

Abstract
Phytanic acid (3,7,11,15-tetramethylhexadecanoic acid) accumulates at high levels throughout the body in the adult form of Refsum disease, a peroxisomal genetic disorder. However, it is still unclear why increased levels of phytanic acid have cytotoxic effects. In the present study, we examined the influence of non-esterified phytanic acid on energy-related functions of mitochondria from adult rat brain. Phytanic acid at low concentrations (5–20 μM, i.e. 5–20 nmol/mg of mitochondrial protein) de-energized mitochondria, as indicated by depolarization, stimulation of non-phosphorylating oxygen uptake and inhibition of the reduction of the tetrazolium dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide. The unbranched homologue palmitic acid exerted much smaller effects. In addition, phytanic acid reduced state 3 respiration, which was partly due to inhibition of the ADP/ATP carrier. Phytanic acid decreased the rate of adenine nucleotide exchange and increased the degree of control, which the ADP/ATP carrier has on state 3 respiration. Important for functional consequences is the finding that mitochondria, which are preloaded with small amounts of Ca2+ (100 nmol/mg of protein), became highly sensitized to rapid permeability transition even when only low concentrations of phytanic acid (below 5 μM) were applied. In conclusion, the incorporation of phytanic acid into the inner mitochondrial membrane increases the membrane H+ conductance and disturbs the protein-linked functions in energy coupling. This is most probably essential for the short-term toxicity of phytanic acid. Thus in neural tissue, which becomes enriched with phytanic acid, the reduction in mitochondrial ATP supply and the facilitation of the opening of the permeability transition pore are two major mechanisms by which the branched-chain fatty acid phytanic acid induces the onset of degenerative processes.
Keywords: ADP/ATP carrier, branched-chain fatty acid, mitochondria, neurodegeneration, permeability transition, respiratory chain, synaptosome
Abbreviations: AAC, ADP/ATP carrier; CAT, carboxyatractyloside; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; PT, permeability transition; PTP, permeability transition pore; RBM, rat brain mitochondria; ΔtPT50, time of half-maximal permeability transition
INTRODUCTION
Phytanic acid (3,7,11,15-tetramethylhexadecanoic acid; see structure in Figure 1) belongs to the group of 3-methyl-branched fatty acids and is a regular constituent of dairy products. 3-Methyl-branched fatty acids are not directly amenable to β-oxidation due to the methyl group in the β-position. These fatty acids are degraded in mammalian cells by joint peroxisomal and mitochondrial β-oxidation [1–3]. Phytanic acid enters the peroxisome with the help of the sterol carrier protein-2. The degradation inside the peroxisomes starts with shortening by one carbon atom via the α-oxidation process to pristanic acid (2,6,10,14-tetramethylpentadecanoic acid). In humans and in rats, α-oxidation is catalysed by an acyl-CoA synthetase (producing phytanol-CoA), a 3-methylacyl-CoA 2-hydroxylase (producing α-hydroxy-phytanol-CoA) and 2-hydroxy-3-methylacyl-CoA lyase that cleaves α-hydroxy-phytanol-CoA into pristanal and formyl-CoA. Interestingly, the α-oxidation pathway is able to shorten the two stereoisomers (3R,7R,11R,15 and 3S,7R,11R,15), which occur in natural phytanic acid. Oxidation of pristanal generates a 2-methyl-branched pristanic acid, which after activation to the CoA-derivative is degraded by normal peroxisomal β-oxidation to 4,8-dimethylnonanoyl-CoA. Before β-oxidation starts, (2R)-stereoisomers of 2-methyl-branched fatty acyl-CoAs are converted into the (2S)-stereoisomers by α-methylacyl-CoA racemase, whereas the β-oxidation pathway accepts only this stereoisomer [4,5]. Furthermore, 4,8-dimethylnonanoyl-CoA is exported to mitochondria as a carnitine derivative, where it is further oxidized by the mitochondrial β-oxidation. In summary, one molecule of pristanoyl-CoA is degraded to three molecules of acetyl-CoA, three molecules of propionyl-CoA and one molecule of isobutyryl-CoA.
Figure 1. Effect of phytanic acid on safranine accumulation by synaptosomes (membrane potential).

Synaptosomes (Syns; 1 mg of protein/ml) suspended in buffer S were equilibrated with membrane potential probe safranine (10 μM). The concentration of phytanic acid (Phyt) added was 50 and 20 μM (traces a and b), and that of Phytol was 50 μM. FCCP was added in two steps to reach a final concentration of 50 nM. Increase in safranine fluorescence was due to release of accumulated safranine to the medium. Antimycin A (AA; 2 μM) was applied to induce finally complete depolarization of mitochondria under in situ conditions. Inset: calibration of changes in the safranine fluorescence, as described in the Experimental section.
