

Abstract
Nonindigenous members of the Daphnia pulex complex have been found in many lakes in New Zealand (NZ) in the past 20 years, suggesting a recent invasion. However, very little is known about the precise phylogenetic origin of invasive Daphnia, whether each lake is invaded by a single clone or multiple clones, the lineage of the invasive clones, and whether they are obligately asexual clones. Furthermore, the source and time of arrival of the invasive genotype(s) are unclear. We address these questions by genomic sequencing of Daphnia populations from 13 lakes in the South Island and 1 lake in the North Island, NZ. All biallelic sites in these NZ populations have similar numbers of reads for the two parental alleles, suggesting each NZ population originates from a single asexual clone. Based on 25,643 monomorphic lineage-specific markers, the invasive Daphnia in the South Island were found to be Daphnia pulicaria Forbes, while those in the North Island are hybrids of D. pulicaria Forbes and D. cf. pulex sensu Hebert. Both the South and North Island Daphnia are phylogenetically clustered with North American Daphnia, thereby suggesting their North American origins. We found also that all South Island clones contain identical mitochondrial genomes, suggesting the origin and proliferation from a single founder clone, which we experimentally verified to be an obligate asexual. Estimates from molecular data imply a colonization time for the South Island clones of ~60 years ago, with a likely invasion route associated with the introduction of salmonids from North America.
Daphnia are small crustaceans commonly found in small lakes and ponds (Benzie 2005). Molecular data revealed that Daphnia pulex is a polyphyletic species complex containing at least eight genetically distinct lineages, which can be assigned to three major clades: the European D. pulex clade (Daphnia pulex Leydig, 1860), the Daphnia tenebrosa Sars, 1898 and European Daphnia pulicaria clade (D. cf. pulicaria sensu Alonso, 1996), and the panarctic D. pulex (D. cf. pulex sensu Hebert, 1995), Daphnia melanica Hebert, 1995, Daphnia middendorffiana Fisher, 1851, and North American D. pulicaria clade (D. pulicaria Forbes, 1893) (Colbourne et al. 1998). Two of the lineages, D. cf. pulex sensu Hebert and D. pulicaria Forbes, can still hybridize and form viable hybrids (Hebert et al. 1989; Crease et al. 2011), and their hybrids have been found to be invasive. For example, a D. cf. pulex sensu Hebert × D. pulicaria Forbes hybrid clone invaded Africa after the 1920s and has spread throughout the range of native sexual D. “pulex” (clones that have been morphologically or genetically determined to be members of the D. pulex species complex but with unclear lineage affiliations), displacing the sexual population (Mergeay et al. 2006). In addition, four hybrid Daphnia clones invaded Japan between 680 and 3400 years ago (So et al. 2015). Recently, nonindigenous Daphnia species morphologically determined to be from the D. pulex complex have been found in several lakes in New Zealand (NZ) (Duggan et al. 2012). Despite the extreme success of Daphnia introductions into several continents, studies on the invasive D. “pulex” are scarce and somewhat ambiguous due to small sample size in certain localities, incorrect lineage identification caused by hybridization, and the lack of testing for obligate asexuality (Duggan et al. 2012; So et al. 2015).
First, most of the previous studies on invasive D. “pulex” were based on just a few clones from each lake or pond (Mergeay et al. 2005; Duggan et al. 2012; So et al. 2015), and when clones within a lake or pond shared a single haplotype, it was concluded that the population consisted of a single Daphnia clone. Because sexual and obligate asexual, indigenous, and nonindigenous D. “pulex” clones can coexist in the same ponds (Mergeay et al. 2006; Lehto and Haag, 2010), and the coexisting clones are often morphologically indistinguishable, by sequencing just a few clones, one or more coexisting genotypes could easily be missed, thus leading to incorrect conclusions.
Second, lineage identification was often based on a single marker gene in previous studies, which may be confounded by hybrids. D. cf. pulex sensu Hebert, D. pulicaria Forbes, and their hybrids are not easily distinguished morphologically, and typically are identified by allozyme electrophoresis of the lactate dehydrogenase (Ldh) locus, with homozygous D. pulicaria Forbes having two “fast” alleles, and homozygous D. cf. pulex sensu Hebert having two “slow” alleles (Hebert et al. 1989; Crease et al. 2011). Hybrids are generally found in disturbed ponds or deforested areas and have a heterozygous slow/fast genotype (Hebert et al. 1989). However, due to the frequent gene flow between D. cf. pulex sensu Hebert and D. pulicaria Forbes (Hebert et al. 1989; Cristescu et al. 2012), some hybrids are also homozygous for the Ldh locus, presumably resulting from backcrossing with either D. cf. pulex sensu Hebert or D. pulicaria Forbes (Xu et al. 2013). Therefore, homozygosity of “fast” or “slow” alleles does not guarantee a specific D. cf. pulex sensu Hebert or D. pulicaria Forbes origin of the invasive Daphnia. In addition to the Ldh locus, the mitochondrial cytochrome c oxidase subunit 1 (CO1) gene has also been used to distinguish invasive D. cf. pulex sensu Hebert and D. pulicaria Forbes (Mergeay et al. 2005; Duggan et al. 2012). Because mitochondrial DNA (mtDNA) is maternally inherited and hybrids could have mtDNA from either D. cf. pulex sensu Hebert or D. pulicaria Forbes (Cristescu et al. 2012; Marková et al. 2013), these hybrids may be erroneously identified as D. cf. pulex sensu Hebert or D. pulicaria Forbes, thus biasing lineage identification.
Third, a number of previous studies did not thoroughly test whether the invasive D. “pulex” clones are obligate parthenogens (OP). Both D. cf. pulex sensu Hebert and D. pulicaria Forbes can reproduce by cyclical parthenogenesis (CP), with extended periods of parthenogenesis interspersed with sexual resting-egg production (Hebert 1978; Hebert et al. 1993). However, some clones in each lineage have lost the ability to engage in meiosis and have become OP (Hebert and Crease 1980; Hebert et al. 1993; Paland et al. 2005). Although OP clones are thought to have much reduced or eliminated male production compared to CP clones, some OP clones can still produce functional males bearing haploid sperm (Innes et al. 2000; Tessier and Cáceres 2004). Such males from OP D. cf. pulex sensu Hebert can mate and form viable hybrids with females from CP D. cf. pulex sensu Hebert, ~40% of which have the OP phenotype (Innes and Hebert 1988), indicating that partially dominant meiosis-suppressing elements are carried by OP males. Further analysis has shown that all OP D. cf. pulex sensu Hebert share common haplotypes on chromosomes VIII and IX that arose by introgression from D. pulicaria Forbes (Lynch et al. 2008; Xu et al. 2015) and are transmitted through OP males without recombination (Tucker et al. 2013). Because CP and OP D. cf. pulex sensu Hebert clones have different traits and evolutionary histories, false designation of the two may lead to incorrect estimation of times and routes of invasion.
