

Abstract

Among all amino acids, the three aromatic systems including phenylalanine, tyrosine, and tryptophan are known to manifest UV light absorption at the higher wavelengths. Their fluorescent properties are important for protein detection and determination of the spatial structure. Based on ab initio quantum chemical calculations, the absorption spectra in the 0–12 eV UV range of these aromatic amino acids were interpreted, and the photochemistry of the lowest-lying excited states was studied. For that, molecular ground-state geometries were determined using both the coupled-cluster single and double (CCSD) and complete active space self-consistent field (CASSCF) methods. The vertical electronic transition energies, associated oscillator strengths, and electric dipole moments of the lowest excited states were computed at the CASPT2//CASSCF level. The decay mechanisms of the lowest-energy excited states were investigated by performing minimum energy path (MEPs) computations. The results showed that the CCSD and CASSCF computations yielded similar structures. The lowest-energy S1 state in phenylalanine and tyrosine, and S1 and S2 in tryptophan are mainly described by the π → π* excitation localized in the aromatic ring. Upon light absorption, the main photoresponse of the molecules is driven by the benzene, phenol, and indole chromophoric units.
Introduction
The photophysics and photochemistry1−4 of sunlight-absorbing aromatic amino acids are essential in photobiology, biophysics, and practical applications. The 20 amino acids5,6 constitute the building blocks of proteins which are complex systems built from more than 100 amino acids through the formation of peptide bonds. Proteins possess binding sites known as regions crucial for interacting with specific substrates. To understand the biological role they play, the determination of their three-dimensional structures is crucial, which is particularly achieved by X-ray diffraction.7 However, this method is limited to crystallized proteins with a spatial conformation that may differ significantly from that of the native form. An alternative method to determine the spatial arrangement, which applies directly to biological systems, is by analyzing the fluorescence features of proteins.3,4,8,9 This strategy is based on the sensitivity of the chromophoric parts to microenvironmental conditions, thereby facilitating the correlation of the photophysical properties of proteins with the three-dimensional structure.
The three amino acids studied herein are considered the main aromatic chromophoric constituents of proteins: phenylalanine, tyrosine, and tryptophan (Figure 1).1 They include benzene, phenol, and indole, which are the moieties responsible for UV light absorption. The three aromatic amino acids show characteristic ultraviolet absorptions in the region of 220 to 190 nm (5.64–6.53 eV) and around 280 nm (4.43 eV).10,11 After UV light absorption, they re-emit the absorbed light in the form of fluorescence. Tryptophan, with its indole component present in other biologically relevant molecules, such as the neurotransmitter serotonin, is the most significant emissive source among aromatic amino acids. Indole is an π conjugated heterocyclic organic molecule with a bicyclic structure, composed of a benzene ring fused with a pyrrole ring. Measurements of the absorption and fluorescence spectra serve as valuable methods for amino acid identification and protein detection. Therefore, it is crucial to know with accuracy the electronic structure characteristics and geometrical properties related to the absorption and emission phenomena in such amino acids.
Figure 1.

Structures of aromatic amino acids, (a) phenylalanine, (b) tyrosine, and (c) tryptophan.
The geometries of phenylalanine and tyrosine were elucidated experimentally in 1970.12 Slight variations in crystal structures were identified among d-, l-, and dl-tyrosine. Khawas12,13 reported the unit cell dimensions and space groups for both tyrosine and phenylalanine. Mostad et al.14,15 conducted a single crystal X-ray analysis of l-tyrosine which showed unit cell dimensions half the size of those stated by Khawas.12,13 In a subsequent study, Mostad and Rømming reported the same data for dl-tyrosine.14
Frey et al.16 presented a precision neutron diffraction structure determination of l-tyrosine and l-tyrosine hydrochloride while evaluating all previous experimental data on tyrosine. The results represent the most accurate and reliable experimental information about the geometrical parameters of tyrosine, serving as the basis for comparison with the current theoretical data.
Davi Creed conducted a comprehensive exploration of the photophysics and photochemistry of near-UV absorbing amino acids in tyrosine and some simple analogs.17 Additionally, the mechanistic studies of direct tyrosine and phenol photolysis focused on specific details of the photoionization and OH bond homolysis. The obtained results regarding possible electron transfer during intramolecular quenching of fluorescence in the phenolic chromophore by the side chain in tyrosine or its derivatives remained inconclusive. Davi Creed also reported that the phenolic forms of tyrosine and its simple derivatives showed absorption maxima in water at around 220 and 275 nm. Weber et al. suggested these two bands arise as a consequence of well-separated π → π* transitions.18
A previous study1 investigated the indole main decay path based on the complete-active-space second-order perturbation theory // complete-active-space self-consistent field (CASPT2//CASSCF), that is, CASPT2 energy profiles computed on top of CASSCF geometries. The findings showed a conical intersection (CI) between the initially populated 1(La, π → π*) state and the 1(Lb, π → π*) state accessible through a barrier-free path from the Franck–Condon region. At this CI, a population portion changes from 1(La, π → π*) to 1(Lb, π → π*), and the system evolves to the minimum structure of the latter, from where fluorescence occurs. The relatively low emission yield was attributed to a nonradiative mechanism from 1(Lb, π → π*) to the ground state via an ethene-like CI.
