

Abstract
Cyclophilin-D is a peptidylprolyl cis–trans isomerase of the mitochondrial matrix. It is involved in mitochondrial permeability transition, in which the adenine nucleotide translocase of the inner membrane is transformed from an antiporter to a non-selective pore. The permeability transition has been widely considered as a mechanism in both apoptosis and necrosis. The present study examines the effects of cyclophilin-D on the permeability transition and lethal cell injury, using a neuronal (B50) cell line stably overexpressing cyclophilin-D in mitochondria. Cyclophilin-D overexpression rendered isolated mitochondria far more susceptible to the permeability transition induced by Ca2+ and oxidative stress. Similarly, cyclophilin-D overexpression brought forward the onset of the permeability transition in intact cells subjected to oxidative stress. In addition, in the absence of stress, the mitochondria of cells overexpressing cyclophilin-D maintained a lower inner-membrane potential than those of normal cells. All these effects of cyclophilin-D overexpression were abolished by cyclosporin A. It is concluded that cyclophilin-D promotes the permeability transition in B50 cells. However, cyclophilin-D overexpression had opposite effects on apoptosis and necrosis; whereas NO-induced necrosis was promoted, NO- and staurosporine-induced apoptosis were inhibited. These findings indicate that the permeability transition leads to cell necrosis, but argue against its involvement in apoptosis.
Keywords: apoptosis, cyclophilin-D, mitochondria, necrosis, permeability transition
Abbreviations: AFC, 7-amino-4-trifluoromethylcoumarin; ANT, adenine nucleotide translocase; CCCP, carbonyl cyanide m-chlorophenylhydrazone; CSA, cyclosporin A; CyP, cyclophilin; CyP-D(+) cells, stable cell line overexpressing CyP-D; GST, glutathione S-transferase; mCyP-D, mature CyP-D; pCyP-D, precursor CyP-D; PPIase, peptidylprolyl cis–trans isomerase; PT, permeability transition; TMRE, tetramethylrhodamine ethylester; TPP+, tetraphenylphosphonium ion; VDAC, voltage-dependent anion channel; ΔψM, mitochondrial inner-membrane potential; H95Q etc., His95→Gln substitution etc
INTRODUCTION
Cyclophilins (CyPs) form a family of enzymes that catalyse the rotation of Xaa-Pro bonds in target proteins. The PPIase (peptidylprolyl cis–trans isomerase) activity is blocked by CSA (cyclosporin A) which occludes the active site. CyPs are located throughout the cell. CyP-D is the isoform found in mitochondria where it is restricted to the intramitochondrial (matrix) compartment [1]. The physiological role of CyP-D is obscure, although it may catalyse the folding of newly imported proteins [2]. ‘Pulldown’ experiments have revealed that CyP-D can bind to the ANT (adenine nucleotide translocase) of the mitochondrial inner membrane and also to the complex between ANT and the VDAC (voltage-dependent anion channel) of the outer membrane [3,4]. In isolated mitochondria, subjected to Ca2+ overload and oxidative stress, the interaction between CyP-D and ANT leads to the transformation of ANT from a selective ADP/ATP antiporter to a non-selective pore, termed the permeability transition (PT) pore (reviewed in [5,6]). PT pore formation is blocked by CSA. Reconstitutions of Ca2+-activated, CSA-inhibited PT pore activity from purified ANT, VDAC and CyP-D in liposomes [3,6] and black lipid membranes [7] have confirmed that these components form the PT pore. Whether ANT has an obligatory pore-forming role has been questioned [8]; nevertheless, the pulldown and reconstitution experiments (as descibed above) indicate that CyP-D binds to ANT. It is not known whether the binding of CyP-D to ANT is contingent on high [Ca2+] and/or oxidative stress, or whether the interaction occurs normally, but only becomes apparent under conditions that transform ANT into a non-selective pore.
From flux measurements, the internal diameter of the open PT pore in mitochondria has been estimated to be about 2 nm [9,10], rendering the inner membrane freely permeable to metabolites and inorganic ions, and uncoupling mitochondrial energy transduction. On these grounds, it was first proposed that PT pore formation is a pathological phenomenon, and that it may be a critical step in cell necrosis brought about by cellular Ca2+ overload and oxidative stress [9–11]. This model is supported by demonstrations that the mitochondrial inner membrane does become non-selectively permeable in cells subjected to Ca2+ overload and oxidative stress [6,12–14]. It remains unclear, however, to what extent PT pore opening is instrumental in lethal cell injury or is merely a consequence of other pathogenic processes.
Mitochondria provide essential steps in the apoptotic signalling pathway by releasing cytochrome c, apoptosis-inducing factor, Smac/DIABLO [direct IAP (inhibitor of apoptosis protein) binding protein], and other apoptogenic proteins from the intermembrane space to the cytosol, where they amplify the caspase cascade (reviewed in [15]). Release of these proteins is controlled by Bcl-2 family proteins, some of which promote the release (e.g. Bax and Bid), whereas others inhibit release (e.g. Bcl-2). PT pore opening has been widely considered in this context, either as a mechanism for mitochondrial swelling and physical rupture of the outer membrane (reviewed in [16]), or by more subtle ultrastructural changes facilitating cytochrome c diffusion to the outer membrane [17]. Bax action was reported to be promoted by PT pore opening, and Bax can bind to VDAC [18–22]. Bid was also reported to induce PT pore opening [17]. On the other hand, Bax-permeabilized mitochondria show no signs of permanent outer membrane rupture [23], and Bax and Bid synergistically permeabilize liposomes to large molecules in the absence of VDAC, ANT or CyP-D [24]. It remains unclear, therefore, whether PT pore formation has a role in apoptosis.
The role of CyP-D and the PT in apoptosis and cell necrosis cannot readily be investigated using CSA as inhibitor of CyP-D and the PT pore, since CSA interacts with other CyPs. In the present study, we address these questions by overexpressing CyP-D in a B50 (neuronal) cell line. We show that the overexpression leads to PT pore opening in the absence of any pathogenic insult, indicating that CyP-D is able to interact with ANT in viable cells. We further show that the overexpression has opposite effects on apoptosis (attenuated) and necrosis (promoted), which is in line with PT pore opening in necrotic, but not apoptotic, cell death.
