 342 | Mean Difference (IV, Fixed, 95% CI) | ‐1.34 [‐3.24, 0.56] |
| 5.3 PANSS—total | 1 | 156 | Mean Difference (IV, Fixed, 95% CI) | 29.60 [20.84, 38.37] |
| 5.4 PANSS—positive subscale | 1 | 156 | Mean Difference (IV, Fixed, 95% CI) | 0.67 [‐1.98, 3.32] |
| 5.5 PANSS—negative subscale | 1 | 156 | Mean Difference (IV, Fixed, 95% CI) | 1.83 [‐1.55, 5.22] |
| 6 Mental state: 2b. Mean end point scores (high score = poor, skewed data) | Other data | No numeric data | ||
| 6.1 SAPS | Other data | No numeric data | ||
| 6.2 SANS | Other data | No numeric data | ||
| 7 Adverse effects: 1. Anticholinergic adverse effects (TESS) | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Total | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.05, 0.80] |
| 7.2 Blood pressure—low | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 4.00] |
| 7.3 Dizziness | 2 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.05, 1.60] |
| 7.4 Constipation | 2 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.02, 1.12] |
| 7.5 Saliva—dry mouth | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.10, 2.53] |
| 7.6 Saliva—hypersalivation | 2 | 81 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.49, 3.08] |
| 7.7 Tachycardia (resting heart rate ≥ 100 beats/min) | 1 | 21 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.72, 3.31] |
| 8 Adverse effects: 2a. Extrapyramidal adverse effects (TESS) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Any | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.20, 0.68] |
| 8.2 Restlessness | 2 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.24, 2.10] |
| 8.3 Tremor | 2 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.21, 1.04] |
| 9 Adverse effects: 2b. Extrapyramidal adverse effects (mean end point scores) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 AIMS (high score = poor) | 1 | 21 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐3.72, 3.52] |
| 9.2 S‐ANRS (high score = poor) | 1 | 21 | Mean Difference (IV, Fixed, 95% CI) | ‐1.90 [‐4.19, 0.39] |
| 10 Adverse effects: 3a. Other significant adverse effects | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Endocrine—mean end point serum prolactin concentration (mcg/L) | 1 | 21 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.45 [0.29, 101.55] |
| 10.2 Haematology—drop in absolute neutrophil count below 1500 mm3 | 1 | 21 | Risk Ratio (M‐H, Fixed, 95% CI) | 12.00 [0.75, 192.86] |
| 10.3 Central nervous system—somnolence/drowsiness (TESS) | 2 | 81 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.55, 2.55] |
| 11 Adverse effects: 3b. Other significant adverse effects (mean end point) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.1 Metabolic—body weight (kg) | 1 | 156 | Mean Difference (IV, Fixed, 95% CI) | 1.71 [‐4.69, 8.11] |
| 11.2 Metabolic—serum cholesterol concentration (mg/dL) | 1 | 156 | Mean Difference (IV, Fixed, 95% CI) | 11.88 [‐2.00, 25.76] |
| 11.3 Endocrine—serum prolactin concentration (mcg/L) | 1 | 156 | Mean Difference (IV, Fixed, 95% CI) | 1.71 [‐4.69, 8.11] |
| 12 Leaving study early: various reasons | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 For any reason | 3 | 187 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.39, 0.97] |
| 12.2 Adverse effects—unspecified | 3 | 187 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.36, 1.15] |
| 12.3 Adverse effects—drop in neutrophil count | 1 | 21 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.45 [0.29, 101.55] |
| 12.4 Adverse effects—neuroleptic malignant syndrome | 1 | 21 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.02, 8.03] |
| 12.5 'Inadequate efficacy' or ≤ 20% reduction in CGI‐I score | 3 | 421 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.43, 0.82] |
2.1. Analysis.
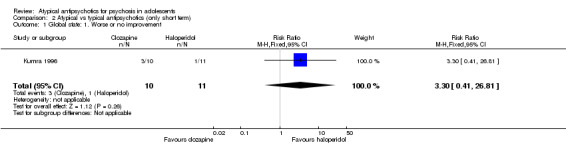
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 1 Global state: 1. Worse or no improvement.
2.2. Analysis.
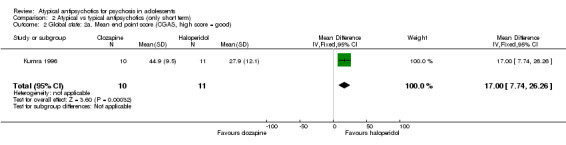
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 2 Global state: 2a. Mean end point score (CGAS, high score = good).
2.3. Analysis.

Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 3 Global state: 2b. Mean end point score (CGI‐I, high score = poor).
2.4. Analysis.
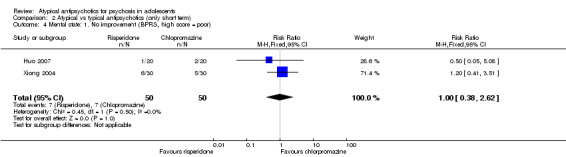
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 4 Mental state: 1. No improvement (BPRS, high score = poor).
2.5. Analysis.
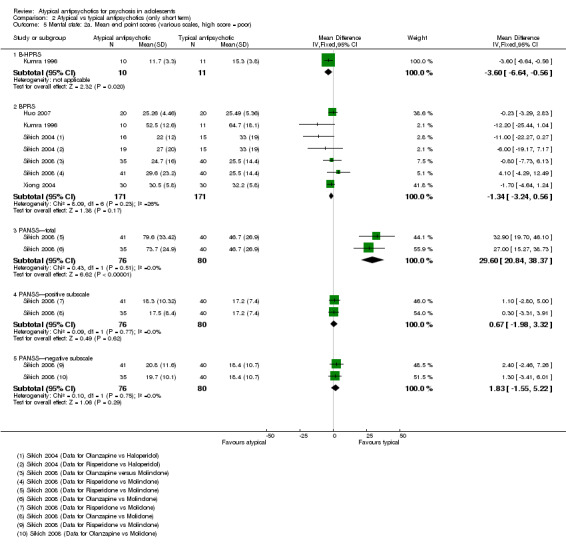
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 5 Mental state: 2a. Mean end point scores (various scales, high score = poor).
2.6. Analysis.
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 6 Mental state: 2b. Mean end point scores (high score = poor, skewed data).
| Mental state: 2b. Mean end point scores (high score = poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Treatment | Mean | SD | N |
| SAPS | ||||
| Kumra 1996 | Clozapine | 19.1 | 11.7 | 10 |
| Kumra 1996 | Haloperidol | 35.9 | 15.6 | 11 |
| SANS | ||||
| Kumra 1996 | Clozapine | 46.0 | 30.3 | 10 |
| Kumra 1996 | Haloperidol | 72.2 | 24.7 | 11 |
2.7. Analysis.
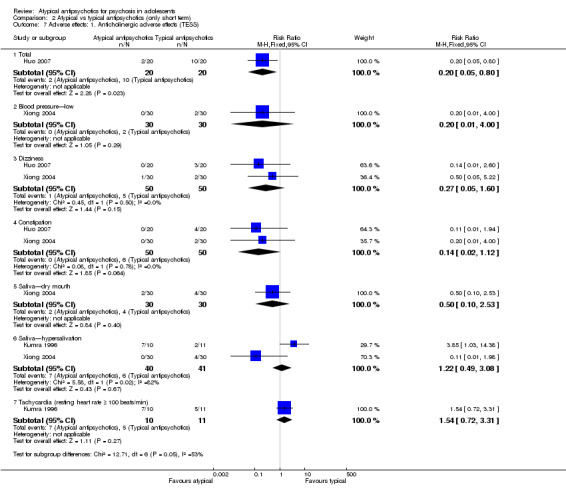
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 7 Adverse effects: 1. Anticholinergic adverse effects (TESS).
2.8. Analysis.
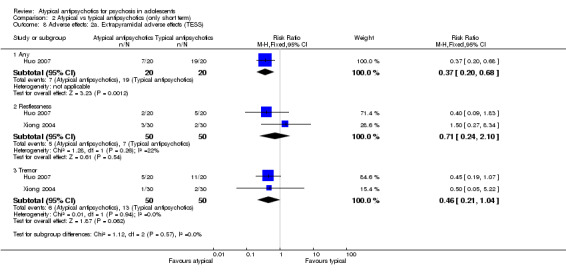
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 8 Adverse effects: 2a. Extrapyramidal adverse effects (TESS).
2.9. Analysis.
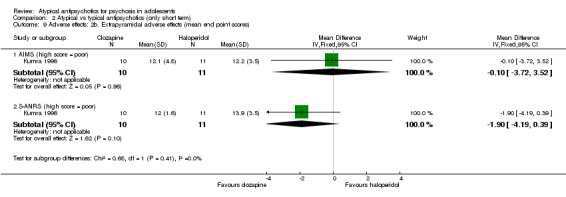
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 9 Adverse effects: 2b. Extrapyramidal adverse effects (mean end point scores).
2.10. Analysis.
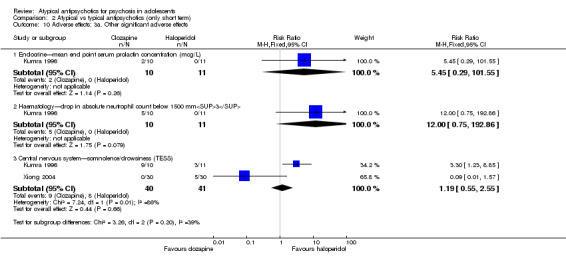
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 10 Adverse effects: 3a. Other significant adverse effects.
2.11. Analysis.
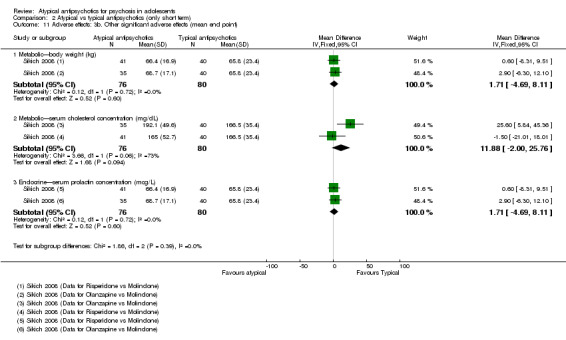
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 11 Adverse effects: 3b. Other significant adverse effects (mean end point).
2.12. Analysis.
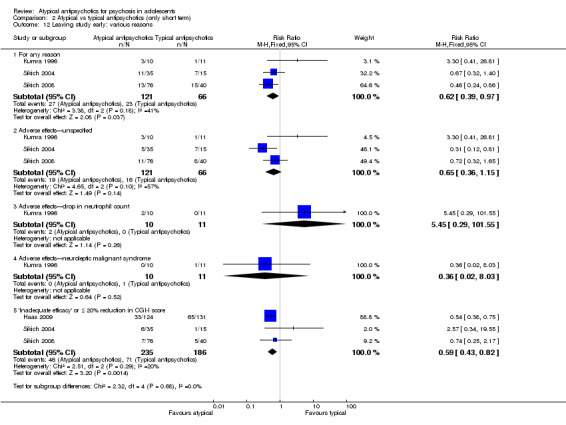
Comparison 2 Atypical vs typical antipsychotics (only short term), Outcome 12 Leaving study early: various reasons.
Comparison 3. Atypical antipsychotics vs atypical antipsychotics (only short term).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. No improvement or no response | 4 | 172 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.65, 1.23] |
| 1.1 No improvement in CGI‐S (risperidone vs olanzapine) | 2 | 111 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.70, 1.54] |
| 1.2 No improvement in CGI (clozapine vs olanzapine) | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.5 [0.24, 1.03] |
| 1.3 No response (< 30% reduction in CGI‐S score) | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.2 [0.52, 2.79] |
| 2 Global state: 2. Mean end point scores (high score = good) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 CGI‐I | 1 | 39 | Mean Difference (IV, Fixed, 95% CI) | ‐1.07 [‐1.92, ‐0.22] |
| 2.2 CGAS | 1 | 39 | Mean Difference (IV, Fixed, 95% CI) | 4.10 [‐6.71, 14.91] |
| 3 Mental state: 1. No response | 3 | Risk Difference (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Clozapine vs olanzapine (≤ 30% reduction in BPRS score) | 1 | 39 | Risk Difference (M‐H, Fixed, 95% CI) | ‐0.44 [‐0.72, ‐0.17] |
| 3.2 Olanzapine vs quetiapine (≤ 40% reduction in PANSS score) | 1 | 20 | Risk Difference (M‐H, Fixed, 95% CI) | ‐0.20 [‐0.62, 0.22] |
| 3.3 Quetiapine vs risperidone (≤ 30% reduction in PANSS score at 6 weeks) | 1 | 22 | Risk Difference (M‐H, Fixed, 95% CI) | 0.18 [‐0.21, 0.58] |
| 3.4 Risperidone vs other atypical antipsychotics (≤ 40% improvement in PANSS total score) | 1 | 29 | Risk Difference (M‐H, Fixed, 95% CI) | ‐0.27 [‐0.64, 0.11] |
| 3.5 Risperidone vs olanzapine (≤ 40% reduction in PANSS score) | 1 | 19 | Risk Difference (M‐H, Fixed, 95% CI) | ‐0.17 [‐0.60, 0.27] |
| 3.6 Risperidone vs quetiapine (≤ 40% reduction in PANSS score) | 1 | 19 | Risk Difference (M‐H, Fixed, 95% CI) | ‐0.37 [‐0.79, 0.05] |
| 4 Mental state: mean end point scores (various scales, high score = poor) | 3 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 BPRS total | 2 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 PANSS total | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 SANS total | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Mental state: mean end point score (BPRS, data skewed, high score = poor) | Other data | No numeric data | ||
| 6 Adverse effects: 1a. Different adverse effects (clozapine vs olanzapine) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Anticholinergic—salivation increase | 1 | 39 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.0 [1.09, 33.02] |
| 6.2 Anticholinergic—sweating | 1 | 39 | Odds Ratio (M‐H, Fixed, 95% CI) | 9.5 [1.69, 53.33] |
| 6.3 Central nervous system—drowsiness | 1 | 39 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.74 [0.21, 105.54] |
| 6.4 Metabolic—appetite increase | 1 | 39 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.26, 3.80] |
| 6.5 Metabolic—drug‐induced diabetes | 1 | 39 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.69 [0.14, 96.22] |
| 6.6 Metabolic—weight gain ≥ 7% of baseline body weight | 1 | 39 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.90 [0.28, 12.87] |
| 6.7 Unspecified—"use of other antipsychotics" | 1 | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.04, 5.77] |
| 7 Adverse effects: 1b. i. Different adverse effects (risperidone vs olanzapine) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Extrapyramidal—muscle stiffness/akathisia | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.22 [0.53, 9.37] |
| 7.2 Metabolic—weight gain ≥ 7% of baseline body weight | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.48 [0.85, 2.58] |
| 8 Adverse effects: 1b.ii. Different adverse effects—means at end of study (risperidone vs olanzapine, high score = poor) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 Endocrine—prolactin (mcg/L) | 1 | 76 | Mean Difference (IV, Fixed, 95% CI) | ‐2.30 [‐9.97, 5.37] |
| 9 Adverse effects: 1b.iii. Different adverse effects—mean change (risperidone vs olanzapine, data skewed, high score = poor) | Other data | No numeric data | ||
| 9.1 Cardiac—QTc (ms) | Other data | No numeric data | ||
| 9.2 Endocrine—prolactin (mcg/L) | Other data | No numeric data | ||
| 10 Adverse effects: 1c. Different adverse effects (risperidone vs quetiapine) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Endocrine—prolactin elevation | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 10.0 [1.53, 65.41] |
| 10.2 Extrapyramidal—akathisia—BARS | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.40, 2.50] |
| 10.3 Extrapyramidal—general—AIMS | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.37, 24.58] |
| 10.4 Extrapyramidal—general—SAS | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.66, 6.04] |
| 10.5 Extrapyramidal—muscle stiffness/akathisia | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.44 [0.60, 32.77] |
| 10.6 Extrapyramidal—use of anticholinergic medication | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.69, 36.13] |
| 10.7 Metabolic—weight gain ≥ 7% of baseline body weight | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.78 [0.92, 3.44] |
| 11 Adverse effects: 2a. Metabolic syndrome measures—means at end of study (clozapine vs olanzapine) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11.1 BMI (kg/m2, high score = poor) | 1 | 38 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐3.35, 2.35] |
| 11.2 Cholesterol (mg/dL, high score = poor) | 1 | 38 | Mean Difference (IV, Fixed, 95% CI) | ‐10.40 [‐31.59, 10.79] |
| 11.3 Glucose (mg/dL, high score = poor) | 1 | 38 | Mean Difference (IV, Fixed, 95% CI) | 10.10 [1.46, 18.74] |
| 11.4 Triglycerides (mg/dL, high score = poor) | 1 | 38 | Mean Difference (IV, Fixed, 95% CI) | 20.20 [‐26.19, 66.59] |
| 12 Adverse effects: 2b.i. Metabolic syndrome measures—means at end of study (risperidone vs olanzapine) | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.1 BMI (kg/m2, high score = poor) | 1 | 34 | Mean Difference (IV, Fixed, 95% CI) | ‐1.40 [‐5.74, 2.94] |
| 12.2 Body weight (kg, high score = poor) | 1 | 76 | Mean Difference (IV, Fixed, 95% CI) | ‐2.30 [‐9.97, 5.37] |
| 12.3 Cholesterol (mg/dL, high score = poor) | 1 | 76 | Mean Difference (IV, Fixed, 95% CI) | ‐27.10 [‐50.13, ‐4.07] |
| 13 Adverse effects: 2b. ii. Metabolic syndrome measures—mean changes (risperidone vs olanzapine, data skewed, high score = poor) | Other data | No numeric data | ||
| 13.1 BMI (kg/m2) | Other data | No numeric data | ||
| 13.2 Cholesterol (mg/dL) | Other data | No numeric data | ||
| 13.3 Glucose (mg/dL) | Other data | No numeric data | ||
| 13.4 Weight gain (kg) | Other data | No numeric data | ||
| 14 Leaving study early: 1a. Clozapine vs olanzapine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 For any reason | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.23, 1.91] |
| 14.2 Because of neutropaenia (absolute neutrophil count = 1200) | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.02, 8.93] |
| 14.3 Because of non‐response | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.03, 1.47] |
| 14.4 Because of weight gain | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.79 [0.30, 113.26] |
| 15 Leaving the study early: 1b. Olanzapine vs quetiapine | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 For any reason | 2 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.55, 1.92] |
| 15.2 Because of non‐response or lack of efficacy or worsening of clinical condition | 2 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.13, 1.74] |
| 15.3 Because of weight gain | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.14, 65.90] |
| 16 Leaving study early: 1c. Risperidone vs olanzapine | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 For any reason | 3 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.52, 1.33] |
| 16.2 Because of non‐response | 3 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.44, 3.04] |
| 16.3 Because of weight gain | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.02, 8.01] |
| 16.4 Because of adverse events | 3 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.21 [0.51, 2.87] |
| 17 Leaving the study early: 1d. Risperidone vs quetiapine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 For any reason | 1 | 20 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.03, 1.86] |
| 17.2 Because of non‐response | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.01, 4.05] |
| 17.3 Because of weight gain | 1 | 19 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
