

Abstract
The development of renin inhibitors for the treatment of hypertension requires highly sensitive substrates to evaluate potency and to characterize the mechanism of tight-binding inhibitors. A series of intramolecularly quenched fluorogenic renin substrates, based on the N-terminal tetradecapeptide sequence of human angiotensinogen (hTDP), was synthesized using a solid-phase technique. Incorporation of the fluorescent amino acid L-Amp [L-2-amino-3-(7-methoxy-4-coumaryl)propionic acid] and the DNP (2,4-dinitrophenyl) group at various positions resulted in >90% quenching efficiency and strong product fluorescence. Shortening the hTDP sequence to an octapeptide from histidine in P5 to histidine in P3′ (substrate 3) resulted in an acceptable kcat/Km (41000 M−1·s−1) and further systematic variation gave substrate 9, DNP-Lys-His-Pro-Phe-His-Leu-Val-Ile-His-L-Amp, with a kcat/Km value of 350000 M−1·s−1 and 94% quenching efficiency. The free side chain of lysine, replacing the isoleucine residue at P6 position in the angiotensinogen sequence, contributed to the increased value for kcat. The pH dependence of kcat/Km for renin and substrate 9 showed that the optimal pH is at pH 6–7. It also showed two titrating groups on the acidic side of the pH optimum, and one titrating group with a pKa of 7.8 on the alkaline side. The combination of good kinetic and spectroscopic properties resulted in a >20-fold improvement in the sensitivity of renin assay, compared with the commercial substrate Arg-Glu(EDANS)-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-Thr-Lys(DABCYL)-Arg {where EDANS is 5-[(2-aminoethyl)amino]naphthalene-1-sulphonic acid and DABCYL is 4-(4-dimethylaminophenylazo)benzoic acid} (kcat/Km=268000 M−1· s−1, quenching efficiency <80%). The detection limit in a microplate renin assay was 60 pM, making substrate 9 well suited for the evaluation of inhibitors at picomolar concentrations.
Keywords: fluorogenic substrate, fluorometric determination, kinetic parameter, pH-dependence, renin, solid-phase synthesis
Abbreviations: L-Amp, L-2-amino-3-(7-methoxy-4-coumaryl)propionic acid; ACE, angiotensin-converting enzyme; Ang I, angiotensin I; Ang II, angiotensin II; AT1 receptor, Ang II subtype 1 receptor; AT2 receptor, Ang II subtype 2 receptor; DABCYL, 4-(4-dimethylaminophenylazo)benzoic acid; Dap, α,β-diaminopropionic acid; DIEA, di-isopropylethylamine; DMF, N,N-dimethylformamide; DNP, 2,4-dinitrophenyl; EDANS, 5-[(2-aminoethyl)amino]naphthalene-1-sulphonic acid; Fmoc, fluoren-9-ylmethoxycarbonyl; EDDnp, N-(2,4-dinitrophenyl)ethylenediamine; hTDP, N-terminal tetradecapeptide of human angiotensinogen; IQFS, intramolecularly quenched fluorogenic substrate; Nva, norvaline; RAS, renin–angiotensin system; TFA, trifluoroacetic acid
INTRODUCTION
The renin–angiotensin system (RAS) plays a central role in the regulation of blood pressure and electrolyte homoeostasis [1]. At the first and rate-limiting step of the RAS cascade, renin (EC 3.4.23.15), a highly specific aspartyl protease, cleaves angiotensinogen, a glycoprotein synthesized in the liver. The product Ang I (angiotensin I) is converted further into Ang II (angiotensin II) by ACE (angiotensin-converting enzyme). Ang II binds to two receptor subtypes, AT1 and AT2 [2,3], in various tissues and organs. Excessive secretion of renin can cause hypertension and exacerbate the abnormalities that occur in other diseases, such as congestive heart failure. Blockade of Ang II production and/or receptor binding are validated treatment approaches for hypertension. This is currently achieved by the use of AT1RAs (AT1 receptor antagonists) and inhibitors of ACE. However, there are drawbacks to the current therapy of hypertension: ACE inhibitors do not only block the conversion of Ang I into Ang II, but also inhibit the degradation of other peptides, including bradykinin, which may be responsible for side effects, such as cough and angioedema [4]. Also, during ACE inhibition, Ang II can still be produced by chymase and other serine proteases. On the other hand, blockade of AT1 receptors in the presence of elevated Ang II levels may result in the stimulation of AT2 receptors, whose role is not clearly defined [5].
Inhibition of renin, which exclusively cleaves angiotensinogen and is not involved in alternative pathways, would surpass the above drawbacks and provide an advantageous way of therapy for the treatment of hypertension without side effects [6]. In the past decade, a considerable number of structurally different synthetic renin inhibitors of excellent (sub-nanomolar) potency and selectivity has been described [7,8]. To guide the drug-discovery process and to perform automated inhibitor screening, a continuous assay at picomolar sensitivity is needed.
Various methods have been introduced for the quantification of renin activity. The radioimmunoassay of Ang I [9] using angiotensinogen or a synthetic tetradecapeptide as substrate has been widely used. Assays based on HPLC separation and quantification of fluorogenic [10] or unlabelled substrates [11] and their hydrolysis products were also employed. Fluorescence assays based on a C-terminal amide of β-naphthylamine [12] or 7-amino-4-methylcoumarin [13] have been developed. The hydrolysis of such substrates generates a C-terminal fragment, which requires sequential digestion by an aminopeptidase to liberate the fluorescent label. The above-mentioned methods are discontinuous and have a low throughput. To overcome this limitation, IQFSs (intramolecularly quenched fluorogenic substrates) for renin have been introduced. In general, IQFSs have been widely used for proteases that require substrates having amino acids on both sides of the scissile bond (P and P′ positions; nomenclature according to Schechter and Berger [14]). The fluorescence signal is suppressed by resonance energy transfer or a collisional mechanism [15], which enables a direct continuous assay of protease activity. In most cases, fluorophore and quencher are located at the N- and C-terminal ends of a hexa- to deca-peptide sequence, and the fluorescence signal increases 5–50-fold upon complete hydrolysis. However, the practical range in most enzymic assays is considerably smaller, since rate measurements are linear only if not more than 5–10% of the substrate is consumed. At low quenching efficiency (<90%, corresponding to a <10-fold increase in total fluorescence upon complete hydrolysis), the increase in fluorescence signal will be only a fraction of the background fluorescence, resulting in an assay of low sensitivity. Therefore the suitability of an IQFS for an enzymic assay is dependent not only on the kinetic parameters of hydrolysis, but also on the strength of the fluorescence signal that is produced. The latter depends on the fluorescence intensity of the donor and the quenching efficiency of the fluorophore/quencher pair, which in turn is a function of the spectral overlap and the spatial distance of the two labels. The IQFSs described for renin to date are based on the sequence of angiotensinogen and carry the low-fluorescent EDANS {5-[(2-aminoethyl)amino]naphthalene-1-sulphonic acid}/DABCYL [4-(4-dimethylaminophenylazo)benzoic acid] [16] or anthranilic acid/EDDnp [N-(2,4-dinitrophenyl)ethylenediamine] [17] label pairs.