EXPERIMENTAL
Expression vectors
Plasmid constructs were prepared encoding (a) pCyP-D (precursor CyP-D) comprising the entire coding sequence for CyP-D, including the mitochondrial targeting sequence, and (b) mCyP-D (mature CyP-D) without the targeting sequence. Both sequences were PCR amplified from rat CyP-D cDNA [1], with the addition of BamH1 and EcoR1 restriction sites. The following primers were used: forward (mCyP-D), 5′-CGCGGGATCCAGTGCAGCGACGGCGGAGCCCG; forward (pCyP-D), 5′-CATAGGATCCAGATGCTAGCTCTGCGCTGCGC; and reverse (for both), 5′-CGCGGAATTCTTAGCTCAACTGGCCACAGT. The PCR products were cloned between the same restriction sites of the prokaryotic expression vector pGEX-3X (Amersham) for production of pCyP-D and mCyP-D fusions to the C-terminus of GST (glutathione S-transferase). The pCyP-D sequence was also cloned between the BamH1 and EcoR1 sites of the eukaryotic expression vector pcDNA3.1 (Invitrogen) for transfection of B50 cells. Point mutations were introduced into CyP-D by PCR overlap extension mutagenesis using the following primers: for H95Q (His95→Gln), 5′-CTGGGATGACCCTCTGGAAGGTGGAGCCTTTG (forward) and 5′-CAAAGGCTCCACCTTCCAGAGGGTCATCCCAG (reverse); for F154A, 5′-GTCTTTATTGTGCAGATAGCGAACTGAGAGCC (forward) and 5′-GCCTCTCAGTTCGCTATCTGCACAATAAAGAC (reverse). The sequences were validated. All plasmids were propagated in Escherichia coli strain DH5α.
Purification of pCyP-D and mCyP-D
pCyP-D and mCyP-D were prepared for use as markers on SDS/PAGE. E. coli cells transformed with the recombinant pGEX vectors were cultured at 30 °C and induced as described previously [3]. Cells were extracted 4 h after induction by sonication in 100 mM NaCl/10 mM Tris/HCl (pH 7.2)/0.5 mM EDTA/1 mM PMSF. The GST-fusion proteins were bound to GSH–agarose, washed with extraction medium (above), and then treated with Factor XA according to the manufacturer's instructions (Amersham) to release CyP-D. The pCyP-D and mCyP-D were further purified by FPLC on Mono-S and Superdex-75 columns (Pharmacia) to single bands on SDS/PAGE (see Figure 1 below).
Figure 1. Expression, processing and quantification of CyP-D in transfected B50 cells.

(A and B) Processing of transfected CyP-D. B50 cells were transiently transfected with wild-type CyP-D (A, lane 1), CyP-D(H95Q) (B, lane 3) or CyP-D(F154A) (B, lane 4), or they were isolated as a clone [CyP-D(+)] stably transfected with wild-type CyP-D (A, lanes 5 and 6). Control cells were not transfected. Mitochondria (mito) were isolated from CyP-D(+) cells (A, lane 6) or control cells (A, lane 7). Transiently transfected cells were extracted 2 days after transfection. The pCyP-D and mCyP-D markers were prepared by cleavage of GST-fusion proteins. The positions of 28 kDa and 20 kDa markers are indicated. Bands were developed using peroxidase-conjugated secondary antibodies. (C) Quantification of CyP-D in mitochondria isolated from control and CyP-D(+) cells. The standard curve (continuous line) was obtained using 50–300 fmol of recombinant CyP-D (inset). Mitochondria isolated from control (CON) and the CyP-D(+)clone (CL) were analysed in parallel with the standards using 25 μg (CON) and 4 μg (CL) of total mitochondrial protein (inset). Bands were developed using fluorescent secondary antibodies.
B50 cell culture and isolation of stable cell line overexpressing CyP-D [CyP-D(+) cells]
B50 cells from a rat neuronal cell line (European Collection of Cell Cultures, Salisbury, Wilts, U.K.) were maintained under CO2/air (1:19) at 37 °C in DMEM (Dulbecco's minimal essential medium) with 10% (w/v) foetal calf serum, and 50 μg/ml gentamycin. At 80–90% confluency, cells on each plate (6 cm diameter) were transfected with 10 μg of recombinant pcDNA plus 40 μl of Lipofectamine 2000 reagent (Invitrogen) in Opti-Mem (Gibco). After 72 h of transfection, cells were washed with serum-free medium and passaged at 1:10 dilution into fresh culture medium. Transfectants were selected in medium containing 600–1000 μg/ml geneticin (Gibco), which was changed every 24 h. After 3 weeks, 21 resistant clones were isolated from 60 plates. The clones were expanded and tested positive for CyP-D overexpression.
Isolation of mitochondria from B50 cells and measurement of TPP+ (tetraphenylphosphonium ion) uptake
B50 cells (approx. 109) were scraped into 10 ml of 225 mM mannitol/75 mM sucrose/0.5 mM EGTA/10 mM Hepes (pH 7.4)/0.1% (w/v) BSA/100 μM PMSF, and extracted with a tight-fitting Potter–Elvehjem homogenizer. The homogenate was centrifuged at 600 g for 5 min. The supernatant was then centrifuged at 8000 g for 8 min to sediment the mitochondria. The mitochondria were washed once in 395 mM sucrose/10 mM EGTA by resuspension/sedimentation, and suspended finally in the same medium. Mitochondrial uptake of TPP+ was measured with a home-made PVC membrane electrode [25]. Mitochondria (0.5 mg of protein) were suspended in 0.5 ml of 120 mM KCl/10 mM Hepes (pH 7.4)/1 μg of rotenone/1.4 μM TPP+/10 μM EGTA/0.5 mM KH2PO4. TPP+ uptake was started with 2 mM succinate as respiratory substrate. Further additions are noted in the legend to Figure 2 (below).
Figure 2. Effect of CyP-D overexpression on TPP+ uptake by isolated mitochondria.

Mitochondria isolated from control B50 cells (A, C) or cloned CyP-D(+) cells (B, D) were preincubated for 7 min in the presence of rotenone and 10 μM EGTA as described in the Experimental section. The following were added: 500 nM CSA (CSA), 2 mM succinate (Succ) and 20 μM CaCl2 (Ca), either alone or with 100 μM t-butylhydroperoxide (TB).
Detection and assay of CyP-D
Cell extracts were analysed in Western blots using a polyclonal antibody raised (in this laboratory) against the peptide CSDGGARGANSSSQNP corresponding to the N-terminus of mCyP-D. Bands were detected either with peroxidase-conjugated antirabbit IgG and the enhanced chemiluminescence reagent (Amersham) or with fluorescent anti-rabbit IgG (Alexa fluor 680 allophycocyanine; Molecular Probes, Eugene, OR, U.S.A.) and the Odyssey 9201 imaging system (Li-Cor Biosciences, Lincoln, NE, U.S.A.). The PPIase activity of CyP-D was determined as described previously [25], with N-succinyl-alanyl-alanyl-prolyl-phenylalanyl-4-nitroanilide as test peptide.