To solve the above issues and understand the origin of the invasive D. “pulex” in NZ, we performed whole-genomic sequencing of D. “pulex” populations from 13 lakes across the South Island and one lake in the North Island, NZ. To determine if each lake contains more than one D. “pulex” genotype, we pooled 50–200 individuals from each lake for whole-genomic sequencing and then performed binomial tests on all biallelic sites to check if the numbers of reads for the two parental alleles deviated from the 1 : 1 ratio expected for a fixed heterozygote. The lineage identifications and likely sources of origin of the invasive D. “pulex” were determined via a survey of D. pulicaria Forbes-specific markers and a whole-genomic phylogenetic tree that includes D. “pulex” clones collected across North America, Europe, and China. A combination of experiments and genetic markers implies invasion by OP clones. With a clear lineage identification and whole-genomic molecular data, we were able to estimate the colonization time and predict the likely route of the D. “pulex” invasion in NZ.
Materials and methods
Nomenclatural remarks
There are extensive nomenclature issues within the D. pulex species complex. D. cf. pulex sensu Hebert, 1995 is a lineage in the complex commonly found in Panarctic regions, which is morphologically indistinguishable but genetically different from its European counterpart D. pulex Leydig, 1860. However, both of these two lineages continue to be referred as D. pulex (Hebert et al. 1989; Crease et al. 2011). Likewise, although North American D. pulicaria Forbes, 1893 and the European D. cf. pulicaria sensu Alonso, 1996 have been assigned to different clades in the species complex (Colbourne et al. 1998), they are still both referred as D. pulicaria (Dufresne et al. 2011). In addition, Daphnia clones that have been morphologically or genetically determined to be members of the D. pulex species complex but with unclear lineage affiliations have been referred as D. pulex (So et al. 2015; Geng et al. 2016). To avoid confusion in this study, we use full taxonomic description names (i.e., D. cf. pulex sensu Hebert, 1995) for Daphnia clones from known lineages. For Daphnia clones with undetermined or mixed lineages, we use a geographical prefix and quotation markers to describe them (i.e., South American D. “pulex”).
Sampling and sequencing
Daphnia populations were collected from 13 lakes in the South Island and 1 lake in the North Island, NZ in March and October 2019 (Fig. 1; Supporting information Table S1). In each location, Daphnia were sampled by tows of a conical plankton net and preserved in DNA/RNA Shield. DNA from each pooled population sample (50–200 individuals) was extracted with Zymo DNA Clean and Concentrator-25 Kit and sequencing libraries were prepared using NEXTFLEX® Rapid DNA-Seq Kit 1.0. The 150-bp paired-end reads were sequenced using Highseq 2500 platform at Center for Genomics and Bioinformatics, Indiana University.
Fig 1.
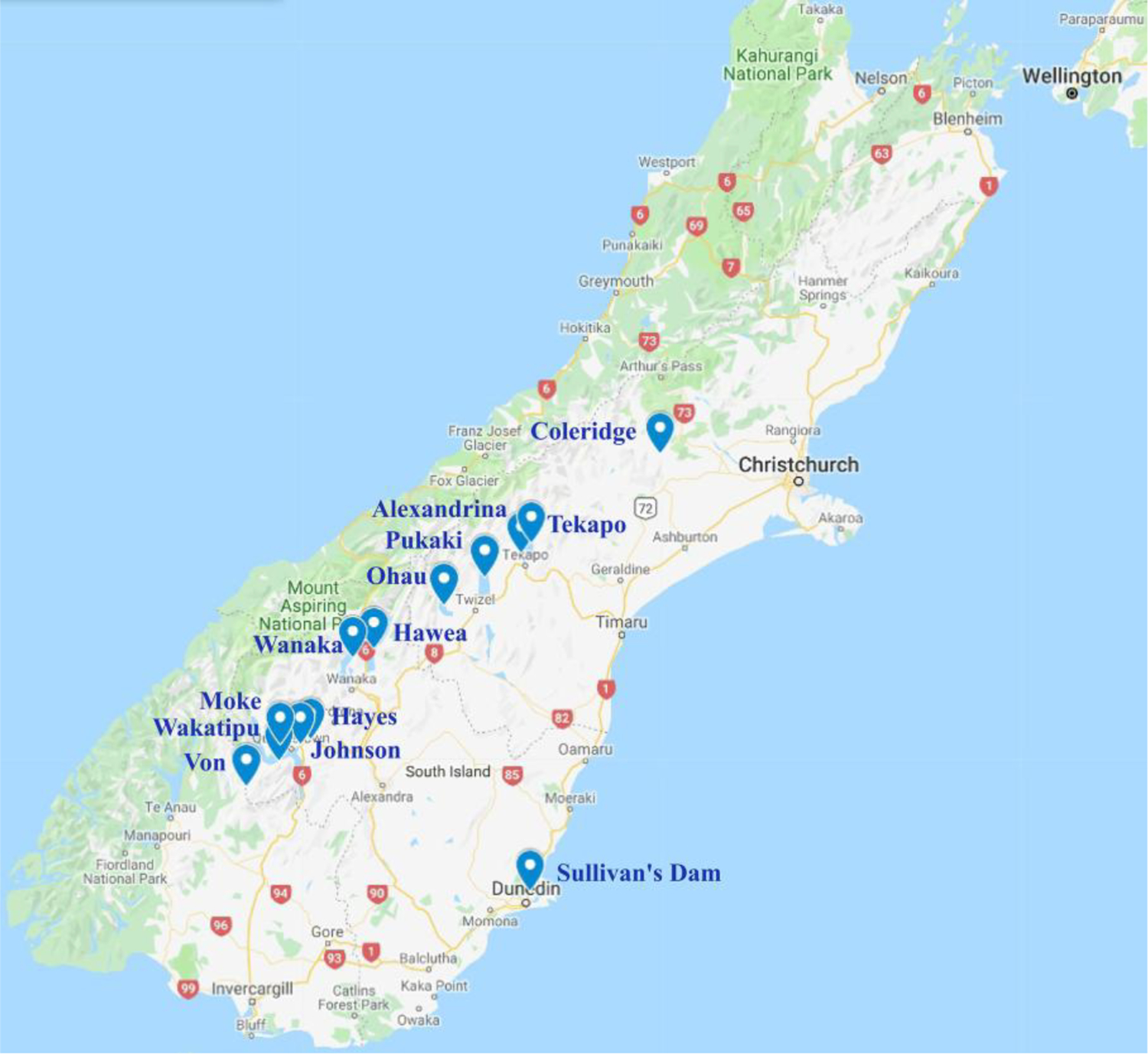
Location of lakes in the South Island, New Zealand, from which the Daphnia in this study were collected (South Island, New Zealand, Google Maps, 16 October 2019). Lake names are highlighted in blue. Lake Kapoai, North Island, not shown.
Reads mapping and SNP calling
First, we trimmed adapter sequences from the sequence reads by applying Trimmomatic (Bolger et al. 2014) to the FASTQ files. Then, we mapped the adapter-trimmed sequence reads to the D. cf. pulex sensu Hebert reference genome (PA42 version 4.1) using Burrows-Wheeler Alignment tool (BWA) (Li and Durbin 2009). Coverage for each site was calculated by counting the total mapped reads, and coverage for each sample is calculated as the total mapped bases divided by the mapped genomic regions. We examined the genomic distribution of the sites coverage and set minimum and maximum sample-coverage cutoffs to avoid analyzing problematic sites (Table 1). Single nucleotide polymorphisms (SNPs) were identified by Samtools (Li et al. 2009) with “samtools mpileup -uf ref.fasta sorted.bam | bcftools call -mv > raw.vcf” and “bcftools filter -s LowQual -e ‘%QUAL<20’ raw.vcf > flt.vcf”.
Table 1.