Roos and co-workers studied benzene and phenol valence excited states using also CASPT2//CASSCF.19 The absorption spectra were consistent with experimental data, showing an average error of approximately 0.1 eV, and a π → π* nature was observed for the lowest-lying states.
The previous experimental and theoretical studies provide valuable knowledge of the spectroscopical, photophysical, and photochemical properties of the aromatic amino acids. Nevertheless, additional data are useful regarding the analysis of all types of orbital excitations in the UV region and the structural changes that occur during the main decay of the molecules following light absorption at the lowest-lying excited states. Therefore, this study aims at exploring such electronic structures and geometrical features. Emphasis is also placed on verifying if the main photoresponse is similar or not to that of the benzene, phenol, and indole1 chromophoric units using the same methodological approach at the CASPT2//CASSCF level for an accurate comparison.
Methods
The ground-state (S0) geometries were determined with the CCSD and CASSCF methods and with the cc-pVDZ and ANO-L-VDZP basis sets, respectively. Conformer structures of the aromatic amino acids are consistent with the previous studies.2,20 Meanwhile, the three amino acid excited state (S1) geometries and minimum energy paths were obtained with the CASSCF method. No symmetry constraints were used in these calculations. Next, dynamical electron correlations were accounted for in the energies by using the CASPT2 method with the ANO basis set of L-type contracted to C, O, N [4s, 3p,1d]/H[2s,1p]. CASSCF/CASPT2 and CCSD computations were conducted with MOLCAS 7.721 and Gaussian 0922 programs, respectively.
In the absorption spectra study, the nCO, nring, πring, πCO, π*ring, and π*CO orbitals were included in the CASSCF/CASPT2 active space. This corresponds to active spaces with 12 electrons in 10 orbitals for phenylalanine (12e, 10o), 12 electrons in 10 orbitals for tyrosine (12e, 10o), and 14 electrons in 12 orbitals for tryptophan (14e, 12o). State-average CASSCF calculations were performed with 30 roots. For the determination of the fluorescence properties, which are only determined by the π-conjugated system, only the π and π* orbitals plus the lone pair of the nitrogen of the indole ring, which is conjugated with the π and π* orbitals, were included in the active space: (8e, 8o), (8e, 8o), and (12e, 11o) for phenylalanine, tyrosine, and tryptophan, respectively. Four roots were state-averaged in CASSCF calculations.
In the CASPT2 computations, a 0.2 au imaginary level-shift correction was applied to minimize the effect of potential intruder states, and the standard zeroth-order Hamiltonian was used as originally implemented.23 Its validity has been well-established previously on organic molecules, giving accurate descriptions and interpretations as compared photophysical experimental data.24,25
Figure 2 presents the spectroscopically relevant orbitals in tyrosine, which include 6 π and π* orbitals of the aromatic ring, 2 π and π* orbitals of the CO double bond, 1 lone pair with two nonbonding electrons of the oxygen atom for the CO group, and a −NH2 group lone pair. The analogous active space is used for phenylalanine. In tryptophan, Figure 3 displays the important orbitals for tryptophan, which consists of 9 π and π* orbitals of the aromatic ring, 2 π and π* orbitals of the CO double bond, and the oxygen lone pair of this carbonyl group.
Figure 2.

State-averaged CASSCF molecular orbitals in the active space chosen for tyrosine. The analogous orbitals are chosen for phenylalanine.
Figure 3.

State-average CASSCF molecular orbitals in the active space chosen for tryptophan.
Results and Discussion
Ground State (S0) and Excited State (S1) Equilibrium Structures
CCSD and CASSCF Ground-State (S0) Geometries
The stable conformations of the three aromatic systems were inferred from the previous studies by Dehareng et al. and Gindensperger et al.2,20 To assess the suitability of the CASSCF method for optimizing the ground-state geometries, a comparison was performed between the ground-state geometries obtained at the CASSCF/ANO-L-VDZP level and those obtained at the CCSD/cc-pVDZ level (Table 1). The labels of the backbone and aromatic ring are presented in Figure 4.
Table 1. Range of Geometrical Deviations for the S0 Geometry Obtained with CCSD and CASSCF.
| phenylalanine | tyrosine | tryptophan | |
|---|---|---|---|
| Bond lengths (Å) | 0.005–0.03 | 0.005–0.03 | 0.005–0.03 |
| Bond angle (deg) | 0.002–4 | 0.0005–4 | 0.009–4 |
| Dihedral angle (deg) | 0.012–5 | 0.03–5 | 0.003–7 |
Figure 4.