Caspase assays
Cells were extracted in 20 mM Hepes (pH 7.2)/2 mM EDTA/5% (w/v) CHAPS/5 mM dithiothreitol/1 mM PMSF and 10 μg/ml each of pepstatin, leupeptin and aprotinin. Caspases were assayed in 20 mM Pipes (pH 7.4)/1 mM EDTA/0.1% (w/v) CHAPS/1 mM dithiothreitol and the fluorescent AFC (7-amino-4-trifluoromethylcoumarin) derivatives of the caspase-selective substrates Ac-DEVD-AFC (N-acetyl-Asp-Glu-Val-Asp-AFC; caspase-3) and Ac-LEHD-AFC (N-acetyl-Leu-Glu-His-Asp-AFC; caspase-9). Protein was determined with the Bradford assay using BSA as standard.
Fluorescence imaging
For microscopy, cells seeded on to laminin-coated coverslips were incubated at 21–23 °C in 100 mM NaCl/4 mM KCl/24 mM Hepes (pH 7.4)/1 mM MgSO4/1 mM CaCl2/1 mM KH2PO4/11 mM glucose. Sodium nitroprusside was added as source of NO. Nuclei were stained with cell-permeant Hoechst 33258 for the measurement of the total number of nuclei and the detection of nuclei with apoptotic morphology, and with plasma membrane-impermeant ethidium homodimer for the detection of necrotic nuclei. Mitochondria were stained with TMRE (tetramethylrhodamine ethylester). TMRE fluorescence was measured at 530 nm (excitation) and >595 nm (emission), using the cell boundary from brightfield images to obtain total cell fluorescence. Images were obtained with an Olympus IX-70 fluorescence microscope with ×60 oil objective and cooled charge-coupled-device camera (Micromax 1401E, Princeton Instruments), and were processed with Metamorph software (Universal Imaging).
RESULTS
Stable cell lines overexpressing CyP-D
CyP-D is a 206 residue (18 kDa) isoform located in mitochondria [26,27]. The protein was just visible by chemiluminescence in Western blots of B50 cell mitochondria (Figure 1A, lane 7), but, due to the protein-loading limit of the gels, not in extracts of whole cells (Figure 1A, lane 2). Transfection with the CyP-D construct (see the Experimental section) increased the B50 cell content of CyP-D substantially 1–2 days after transfection (Figure 1A, lane 1).
The physiological effects of CyP-D were investigated by generating a stable B50 cell line overexpressing CyP-D [henceforth termed CyP-D(+) cells]. First, we ensured that the overexpressed CyP-D was processed normally. Rat CyP-D is synthesized in the cytoplasm as pCyP-D containing an N-terminal sequence of 29 amino acids that targets the protein to mitochondria. After import into mitochondria, the targeting sequence is cleaved off to yield the mature protein (mCyP-D) in the mitochondrial matrix [1]. Both aspects, N-terminal processing and mitochondrial targeting, were checked. In order to discriminate clearly between pCyP-D and mCyP-D these were expressed as GST-fusion proteins in E. coli, cleaved from GST, purified, and used as markers. Figure 1(A) shows that the overexpressed CyP-D in the CyP-D(+) cells (lane 5) aligned with mCyP-D, rather than pCyP-D, and that the protein was recovered in the mitochondria (lane 6). This is consistent with the overexpressed CyP-D being imported into mitochondria and N-terminally processed to the mature form.
The importance of confirming that normal processing actually occurs is underlined by Figure 1(B), where B50 cells were transfected with CyP-Ds carrying mutations in the PPIase active site. The active site of the cytosolic isoform, CyP-A, has been defined (X-ray studies [28]), and the same residues, including the two mutated ones, are found in CyP-D. To determine catalytic efficiencies, the H95Q and F154A mutants were obtained by Factor XA cleavage of GST-fusion proteins produced in E. coli. The H95Q and F154A mutants retained about 20% and 2% of the catalytic efficiency of wild-type CyP-D respectively (kcat/KM, 7 μM−1·s−1 at 14 °C; results not shown). These values compare well with the 15% and 3% residual activities in the corresponding mutants of CyP-A (H54Q and F113A respectively [29]). As shown in Figure 1(B), both H95Q and F154A mutants displayed defective processing in B50 cells. Both were expressed in relatively high quantities, but both aligned with pCyP-D on SDS/PAGE, rather than mCyP-D, indicating an absence of N-terminal processing. The uncleaved H95Q and F154A CyPs were recovered in both mitochondrial and cytosolic fractions (results not shown).
Purification of CyP-D from rat liver can produce two proteins, one with residue 30 at the N-terminus (mCyP-D in the present study) and a shorter form lacking a further 10 N-terminal amino acids [30]. It was suggested that pCyP-D may be cleaved in two ways to yield two forms of mCyP-D [30]. However, proteolysis of mCyP-D during purification could not be ruled out. In addressing this question, we observed that [35S]pCyP-D was processed to a single form of mCyP-D following import into mitochondria, consistent with a single mCyP-D species [1]. The recovery of a single band of overexpressed CyP-D (Figure 1) substantiates this conclusion.
CyP-D was quantified in Western blots by fluorescence imaging as reported in Figure 1(C). Control mitochondria contained 70 fmol of CyP-D/25 μg of total mitochondrial protein (2.8 pmol of CyP-D/mg of protein) and CyP-D(+) mitochondria 226 fmol of CyP-D/4 μg of protein (57 pmol of CyP-D/mg of protein). Thus mitochondrial CyP-D was increased about 20-fold in the CyP-D(+) cells. The actual content of CyP-D in the CyP-D(+) clone (57 pmol of CyP-D/mg of protein) is similar to that of normal rat liver mitochondria (about 50 pmol/mg [31]).
Effect of CyP-D overexpression on PT in isolated mitochondria
CyP-D is an essential component of the PT pore ([3,4,7] and references therein). The effects of CyP-D overexpression on the PT pore were first tested in isolated mitochondria. PT pore opening allows free diffusion of ions across the inner membrane and dissipates ΔψM (mitochondrial inner-membrane potential). Here, PT pore opening was measured using TPP+, which is accumulated electrophoretically by mitochondria in response to ΔψM and is released on PT pore opening [32].
Figure 2(A) records TPP+ uptake by normal B50 cell mitochondria. The mitochondria were preincubated with rotenone to inhibit NAD+-linked oxidation of endogenous substrates and to dissipate ΔψM {other experiments established that addition of the uncoupling agent CCCP (carbonyl cyanide m-chlorophenylhydrazone) at this point to abolish any residual ΔψM caused no detectable change in extramitochondrial [TPP+], confirming that the inner membrane was fully depolarized after incubation with rotenone}. Low [EGTA] was also included to remove trace amounts of Ca2+. The introduction of succinate as respiratory substrate generated ΔψM, as shown by the rapid uptake of TPP+. Ca2+ was then added, either alone, or together with t-butylhydroperoxide. Ca2+ alone caused a transient depolarization. This is typically observed on addition of Ca2+ to mitochondria, as Ca2+ is accumulated electrophoretically across the inner membrane [33], so that ΔψM is depressed during rapid Ca2+ uptake and restored when Ca2+ uptake is complete [11,32]. In the present study, rather more TPP+ was taken up following Ca2+ accumulation, which may reflect a small increase in mitochondrial volume. However, co-addition of Ca2+ and peroxide brought about a progressive loss of ΔψM (Figure 2A), which was not observed with peroxide alone (results not shown; the trace was similar to that with Ca2+ alone). Thus Ca2+ and peroxide synergistically induced PT pore opening in B50 cell mitochondria, as they do in mitochondria from other sources (e.g. liver and heart [9,34]).
