

Abstract
In the RAS (renin–angiotensin system), Ang I (angiotensin I) is cleaved by ACE (angiotensin-converting enzyme) to form Ang II (angiotensin II), which has effects on blood pressure, fluid and electrolyte homoeostasis. We have examined the kinetics of angiotensin peptide cleavage by full-length human ACE, the separate N- and C-domains of ACE, the homologue of ACE, ACE2, and NEP (neprilysin). The activity of the enzyme preparations was determined by active-site titrations using competitive tight-binding inhibitors and fluorogenic substrates. Ang I was effectively cleaved by NEP to Ang (1–7) (kcat/Km of 6.2×105 M−1·s−1), but was a poor substrate for ACE2 (kcat/Km of 3.3×104 M−1·s−1). Ang (1–9) was a better substrate for NEP than ACE (kcat/Km of 3.7×105 M−1·s−1 compared with kcat/Km of 6.8×104 M−1·s−1). Ang II was cleaved efficiently by ACE2 to Ang (1–7) (kcat/Km of 2.2×106 M−1·s−1) and was cleaved by NEP (kcat/Km of 2.2×105 M−1·s−1) to several degradation products. In contrast with a previous report, Ang (1–7), like Ang I and Ang (1–9), was cleaved with a similar efficiency by both the N- and C-domains of ACE (kcat/Km of 3.6×105 M−1·s−1 compared with kcat/Km of 3.3×105 M−1·s−1). The two active sites of ACE exhibited negative co-operativity when either Ang I or Ang (1–7) was the substrate. In addition, a range of ACE inhibitors failed to inhibit ACE2. These kinetic data highlight that the flux of peptides through the RAS is complex, with the levels of ACE, ACE2 and NEP dictating whether vasoconstriction or vasodilation will predominate.
Keywords: active-site titration, angiotensin-converting enzyme, cardiovascular disease, neprilysin, renin–angiotensin system, vasopeptidase
Abbreviations: ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; C-ACE, C-domain of human ACE; FL-ACE, full-length somatic human ACE; N-ACE, N-domain of human ACE; AMC, 7-amido-4-methylcoumarin; Ang, angiotensin; CHO, Chinese-hamster ovary; Dnp, 2,4-dinitrophenyl; Dpa, 3-(2,4-dinitrophenyl)-L-2,3-diaminopropionic acid; fMLP, N-formylmethionyl-leucylphenylalanine; Mca, (7-methoxycoumarin-4-yl)acetyl; MI, myocardial infarction; NEP, neprilysin; RAS, renin–angiotensin system; Suc, succinoyl
INTRODUCTION
The RAS (renin–angiotensin system) is a key regulator of blood pressure and fluid and electrolyte homoeostasis. ACE (angiotensin-converting enzyme) generates the physiologically active peptide Ang II (angiotensin II) by cleaving the C-terminal dipeptide His–Leu from Ang I (angiotensin I) [1]. The binding of Ang II to the Ang II type 1 receptor mediates regional blood flow, hyperplastic and hypertrophic vascular smooth muscle cell proliferation and migration, and the regulation of local sympathetic activity, pressor and tachycardic responses [2]. Ang II may also be involved in platelet activation and aggregation [3] and maintenance of cardiovascular structure and repair [4,5]. ACE inhibitors are an effective first-line treatment against essential hypertension [6] and reduce mortality from congestive heart failure and in asymptomatic left-ventricular systolic dysfunction after MI (myocardial infarction) [7].
The traditional view that Ang II is the key product of the RAS has been questioned with the recent discovery of a novel carboxypeptidase, ACE2 (ACE homologue) [8,9], as well as growing evidence for a physiological role for Ang (1–7). In vitro, it has been reported that ACE2 cleaves Ang I to form Ang (1–9) and cleaves Ang II to form Ang (1–7) [8,10]. Ang (1–7) opposes the cardiovascular actions of Ang II, acting as a vasodilator with antiproliferative effects, and is protective against MI [11,12]. ACE2 levels may therefore confound the relationship between ACE and atherothrombotic disorders by reducing levels of the vasoconstrictor Ang II and increasing levels of the vasodilator Ang (1–7). In addition, or alternatively, ACE2 may compete with ACE for Ang I, forming Ang (1–9), the biological properties of which are not known. For a recent review on the structure and function of ACE and ACE2, see [13].
The gene encoding human ACE has been localized to chromosome locus 17q23 [14] and comprises 26 exons [15]. There are two forms of ACE: somatic ACE, which is transcribed from exons 1–26, excluding exon 13, which is removed by splicing during translation, and testicular ACE, which is transcribed from exons 13–26 [15]. Somatic ACE has two active-site domains, designated the N- and C-domains, while testicular ACE contains only the C-domain active site [15,16]. The two active sites of ACE have similar, but not identical, substrate specificities and catalytic activities [15,17]. ACE2 has 42% sequence identity and 61% sequence similarity with ACE [8]. However, in contrast with ACE, it has a single His-Glu-Xaa-Xaa-His (HEXXH) active-site motif [8]. ACE2 differs in substrate specificity from ACE in that it functions exclusively as a carboxy-peptidase [10].
Vasopeptidase inhibitors, such as omapatrilat, simultaneously inhibit ACE and NEP (neprilysin; EC 3.4.24.11) [18]. In patients with hypertension, omapatrilat produced greater decreases in systolic, diastolic and pulse pressure than ACE inhibition alone. NEP cleaves a variety of physiologically relevant substrates, including enkephalins, substance P, atrial natriuretic peptide and angiotensins [19,20]. NEP is a zinc metallopeptidase with a single HEXXH active-site motif [19,20] with a preference for cleaving on the N-terminal side of hydrophobic residues, displaying mainly, but not exclusively, endopeptidase activity.
There are conflicting reports in the literature as to the role of ACE2 in the metabolism of Ang I and Ang II, and little information relating to the metabolism of angiotensin peptides by NEP. In addition, previous studies on the metabolism of angiotensin peptides by ACE have been carried out with limited enzyme preparations and/or with few angiotensin peptides being examined and under different reaction conditions. Therefore we decided to rigorously examine the kinetics of angiotensin peptide metabolism by these three peptidases under a defined set of experimental conditions. We have compared the kinetics of hydrolysis of Ang I, Ang II, Ang (1–9) and Ang (1–7) by human somatic ACE containing both active sites, by the individual N- and C-domain active sites of ACE, and by NEP and ACE2 in order to provide a biochemical basis for the potential role of each enzyme in the RAS.