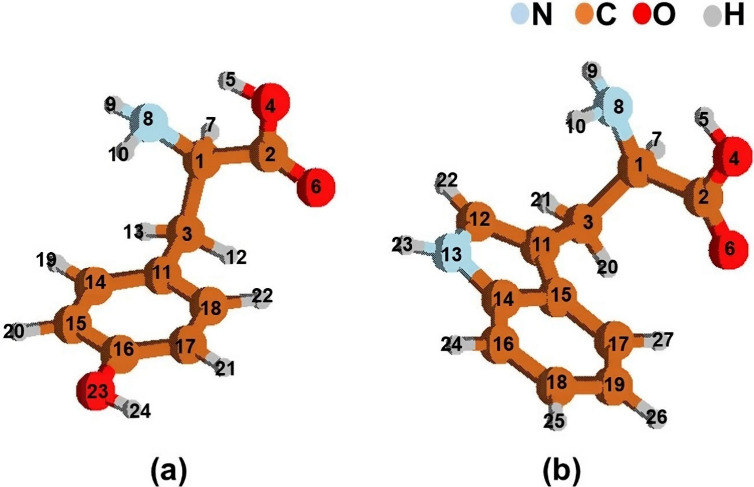
Structures with the atom labels of (a) phenylalanine/tyrosine and (b) tryptophan in their ground state geometries.
Table 1 shows that the largest differences in bond lengths, bond angles, and dihedral angles of the aromatic amino acids are 0.03 Å, 4°, and 7°, respectively, and are primarily located in the hydroxyl and amino groups of the molecular backbone. The larger differences appear in the bond lengths (4,5), (8,10), and (8,9); in the bond angles (2,4,5) and (1,8,10); and in the dihedral angles (2,1,8,9), (7,1,8,9), (7,1,2,6), (3,1,8,9), and (8,1,2,6). The differences are not very big in the aromatic ring, where the excitation is localized, so we conclude that CASSCF optimization provides accurate ground state geometries for the system here studied.
The CCSD method considerably overestimates the values of the excitation energies. This is due to the highly multiconfigurational characters of the excited states, which are not correctly treated at this level of theory. It is known that the CASSCF method with which the geometries were optimized may suffer from a lack of dynamic correlation. However, the cheaper CASSCF method can be used to determine geometries, and then CASPT2 calculations are carried out to add the dynamic electron correlation contributions yielding accurate estimates for the energies. Therefore, we used the CASSCF method to obtain the excited state geometries.
The CASSCF S0 geometries were also compared with tyrosine experimental data16 in Tables S1, S2, S3, S4a, and S5a, and with experimental data corresponding to the aromatic ring of tryptophan (indole)26 in Tables S4b and S5b, for further benchmarking our results. Tables S1–S3 show bond lengths and angles as well as dihedral angles for the backbone of the aromatic amino acids, showing a good agreement with the experiment.16Tables S4 and S5 show the S0 bond lengths and angles for the aromatic rings of phenylalanine, tyrosine, and tryptophan. Tyrosine and phenylalanine show slight differences in the C16 vicinity, where tyrosine has the hydroxyl group instead of hydrogen. In the aromatic ring, bond lengths and angles were almost identical. Moreover, geometries of tryptophan (Figure 5b) ring closely matched the experimental data reported by Takigawa et al.26 based on the measured bond lengths and angles of the indole unit. Overall, differences between calculated and experimental data in tyrosine for bond lengths are on average 0.015 Å. The largest difference is related to the carboxyl group (0.05 Å of the discrepancy). Additionally, average differences on calculated and experimental bond angles were 2°, and the largest deviation was 5° and related to the amino group. For tyrosine and phenylalanine, it is also worth analyzing the differences in the bond lengths of both molecules from experimental and theoretical viewpoints. Tables S4a and S5a indicate that the average calculated difference is 0.014 Å, and the experimental one is 0.03 Å. The largest differences appear once more at the C16 vicinity. The results for tryptophan (Tables S4b and S5b) show that the calculated geometrical features deviate only slightly from the experimental ones. Here, the average differences were not calculated because only the indole related bond lengths and angles were experimentally determined. From our results, we consider that the agreement between the computed and the experimental results is satisfactory, except for the carboxyl and amino groups where the discrepancies are somewhat larger.
Figure 5.

Computed gas phase absorption spectra of (a) phenylalanine and (b) tyrosine.
CASSCF Lowest-Lying Singlet Excited State Structure
Generally, aromatic amino acid molecules have planar geometries in the aromatic ring for S1 and S0. Table 2 shows the geometrical differences between S1 and S0 found at the CASSCF/ANO-L-VDZP level for the three amino acids. Changes in bond lengths and angles as well as dihedral angles are of key interest to understanding the structural photoresponse to light absorption. The observed changes on S1 and S0 are small in all cases. In Table 2, the largest differences of the bond lengths are 0.04 Å (phenylalanine and tyrosine) and 0.06 Å (tryptophan). The largest differences for all amino acids in the bond and dihedral angles are 0.3–4° and 1.3–3.5°. The largest difference in bond lengths appears in the aromatic ring. S1 has larger bond lengths in both phenylalanine and tyrosine rings. For tryptophan, bond lengths increase in S1 except for the N13–C14 and C11–C15 bonds of the pyrrole ring (see labeling in Figure 4b), which decrease. This suggests electron localization occurring in S1, which was also observed in indole.2 The larger changes of bond and dihedral angles from S0 to S1 appear in the backbone (Figure 4a–b), except for tyrosine, in which the hydroxyl group shows big changes in the bond and dihedral angles.