3.1. Analysis.
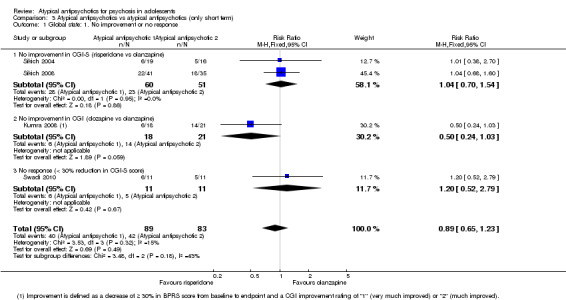
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 1 Global state: 1. No improvement or no response.
3.2. Analysis.
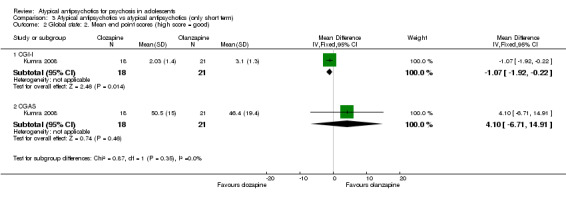
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 2 Global state: 2. Mean end point scores (high score = good).
3.3. Analysis.

Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 3 Mental state: 1. No response.
3.4. Analysis.
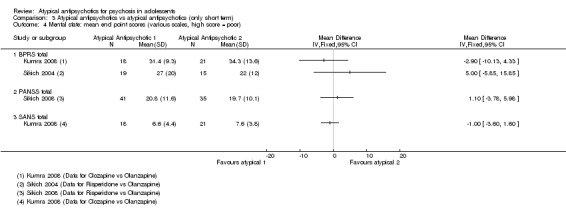
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 4 Mental state: mean end point scores (various scales, high score = poor).
3.5. Analysis.
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 5 Mental state: mean end point score (BPRS, data skewed, high score = poor).
| Mental state: mean end point score (BPRS, data skewed, high score = poor) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Sikich 2004 | Risperidone | 27 | 20 | 19 |
| Sikich 2004 | Olanzapine | 22 | 12 | 16 |
| Sikich 2008 | Risperidone | 29.6 | 23.2 | 41 |
| Sikich 2008 | Olanzapine | 24.7 | 16 | 35 |
3.6. Analysis.
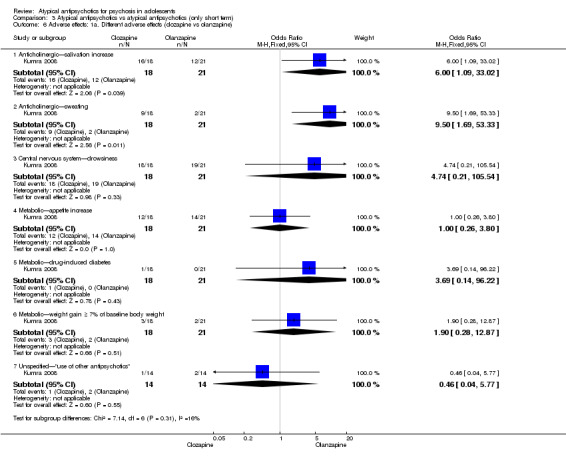
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 6 Adverse effects: 1a. Different adverse effects (clozapine vs olanzapine).
3.7. Analysis.
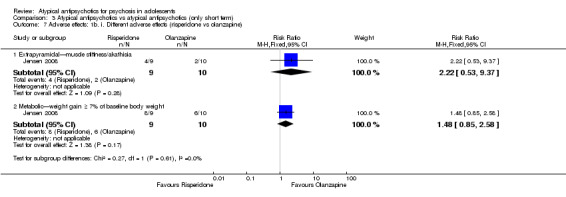
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 7 Adverse effects: 1b. i. Different adverse effects (risperidone vs olanzapine).
3.8. Analysis.
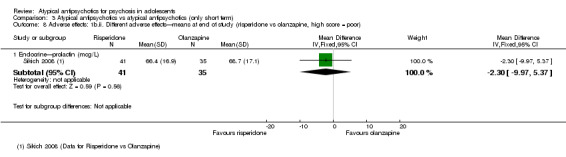
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 8 Adverse effects: 1b.ii. Different adverse effects—means at end of study (risperidone vs olanzapine, high score = poor).
3.9. Analysis.
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 9 Adverse effects: 1b.iii. Different adverse effects—mean change (risperidone vs olanzapine, data skewed, high score = poor).
| Adverse effects: 1b.iii. Different adverse effects—mean change (risperidone vs olanzapine, data skewed, high score = poor) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Cardiac—QTc (ms) | ||||
| Sikich 2008 | Riesperidone | 0.5 | 29.5 | 41 |
| Sikich 2008 | Olanzapine | 11.2 | 16.8 | 35 |
| Endocrine—prolactin (mcg/L) | ||||
| Sikich 2008 | Risperidone | 19.5 | 21.5 | 41 |
| Sikich 2008 | Olanzapine | ‐1.5 | 20.2 | 35 |
3.10. Analysis.
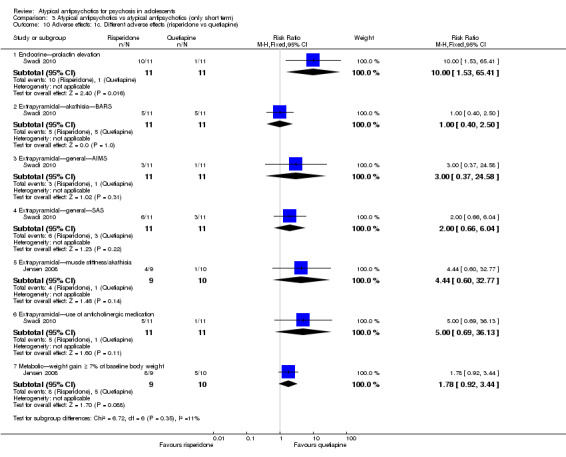
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 10 Adverse effects: 1c. Different adverse effects (risperidone vs quetiapine).
3.11. Analysis.
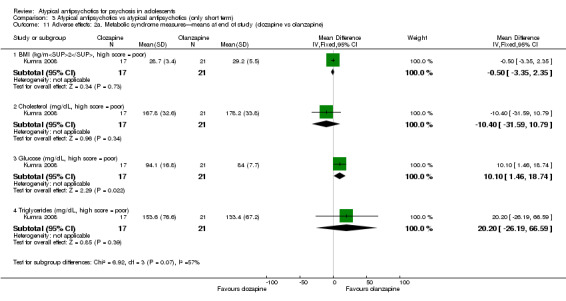
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 11 Adverse effects: 2a. Metabolic syndrome measures—means at end of study (clozapine vs olanzapine).
3.12. Analysis.
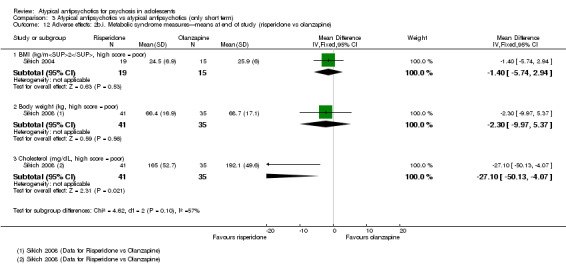
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 12 Adverse effects: 2b.i. Metabolic syndrome measures—means at end of study (risperidone vs olanzapine).
3.13. Analysis.
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 13 Adverse effects: 2b. ii. Metabolic syndrome measures—mean changes (risperidone vs olanzapine, data skewed, high score = poor).
| Adverse effects: 2b. ii. Metabolic syndrome measures—mean changes (risperidone vs olanzapine, data skewed, high score = poor) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| BMI (kg/m2) | ||||
| Sikich 2008 | Risperidone | 1.3 | 1.5 | 41 |
| Sikich 2008 | Olanzapine | 2.2 | 1.2 | 35 |
| Cholesterol (mg/dL) | ||||
| Sikich 2008 | Risperidone | ‐10.2 | 26.7 | 41 |
| Sikich 2008 | Olanzapine | 19.9 | 23.9 | 35 |
| Glucose (mg/dL) | ||||
| Sikich 2008 | Risperidone | 1.2 | 7.3 | 41 |
| Sikich 2008 | Olanzapine | 0.6 | 15.7 | 35 |
| Weight gain (kg) | ||||
| Sikich 2008 | Risperidone | 3.6 | 4 | 41 |
| Sikich 2008 | Olanzapine | 6.1 | 3.6 | 35 |
3.14. Analysis.
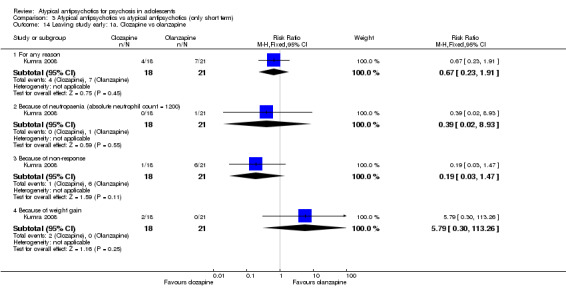
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 14 Leaving study early: 1a. Clozapine vs olanzapine.
3.15. Analysis.
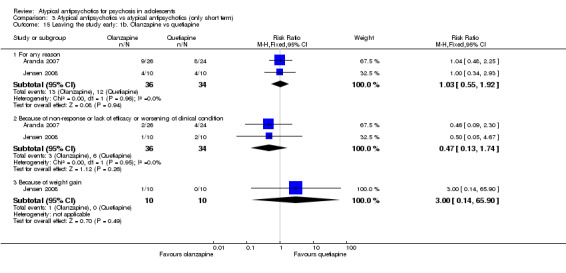
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 15 Leaving the study early: 1b. Olanzapine vs quetiapine.
3.16. Analysis.
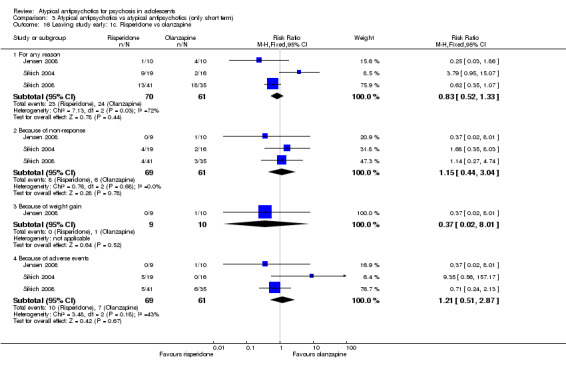
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 16 Leaving study early: 1c. Risperidone vs olanzapine.
3.17. Analysis.
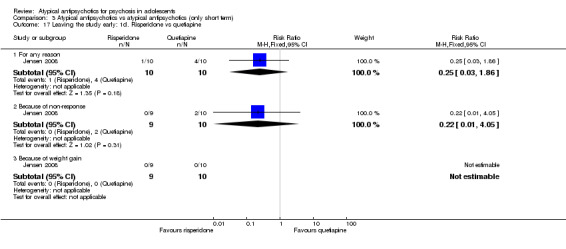
Comparison 3 Atypical antipsychotics vs atypical antipsychotics (only short term), Outcome 17 Leaving the study early: 1d. Risperidone vs quetiapine.