MATERIALS AND METHODS
Materials
Ang (1–9) was synthesized by Severn Biotech (Kidderminster, Worcestershire, U.K.). Leu-Ile-Tyr was synthesized by Pepceuticals (Leicester, U.K.). All other peptides were purchased from Sigma Aldrich Co., Calbiochem or Bachem Bioscience. Recombinant human ACE2 and NEP were obtained from R & D Systems (Europe) (Oxford, U.K.). ACE inhibitors were provided as stated previously [21]. Fluorogenic peptides Mca-ASDK-Dpa [(7-methoxycoumarin-4-yl)acetyl-Ala-Ser-Asp-Lys-3-(2,4-dinitrophenyl)-L-2,3-diaminopropionic acid] and Mca-APK-Dnp (Mca-Ala-Pro-Lys-2,4-dinitrophenyl) were synthesized by Dr G. Knight (University of Cambridge, Cambridge, U.K.).
Expression and purification of human FL-ACE (full-length ACE), N-ACE (N-domain of ACE) and C-ACE (C-domain of ACE)
The cDNAs for human FL-ACE and C-ACE were kindly provided by Professor P. Corvol (Inserm U36, College de France, Paris, France) in the vector pECE [17,22]. The cDNAs were digested out of pECE using EcoRI and ligated into the expression vectors pIRESneo and pIREShyg (Clontech Laboratories) respectively. The cDNA encoding N-ACE was constructed by amplifying bp 9–1963 of FL-ACE using the primers 5′-CTCGGATCGATTTCTGCG-3′ and 5′-TTATATATTTTAACCACCAAATTGAGGATGTCTCCAAGCAGCCTCATCAGTCAC-3′. The PCR product was then cloned into the expression vector pcDNA3.1/V5-His TOPO (Invitrogen Life Technologies).
CHO (Chinese-hamster ovary) cells were cultured in Ham's F-12 nutrient mix (BioWhittaker, Wokingham, Berkshire, U.K.), supplemented with 2 mM L-glutamine, 10% (v/v) foetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 2.5 mg/ml amphotericin B, at 37 °C with 5% CO2. HEK-293 cells were cultured in Dulbecco's modified Eagle's medium (BioWhittaker) supplemented with 4.5 g/l glucose, 2 mM L-glutamine, 10% (v/v) foetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 2.5 mg/ml amphotericin B and 1% (v/v) non-essential amino acids, at 37 °C with 5% CO2. For stable transfections, 30 μg of DNA was introduced to cells by electroporation, and selection was performed in normal growth medium containing 1 mg/ml G418 (PAA Laboratories, Pasching, Austria) or hygromycin B (0.5 mg/ml) (Invitrogen) in PBS. FL-ACE and C-ACE were stably transfected into CHO cells, and N-ACE into HEK-293 cells. Stably transfected cells were grown to confluence, then cultured for 5 days in OPTIMEM-1 (Life Technologies) supplemented with 2.4 g/l GlutaMAX™-1, 100 units/ml penicillin and 100 μg/ml streptomycin, at 37 °C with 5% CO2, harvesting the medium every 24 h. The pooled media were then subjected to lisinopril–2.8 nm-Sepharose chromatography to purify the ACE, as described previously [23].
Protein assay and SDS/PAGE
Protein concentrations were determined using the bicinchoninic acid assay with BSA as standard [24]. To assess the purity of the ACE preparations, 5 μg of protein was separated by SDS/PAGE, using a 7–17% (w/v) polyacrylamide gradient resolving gel, and stained with Coomassie Blue.
Fluorogenic substrate assays
ACE and ACE2 were assayed with the fluorogenic substrates Mca-ASDK-Dpa [25] and Mca-APK-Dnp [10] respectively, in 50 mM Hepes/NaOH, 200 mM NaCl and 10 μM ZnCl2, pH 6.8, at room temperature (22 °C) unless otherwise stated. NEP was assayed with the fluorogenic substrate Suc-Ala-Ala-Phe-AMC (succinoyl-Ala-Ala-Phe-7-amido-4-methylcoumarin) using a coupled assay as described previously [26]. Reactions were followed kinetically for 1 h using a Biotek Synergy HT plate reader (λexcitation=320 nm and λemission=405 nm). Data were fitted and plotted using Grafit v.4.0 (Sigma-Aldrich).
Active-site titrations
ACE preparations were pre-incubated with 0.05–6.25 equivalents of the competitive inhibitor lisinopril for 20 min at room temperature before the addition of 50 μM Mca-ASDK-Dpa. NEP was pre-incubated with 0.01–12 equivalents of the competitive inhibitor thiorphan for 20 min before the addition of Suc-Ala-Ala-Phe-AMC using a coupled assay as described previously [26]. In order to characterize the two active sites of somatic ACE, lisinopril and Mca-ASDK-Dpa, with near identical affinities for both active sites, were used. The enzyme concentration determined represents the combined concentrations of the two active sites and was therefore divided by 2 to give the average active concentration across both active sites. Ki determinations from the tight-binding inhibition were (1.0±0.2)×10−10 M for ACE with lisinopril and (3.5±1.2)×10−9 M for NEP with thiorphan, in agreement with data published previously [27,28]. E0 was estimated by least-squares fit to the equation for competitive tight-binding inhibition [29]:
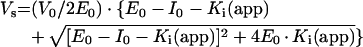 |
where Ki(app) is equal to Ki·(1+[S]/Km). The following molecular masses were used: 174 kDa (FL-ACE), 90 kDa (C-ACE and N-ACE), 120 kDa (ACE2) and 90 kDa (NEP).
Kinetic assays
Substrate concentrations from 0.2 to 5 times reported Km were used. Kinetic assays were optimized to ensure that <10% of the substrate was hydrolysed. Under these conditions, product formation was linear with respect to time over the duration of the incubation. All reactions were carried out in 50 mM Hepes/NaOH, 150 mM NaCl, 10 μM ZnCl2, pH 7.4, at 37 °C. Initial velocities (V0) were plotted against substrate concentration and fitted to the Michaelis–Menten equation (V=Vmax[S]/Km+[S]). Catalytic-centre activities (kcat) were calculated from the equation kcat=Vmax/[E].