Our strategy to obtain IQFSs of high sensitivity for measuring renin activity was based on the following considerations: (i) to use the 7-methoxycoumarin/DNP (2,4-dinitrophenyl) as fluorophore/quencher pair, which is known to combine high fluorescence intensity with good quenching efficiency [18,19], (ii) to introduce the 7-methoxycoumarin group via L-Amp {L-2-amino-3-(7-methoxy-4-coumaryl)propionic acid} [20], which is compatible with solid-phase peptide-synthesis methodology and, moreover, allows the easy variation of its position within the peptide sequence, (iii) to place the DNP quencher either at the N-terminal amino acid or the side chain of appropriate amino acids, (iv) to search for the shortest possible substrate sequence and monitor both cleavage rates (kcat/Km) and quenching efficiency, and (v) to optimize this substrate by systematic variations at individual positions. Starting from the N-terminal tetradecapetide sequence of angiotensinogen (hTDP), we have synthesized 16 IQFS, as well as their putative fluorescent products (Table 1). Our strategy for the parallel optimization of kinetic and spectroscopic properties led to the identification of a renin substrate of excellent sensitivity. Synthesis, kinetic and spectroscopic parameters of the new substrates are reported in the present paper.
Table 1. Amino acid sequence of hTDP, synthesized peptides and commercial substrate 20.
Calculated and found Mr and HPLC retention times (ret. time) of synthesized compounds. Bold residues indicate the hydrolysed bond.
| P10-P9-P8-P7-P6-P5-P4-P3-P2-P1- | P1′-P2′-P3′-P4′ | Ret. time (min) | Calculated Mr | Found Mr | |
|---|---|---|---|---|---|
| hTDP | Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu- | Val-Ile-His-Asn | |||
| 1 | DNP-Phe-His-Leu- | Val-Ile-His-L-Amp | 26.7 | 1175.8 | 1175.5 |
| 2 | DNP-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 26.3 | 1272.8 | 1272.6 |
| 3 | DNP-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 21.7 | 1409.9 | 1409.6 |
| 4 | DNP-Ile-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 25.9 | 1523.0 | 1522.7 |
| 5 | DNP-Tyr-Ile-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 25.6 | 1686.0 | 1685.8 |
| 6 | DNP-Val-Tyr-Ile-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 26.8 | 1785.1 | 1784.9 |
| 7 | DNP-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 23.2 | 1941.2 | 1941.0 |
| 8 | DNP-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 22.1 | 2056.2 | 2056.0 |
| 9 | DNP-Lys-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 17.7 | 1538.0 | 1537.7 |
| 10 | Lys(DNP)-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 20.0 | 1538.0 | 1537.7 |
| 11 | Ac-Lys(DNP)-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 23.0 | 1580.0 | 1579.7 |
| 12 | DNP-Dap-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 17.0 | 1495.9 | 1495.7 |
| 13 | Dap(DNP)-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 18.1 | 1495.9 | 1495.7 |
| 14 | Ac-Dap(DNP)-His-Pro-Phe-His-Leu- | Val-Ile-His-L-Amp | 21.0 | 1537.9 | 1537.7 |
| 15 | His-Pro-Amp-His-Leu- | Val-Ile-His-Lys(DNP) | 18.0 | 1390.8 | 1390.6 |
| 16 | Amp-His-Pro-Phe-His-Leu- | Val-Ile-His-Lys(DNP) | 20.8 | 1538.0 | 1537.7 |
| 17 | Val-Ile-His-L-Amp | 6.2 | 612.4 | 612.3 | |
| 18 | His-Pro-Amp-His-Leu | 6.6 | 747.5 | 747.3 | |
| 19 | Amp-His-Pro-Phe-His-Leu | 11.2 | 894.6 | 894.4 | |
| 20 | Arg-Glu(EDANS)-Ile-His-Pro-Phe-His-Leu- | Val-Ile-His-Thr-Lys(DABCYL)-Arg |
EXPERIMENTAL
Materials
Buffer substances and biochemicals were obtained from Fluka (Buchs, Switzerland) and were of the highest purity available. Chemical reagents and solvents were purchased from Aldrich, BSA (catalogue number 126593) from Calbiochem/Merck (Darmstadt, Germany), ε-DNP-Lys-OH from Sigma (St. Louis, MO, U.S.A.), DNP-Pro-OH, DNP-Nva-OH (where Nva is norvaline), Fmoc-Lys(DNP)-OH (where Fmoc is fluoren-9-ylmethoxycarbonyl) and Fmoc-Dap(DNP)-OH (where Dap is α,β-diaminopropionic acid) from Bachem (Bubendorf, Switzerland). All other Fmoc-amino acids were purchased from CBL-Patras (Patra, Greece). All amino acids used were of L-configuration. (2R)-N-{N-[Bis (methylthio) methylidene] glycyl} bornane-10, 2-sultam, was a gift from Dr Graham Knight (Department of Cell Adhesion and Signalling, Strangeways Research Laboratory, Cambridge, U.K.). The aminomethyl-polystyrene resin and the (RS)-2-chloro-4′-carboxytriphenylmethanol handle was a gift from Dr Nikos Ferderigos (Department of Chemistry, University of Athens, Athens, Greece). The renin substrate Arg-Glu(EDANS)-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-Thr-Lys (DABCYL)-Arg was purchased from Molecular Probes (Eugene, OR, U.S.A.).
Enzymes
Recombinant human pro-renin was expressed in CHO (Chinese-hamster ovary) cells, and clarified supernatants were loaded on to a concanavalin A–Sepharose Fast Flow column. After washing, bound material was eluted with 0.5 M methyl α-D-glucopy-ranoside and 0.5 M methyl α-D-mannopyranoside. Pro-renin-containing fractions were supplemented with 2 M NaCl, applied to a butyl-Sepharose Fast-Flow column and were eluted from the column with 1 M NaCl. Fractions containing pro-renin were pooled, concentrated, and activated by trypsin treatment (0.5 mg/ml trypsin for 1 h at 25 °C). Activated renin was directly applied to a Superdex 75 size-exclusion column. Renin-containing fractions were analysed by SDS/PAGE, pooled, dipped in liquid nitrogen and stored at −80 °C.