Comparison 4. Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. No response—not sustained response at end of 8 weeks | 1 | 255 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.45, 0.77] |
| 2 Global state: 2a. Mean end point score (CGI‐I, high score = poor) | 3 | 468 | Mean Difference (IV, Fixed, 95% CI) | ‐0.34 [‐0.55, ‐0.13] |
| 3 Global state: 2b. Mean scores—at 6 weeks (aripiprazole 30 mg vs 10 mg) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 Change at CGAS score | 1 | 198 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.29, 0.49] |
| 3.2 Change in CGI‐S score | 1 | 196 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.13, ‐0.07] |
| 3.3 Mean end point CGI‐I score | 1 | 196 | Mean Difference (IV, Fixed, 95% CI) | ‐0.20 [‐0.23, ‐0.17] |
| 4 Mental state: 1. No response | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 ≤ 30% reduction in PANSS score | 1 | 255 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.59, 1.03] |
| 4.2 ≤ 40% improvement in PANSS score | 1 | 255 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.68, 0.98] |
| 4.3 Not achieving remission at 6 weeks on PANSS (aripiprazole 30 mg vs 10 mg) | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.66, 1.25] |
| 5 Mental state: 2. Mean end point scores (high score = poor) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 BPRS | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐4.40 [‐19.20, 10.40] |
| 5.2 PANSS | 2 | 451 | Mean Difference (IV, Fixed, 95% CI) | ‐3.49 [‐7.26, 0.28] |
| 6 Adverse effects: 1. Endrocrine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Prolactin—elevation above normal | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.04, 1.73] |
| 6.2 Prolactin—elevation > 100 ng/mL | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 46.46 [6.50, 332.17] |
| 6.3 Prolactin—symptomatic hyperprolactinaemia | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.70 [0.78, 17.45] |
| 7 Adverse effects: 2. Extrapyramidal | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Any | 1 | 254 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.31 [1.86, 5.87] |
| 7.2 Akathisia | 2 | 459 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.31 [1.46, 7.53] |
| 7.3 Dystonia | 2 | 459 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [1.12, 4.18] |
| 7.4 Oculogyric crisis | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.17 [0.13, 77.01] |
| 7.5 Parkinsonism | 2 | 459 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.32 [1.36, 3.98] |
| 7.6 Tremor | 2 | 459 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.27 [1.78, 10.24] |
| 7.7 Use of antiparkinsonian medications | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.86 [1.91, 12.38] |
| 8 Adverse effects: 3. Metabolic | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Weight gain—any | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [1.07, 1.44] |
| 8.2 Weight gain—moving from overweight to obese | 1 | 42 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.96 [0.50, 7.67] |
| 8.3 Weight gain—"experiencing weight gain as an adverse effect" | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.32 [1.47, 7.49] |
| 9 Adverse effects: 4. Others | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 Central nervous system—somnolence | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.17 [1.68, 5.99] |
| 9.2 Non‐specific—self‐injury and aggression | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.04, 3.34] |
| 9.3 Non‐specific—treatment‐emergent adverse effects | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.97, 1.34] |
| 9.4 Non‐specific—worsening of psychiatric symptoms | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.36, 4.80] |
| 10 Adverse effects: 5. Others (particular to the aripiprazole 30 mg vs 10 mg comparison) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Central nervous system—somnolence | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.96 [1.00, 3.83] |
| 10.2 Extrapyramidal—any adverse effects | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [0.89, 3.11] |
| 10.3 Extrapyramidal—akathisia | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.96 [0.77, 5.02] |
| 10.4 Extrapyramidal—dystonia | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.09, 2.62] |
| 10.5 Extrapyramidal—parkinsonism (SAS) | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.03 [1.17, 3.52] |
| 10.6 Extrapyramidal—tremor | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.88 [1.35, 25.62] |
| 10.7 Metabolic—weight gain of ≥ 5% of body weight | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.36, 1.93] |
| 11 Adverse effects: 6. Laboratory tests (particular to the aripiprazole 30 mg vs 10 mg comparison) | Other data | No numeric data | ||
| 11.1 Change in serum cholesterol concentration (data skewed, high score = poor) | Other data | No numeric data | ||
| 11.2 Change in serum prolactin concentration (data skewed, high score = poor) | Other data | No numeric data | ||
| 12 Leaving study early: 1. Various reasons | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 For any reason | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.52, 1.06] |
| 12.2 Adverse effects | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.28, 2.81] |
| 12.3 Insufficient response | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.45, 1.32] |
| 12.4 Other reasons | 1 | 257 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.18, 3.47] |
| 13 Leaving study early: 2. Ziprasidone 160 mg vs 80 mg | 1 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.22 [0.58, 8.44] |
| 14 Leaving study early: 3. Aripiprazole 30 mg vs aripiprazole 10 mg | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 For any reason | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.60, 2.04] |
| 14.2 Adverse effects | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.17, 1.85] |
| 14.3 Lack of efficacy | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.02, 1.65] |
| 15 Quality of life: mean end point score—at 6 weeks (PQ‐LES‐Q, aripiprazole 30 mg vs aripiprazole 10 mg, data skewed, high score = good) | Other data | No numeric data |
4.1. Analysis.
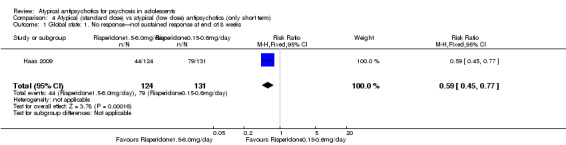
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 1 Global state: 1. No response—not sustained response at end of 8 weeks.
4.2. Analysis.
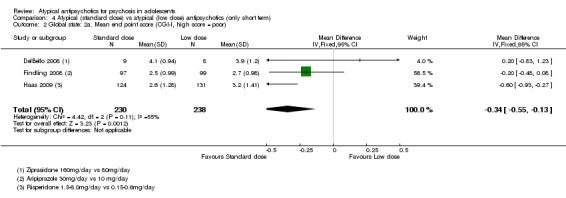
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 2 Global state: 2a. Mean end point score (CGI‐I, high score = poor).
4.3. Analysis.
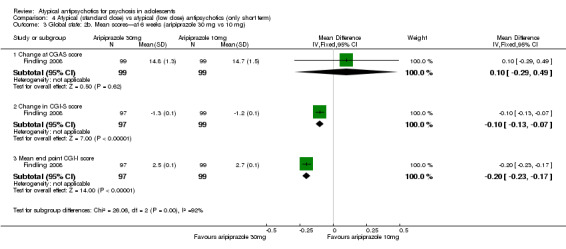
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 3 Global state: 2b. Mean scores—at 6 weeks (aripiprazole 30 mg vs 10 mg).
4.4. Analysis.
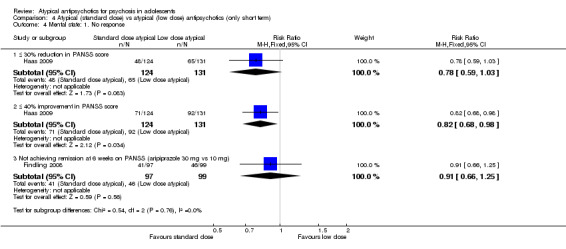
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 4 Mental state: 1. No response.
4.5. Analysis.
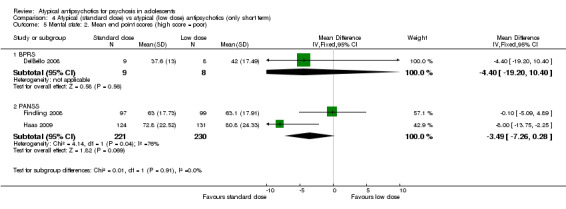
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 5 Mental state: 2. Mean end point scores (high score = poor).
4.6. Analysis.
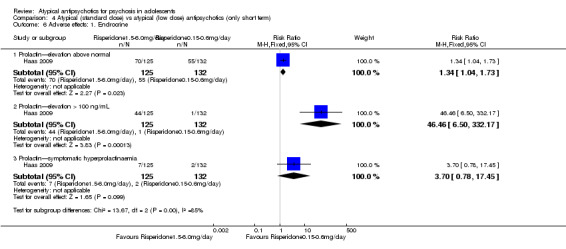
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 6 Adverse effects: 1. Endrocrine.
4.7. Analysis.
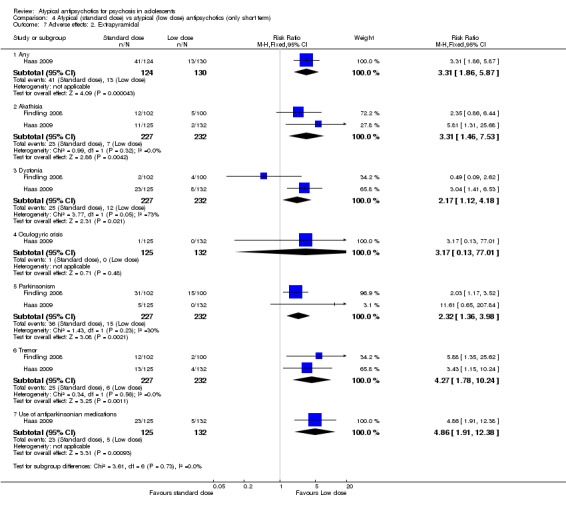
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 7 Adverse effects: 2. Extrapyramidal.
4.8. Analysis.
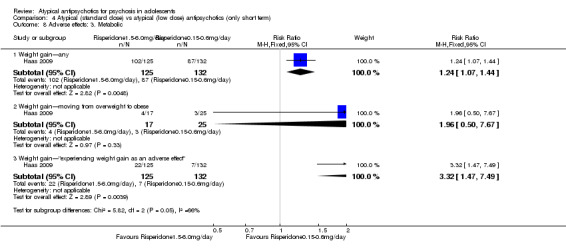
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 8 Adverse effects: 3. Metabolic.
4.9. Analysis.
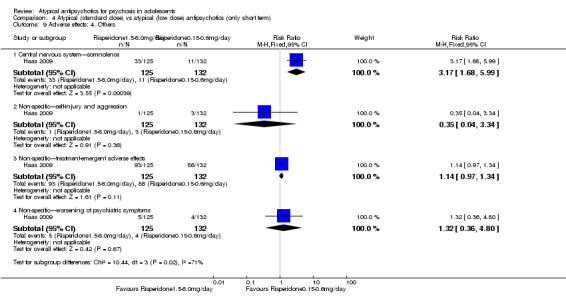
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 9 Adverse effects: 4. Others.
4.10. Analysis.
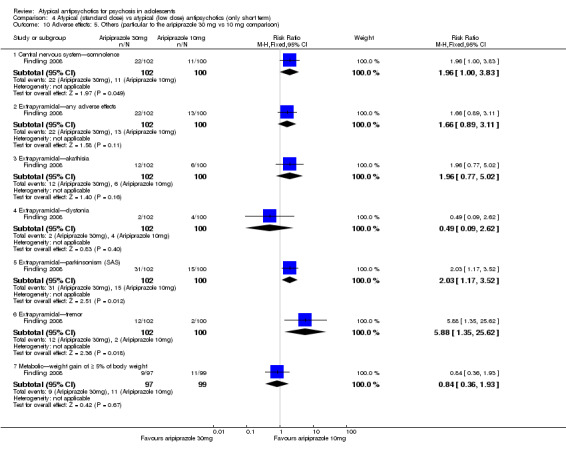
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 10 Adverse effects: 5. Others (particular to the aripiprazole 30 mg vs 10 mg comparison).
4.11. Analysis.
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 11 Adverse effects: 6. Laboratory tests (particular to the aripiprazole 30 mg vs 10 mg comparison).
| Adverse effects: 6. Laboratory tests (particular to the aripiprazole 30 mg vs 10 mg comparison) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Change in serum cholesterol concentration (data skewed, high score = poor) | ||||
| Findling 2008 | Aripiprazole 30mg | ‐5.01 | 23.28 | 95 |
| Findling 2008 | Aripiprazole 10mg | ‐7.43 | 27.99 | 98 |
| Change in serum prolactin concentration (data skewed, high score = poor) | ||||
| Findling 2008 | Aripiprazole 30mg | ‐15.14 | 26.87 | 92 |
| Findling 2008 | Aripiprazole 10mg | ‐11.93 | 23.29 | 98 |
4.12. Analysis.
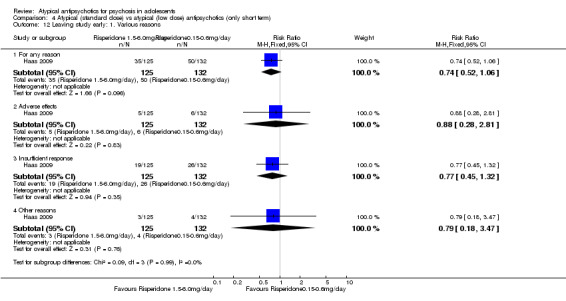
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 12 Leaving study early: 1. Various reasons.
4.13. Analysis.
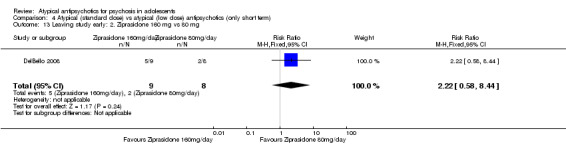
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 13 Leaving study early: 2. Ziprasidone 160 mg vs 80 mg.
4.14. Analysis.
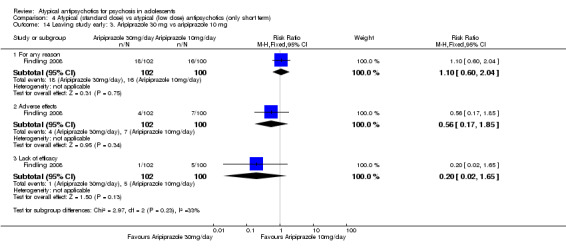
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 14 Leaving study early: 3. Aripiprazole 30 mg vs aripiprazole 10 mg.
4.15. Analysis.
Comparison 4 Atypical (standard dose) vs atypical (low dose) antipsychotics (only short term), Outcome 15 Quality of life: mean end point score—at 6 weeks (PQ‐LES‐Q, aripiprazole 30 mg vs aripiprazole 10 mg, data skewed, high score = good).
| Quality of life: mean end point score—at 6 weeks (PQ‐LES‐Q, aripiprazole 30 mg vs aripiprazole 10 mg, data skewed, high score = good) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Findling 2008 | Aripiprazole 30mg | 50.2 | 90 | 97 |
| Findling 2008 | Aripiprazole 10mg | 50.1 | 90.9 | 99 |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Aranda 2007.
| Methods | Allocation: randomised. Blindness: open. Duration: 6 months. Design: single centre. Country: Spain. | |
| Participants | Diagnosis: psychosis (DSM‐IV). N = 50. Age, years: range 12 to 18, mean age for quetiapine group 16.3 and olanzapine group 15.6. Sex: not stated. Setting: inpatient. | |
| Interventions | 1. Quetiapine mean dose 532.8 mg/d. N = 24. 2. Olanzapine mean dose 9.7 mg/d. N = 26. |
|
| Outcomes | Leaving the study early. Unable to use: Global state: CGI‐S (no SD). Mental state: PANSS (no SD). Neuropsychological test for cognition: WCST (no usable data). Adverse effects (no usable data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described in the report. |
| Allocation concealment (selection bias) | Unclear risk | Not described in the report. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was an open‐label study. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This was an open‐label study. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The issue was partially addressed. |
| Selective reporting (reporting bias) | Unclear risk | The study reported multiple outcomes but partially (e.g. no SD for mean end point PANSS score). |
| Other bias | Unclear risk | This seems to be a well‐designed study, but not all results are reported. |
DelBello 2008.