HPLC analysis of Ang cleavage products
Peptide hydrolysis products were separated using reverse-phase HPLC (μBondapak C-18 reverse-phase column, Phenomenex) with UV detection at 214 nm. All separations were carried out at room temperature with a flow rate of 1.5 ml/min. Mobile phase A consisted of 0.08% (v/v) orthophosphoric acid and mobile phase B consisted of 30% (v/v) acetonitrile in 0.08% (v/v) orthophosphoric acid. A linear solvent gradient of 4.5% B to 100% B over 15 min followed by 5 min at final conditions, and 8 min re-equilibration was used. The peak area corresponding to the product was integrated to calculate product formation, and compared with standard product curves.
Determination of inhibition constants
Assays were carried out as described above with a substrate concentration of 100 μM. Inhibitors were pre-incubated with the enzyme at 37 °C for 10 min, before the addition of substrate. The reactions were carried out for the same incubation period as for the kinetic assays, and the products formed were separated and quantified by reverse-phase HPLC.
ACE2 inhibition
Assays were carried out in a total volume of 100 μl containing 50 mM Hepes/NaOH, 150 mM NaCl, pH 7.4, 16 or 10 ng of ACE2 protein (with Ang I or Ang II as substrate respectively), 100 μM substrate and 100 μM inhibitor. Inhibitors were pre-incubated with the enzyme at 37 °C for 10 min before addition of substrate. Reactions were carried out for 3 h (Ang I) or 1 h (Ang II).
ACE2 substrate cleavage
Assays were carried out in a total volume of 100 μl containing 50 mM Hepes/NaOH, 150 mM NaCl, pH 7.4, 10 ng of ACE2 protein and 100 μM furylacryloyl-Phe-Gly-Gly, 1 mM fMLP (N-formylmethionyl-leucylphenylalanine), 1 mM hippuryl-Phe-Arg or 1 mM benzyloxycarbonyl-Phe-His-Leu. Reactions were carried out for 2 or 18 h (fMLP), and reaction products were separated and quantified by reverse-phase HPLC as described above.
RESULTS
Purification of ACE and active-site titration of ACE and NEP
The constructs of human FL-ACE, C-ACE and N-ACE (Figure 1) were expressed in mammalian cells and then purified. Although FL-ACE and C-ACE are expressed as membrane-bound proteins, they are shed into the conditioned medium by the action of the endogenous ACE secretase [30]. N-ACE lacks a membrane-anchoring domain and is secreted directly into the cell medium. The constructs were purified from the conditioned medium by chromatography on lisinopril–2.8 nm-Sepharose [23]. All preparations were purified to apparent homogeneity as assessed by SDS/PAGE (Figure 2). Recombinant FL-ACE migrated with an apparent molecular mass of 174 kDa, while the recombinant N- and C-domains migrated with an apparent molecular mass of 90 kDa each.
Figure 1. Schematic diagram of the ACE constructs and ACE2.

Human FL-ACE, C-ACE and N-ACE constructs are shown with their amino acid numbering. ACE2 is shown for comparison. Signal peptides are indicated by diagonal lines, transmembrane domains by a black box, and the bridge region of somatic ACE by an open box. The unique C-terminal domain of ACE2 is represented by a dotted box.
Figure 2. SDS/PAGE of the ACE preparations.

The affinity-purified preparations of recombinant human ACE (5 μg of protein each) were analysed by SDS/PAGE as described in the Materials and methods section. Lane 1, FL-ACE; lane 2, C-ACE; lane 3, N-ACE. Molecular-mass markers (kDa) are indicated on the right.
In contrast with previous studies where the assumption was that the enzyme preparations were fully active, we undertook active-site titrations using appropriate competitive tight-binding inhibitors. To determine the active proportion of each ACE preparation, active-site titrations were carried out using the fluorogenic substrate Mca-ASDK-Dpa [25] and the inhibitor lisinopril [31] as described in the Materials and methods section. The active-site titrations revealed that 53.1% of FL-ACE, 49.2% of N-ACE and 11.1% of the C-ACE preparations were active. Several preparations of C-ACE were purified to apparent homogeneity as assessed by SDS/PAGE and each gave a similar result. The reason for this difference in activity between the different preparations is not readily apparent. There are two potential possibilities for the binding of lisinopril and Mca-ASDK-Dpa to the two active sites in somatic ACE: (i) the two active sites bind lisinopril and Mca-ASDK-Dpa independently or (ii) the two sites are not independent and exhibit negative co-operativity. If we assumed that the two sites are not independent and exhibit negative co-operativity, then from the active-site titration experiment, the proportion of active enzyme determined would be greater than the total amount of enzyme added to the reaction, leading to a calculation of >100% activity. We therefore considered that the former possibility was the more likely. Active-site titration using the fluorogenic substrate Suc-Ala-Ala-Phe-AMC with the inhibitor thiorphan [26] revealed that the NEP preparation was 85.8% active. These values were taken into account for all subsequent kinetic analyses. Owing to the lack of availability of a tight-binding competitive inhibitor of ACE2, it was not possible to active-site titrate this enzyme.
Kinetic constants were calculated for each preparation of ACE using the fluorogenic peptide Mca-ASDK-Dpa (Table 1). The separate N- and C-domains of ACE cleaved Mca-ASDK-Dpa with very similar kinetic parameters, in agreement with those reported previously for constructs of human ACE containing only a functional N-terminal active site or a functional C-terminal active site [32]. ACE2 cleaved the fluorogenic substrate Mca-APK-Dnp with a Km of 76.6 μM, a kcat of 60.9 s−1 and a kcat/Km of 7.9×105 M−1·s−1, virtually identical with the kcat/Km of 7.7×105 M−1·s−1 that was reported previously [10]. NEP cleaved the fluorogenic substrate Suc-Ala-Ala-Phe-AMC with a Km of 36.5 μM, a kcat of 1.4 s−1 and a kcat/Km of 3.8×104 M−1·s−1.
Table 1. Kinetic constants for human FL-ACE, N-ACE and C-ACE with the fluorogenic peptide Mca-ASDK-Dpa.