Summary of sample information. Coverage denotes the mean depth of genomic coverage and chosen coverage range (in square brackets) of sites analyzed in each population. Mapped sites: Sites covered by at least one read. Deviating sites are biallelic sites (minor allele frequency > 0.05) with read numbers for the two parental alleles deviating significantly from 1 : 1 after Benjamini–Hochberg procedure (FDR = 0.05). Sites with more than two alleles are not used in our analysis.
| Samples | Mapped sites | Coverage (X) | Biallelic sites | Deviating sites |
|---|---|---|---|---|
| Alexandrina | 168,224,903 | 98 [40–200] | 1,540,667 | 288 |
| Coleridge | 166,329,006 | 84 [20–150] | 1,491,775 | 252 |
| Hawea | 165,334,107 | 51 [20–120] | 1,416,048 | 35 |
| Hayes | 168,826,070 | 139 [50–250] | 1,538,123 | 667 |
| Johnson | 168,204,869 | 125 [50–250] | 1,523,441 | 572 |
| Ohau | 168,392,915 | 102 [50–200] | 1,539,681 | 340 |
| Pukaki | 168,109,981 | 94 [50–200] | 1,536,485 | 238 |
| Sullivan’s Dam | 167,880,110 | 88 [50–200] | 1,525,683 | 224 |
| Moke | 164,531,082 | 43 [20–100] | 1,509,765 | 41 |
| Tekapo | 166,918,220 | 78 [30–150] | 1,494,053 | 175 |
| Von | 166,794,759 | 82 [30–150] | 1,488,926 | 196 |
| Wakatipu | 167,805,336 | 107 [50–200] | 1,513,257 | 411 |
| Wanaka | 164,530,961 | 42 [20–100] | 1,370,378 | 14 |
| Kapoai | 175,278,062 | 48[20–100] | 2,059,905 | 147 |
If each NZ population represents a single clone, all of its biallelic sites should have equal numbers of reads mapped to the two parental alleles. To check this, we searched for sites with reads for the two parental alleles deviating from a 1 : 1 ratio. To minimize mapping bias, (1) we remapped the reads using bowtie with “-q -m 1 -v 3 -best” to obtain unique mapped reads (Stevenson et al. 2013); (2) we removed regions with > three SNPs within 100 bp because additional differentiating sites will interfere with read alignment (Stevenson et al. 2013); and (3) to reduce the mapping bias toward the reference alleles (Degner et al. 2009), we generated an assembly by masking all of the biallelic sites with “N” in the PA42 4.1 reference. Only biallelic sites with reads for the two alleles deviating from a 1 : 1 ratio from both the original and masked assemblies are considered true deviating sites.
Characterizing D. pulicaria Forbes-specific markers
To identify D. pulicaria Forbes-specific markers, we first searched for homozygous SNPs shared by all 14 D. pulicaria Forbes clones (Supporting Information Table S2). Then, we checked these SNPs in 30 D. cf. pulex sensu Hebert clones collected across North American and eliminated those that appeared in any of the D. cf. pulex sensu Hebert clones. For the remaining SNPs, we checked the corresponding loci in the genome in each of the D. cf. pulex sensu Hebert clones and only kept those that are homozygous for the same two bases across all D. cf. pulex sensu Hebert clones.
Origin and colonization time of the NZ populations
To estimate the origin of the South Island D. pulicaria Forbes clones, mitochondrial CO1 gene sequences for D. “pulicaria” clones sampled across North America, South America, Europe, Asia, NZ, and the north polar region are downloaded from NCBI or generated from raw reads using Samtools (Li et al. 2009) with the command: samtools mpileup -uf ref.fa aln.bam | bcftools call -c | vcfutils.pl vcf2fq > cns.fq. CO1 sequences were aligned with ClustalW program from MEGA 7 and the maximum-likelihood tree based on CO1 gene sequences was constructed using MEGA 7 with 100 Bootstrap (Kumar et al. 2016).
To estimate when the South Island genotype diverged from North American D. pulicaria Forbes, we extracted coding sequence from the reference genome (PA42 4.1). After mapping the reads from each clone to the coding sequences, we generated consensus sequences for each clone using Samtools (Li et al. 2009). Custom scripts were then used to count the number of synonymous/nonsynonymous changes and sites in the coding sequences.
Phylogenetic analysis
To generate clean mitochondrial and nuclear genomes for the phylogenetic analysis, we removed regions that are subjected to transposition of cytoplasmic mitochondrial DNA into the nuclear genome in the historical past. We discarded read pairs for which two reads in the pair mapped to both mitochondrial genome and nuclear genome. First, trimmed reads were mapped to the mitochondrial reference genome using BWA (Li and Durbin 2009) with default parameters. We extracted paired-end reads that aligned to the mitochondrial genome from each BAM file. GSNAP (Wu and Watanabe 2005) was used to realign these reads to the mitochondrial reference genome and nuclear genome separately following the pipeline from MToolBox (Calabrese et al. 2014). Read pairs that mapped to both the mitochondrial and nuclear genomes were removed from downstream analysis.
We generated phylogenetic trees using nuclear and mitochondrial genomes, separately. For the nuclear genome, a phylogenetic tree was constructed using the consensus genomic sequences for each NZ Daphnia population, D. pulicaria Forbes, and D. cf. pulex sensu Hebert clones. Consensus sequences for each population/clone were generated using Samtools (Li et al. 2009) with command: samtools mpileup -uf ref.fa aln.bam | bcftools call -c | vcfutils.pl vcf2fq > cns.fq. We randomly selected 1% of the consensus sequences and repeated this 1000 times, constructing maximum-likelihood trees for each subset of data using iq-tree with GTR + I model and ultrafast bootstrap (Nguyen et al. 2014; Hoang et al. 2017). The consensus tree was generated using the Consense program, and branch lengths estimated by the Dnaml program in the PHYLIP package (Felsenstein 1993). A separate phylogenetic tree based on mitochondrial data was constructed with the maximum likelihood method using MEGA 7 with 100 Bootstrap (Kumar et al. 2016).
Phenotypic tests
The reproductive modes of NZ Daphnia clones were determined using a sexuality test. Because sexual Daphnia need sperm fertilization to produce diapausing embryos, the sexuality test is to check whether, in the absence of males, diapausing embryos are deposited into ephippia by females. The consistent presence of embryos in ephippia in the absence of males implies an obligately asexual clone, whereas consistent results of no ephippial embryos from at least three consecutive rounds of tests were used to infer a male-requiring CP. We decapsulated a total of 30 ephippial embryos produced by clones from Lake Alexandrina in the absence of males. All 30 contained embryos and some of these hatched, consistent with the clones from this lake being obligate asexuals.
Male production was induced by adding the hormone methyl farnesoate (MF) to the medium. Adult females for each clone were isolated and placed into 50-mL tubes containing MF at a concentration of 400 nM (changed daily). We examined the sex of offspring using a dissection microscope. Males can be visually distinguished from females based on the enlarged antennules and flattened ventral carapace margin. Successful production of males by ≥ 5 individuals was taken to be evidence for a male-producing clone.
Results
Testing if each lake consists of a single clone
To understand the Daphnia invasion in NZ, we sampled Daphnia populations from 13 lakes in the South Island and one in the North Island, NZ (Fig. 1; Supporting Information Table S1). Daphnia populations from each lake were determined to be D. pulex sensu lato based on morphological features (Benzie 2005; Ebert 2005). Each population was sequenced to an average depth of sequencing coverage of ×86 (Table 1), involving more than 100 million 150-bp reads per population. On average, 1.5 million biallelic sites were identified for each population (Table 1).