| Methods | Allocation: randomised. Blindness: open label. Duration: 3 weeks. Design: multi‐centre. Country: USA. | |
| Participants | Diagnosis: schizophrenia and schizoaffective disorder (DSM‐IV‐TR). N = 17. Age, years: range 10 to 17, mean age 14.6. Sex: 11 male, 6 female. Inclusion criteria: diagnosis of schizophrenia or schizoaffective disorder as defined by the DSM‐IV‐TR; diagnosis confirmed by KID‐SCID; BPRS score ≥ 35 with a score ≥ 4 on at least one of the items—unusual thought content, suspiciousness, hallucinations or conceptual disorganisation. Only participants with a body mass index (BMI) between the 5th and 95th percentiles were included. Exclusion criteria: currently on stable well‐tolerated treatment; suspected or established substance‐induced psychotic disorder; treatment with clozapine in the last 12 weeks; a depot antipsychotic in the last 4 weeks or a monoamine oxidase inhibitor (MAOI) in the past 2 weeks; imminent risk of suicide or homicide; IQ ≤ 70; autism or other pervasive developmental disorder; pregnancy, breast‐feeding or unwillingness to use contraceptions; any serious medical or neurological illness; any screening laboratory value that deviated significantly from the reference range; history of cardiac problems, QTc prolongation ≥ 460 ms; or DSM‐IV‐TR‐defined psychoactive substance abuse or dependence within the preceding month. Setting: unclear. | |
| Interventions | 1. Ziprasidone 80 mg/d. N = 8. 2. Ziprasidone 160 mg/d. N = 9. |
|
| Outcomes | Leaving the study. Global state: CGI‐S. Mental state: BPRS‐A. Unable to use: Adverse effects: SARS, AIMS, BARS (no usable data). Laboratory tests (no usable data). |
|
| Notes | The study was divided into two periods: fixed doses of ziprasidone—three weeks; flexible doses—24 weeks. Only outcome data after a fixed‐dose trial were usable. The study was funded by Pfizer Inc. The authors reported separate data for bipolar disorder and schizophrenia/schizoaffective disorder. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details of sequence generation were not mentioned. Because the study population had a narrow age range, the different groups were comparable. |
| Allocation concealment (selection bias) | Unclear risk | Not described in the report. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was an open‐label trial. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This was an open‐label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Authors reported the number of participants who had to discontinue with the study. The primary aim of the study was to test the tolerability of high‐ vs low‐dose regimens of ziprasidone. Measuring efficacy was not the primary aim of this study. Adverse effects experienced by study participants were reported in sufficient detail. |
| Selective reporting (reporting bias) | Low risk | Authors reported many outcome measures, especially adverse effects, in sufficient detail. |
| Other bias | Unclear risk | Study participants were selected from 10 centres and met fairly broad inclusion criteria. This could lead to inconclusive evidence of a particular condition. Authors reported findings separately for bipolar disorder and schizophrenia/schizoaffective disorder. |
Findling 2008.
| Methods | Allocation: randomly assigned. Blindness: double. Duration: 6 weeks. Design: multi‐centre. Country: USA, Europe, South America, Asia, the Carribean, South Africa. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 302. Age, years: range 13 to 17, mean age 15.4 + 1.4. Sex: 171 male, 131 female. Inclusion criteria: diagnosis of schizophrenia as defined by the DSM‐IV; diagnosis of schizophrenia confirmed by an adequately trained clinician (e.g. child psychiatrist) by means of the K‐SADS‐PL, PANSS score ≥ 70. Exclusion criteria: psychiatric comorbidity requiring pharmacotherapy, any evidence of suicide risk. Current or past history of schizoaffective disorder, major depression, mental retardation, neuroleptic malignant syndrome, any neurological disorder except Tourette's syndrome, severe head trauma or unstable medical disorder. Participants with resistant illness to two different antipsychotic drugs, sexually active adolescent boys or girls who did not agree to use contraceptives, positive screens for illegal drugs within 3 months of baseline. Setting: inpatient and outpatient. | |
| Interventions | 1. Placebo. N = 100. 2. Aripiprazole 10 mg/d. N = 100. 3. Aripiprazole 30 mg/d. N = 102. |
|
| Outcomes | Leaving the study. Global state: CGI‐S, CGI‐I, CGAS. Mental state: PANSS. Quality of life: P‐QLES‐Q. Adverse effects: AIMS, BARS, SAS. Biochemistry. Unable to use: Vital signs (no usable data). Electrocardiogram (ECG) (no usable data). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation was stratified and was done separately across three geographical categories. |
| Allocation concealment (selection bias) | Unclear risk | Not described in the report. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This was a double‐blind placebo‐controlled trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This was a double‐blind placebo‐controlled trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Incomplete outcome data were analysed. |
| Selective reporting (reporting bias) | Low risk | Authors reported multiple outcome measures. |
| Other bias | Low risk | This was a well‐designed RCT. |
Haas 2009.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 weeks. Design: multi‐centre. Country: Belgium, Bulgaria, Czech Rep, Estonia, Germany, Poland, Romania, USA. | |
| Participants | Diagnosis: catatonic, disorganised, paranoid, residual, undifferentiated schizophrenia (DSM‐IV), acute episode. N = 257. Age, years: range 13 to 17, mean age 15.6. Sex: 145 male, 112 female. Inclusion criteria: diagnosis of schizophrenia as defined by the DSM‐IV, currently hospitalised with an acute episode (PANSS score 60 to 120). Negative serum pregnancy test. Exclusion criteria: schizophreniform disorder, significant suicidal risk or risk of violence, past history of neuroleptic malignant syndrome, tardive dyskinesia, known or suspected seizure disorder BMI ≤ 5th percentile or BMI > 95th percentile and administration of more than two doses of drug in the drug‐free washout period. Setting: inpatient. | |
| Interventions | 1. Risperidone dose range 1.5 to 6.0 mg/d. N = 125. 2. Risperidone dose range 0.15 to 0.6 mg/d. N = 132. In association with psychotherapy and psychoeducation. |
|
| Outcomes | Leaving the study. Global state: CGI‐S, CGI‐I. Mental state: PANSS. Adverse effects: SAS, AIMS, BARS. Unable to use: Laboratory tests (no usable data). |
|
| Notes | Investigators were required as per protocol to adjust medications up to the maximum tolerated dose over a period of 12 days to ensure that a full dose range would be explored for safety. The dose was to remain stable during the last 4 weeks of the double‐blind period. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated schedule. |
| Allocation concealment (selection bias) | Unclear risk | Not described in the report. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This was a double‐blind study. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This was a double‐blind study. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Incomplete data were accounted for by using intention‐to‐treat analysis. |
| Selective reporting (reporting bias) | Low risk | Supplementary data in journal website gives details of many of the outcome measures. |
| Other bias | Low risk | This was a well‐designed RCT. |
Huo 2007.
| Methods | Allocation: randomised. Blindness: unclear. Duration: 8 weeks. Design: single centre. Country: China. | |
| Participants | Diagnosis: schizophrenia (CCMD‐3). N = 40. Age, years: mean age 14. Sex: 21 male, 19 female. Inclusion criteria: diagnosis of schizophrenia, age ≤ 16 years. Exclusion criteria: patients with organic diseases. Setting: inpatient and outpatient. | |
| Interventions | 1. Risperidone mean dose 3.18 ± 0.66 mg/d, range 2 to 4 mg/d. N = 20. 2. Perphenazine mean dose 16.2 ± 6.4 mg/d, range 10 to 24 mg/d. N = 20. |
|
| Outcomes | Mental state: BPRS. Adverse effects: TESS. | |
| Notes | Average length of illness: 2 to 8 months. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described in the report. |
| Allocation concealment (selection bias) | Unclear risk | Not described in the report. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described in the report. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described in the report. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The results accounted for all participants who started the study. |
| Selective reporting (reporting bias) | Low risk | The study reported all outcomes it had intended to report at the beginning in the study. |
| Other bias | Low risk | Although the study was in Chinese, we found after translation that reporting was done in a clear and easy to understand way. |
Jensen 2008.
| Methods | Allocation: randomised. Blindness: open. Duration: 12 weeks. Design: single centre. Country: USA. | |
| Participants | Diagnosis: schizophrenia, schizoaffective disorder, schizophreniform disorder, psychotic disorder NOS (DSM‐IV). N = 30. Age, years: range 10 to 18, mean age 15.2 ± 2.1. Sex: 20 male, 10 female. Inclusion criteria: boys and girls aged 10 to 18 years, inclusive, with a diagnosis of schizophrenia/schizoaffective disorder, schizophreniform disorder, or psychotic disorder NOS. At least one positive or negative symptom associated with schizophrenia of moderate or greater severity on PANSS that had been present throughout the past 2 weeks. Exclusion criteria: diagnosis of mental retardation, affective disorder (i.e. major depressive disorder or bipolar disorder) with psychotic features, current alcohol or drug dependence or abuse, history of serious adverse reactions or non‐response to an adequate trial of any of the proposed treatments, pregnant or refused to practice contraception, serious and unstable medical condition. Setting: inpatient and outpatient. | |
| Interventions | 1. Risperidone mean dose 3.4 ± 1.5 mg/d, range 1 to 6 mg/d. N = 10. 2. Olanzapine mean dose 14.6 ± 4.6 mg/d, range 5 to 20 mg/d. N = 10. 3. Quetiapine mean dose 611 ± 253.4 mg/d, range 100 to 800 mg/d. N = 10. |
|
| Outcomes | Leaving the study early. Global state: CGI‐S, CGAS. Mental state: PANSS. Adverse effects: AIMS, SAS. Unable to use: Laboratory tests (no usable data). |
|
| Notes | Of 30 participants 27 were inpatients. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random sequence was performed using computer‐generated randomisation schedule. |
| Allocation concealment (selection bias) | Unclear risk | Not described in the report. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was an open‐label study. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This was an open‐label study. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat analysis was done using the 'last observation carried forward' method for participants who did not complete the study. |
| Selective reporting (reporting bias) | Low risk | Outcomes were reported in sufficient detail. |
| Other bias | High risk | As the study was open label, it was probably subjected to interviewer's bias or researcher's bias. |
Kryzhanovskaya 2009.
| Methods | Allocation: randomised. Blindness: double. Duration: 6 weeks. Design: multi‐centre. Country: USA, Russia. | |
| Participants | Diagnosis: paranoid, disorganised, catatonic, undifferentiated, residual schizophrenia (DSM‐IV‐TR). N = 107. Age, years: range 13 to 17, mean age 16. Sex: 75 male, 32 female. Inclusion criteria: diagnosis confirmed by the K‐SADS‐PL. Baseline score ≥ 35 on the anchored version of the BPRS‐C (21 items), with a score of 3 or higher on at least one of the following BPRS‐C items at enrolment and randomisation: hallucinations, delusions or peculiar fantasies. Exclusion criteria: previous participation in a clinical trial of oral olanzapine. Treatment within 30 days of the trial with a drug without regulatory approval for any indication. Previous non‐response to an adequate dose/duration of olanzapine treatment. Pregnancy, nursing, or refusal to practise contraception (for females). Acute or unstable medical condition. Prolactin levels > 200 ng/L at randomisation. DSM‐IV‐TR substance dependence within 30 days. Current diagnosis of a comorbid psychiatric or developmental disorder. Setting: inpatient and outpatient. | |
| Interventions | 1. Olanzapine mean dose 11.1 ± 4.0 mg/d, range 2.5 to 20 mg/d. N = 72. 2. Placebo. N = 35. |
|
| Outcomes | Leaving the study early. Global state: CGI‐I, CGI‐S. Mental state: BPRS‐C, PANSS, OAS. Adverse effects: SAS, BARS, AIMS. Unable to use: Laboratory tests (no usable data). |
|
| Notes | Mean age at onset of illness ∽ 13 years. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described in the report. |
| Allocation concealment (selection bias) | Unclear risk | Not described in the report. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | There was a double‐blind phase followed by an open‐label phase. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | There was a double‐blind phase followed by an open‐label phase. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Appropriate intention‐to‐treat analysis was performed. |
| Selective reporting (reporting bias) | Low risk | Multiple outcome measures were reported in detail. |
| Other bias | Low risk | This was a well‐designed RCT. |
Kumra 1996.
| Methods | Allocation: randomised. Blindness: double. Duration: 6 weeks Design: single centre. Country: USA. | |
| Participants | Diagnosis: disorganised, undifferentiated, paranoid schizophrenia (DSM‐III‐R). N = 21. Age, years: range 6 to 18, mean 14 ± 2.3 . Sex: 11 male, 10 female. Inclusion criteria: diagnosis of schizophrenia as defined by the DSM‐III‐R, with documented psychotic symptoms by the age of 12 years; intolerance, non‐response or both to at least two different neuroleptic drugs; IQ of 70 or greater. In other words, this study included only adolescents with treatment‐resistant schizophrenia. Exclusion criteria: neurological or medical disease. Setting: inpatient. | |
| Interventions | 1. Clozapine mean dose 176 ± 149 mg/d, range 25 to 525 mg/d. N = 10. 2. Haloperidol mean dose 16 ± 8 mg/d, range 7 to 27 mg/d. N = 11. |
|
| Outcomes | Leaving the study early. Global state: CGI, CGAS. Mental state: BPRS, SANS, SAPS, B‐HPRS). Adverse effects: SAS, TESS, AIMS. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation sequence was done using table of random numbers. |
| Allocation concealment (selection bias) | Unclear risk | Not described in the report. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Raters, treating physicians and nurses were blind to the study status. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Raters were blind to the study status. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Appropriate intention‐to‐treat analysis was conducted. |
| Selective reporting (reporting bias) | High risk | Positive findings were highlighted in the 'Results' section. The high incidence of neutropenia in the clozapine group has been reported but not adequately discussed. |
| Other bias | High risk | This is a well‐designed RCT for treatment‐resistant schizophrenia in childhood, but the total number of children recruited is small. |
Kumra 2008.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks. Design: multi‐centre. Country: USA. | |
| Participants | Diagnosis: treatment‐resistant schizophrenia, schizoaffective disorder (DSM‐IV). N = 39. Age, years: range 10 to 18, mean age 15.6 (2.1). Sex: 21 male, 18 female. Inclusion criteria: diagnosis of schizophrenia or schizoaffective disorder based on a structured interview, non‐response to at least two different neuroleptic drugs, baseline BPRS ≥ 35. In other words, this study included only adolescents with treatment‐resistant schizophrenia. Exclusion criteria: premorbid diagnosis of mental retardation (IQ < 70); history of serious adverse reactions to proposed treatments; pregnancy; serious or unstable medical condition; failure to respond to adequate trials of clozapine (≥ 300 mg/d for 12 weeks) or olanzapine (≥ 20 mg/d for 8 weeks). Setting: inpatient. | |
| Interventions | 1. Clozapine mean dose 403.1 ± 201.8 mg/d, range 50 to 700 mg/d. N = 18. 2. "High‐dose" olanzapine mean dose 26.2 ± 6.5 mg/d, range 10 to 30 mg/d. N = 21. |
|
| Outcomes | Leaving the study early. Global state: CGI, CGAS. Mental state: BPRS, SANS. Adverse effects: TESS, AIMS. Laboratory tests. |
|
| Notes | Age of onset of psychosis, years: 12.7 ± 2.4 (clozapine group); 11.75 ± 3.2 (olanzapine group). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation sequence was followed. |
| Allocation concealment (selection bias) | Low risk | Adequate attempts were made to maintain allocation concealment (using numbered containers, centralised telephone, etc). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This was a double‐blind RCT, but the details of blinding and how it was maintained had not been described in the report. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This was a double‐blind RCT, but the details of blinding and how it was maintained had not been described in the report. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Appropriate intention‐to‐treat analysis was performed. All participants who had been randomly assigned were included. in the analysis |
| Selective reporting (reporting bias) | Low risk | Adequate numbers of outcome measures were reported. |
| Other bias | Low risk | This was a well‐designed RCT. |
Sikich 2004.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 weeks. Design: multi‐centre. Country: North America. | |
| Participants | Diagnosis: psychotic disorder (K‐SADS‐PL,SCID). N = 51. Age, years: range 8 to 19; mean age 14 years 8 months. Sex: 30 male, 20 female. Inclusion criteria: at least one positive psychotic symptom of moderate or greater severity on the BPRS‐C, present for two weeks, IQ > 69. Exclusions: acute substance intoxication or withdrawal; history of adverse reactions or non‐response to study medications; prior diagnosis of a pervasive developmental disorder; serious medical illness; pregnancy or refusal to practise contraception; imminent risk of harm to self or others. Setting: inpatient and outpatient. | |
| Interventions | 1. Risperidone mean dose 3.3 mg/d. N = 19. 2. Olanzapine mean dose 12.3 mg/d. N = 16. 3. Haloperidol mean dose 5.3 mg/d. N = 15. |
|
| Outcomes | Leaving the study early. Global state: CGI, CGI‐S. Mental state : BPRS, CPRS. Adverse effects: AIMS, SAS. Unable to use: Laboratory tests (no usable data). |
|
| Notes | 36% discontinuation rate. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation schedule, stratified by age. |
| Allocation concealment (selection bias) | Unclear risk | Not adequately described in the study. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This was a double‐blind study, but the details of how blinding was maintained were not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | As the study compares drugs from different classes, the raters could have made a guess about young people being on haloperidol. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who were randomly assigned were included in the analysis. |
| Selective reporting (reporting bias) | Unclear risk | Many different outcome measures were reported. |
| Other bias | High risk | The analysis did not use correction techniques to take into account multiple analyses on a small sample. The authors justified this by stating that these analyses were 'exploratory' rather than 'inferential'. |
Sikich 2008.