Table 2. CASSCF S1 and S0 Range of Geometrical Differences (S1–S0).
| phenylalanine | tyrosine | tryptophan | |
|---|---|---|---|
| Lengths (Å) | 0–0.04 | 0–0.04 | 0–0.06 |
| Angle (deg) | 0.0–0.3 | 0.0–1.7 | 0.08–4 |
| Dihedral angle (deg) | 0.03–1.3 | 0.02–1.4 | 0.03–3.5 |
Absorption and Emission Spectra
Phenylalanine and Tyrosine
CASPT2 (12e, 10a) vertical excitation energies, oscillator strengths, and electric dipole moments can be found in Tables 3 and 4 and Figure 5. The lowest energies on electronic excitation were located vertically at 4.66 eV (∼266 nm) in phenylalanine and 4.49 eV (∼276 nm) in tyrosine. These transitions are attributed to S1 (π, π*) and correspond to an excitation from a bonding π orbital to an antibonding π* orbital. Additionally, there are 29 states located vertically between 4.4 eV (∼282 nm) and 12 eV (∼103 nm) above the ground state. Some trends are observed from the overall data. Thus, the most intense band (π, π*) in phenylalanine and tyrosine peaks at 6 eV. The (π, π*) transitions have the largest oscillator strengths, which are caused by a higher transition probability. The charge transfer excitation of (n, π*) nature corresponding to the excitation from the nNH2 orbital to the antibonding π* orbital of the aromatic ring appears at high energies in tyrosine. Localized excitations (π and π*) appear at low energies in both molecules. The S2 is identified as (n, π*) and corresponds to the excitation from nCO to π* in the carbonyl group. The S1, S3, and S4 states are (π, π*) related to the aromatic ring. By comparing phenylalanine and tyrosine, the −OH group of tyrosine increases the π orbitals energies of the aromatic ring, stabilizing the excited states and increasing the S1 associated oscillator strength. Hence, tyrosine can absorb much more solar radiation at long wavelengths. The electric dipole moments of phenylalanines S1 and S0 are larger than those of tyrosine. In each molecule, the electric dipole moment does not change between the ground and excited state, which indicates that polar solvent effects should not affect these spectroscopic properties. The lowest S1 states of both molecules are those mainly responsible for light absorption in the lowest energy part of the spectrum. The absorption energies are consistent with experimental data.27,28
Table 3. Computed Nature, Excitation Energies (ΔE, in eV), Oscillator Strengths (f), and Electric Dipole Moments (μ, in D) for the Lowest Electronic States in Phenylalanine.
| state | nature | ΔECASPT2 | f | μ |
|---|---|---|---|---|
| S0 | 85.8%... | ... | ... | 4.85 |
| S1 | 67.2% πr πr* | 4.66 (4.64)27 | 0.000053 | 4.92 |
| S2 | 71.6% nCOπCO* | 5.62 | 0.00186 | 3.67 |
| S3 | 78.3% πr πr* | 5.67 | 0.00265 | 4.35 |
| S4 | 80.4% πr πr* | 6.12 | 0.784 | 4.64 |
Table 4. Computed Nature, Excitation Energies (ΔE, in eV), Oscillator Strengths (f), and Electric Dipole Moments (μ, in D) for the Lowest Electronic States in Tyrosine.
| state | nature | ΔECASPT2 | f | μ |
|---|---|---|---|---|
| S0 | 85.6%... | ... | ... | 3.28 |
| S1 | 75.4% πr πr* | 4.49(4.40)27 | 0.0056 | 3.27 |
| S2 | 81.0% nCO πCO* | 5.54 | 0.0019 | 2.18 |
| S3 | 65.5% πr πr* | 5.67 | 0.006 | 4.01 |
| S4 | 79.1% πr πr* | 5.98 | 0.759 | 3.36 |
After UV irradiation and population of the lowest-lying state, S1, the MEP computations on this excited state show a free-barrier path leading directly from the Franck–Condon region to the S1 minimum. In the Supporting Information, Figure S1a–b depicts the S1 states energy evolution through the steepest descent path toward the equilibrium S1 geometry for phenylalanine and tyrosine. Here, the molecule is trapped on the excited state surface, and emission will take place.
For the S1 state, the computed vertical emission energies (EVE) and band origin (Te), together with the vertical absorption energy (EVA), are compiled in Table 5 and Table 6 (see scheme for the definition of this magnitude in Figure S1c). The findings agree with the reported experimental data.27,28 This means that the structure found in the excited-state minimum is responsible for the light emission. We also compared our results of phenylalanine with those of benzene and tyrosine with those of phenol. The CASPT2 values of benzene and phenol are 4.84 and 4.53 eV, respectively. The experimental values of benzene and phenol are 4.9 and 4.51 eV, respectively.19 Overall, we conclude that the photochemical properties of phenylalanine are controlled by benzene, and the photochemical properties of tyrosine are controlled by phenol.