Assays were performed as described in the Materials and methods section. Data shown are the means±S.E.M. of three determinations.
| Kinetic constant | FL-ACE | N-ACE | C-ACE |
|---|---|---|---|
| Km (μM) | 31.7±2.7 | 44.1±6.1 | 55.6±6.4 |
| kcat (s−1) | 9.3±0.3 | 13.6±0.6 | 10.2±0.4 |
| kcat/Km (M−1·s−1) | (2.9±0.3)×105 | (3.1±0.5)×105 | (1.8±0.3)×105 |
Kinetics of hydrolysis of angiotensin peptides by FL-ACE, ACE2 and NEP
In order to determine the catalytic efficiencies of ACE, ACE2 and NEP with a number of angiotensin peptides, experiments were carried out to calculate Km, kcat and kcat/Km for each enzyme with each substrate. The catalytic efficiencies of ACE, ACE2 and NEP for Ang I, Ang (1–9), Ang II and Ang (1–7) are shown in Table 2. Although FL-ACE and NEP hydrolysed Ang I efficiently (kcat/Km of 1.8×105 M−1·s−1 and 6.2×105 M−1·s−1 respectively), ACE2 hydrolysed Ang I very slowly with a kcat/Km of 3.3×104 M−1·s−1. The kcat of 3.5 s−1 for FL-ACE with Ang I is similar to that reported previously [33,34].
Table 2. Kinetic constants for the metabolism of angiotensin peptides catalysed by ACE, ACE2 and NEP.
The metabolism of the various angiotensin peptides was assessed as described in the Materials and methods section. Data shown are the means±S.E.M. of three determinations. N.C., not cleaved.
| Substrate | Kinetic constant | FL-ACE | ACE2 | NEP | N-ACE | C-ACE |
|---|---|---|---|---|---|---|
| Ang I | Km (μM) | 19.0±3.9 | 86.8±12.7 | 55.1±6.5 | 17.3±4.0 | 17.2±1.3 |
| kcat (s−1) | 3.5±0.3 | 2.9±0.2 | 34.1±1.4 | 3.9±0.3 | 2.7±0.1 | |
| kcat/Km (M−1·s−1) | (1.8±0.3)×105 | (3.3±0.2)×104 | (6.2±1.0)×105 | (2.2±0.4)×105 | (1.6±0.1)×105 | |
| Ang (1–9) | Km (μM) | 16.9±4.1 | N.C. | 111.4±10.0 | 33.5±9.9 | 19.7±3.4 |
| kcat (s−1) | 1.1±0.1 | N.C. | 41.8±1.4 | 1.7±0.2 | 2.7±0.2 | |
| kcat/Km (M−1·s−1) | (6.8±1.4)×104 | N.C. | (3.7±1.4)×105 | (5.0±1.3)×104 | (1.4±0.2)×105 | |
| Ang II | Km (μM) | N.C. | 5.7±1.0 | 179.0±28.6 | N.C. | N.C. |
| kcat (s−1) | N.C. | 12.8±0.6 | 40.0±2.8 | N.C. | N.C. | |
| kcat/Km (M−1·s−1) | N.C. | (2.2±0.4)×106 | (2.2±1.0)×105 | N.C. | N.C. | |
| Ang (1–7) | Km (μM) | 7.9±0.8 | N.C. | * | 8.4±1.4 | 4.4±1.4 |
| kcat (s−1) | 2.8±0.1 | N.C. | * | 3.0±0.1 | 1.4±0.1 | |
| kcat/Km (M−1·s−1) | (3.5±0.3)×105 | N.C. | * | (3.6±0.5)×105 | (3.3±1.2)×105 |
* Km for Ang (1–7) hydrolysis by NEP >500 μM, therefore making it impossible to accurately determine kcat and kcat/Km for this cleavage.
Ang (1–9) was cleaved relatively poorly by FL-ACE (kcat/Km 6.8×104 M−1·s−1), but was not hydrolysed by ACE2 even after prolonged incubation. Ang (1–9) was cleaved by NEP with a kcat/Km of 3.7×105 M−1·s−1. Although FL-ACE had a higher affinity for Ang (1–9) than NEP (Km of 16.9 compared with 111.4 μM respectively), NEP cleaved Ang (1–9) with a higher catalytic-centre activity than FL-ACE (41.8 compared with 1.1 s−1 respectively). Although Ang II is not hydrolysed by ACE, it was hydrolysed very efficiently by ACE2, giving a kcat/Km of 2.2×106 M−1·s−1, the highest catalytic efficiency seen for any of the angiotensin peptides with ACE, ACE2 or NEP. NEP cleaved Ang II with a kcat/Km of 2.2×105 M−1·s−1 to several degradation products. Ang (1–7) was cleaved efficiently by ACE (kcat/Km of 3.5×105 M−1·s−1), but was cleaved very poorly by NEP. The Km for hydrolysis of Ang (1–7) by NEP was >500 μM, therefore it was impossible to accurately determine kcat and kcat/Km values for this cleavage.
Kinetics of hydrolysis of angiotensin peptides by the two domains of ACE
We also determined the kinetic constants for the hydrolysis of the angiotensin peptides by the two separate domains of ACE (Table 2). C-ACE cleaved Ang I with a very similar kcat/Km as observed with human FL-ACE (1.6×105 M−1·s−1 compared with 1.8×105 M−1·s−1), whereas N-ACE cleaved Ang I slightly more efficiently. The Km values for all three enzyme preparations were virtually identical, and the difference seen between the two domains was due to the slightly higher kcat for N-ACE. Interestingly, Ang (1–9) was preferentially cleaved by C-ACE when compared with N-ACE, with the difference being predominantly accounted for by the higher affinity of the peptide for C-ACE. C-ACE cleaved Ang (1–9) more efficiently than FL-ACE. Ang (1–7) was cleaved to a similar extent by both N- and C-ACE, with N-ACE having a lower affinity, but a higher catalytic-centre activity, for Ang (1–7) than that of C-ACE.