If each NZ Daphnia population represents a single clone, assuming no mutation or gene conversion, all of its biallelic sites should have similar numbers of reads mapped to the two parental alleles, as expected for a fixed heterozygote. To test this, we performed binomial tests on all biallelic sites to check for deviations from a 1 : 1 ratio. Over all 14 samples, with an average of 1.5 million significant biallelic sites per sample, only 14–667 sites in each population have unequal numbers of reads mapped to the two parental alleles after correcting for multiple comparisons (Table 1), while a typical sexually reproducing D. cf. pulex sensu Hebert population from North America has > 98% of heterozygous sites with minor-allele frequencies significantly deviating from 0.5, and a strong skew of the site-frequency spectrum toward low-frequency alleles (Lynch et al. 2008). Therefore, our results suggest that each NZ population is a single clone. In principle, the tiny fraction of sites deviating from a 1 : 1 ratio might have arisen from an inherent bias toward more effective mapping of one allele (Degner et al. 2009), although this seems very unlikely, as it would have to occur in population-specific samples. A more likely explanation is the low level of mutation and gene conversion operating in all populations, including those that are obligately asexual.
Lineage identification of Daphnia from NZ lakes
Because traditional lineage identifications using the mitochondrial genome and/or the nuclear-encoded Ldh locus may be biased due to introgression between D. cf. pulex sensu Hebert and D. pulicaria Forbes (Omilian and Lynch 2009; Cristescu et al. 2012; Marková et al. 2013), we relied on whole-genomic information to distinguish D. cf. pulex sensu Hebert, D. pulicaria Forbes, and their hybrids. To obtain whole-genomic information, we collected 30 sexual D. cf. pulex sensu Hebert clones from temporary ponds across North America, 6 D. “pulex” from Oregon, USA, 3 D. pulex Leydig from Europe, and 3 D. “pulex” from China (Supporting Information Table S2). Each clone was sequenced to an average depth of sequencing coverage of ×10. By comparing whole-genomic information between these 30 D. cf. pulex sensu Hebert clones and 14 D. pulicaria Forbes (Xu et al. 2015), we ientified 25,643 informative sites, in which all D. pulicaria Forbes are monomorphic for one nucleotide and all D. cf. pulex sensu Hebert are monomorphic for another nucleotide (Supporting Information File S2). These 25,643 informative sites span 11 of the 12 chromosomes in D. cf. pulex sensu Hebert (Supporting Information Fig. S1). We found that all South Island Daphnia have > 23,867 (93%) sites homozygous for D. pulicaria Forbes-specific nucleotides, while only 807 (3.3%) sites are homozygous in the North Island clone. This suggests a D. pulicaria Forbes origin of the South Island Daphnia clones, and a hybrid origin of the North Island clone.
To further confirm the lineages of Daphnia clones in NZ, we constructed a consensus maximum-likelihood (ML) tree using whole-genomic coding sequences from 14 NZ clones, 14 D. pulicaria Forbes, 30 D. cf. pulex sensu Hebert clones, 3 D. pulex Leydig from Europe, and 3 D. “pulex” from China. Heterozygous sites in each sequence were represented using IUPAC ambiguity code. Within this tree, the South Island clones formed a monophyletic clade with all North American D. pulicaria Forbes clones, further supporting the view that the South Island clones are D. pulicaria Forbes (Fig. 2). The North Island clone is in a clade with D. pulicaria Forbes and D. cf. pulex sensu Hebert, but clusters with neither D. pulicaria Forbes nor D. cf. pulex sensu Hebert (Fig. 2), confirming its hybrid origin. Unlike the situation for nuclear data, for the ML tree based on full-length mitochondrial genomes, D. cf. pulex sensu Hebert and D. pulicaria Forbes clones interlaced outside of the NZ clones (Supporting Information Fig. S2). This discordant phylogeny between the mitochondrial and nuclear genomes may represent historical hybridization between D. cf. pulex sensu Hebert and D. pulicaria Forbes (Marková et al. 2013).
Fig 2.
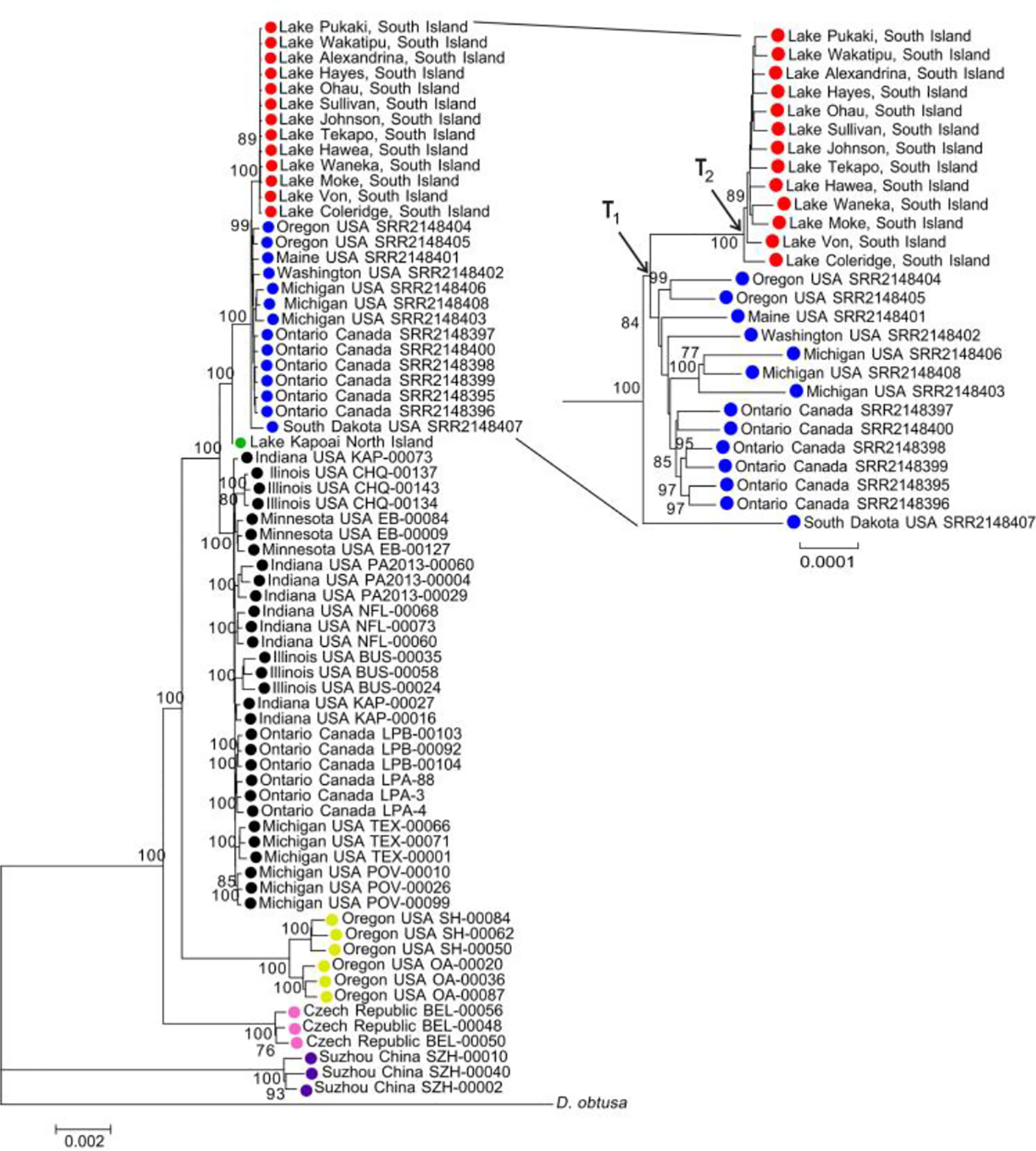
Maximum likelihood tree using nuclear genome sequences. South Island, New Zealand (NZ) Daphnia samples (red circles); North Island, NZ Daphnia sample (green circle); Daphnia cf. pulex sensu Hebert (black circles); Oregon D. “pulex” (yellow circles); D. pulex Leydig (pink circles), and Chinese D. “pulex” samples (purple circles); Daphnia pulicaria Forbes samples are marked with blue circles. Inset on the right shows the phylogeny of the South Island and North American D. pulicaria Forbes in more detail. Bootstrap values > 75/100 are shown. T1 is the divergence time between the north American and the south Island D. pulicaria Forbes. T2 is the expansion time of South Island D. pulicaria Forbes.