Phytanic acid accumulates throughout the body as a consequence of a number of peroxisomal genetic defects, most prominently in adult Refsum disease [2]. In this disease, generally called ‘classical Refsum disease’, accumulation of phytanic acid is due to mutations in the structural gene encoding the phytanoyl-CoA hydroxylase. In other disorders of peroxisomal fatty acid oxidation, such as in infantile Refsum disease, Zellweger syndrome and neonatal adrenoleucodystrophy, moderate accumulation of phytanic acid is accompanied by accumulation of pristanic acid and of very long-chain fatty acids [6]. In addition, in the ‘Refsum-like’ α-methylacyl-CoA racemase deficiency, accumulation of pristanic acid dominates and increased levels of phytanic acid are secondary to an impaired oxidation of pristanic acid [7].
In patients suffering from ‘classical Refsum disease’, the total plasma concentration of phytanic acid may increase to values as high as 1000–5000 μM, from a normal level of approx. 5 μM [2]. Clinical features of Refsum disease, such as cardiac malfunctions and those in the olfactory and auditory nerves, suggest that the supraphysiological concentration of phytanic acid exerts cytotoxic activities, which are most prominent in tissues with a high oxidative ATP generation, such as brain and heart [2].
For phytanic acid, the following peculiarities characteristic for branched-chain fatty acids are known: first, the metabolism of phytanic acid differs from that of its unbranched homologue, palmitic acid. Degradation of phytanoyl-CoA, the activated form of phytanic acid, is initiated by peroxisomal α- and β-oxidation [1,2]. Secondly, the hydrocarbon tail of phytanic acid has a crosssectional area twice as large as that of palmitic acid [8]. Consequently, incorporation of esterified phytanic acid into membranes will distort the arrangement of membrane constituents and their functional interactions [9,10]. The bulky hydrocarbon tail suggests that the interaction of phytanic acid with membrane constituents differs from that of unbranched, long-chain fatty acids, e.g. palmitic acid [11,12]. Thirdly, intracellular fatty acid-binding proteins promote to a lesser extent the esterification and oxidation of phytanic acid when compared with that of palmitic acid. This implies that non-esterified phytanic acid may increasingly accumulate to high intracellular levels enhancing its potential cytotoxicity [13]. Finally, phytanic acid modulates gene expression via interaction with the retinoid-X-receptor or with members of peroxisome-proliferator-activated receptor family [14,15]. Since activation of members of the peroxisome-proliferator-activated receptor family promotes the expression of enzymes of mitochondrial and peroxisomal β-oxidation pathway, their increase by phytanic acid could change the balance of the cellular metabolism of fatty acids [16].
Recently, phytanic acid was found to promote the expression of various proteins, which are potential modulators of mitochondrial ATP production [14,15,17]. Nevertheless, the short-term, direct effects of non-esterified phytanic acid on the mitochondrial energy transduction system have not yet been investigated. Therefore, in the present study, we have characterized the influence of phytanic acid on energy-dependent mitochondrial functions in synaptosomes (nerve endings) and in isolated RBM (rat brain mitochondria). Brain mitochondria are in the focus of current research, because several neurodegenerative diseases have been clearly associated with a partly impaired mitochondrial ATP generation [18–20]. In the present study, particular attention was given to the interaction of phytanic acid with the AAC (ADP/ATP carrier). This transport protein, which is a main rate-limiting step for the mitochondrial ATP supply [21,22], enhances uncoupling by non-esterified fatty acid [23,24]. Moreover, the AAC has been regarded as a component or modulator of the PTP (permeability transition pore) in the inner mitochondrial membrane [25–27].
EXPERIMENTAL
Materials
Phytanic acid was from ULTRA Scientific (North Kingstown, RI, U.S.A.). If not otherwise indicated, chemicals were from Sigma (Deisenhofen, Germany) and were of analytical grade. [3H]-Tetraphenylphosphonium bromide, [14C]sucrose and [14C]ADP were obtained from NEN Life Science Products (Zaventem, Belgium).
Preparation of synaptosomes and mitochondria
Synaptosomes were isolated from adult rat brain as described in [28]. Mitochondria were prepared as described in [29]. Protein contents in the stock suspensions were measured by biuret method. For measurements, synaptosomes were suspended in buffer S (122 mM NaCl, 3.1 mM KCl, 0.4 mM KH2PO4, 5 mM NaHCO3, 1.2 mM MgCl2, 20 mM Hepes, 50 μM Ca2+, 10 mM glucose, 5 mM pyruvate and 5 mM malate, pH 7.4) as described in [30]. Mitochondria were suspended in buffer M (110 mM mannitol, 60 mM KCl, 60 mM Tris, 10 mM KH2PO4, 0.5 mM EGTA, 5 mM pyruvate and 5 mM malate, pH 7.4).
Safranine fluorescence
Alteration in energization of synaptosomes or of mitochondria was monitored fluorimetrically by recording the release of the membrane potential probe safranine, which accumulates as a permeant cation in polarized synaptosomes or mitochondria. Fluorescence was determined at 495 nm excitation and 586 nm emission using PerkinElmer Luminescence Spectrometer LS 50B [31]. Safranine fluorescence was calibrated by the treatment of synaptosomes or mitochondria suspended in substrate-free buffers S and M, supplemented with succinate (5 and 1 mM respectively). Energization of synaptosomes or mitochondria was manipulated by the addition of various concentrations of n-butylmalonate (a competive inhibitor of the dicarboxylate carrier). At this incubation condition, safranine fluorescence was measured and, in addition, the membrane potential via accumulation of [3H]tetraphenylphosphonium bromide (see below). Membrane potentials were estimated using a synaptosomal volume of 3.6 μl/mg of protein [32] and a mitochondrial volume of 1 μl/mg of protein [33].