In the absence of Ca2+, mitochondria from CyP-D(+) cells (Figure 2B) accumulated about the same amount of TPP+ as those from control cells; in 15 such experiments, each with different mitochondrial preparations, TPP+ uptake by control and CyP-D(+) mitochondria was not significantly different (0.70±0.03 and 0.66±0.03 nmol/mg of protein respectively). However, addition of Ca2+ and, in particular, of Ca2+ plus peroxide, brought about a rapid collapse of ΔψM. Both the Ca2+-induced (results not shown) and the Ca2+-plus-peroxide-induced loss of ΔψM were prevented by the PT pore inhibitor CSA (Figure 2D), such that the TPP+ accumulation approached that of control mitochondria (Figure 2C). The inhibition by CSA confirms that the loss of ΔψM in CyP-D(+) cells was due to PT pore opening. It appears then that CyP-D overexpression sensitizes ANT to agents (Ca2+ and oxidants) that transform it into the PT pore.
Effect of CyP-D overexpression on PT in intact cells
TMRE and other rhodamines are lipid-soluble cations accumulated in mitochondria according to the magnitude of ΔψM and are widely used to monitor ΔψM changes in intact cells (for example, [35]). TMRE accumulation in B50 cell mitochondria is shown below (see Figure 4A). Figure 3(A) shows that control B50 cells accumulated more TMRE than the CyP-D(+) cells. Addition of CSA increased TMRE uptake by CyP-D(+) cells, but not detectably by the control cells. From the relative increases in fluorescence, the CyP-D(+) cells accumulated only 25% of that accumulated by control cells, but this difference was largely abolished by CSA (Figure 3B). The most direct interpretation is that CyP-D overexpression brings about a degree of PT pore opening in intact cells (as it does in isolated mitochondria, Figure 2) sufficient to dissipate partially ΔψM, and that CSA restores ΔψM by inhibiting PT pore opening.
Figure 4. Changes in cellular TMRE distribution following dissipation of ΔψM with the uncoupling agent CCCP.

A control B50 cell was preincubated with 500 nM TMRE for 10 min, washed in standard suspension medium (see the Experimental section) and then imaged at various times after addition of 50 nM CCCP. (A–D) Images were taken at time zero (A, D), 5 min (B) and 60 min (C) after CCCP addition. Bottom panel: Cel, mean cellular TMRE fluorescence; Nuc, mean nuclear TMRE fluorescence [from the area designated ‘n’ in (D)]; Mit, mean mitochondrial TMRE fluorescence in excess of cytosolic fluorescence. Mit was calculated as the difference between the mean cytoplasmic fluorescence [area ‘c’ in (D)] and the mean cytosolic fluorescence (taken to be equal to mean nuclear fluorescence).
Figure 3. Effect of CyP-D overexpression on TMRE uptake by B50 cells.

(A) TMRE (40 nM) was added to control B50 cells (•) or to cloned CyP-D(+) cells (○) at time zero. In each case, the increase in TMRE fluorescence of 5 cells was measured with time. CSA (500 nM) was added when indicated. (B) The increase in cell TMRE fluorescence was measured after 1 h exposure to 40 nM TMRE in the presence and absence of CSA. The results are expressed as the means±S.E.M. (30 cells from 6 coverslips). Abbreviations: CON, control B50 cells; CL, cloned CyP-D(+) cells.
We also checked whether CyP-D overexpression sensitized mitochondria in intact cells to an added stimulus (hydroperoxide; see Figure 5 below) for PT pore opening. PT pore opening was monitored from the collapse in ΔψM as indicated by the release of mitochondrially accumulated TMRE. The release was most conveniently measured as an increase in nuclear fluorescence, after confirming the nuclear region from brightfield images. This procedure was first checked using the proton ionophore CCCP to collapse ΔψM, as shown in Figure 4. Before addition of CCCP, TMRE was largely sequestered within mitochondria, with the nuclear (mitochondria-free) region relatively TMRE free (Figure 4A). Addition of CCCP induced TMRE loss from mitochondria to the cytosol and nucleus (Figure 4B). Subsequently, TMRE was lost from the cell (Figure 4C). Figure 4 (bottom panel) quantifies these changes from the nuclear and cytoplasmic areas designated in Figure 4(D). TMRE fluorescence becomes self quenched at high concentrations [36,37]. Consequently, on release from mitochondria (dilution), total cellular TMRE fluorescence increased, along with that of the nucleus. Thereafter, total TMRE fluorescence decreased as the dye effluxed from the cell. In order to confirm that the increase in nuclear fluorescence corresponded to loss from mitochondria, the mean mitochondrial TMRE fluorescence (in excess of the cytosolic fluorescence) was calculated. Since TMRE is diffusible, the mean cytosolic TMRE fluorescence was taken to be the same as the mean nuclear TMRE fluorescence (area ‘n’, Figure 4D); this value was then subtracted from the mean cytoplasmic TMRE fluorescence (area ‘c’, Figure 4D) to give the mean mitochondrial TMRE fluorescence in excess of cytosolic. The calculated data confirm that TMRE was lost from the mitochondria to the cytosol, and that the transient rise in nuclear fluorescence may be used as an index of ΔψM dissipation.
Figure 5. Effect of CyP-D overexpression on the release of mitochondrial TMRE induced by t-butylhydroperoxide and CCCP.

Loss of mitochondrial TMRE to the nuclear region of control B50 cells (control) or cloned CyP-D(+) cells (clone) was measured as in Figure 4 (mean nuclear fluorescence). Either 100 μM t-butylhydroperoxide (TB) or 500 nM CCCP were added at time zero, as indicated by the arrow. When present, 500 nM CSA was introduced 5 min before TB or CCCP. Each figure shows data from 6 cells. The arbitrary fluorescence units are the same for all Figure parts, but (C) and (F) are drawn to a smaller scale.