Table 5. Computed Vertical Absorption Energies (EVA), Vertical Emission Energies (EVE), and Band Origin (Te) for Phenylalaninea.
|
EVA |
EVE |
Te |
||||
|---|---|---|---|---|---|---|
| S1 state | eV | nm | eV | Nm | eV | nm |
| CASSCF | 4.87 | 255 | 4.46 | 278 | 4.64 | 267 |
| CASPT2 | 4.66 | 266 | 4.30 | 288 | 4.44 | 279 |
| Experimental27,28 | 4.64 | 267 | 4.39 | 282 | - | - |
Available experimental data are also included.
Table 6. Computed Vertical Absorption Energies (EVA), Vertical Emission Energies (EVE), and Band Origin (Te) for Tyrosinea.
|
EvA |
EvE |
Te |
||||
|---|---|---|---|---|---|---|
| S1 state | eV | nm | eV | Nm | eV | nm |
| CASSCF | 4.83 | 257 | 4.42 | 281 | 4.59 | 270 |
| CASPT2 | 4.49 | 276 | 4.15 | 299 | 4.28 | 290 |
| Experimental27,28 | 4.402 | 282 | 4.09 | 304 | - | - |
Available experimental data are also included.
Tryptophan
Table 7 and Figure 6 show the CASPT2 vertical excitation energies, oscillator strengths, and electric dipole moments for tryptophan. The lowest state is vertically located at 4.33 eV (∼286 nm). This transition is attributed to S1 (π, π*) corresponding to an excitation from a bonding π orbital to an antibonding π* orbital in the aromatic ring. The analysis of the 29 states located vertically between 4.2 eV (∼296 nm) and 12 eV (∼103 nm) shows the following trends. Due to tryptophan containing the largest ring size among the aromatic amino acids, it shows more intense bands related to (π, π*) excitations than phenylalanine and tyrosine. S3 and S4 states of tryptophan have (n, π*) nature, corresponding to excitations from the carbonyl lone pair orbital (nCO) to the antibonding π* orbital. Moreover, the S1 and S2 states have a (π, π*) nature showing localization in the aromatic ring, consistent with the CASPT2 energy position for the Lb and La states of indole,1 which were found at 4.36 and 4.77 eV, respectively. La is characterized by a higher electric dipole moment, 9.68 D, compared to the Lb and S0 states, with values of 6.02 and 6.62 D, respectively. This indicates a slight decrease in the energy of La with respect to that of Lb and S0 in polar solvents. Considering the nearby energies of S1 and S2, the two states absorb sunlight in the low energy part of the spectrum.
Table 7. Computed Nature, Excitation Energies (ΔE, in eV), Oscillator Strengths (f), and Electric Dipole Moments (μ, in D) for the Lowest Electronic States in Tryptophan.
| State | Nature | ΔECASPT2 | f | μ |
|---|---|---|---|---|
| S0 | 83.3%... | ... | ... | 6.627 |
| S1 | 66.8% πr πr* | 4.3324 | 0.0177 | 6.023 |
| 4.42 | ||||
| S2 | 58.1%πr πr* | 4.77 | 0.0672 | 9.684 |
| S3 | 46.8% nCOπCO* | 5.71 | 0.0358 | 5.808 |
| S4 | 69.8% nCOπCO* | 5.71 | 0.002 | 5.039 |
Figure 6.

Computed gas phase absorption spectra of tryptophan.
Based on the experience of some of the authors of this work and other groups on distinct type of excited states,2,18,29−35 we can state that the two lowest excited states examined in the present study are pure valence states. Only the excited-state evolution of these two low-lying singlet valence excited states, Lb and La, will be further analyzed.
By considering the oscillator strengths, it is possible to assert that most of the molecules will reach the La state after UV irradiation. From the FC geometry, the MEP on the La state shows a barrierless path leading directly to a CI involving Lb and La, placed at 4.44 eV with respect to the ground-state minimum (Figure S2). Such a degenerate region is a converged point on the obtained MEP, and it is not the lowest-energy point of the crossing seam but the first funnel that is reached by the excited molecule. Consequently, it is the most photophysically relevant crossing point. The CI has a basically planar geometry, and the single and double bonds are interchanged with respect to the S0 equilibrium structure. The CI was previously characterized by Brand and co-workers36,37 at the DFT/MRCI level. An efficient internal conversion (IC) process occurs at the CI bringing the molecule from La to Lb. Most MEP points in the present calculations show nearby energies between Lb and La, suggesting a high probability of populating Lb along the decay path of the La state from FC. However, the La equilibrium structure has not been determined. The previous study on indole1 at the CASPT2//CASSCF level reported an equilibrium structure for La,. Such an La minimum in indole was situated only at 0.12 eV below Lb, and it is nearly degenerate with the CI point, therefore mediating ultrafast relaxation to the Lb state. In tryptophan, where there is even less evidence of the La minimum, this state is not expected to contribute to the fluorescence phenomenon, contrasting with the early suggested dual fluorescence in polar and nonpolar solvents based on polarization measurements. Additionally, our study seems to be more in agreement with the later experimental results suggesting that while two different states seem to contribute to the lowest-energy absorption band only one produces emission. Figure S2c shows the MEP from the CI, where Lb evolved through the steepest descent path to Lb equilibrium geometry. This structure is the same as that obtained by MEP computations directly from the FC region on the Lb state (Figure S2a–b). From the Lb minimum geometry, emissions will occur. For this state (Lb), the computed CASPT2 EVA, EVE, and Te are 4.33, 3.98, and 4.09 eV, respectively, as compiled in Tables 8, which are consistent with previous experimental data.27,28
Table 8. Computed Vertical Absorption Energies (EVA), Vertical Emission Energies (EVE), and Band Origin (Te) for Tryptophana.