Inhibition of ACE and ACE2 by angiotensin peptides
We examined the ability of the angiotensin peptides to inhibit cleavage of Ang I, Ang (1–9) and Ang (1–7). The cleavage of Ang (1–9) and Ang (1–7) by ACE, and the cleavage of Ang II by ACE2, were not inhibited by Ang I, Ang (1–9), Ang II, Ang (1–7) or Ang (1–5). However, cleavage of Ang I by either N-ACE or C-ACE was inhibited by Ang (1–7) (86.7±7.6 μM and 33.3±7.2 μM respectively) and Ang (1–9) (29.3±5.8 μM and 198.0±6.5 μM respectively), with Ang (1–9) being more potent against N-ACE, whereas Ang (1–7) was more potent against C-ACE.
Inhibition and substrate specificity of ACE2
Previously, we reported that the ACE inhibitors captopril, enalaprilat and lisinopril did not inhibit ACE2 [8]. A number of other ACE inhibitors, including the carboxylalkyl compounds cilazaprilat, indolaprilat, perindoprilat, quinaprilat and spiraprilat, the thiol compounds rentiapril and zofenapril, and the phosphoryl compounds ceranopril and fosinoprilat [21], were assessed for their ability to inhibit ACE2. All of them failed to inhibit the hydrolysis of either Ang I or Ang II by ACE2 at concentrations that abolished ACE activity (results not shown). The tripeptide Leu-Ile-Tyr (acein-2) has been reported to inhibit ACE in a non-competitive manner with an IC50 of 0.82 μM [35]. However, the activity of ACE2 was not affected by this peptide at concentrations that inhibited ACE completely (results not shown). Numerous synthetic peptide substrates have been used to measure the enzymic activity of ACE in biological samples including furylacryloyl-Phe-Gly-Gly, fMLP, hippuryl-Phe-Arg and benzyloxycarbonyl-Phe-His-Leu. However, under conditions where they were efficiently cleaved by ACE, no hydrolytic activity was observed with ACE2 (results not shown).
DISCUSSION
In the present study, we have rigorously examined the kinetics of angiotensin peptide metabolism by ACE, ACE2 and NEP. For the first time, we have investigated the metabolism of a range of angiotensin peptides under a single set of experimental conditions by both full-length two-domain ACE and by the isolated N- and C-domains of ACE, allowing peptide metabolism by the two active sites of ACE to be compared directly. In addition, we have examined the hydrolysis of angiotensin peptides by the recently discovered homologue of ACE, ACE2, and by the related zinc metallopeptidase, NEP [36]. As with ACE, Cl− ions affect the activity of ACE2 in a substrate- and pH-dependent manner [37]. Therefore we chose to perform all the kinetic analyses under identical conditions that closely mimic the salt concentration and pH of the extracellular environment (i.e. 150 mM NaCl and pH 7.4). Furthermore, we have determined the amount of active enzyme in each preparation by active-site titration with appropriate competitive tight-binding inhibitors and have taken this into account in the kinetic analyses.
In order to determine the catalytic efficiencies of ACE, ACE2 and NEP with a number of angiotensin peptides, experiments were carried out to calculate Km, kcat and kcat/Km for each enzyme with each substrate. The key findings are summarized here and in Figure 3. Ang I was cleaved efficiently by both FL-ACE and NEP, although it was a slightly better substrate for NEP. The affinity was higher for FL-ACE than NEP, but the catalytic-centre activity was higher for NEP. In contrast, Ang I was hydrolysed only very slowly by ACE2. As with Ang I, Ang (1–9) was cleaved preferentially by NEP rather than ACE, despite the higher affinity of Ang (1–9) for ACE. Ang (1–9) was not hydrolysed by ACE2 even after prolonged incubation. Although Ang II is not hydrolysed by ACE, it was hydrolysed very efficiently by ACE2, giving a kcat/Km of 2.2×106 M−1·s−1, the highest catalytic efficiency seen for any of the angiotensin peptides with ACE, ACE2 or NEP. It was also hydrolysed efficiently by NEP to several degradation products. Ang (1–7) was cleaved efficiently by ACE, but was hydrolysed very poorly by NEP. Ang (1–7) was not cleaved by ACE2.
Figure 3. Schematic diagram of the RAS.

The role of ACE, ACE2 and NEP in the metabolism of the various angiotensin peptides is shown with the kcat/Km for each reaction. The thickness of the arrows is representative of the catalytic efficiency of each reaction.
Traditionally, it was considered that the RAS would act to produce Ang II, which would then bind to its receptors or be broken down further to Ang III and Ang IV. However, the identification of a novel member of the RAS, ACE2, adds another layer of complexity to the system. Ang I can be cleaved either by ACE to Ang II or by other enzymes to Ang (1–9) and by NEP to Ang (1–7). However, the poor kinetics observed with ACE2 suggest that it is unlikely that this enzyme has a major role to play in the metabolism of Ang I to Ang (1–9). Ang (1–7) can be generated by the action of ACE2 on Ang II or by the metabolism of Ang (1–9) by ACE and NEP. Thus, under certain conditions and depending on the local concentrations of the enzymes and substrates, the RAS might favour Ang (1–7) rather than Ang II production, and thus vasodilatory forces will prevail.
Recently, it was reported that the two active sites within bovine somatic ACE exhibit strong negative co-operativity [38]. This was based on the observation that the kcat values obtained for the somatic enzyme with a range of tripeptide substrates were not the sum of the kcat values for individual domains, but, in every case, represented the means of the values of the corresponding catalytic constants obtained for single-domain forms. From the data in Tables 1 and 2, a similar phenomenon is observed with both Ang I and Ang (1–7) acting as substrates of FL-ACE, N-ACE and C-ACE. Here the kcat values for both of these substrates with FL-ACE are near the means of the values obtained for the individual domains. However, such negative co-operativity was not seen for the hydrolysis of Ang (1–9) by the individual domains of ACE and FL-ACE (Table 2), suggesting that Ang (1–9), like bradykinin [39], is possibly metabolized by both active sites. The hydrolysis of Ang (1–9) by ACE is complicated further by the observation that this peptide also inhibits preferentially the N-domain (see below), and thus the kinetics of the reaction may deviate from classic Michaelis–Menten kinetics.