Control B50 cells exposed to t-butylhydroperoxide as PT pore activator showed a transient increase in nuclear TMRE fluorescence after a lag of about 20 min (Figure 5A) indicating dissipation of ΔψM. This was abolished by CSA (Figure 5B), in line with PT pore opening being the cause of the decrease in ΔψM. Equivalent experiments with CyP-D(+) cells displayed a much smaller (Figure 5D), but still CSA sensitive (Figure 5E), temporary rise in nuclear fluorescence. A smaller increase in nuclear fluorescence in the CyP-D(+) cells would be anticipated, since they take up less TMRE (Figure 3). In agreement, rapid collapse of ΔψM with 500 nM CCCP produced a greater increase in nuclear TMRE fluorescence in control cells (Figure 5C) than in CyP-D(+) cells (Figure 5F). However, peroxide induced the dissipation of ΔψM in CyP-D(+) cells without a detectable lag phase (Figure 5D), unlike in the control cells (Figure 5A). This substantiates the conclusion that the tendency of ANT to deform into the PT pore is enhanced by the overexpression of CyP-D.
Effects of CyP-D overexpression on cell death
CyPs have been widely implicated in necrotic and apoptotic cell death, although the identity(s) of the CyP is controversial (see the Discussion). In the present study, CyP-D(+) cells were used to examine specifically the relevance of CyP-D. The NO donor nitroprusside was used as lethal insult. NO is believed to be an important component of excitotoxic cell death in neurons, brought about by a period of ischaemia. Under these conditions, glutamate accumulates and becomes neurotoxic by inducing excessive Ca2+ influx which, in turn, activates NO synthase [38,39]. NO brings about both apoptosis and necrosis according to the intensity of insult, possibly by inhibiting respiration, since it competes with O2 at cytochrome oxidase and can also nitrosylate and inactivate complex I [40,41]. Exposure of B50 cells to 100 μM nitroprusside for a period of about 30 min leads mainly to apoptosis [42], whereas prolonged exposure produces cell necrosis (see below).
In Figure 6, cells were incubated continuously with nitroprusside. Cell necrosis was quantified by entry into the cell of the nuclear stain ethidium homodimer, which enters dead, but not live, cells. Stained nuclei had normal morphologies, consistent with necrosis (rather than the condensed morphologies of apoptotic nuclei shown in Figure 7 below). CyP-D(+) cells underwent necrosis sooner than control cells; for example, at 2 h after exposure to NO there were about 4-fold more necrotic CyP-D(+) cells than control cells. CSA retarded necrosis in both types of cell. These data are representative of five such experiments and indicate that the PT pore promotes NO-induced cell necrosis.
Figure 6. CyP-D overexpression promotes NO-induced necrosis of B50 cells.

Control B50 cells (Con) and CyP-D(+) cells (Clone) were incubated with 100 μM nitroprusside as NO donor in the presence and absence of 500 nM CSA. Cell necrosis was quantified by nuclear staining with the live-cell impermeant ethidium homodimer. About 400 cells were analysed per time point under each condition. For clarity, the CyP-D(+) data without CSA are reproduced in (B).
Figure 7. CyP-D overexpression inhibits NO- and staurosporine-induced apoptosis of B50 cells.

Control B50 cells (open columns) and CyP-D(+) cells (closed columns) were treated with either 100 μM nitroprusside for 40 min or with 400 nM staurosporine continuously, or were left untreated. The following day cells were stained with cell-permeant Hoechst 33258 for counting of apoptotic (APO) nuclei and total nuclei, and with live-cell impermeant ethidium homodimer for counting nuclei of necrotic cells (NEC). Examples of nuclei with normal and apoptotic morphology stained with Hoechst 33258 are shown in (D). Nuclei of necrotized cells were morphologically similar to normal nuclei, but stained with ethidium homodimer. In (A–C), the results are expressed as the means±S.E.M. for 5 separate experiments (>500 cells analysed in each experiment).
Apoptosis was induced by 40 min exposure to nitroprusside, and was assayed the following day after staining with the cell-permeable Hoechst 33258. Apoptotic nuclei were distinguished as condensed and fragmented (Figure 7D) and were largely absent from untreated cultures of both control and CyP-D(+) cells (Figure 7A). NO induced about 17% apoptosis of control cells, but was without effect on the CyP-D(+) cells (Figure 7B). Since the CyP-D(+) cells were more susceptible to necrosis than control cells (Figure 6), and cultures of CyP-D(+) cells subjected to 40 min of nitroprusside contained a small fraction of necrotic nuclei the following day (about 5%, Figure 7B), it seemed possible that some CyP-D(+) cells, otherwise destined for apoptosis, may have succumbed to necrosis, i.e. that, in part, the lack of apoptosis reflected the promotion of necrosis, rather than inhibition of apoptosis. Accordingly, we also tested staurosporine as an apoptotic stimulus. Staurosporine can also induce necrosis under certain conditions [43], but in the present study there were negligible necrotic cells the following day after addition of staurosporine to either control or Cyp-D(+) cultures (Figure 7C). Figure 7(C) shows that staurosporine-induced apoptosis was also substantially reduced in CyP-D(+) cells (28%) with respect to control cells (85%), as judged from nuclear morphologies.
As another marker of apoptosis, we measured caspase activation. Caspases are expressed constitutively as inactive proenzymes and become activated after proteolytic cleavage. Caspase activation shows a hierarchy in which the ‘effector’ caspases, which degrade a variety of cellular substrates, are cleaved and activated by ‘initiator’ caspases. In the present study we assayed the ‘effector’ caspase-3 and the ‘initiator’ caspase-9, which is activated at the apoptosome. Transient exposure to nitroprusside brought about a 6–8-fold activation of both caspases within 4–6 h (Figure 8). In contrast, there was no detectable activation of either caspase-3 or caspase-9 by nitroprusside in CyP-D(+) cells. This correlates with the formation of apoptotic nuclei in control cells, but not CyP-D(+) cells, following exposure to nitroprusside (Figure 7B). Staurosporine was more effective in activating both caspases (Figure 8). But again, activation of both caspase-9 and caspase-3 was substantially reduced in CyP-D(+) cells with respect to control cells. In summary, from the data of Figures 6–8, we concluded that CyP-D has opposite effects on apoptosis and necrosis in B50 cells.
Figure 8. CyP-D overexpression inhibits caspase activation induced by nitroprusside and by staurosporine.

Control B50 cells and CyP-D(+) cells (clone) were either incubated for 40 min with 100 μM nitroprusside or continuously with 400 nM staurosporine. Cells were extracted at the times indicated after removal of nitroprusside or the addition of staurosporine, and assayed for caspase-3 and caspase-9 as stated. The results are expressed as the means±S.E.M. for 5 separate experiments.
DISCUSSION
To date, the question has remained open of whether CyP-D can interact with ANT normally, implying a physiological perspective to the interaction, or only under pathological conditions of high [Ca2+] and oxidative stress (see the Introduction). The present data provide the first indication that CyP-D can interact with ANT under physiological conditions. Thus PT pore opening was enhanced in CyP-D(+) cells in the absence of insult (Figure 3). It follows that overexpressed CyP-D interacts with ANT in healthy unstressed cells. Since ΔψM in control cells was unaffected by CSA (Figure 3), the endogenous amount of CyP-D in control cells is evidently insufficient to cause measurable ΔψM dissipation at physiological [Ca2+]. Nevertheless, if overexpressed CyP-D binds to ANT under normal conditions, then the same will be true for endogenous CyP-D in control cells, albeit to a lesser extent.