|
EVA |
EVE |
Te |
||||
|---|---|---|---|---|---|---|
| S1 state | eV | nm | eV | nm | eV | nm |
| CASPT2 | 4.33 | 286 | 3.96 | 313 | 4.08 | 304 |
| Experimental27,28 | 4.42 | 281 | 3.56 | 348 | - | - |
Available experimental data are also included.
According to the previous description, both La and Lb states populated after light absorption decay to the Lb minimum, which is the emissive structure. The comparative analysis with indole1 shows that tryptophan absorption and emission energies and indole had significant similarities. Therefore, the main photochemical properties of tryptophan are driven mainly by indole.
Conclusions
In conclusion, this study shows that the optimization of the ground state geometries of aromatic amino acids using CCSD and CASSCF gives similar structural features. The largest differences in the ground state geometries appear in the backbone and not in the aromatic ring; therefore, we can consider that the CASSCF method is accurate enough for optimizing the excited state geometries. The theoretical results of the optimization of the ground and excited state geometries of phenylalanine, tyrosine, and tryptophan using CASSCF show that structural changes are mainly localized on the aromatic ring. The hydroxyl group of the aromatic ring of tyrosine decreases the excitation energies and increases the absorption intensity at long wavelengths. The states absorbing at the lowest energy part of the spectrum are S1 for phenylalanine and tyrosine and S1 and S2 for tryptophan. These states are πr → πr* excitations localized in the aromatic ring. The photoresponse of the amino acids when these states are populated is to drive the molecules toward the equilibrium geometry of S1, from where they will emit fluorescence light. The main emission properties are totally driven by benzene, phenol, and indole in phenylalanine, tyrosine, and tryptophan, respectively.
Acknowledgments
The authors are grateful for the support received through the EM-TCCM program, RISPRO LPDP No. 123/E4.1/AK.04.RA/2021, and a part of DPPM UII No. 004/Dir/DPPM/70/Pen.Pemula/XII/2023. We extend our sincere gratitude to Minori Abe and Masahiko Hada for their invaluable support in facilitating the calculations needed for the revisions of this paper. S. is also thankful to Manuela Merchán, Ria Broer, Remco Havenith, Claudiu Sergentu, Angelo Giussani, and Antonio Francés-Monerris for the valuable discussions and assistance provided in the Molcas program as well as to Arif Darmawan who helped with Mendeley referencing. D.R.-S. acknowledges support by the Spanish Agencia Estatal de Investigación of the Ministerio de Ciencia e Innovación (MICINN) and the European Regional Development Fund (FEDER) through project No. PID2021-127199NB-I00 and by the Conselleria d’Innovació, Universitats, Ciència i Societat Digital through project No. CIAICO/2022/121.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c00875.