Testing a single-clone origin hypothesis for all South Island Daphnia clones
Because all South Island Daphnia clones are D. pulicaria Forbes, we wanted to test if they all derive from a single D. pulicaria Forbes clone. We compared coding sequences of the 13 South Island Daphnia clones and found that the average number of synonymous substitutions among clones is < 24 (over a total of 7.05 million synonymous sites), implying that all South Island Daphnia originated from single founder clone. In further support of the single-clone origin hypothesis, we found that all 13 South Island Daphnia populations have identical mitochondrial genomes. Moreover, six of the seven clones from four different South Island lakes collected > 7 years ago (Duggan et al. 2012) have identical sequence for the CO1 gene as our South Island samples. The only exception is a clone collected from Lake Wanaka by Duggan et al. (2012) with one unique SNP, which is likely a de novo mutation. To understand if the South and North Island Daphnia clones have the same maternal origin, we compared their 15-kb mtDNA genome sequences, and found 54 nucleotide differences, thereby ruling out origination from a single clone.
To check if the South Island clones are OP, we experimentally examined their ability to produce diapausing embryos without fertilization. CP Daphnia produce haploid diapausing eggs that require fertilization by sperm, while obligately asexual Daphnia are capable of producing diploid diapausing eggs in the absence of males. We found that in the absence of males, the South Island Daphnia (from Lake Alexandrina) can generate ephippia containing embryos, indicating that the life cycle can be completed without sex. We also tested the male-producing ability for the Daphnia from Lake Alexandrina and found consistent male production. These results suggest that the Daphnia invasion of the South Island involved a single obligately asexual clone capable of (but not requiring) male production.
Due to the lack of available live animals in the laboratory, we were unable to experimentally examine the reproductive mode and male-producing ability in the North Island Daphnia. Instead, we used informative markers to predict their traits. Chromosomes VIII and IX in D. cf. pulex sensu Hebert contain > 30,000 markers that are informative with respect to the origin of asexuality (Tucker et al. 2013; Xu et al. 2015). All of these markers are heterozygous in obligate asexual hybrids but are homozygous in CP D. cf. pulex sensu Hebert or D. pulicaria Forbes. The North Island Daphnia carry the heterozygous markers at > 87% of these reference sites, suggesting obligate asexuality. We also examined the male-producing ability in the North Island Daphnia by reference to markers identified by Ye et al. (2019), finding a complete absence of markers associated with the loss of male-producing ability, thereby suggesting a capacity for male production. Thus, our results indicate that the Daphnia invasion of North Island also involved an OP clone, capable of producing males.
Origin and colonization time of the NZ Daphnia clones
To infer the geographic origin of the D. pulicaria Forbes that invaded South Island, genetic data are required from throughout the natural range of this species. Although whole nuclear and mitochondrial genomes are available for samples from North America and NZ, the only data available for D. “pulicaria” from other continents are mitochondrial CO1 gene sequences. For the maximum-likelihood tree using mitochondrial CO1 gene sequences collected from D. “pulicaria” across North America, South America, Europe, Asia, NZ, and the north polar region, the South Island Daphnia clustered with a subset of North American clones and a few clones from South America (Fig. 3). Because South American D. “pulicaria” are thought to have been introduced from North America (Crease et al. 2012), the phylogeny suggests that the South Island Daphnia ultimately came from North America.
Fig 3.
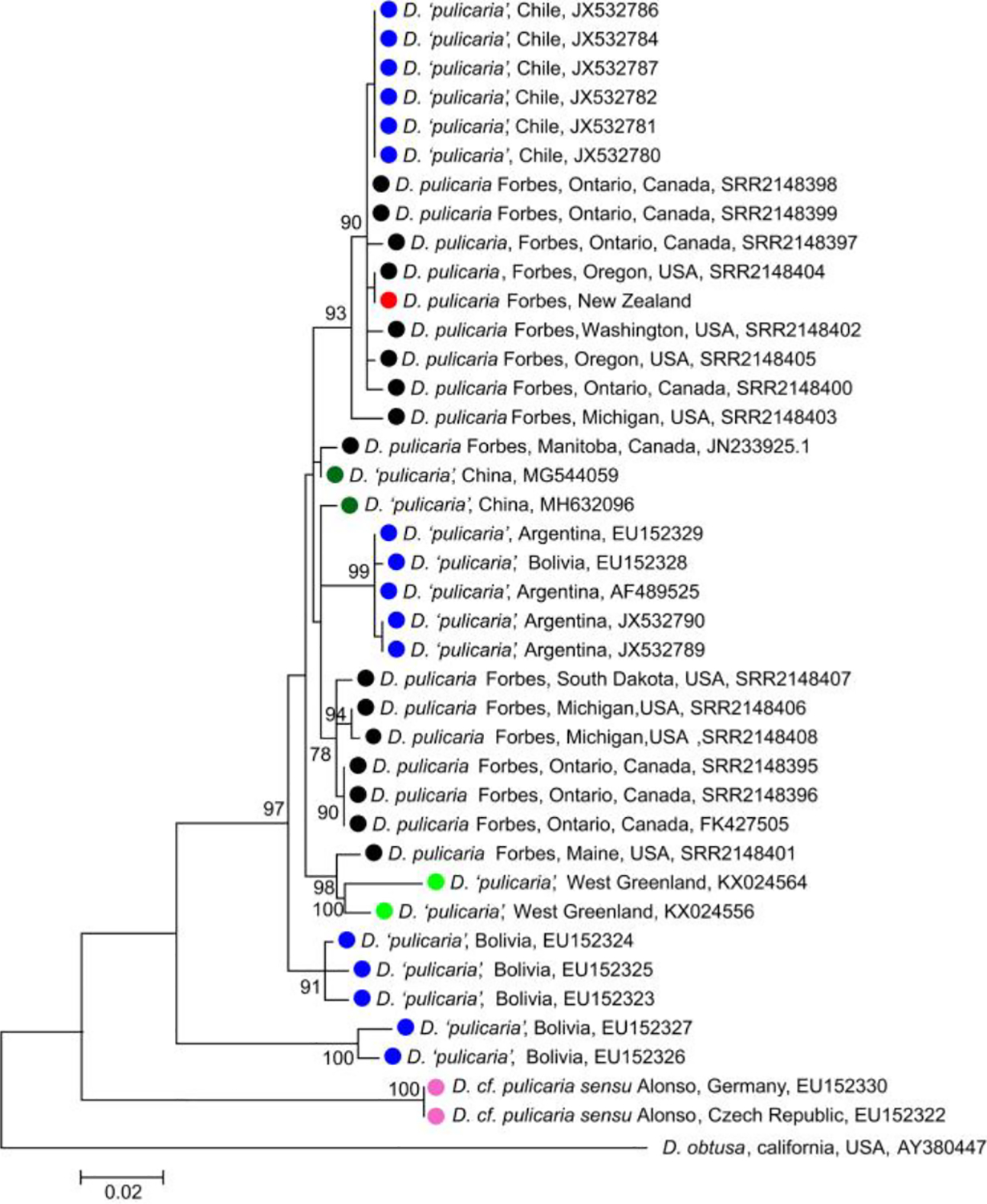
Maximum-likelihood tree using partial nucleotide sequences from the COI gene. Clones from different continents are color coded: South Island, New Zealand (red), Asia (dark green), South America (blue), North America (black), Europe (pink), and Greenland (light green). The codes following each sample are accession numbers from GenBank or Sequence Read Archive. We used geographical location and quotation markers to describe clones without clear lineage identification (i.e., D. “pulicaria,” China).