Ang (1–7) has been reported to be cleaved 100-fold more efficiently by the N-domain of ACE than by the C-domain [11]. Surprisingly, we were unable to confirm this observation: Ang (1–7) was cleaved to a similar extent by both the N- and C-domains of ACE, with N-ACE having a lower affinity, but a higher catalytic-centre activity for Ang (1–7) than did C-ACE. One possibility that may account for this difference is the relatively small amount of C-ACE that we found to be active (approx. 10%) as determined by active-site titration with lisinopril. Previously, the quantity of active enzyme was not taken into account, which could explain the apparently poor kinetics obtained for the hydrolysis of Ang (1–7) by C-ACE [11]. Alternatively, this difference may be due to the preparations of ACE used. Whereas we used recombinant forms of isolated N- and C-domains of human ACE, the previous study used an N-domain fragment of ACE purified from human ileal fluid and rabbit testicular ACE for the C-domain [11]. Differences in folding and/or glycosylation of the various forms of the enzyme may influence binding of this peptide to the active sites, which is supported by the observation that addition of the first 141 amino acids from the N-domain of ACE to the C-domain resulted in a construct that was able to efficiently cleave Ang (1–7) [40]. We also studied inhibition of N-ACE and C-ACE by Ang (1–9) and Ang (1–7). We used the same substrate concentrations (0.1 mM) as used in a previous study of the inhibition of ACE by Ang (1–7) [11]. However, although Ang (1–7) was a better inhibitor of the C-domain (33.3±7.2 μM as compared with 86.7±7.6 μM for the N-domain), as with the kinetics of peptide hydrolysis, we did not find the same magnitude of difference in inhibition between the two domains noted previously [11].
Interestingly, Ang (1–9) was preferentially cleaved by C-ACE when compared with N-ACE; the difference being predominantly accounted for by the higher affinity of the peptide for the C-domain active site. We also found that Ang (1–9) was a significantly better inhibitor of the N-domain as compared with the C-domain when Ang I was substrate (29.3±5.8 μM compared with 198.0±6.5 μM). Ang (1–9) is present in human serum [41] and has a physiological role in potentiating the effects of bradykinin activity through the BK2 receptor [42]. However, the precise role of Ang (1–9) in the RAS is not clear. Although it has been widely suggested that ACE2 may have a role in the production of Ang (1–9) [9,43–46], the kinetics for the hydrolysis of Ang I by ACE2 are extremely poor, suggesting that the enzyme is unlikely to participate in this reaction in vivo. Both cathepsin A [47] and carboxypeptidase A-like activity [48] are more likely to be involved in the in vivo conversion of Ang I into Ang (1–9). In the present study, we have shown that Ang (1–9) is a substrate for ACE, being cleaved preferentially by the C-domain with kinetic parameters very similar to those for the hydrolysis of Ang I, implying that once Ang (1–9) has been formed through the action of another activity, this may be an important pathway in the RAS.
The lack of inhibition of ACE2 by any of the ACE inhibitors is entirely consistent with the recently described model of the active site of ACE2 in which the S2′ pocket is smaller than the corresponding pocket in ACE [37]. This modification blocks access of the proline residue of lisinopril and explains the difference in substrate specificity between ACE2 and ACE (carboxypeptidase compared with dipeptidyl carboxypeptidase respectively). The experimental data presented in the present study clearly indicate that the clinical use of ACE inhibitors will not have any detrimental effect on ACE2 activity in vivo.
Studies of ace2-knockout mice have provided evidence that ACE2 has a role in cardiovascular regulation [49]. The ace2-knockout mice demonstrated a severe reduction in cardiac contractility, and increased kidney, heart and plasma Ang II in the absence of any alterations in blood pressure [49]. Two recent studies provide evidence for a role for ACE2 in the human heart [50,51]. Ang (1–7) formed in the intact human myocardial circulation was decreased markedly when Ang II formation was suppressed, suggesting that Ang (1–7) is likely to be formed from Ang II by the action of ACE2 [51]. In ventricular membrane preparations from failing human heart ventricles from subjects with primary pulmonary hypertension or idiopathic dilated cardiomyopathy, ACE2 was found to be up-regulated. In this system, the Ang II formed was efficiently hydrolysed to Ang (1–7) by ACE2, and again Ang (1–7) formation was dependent on the availability of Ang II [50]. These data provide in vivo evidence for a role for ACE2 in the human heart and reinforce the findings of the present study that ACE2 has a key role to play in reducing levels of the vasoconstrictor Ang II and in increasing levels of the vasodilator Ang (1–7).
Acknowledgments
We thank the British Heart Foundation for financial support. D.A.T. was in receipt of a Biotechnology and Biological Sciences Research Council CASE (Co-operative Awards in Science and Engineering) studentship.