The molar concentration of active renin was determined by active-site titration with the renin inhibitor CGP 38′560 [21]. Inhibitor (from 3.5 nM to 69.5 nM) was incubated with 0.5 μg/ml renin for 1 h at 37 °C. Substrate 3 (2.1 μM final concentration) was added and the residual activity, v, was measured. The {inhibitor concentration I; activity v} data pairs were fitted to eqn (1), and dissociation constant, Ki, and enzyme concentration, E0, were calculated. The activity of the control experiment without inhibitor was denoted v0.
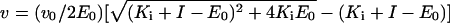 |
(1) |
Peptide synthesis
The fluorescent marker L-Amp was synthesized according to the procedure described in the literature by using (2R)-N-{N-[bis(methylthio)methylidene]glycyl}bornane-10,2-sultam and 4-bromomethyl-7-methoxycoumarin [20]. Fmoc-L-Amp was prepared by a standard procedure using N-(Fmoc-oxy)succinimide. All the peptides were synthesized manually using a solid-phase technique, using the Fmoc methodology, on a trityl-type resin prepared from aminomethyl-polystyrene resin (1 mmol/g) and an (RS)-2-chloro-4′-carboxytriphenylmethanol handle [22]. The resin was activated by treatment with acetylbromide for 3 h. The first Fmoc-amino acid (two equivalent amounts) in anhydrous dichloromethane was treated with DIEA (di-isopropylethylamine) (eight equivalent amounts) and added to the activated resin. De-protection of the Fmoc group was achieved by repetitive treatment with 20% piperidine in DMF (N,N-dimethylformamide). Elongation of the peptide chain was achieved using four equivalent amounts of Fmoc-amino acid derivatives utilizing the DIC (di-isopropylcarbodi-imide)/HOBt (N-hydroxybenzotriazol) method in DMF. For the direct dinitrophenylation [23] of the free N-terminal amino acid of the resin-bound peptides, DIEA (15 equivalent amounts) in DMF was added, followed by the addition of DNFB (2,4-dinitrofluorobenzene) (ten equivalent amounts) in DMF, and allowed to react for 10 min. The side-chain-protecting groups and resin cleavage was performed in a single step by treatment with a mixture of TFA (trifluoroacetic acid)/water (9:1, v/v) for 3 h. The crude peptides precipitated by the addition of ether were collected and dried. The final de-protected peptides were purified by semi-preparative HPLC performed on an Agilent 1100 series system running under Chemstation for LC Rev.A.08.03 (Agilent Technologies software), using a C18 Nucleosil column (8 mm×250 mm; Machery Nagel, Düren, Germany) and a two-step solvent system: (A) TFA/water (1:1000, v/v) and (B) TFA/acetonitrile/water (0.8:900:100, by vol.). The column was eluted at a flow rate of 4 ml·min−1 with a gradient of 0 to 100% solvent B within 40 min. Analytical HPLC was performed on a Thermo Separation Products system consisting of P1000 pump, AS3000 autosampler, UV1000 and F3000 detector, running under ChromQuest 3.0 software, using a C18 Nucleosil column (3 mm×125 mm, Machery Nagel) and a two-step solvent system: (A) TFA/water (1:1000, v/v) and (B1) TFA/acetonitrile/water (0.85:700:300, by vol.). The column was eluted at a flow rate of 0.5 ml·min−1 with a gradient of 30–100% solvent B1 within 7.5 or 30 min. Absorbance of eluted compounds was monitored at 360 nm, the settings for fluorescence detection were λexcitation, 328 nm and λemission, 388 nm. Amino acid analyses were carried out using the dabsyl chloride (4-dimethylaminoazobenzene-4′-sulphonyl chloride) method [24] and were in agreement with the proposed amino acid contents. The molecular mass and purity were checked by liquid chromatography–electrospray ionization MS (Q-TOF Micromass, service by NIBR Basel Central Technologies). Calculated and found Mr values are listed in Table 1. Purity for all substrates was >98%, except for substrate 2, which was 79%, due to incomplete dinitrophenylation. The stock solutions of the peptides were prepared in DMSO, and their concentrations were determined by amino acid analysis or were measured spectrophotometrically based on the absorption of the DNP group. Molar absorption coefficients at 410 nm were determined in solutions made from commercially available DNP-Nva-OH, DNP-Pro-OH and H-Lys(DNP)-OH and found to be 7753, 14370 and 7953 M−1·cm−1 respectively. The first value was used to calculate the concentration of substrates 1, 3–9 and 12, the second value for substrate 2, and the latter for substrates 10, 11 and 13–16.
Enzyme assays
Buffer conditions for kinetic determinations were: 50 mM Mops/NaOH, pH 7.0, 2 mM EDTA, 0.5% BSA, 0.1% NaN3 and 1% DMSO at 37 °C. Hydrolysis of the fluorogenic substrates 1–16 was monitored in a 1 ml reaction volume by measuring the fluorescence increase at λex, 328 nm and λem, 388 nm in a LS 50B spectrofluorimeter (PerkinElmer). The commercial substrate 20 (Table 1) was measured at λex, 340 nm and λem, 485 nm. Specificity constants (kcat/Km) were determined under pseudo-first-order conditions, via the ‘progress curve method’ using a substrate concentration (0.2 or 0.5 μM) far below Km, and a final renin concentration of 27.3 or 8.2 nM. Product formation was monitored for at least five half-lives. The {time; fluorescence} data pairs were fitted to eqn (2), and the apparent first-order rate constant, kobs, was calculated. The second-order rate constant, kcat/Km, was calculated according to eqn (3). Fluorescence increase and quenching efficiency were determined according to eqns (4a) and (4b) respectively, where F0 is the fluorescence of the intact substrate and F1 is the fluorescence of the product after complete hydrolysis. F0 and F1 derived after the subtraction of buffer autofluorescence (Fbuffer) from both, the initial fluorescence (Finit) and the final fluorescence (Fmax) calculated by fitting the data to eqn (2). All non-linear regression was performed using Origin 7.0 software, and r2 was greater than 0.98 in all cases.