To estimate when the South Island genotype diverged from North American D. pulicaria Forbes, we calculated the nuclear genome-wide divergence between synonymous sites for these two groups. The divergence time between the South Island genotype and North American D. pulicaria Forbes (T1, Fig. 2) can be estimated by the average pairwise distance of synonymous sites (ds) between North American D. pulicaria Forbes and the South Island, NZ haplotypes (ds = 0.00023). Letting T1 = ds/2 μ, with μ = 5.85 × 10−9 being the mutation rate per site per generation (obtained from a mutation-accumulation experiment; Keith et al. 2016), and assuming ~5 generations per year, we estimate that the South Island genotype diverged from other North American D. pulicaria Forbes ~3400 years ago. Because the North Island Daphnia have a hybrid origin, and we do not have genomic sequences available for North American hybrids, the divergence time between the North Island genotype and North American hybrids cannot be estimated by this means.
We also estimated the expansion time of the South Island clones (T2, Fig. 2). Assuming these clones descended from a single primary colonization event, then the nucleotide diversity among South Island haplotypes would have been acquired after the colonization, and the expansion time can be estimated from the average synonymous nucleotide divergence between the most distant population (Lake Coleridge) and the remaining populations. We found that the Daphnia population from Lake Coleridge has an average of 24 synonymous substitutions (over a total of 7.05 million synonymous sites) compared to the remaining populations, implying an expansion time of T2 = ds/(2 ·μ) = 3.4 × 10−6/(2× 5.85 × 10−9/site/generation × 5 generations/year) = ~58 years. This calculation is supported by the presence of characteristic ephippia of D. “pulex” in a sediment-dated core taken in Lake Hayes that suggested appearance of this species in the lake in the late 1950s or early 1960s (Samiulah Khan pers. comm.).
Discussion
Determining if each lake consists of a single Daphnia clone using pooled sequencing
Most of the previous studies on Daphnia invasions only sequenced a few genes from a few clones from each lake or pond (Mergeay et al. 2005; Duggan et al. 2012; So et al. 2015), possibly leading to undersampling of coexisting genotypes. In Africa, the invasive D. cf. pulex sensu Hebert and native D. “pulex” are thought to have coexisted for ~60 years before the native D. “pulex” was completely displaced (Mergeay et al. 2005). Because invasive D. cf. pulex sensu Hebert and native D. “pulex” in Africa are not easily distinguished morphologically and could only be identified by sequencing dozens of clones, which may require extensive lab work to check the genotype of each clone. Our study provided a relatively easy solution for checking for potential coexistence by pooling 50–200 individuals for whole-genomic sequencing. If all individuals in a single sample derived from a single clone, all polymorphic sites should have fixed heterozygosity and similar numbers of reads mapped to the two parental alleles.
Although the NZ lakes in our study have historically been inhabited by native NZ species of Daphnia (Burns et al. 2017), we found each lake sample to consist of just a single non-native Daphnia clone, owing to complete displacement by just two invading clones from North America (one on each island). Genome-wide sequence analyses did not reveal any evidence of adaptive divergence within the invasive clones, but this does not rule out fixation of substantial numbers of mutations in the founder population before it spread across the landscape.
Distinguishing D. cf. pulex sensu Hebert and D. pulicaria Forbes
North American D. cf. pulex sensu Hebert and D. pulicaria Forbes are thought to have recently diverged from a common ancestor and adapted to distinct environments while still experiencing introgression (Omilian and Lynch 2009; Cristescu et al. 2012). Such introgression may cause their hybrids to have mtDNA from either D. cf. pulex sensu Hebert or D. pulicaria Forbes, thus biasing inferences based on trees derived solely from mtDNA data (Marková et al. 2013). Although the nuclear-encoded Ldh locus has been used to discriminate D. cf. pulex sensu Hebert and D. pulicaria Forbes, inferring lineages solely based on the Ldh locus can still be misinformative due to hybridization and backcrossing (Xu et al. 2013). For example, an Ldh Slow-Slow Daphnia genotype derived from crossing between Slow-Fast hybrids and Slow-Slow D. cf. pulex sensu Hebert may be erroneously inferred as a non-hybrid clone. To eliminate the limitations of a single nuclear marker to derive inferences, we generated a set of 25,643 D. pulicaria Forbes-specific nuclear markers that are monomorphic in every nonhybrid D. pulicaria Forbes, and unique with respect to D. cf. pulex sensu Hebert. These markers will be useful for diagnostic purposes in other future studies involving these two lineages, including the identification of hybrids. They may also serve as a resource for studying genes involved in potential adaptive divergence between D. cf. pulex sensu Hebert and D. pulicaria Forbes.
Invasive D. “pulex” in NZ
In our study, we identified independent invasions of D. “pulex” on the South and North Islands of NZ. Phylogenetic data from mitochondrial genomes revealed that both invasive D. “pulex” originated from North America. Intriguingly, all 13 lakes in the South Island were invaded by a single D. pulicaria Forbes clone, suggesting higher fitness of the invasive Daphnia clones compared to the NZ-native Daphnia clones. Consistent with this hypothesis, Burns (2013) found that the invasive Daphnia could displace the native Daphnia when water temperature and nutrition level are relatively high.
The invasive Daphnia on both islands are obligate asexuals. The origin of asexuality for the North Island Daphnia is likely caused by hybridization between D. cf. pulex sensu Hebert and D. pulicaria Forbes prior to the arrival in NZ (Innes and Hebert 1988), while that for the South Island D. pulicaria Forbes is still not clear. Obligately asexual D. cf. pulex sensu Hebert × D. pulicaria Forbes hybrids are notoriously invasive, colonizing, and spreading throughout many African lakes within just a few decades (Mergeay et al. 2006), and invading Japan between 680 and 3400 years ago (So et al. 2015). The Daphnia invasion time in Japan overlapped with the estimated invasion time of South Island Daphnia clone in NZ, suggesting a potential clone transfer from Japan or origination from the same event.