Recently, it was proposed that CyP-D binds only to ANT that has been partially misfolded, and that it acts as chaperone, preventing further misfolding into the PT pore [14,44]. However, it seems unlikely that unstressed B50 cells contain resident misfolded ANT. On the contrary, the interaction between ANT and CyP-D in viable cells occurs presumably with ANT in its native state. In addition, the chaperone model requires that CyP-D blocks the transformation of ANT into the PT pore. However, CyP-D overexpression promoted PT pore formation in both stressed (Figure 5) and unstressed (Figure 3) B50 cells, and in isolated mitochondria (Figure 2). These findings agree with other indications that CyP-D activates the PT. In particular, activation by CyP-D has been demonstrated directly by the formation of PT pores from purified ANT in black lipid membranes [7] and liposomes [6] on addition of CyP-D. Also, the CyP inhibitor, CSA, blocks the PT pore at all levels of investigation, i.e. in reconstituted systems [3,6,7], in isolated mitochondria [11] and in intact cells [35], again indicating a positive effect of CyP-D on the PT pore. CSA also reversed PT pore activation by CyP-D overexpression in cells (Figures 3 and 5) and in isolated mitochondria (Figure 2). We conclude, therefore, that CyP-D associates with native-state (antiporter) ANT and promotes its conversion to the PT pore.
The CyP-D(+) cells acquired only 25% of the TMRE fluorescence of control cells (Figure 3). At the low [TMRE] in these experiments (40 nM), we obtained no evidence for significant quenching of intramitochondrial TMRE fluorescence; that is, whereas with higher [TMRE] (500 nM) there was an increase in total cell fluorescence on release of mitochondrial TMRE with CCCP (Figure 4). This was not seen under the low-TMRE-loading conditions of Figure 3 (results not shown). From this it appears that the reduced fluorescence of CyP-D(+) cells reflects a similarly reduced uptake of TMRE (any quenching would reduce this further). The estimated 4-fold decrease in mitochondrial TMRE uptake in CyP-D(+) cells agrees with the fact that the increase in nuclear fluorescence on addition of 500 nM CCCP was about 4-fold larger in control than in CyP-D(+) cells (Figures 5C and 5F). A 4-fold decrease in mitochondrial TMRE corresponds to a decrease in ΔψM of 36 mV (Nernst equation) with respect to control cells. The H+ electrochemical gradient (ΔμH+) across the mitochondrial inner membrane in neuronal cells is believed to comprise ΔψM of at least 150 mV and a pH gradient of about 0.5 unit, giving a total gradient of about 17 kJ/mol [45]:
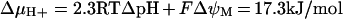 |
In CyP-D(+) cells, ΔμH+ becomes >14 kJ/mol. The consequences can be evaluated from the H+/ATP stoichiometry for mitochondrial ATP synthesis. The H+-driven rotor of the synthase contains 10 subunits, which indicates that 10 H+ flow in per complete rotation synthesizing 3 molecules of ATP [46]. In addition, the equivalent of 1 H+ enters electrogenically during the entry of Pi and ADP into mitochondria, and the exit of ATP, i.e. with the transport process of Pi−/H+ symport and electrogenic ADP3−/ATP4− exchange between cytosol and mitochondria [47]. From these values, the synthesis of cytosolic ATP by mitochondria is driven by the influx of 4.3 H+/ATP down a gradient of >17 kJ/mol. This yields a value for the free energy available from H+ influx (−ΔGnH+) of >74 kJ/mol ATP (cytosolic) in control cells. This value clearly exceeds the estimated energy required for the synthesis of cytosolic ATP (−ΔGATP) of 59–61 kJ/mol in neuronal and other cells [48]. However, the same calculation for CyP-D(+) cells yield a −ΔGnH+ value of >59 kJ/mol ATP (cytosolic), i.e. bordering on the estimated normal −ΔGATP value. It appears then that ΔψM in CyP-D(+) cells may be barely sufficient to maintain normal cytosolic phosphorylation potential. Yet the cells are perfectly viable. It had been predicted [6,19] that PT pore opening that was insufficient to produce necrosis would trigger apoptosis. However, the CyP-D(+) cells showed no increased tendency (with respect to control cells) to undergo apoptosis in the absence of insult (Figure 7A).
The mitochondrial PT has attracted considerable attention as a potential mechanism of lethal cell injury. CSA has been widely used as a test for the PT and attenuates apoptotic and necrotic cell death caused by ischaemia and reperfusion in heart, liver, brain and other tissues (reviewed in [5,6,44]). However, the cytoprotective target of CSA cannot be defined by the use of CSA alone, since cells contain numerous CyPs (>8 in humans [49]), many of which bind CSA tightly. Clearly, additional means, not reliant on CSA, are needed to investigate the pathogenic role of the PT. In the present study, CyP-D overexpression was used as a means of examining the consequences of PT pore opening on B50 cell viability when exposed to NO.
Continuous exposure to NO induced necrosis more effectively in CyP-D(+) cells than control cells, and the induced necrosis was partially suppressed by CSA (Figure 6). This is in line with the hypothesis [9,11] that PT pore opening is a factor in the pathogenesis of necrotic cell death. According to this, PT pore-induced uncoupling of mitochondrial energy transduction that is sufficient to make −ΔGnH+ less than −ΔGATP would allow the ATP synthase to reverse, so that mitochondria hydrolyse ATP, rather than synthesize it, thereby accelerating the onset of cell death (reviewed in [5]). PT pore opening in ischaemia/reperfusion has been detected by several approaches, notably the entry from the cytosol into mitochondria of the otherwise impermeant 2-deoxyglucose (reviewed in [6]) and calcein (reviewed in [44]), and the loss of ΔψM [35]. It remained unclear, however, whether PT pore opening actually contributes to the progression of the injury or merely occurs when cells are already irreversibly compromised, since, although CSA is protective, the cytoprotective target of CSA is unresolved (see above). In the present study, we show that an increased tendency for PT pore opening hastens the onset of cell necrosis, i.e. that the PT does cause cell necrosis.