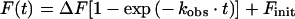 |
(2) |
where ΔF=Fmax−Finit
 |
(3) |
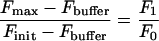 |
(4a) |
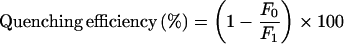 |
(4b) |
The kinetic parameters kcat and Km were derived using initial-rate methods. The enzyme concentration for initial-rate determination was chosen to hydrolyse less than 10% of the substrate present. Initial estimates of Km were made using a wide range of substrate concentrations. More accurate Km and Vmax values were determined using five different substrate concentrations in the Km range 0.5–3. After the addition of renin at 37 °C, in a 1 ml vial containing buffer and substrate, aliquots withdrawn at 10, 20, 30 and 40 min were added to inhibitor CGP 38′560 solutions (final concentration 5 μM) to stop the reaction. The solutions were analysed by HPLC, and concentrations were calculated from the product peaks using a calibration curve for the respective reaction product. Initial reaction velocities were calculated as the concentration of the fluorescent products produced per min. The kinetic parameters Vmax and Km were determined by fitting the {substrate concentration; velocity} pairs to the Michaelis–Menten equation. The errors were <5% for any kinetic parameter value obtained. The cleavage sites were confirmed by comparison of the HPLC retention times of the products with that of the fluorescent reference peptides (listed in Table 1).
For pH-dependence studies, 50 mM buffers containing 0.5% BSA, 2 mM EDTA and 0.1% NaN3 were prepared from sodium acetate (pH 4.5, 4.9 and 5.4), Mes (pH 5.5, 5.8 and 6.2), Mops (pH 6.7, 7.1, 7.7 and 8.1) and Taps (8.2, 8.7, 9.1 and 9.6) using correction factors for the change of pKa values with temperature [25]. The final renin concentration was 8.2–41 nM. Dissociation constants for ionizable groups were calculated by fitting data for kcat/Km at the different pH values to the exponential forms of eqn (5), describing an enzyme with three ionizable groups with only the monoprotonated form being active or eqn (6), describing a system with two ionizable groups on the enzyme (with the monoprotonated form being active) and one ionizable group on the substrate [26].
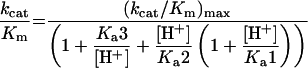 |
(5) |
 |
(6) |
For experiments on microplates, a Spectramax Gemini XS fluorescence plate reader (Molecular Devices, Sunnyvale, CA, U.S.A.) and black 96-well microplates (Costar) were used. The reader was set to medium sensitivity, and rates were calculated using the SoftMaxPro4.1 software package.
RESULTS AND DISCUSSION
Commercial renin substrates are based on the EDANS/DABCYL pair, which was shown to provide only limited sensitivity due to the low fluorescence intensity of the dansyl group [19]. In contrast, substrates for metalloproteases using the 7-methoxycoumaryl/DNP pair offer good quenching efficiency and approx. 10-fold increased fluorescence intensity upon hydrolysis [27,28]. An elegant approach to incorporate the 7-methoxycoumaryl moiety into IQFSs via L-Amp has been described [20], offering the advantage of introducing the fluorophore into any position of the peptide chain. We have developed a method for the solid-phase synthesis of IQFS based on the direct dinitrophenylation to the N-terminal amino acid of the resin-bound peptides [23]. The combination of L-Amp as fluorescent marker and DNP as quencher was applied for the solid-phase synthesis of a series of 16 renin IQFS, based on the sequence of hTDP (see Table 1 for sequences of substrates 1–16, reference products 17–19 and reference substrate 20).
In contrast with most studies on renin substrates in the literature, we determined the absolute molar concentration of our renin solution by active-site titration (results not shown). Furthermore, the kcat/Km values of all substrates were determined directly under pseudo-first-order conditions, via the ‘progress curve method’, as described in the Experimental section. In terms of precision, this direct determination of kcat/Km was preferred over the calculation derived from independently determined kcat and Km values [29]. Accurate values for kcat/Km are obtained by using very low substrate concentrations, and common problems (substrate concentration not known exactly, limited substrate solubility and product inhibition) could be avoided. Furthermore, the values for kcat/Km obtained by this method were not influenced by the strength of the optical signal (when comparing substrates of different quenching efficiencies, or with different fluorophores). The increase in fluorescence intensity upon hydrolysis was indicated by the ratio F1/F0, where F1 is the fluorescence intensity after complete hydrolysis and F0 is the fluorescence of the intact substrate (eqn 4a). The individual values for kcat and Km were obtained by applying the initial-rate method (shown for substrate 9 in Figure 1). To avoid inner filter effects at high substrate concentrations, the measurements of initial rates were performed by HPLC. Values for kcat/Km, calculated from the HPLC data, correlated well to the results from the pseudo-first-order experiments.
Figure 1. Determination of Michaelis–Menten parameters for the renin-catalysed hydrolysis of substrate 9 by the initial-rate method.

The initial-rate assay was performed at the following conditions: renin concentration, 0.27 nM; five substrate concentrations over the Km range 0.5–3; assay buffer, 50 mM Mops/NaOH, pH 7.0, 2 mM EDTA, 0.5% BSA, 0.1% NaN3 and 1% DMSO; excitation/emission wavelengths 328/388 nm; and temperature, 37 °C. Product concentration was determined by HPLC. Data were fitted to the Michaelis–Menten equation.
Effect of substrate length on kinetic parameters and quenching efficiency of substrates 1–8
From previous data [17,30], there is no consensus on the optimal length of peptide renin substrates. The hexapeptide Abz (aminobenzoic acid)-Phe-His-Leu-Val-Ile-His-EDDnp (occupying position P3–P3′) had a Vmax of 17 nmol of product/Goldblatt unit of renin per min (corresponding to a kcat of 11 s−1, given that 1 Goldblatt unit=1 μg of renin=25 pmol of renin) and a Km value of 1.3 μM [17], and kcat/Km would be 8.7×106 M−1·s−1. In contrast, the similar substrate pyridylglycine-Phe-His-Leu-Val-Ile-His-β-Ala-NH2 was found to have a kcat/Km of only 1055 M−1· s−1, and only longer substrates, such as pyridylglycine-Tyr-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-β-Ala-NH2 (kcat/Km=85000 M−1·s−1), showed acceptable turnover rates [30]. Therefore a set of substrates (1–8), with L-Amp at position P4′ and DNP at Nα-terminal position was synthesized, containing the sequence of angiotensinogen from position P3′ to P3 for the shortest (1) and to position P10 for the longest (8) substrate respectively. Substrates 1–8 were subjected to hydrolysis catalysed by human recombinant renin. The kinetic parameters, the F1/F0 values, and the quenching efficiencies are summarized in Table 2.
Table 2. Kinetic parameters for hydrolysis of substrates 1–8 by recombinant human renin, increase of fluorescence intensities and quenching efficiencies.