References
- 1.Soubrier F., Hubert C., Testut P., Nadaud S., Alhenc-Gelas F., Corvol P. Molecular biology of the angiotensin I converting enzyme: I. biochemistry and structure of the gene. J. Hypertens. 1993;11:471–476. doi: 10.1097/00004872-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Rice G. I., Foy C. A., Grant P. J. Angiotensin converting enzyme and angiotensin II type 1-receptor gene polymorphisms and risk of ischaemic heart disease. Cardiovasc. Res. 1999;41:746–753. doi: 10.1016/s0008-6363(98)00246-6. [DOI] [PubMed] [Google Scholar]
- 3.Dzau V. J. Cell biology and genetics of angiotensin in cardiovascular disease. J. Hypertens. 1994;12:S3–S10. [PubMed] [Google Scholar]
- 4.Brown N. J., Vaughan D. E. The renin–angiotensin and fibrinolytic systems: co-conspirators in the pathogenesis of ischemic cardiovascular disease. Trends Cardiovasc. Med. 1996;6:239–243. doi: 10.1016/S1050-1738(96)00091-6. [DOI] [PubMed] [Google Scholar]
- 5.Fluharty S. J., Reagan L. P., Yee D. K. The angiotensin type 1 and type 2 receptor families: siblings or cousins? In: Mukhopadhyay A. K., Raizada M. K., editors. Tissue Renin–Angiotensin Systems. New York: Plenum Press; 1995. pp. 193–211. [DOI] [PubMed] [Google Scholar]
- 6.Carretero O. A., Oparil S. Essential hypertension. Part II: treatment. Circulation. 2000;101:446–453. doi: 10.1161/01.cir.101.4.446. [DOI] [PubMed] [Google Scholar]
- 7.Fonarow G. C. Heart failure: recent advances in prevention and treatment. Rev. Cardiovasc. Med. 2000;1:25–33. [PubMed] [Google Scholar]
- 8.Tipnis S. R., Hooper N. M., Hyde R., Karran E., Christie G., Turner A. J. A human homolog of angiotensin-converting enzyme: cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 9.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robinson K., Jeyaseelan R., et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87:e1–e9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 10.Vickers C., Hales P., Kaushik V., Dick L., Gavin J., Tang J., Godbout K., Parsons T., Baronas E., Hsieh F., et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 11.Deddish P. A., Marcic B., Jackman H. L., Wang H.-Z., Skidgel R. A., Erdös E. G. N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin-(1–7) and keto-ACE. Hypertension. 1998;31:912–917. doi: 10.1161/01.hyp.31.4.912. [DOI] [PubMed] [Google Scholar]
- 12.Ferrario C. M., Chappell M. C., Tallant E. A., Brosnihan K. B., Diz D. I. Counterregulatory actions of angiotensin-(1–7) Hypertension. 1997;30:535–541. doi: 10.1161/01.hyp.30.3.535. [DOI] [PubMed] [Google Scholar]
- 13.Turner A. J., Hooper N. M. The angiotensin-converting enzyme gene family: genomics and pharmacology. Trends Pharmacol. Sci. 2002;23:177–183. doi: 10.1016/s0165-6147(00)01994-5. [DOI] [PubMed] [Google Scholar]
- 14.Mattei M.-G., Hubert C., Alhenc-Gelas F., Roeckel N., Corvol P., Soubrier F. Angiotensin-I converting enzyme gene is on chromosome 17. Cytogenet. Cell Genet. 1989;51:1041. [Google Scholar]
- 15.Hubert C., Houot A.-M., Corvol P., Soubrier F. Structure of the angiotensin I-converting enzyme gene: two alternative promoters correspond to evolutionary steps of a duplicated gene. J. Biol. Chem. 1991;266:15377–15383. [PubMed] [Google Scholar]
- 16.Soubrier F., Alhenc-Gelas F., Hubert C., Allegrini J., John M., Tregear G., Corvol P. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc. Natl. Acad. Sci. U.S.A. 1988;85:9386–9390. doi: 10.1073/pnas.85.24.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei L., Alhenc-Gelas F., Corvol P., Clauser E. The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J. Biol. Chem. 1991;266:9002–9008. [PubMed] [Google Scholar]
- 18.Campbell D. J. Vasopeptidase inhibition: a double-edged sword? Hypertension. 2003;41:383–389. doi: 10.1161/01.HYP.0000054215.71691.16. [DOI] [PubMed] [Google Scholar]
- 19.Turner A. J., Isaac R. E., Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. BioEssays. 2001;23:261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 20.Erdös E. G., Skidgel R. A. Neutral endopeptidase 24.11 (enkephalinase) and related regulators of peptide hormones. FASEB J. 1989;3:145–151. [PubMed] [Google Scholar]
- 21.Hooper N. M., Hryszko J., Oppong S. Y., Turner A. J. Inhibition by converting enzyme inhibitors of pig kidney aminopeptidase P. Hypertension. 1992;19:281–285. doi: 10.1161/01.hyp.19.3.281. [DOI] [PubMed] [Google Scholar]
- 22.Wei L., Alhenc-Gelas F., Soubrier F., Michaud A., Corvol P., Clauser E. Expression and characterization of recombinant human angiotensin I-converting enzyme: evidence for a C-terminal transmembrane anchor and for a processing of the secreted recombinant and plasma enzymes. J. Biol. Chem. 1991;266:5540–5546. [PubMed] [Google Scholar]
- 23.Hooper N. M., Keen J., Pappin D. J. C., Turner A. J. Pig kidney angiotensin converting enzyme: purification and characterization of amphipathic and hydrophilic forms of the enzyme establishes C-terminal anchorage to the plasma membrane. Biochem. J. 1987;247:85–93. doi: 10.1042/bj2470085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 25.Dive V., Cotton J., Yiotakis A., Michaud A., Vassiliou S., Jiracek J., Vazeux G., Chauvet M.-T., Cuniasse P., Corvol P. RXP 407, a phosphinic peptide, is a potent inhibitor of angiotensin I converting enzyme able to differentiate between its two active sites. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4330–4335. doi: 10.1073/pnas.96.8.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mumford R. A., Strauss A. W., Powers J. C., Pierzchala P. A., Nishino N., Zimmerman M. A zinc metalloendopeptidase associated with dog pancreatic membranes. J. Biol. Chem. 1980;255:2227–2230. [PubMed] [Google Scholar]
- 27.Bull H. G., Thornberry N. A., Cordes M. H. J., Patchett A. A., Cordes E. H. Inhibition of rabbit lung angiotensin-converting enzyme by Nα-[(S)-1-carboxy-3-phenylpropyl]L-alanyl-L-proline and Nα-[(S)-1-carboxy-3-phenylpropyl]L-lysyl-L-proline. J. Biol. Chem. 1985;260:2952–2962. [PubMed] [Google Scholar]