| Methods | Allocation: randomised. Blindness: double. Duration: 8 weeks. Design: multi‐centre. Country: North America. | |
| Participants | Diagnosis: schizophrenia, schizoaffective disorder or schizophreniform disorder (DSM‐IV). N = 116. Age, years: 8 to 19. Sex: 75 male, 41 female. Inclusion criteria: current positive psychotic symptoms of at least moderate intensity on PANSS. Exclusions: prior evidence of mental retardation; current major depressive episode; active substance abuse; acute substance intoxication or withdrawal; history of intolerance or non‐response to study medications; history of an adequate trial of any study medications; imminent risk of harm to self or others Setting: inpatient and outpatient. | |
| Interventions | 1. Molindone mean dose 59.9 ± 33.5 mg/d. N = 40. 2. Olanzapine mean dose 11.4 ± 5.0 mg/d. N = 35. 3. Risperidone mean dose 2.8 ± 1.4 mg/d. N = 41. |
|
| Outcomes | Leaving the study early. Global state: CGI, Adult and Child Functional Assessment Scale. Mental state: PANSS, BPRS‐C. Adverse effects: SARS, BARS, AIMS. Laboratory tests. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not adequately described in the study. |
| Allocation concealment (selection bias) | Unclear risk | Not adequately described in the study. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This was a double‐blind study, and blinding was maintained even after discontinuation of one of the arms (olanzapine group) of the study for ethical reasons. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding was maintained even after discontinuation of one of the arms of the study. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who had been randomly assigned were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | Multiple outcome measures are described in sufficient detail. |
| Other bias | Low risk | This is a well‐designed RCT. |
Swadi 2010.
| Methods | Allocation: randomised. Blindness: single blind (open label with blind midpoint and end point assessments). Duration: 6 weeks. Design: single centre. Country: New Zealand. | |
| Participants | Diagnosis: first‐onset psychotic disorder or mood disorder with psychotic features (DSM‐IV). N = 22. Age, years: range 15 to 18. Sex: 13 male, 9 female. Inclusion criteria: individuals younger than 19 years of age, with a first‐onset psychotic disorder or a mood disorder with psychotic features according to DSM‐IV. Exclusion criteria: individuals with alcohol or substance dependence not in full remission and persons who had received earlier treatment with atypical antipsychotic drugs. Setting: inpatient. | |
| Interventions | 1. Risperidone mean dose 2.9 mg/d, range 1.5 to 5 mg/d. N = 11. 2. Quetiapine mean dose 607 mg/d, range 100 to 800 mg/d. N = 11. |
|
| Outcomes | Global state: CGI‐S. Mental state: PANSS, BPRS, HAM‐D, YMRS. Adverse effects: AIMS, SARS, BARS. Laboratory tests. | |
| Notes | This is a well‐designed study involving a small number of participants from a single centre. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation process, four strata. |
| Allocation concealment (selection bias) | Unclear risk | Not adequately described in the study. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study with blind midpoint and end point assessments. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label study with blind midpoint and end point assessments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Clear flow chart for study patients accounting for all participants. |
| Selective reporting (reporting bias) | Low risk | No source of selective reporting could be found. |
| Other bias | Unclear risk | Lack of double blindness puts the study under suspicion of further biases. |
Xiong 2004.
| Methods | Allocation: randomised. Blindness: not clear. Duration: 8 weeks. Design: single centre. Country: China. | |
| Participants | Diagnosis: childhood‐onset schizophrenia (CCMD‐2‐R). N = 60. Age, years: 7 to 16, mean age ∽ 14. Sex: 34 male, 26 female. Inclusion criteria: children with the diagnosis of schizophrenia according to CCMD‐2‐R should be between 7 and 16 years of age with no physical problems and no organic neurological disease. Setting: inpatient. | |
| Interventions | 1. Risperidone dose range 0.5 to 5 mg/d. N = 30. 2. Chlorpromazine dose range 50 to 400 mg/d. N = 30. |
|
| Outcomes | Mental state: BPRS. Adverse effects: TESS. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not adequately described in the study. |
| Allocation concealment (selection bias) | Unclear risk | Not adequately described in the study. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding not clear. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding not clear. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data reported for all participants. |
| Selective reporting (reporting bias) | Low risk | The study reported all of the outcomes that it had intended to report at the start of study. |
| Other bias | Unclear risk | Unclear blinding makes this study prone to further biases. |
AIMS—Abnormal Involuntary Movement Scale. BARS—Barnes Akathisia Rating Scale. B‐HPRS—Bunney‐Hamburg Psychosis Rating Scale. BPRS—Brief Psychiatric Rating Scale. BPRS‐C—Brief Psychiatric Rating Scale for Children. CBCL—Child Behaviour Check List.
CCMD‐2‐R—Chinese Classification of Mental Disorders, Second Edition, Revised.
CCMD‐3—Chinese Classification of Mental Disorders, Third Edition. CGAS—Children's Global Assessment Scale. CGI—Clinical Global Impressions scale.
DSM‐III‐R—Diagnostic and Statistical Manual of Mental Disorders, Third Edition, Revised.
DSM‐IV—Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition.
DSM‐IV‐TR—Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. GAS—Global Assessment of Functioning. K‐SADS‐P—Schedule for Affective Disorders and Schizophrenia for School‐Age Children.
K‐SADS‐PL—Schedule for Affective Disorders and Schizophrenia for School‐Age Children—Present and Lifetime Version.
KID‐SCID—Childhood Disorders Version of the Structured Clinical Interview for DSM‐IV Disorders. NOSIE—Nurses' Observation Scale for Inpatient Evaluation. OAS—Overt Agression Scale. PANSS—Positive and Negative Syndrome Scale. P‐QLES‐Q—Paediatric Quality of Life Enjoyment and Satisfaction Questionnaire. SAS—Simpson Angus Scale. S‐ANRS—Simpson‐Angus Neurological Rating Scale. S‐AEPS—Simpson‐Angus Extrapyramidal Symptoms Scale. SANS—Scale for the Assessment of Negative Symptoms. SAPS—Scale for the assessment of Positive Symptoms. SCID—Structured Clinical Interview for DSM‐IV. STESS—Subjective Treatment Emergent Symptoms Scale. TESS—Treatment Emergent Symptoms Scale. WCST—Wisconsin Card Sorting Test.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Amminger 2006 | Allocation: randomised. Participants: individuals (13 to 24 years) at ultra‐high risk (UHR) for psychosis Interventions: omega‐3 fatty acids vs placebo. Outcomes: no separate data reported for 13 to 17 years age group. |
| Antropov 1981 | Allocation: not randomised, open‐label study. |
| Berger 2007 | Allocation: randomised. Participants: people in first psychotic episode, drug naive, age range 15 to 25 years. Interventions: 200 mg/d vs 400 mg/d quetiapine fumarate. Outcomes: no separate data reported for 15 to 17 years age group. |
| Bertelsen 2005 | Allocation: randomised. Participants: people with first episode of schizophrenia or psychosis, age range 18 to 45 years. |
| Buchsbaum 2007 | Allocation: randomised. Participants: people with a diagnosis of psychosis NOS (DSM‐IV), age range 13 to 21 years. Interventions: olanzapine vs haloperidol. Outcomes: no usable data. |
| Chen 2007 | Allocation: randomised. Participants: children and adolescents with first episode of schizophrenia, most participants younger than 13 years of age. |
| Davidson 2004 | Allocation: randomised. Participants: people with first episode and early psychosis, no age range given. |
| Gao 2007 | Allocation: randomised. Participants: children and adolescents with schizophrenia, most participants younger than 13 years of age. |
| Jenner 2004 | Allocation: not randomised, retrospective study. |
| Johnson 2004 | Allocation: randomised. Participants: adolescents with diagnosis of schizophrenia (DSM‐IV), age range 13 to 17 years. Interventions: risperidone vs placebo. Outcomes: no data provided, no usable data. |
| Killackey 2006 | Allocation: randomised. Participants: people with first episode of psychosis, age range not clear. |
| Klier 2005 | Allocation: randomised. Participants: people with diagnosis of "at‐risk‐mental‐state" for psychosis, age range 13 to 25 years. Interventions: omega‐3 fatty acids and standard care vs standard care. Outcomes: no usable data. |
| Leblanc 2006 | Allocation: randomised. Participants: people with schizophrenia or related psychosis, age range not clear. |
| Leclerc 2006 | Allocation: randomised. Participants: people with first psychotic episode, age range not clear. |
| Liang 2003 | Allocation: not randomised, blinding unclear. |
| Linszen 2006 | Allocation: randomised. Participants: people with schizophrenia, mean age at onset of psychosis 19.3 years, focus on young adults rather than adolescents. |
| Lv 2004 | Allocation: randomised. Participants: adolescents with schizophrenia. Intervention: clozapine vs risperidone. Outcomes: insufficient data, unable to use data. |
| Malik 1980 | Allocation: randomised. Participants: adolescents with a diagnosis of acute, catatonic, paranoid or simple schizophrenia; age range 14 to 19 years (mean age 17 years). Interventions: loxapine vs trifluoperazine. Reason for exclusion: This study will be considered for the other review by the authors on typical antipsychotic medications. We excluded this study, done in the 1970s, from the current review, as this study did not provide any atypical antipsychotic medication in any of the arms. |
| Mathai 2004 | Allocation: randomised. Participants: children with psychiatric disorders, no specific diagnosis, age range 4 to 14 years (mean age 9 years). |
| McConville 2003 | Allocation: not randomised, open‐label study. |
| McGlashan 2003 | Allocation: randomised. Participants: people meeting criteria of schizophrenia prodrome, most study participants not within 13 to 17 years age range. |
| McGorry 2007 | Allocation: randomised. Participants: people meeting ultra‐high risk (UHR) criteria for psychotic disorder, mean age 18.36 years, most study participants not within 13 to 17 years age range. |
| Newton 2005 | Allocation: not randomised, open‐label study. |
| Otsuka 2005 | Allocation: randomised. Participants: people with diagnosis of schizophrenia spectrum disorder or bipolar spectrum disorder, age range 13 to 17 years. Interventions: aripiprazole. Outcomes: a phase II study, insufficient data, unable to use data. |
| Power 2004 | Allocation: randomised. Participants: people with first episode of psychosis, mean age 26.3 ± 6.2 years, most participants not within 13 to 17 years age range. |
| Schepp 1999 | Allocation: randomised. Participants: people with a DSM‐IV diagnosis of schizophrenia, age range 15 to 19 years. Interventions: behavioural self‐management study. Outcomes: insufficient data, unable to use data. |
| Sela 2003 | Allocation: not randomised. |
| Stain 2006 | Allocation: randomised. Participants: youths at risk for psychosis, age range not clear. |
| Tandon 2005 | Allocation: randomised. Participants: hospitalised children and adolescents with a diagnosis of acute or transient psychotic disorder. Interventions: risperidone and placebo. Outcomes: no data comparing groups available from abstracts, no response from corresponding author to our request for full paper. |
| Ueland 2004 | Allocation: randomised. Participants: individuals with a diagnosis of schizophrenia, schizoaffective disorder, schizotypal personality disorder, bipolar disorder, psychotic disorder NOS, major depressive disorder (DSM‐IV); age range 12 to 18 years (mean 15.3 years). Interventions: cognitive remediation programme (CRP) + psychoeducational treatment programme (PTP) vs PTP. |
| van Bruggen 2003 | Allocation: randomised. Participants: adolescents and young adults with first or second episode of schizophrenia, schizophreniform or schizoaffective disorder (DSM‐IV), age range 16 to 28 years. Interventions: olanzapine vs risperidone. Outcomes: no separate data reported for 16 to 17‐year‐olds. Corresponding author could not be contacted. |
| van Nimwegen 2006 | Allocation: randomised. Participants: people with schizophrenia, schizophreniform or schizoaffective disorder (DSM‐IV), mean age ∼ 25 years. |
| Versiani 1978 | Allocation: randomised. Participants: adolescents with a diagnosis of schizophrenia (DSM‐II), age range 13 to 18 years (mean age 16.1 years). Interventions: loxapine vs haloperidol. Reason for exclusion: This study will be considered by the authors for the other review on typical antipsychotic medications. We excluded this study, done in the 1970s, from the current review, as this study did not provide any atypical antipsychotic medication in any of the arms. |
| Wang 2007 | Allocation: randomised. Participants: children and adolescents with schizophrenia, most participants younger than 13 years. |
| Wykes 2007 | Allocation: randomised. Participants: adolescents and adults with a diagnosis of schizophrenia (DSM‐IV), age range 14 to 22 years (mean age 18.2 years). Interventions: cognitive remediation therapy (CRT) vs treatment as usual, details of treatment as usual not given. |
| Xiu 2004 | Allocation: randomised. Participants: people with schizophrenia, mean age ∼ 27 years. |
| Yang 2007 | Allocation: randomised. Participants: people with schizophrenia, mean age ∼ 11 years. |
| Yao 2003 | Allocation: randomised. Participants: children with schizophrenia, mean age ∼ 11 years. |
| Yi 2006 | Allocation: randomised. Participants: children and adolescents with schizophrenia, most study participants younger than 13 years. |
| Zhang 2007 | Allocation: randomised. Participants: children with mean age 10.07 ± 2.35 years. |
| Zhou 2007 | Allocation: randomised. Participants: children and adolescents with schizophrenia, most study participants younger than 13 years. |
| 杨玲 2004 | Allocation: randomised. Participants: children and adolescents with schizophrenia, age range 6 to 14 years. Interventions: chlorpromazine vs clozapine vs risperidone. Outcomes: no separate data given for 13 to 14 years age group. |
| 谭友果 2002 | Allocation: not randomised, blinding not stated. |
Characteristics of ongoing studies [ordered by study ID]
Alaghband‐rad 2006.
| Trial name or title | Management of first episode of psychoses in Iran: unique features and challenges. |
| Methods | Randomised. |
| Participants | Drug‐naive inpatients, aged 15 to 60 years, who were diagnosed with first episode of psychosis . |
| Interventions | Treatment as usual (TAU) vs standard telephone follow‐up (ST‐TF) vs standard‐home visit (ST‐HV). |
| Outcomes | Comprehensive batteries of clinical ratings, cognitive and neuropsychological tests. |
| Starting date | 2006. |
| Contact information | rad@ams.ac.ir |
| Notes | Corresponding author did not respond to our enquiry about the study Another study under the same study ID includes adolescents (15 to 18 years) as participants. It seems highly likely that this is part of the main study, the characteristics of which are mentioned above. |
AstraZeneca 2004.