Hydrolysis conditions: 50 mM Mops/NaOH, pH 7.0, 2 mM EDTA, 0.5% BSA, 0.1% NaN3 and 1% DMSO at 37 °C. Fluorescence was measured at excitation and emission wavelengths of 328 nm and 388 nm respectively. q.e., quenching efficiency.
| Substrate | kcat/Km (M−1·s−1)* | F1/F0 | q.e. (%) | kcat (s−1)† | Km (μM)† |
|---|---|---|---|---|---|
| 1 | No hydrolysis | − | − | − | − |
| 2 | 16000 | –‡ | − | 0.02 | 2.30 |
| 3 | 41000 | 12 | 92 | 0.20 | 4.45 |
| 4 | 29000 | 18 | 94 | 0.11 | 4.56 |
| 5 | 64000 | 14 | 93 | 0.06 | 1.22 |
| 6 | 22000 | 10 | 90 | 0.02 | 1.49 |
| 7 | 96000 | 6 | 83 | 0.08 | 0.55 |
| 8 | 181000 | 5 | 80 | 0.13 | 0.82 |
* Determined via the progress-curve method under pseudo-first-order conditions.
† Determined via the initial-rate method.
‡ This value was not determined because of the high fluorescence of the intact substrate solution, caused by impurities.
Substrate 1 was completely resistant to hydrolysis; substrates 2–8 were cleaved completely by renin at one single bond. In analytical HPLC, the retention time of the fluorescent product was identical with that of the reference peptide 17, showing that all substrates were cleaved at the Leu–Val bond. The hydrolysis of substrate 2 required high renin concentrations and prolonged reaction times. Further elongation of the peptide chain with histidine at position P5 (substrate 3, P5-P4′) increased the kcat/Km value considerably. The introduction of isoleucine at position P6, tyrosine at P7 and valine at P8 (substrates 4–6) did not result in a significant change of kcat/Km values. The introduction of arginine at P9 (substrate 7) and aspartate at P10 position (8) improved both kcat and Km values. Both substrates showed a high affinity to the enzyme, having the lowest (submicromolar) Km values reported to date for synthetic renin substrates. Substrate 8 has the identical sequence from position P3′ to P10 as hTDP, but the asparagine at position P4′ was substituted by L-Amp and its terminal amino group is blocked by the DNP group. The Km values for hTDP were found to be 13.3±6.2 μM [31] and 20.7±7 μM [32]. High Km values have been also found when the asparagine residue was replaced by serine (Km=8.4 μM) [11] or threonine (Km=27.0 μM) [32], whereas the Km values reported for angiotensinogen are low, varying from 0.4 to 5.7 μM [33]. The modification of peptide substrates for proteases with bulky hydrophobic groups can alter their kinetic behaviour. Interaction of the N-terminal DNP at position P6 (substrate 3) or P8 (5) with a renin-binding site may explain the higher kcat/Km values of these substrates. In contrast, the S9 subsite obviously cannot accommodate the hydrophobic DNP group resulting in a poor kcat/Km value (substrate 6). This is also indicated by a good kcat/Km value obtained when position P9 was occupied by the hydrophilic residue arginine in substrate 7. Position P10 also seems to prefer a hydrophilic residue, since the replacement of DNP at this position (substrate 7) by the aspartyl group (substrate 8) increased the kcat/Km value.
In agreement with the theory on fluorescence resonance energy transfer, the quenching efficiency of the examined IQFSs was reduced with increasing length of the peptide chain. The 12–18-fold increase in fluorescence upon hydrolysis of the shorter peptides (3–5) was in the acceptable range, whereas substrate 8 (F1/F0=5, corresponding to 80% quenching) was classified as rather insensitive, despite its excellent kinetic parameters. For this reason, we went through another round of modification starting from substrate 3, which represents the shortest sequence with acceptable kinetic constants.
Kinetic parameters and quenching efficiency of modified substrates 9–16
The data presented in Table 2 clearly showed that the sequence His-Pro-Phe-His-Leu-Val-Ile-His-L-Amp was important for interaction with renin, while the presence of the hydrophobic amino acid isoleucine at position P6 did not improve the kinetic parameters. To replace isoleucine at P6, we designed a series of six substrates with the sequence Xaa-His-Pro-Phe-His-Leu-Val-Ile-His-L-Amp, where Xaa is DNP-Lys, H-Lys(DNP), acetyl-Lys(DNP), DNP-Dap, H-Dap(DNP) and acetyl-Dap(DNP) (substrates 9–14). The hydrophilic amino acids lysine and Dap could carry DNP either at the α- or side-chain-amino group. A bathochromic shift of about 10 nm was seen in the UV-absorption spectra of ε-DNP-Lys and β-DNP-Dap derivatives when compared with those of α-DNP-derivatives (results not shown), and this should result in a better overlap with the emission spectrum of L-Amp and better quenching efficiency. The kinetic parameters, the F1/F0 values, and the quenching efficiencies of substrates 9–14 are listed in Table 3.
Table 3. Kinetic parameters for hydrolysis of substrates 9–16 by recombinant human renin, increase of fluorescence intensities and quenching efficiencies.
Hydrolysis conditions: 50 mM Mops/NaOH, pH 7.0, 2 mM EDTA, 0.5% BSA, 0.1% NaN3 and 1% DMSO at 37 °C. Fluorescence was measured at excitation and emission wavelengths of 328 nm and 388 nm respectively. q.e., quenching efficiency.
| Substrate | kcat/Km (M−1·s−1)* | F1/F0 | q.e. (%) | kcat (s−1)† | Km (μM)† |
|---|---|---|---|---|---|
| 9 | 352000 | 16 | 94 | 1.90 | 5.50 |
| 10 | 111000 | 10 | 90 | 0.14 | 1.24 |
| 11 | 117000 | 12 | 92 | 0.15 | 1.94 |
| 12 | 43000 | 6 | 83 | 0.12 | 2.96 |
| 13 | 108000 | 7 | 86 | 0.26 | 3.40 |
| 14 | 108000 | 7 | 86 | 0.27 | 2.94 |
| 15 | No hydrolysis | − | − | − | − |
| 16 | 122000 | 7 | 86 | 0.55 | 3.70 |
* Determined via the progress-curve method under pseudo-first-order conditions.
† Determined via the initial-rate method.
The replacement of isoleucine at position P6 (substrate 4) with lysine (substrate 9) increased the rate of hydrolysis 16-fold without a significant change in the Km value. When the shorter Dap was used at the same position (substrate 12), the rate of hydrolysis and kcat/Km value hardly changed, compared with those of substrate 4. Shifting the position of the DNP group to the side-chain-amino group of either N-terminal free (substrates 10 and 13) or N-terminal acetylated lysine and Dap at the P6 position (substrates 11 and 14) gave substrates with kcat/Km values of approx. 100000 M−1·s−1. The kinetic data for substrate 9, which had an excellent kcat/Km of 350000 M−1·s−1, made a direct contribution of the side-chain-amino group of the lysine in position P6 to renin activity very likely. This improvement in catalytic efficiency was not due to better binding (Km values of the P6 isoleucine substrate 4 and the P6 lysine substrate 9 were comparable), but had to be attributed to a higher kcat value. It has been shown for other endoproteases that improved subsite occupation could affect kcat more than Km, and resulted in an activation of the catalytic machinery of the enzyme [34].