- 28.Rose C., Voisin S., Gros C., Schwartz J.-C., Ouimet T. Cell-specific activity of neprilysin 2 isoforms and enzymic specificity compared with neprilysin. Biochem. J. 2002;363:697–705. doi: 10.1042/0264-6021:3630697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams J. W., Morrison J. F. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- 30.Woodman Z. L., Oppong S. Y., Cook S., Hooper N. M., Schwager S. L. U., Brandt W. F., Ehlers M. R. W., Sturrock E. D. Shedding of somatic angiotensin-converting enzyme (ACE) is inefficient compared with testis ACE despite cleavage at identical stalk sites. Biochem. J. 2000;347:711–718. [PMC free article] [PubMed] [Google Scholar]
- 31.Bull H. G., Thornberry N. A., Cordes E. H. Purification of angiotensin-converting enzyme from rabbit lung and human plasma by affinity chromatography. J. Biol. Chem. 1985;260:2963–2972. [PubMed] [Google Scholar]
- 32.Cotton J., Hayashi M. A. F., Cuniasse P., Vazeux G., Ianzer D., De Camargo A. C. M., Dive V. Selective inhibition of the C-domain of angiotensin I converting enzyme by bradykinin potentiating peptides. Biochemistry. 2002;41:6065–6071. doi: 10.1021/bi012121x. [DOI] [PubMed] [Google Scholar]
- 33.Bunning P., Budek W., Escher R., Schonherr E. Characteristics of angiotensin converting enzyme and its role in the metabolism of angiotensin I by endothelium. J. Cardiovasc. Pharmacol. 1986;8:S52–S57. doi: 10.1097/00005344-198600101-00011. [DOI] [PubMed] [Google Scholar]
- 34.Liu X., Fernandez M., Wouters M. A., Heyberger S., Husain A. Arg1098 is critical for the chloride dependence of human angiotensin I-converting enzyme C-domain catalytic activity. J. Biol. Chem. 2001;276:33518–33525. doi: 10.1074/jbc.M101495200. [DOI] [PubMed] [Google Scholar]
- 35.Nakagomi K., Yamada R., Ebisu H., Sadakane Y., Akizawa T., Tanimura T. Isolation of acein-2, a novel angiotensin-I-converting enzyme inhibitory peptide derived from a tryptic hydrolysate of human plasma. FEBS Lett. 2000;467:235–238. doi: 10.1016/s0014-5793(00)01163-7. [DOI] [PubMed] [Google Scholar]
- 36.Gafford J. T., Skidgel R. A., Erdös E. G., Hersh L. B. Human kidney “enkephalinase”, a neutral metalloendopeptidase that cleaves active peptides. Biochemistry. 1983;22:3265–3271. doi: 10.1021/bi00282a035. [DOI] [PubMed] [Google Scholar]
- 37.Guy J. L., Jackson R. M., Acharya K. R., Sturrock E. D., Hooper N. M., Turner A. J. Angiotensin-converting enzyme-2 (ACE2): comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry. 2003;42:13185–13192. doi: 10.1021/bi035268s. [DOI] [PubMed] [Google Scholar]
- 38.Binevski P. V., Sizova E. A., Pozdnev V. F., Kost O. A. Evidence for the negative cooperativity of the two active sites within bovine somatic angiotensin-converting enzyme. FEBS Lett. 2003;550:84–88. doi: 10.1016/s0014-5793(03)00825-1. [DOI] [PubMed] [Google Scholar]
- 39.Georgiadis D., Beau F., Czarny B., Cotton J., Yiotakis A., Dive V. Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin. Circ. Res. 2003;93:148–154. doi: 10.1161/01.RES.0000081593.33848.FC. [DOI] [PubMed] [Google Scholar]
- 40.Marcic B., Deddish P. A., Jackman H. L., Erdös E. G., Tan F. Effects of the N-terminal sequence of ACE on the properties of its C-domain. Hypertension. 2000;36:116–121. doi: 10.1161/01.hyp.36.1.116-a. [DOI] [PubMed] [Google Scholar]
- 41.Lawrence A. C., Evin G., Kladis A., Campbell D. J. An alternative strategy for the radioimmunoassay of angiotensin peptides using amino-terminal-directed antisera: measurement of eight angiotensin peptides in human plasma. J. Hypertens. 1990;8:715–724. doi: 10.1097/00004872-199008000-00005. [DOI] [PubMed] [Google Scholar]
- 42.Erdös E. G., Jackman H. L., Brovkovych V., Tan F., Deddish P. A. Products of angiotensin I hydrolysis by human cardiac enzymes potentiate bradykinin. J. Mol. Cell. Cardiol. 2002;34:1569–1576. doi: 10.1006/jmcc.2002.2080. [DOI] [PubMed] [Google Scholar]
- 43.Sibinga N. E. S., Ware J. A. A pair of ACEs, for openers? Circ. Res. 2000;87:523–525. doi: 10.1161/01.res.87.7.523. [DOI] [PubMed] [Google Scholar]
- 44.Bernstein K. E. Two ACEs and a heart. Nature (London) 2002;417:799–802. doi: 10.1038/417799a. [DOI] [PubMed] [Google Scholar]
- 45.Oudit G. Y., Crackower M. A., Backx P. H., Penninger J. M. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc. Med. 2003;13:93–101. doi: 10.1016/s1050-1738(02)00233-5. [DOI] [PubMed] [Google Scholar]
- 46.Yagil Y., Yagil C. Hypothesis: ACE2 modulates blood pressure in the mammalian organism. Hypertension. 2003;41:871–873. doi: 10.1161/01.HYP.0000063886.71596.C8. [DOI] [PubMed] [Google Scholar]
- 47.Jackman H. L., Massad M. G., Sekosan M., Tan F., Brovkovych V., Marcic B. M., Erdös E. G. Angiotensin 1–9 and 1–7 release in human heart: role of cathepsin A. Hypertension. 2002;39:976–981. doi: 10.1161/01.hyp.0000017283.67962.02. [DOI] [PubMed] [Google Scholar]
- 48.Kokkonen J. O., Saarinen J., Kovanen P. T. Regulation of local angiotensin II formation in the human heart in the presence of interstitial fluid: inhibition of chymase by protease inhibitors of interstitial fluid and of angiotensin-converting enzyme by Ang-(1–9) formed by heart carboxypeptidase A-like activity. Circulation. 1997;95:1455–1463. doi: 10.1161/01.cir.95.6.1455. [DOI] [PubMed] [Google Scholar]
- 49.Crackower M. A., Sarao R., Oudit G. Y., Yagil C., Kozieradzki I., Scanga S. E., Oliveira-dos-Santos A. J., Costa J. D., Zhang L., Pei Y., et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature (London) 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 50.Zisman L. S., Keller R. S., Weaver B., Lin Q., Speth R., Bristow M. R., Canver C. C. Increased angiotensin-(1–7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme homologue ACE2. Circulation. 2003;108:1707–1712. doi: 10.1161/01.CIR.0000094734.67990.99. [DOI] [PubMed] [Google Scholar]
- 51.Zisman L. S., Meixell G. E., Bristow M. R., Canver C. C. Angiotensin-(1–7) formation in the intact human heart: in vivo dependence on angiotensin II as substrate. Circulation. 2003;108:1679–1681. doi: 10.1161/01.CIR.0000094733.61689.D4. [DOI] [PubMed] [Google Scholar]