| Trial name or title | A 6‐week, multi‐centre, randomised, double‐blind, parallel‐group, placebo‐controlled, phase 3b study of the efficacy and safety of quetiapine fumarate (Seroquel™) immediate‐release tablets compared with placebo in adolescents with schizophrenia. |
| Methods | Randomised. |
| Participants | Males and females aged 13 to 17 years with documented clinical diagnosis of schizophrenia. |
| Interventions | Quetiapine fumarate (Seroquel™) vs placebo. |
| Outcomes | PANSS, level of functioning, safety, efficacy, tolerability, hostility, aggression. |
| Starting date | July 2004. |
| Contact information | ClinicalTrials.gov identifier NCT00090324. AstraZeneca Information Center (8 AM to 7 PM EST). Tel 001‐800‐236‐9933. |
| Notes |
AstraZeneca 2005.
| Trial name or title | A 26‐week, multi‐centre, open‐label, phase 3b study of the safety and tolerability of quetiapine fumarate (Seroquel™) immediate‐release tablets in daily doses of 400 mg to 800 mg in children and adolescents with bipolar I disorder and in adolescents with schizophrenia. |
| Methods | Randomised. |
| Participants | Males and females aged 10 to 17 years with documented clinical diagnosis of schizophrenia or bipolar I disorder. |
| Interventions | Quetiapine fumarate (Seroquel™) 400 mg/d versus 800 mg/d. |
| Outcomes | Adverse events (AEs); rate of participant withdrawal due to AEs; changes in clinical laboratory test results and SARS, BARS and AIM scores; changes in menses for female participants; changes in weight and BMI; changes in CGAS. |
| Starting date | July 2004. |
| Contact information | ClinicalTrials.gov identifier NCT00227305. AstraZeneca Information Center (8 AM to 7 PM EST). Tel 001‐800‐236‐9933. |
| Notes |
Bechdolf 2007.
| Trial name or title | Development and pilot evaluation of modified cognitive behavioural therapy for adolescents with early‐onset psychosis. |
| Methods | Randomised. |
| Participants | Males and females aged 14 to 18 years with DSM‐IV diagnosis of schizophrenia, schizophreniform or schizoaffective disorder, delusional disorder. |
| Interventions | Modified cognitive behavioural therapy (mCBT) + treatment as usual (TAU) vs TAU. |
| Outcomes | PANSS, social functioning (GAF), suicide, suicide attempts, rehospitalisation, severe depressive symptom exacerbation, quality of life. |
| Starting date | May 2007. |
| Contact information | ClinicalTrials.gov identifier NCT00465920. Andreas Bechdolf, PD, Dr; +49 221 478 3869; andreas.bechdolf@uk‐koeln.de. Bettina Pohlmann, Dr; + 49 221 478 3870; bettina.pohlmann@uk‐koeln.de. |
| Notes | mCBT is an individual outpatient treatment consisting of 20 sessions and 5 psychoeducational sessions with parents. |
Pfizer 2005.
| Trial name or title | Six‐week, double‐blind, placebo‐controlled phase III trial evaluating the efficacy, safety and pharmacokinetics of flexible doses of oral ziprasidone in adolescent participants with schizophrenia. |
| Methods | Randomised. |
| Participants | Males and females aged 13 to 17 years with DSM‐IV diagnosis of schizophrenia. |
| Interventions | Ziprasidone vs placebo. |
| Outcomes | BPRS, PANSS, CGI. |
| Starting date | April 2006. |
| Contact information | ClinicalTrials.gov Identifier: NCT00257192. United States: Food and Drug Administration. |
| Notes |
Differences between protocol and review
Addition of text for risk ratio calculation, to reflect changes in the template methodology of the Cochrane Schizophrenia Group
Updated approach to missing data section, reflecting new methods in the CSG template and the methods we employed when we extracted and analysed the data. Previous text for comparison is provided in Appendix 1
Contributions of authors
Ajit Kumar—contributed to writing the protocol, screening various studies, extracting data and writing the review.
Soumitra S Datta—contributed to writing the protocol, screening various studies, extracting data and writing the review.
Stephen D Wright—contributed to writing the protocol.
Vivek A Furtado—contributed to writing the protocol and extracting data.
Paul SS Russell—contributed to writing the protocol.
Sources of support
Internal sources
-
South London and Maudsley NHS Foundation Trust, London, UK.
Soumitra Shankar Datta worked as a substantive consultant child psychiatrist at the South London and Maudsley NHS Foundation Trust when the work on this review was completed. Soumitra acknowledges the support provided by his employers (South London and Maudsley NHS Foundation Trust) in terms of freeing up protected time for research.
-
Institute of Psychiatry, King's College, London, UK.
Soumitra Shankar Datta has an honorary research position with the Institute of Psychiatry, London. This was of immense benefit in terms of gaining access to various published literature on the topic.
External sources
No sources of support supplied
Declarations of interest
None known.
New
References
References to studies included in this review
Aranda 2007 {published data only}
- Aranda OR, Zabala A, Bombn I, Burdalo M, Sancho AR, Moreno D, et al. Quetiapine and olanzapine cognitive efficacy in adolescents with early onset psychosis. Schizophrenia Bulletin 2007;33(2):456. [Google Scholar]
- Parellada MJ, Moreno D, Ruiz‐Sancho A, Medina O, Arango C. Open‐label randomized trial comparing the efficacy and tolerability of quetiapine and olanzapine in first episode psychosis in adolescents. Journal of the European College of Neuropsychopharmacology 2006;16(Suppl 4):S377. [P.3.a.025] [Google Scholar]
- Robles O, Zabala A, Parellada MJ, Ruiz A, Moreno MD, Burdalo MT, et al. Cognitive efficacy of quetiapine and olanzapine in early onset first episode psychosis. Journal of the European College of Neuropsychopharmacology 2006;16(Suppl 4):S381. [P.3.a.032] [Google Scholar]
DelBello 2008 {published data only}
- DelBello MP, Versavel M, Ice K, Keller D, Miceli J. Tolerability of oral ziprasidone in children and adolescents with bipolar mania, schizophrenia, or schizoaffective disorder. Journal of Child and Adolescent Psychopharmacology 2008;18(5):491‐9. [PUBMED: 18928413] [DOI] [PubMed] [Google Scholar]
- DelBello MP, Versavel M, Ice K, Keller D, Miceli J. Ziprasidone dosing study in pediatric patients with bipolar disorder, schizophrenia or schizoaffective disorder. 13th Biennial Winter Workshop on Schizophrenia Research, 2006 February 4‐10, Davos, Switzerland. [8894]
Findling 2008 {published data only}
- Carson WH, Nyilas M, Forbes RA, Pikalov A, McQuade RD, Owen R, et al. A 6‐week study of the efficacy and tolerability of aripiprazole in pediatric patients with schizophrenia. Schizophrenia Research 2008;98:41‐2. [8894] [Google Scholar]
- Carson WH, Nyilas M, Forbes RA, Pikatov A, McQuade RD, Owen R, et al. A 6‐week study of the efficacy and tolerability of aripiprazole in pediatric patients with schizophrenia. 14th Biennial Winter Workshop on Schizophrenia and Bipolar Disorders, 2008 February 3‐7, Montreux, Switzerland. [8894]
- Findling RL, Robb A, Nyilas M, Forbes RA, Jin N, Ivanova S, et al. A multiple‐center, randomized, double‐blind, placebo‐controlled study of oral aripiprazole for treatment of adolescents with schizophrenia. The American Journal of Psychiatry 2008;165(11):1432‐41. [PUBMED: 18765484] [DOI] [PubMed] [Google Scholar]
- Nyilas M, Findling RL, Johnson B, Forbes RA, Pikalov A, Marcus R, et al. Efficacy and tolerability of aripiprazole in adolescents with schizophrenia. 46th Annual Meeting of the American College of Neuropsychopharmacology, 2007 December 9‐13, Boca Raton, Florida, USA. [8894]
- Otsuka Pharmaceutical Development & Commercialization Inc. Aripiprazole in adolescents with schizophrenia, 2005. http:www.clinicaltrials.gov.
Haas 2009 {published data only}
- Haas M, Eerdekens M, Kushner S, Singer J, Augustyns I, Quiroz J, et al. Efficacy, safety and tolerability of two dosing regimens in adolescent schizophrenia: double‐blind study. The British Journal of Psychiatry 2009;194(2):158‐64. [PUBMED: 19182179] [DOI] [PubMed] [Google Scholar]
Huo 2007 {published data only}
- Huo W‐H, Ma Y‐B, Li G‐Z. A controlled study of risperidone in child schizophrenia [利培酮治疗儿童少年精神分裂症的对照研究]. Medical Journal of Chinese People's Health [中国民康医学] 2007;19(11):472‐3. [CHINESE: Academic Journals] [Google Scholar]
Jensen 2008 {published data only}
- Jensen J, Sculz SC. A comparative study of new medications for psychosis in adolescents, 2005. http:www.clinicaltrials.gov.
- Jensen JB, Kumra S, Leitten W, Oberstar J, Anjum A, White T, et al. A comparative pilot study of second‐generation antipsychotics in children and adolescents with schizophrenia‐spectrum disorders. Journal of Child and Adolescent Psychopharmacology 2008;18(4):317‐26. [PUBMED: 18759641] [DOI] [PubMed] [Google Scholar]
- Jensen JB, Leitten W, Wozniak J, Anjum A, White T, Oberstar J, et al. Efficacy and tolerability of atypical antipsychotics in adolescents with psychosis. Schizophrenia Bulletin 2007;33(2):434. [Google Scholar]
Kryzhanovskaya 2009 {published data only}
- Dittmann RW, Krzyhanovskaya L, Schulz C, McDougle CJ, Frazier JA, Robertson‐Plouch C, et al. Results from a double‐blind, placebo‐controlled study of olanzapine in adolescents with schizophrenia. Schizophrenia Research 2006;86(Suppl 1):S133. [Google Scholar]
- Eli Lilly, Company. Olanzapine versus placebo in the treatment of adolescents with schizophrenia. Eli Lilly and Company Clinical Trial Registry, 2002. [4066]
- Eli Lilly, Company. Olanzapine versus placebo in the treatment of adolescents with schizophrenia, 2003. http:www.clinicaltrials.gov.
- Joan S, Earley WR, Kryzhanovskaya L, Miner C, Dittmann RW. Olanzapine in the treatment of schizophrenia in adolescents: an ongoing double‐blind placebo‐controlled study. Schizophrenia Bulletin 2005;31:489. [117756] [Google Scholar]
- Kryzhanovskaya L, Schulz C, McDougle CJ, Frazier JA, Dittmann RW, Robertson‐Plouch C, et al. A double‐blind, placebo‐controlled study of olanzapine in adolescents with schizophrenia. 13th Biennial Winter Workshop on Schizophrenia Research, 2006 February 4‐10, Davos, Switzerland. [8894]
- Kryzhanovskaya L, Schulz C, McDougle CJ, Frazier JA, Dittmann RW, Robertson‐Plouch C, et al. Results from a double‐blind, placebo‐controlled study of olanzapine in adolescents with schizophrenia. 5th International Conference on Early Psychosis, 2006 October 4‐6, Birmingham, United Kingdom. [5985]
- Kryzhanovskaya L, Schulz C, McDougle CJ, Frazier JA, Dittmann RW, Tohen MF. A double‐blind, placebo‐controlled study of olanzapine in adolescents with schizophrenia. 159th Annual Meeting of the American Psychiatric Association, 2006 May 20‐25, Toronto, Canada. [APA2006L1_5100]
- Kryzhanovskaya L, Schulz C, McDougle CJ, Frazier JA, Shen J, Dittman R, et al. A double‐blind, placebo‐controlled study of olanzapine in adolescents with schizophrenia. Neuropsychopharmacology 2005;30(Suppl 1):S258‐9. [Google Scholar]
- Kryzhanovskaya L, Schulz SC, McDougle C, Frazier J, Dittmann R, Robertson‐Plouch C, et al. Olanzapine versus placebo in adolescents with schizophrenia: a 6‐week, randomized, double‐blind, placebo‐controlled trial. Journal of the American Academy of Child and Adolescent Psychiatry 2009;48(1):60‐70. [PUBMED: 19057413] [DOI] [PubMed] [Google Scholar]
Kumra 1996 {published data only}
- Frazier JA, Gordon CT, McKenna K, Lenane MC, Jih D, Rapoport JL. An open trial of clozapine in 11 adolescents with childhood onset schizophrenia. Journal of the American Academy of Child and Adolescent Psychiatry 1994, issue 5:658‐63. [MEDLINE: ; PMID: 8056728] [DOI] [PubMed]
- Gordon CT, Frazier JA, McKenna K, Giedd J, Zametkin A, Zahn T, et al. Childhood‐onset schizophrenia: an NIMH study in progress. Schizophrenia Bulletin 1994, issue 4:697‐712. [MEDLINE: ; PMID: 7701277] [DOI] [PubMed]
- Kumra S, Frazier JA, Jacobsen LK, McKenna K, Gordon CT, Lenane MC, et al. Childhood‐onset schizophrenia: a double‐blind clozapine‐haloperidol comparison. Archives of General Psychiatry 1996;53(12):1090‐7. [MEDLINE: ; PMID 8956674] [DOI] [PubMed] [Google Scholar]
- Kumra S, Jacobsen LK, Rapoport JL. Childhood‐onset schizophrenia—a double‐blind clozapine trial. 149th Annual Meeting of the American Psychiatric Association, 1996 May 4‐9, New York, NY, USA. [MEDLINE: ; PMID: 8956674] 8956674
- Piscitelli SC, Frazier JA, McKenna K, Albus KE, Grothe DR, Gordon CT, et al. Plasma clozapine and haloperidol concentrations in adolescents with childhood‐onset schizophrenia: association with response. Journal of Clinical Psychiatry 1994, issue Suppl B:94‐7. [MEDLINE: ; PMID: 7961584] [PubMed]
Kumra 2008 {published data only}
- Kumra S, Kranzler H, Gerbino‐Rosen G, Kester HM, DeThomas C, Kafantaris V, et al. Clozapine and "high‐dose" olanzapine in refractory early‐onset schizophrenia: a 12‐week randomized and double‐blind comparison. Biological Psychiatry 2008;63(5):524‐9. [EMBASE: 2008071168; MEDLINE: ; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
- Kumra S, Kranzler H, Moise F, McCarthy J, Kafantaris V, Kane JM. Treating refractory childhood schizophrenia, 2002. http:www.clinicaltrials.gov.
Sikich 2004 {published data only}
- Sikich L, Hamer RM, Bashford RA, Sheitman BB, Lieberman JA. A pilot study of risperidone, olanzapine, and haloperidol in psychotic youth: a double‐blind, randomized, 8‐week trial. Neuropsychopharmacology 2004;29(1):133‐45. [EMBASE: 2004025276; 14583740 PMID] [DOI] [PubMed] [Google Scholar]
- Sikich L, Hooper SR, Malekpour AH, Sheitman BB, Lieberman JA. A double blind comparison of typical versus atypical antipsychotic agents on selected neurocognitive functions in children and adolescents with psychotic disorders. Schizophrenia Research 2001, issue 1,2:245. [MEDLINE: ; PMID: 11525078] 11525078
- Sikich L, Horrigan JP, Lieberman JA, Barnhill LJ Jr, Sheitman BB, Courvoisie HE. Comparative use of olanzapine and risperidone in psychotic youth. 155th Annual Meeting of the American Psychiatric Association, 2002 May 18‐23, Philadelphia, PA, USA. [NO.: 48D]
- Sikich L, Horrigan JP, Lieberman JA, Barnhill LJ, Sheitman BB, Courvoisie HE. Comparative use of olanzapine and risperidone in psychotic youth. 154th Annual Meeting of the American Psychiatric Association, 2001 May 5‐10, New Orleans, LA, USA. [MEDLINE: ; PMID: 11290639] 11290639
- Sikich L, Williamson K, Malekpour A, Bashford RA, Hooper S, Sheitman B, et al. Interim results of a randomized controlled trial of haloperidol, risperidone, and olanzapine in psychotic youth. 38th Annual Meeting of the American College of Neuropsychopharmacology, 1999 December 12‐16, Acapulco, Mexico. [LMD152419]
Sikich 2008 {published data only}
- Sikich L. Treatment of schizophrenia and related disorders in children and adolescents, 2003. http:www.clinicaltrials.gov.