For the first time, the data showed that highly conserved amino acids in the parent angiotensinogen sequence could be modified, resulting in improved human renin substrates and suggesting that further modifications of the angiotensinogen sequence could provide important information about specificity. Wang and Liang [35] prepared modified porcine renin substrates bearing a tryptophan or a p-nitrophenylalanine residue at position P3, instead of phenylalanine, and found only slightly decreased values for kcat/Km, relative to the parent sequence. Since L-Amp has some structural similarity to tryptophan, having a coumarin ring instead of indole ring, substrate 15 with L-Amp at P3 and Lys(DNP) at position P4′ was synthesized, to reduce the distance between fluorophore and quencher and to obtain a higher F1/F0 value. However, in contrast with Wang and Liang's substrate, substrate 15 was completely resistant to hydrolysis. One may speculate that the substrate specificity of porcine renin with respect to position P3 was more relaxed, compared with the human enzyme.
Substrate 16 is an analogue of our best compound (substrate 9), with the positions of fluorophore and quencher exchanged. It showed a specificity constant, kcat/Km, of 122000 M−1·s−1, indicating that the resistance of 15 to hydrolysis was due to the presence of L-Amp at position P3 and that Lys(DNP) was well tolerated at the P4′ position. The presence of Lys(DNP) or L-Amp at position P4′ resulted in good kinetic parameters, showing that subsite S4′ could accommodate variable bulky side chains. The kinetic constants of substrates 10 and 13, which are analogues of substrate 16 (all covering positions P6–P4′, with a free N-terminus and a bulky non-charged residue at P6), were similar.
Although the F1/F0 values of modified peptides 9–16 were not higher than those of substrate 4, this set of compounds gave renin substrates with very interesting kinetic parameters. Substrate 9 has a very high kcat/Km value and a satisfactory (16-fold) increase of fluorescence intensity upon hydrolysis that makes it suitable for renin determination of high sensitivity (see below). Comparison of the kcat and kcat/Km values with data from the literature is difficult, because previous studies did, in most cases, not give the molar concentration of renin used.
Comparison of substrate 9 and EDANS/DABCYL substrate 20
The IQFSs described so far for measuring renin activity were based on the combination of EDANS and DABCYL groups. We compared our best substrate 9 with Arg-Glu(EDANS)-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-Thr-Lys(DABCYL)-Arg (20). Figure 2 shows the progress curves of substrate 9 and substrate 20. A considerable more sensitive setting of the instrument was required to determine kobs for substrate 20 (inset in Figure 2) and a kcat/Km value of 268000 M−1·s−1 and a value of 4.9 for F1/F0 were obtained. On a microplate fluorescence reader, we compared the performance of substrate 9 and substrate 20 (both at 5 μM) directly in a range of practical renin concentrations (0.03–2 nM, Figure 3). On this instrument, and using a 10 min assay time, substrate 9 had a limit of detection of 0.06 nM renin, whereas substrate 20 was useful only for the determination of renin concentrations >1 nM. The specificity constants of the two substrates differed by less than a factor of 2, and therefore this >20-fold improvement in assay sensitivity had to be attributed mainly to the higher fluorescence intensity of the L-Amp, compared with dansyl, and to the better quenching efficiency, caused by the reduced distance of fluorophore and quencher in substrate 9.
Figure 2. Renin-catalysed hydrolysis of substrate 9 (▪) and substrate 20 (○).

Progress curves under pseudo-first-order conditions: renin concentration, 8.2 nM; substrate concentration 0.2 μM; assay buffer, 50 mM Mops/NaOH, pH 7.0, 2 mM EDTA, 0.5% BSA, 0.1% NaN3 and 1% DMSO; excitation/emission wavelengths, 328/388 nm for substrate 9 and 340/485 nm for substrate 20; and temperature, 37 °C. The inset shows hydrolysis of substrate 20 using a 7-fold-increased sensitivity of the spectrofluorimeter.
Figure 3. Determination of renin activity using substrates 9 (▪) and 20 (○) in a 96-well microplate.

Conditions: renin concentration, 0.03–2 nM; substrate concentration, 5 μM; assay buffer, 50 mM Mops/NaOH, pH 7.0, 2 mM EDTA, 0.5% BSA, 0.1% NaN3 and 1% DMSO; excitation/emission wavelengths, 328/388 nm for substrate 9 and 340/485 nm for substrate 20; and temperature, 37 °C. Assay time was 10 min at 37 °C, the plate was read at 30 s intervals and slopes were calculated. Results are means±S.D. for three wells. RFU, relative fluorescence units.
pH dependence of kcat/Km for substrate 9
The pseudo-first-order rate constant kcat/Km was determined in a series of buffers ranging from pH 4.5 to 9.6. The plot of log (kcat/Km) against pH for substrate 9 showed an asymmetrical bell-shaped dependence on pH with limiting slopes of 2 (ascending limb) and −1 (descending limb), and a pH optimum between pH 6 and 7 (Figure 4). As the pH-dependence of kcat/Km describes the ionization of free substrate and free enzyme, this pH-response indicated that two ionizable groups had to be deprotonated and one had to be protonated for optimal activity. However, the data did not allow distinguishing between a kinetic model that had three ionizable groups at the enzyme (described by eqn 5), and the competing model that had two ionizable groups at the enzyme (Asp38 and Asp226, with the monoprotonated form being active) and one titrating group at the substrate (described by eqn 6). The data could be fitted to both eqns (5) and (6) with similar quality, and the pKa value for the group titrating at the alkaline limb was found to be 7.8 in both models. The pKa values at the acidic limb could not be separated from each other; the fit to both models gave a value of 5.4 for this combined dissociation constant.
Figure 4. pH-dependence of kcat/Km of renin-catalysed hydrolysis of substrate 9.

Buffers (50 mM) containing 0.5% BSA, 2 mM EDTA and 0.1% NaN3 were prepared from sodium acetate (pH 4.5, 4.9 and 5.4), Mes (pH 5.5, 5.8 and 6.2), Mops (pH 6.7, 7.1, 7.7 and 8.1) and Taps (8.2, 8.7, 9.1 and 9.6) using correction factors for the change of pKa values with temperature. The final renin concentration was 8.2–41 nM. Values for kcat/Km were determined by the progress-curve method at 0.2 μM substrate concentrations and fitted to eqns (5) (continuous line) and (6) (dotted line). Results from two experiments are shown.