The involvement of the PT pore in apoptosis is controversial. According to one widely considered model, PT-induced swelling of the matrix space and resultant physical rupture of the mitochondrial outer membrane provide the root mechanism for the release of apoptogenic proteins from the mitochondrial intermembrane space to the cytosol (reviewed in [16]). A related model has been suggested in relation to apoptosis provoked by ischaemia and reperfusion, since this is associated with cellular Ca2+ overload and oxidative stress, the two basic triggers of the PT [6,19]. In a third model, it was proposed that more subtle ultrastructural changes to mitochondria, brought about by transient PT pore opening, facilitate the diffusion of cytochrome c from the intercristal spaces to the intermembrane space, from where it is released to the cytosol [17]. All of these models lead to the prediction that apoptosis would be enhanced in CyP-D(+) cells, where PT pore opening is facilitated. Yet, the opposite was observed (Figures 7 and 8).
Two recent studies [50,51] have also reported inhibition of apoptosis in CyP-D-overexpressing cells. Both attributed CyP-D inhibition of apoptosis to CyP-D inhibition of the PT pore. However, it is clear that CyP-D activates the PT pore in B50 cells (Figures 2, 3 and 5), in line with CSA inhibition of the PT pore and other evidence (see above), and this fact, together with the suppressed apoptosis in CyP-D(+) cells (Figures 7 and 8), argues against a role of the PT in apoptosis. But why is CyP-D anti-apoptotic? Lin and Lechleiter [50], and Schubert and Grimm [51], analysed the capacity of active site (PPIase) mutants of CyP-D to prevent apoptosis, but with contrasting conclusions. We were unable to tackle this question, since the two active site mutants tested were incorrectly processed by the B50 cells (Figure 1B). Thus whether CyP-D protects via its PPIase activity remains unknown. In the present study, we do provide evidence that CyP-D interacts with ANT in healthy cells. In this connection, Schubert and Grimm [51] identified ANT-1, but not ANT-2, as having a pro-apoptotic role. ANT-1 overexpression induced spontaneous apoptosis [51]. Along these lines, CyP-D in high concentrations may bind sufficiently to ANT-1 to negate its apoptotic function. According to our reasoning, however, CyP-D facilitates conversion of ANT from antiporter to non-selective pore, and any apoptotic function of ANT is unlikely to involve this conversion.
Finally, it should be stressed that whereas CyP-D has opposite effects on apoptosis and necrosis in B50 cells (present study), both apoptosis [42] and necrosis (Figure 6) in B50 cells are inhibited by CSA. Thus CSA inhibits the two forms of cell death by interacting with different CyPs; if CyP-D is the cytoprotective target of CSA when it inhibits necrosis (as reasoned above), then CyP-D is not the target of CSA when it blocks apoptosis. This is in line with our previous conclusion [42], from antisense experiments, that CyP-A is involved in B50 cell apoptosis induced by staurosporine and NO, and provides a target for cytoprotection by CSA.
Acknowledgments
This work was supported by the Medical Research Council (G9901348) and the Wellcome Trust.
References
- 1.Johnson N., Khan A., Virji S., Ward J. M., Crompton M. Import and processing of heart mitochondrial cyclophilin D. Eur. J. Biochem. 1999;263:353–359. doi: 10.1046/j.1432-1327.1999.00490.x. [DOI] [PubMed] [Google Scholar]
- 2.Matoushek A., Rospert S., Schmidt K., Glick B. S., Schatz G. Cyclophilin catalyses protein folding in yeast mitochondria. Proc. Natl. Acad. Sci. U.S.A. 1995;92:6319–6323. doi: 10.1073/pnas.92.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crompton M., Virji S., Ward J. M. Cyclophilin D binds strongly to complexes of the voltage dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 4.Woodfield K., Ruck A., Brdiczka D., Halestrap A. P. Direct demonstration of a direct interaction between cyclophilin D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 6.Halestrap A. P., McStay G. P., Clarke S. J. The permeability transition pore complex: another view. Biochimie. 2002;84:153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- 7.Brustovetsky N., Tropschug M., Heimpel S., Heidkaemper D., Klingenberg M. A large Ca2+-dependent channel formed by recombinant ADP/ATP carrier from Neurospora crassa resembles the mitochondrial permeability transition pore. Biochemistry. 2002;41:11804–11811. doi: 10.1021/bi0200110. [DOI] [PubMed] [Google Scholar]
- 8.Kokoszka J. E., Waymire K. G., Levy S. E., Sligh J. E., Cal J. Y., Jones D. P., MacGregor J. R., Wallace D. C. The ADP/ATP translocator is not essential for the permeability transition pore. Nature (London) 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crompton M., Costi A. Kinetic evidence for a heart mitochondrial pore activated by calcium, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular calcium overload. Eur. J. Biochem. 1988;178:489–501. doi: 10.1111/j.1432-1033.1988.tb14475.x. [DOI] [PubMed] [Google Scholar]
- 10.Crompton M., Costi A. A heart mitochondrial calcium-dependent pore of possible relevance to reperfusion-induced injury. Biochem. J. 1990;266:33–39. doi: 10.1042/bj2660033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crompton M., Ellinger H., Costi A. Inhibition by cyclosporin A of a calcium dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 12.Nieminen A., Saylor A. K., Tesfai S. A., Herman B., Lemasters J. J. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochem. J. 1995;307:99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffiths E. J., Halestrap A. P. Mitochondrial non-specific pores remain closed during cardiac ischaemia but open during reperfusion. Biochem. J. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waldmeier P. C., Feldtrauer J. J., Qian T., Lemasters J. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol. Pharm. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- 15.Martinou J. C., Green D. R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001;2:63–67. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 16.Zamzami N., Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nat. Rev. Mol. Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 17.Scorrano L., Ashiya M., Buttle K., Weiler S., Oakes S. A., Mannella C. A., Korsmeyer S. J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 18.Narita M., Shimizu S., Ito T., Chittenden T., Lutz R. J., Matsuda H., Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crompton M., Barksby E., Johnson N., Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie. 2002;84:143–152. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- 20.Capano M., Crompton M. Biphasic translocation of bax to mitochondria. Biochem. J. 2002;367:169–178. doi: 10.1042/BJ20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pastorino J. G., Sulga N., Hoek J. B. Mitochondrial binding of hexokinase II inhibits bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 22.Vyssokikh M. Y., Zorova L., Zorov D., Heimlich G., Jurgensmeier J., Schreiner D., Brdiczka D. The intra-mitochondrial cytochrome c distribution varies correlated to the formation of a complex between VDAC and the adenine nucleotide translocase: this affects Bax-dependent cytochrome c release. Biochim. Biophys. Acta. 2004;1644:27–36. doi: 10.1016/j.bbamcr.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 23.von Ahsen O., Renken C., Perkins G., Kluck R. M., Bossy-Wetzel E., Newmeyer D. D. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cytochrome c release. J. Cell Biol. 2000;150:1027–1036. doi: 10.1083/jcb.150.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuwana T., Mackey M. R., Perkins G., Ellsiman M. H., Latterich M., Schneider R., Green D. R., Newmeyer D. D. Bid, Bax and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 25.McGuinness O., Yafei N., Costi A., Crompton M. The presence of two classes of high affinity cyclosporin A binding sites in mitochondria. Eur. J. Biochem. 1990;194:671–679. doi: 10.1111/j.1432-1033.1990.tb15667.x. [DOI] [PubMed] [Google Scholar]