- Sikich L, Frazier JA, McClellan J, Findling RL, Vitiello B, Ritz L, et al. Double‐blind comparison of first‐ and second‐generation antipsychotics in early‐onset schizophrenia and schizo‐affective disorder: findings from the treatment of early‐onset schizophrenia spectrum disorders (TEOSS) study. The American Journal of Psychiatry 2008;165(11):1420‐31. [PUBMED: 18794207] [DOI] [PubMed] [Google Scholar]
Swadi 2010 {published data only}
- Swadi HS, Craig BJ, Pirwani NZ, Black VC, Buchan JC, Bobier CM. A trial of quetiapine compared with risperidone in the treatment of first onset psychosis among 15‐ to 18‐year‐old adolescents. International Clinical Psychopharmacology 2010;25(1):1‐6. [PUBMED: 19809337] [DOI] [PubMed] [Google Scholar]
Xiong 2004 {published data only}
- Xiong Y. Comparison study of childhood schizophrenia treated with risperidone and chlorpromazine. Guizhou Medical Journal 2004;28(8):697‐8. [MEDLINE: ] [Google Scholar]
References to studies excluded from this review
Amminger 2006 {published data only}
- Amminger GP, Schafer MR. Indicated prevention with omega‐3 fatty acids in adolescents at ultra‐high risk for psychosis—rationale, methods, and 3‐months outcome. Schizophrenia Research 2006;86(Suppl 1):S97‐8. [Google Scholar]
- Amminger GP, Schafer MR. Is it feasible to conduct a RCT in ultra‐high risk individuals at a child and adolescent psychiatric service?. Schizophrenia Research 2006;86(Suppl 1):S98. [Google Scholar]
Antropov 1981 {published data only}
- Antropov IUF. Various methods of overcoming resistance to therapy in childhood and adolescent schizophrenia [O nekotorykh metodakh preodoleniia terapevticheskoi rezistentnosti pri shizofrenii u detei i podrostkov]. Zhurnal Nevropatologii i Psikhiatrii imeni S.S. Korsakova 1981, issue 10:1522‐6. [MEDLINE: ; PMID: 6118983] [PubMed]
Berger 2007 {published data only}
- Berger G. A naturalistic, prospective, single centre, double blinded, fixed dose, randomised, four week comparison study investigating efficacy, tolerability and safety of 200 mg per day versus 400 mg per day quetiapine fumarate in 200 drug naïve first episode psychosis patients aged 15 to 25 years, 2007. http:www.clinicaltrials.gov. [PubMed]
Bertelsen 2005 {published data only}
- Bertelsen M. Randomized controlled trial of two years integrated treatment versus standard treatment of patients with first episode of schizophrenia or psychosis, five years follow‐up. The OPUS trial. Schizophrenia Bulletin 2005;31:519‐20. [112412] [Google Scholar]
Buchsbaum 2007 {published data only}
- Buchsbaum MS, Haznedar MM, Aronowitz J, Brickman AM, Newmark RE, Bloom R, et al. FDG‐PET in never‐previously medicated psychotic adolescents treated with olanzapine or haloperidol. Schizophrenia Research 2007;94(1‐3):293‐305. [MEDLINE: ; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
- Buchsbaum MS, Haznedar MM, Hazlett E. Antipsychotic response prediction with FDG‐PET in schizophrenia. Thematic Conference of the World Psychiatric Association on "Treatments in psychiatry: an update," 2004 November 10‐13, Florence, Italy. [WO32.1]
Chen 2007 {published data only}
- Chen P. [Title available only in Chinese characters] [利培酮与奋乃静治疗少儿精神分裂症对照观察]. Linchuang Jingshen Yixue Zazhi [临床精神医学杂志] 2007;17(1):54. [CHINESE: Academic Journals] [Google Scholar]
Davidson 2004 {published data only}
- Davidson M. Reducing the risk of early transition to psychosis: using long‐acting atypical antipsychotics in young patients. Thematic Conference of the World Psychiatric Association on "Treatments in psychiatry: an update," 2004 November 10‐13, Florence, Italy. [SAS5.2.]
Gao 2007 {published data only}
- Gao C. A comparative study of risperidone and perphenazine in the treatment of child schizophrenia [利培酮和奋乃静治疗儿童精神分裂症对照研究]. Chinese Journal of Health Psychology [中国健康心理学杂志] 2007;15(10):950‐1. [CHINESE: Academic Journals] [Google Scholar]
Jenner 2004 {published data only}
- Jenner J, Willige G. Results of integrated treatment (HIT) in early psychoses: a pilot study. Schizophrenia Research 2004;70(1):16. [ISI:000224551100038] [Google Scholar]
Johnson 2004 {published data only}
- Johnson & Johnson Pharmaceutical Research & Development LLC. A randomized, double‐blind, placebo‐controlled clinical study of the efficacy and safety of risperidone for the treatment of schizophrenia in adolescents, 2004. http:www.clinicaltrials.gov.
Killackey 2006 {published data only}
- Killackey E, Jackson H, McGorry P. Vocational intervention in a specialist early psychosis service: first results of a pilot randomised controlled trial. Schizophrenia Research 2006;86(Suppl 1):S28‐9. [Google Scholar]
Klier 2005 {published data only}
- Klier C, Hollmann M, Schlögelhofer M, Mossaheb N, Friedrich M, Amminger PG. Indicated prevention with omega‐3 fatty acids (EPA/DHA) in adolescents with "at‐risk‐mental‐state" for psychosis. XIII World Congress of Psychiatry, 10‐15 September 2005, Cairo, Egypt. [P11.P005]
Leblanc 2006 {published data only}
- Leblanc J, Letourneau K, Demers MF, Bouchard RH, Roy MA. The impact of modafinil as an adjunct to a second generation antipsychotic on cognitive functioning in patients with first psychotic episode. Schizophrenia Research 2006;86(Suppl 1):S131. [Google Scholar]
Leclerc 2006 {published data only}
- Leclerc C, Gauvin D, Lecomte T. CBT group to maintain and improve relationships and intimacy abilities of young adults with a first psychotic episode—a pilot study. Schizophrenia Research 2006;86(Suppl 1):S138‐9. [Google Scholar]
Liang 2003 {published data only}
- Liang Ligui. A clinical trial of risperidone in treatment of childhood patients with first‐episode schizophrenia. Journal of Clinical Psychological Medicine 2003, issue 01. [MEDI0306]
Linszen 2006 {published data only}
- Linszen D, Haan L, Dingemans P, Lenior R, Amelsvoort T, Wouters L. The Amsterdam critical period intervention in the early phase of schizophrenia‐like psychoses. Schizophrenia Research 2006;86(Suppl 1):S61. [Google Scholar]
Lv 2004 {published data only}
- Lv L‐X, Guo S‐Q, Chen W, Li Q, Cheng J, Zhang H‐Y, et al. Effects of clozapine and risperidone on serum interleukin‐2,‐10,‐18 in adolescent schizophrenia patients. Journal of Applied Clinical Pediatrics 2004;19(11):983‐6. [EMBASE: 2004428997] [Google Scholar]
Malik 1980 {published data only}
- Malik SC, Kumar K. Loxapine in adolescent schizophrenia—a comparative study with trifluoperazine. Current Therapeutic Research Clinical and Experimental 1980;28(3):432‐46. [MEDLINE: ; PMID 3381314] 3381314 [Google Scholar]
Mathai 2004 {published data only}
- Mathai J, Bourne A. Pilot study investigating the effect of intercessory prayer in the treatment of child psychiatric disorders. Australasian Psychiatry 2004;12(4):386‐9. [116287] [DOI] [PubMed] [Google Scholar]
McConville 2003 {published data only}
- McConville B, Carrero L, Sweitzer D, Pott Larry, Chaney R, Foster K, et al. Long‐term safety, tolerability, and clinical efficacy of quetiapine in adolescents: an open‐label extension trial. Journal of Child & Adolescent Psychopharmacology 2003, issue 1:75‐82. [PSYCINFO: 2003‐05060‐010] [DOI] [PubMed]
McGlashan 2003 {published data only}
- Hawkins KA, Addington J, Keefe RS, Christensen B, Woods SW, Zipursky RB, et al. Effect of olanzapine vs. placebo on the neuropsychological status of prodromal subjects. 12th Biennial Winter Workshop on Schizophrenia, 2004 February 7‐13, Davos, Switzerland. [PsycINFO]
- Hoffman RE, Woods SW, Hawkins KA, Pittman B, Tohen M, Preda A, et al. Extracting spurious messages from noise and risk of schizophrenia‐spectrum disorders in prodromal population. British Journal of Psychiatry 2007;191(2):355‐6. [PsycINFO] [DOI] [PubMed] [Google Scholar]
- McGlashan TH, Miller TJ, Zipursky RB, Woods SW, Perkins DO, Hawkins KA, et al. Intervention in the schizophrenic prodrome: the prevention through risk identification, management, and education initiative. 156th Annual Meeting of the American Psychiatric Association, 2003 May 17‐22, San Francisco, CA, USA.
- McGlashan TH, Zipursky RB, Perkins D, Addington J, Miller TJ, Woods SW, et al. The PRIME North America randomized double‐blind clinical trial of olanzapine versus placebo in patients at risk of being prodromally symptomatic for psychosis. I. Study rationale and design. Schizophrenia Research 2003;61(1):7‐18. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- McGlashan TH, Zipursky RB, Perkins DO, Addington J, Woods SW, Miller TJ, et al. Olanzapine versus placebo treatment of the schizophrenia prodrome: one year results. Schizophrenia Research 2003;60:295. [Google Scholar]
- Miller TJ, Zipursky RB, Perkins D, Addington J, Woods SW, Hawkins KAHR, et al. The PRIME North America randomized double‐blind clinical trial of olanzapine versus placebo in patients at risk of being prodromally symptomatic for psychosis. II. Baseline characteristics of the "prodromal" sample. Schizophrenia Research 2003;61(1):19‐30. [MEDLINE: ; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Woods SW, Breier A, Zipursky RB, Perkins DO, Addington J, Miller TJ, et al. Olanzapine versus placebo for prodromal symptoms. Schizophrenia Research 2003;60:306‐7. [DOI] [PubMed] [Google Scholar]
- Woods SW, Breier A, Zipursky RB, Perkins DO, Addington J, Miller TJ, et al. Randomized trial of olanzapine versus placebo in the symptomatic acute treatment of the schizophrenic prodrome. Biological Psychiatry 2003;54(4):453‐64. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Woods SW, Breier A, Zipursky RB, Perkins DO, Addington J, Miller TJ, et al. Randomized trial of olanzapine versus placebo in the symptomatic acute treatment of the schizophrenic prodrome. [erratum appears in Biological Psychiatry 2003 Aug 15;54(4):497]. Biological Psychiatry 2003, issue 4:453‐64. [PMID: 12915290] [DOI] [PubMed]
McGorry 2007 {published data only}
- McGorry PD, Phillips LJ, Nelson B, Leicester S, Baker K, Krstev H, et al. A double blind, placebo‐controlled randomized trial of low‐dose risperidone, cognitive‐behaviour therapy, and befriending in young people with subthreshold symptoms at incipient risk of psychotic disorder: six month outcome data. Schizophrenia Bulletin 2007;33(2):446. [Google Scholar]
- Nelson B, Phillips LJ, Yung AR, Francey SM, Leicester S, Baker K, et al. A double blind, placebo‐controlled randomized trial of low‐dose risperidone, intensive psychological treatment and supportive therapy in young people with subthreshold symptoms at incipient risk of psychotic disorder: baseline characteristics of the sample. Schizophrenia Bulletin 2007;33(2):450. [Google Scholar]
Newton 2005 {published data only}
- Newton E, Landau S, Smith P, Monks P, Shergill S, Wykes T. Early psychological intervention for auditory hallucinations: an exploratory study of young people's voices groups. Journal of Nervous and Mental Disease 2005;193(1):58‐61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Otsuka 2005 {published data only}
- Otsuka Pharmaceutical Development & Commercialization Inc. A phase II study to test PK tolerability in children and adolescents, 2005. http:www.clinicaltrials.gov.
Power 2004 {published data only}
- Power P, Craig T, Garety P, Rahaman N, Colbert S, Fornells‐Ambrojo M. Lambeth early onset (LEO) trial: a randomised controlled trial of assertive community follow‐up in early psychosis: initial 6 month data. Unknown source 1994. [MEDLINE: ]
- Power P, Craig T, Mcguire P, Iacoponi E, Garety P, Russell M. A randomised controlled trial of an early detection team in first episode psychosis: the LEO cat trial. Schizophrenia Research 2004;67(1):36. [ISI: 000188788100082] [Google Scholar]
Schepp 1999 {published data only}
- Schepp KG. Self‐management therapy for youth with schizophrenia, 1999. http:www.clinicaltrials.gov.