The pH-dependence of renin activity had been investigated earlier [36], and the authors calculated Vmax/Km values from the individual constants Vmax and Km for the porcine tetradecapeptide substrate Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-Leu-Val-Tyr-Ser (covering positions P10–P4′). A symmetrical bell-shaped pH-dependence with pKa values of 6.3 and 6.9 and slopes of +1 and −1 was found. When using a corresponding peptide with P2 histidine replaced by glutamine, the Vmax/Km value was lower and almost pH-independent. From these data and from data of solvent isotope effects, the authors concluded that the protonated histidine in position P2 of the substrate formed an ion pair with the catalytic residue Asp226, thereby lowering its pKa value and allowing it to function as an acid at neutral pH. However, in an X-ray study of mouse renin with an inhibitor spanning the P6–P4′ binding sites [37], the distance between P2 histidine and Asp226 was found to be too long for such an interaction.
Our data do not exclude that the protonation status of the P2 histidine (in addition to the catalytic residues Asp38 and Asp226) may be important for optimal renin activity, but suggest that this histidine (or another group titrating around pH 6) has to be deprotonated. The observed pH-dependence supports the idea that the protonation status of the substrate may be an important factor to adjust the pH optimum for renin to a narrow neutral range. The three histidine residues in positions P5, P2 and P3′ of substrate 9 may all titrate with pKa values of approx. 6, and it is currently unknown whether the observed steep response to lower pH values is due to protonation of one single residue or is a combination of weak effects when two or three histidine residues become protonated.
In conclusion, this work proves that the combination of L-Amp and DNP for solid-phase synthesis of IQFSs was very advantageous as providing flexibility to the design of the substrates and gave a satisfying increase in fluorescence intensity upon hydrolysis. The majority of the synthesized IQFSs was good renin substrates (kcat/Km>100000 M−1·s−1) with >90% quenching efficiency. Substrate 9, with the sequence DNP-Lys-His-Pro-Phe-His-Leu-Val-Ile-His-L-Amp, had the best kinetic parameters and good quenching efficiency, giving a significant increase in fluorescence intensity when using picomolar renin concentrations. It will be a very useful tool for automated renin inhibitor screening, but also, and more importantly, for detailed kinetic characterization of tight-binding renin inhibitors.
Acknowledgments
Many thanks are expressed to Dr Graham Knight (Department of Cell Adhesion and Signalling, Strangeways Research Laboratory, Cambridge, U.K.) for kindly providing (2R)-N-{N-[bis(methylthio)methylidene]glycyl}bornane-10,2-sultam and to Dr Nikos Ferderigos (Department of Chemistry, University of Athens, Athens, Greece) for kindly supplying the aminomethyl-polystyrene resin and the (RS)-2-chloro-4′-carboxytriphenylmethanol handle. We also thank Dr S. Geisse, Mrs A. Patoux, Mr P. Graff and Mr R. Knecht (NIBR Central Technologies, Basel, Switzerland) for fermentation of pro-renin-expressing cells, performing the HPLC/MS and amino acid analyses. This work was supported by funds from the ‘Special Account for Research Grants’ of the National and Kapodistrian University of Athens.
References
- 1.He F. J., MacGregor G. A. Salt, blood pressure and the renin–angiotensin system. J. Renin Angiotensin Aldosterone Syst. 2003;4:11–16. doi: 10.3317/jraas.2003.001. [DOI] [PubMed] [Google Scholar]
- 2.De Gasparo M., Catt K. J., Inagami T., Wright J. W., Unger T. International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- 3.Kaschina E., Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Pressure. 2003;12:70–88. doi: 10.1080/08037050310001057. [DOI] [PubMed] [Google Scholar]
- 4.Waeber B., Nussberger J., Brunner H. R. Angiotensin-converting enzyme inhibitors in hypertension. In: Laragh J. H., Brenner B. M., editors. Hypertension: Pathophysiology, Diagnosis and Management. 2nd edn. New York: Raven Press; 1995. pp. 2861–2876. [Google Scholar]
- 5.Taal M. W., Brenner B. M. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int. 2000;57:1803–1817. doi: 10.1046/j.1523-1755.2000.00031.x. [DOI] [PubMed] [Google Scholar]
- 6.Hollenberg N. K. Pharmacologic interruption of the renin–angiotensin system and the kidney: differential responses to angiotensin-converting enzyme and renin inhibition. J. Am. Soc. Nephrol. 1999;10(Suppl. 11):S239–S242. [PubMed] [Google Scholar]
- 7.Wood J. M., Maibaum J., Rahuel J., Grütter M. G., Cohen N.-C., Rasetti V., Rüger H., Göschke R., Stutz S., Fuhrer W., et al. Structure-based design of aliskiren, a novel orally effective renin inhibitor. Biochem. Biophys. Res. Commun. 2003;308:698–705. doi: 10.1016/s0006-291x(03)01451-7. [DOI] [PubMed] [Google Scholar]
- 8.Gupta S. P. Quantitative structure–activity relationships of renin inhibitors. Mini Rev. Med. Chem. 2003;3:315–321. doi: 10.2174/1389557033488051. [DOI] [PubMed] [Google Scholar]
- 9.Haber E., Koerner T., Page L. B., Kliman B., Purnode A. Application of a radioimmunoassay for angiotensin I to the physiologic measurements of plasma renin activity in normal human subjects. J. Clin. Endocrinol. Metab. 1969;29:1349–1355. doi: 10.1210/jcem-29-10-1349. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura-Imajo N., Satomura S., Matsuura S., Murakami K. Renin assay using a fluorogenic substrate and high performance liquid chromatography. Clin. Chim. Acta. 1992;211:47–57. doi: 10.1016/0009-8981(92)90104-x. [DOI] [PubMed] [Google Scholar]
- 11.Poe M., Wu J. K., Lin T.-Y., Hoogsteen K., Bull H. G., Slater E. E. Renin cleavage of a human kidney renin substrate analogous to human angiotensinogen, H-Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-Ser-OH, that is human renin specific and is resistant to cathepsin D. Anal. Biochem. 1984;140:459–467. doi: 10.1016/0003-2697(84)90194-5. [DOI] [PubMed] [Google Scholar]