The bond lengths (Å), bond angles (deg), and dihedral angles (deg) of the backbone in the ground state geometries. The bond length (Å) and bond angles (deg) of the aromatic ring in the ground state geometries. Computed absorption spectrum of tyrosine and phenylalanine in 30 roots. Evolution of the S1 MEP of phenylalanine and tyrosine. Scheme for the definition of vertical absorption energy (EVA), the electronic band origin (Te), and vertical emission energy (EVE). Evolution of the S1 MEP of tryptophan and evolution of S2 MEP. XYZ Cartesian coordinates of relevant structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Giussani A.; Merchán M.; Roca-Sanjuán D.; Lindh R. Essential on the Photophysics and Photochemistry of the Indole Chromophore by Using a Totally Unconstrained Theoretical Approach. J. Chem. Theory Comput 2011, 7, 4088–4096. 10.1021/ct200646r. [DOI] [PubMed] [Google Scholar]
- Gindensperger E.; Haegy A.; Daniel C.; Marquardt R. Ab initio study of the electronic singlet excited-state properties of tryptophan in the gas phase: The role of alanyl side-chain conformations. Chem. Phys. 2010, 374, 104–110. 10.1016/j.chemphys.2010.07.004. [DOI] [Google Scholar]
- Liu H.; Zhang H.; Jin B. Fluorescence of tryptophan in aqueous solution. Spectrochim Acta - Part A Mol. Biomol Spectrosc 2013, 106, 54–59. 10.1016/j.saa.2012.12.065. [DOI] [PubMed] [Google Scholar]
- Callis P. R. Binding phenomena and fluorescence quenching. II: Photophysics of aromatic residues and dependence of fluorescence spectra on protein conformation. J. Mol. Struct. 2014, 1077, 22–29. 10.1016/j.molstruc.2014.04.051. [DOI] [Google Scholar]
- Young T. S.; Schultz P. G. Beyond the canonical 20 amino acids: expanding the genetic lexicon. J. Biol. Chem. 2010, 285, 11039–44. 10.1074/jbc.R109.091306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton H.Principles of Biochemistry; Prentice Hall, 2002; Vol. 3. [Google Scholar]
- Klug H. P.; Alexander L. E.. X-ray Diffraction Procedures: for Polycrystalline and Amorphous Materials, 2nd ed.; Wiley, New York (N.Y.), 1974. [Google Scholar]
- Savarese M.; Aliberti A.; De Santo I.; Battista E.; Causa F.; Netti P. A.; Rega N. Fluorescence Lifetimes and Quantum Yields of Rhodamine Derivatives: New Insights from Theory and Experiment. J. Phys. Chem. A 2012, 116, 7491–7497. 10.1021/jp3021485. [DOI] [PubMed] [Google Scholar]
- Rothenberger G.; Fitzmaurice D.; Graetzel M. Spectroscopy of conduction band electrons in transparent metal oxide semiconductor films: optical determination of the flatband potential of colloidal titanium dioxide films. J. Phys. Chem. 1992, 96, 5983–5986. 10.1021/j100193a062. [DOI] [Google Scholar]
- Demchenko A. P. Ultraviolet Spectroscopy of Proteins. 1987 1987, 568–568. 10.1515/9783112581704-020. [DOI] [Google Scholar]
- Wetlaufer D. B. Ultraviolet spectra Of Proteins and Amino Acids. Adv. Protein Chem. 1963, 17, 303–390. 10.1016/S0065-3233(08)60056-X. [DOI] [Google Scholar]
- Khawas B.; The unit cells and space groups of L-methionine, L-β-phenylalanine and DL-tyrosine. Acta Crystallogr. Sect B Struct Crystallogr. Cryst. Chem. 1970, 26, 1919–1922. 10.1107/S0567740870005186. [DOI] [Google Scholar]
- Khawas B.; X-ray study of L-arginine HCl, L-cysteine, DL-lysine and DL-phenylalanine. Acta Crystallogr. Sect B Struct Crystallogr. Cryst. Chem. 1971, 27, 1517–1520. 10.1107/S056774087100431X. [DOI] [Google Scholar]
- Mostad A.; Nissen H. M.; Rømming C.; Olli M.; Pilotti Å Crystal Structure of L-Tyrosine. Acta Chem. Scand. 1972, 26, 3819–3833. 10.3891/acta.chem.scand.26-3819. [DOI] [PubMed] [Google Scholar]
- Mostad A.; Nissen H. M.; Rømming C.; Olli M.; Pilotti A. Crystal structure of L-tyrosine. Acta Chem. Scand. 1972, 26, 3819–3833. 10.3891/acta.chem.scand.26-3819. [DOI] [PubMed] [Google Scholar]
- Frey M. N.; Koetzle T. F.; Lehmann M. S.; Hamilton W. C. Precision neutron diffraction structure determination of protein and nucleic acid components. X. A comparison between the crystal and molecular structures of L-tyrosine and L-tyrosine hydrochloride. J. Chem. Phys. 1973, 58, 2547–2556. 10.1063/1.1679537. [DOI] [Google Scholar]
- Creed D. the Photophysics and Photochemistry of the near-UV Absorbing Amino Acids-II. Tyrosine and Its Simple Derivatives. Photochem. Photobiol. 1984, 39, 563–575. 10.1111/j.1751-1097.1984.tb03891.x. [DOI] [Google Scholar]
- Weber G. Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds. Biochem. J. 1960, 75, 335–345. 10.1042/bj0750335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentzon J.; Malmqvist P-ke; Flscher M.; Roos B. O. A CASPT2 study of the valence and lowest Rydberg electronic states of benzene and phenol. Theor. Chim. Acta 1995, 91, 91–108. 10.1007/BF01113865. [DOI] [Google Scholar]
- Dehareng D.; Dive G. Vertical Ionization Energies of α-L-Amino Acids as a Function of Their Conformation: an Ab Initio Study. Int. J. Mol. Sci. 2004, 5, 301–332. 10.3390/i5110301. [DOI] [Google Scholar]
- Aquilante F.; De Vico L.; Ferre N.; Ghigo G.; Malmqvist P.-a.; Neogrady P.; Pedersen T. B.; Pitonak M.; Reiher M.; Roos B. O.; Serrano-Andres L.; Urban M.; Veryazov V.; Lindh R. MOLCAS 7: The Next Generation. J. Comput. Chem. 2010, 31, 224–247. 10.1002/jcc.21318. [DOI] [PubMed] [Google Scholar]
- Frisch M., Trucks G., Schlegel H., Scuseria G.. Gaussian 09W, Revision A.1; Gaussian: Wallingford, CT, 2009.