Source of invasive Daphnia in NZ
To infer the source of D. “pulex” in NZ, we checked the possible routes of the D. “pulex” invasions. It is thought that Daphnia thomsoni Sars, 1894 (formerly Daphnia carinata, Burns et al. 2017) and Daphnia tewaipounamu Burns et al., 2017 are the only native Daphnia species in NZ, although there are several non-indigenous invasive Daphnia. One recent invader, Daphnia galeata, now common in lakes throughout the North Island of NZ, may have been introduced to the South Island during translocation of farmed Chinese grass carp (Ctenopharyngodon idella) for the purpose of weed control in northern South Island waterways (Duggan and Pullan 2017). The presence of D. “pulex” in some shallow constructed ponds in suburban Auckland, North Island, has also been attributed to introductions of grass carp (Branford and Duggan 2017). However, Chinese grass carp have only been used for weed control on the North Island and the northern tip of the South Island, as water temperatures in southern NZ are generally too cold for these fish. Therefore, it is highly unlikely that grass carp were a major vector for the introduction of D. pulicaria Forbes to the South Island.
Duggan et al. (2012) suggested introduced salmonids (trout, salmon) and associated recreational fishing equipment as potential vectors for D. pulicaria Forbes in the South Island. The most common salmonid in NZ, Chinook salmon (Oncorhynchus tshawytschca), was initially introduced from the Sacramento River, California, USA to South Island, NZ, between 1901 and 1907. Thus, the founder D. pulicaria clone in the South Island might be associated with one of the Chinook salmon introductions. Salmon fishing in NZ has become popular since the 1970s, attracting many international tourists. The D. pulicaria Forbes clone in the South Island has been known to produce diapausing ephippia, which float on the water surface for long periods, adhering to any surfaces they encounter, such as fishing equipment, skin, fur, feathers, and clothing (Burns 2013). The movement of ephippia adhering to recreational equipment, people, and wildlife likely facilitated the spread of D. pulicaria Forbes throughout the South Island. In support of this, the invasion time estimated from the molecular data (58 years) is roughly consistent with transport during this period.
Methods and resources generated in this study
In this study, we provide methods and resources that could be of benefit in future studies of invasive Daphnia species: (1) we provided an easy solution to check for potential coexistence of Daphnia clones that are hard to distinguish morphologically; (2) we identified 25,643 D. pulicaria Forbes-specific nuclear markers that can be used to easily distinguish D. cf. pulex sensu Hebert and D. pulicaria Forbes; (3) we showed that the origin and expansion time of the invasive Daphnia clones could be accurately estimated using whole genomic data.
Supplementary Material
Acknowledgments
We thank Joachim Mergeay the insightful comments and suggestions on dealing with the taxonomy confusion in the Daphnia pulex species complex. We thank Wei-chin Ho for helpful discussions and Samiullah Khan, Marc Schallenberg, and John McCallum for their help in sampling. This work is supported by NIH grant R35-GM122566-01 to Michael Lynch, financial support from Alan Wilson at Otago and the University of Otago to Carolyn Burns.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Conflict of Interest None declared.
Data Availability Statement
NZ population data for this study were deposited to NCBI under BioProject ID PRJNA573527. D. cf. pulex sensu Hebert and D. pulicaria Forbes genomic data can be accessed at BioProject ID PRJNA573529 and accession numbers SAMN03964756-SAMN03964769 (Xu et al. 2015), and the D. obtusa genomic sequences used in this study can be accessed at NCBI under accession number SAMN12816670. The D. cf. pulex sensu Hebert genome assembly PA42 v4.1 is available at GenBank under accession GCA_900092285.2 (Ye et al. unpubl.) and the corresponding annotation file is provided in Supporting Information File S1.
References
- Alonso M 1996. Crustacea, Branchiopoda, Anomopoda. Museum Nacional de Ciencias Naturales. [Google Scholar]
- Benzie JA 2005. Cladocera: The genus Daphnia (including Daphniopsis). Kenobi Productions & Backhuys Publishers, p. 376. [Google Scholar]
- Bolger AM, Lohse M, and Usadel B. 2014. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30: 2114–2120. doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branford SN, and Duggan IC. 2017. Grass carp (Ctenopharyngodon idella) translocations, including hitchhiker introductions, alter zooplankton communities in receiving ponds. Mar. Freshw. Res 68: 2216. doi: 10.1071/mf17051 [DOI] [Google Scholar]
- Burns CW 2013. Predictors of invasion success by Daphnia species: Influence of food, temperature and species identity. Biol. Invasions 15: 859–869. doi: 10.1007/s10530-012-0335-5 [DOI] [Google Scholar]
- Burns CW, Duggan IC, Banks JC, and Hogg ID. 2017. A new, subalpine species of Daphnia (Cladocera, Anomopoda) in the D. carinata species complex, in the South Island, New Zealand. Hydrobiologia 798: 151–169. doi: 10.1007/s10750-016-2702-1 [DOI] [Google Scholar]
- Calabrese C, Simone D, Diroma MA, Santorsola M, Guttà C, Gasparre G, Picardi E, Pesole G, and Attimonelli M. 2014. MToolBox: A highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 30: 3115–3117. doi: 10.1093/bioinformatics/btu483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbourne JK, Crease TJ, Weider LJ, Hebert PD, Duferesne F, and Hobaek A. 1998. Phylogenetics and evolution of a circumarctic species complex (Cladocera: Daphnia pulex). Biol. J. Linn. Soc. Lond 65: 347–365. doi: 10.1111/j.1095-8312.1998.tb01146.x [DOI] [Google Scholar]
- Crease TJ, Floyd R, Cristescu ME, and Innes D. 2011. Evolutionary factors affecting lactate dehydrogenase A and B variation in the Daphnia pulex species complex. BMC Evol. Biol 11: 212. doi: 10.1186/1471-2148-11-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crease TJ, Omilian AR, Costanzo KS, and Taylor DJ. 2012. Transcontinental Phylogeography of the Daphnia pulex species complex. PLoS One 7: e46620. doi: 10.1371/journal.pone.0046620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu ME, Constantin A, Bock DG, Caceres CE, and Crease TJ. 2012. Speciation with gene flow and the genetics of habitat transitions. Mol. Ecol 21: 1411–1422. doi: 10.1111/j.1365-294X.2011.05465.x [DOI] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, and Pritchard JK. 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212. doi: 10.1093/bioinformatics/btp579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufresne F, Marková S, Vergilino R, Ventura M, and Kotlik P. 2011. Diversity in the reproductive modes of European Daphnia pulicaria deviates from the geographical parthenogenesis. PLoS One 6: e20049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan IC, and Pullan SG. 2017. Do freshwater aquaculture facilities provide an invasion risk for zooplankton hitchhikers? Biol. Invas 19: 307–314. doi: 10.1007/s10530-016-1280-5 [DOI] [Google Scholar]