Sela 2003 {published data only}
- Sela B‐A, Stahl Z, Gavendo S, Ruderman V, Belmaker RH, Levine J. Homocysteine reducing strategy in young male schizophrenic patients: effects on symptoms and cognition. Journal of Inherited Metabolic Disease 2003;26(Suppl 1):102. [CN‐00461708] [Google Scholar]
Stain 2006 {published data only}
- Stain HJ, Startup M, Carr V, Baker A, Schall U. The depth project: a multisite RCT for youths at risk for psychosis. Schizophrenia Research 2006;86(Suppl 1):S51‐2. [Google Scholar]
Tandon 2005 {published data only}
- Tandon R, Sitholey P, Katiyar M, Kumar A. Short term tolerability of risperidone in children and adolescents with acute and transient psychotic disorders. XIII World Congress of Psychiatry, 10‐15 September 2005, Cairo, Egypt. [P13.P250]
Ueland 2004 {published data only}
- Ueland T, Rund BR. A controlled randomized treatment study: the effects of a cognitive remediation program on adolescents with early onset psychosis. Acta Psychiatrica Scandinavica 2004, issue 1:70‐4. [PMID: 14674961] [DOI] [PubMed]
- Ueland T, Rund BR. Cognitive remediation for adolescents with early onset psychosis: a 1‐year follow‐up study. Acta Psychiatrica Scandinavica 2005;111(3):193‐201. [PUBMED: 15701103] [DOI] [PubMed] [Google Scholar]
van Bruggen 2003 {published data only}
- Bruggen J, Tijssen J, Dingemans P, Gersons B, Linszen D. Symptom response and side‐effects of olanzapine and risperidone in young adults with recent onset schizophrenia. International Clinical Psychopharmacology 2003;18(6):341‐6. [PSYCINFO: 2004‐16292‐005] [DOI] [PubMed] [Google Scholar]
van Nimwegen 2006 {published data only}
- Nimwegen L, Haan L. Early withdrawal in a double‐blind randomized clinical trial with olanzapine and risperidone performed in adolescents with first psychosis. Psychopathology 2006;39(3):158. [EMBASE: 160407; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Nimwegen L, Haan L, Beveren N, Laan W, Brink W, Linszen D. Obsessive compulsive symptoms in a randomized double blind study with olanzapine or risperidone in young patients with recent‐onset schizophrenia or related disorders. 13th Biennial Winter Workshop on Schizophrenia, 6‐10 February 2006, Davos, Switzerland. [12650950]
- Nimwegen L, Haan L, Beveren N, Laan W, Brink W, Linszen D. Obsessive‐compulsive symptoms in a randomized, double‐blind study with olanzapine or risperidone in young patients with early psychosis. Journal of Clinical Psychopharmacology 2008;28(2):214‐8. [EMBASE: 2008140943; MEDLINE: ; MEDLINE: ; PsycINFO] [DOI] [PubMed] [Google Scholar]
Versiani 1978 {published data only}
- Versiani Marcis, Silva JA, Frota LH, Mundim FD. Double‐blind comparison between loxapine and haloperidol in the treatment of adolescent schizophrenic patients. Current Therapeutic Research 1978;24:559‐66. [PsycINFO 63‐03832] [Google Scholar]
Wang 2007 {published data only}
- Wang H. [Title available only in Chinese characters] [奎硫平和利培酮治疗儿童精神分裂症对照研究]. Linchuang Jingshen Yixue Zazhi [临床精神医学杂志] 2007;17(3):199‐200. [CHINESE: Academic Journals] [Google Scholar]
Wykes 2007 {published data only}
- Newton E, Landau S. Cognitive remediation therapy (CRT): a new psychological treatment for young patients with schizophrenia. Schizophrenia Research 2006;86(Suppl 1):S143‐4. [DOI] [PubMed] [Google Scholar]
- Wykes T. Cognitive remediation for young people with schizophrenia. Current Controlled Trials 2006. [ISRCTN: 75187865]
- Wykes T. Cognitive remediation therapy (CRT)) for young people with schizophrenia. 2nd International Conference on Early Psychosis, 2000 Mar 31‐Apr 2, New York, NY, USA. [EMBASE:: 2004180181]
- Wykes T, Newton E, Landau S, Rice C, Thompson N, Frangou S. Cognitive remediation therapy (CRT) for young early onset patients with schizophrenia: an exploratory randomized controlled trial. Schizophrenia Research 2007;94(1‐3):221‐30. [PUBMED: 17524620] [DOI] [PubMed] [Google Scholar]
- Wykes T, Newton E, Landau S, Rice C, Thompson N, Frangou S. Remediation in adolescent onset results of a randomised control trial. Schizophrenia Bulletin 2007;33(2):610. [Google Scholar]
- Wykes T, Reeder C, Williams C, Corner J, Rice C, Everitt B. Are the effects of cognitive remediation therapy (CRT) durable? Results from an exploratory trial in schizophrenia. Schizophrenia Research 2003;61(2‐3):163‐74. [MEDLINE: ; CN‐00456712.] [DOI] [PubMed] [Google Scholar]
Xiu 2004 {published data only}
- Xiu L, Jie W. Effects of the nursing care of mutual participation model on the rehabilitation of inpatients with early schizophrenia. Chinese Journal of Clinical Rehabilitation 2004;8(24):4958‐9. [117756] [Google Scholar]
Yang 2007 {published data only}
- Yang W, Liu X‐Q, Li X‐W. A ramdomized controlled clinical study of aripiprazole in the treatment of childhood schizophrenia [阿立呱唑治疗儿童精神分裂症多中心随机对照研究]. Hunan Medical Journal 2007;24(6):942‐4. [CHINESE: Academic Journals] [Google Scholar]
Yao 2003 {published data only}
- Yao H. A study of risperidone in the treatment of child schizophrenia. Journal of Clinical Psychological Medicine 2003, issue 02. [MEDI0307]
Yi 2006 {published data only}
- Yi Q‐M, Fan W‐L, Chen Z‐Q. The contrast investigation between quetiapine and perphenazlne in the treatment of first episode of schizophrenia in children [奎硫平与奋乃静治疗儿童首发精神分裂症对照研究]. Journal of the North Sichuan Medical College 2006;2(6):522‐4. [CHINESE: Academic Journals] [Google Scholar]
Zhang 2007 {published data only}
- Zhang Y, Zhang H, Gu W. A control study of aripiprazole in the treatment of childhood schizophrenia [阿立哌唑治疗儿童精神分裂症对照研究]. Journal of Clinical Psychosomatic Diseases [临床心身疾病杂志] 2007;13(2):122‐4. [CHINESE: Academic Journals] [Google Scholar]
Zhou 2007 {published data only}
- Zhou Z‐H, Yuan G‐Z, Zhu H‐M. Effects of olanzapine treatment on the auditory evoked event‐related potential P300 in childhood and adolescent schizophrenia [奥氮平对儿少期精神分裂症患者听觉事件相关电位P_(300)的影响]. Chinese Journal of Behavioral Medical Science [中国行为医学科学] 2007;16(10):906‐7. [CHINESE: Academic Journals] [Google Scholar]
杨玲 2004 {published data only}
- 杨玲, 杨庆南, 侯永花, 刘涛, 严宏力. [only Chinese Title available] [抗精神病药物对精神分裂症患儿心电图的影响]. Journal of Applied Clinical Pediatrics [实用儿科临床杂志] 2004;19(12):1076‐7. [Google Scholar]
谭友果 2002 {published data only}
- 谭友果, 刘成文, 刘勇, 甘枝勤. [only Chinese Title available] [首发青少年精神分裂症药物疗效的纵向比较]. Chinese Journal of Nervous and Mental Diseases 2002;28(5):365‐7. [Google Scholar]
References to ongoing studies
Alaghband‐rad 2006 {published data only}
- Alaghband‐rad J, Shahrivar Z, Mahmoodi J, Salesian N. First episode psychoses among Iranian adolescents. Schizophrenia Research 2006;86(Suppl 1):S65. [Google Scholar]
- Alaghband‐rad J, Sharifi V, Amini H, Shahrivar Z, Mottaghipour Y, Mahmoodi J, et al. Management of first episode psychoses in Iran: unique features and challenges. Schizophrenia Research 2006;86(Suppl 1):S42. [Google Scholar]
AstraZeneca 2004 {published data only}
- AstraZeneca. A 6‐week, multicenter, randomized, double‐blind, parallel‐group, placebo‐controlled, phase 3b study of the efficacy and safety of quetiapine fumarate (Seroquel™) immediate‐release tablets compared with placebo in adolescents with schizophrenia, 2004. http:www.clinicaltrials.gov.
AstraZeneca 2005 {published data only}
- AstraZeneca. A 26‐week, multicenter, open‐label phase 3b study of the safety and tolerability of quetiapine fumarate (Seroquel™) immediate‐release tablets in daily doses of 400 mg to 800 mg in children and adolescents with bipolar I disorder and adolescents with schizophrenia, 2005. http:www.clinicaltrials.gov.
Bechdolf 2007 {published data only}
- Bechdolf A. Development and pilot evaluation of modified cognitive behavioural therapy for adolescents with early onset psychosis, 2007. http:www.clinicaltrials.gov.
Pfizer 2005 {published data only}
- Pfizer. Six week, double‐blind, placebo controlled phase III trial evaluating the efficacy, safety and pharmacokinetics of flexible doses of oral ziprasidone in adolescent subjects with schizophrenia, 2005. http:www.clinicaltrials.gov.
Additional references
Almandil 2011
- Almandil NB, Wong IC. Review on the current use of antipsychotic drugs in children and adolescents. Archives of Disease in Childhood. Education and Practice Edition 2011;96(5):192‐6. [PUBMED: 21771730] [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [OLZ020600] [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 1983
- Andreasen NC. The Scale for the Assessment of Negative Symptoms (SANS). Iowa City: The University of Iowa, 1983.
Andreasen 1984
- Andreasen NC. The Scale for the Assessment of Positive Symptoms, (SAPS). Iowa City: The University of Iowa, 1984.
Andreasen 2010
- Andreasen NC, Pressler M, Nopoulos P, Miller D, Ho BC. Antipsychotic dose equivalents and dose‐years: a standardized method for comparing exposure to different drugs. Biological Psychiatry 2010;67(3):255‐62. [PUBMED: 19897178] [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). 4th Edition. Washington DC: APA, 1994. [Google Scholar]
Ardizzone 2010
- Ardizzone I, Nardecchia F, Marconi A, Carratelli TI, Ferrara M. Antipsychotic medication in adolescents suffering from schizophrenia: a meta‐analysis of randomized controlled trials. Psychopharmacology Bulletin 2010;43(2):45‐66. [PUBMED: 21052042] [PubMed] [Google Scholar]
Ballageer 2005
- Ballageer T, Malla A, Manchanda R, Takhar J, Haricharan R. Is adolescent‐onset first‐episode psychosis different from adult onset?. Journal of the American Academy of Child and Adolescent Psychiatry 2005;44(8):782‐9. [DOI] [PubMed] [Google Scholar]
Barnes 1989
- Barnes TR. A rating scale for drug‐induced akathisia. British Journal of Psychiatry 1989;154:672‐6. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Buchanan 2010
- Buchanan RW, Kreyenbuhl J, Kelly DL, Noel JM, Boggs DL, Fischer BA, et al. Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophrenia Bulletin 2010;36(1):71‐93. [PUBMED: 19955390] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bunney 1963
- Bunney WEJ, Hamburg DA. Methods for reliable longitudinal observation of behavior. Archives of General Psychiatry 1963;9:280‐294. [DOI] [PubMed] [Google Scholar]
Caccia 2011
- Caccia S, Clavenna A, Bonati M. Antipsychotic drug toxicology in children. Expert Opinion on Drug Metabolism & Toxicology 2011;7(5):591‐608. [PUBMED: 21401439] [DOI] [PubMed] [Google Scholar]
Clark 1998
- Clark A, Lewis S. Treatment of schizophrenia in childhood and adolescence. Journal of Child Psychology and Psychiatry 1998;39:1071‐81. [PubMed] [Google Scholar]
Crossley 2010
- Crossley NA, Constante M, McGuire P, Power P. Efficacy of atypical v. typical antipsychotics in the treatment of early psychosis: meta‐analysis. British Journal of Psychiatry 2010;196(6):434‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Datta 2011
- Datta SS, Kumar A, Wright S, Russell P. Typical antipsychotics for psychosis in adolescents. Cochrane Database of Systematic Reviews Protocol published in Jan 2012. [DOI] [PMC free article] [PubMed]
De Hert 2011
- Hert M, Dobbelaere M, Sheridan EM, Cohen D, Correll CU. Metabolic and endocrine adverse effects of second‐generation antipsychotics in children and adolescents: a systematic review of randomized, placebo controlled trials and guidelines for clinical practice. European Psychiatry 2011;26(3):144‐58. [PUBMED: 21295450] [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium, 25‐28 October 2000, Cape Town, Africa.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Endicott 2006
- Endicott J, Nee J, Yang R, Wohlberg C. Pediatric Quality of Life Enjoyment and Satisfaction Questionnaire (PQ‐LES‐Q): reliability and validity. Journal of the American Academy of Child and Adolescent Psychiatry 2006;45(4):401‐7. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gilat 2011
- Gilat Y, Ben‐Dor DH, Magen A, Wolovick L, Vekslerchik M, Weizman A, et al. Trends in prescribing of psychotropic medications for inpatient adolescents in Israel: a 10 years retrospective analysis. European Psychiatry 2011;26(4):265‐9. [PUBMED: 21276716] [DOI] [PubMed] [Google Scholar]
Gogtay 2011
- Gogtay N, Vyas NS, Testa R, Wood SJ, Pantelis C. Age of onset of schizophrenia: perspectives from structural neuroimaging studies. Schizophrenia Bulletin 2011;37(3):504‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy U. ECDEU Assessment Manual for Psychopharmacology. Revised. Rockville: National Institute of Mental Health, 1976. [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery, and Psychiatry 1960;23:56‐62. [PUBMED: 14399272] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane‐handbook.org.
Hodges 1990
- Hodges K. Child and Adolescent Functional Assessment Scale. Ypsilanti, Michigan: Eastern Michigan University, Department of Psychology, 1990.
Hollis 2000a
- Hollis CP. Adolescent schizophrenia. Advances in Psychiatric Treatment 2000;6:83‐92. [Google Scholar]
Hollis 2000b
- Hollis CP. The adult outcomes of child and adolescent‐onset schizophrenia: diagnostic stability and predictive validity. American Journal of Psychiatry 2000;157:1653‐9. [DOI] [PubMed] [Google Scholar]
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Imran 2011
Kapur 2001
- Kapur S, Seeman P. Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics? A new hypothesis. American Journal of Psychiatry 2001;158(3):360‐9. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13:261‐76. [DOI] [PubMed] [Google Scholar]
Kennedy 2007
- Kennedy E, Kumar A, Datta SS. Antipsychotic medication for childhood‐onset schizophrenia. Schizophrenia Bulletin 2007;33(5):1082‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kennedy 2007a
- Kennedy E, Kumar A, Datta SS. Antipsychotic medication for childhood‐onset schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD004027.pub2; PUBMED: 17636744] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kumra 2008a
- Kumra S, Oberstar JV, Sikich L, Findling RL, McClellan JM, Vinogradov S, et al. Efficacy and tolerability of second‐generation antipsychotics in children and adolescents with schizophrenia. Schizophrenia Bulletin 2008;34(1):60‐71. [PUBMED: 17923452] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lieberman 2005
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. The New England Journal of Medicine 2005;353(12):1209‐23. [PUBMED: 16172203] [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams CE, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Owen 2011
- Owen MJ, O'Donovan MC, Thapar A, Craddock N. Neurodevelopmental hypothesis of schizophrenia. British Journal of Psychiatry 2011;198(3):173‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rapoport 2011
- Rapoport JL, Gogtay N. Childhood onset schizophrenia: support for a progressive neurodevelopmental disorder. International Journal of Developmental Neuroscience 2011;29(3):251‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schaffer 1983
- Schaffer D, Gould MS, Brasic J. A Children's Global Assessment Scale (CGAS). Archives of General Psychiatry 1983;40:1228‐31. [DOI] [PubMed] [Google Scholar]
Schubart 2010
- Schubart CD, Gastel WA, Breetvelt EJ, Beetz SL, Ophoff RA, Sommer IE, et al. Cannabis use at a young age is associated with psychotic experiences. Psychological Medicine 2010;Oct 7:1‐10. [DOI] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Seeman 2002
- Seeman P. Atypical antipsychotics: mechanism of action. Canandian Journal of Psychiatry 2002;47(1):27‐38. [PubMed] [Google Scholar]
Simpson 1970
- Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatrica Scandinavica 1970; (Suppl 212) :11‐19. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Werry 1991
- Werry JS, McClellan JM, Chard L. Childhood and adolescent schizophrenic, bipolar, and schizoaffective disorders: a clinical and outcome study. Journal of the American Academy of Child and Adolescent Psychiatry 1991;30(3):457‐65. [DOI] [PubMed] [Google Scholar]
Werry 1992
- Werry JS. Child and adolescent (early onset) schizophrenia: a review in light of DSM‐IIIR. Journal of Autism and Developmental Disorders 1992;22:601‐24. [DOI] [PubMed] [Google Scholar]
WHO 1992
- World Health Organization. ICD‐10. The ICD‐10 Classification of Mental and Behavioural Disorders. Clinical Descriptions and Diagnostic Guidelines. Geneva: WHO, 1992. [Google Scholar]
Woods 2003
- Woods SW. Chlorpromazine equivalent doses for the newer atypical antipsychotics. Journal of Clinical Psychiatry 2003;64(6):663‐7. [PUBMED: 12823080] [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
Young 1978
- Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. The British Journal of Psychiatry 1978;133:429‐35. [PUBMED: 728692] [DOI] [PubMed] [Google Scholar]
Yudofsky 1986
- Yudofsky SC, Silver JM, Jackson W, Endicott J, Williams D. The Overt Aggression Scale for the objective rating of verbal and physical aggression. The American Journal of Psychiatry 1986;143(1):35‐9. [PUBMED: 3942284] [DOI] [PubMed] [Google Scholar]
Zammit 2010
- Zammit S, Lewis G, Dalman C, Allebeck P. Examining interactions between risk factors for psychosis. British Journal of Psychiatry 2010;197(3):207‐11. [DOI] [PubMed] [Google Scholar]