- Roos B. O.; Andersson K.; Fülscher M. P.; Malmqvist P.; Serrano-Andrés L.; Pierloot K.; Merchán M.. Multiconfigurational Perturbation Theory: Applications in Electronic Spectroscopy; John Wiley & Sons, Ltd, 2007; pp 219–331. [Google Scholar]
- Serrano-Andrés L., Merchán M.. Photostability and Photoreactivity in Biomolecules: Quantum Chemistry of Nucleic Acid Base Monomers and Dimers. In Radiation Induced Molecular Phenomena in Nucleic Acids; Springer: Dordrecht, Netherlands, 2008; pp 435–472. [Google Scholar]
- Olivucci M. M.Computational Photochemistry; Elsevier, 2005. [Google Scholar]
- Takigawa T.; Ashida T.; Sasada Y.; Kakudo M. The crystal structures of L-tryptophan hydrochloride and hydrobromide. Bull. Chem. Soc. Jpn. 1966, 39, 2369–78. 10.1246/bcsj.39.2369. [DOI] [PubMed] [Google Scholar]
- Feraud K.; Dunn M. S.; Kaplan J. Spectroscopic Investigations of Amino Acids and Amino Acid Derivatives: I. Ultra-Violet Absorption Spectra of l-Tyrosine, dl-Phenylalanine, and l-Tryptophane. J. Biol. Chem. 1935, 112, 323–328. 10.1016/S0021-9258(18)74990-9. [DOI] [Google Scholar]
- Yang H.; Xiao X.; Zhao X.; Wu Y.. Intrinsic Fluorescence Spectra of Tryptophan, Tyrosine and Phenyloalanine. Proceedings of the 5th International Conference on Advanced Design and Manufacturing Engineering; Springer Nature, 2015; pp 224–233. 10.2991/icadme-15.2015.46 [DOI]
- Ashford D. L.; Glasson C. R. K.; Norris M. R.; Concepcion J. J.; Keinan S.; Brennaman M. K.; Templeton J. L.; Meyer T. J. Controlling Ground and Excited State Properties through Ligand Changes in Ruthenium Polypyridyl Complexes. Inorg. Chem. 2014, 53, 5637–5646. 10.1021/ic500408j. [DOI] [PubMed] [Google Scholar]
- Salmahaminati; Abe M.; Purnama I.; Mulyana J. Y.; Hada M. Density Functional Study of Metal-to-Ligand Charge Transfer and Hole-Hopping in Ruthenium(II) Complexes with Alkyl-Substituted Bipyridine Ligands. ACS Omega 2020, 6, 55–64. 10.1021/acsomega.0c01199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D. W.; Ito A.; Meyer T. J. ligand charge transfer (MLCT) excited states *. Pure Appl. Chem. 2013, 85, 1257–1305. 10.1351/PAC-CON-13-03-04. [DOI] [Google Scholar]
- Féraud G.; Broquier M.; Dedonder C.; Jouvet C.; Grégoire G.; Soorkia S. Excited State Dynamics of Protonated Phenylalanine and Tyrosine: Photo-Induced Reactions Following Electronic Excitation. J. Phys. Chem. A 2015, 119, 5914–5924. 10.1021/jp5065837. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Ai Y.-J.; Luo Y.; Fang W.-H. Nonradiative decay of the lowest excited singlet state of 2-aminopyridine is considerably faster than the radiative decay. J. Chem. Phys. 2009, 130, 144315 10.1063/1.3113664. [DOI] [PubMed] [Google Scholar]
- Hameka H. F.; Jensen J. O. Calculations of the energies and geometries of the ground and first excited states of phenylalanine and tyrosine. J. Mol. Struct THEOCHEM 1993, 288, 9–16. 10.1016/0166-1280(93)90089-T. [DOI] [Google Scholar]
- Salmahaminati N.; Inagaki A.; Hada M.; Abe M. Density Functional Study on the Photopolymerization of Styrene Using Dinuclear Ru-Pd and Ir-Pd Complexes with Naphthyl-Substituted Ligands. J. Phys. Chem. A 2023, 127 (12), 2810–2818. 10.1021/acs.jpca.3c01299. [DOI] [PubMed] [Google Scholar]
- Brand C.; Küpper J.; Pratt D. W.; et al. Vibronic coupling in indole: I. theoretical description of the 1La–1Lb interaction and the electronic spectrum. Phys. Chem. Chem. Phys. 2010, 12, 4968–4979. 10.1039/c001776k. [DOI] [PubMed] [Google Scholar]
- Küpper J.; Pratt D. W.; Leo Meerts W.; Brand C.; Tatchen J.; Schmitt M. Vibronic coupling in indole: II. Investigation of the 1La – 1Lb interaction using rotationally resolved electronic spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 4980–4988. 10.1039/c001778g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.