- Duggan I, Robinson K, Burns C, Banks J, and Hogg I. 2012. Identifying invertebrate invasions using morphological and molecular analyses: North American Daphnia ‘pulex’ in New Zealand fresh waters. Aquat. Invas 7: 585–590. doi: 10.3391/ai.2012.7.4.015 [DOI] [Google Scholar]
- Ebert D 2005. Ecology, epidemiology, and evolution of parasitism in Daphnia. National Library of Medicine (US), National Center for Biotechnology Information. [Google Scholar]
- Felsenstein J 1993. PHYLIP (phylogeny inference package). Univ. of Washington. [Google Scholar]
- Geng X, Cheng R, Xiang T, Deng B, Wang Y, Deng D, and Zhang H. 2016. The complete mitochondrial genome of the Chinese Daphnia pulex (Cladocera, Daphniidae). ZooKeys 615: 47–60. doi: 10.3897/zookeys.615.8581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PD 1978. The population bilogy of Daphnia (Crustacea, Daphnidae). Biol. Rev 53: 387–426. doi: 10.1111/j.1469-185X.1978.tb00860.x [DOI] [Google Scholar]
- Hebert PD, and Crease TJ. 1980. Clonal coexistence in Daphnia pulex (Leydig): Another planktonic paradox. Science 207: 1363–1365. doi: 10.1126/science.207.4437.1363 [DOI] [Google Scholar]
- Hebert PD, Beaton MJ, Schwartz SS, and Stanton DJ. 1989. Polyphyletic origins of asexuality in Daphnia pulex. I. Breeding-system variation and levels of clonal diversity. Evolution 43: 1004–1015. doi: 10.1111/j.1558-5646.1989.tb02546.x [DOI] [PubMed] [Google Scholar]
- Hebert PD, Schwartz SS, Ward RD, and Finston TL. 1993. Macrogeographic patterns of breeding system diversity in the Daphnia pulex group. I. Breeding systems of Canadian populations. Heredity 70: 148–161. doi: 10.1038/hdy.1993.24 [DOI] [PubMed] [Google Scholar]
- Hebert PD 1995. The Daphnia of North America: An illustrated fauna (on CD-ROM). Cyber Natural Software. [Google Scholar]
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, and Vinh LS. 2017. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol 35: 518–522. doi: 10.1093/molbev/msx281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes DJ, and Hebert PD. 1988. The origin and genetic basis of obligate parthenogenesis in Daphnia pulex. Evolution 42: 1024. doi: 10.2307/2408918 [DOI] [PubMed] [Google Scholar]
- Innes DJ, Fox CJ, and Winsor GL. 2000. Avoiding the cost of males in obligately asexual Daphnia pulex (Leydig). Proc. R. Soc. Lond. B 267: 991–997. doi: 10.1098/rspb.2000.1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith N, and others. 2016. High mutational rates of large-scale duplication and deletion in Daphnia pulex. Genome Res. 26: 60–69. doi: 10.1101/gr.191338.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, and Tamura K. 2016. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol 33: 1870–1874. doi: 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehto MP, and Haag CR. 2010. Ecological differentiation between coexisting sexual and asexual strains of Daphnia pulex. J. Anim. Ecol 79: 1241–1250. doi: 10.1111/j.1365-2656.2010.01726.x [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, others, and 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Seyfert A, Eads B, and Williams E. 2008. Localization of the genetic determinants of meiosis suppression in Daphnia pulex. Genetics 180: 317–327. doi: 10.1534/genetics.107.084657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marková S, Dufresne F, Manca M, and Kotlík P. 2013. Mitochondrial capture misleads about ecological speciation in the Daphnia pulex complex. PLoS One 8: e69497. doi: 10.1371/journal.pone.0069497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergeay J, Verschuren D, and De Meester L. 2005. Cryptic invasion and dispersal of an American Daphnia in East Africa. Limnol. Oceanogr 50: 1278–1283. doi: 10.4319/lo.2005.50.4.1278 [DOI] [Google Scholar]
- Mergeay J, Verschuren D, and Meester LD. 2006. Invasion of an asexual American water flea clone throughout Africa and rapid displacement of a native sibling species. Proc. R. Soc. Lond. B 273: 2839–2844. doi: 10.1098/rspb.2006.3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, and Minh BQ. 2014. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol 32: 268–274. doi: 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omilian AR, and Lynch M. 2009. Patterns of intraspecific DNA variation in the Daphnia nuclear genome. Genetics 182: 325–336. doi: 10.1534/genetics.108.099549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paland S, Colbourne JK, and Lynch M. 2005. Evolutionary history of contagious asexuality in Daphnia pulex. Evolution 59: 800–813. doi: 10.1111/j.0014-3820.2005.tb01754.x [DOI] [PubMed] [Google Scholar]
- Sars GO 1894. Contributions to the knowledge of the freshwater Entomostraca of New Zealand as shown by artificial hatching from dried mud. Kongelige Danske Videnskabernes Selskabets Skrifter. I. Mathematisknaturviden Klasse 5: 1–62. [Google Scholar]
- So M, Ohtsuki H, Makino W, Ishida S, Kumagai H, Yamaki KG, and Urabe J. 2015. Invasion and molecular evolution of Daphnia pulex in Japan. Limnol. Oceanogr 60: 1129–1138. doi: 10.1002/lno.10087 [DOI] [Google Scholar]
- Stevenson KR, Coolon JD, and Wittkopp PJ. 2013. Sources of bias in measures of allele-specific expression derived from RNA-seq data aligned to a single reference genome. BMC Genomics 14: 536. doi: 10.1186/1471-2164-14-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier AJ, and Cáceres CE. 2004. Differentiation in sex investment by clones and populations of Daphnia. Ecol. Lett 7: 695–703. doi: 10.1111/j.1461-0248.2004.00627.x [DOI] [Google Scholar]
- Tucker AE, Ackerman MS, Eads BD, Xu S, and Lynch M. 2013. Population-genomic insights into the evolutionary origin and fate of obligately asexual Daphnia pulex. Proc. Natl. Acad. Sci. USA 110: 15740–15745. doi: 10.1073/pnas.1313388110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, and Watanabe CK. 2005. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21: 1859–1875. doi: 10.1093/bioinformatics/bti310 [DOI] [PubMed] [Google Scholar]
- Xu S, Innes DJ, Lynch M, and Cristescu ME. 2013. The role of hybridization in the origin and spread of asexuality in Daphnia. Mol. Ecol 22: 4549–4561. doi: 10.1111/mec.12407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Spitze K, Ackerman MS, Ye Z, Bright L, Keith N, Jackson CE, Shaw JR, and Lynch M. 2015. Hybridization and the origin of contagious asexuality in Daphnia pulex. Mol. Biol. Evol 32: 3215–3225. doi: 10.1093/molbev/msv190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Molinier C, Zhao C, Haag CR, and Lynch M. 2019. Genetic control of male production in Daphnia pulex. Proc. Natl. Acad. Sci. USA 116: 15602–15609. doi: 10.1073/pnas.1903553116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NZ population data for this study were deposited to NCBI under BioProject ID PRJNA573527. D. cf. pulex sensu Hebert and D. pulicaria Forbes genomic data can be accessed at BioProject ID PRJNA573529 and accession numbers SAMN03964756-SAMN03964769 (Xu et al. 2015), and the D. obtusa genomic sequences used in this study can be accessed at NCBI under accession number SAMN12816670. The D. cf. pulex sensu Hebert genome assembly PA42 v4.1 is available at GenBank under accession GCA_900092285.2 (Ye et al. unpubl.) and the corresponding annotation file is provided in Supporting Information File S1.