- 12.Reinharz A., Roth M. Studies on renin with synthetic substrates. Eur. J. Biochem. 1969;7:334–339. doi: 10.1111/j.1432-1033.1969.tb19613.x. [DOI] [PubMed] [Google Scholar]
- 13.Murakami K., Ohsawa T., Hirose S., Takada K., Sakakibara S. New fluorogenic substrates for renin. Anal. Biochem. 1981;110:232–239. doi: 10.1016/0003-2697(81)90140-8. [DOI] [PubMed] [Google Scholar]
- 14.Schechter I., Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 15.Gershkovich A. A., Kholodovych V. V. Fluorogenic substrates for proteases based on intramolecular fluorescence energy transfer (IFETS) J. Biochem. Biophys. Methods. 1996;33:135–162. doi: 10.1016/s0165-022x(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 16.Wang G. T., Chung C. C., Holzman T. F., Krafft G. A. A continuous fluorescence assay of renin activity. Anal. Biochem. 1993;210:351–359. doi: 10.1006/abio.1993.1207. [DOI] [PubMed] [Google Scholar]
- 17.Oliveira M. C. F., Hirata I. Y., Chagas J. R., Boschcov P., Gomes R. A. S., Figueiredo A. F. S., Juliano L. Intramolecularly quenched fluorogenic peptide substrates for human renin. Anal. Biochem. 1992;203:39–46. doi: 10.1016/0003-2697(92)90040-e. [DOI] [PubMed] [Google Scholar]
- 18.Knight C. G. A quenched fluorescent substrate for thimet peptidase containing a new fluorescent amino acid, DL-2-amino-3-(7-methoxy-4-coumaryl)propionic acid. Biochem. J. 1991;274:45–48. doi: 10.1042/bj2740045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knight C. G. Fluorimetric assays of proteolytic enzymes. Methods Enzymol. 1995;248:18–34. doi: 10.1016/0076-6879(95)48004-8. [DOI] [PubMed] [Google Scholar]
- 20.Knight C. G. Stereospecific synthesis of L-2-amino-3-(7-methoxy-4-coumaryl)propionic acid, an alternative to tryptophan in quenched fluorescent substrates for peptidases. Lett. Pept. Sci. 1998;5:1–4. [Google Scholar]
- 21.Rahuel J., Rasetti V., Maibaum J., Rüeger H., Göschke R., Cohen N.-C., Stutz S., Cumin F., Fuhrer W., Wood J. M., Grütter M. G. Structure-based drug design: the discovery of novel nonpeptide orally active inhibitors of human renin. Chem. Biol. 2000;7:493–504. doi: 10.1016/s1074-5521(00)00134-4. [DOI] [PubMed] [Google Scholar]
- 22.Zikos C. C., Ferderigos N. G. (R,S) 2-Fluoro (chloro)-4′-carboxy-triphenyl methanol: novel acid labile trityl type handles for solid phase peptide synthesis. Tetrahedron Lett. 1994;35:1767–1768. [Google Scholar]
- 23.Paschalidou K., Tzougraki C. A general and direct method for the solid phase synthesis of intramolecularly quenched fluorogenic protease substrates using a bifunctional coumarin derivative. Lett. Pept. Sci. 2001;7:249–253. [Google Scholar]
- 24.Knecht R., Chang J. Y. Liquid chromatographic determination of amino acids after gas-phase hydrolysis and derivatization with (dimethylamino)azobenzenesulfonyl chloride. Anal. Chem. 1986;58:2375–2379. doi: 10.1021/ac00125a006. [DOI] [PubMed] [Google Scholar]
- 25.Blanchard J. S. Buffers for enzymes. Methods Enzymol. 1984;104:404–414. doi: 10.1016/s0076-6879(84)04107-0. [DOI] [PubMed] [Google Scholar]
- 26.Segel I. H. New York: John Wiley & Sons Inc.; 1993. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. [Google Scholar]
- 27.Knight C. G., Willenbrock F., Murphy G. A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- 28.Neumann U., Kubota H., Frei K., Ganu V., Leppert D. Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and TACE. Anal. Biochem. 2004;328:166–173. doi: 10.1016/j.ab.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 29.Crompton I. E., Waley S. G. The determination of specificity constants in enzyme-catalysed reactions. Biochem J. 1986;239:221–224. doi: 10.1042/bj2390221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura N., Satomura S., Matsuura S., Murakami K. Identification of the active site of human renin with use of new fluorogenic peptides. J. Biochem. (Tokyo) 1991;109:741–745. doi: 10.1093/oxfordjournals.jbchem.a123450. [DOI] [PubMed] [Google Scholar]
- 31.Pilote L., McKercher G., Thibeault D., Lamarre D. Enzymatic characterization of purified recombinant human renin. Biochem. Cell Biol. 1995;73:163–170. doi: 10.1139/o95-020. [DOI] [PubMed] [Google Scholar]
- 32.Cumin F., Le-Nguyen D., Castro B., Menard J., Corvol P. Comparative enzymatic studies of human renin acting on pure natural or synthetic substrates. Biochim. Biophys. Acta. 1987;913:10–19. doi: 10.1016/0167-4838(87)90226-3. [DOI] [PubMed] [Google Scholar]
- 33.Stammers D. K., Dann J. G., Harris C. J., Smith D. R. Comparison of angiotensinogen and tetradecapeptide as substrates for human renin. Arch. Biochem. Biophys. 1987;258:413–420. doi: 10.1016/0003-9861(87)90362-6. [DOI] [PubMed] [Google Scholar]
- 34.Stein R. L. Catalysis by human leukocyte elastase. 5. Structural features of the virtual transition state for acylation. J. Am. Chem. Soc. 1985;107:7768–7769. [Google Scholar]
- 35.Wang W., Liang T. C. Fluorogenic peptides containing only α-amino acids. Biochem. Biophys. Res. Commun. 1994;201:835–840. doi: 10.1006/bbrc.1994.1776. [DOI] [PubMed] [Google Scholar]
- 36.Green D. W., Aykent S., Gierse J. K., Zupec M. E. Substrate specificity of recombinant human renal renin: effect of histidine in the P2 subsite on pH dependence. Biochemistry. 1990;29:3126–3133. doi: 10.1021/bi00464a032. [DOI] [PubMed] [Google Scholar]
- 37.Dealwis C. G., Frazao C., Badasso M., Cooper J. B., Tickle I. J., Driessen H., Blundell T. L., Murakami K., Miyazaki H., Sueiras-Diaz J., et al. X-ray analysis at 2.0 Å resolution of mouse submaxillary renin complexed with a decapeptide inhibitor CH-66, based on the 4–16 fragment of rat angiotensinogen. J. Mol. Biol. 1994;236:342–360. doi: 10.1006/jmbi.1994.1139. [DOI] [PubMed] [Google Scholar]