- 26.Woodfield K., Price N., Halestrap A. P. cDNA cloning of rat mitochondrial cyclophilin. Biochim. Biophys. Acta. 1997;1351:27–30. doi: 10.1016/s0167-4781(97)00017-1. [DOI] [PubMed] [Google Scholar]
- 27.Bergsma D. L., Eder C., Gross M., Kersten H., Sylvester D., Appelbaum E., Cusimano D., Livi G. P., McLaughlin M. M., Kaysan K., et al. The cyclophilin multigene family of peptidyl-prolyl-isomerases. J. Biol. Chem. 1991;266:23204–23214. [PubMed] [Google Scholar]
- 28.Kallen J., Walkenshaw M. D. The X-ray structure of a tetrapeptide bound to the active site of human cyclophilin A. FEBS Lett. 1992;300:286–290. doi: 10.1016/0014-5793(92)80865-e. [DOI] [PubMed] [Google Scholar]
- 29.Zydowsky L. D., Etzkorn F. A., Chang H. Y., Ferguson S. B., Stolz L. A., Ho S. I., Walsh C. T. Active site mutants of human cyclophilin A separate peptidylprolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Prot. Sci. 1992;1:1092–1099. doi: 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connern C. P., Halestrap A. P. Purification and N-terminal sequencing of peptidylprolyl cis–trans-isomerase from mitochondria reveals the existence of a distinct cyclophilin. Biochem. J. 1992;284:381–385. doi: 10.1042/bj2840381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halestrap A. P., Davidson A. M. Inhibition of calcium-induced swelling of mitochondria by cyclosporin is probably caused by binding to peptidylprolyl cis–trans-isomerase and preventing its binding to the adenine nucleotide translocase. Biochem. J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al Nasser I., Crompton M. The reversible calcium-induced permeabilization of rat liver mitochondria. Biochem. J. 1986;239:19–29. doi: 10.1042/bj2390019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crompton M., Heid I. The cycling of calcium, sodium and protons across the inner membrane of cardiac mitochondria. Eur. J. Biochem. 1978;91:599–608. doi: 10.1111/j.1432-1033.1978.tb12713.x. [DOI] [PubMed] [Google Scholar]
- 34.Crompton M., Costi A., Hayat L. Evidence for the presence of a calcium dependent pore activated by oxidative stress in heart mitochondria. Biochem. J. 1987;245:915–918. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobson J., Duchen M. R. Mitochondrial oxidative stress and cell death in astrocytes – requirement for stored Ca2+ and sustained opening of the permeability transition pore. J. Cell Sci. 2002;115:1175. doi: 10.1242/jcs.115.6.1175. [DOI] [PubMed] [Google Scholar]
- 36.Duchen M. R., Leyssens A., Crompton M. Transient mitochondrial depolarisations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J. Cell Biol. 1998;142:975–988. doi: 10.1083/jcb.142.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emaus R. K., Grunwald R., Lemasters J. J. Rhodamine 123 as a probe of transmembrane potential in isolated rat liver mitochondria. Biochim. Biophys. Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- 38.Dawson V. L., Kizushi V. M., Huang P. L., Snyder S. H., Dawson T. L. Resistance to neurotoxicity in cortical cultures from neuronal NOS-deficient mice. J. Neurosci. 1996;16:2479–2487. doi: 10.1523/JNEUROSCI.16-08-02479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown G. C., Bal-Price A. Inflammatory neurodegeneration is mediated by nitric oxide, glutamate and mitochondria. Mol. Neurobiol. 2003;27:325–355. doi: 10.1385/MN:27:3:325. [DOI] [PubMed] [Google Scholar]
- 40.Clementi E., Brown G. G., Feelisch M., Moncada S. Persistent inhibition of cell respiration by nitric oxide; crucial role of S-nitrosylation of mitochondrial complex 1 and protective action of glutathione. Proc. Natl. Acad. Sci. U.S.A. 1998;99:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borutaite V., Brown G. C. Nitric oxide induces apoptosis via hydrogen peroxide but necrosis via energy and thiol depletion. Free Radical Biol. Med. 2003;35:1457–1468. doi: 10.1016/j.freeradbiomed.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 42.Capano M., Virji S., Crompton M. Cyclophilin A is involved in excitotoxin-induced caspase activation in rat neuronal B50 cells. Biochem. J. 2002;363:29–36. doi: 10.1042/bj3630029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poliseno L., Bianchi L., Citti L., Liberatori S., Mariani L., Salvetti A., Evangelista M., Bini L., Pallini V., Rainaldi G. Bcl2-low-expressing MCF7 cells undergo necrosis rather than apoptosis upon staurosporine treatment. Biochem. J. 2004;379:823–832. doi: 10.1042/BJ20031538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waldmeier P. C., Zimmerman K., Qian T., Tintelnot-Blomley M., Lemasters J. J. Cyclophilin D as a drug target. Curr. Med. Chem. 2003;10:1485–1506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- 45.Nicholls D. G., Ward M. W. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- 46.Stock D., Gibbons C., Leslie A. G., Walker J. E. The rotary mechanism of the ATP synthase. Curr. Opin. Struct. Biol. 2000;10:672–679. doi: 10.1016/s0959-440x(00)00147-0. [DOI] [PubMed] [Google Scholar]
- 47.Pfaff E., Heldt H. W., Klingenberg M. Adenine nucleotide translocase of mitochondria. Eur. J. Biochem. 1969;10:484–493. doi: 10.1111/j.1432-1033.1969.tb00715.x. [DOI] [PubMed] [Google Scholar]
- 48.Veech R. L., Lawson J. W. R., Cornell N. W., Krebs H. A. The cytoplasmic phosphorylation potential. J. Biol. Chem. 1979;254:6538–6547. [PubMed] [Google Scholar]
- 49.Galat A. Variations of sequences and amino acid compositions of proteins that sustain their biological functions: an analysis of the cyclophilin family of proteins. Arch. Biochem. Biophys. 1999;371:149–162. doi: 10.1006/abbi.1999.1434. [DOI] [PubMed] [Google Scholar]
- 50.Lin D. T., Lechleiter J. D. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidylprolyl isomerisation. J. Biol. Chem. 2002;277:31134–31141. doi: 10.1074/jbc.M112035200. [DOI] [PubMed] [Google Scholar]
- 51.Schubert A., Grimm S. Cyclophilin D, a component of the permeability transition pore, is an apoptosis repressor. Cancer Res. 2004;64:85–93. doi: 10.1158/0008-5472.can-03-0476. [DOI] [PubMed] [Google Scholar]