Oxygen uptake
Mitochondria were added to 2 ml of buffer M. Oxygen uptake was measured by oxygraph (Oroboros Oxygraph®; Bioenergetics and Biomedical Instruments, Innsbruck, Austria) at 25 °C. The respiratory control index was >6 with pyruvate plus malate as substrates.
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide] reduction
Reduction of MTT was used to assess the activity of the mitochondrial respiratory chain [34] in mitochondria. First, mitochondria were suspended in 1.5 ml Eppendorf tubes with 1 ml of ice-cold buffer M, and, thereafter, tubes were warmed to 30 °C in the presence of MTT (0.1 mg/ml). After a 5 min incubation period at 30 °C, tubes were centrifuged, the supernatants were removed and the pellets were suspended in 1 ml of pure ethanol. Reduction of MTT is given by measuring absorbance at 595 nm.
Mitochondrial membrane potential
Mitochondria (1 mg of protein) were incubated with 1 ml of buffer M in 1.5 ml Eppendorf tubes. To the incubation mixture M, various concentrations of phytanic acid or FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) together with 10 μl of 100 μM [3H]tetraphenylphosphonium bromide (50 μCi/ml) plus 5 μl of 10 mM [14C]sucrose (50 μCi/ml) were added. [14C]Sucrose was added for estimating the volume of the medium adherent to the mitochondrial pellet after centrifugation (sucrose cannot permeate the inner membrane). After 1 min of equilibration at 25 °C, mitochondria were sedimented by centrifugation. For counting radioactivity, aliquots of supernatant (100 μl) and dissolved pellet (in 200 μl of 2% SDS solution) were transferred to 1 ml of scintillation cocktail. The membrane potential was calculated from triplicate determinations from the radioactivity ([3H]TTP+) trapped in the mitochondrial pellet, and the radioactivity in 1 μl of the supernatant using the Nernst equation. Radioactivity entrapped in the mitochondrial pellet was corrected for radioactivity in the adherent medium, but not for unspecific binding of [3H]TPP+ to mitochondria.
AAC activity
Activity of AAC was measured using the atractyloside-inhibitor stop technique [35]. Briefly, 0.1 ml of [14C]ADP (40 μM) was added to mitochondria (0.3 mg of mitochondrial protein) suspended in 0.3 ml of ice-cold 250 mM sucrose. Uptake of [14C]ADP by mitochondria was terminated after 1 min incubation by the addition of 0.2 ml of 1 mM atractyloside. After centrifugation, washing of the mitochondrial pellet and dissolving in 0.2 ml of 2% SDS solution, the radioactivity was counted.
Permeability transition
Permeability transition was measured as collapse of the membrane potential Δψm (at the inner mitochondrial membrane) of isolated mitochondria. Mitochondria suspended in EDTA-free buffer M (1.0 mg of protein/ml) were preloaded with Ca2+ (100 nmol/mg of protein) before addition of 10 μM safranine as a probe for Δψm. Collapse of Δψm was monitored fluorimetrically from the release of the accumulated safranine at 495 nm excitation and 586 nm emission using Perkin Elmer Luminescence Spectrometer LS 50B.
Flux control coefficient of the AAC
According to the metabolic control theory [36,37], the flux control coefficient of AAC (CAAC) in phosphorylating mitochondria can be defined as the fractional change in respiration (dJO2) in proportion to the fractional change in the amount of the AAC (dAAC), where both entities are given in relation to the absolute values, JO2 and AACmax respectively. Thus CAAC is given by the following equation:
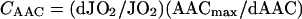 |
JO2 is the maximal phosphorylating respiration (state 3) and AACmax is the total amount of the AAC activity. CAAC was estimated by inhibitor titration, as described in [21] using CAT (carboxyatractyloside) as specific and irreversible inhibitor of the AAC.
RESULTS
Effect of phytanic acid on the energization of synaptosomes and of isolated mitochondria
Additions of phytanic acid (20 and 50 μM) to samples of synaptosomes in buffer S (Figure 1, traces a and b), which were equilibrated with the membrane potential probe safranine, caused release of safranine into the medium, which can be recorded as increase in safranine fluorescence. Final addition of antimycin A (an inhibitor of complex III of the respiratory chain) caused complete release of safranine from synaptosomes. Release of safranine from synaptosomes is also induced by stepwise addition of the protonophore FCCP (trace d). In contrast with phytanic acid, phytol (3,7,11,15-tetramethylhexadec-trans-2-en-1-ol), the side chain of chlorophyll and precursor molecule of phytanic acid formation, did not induce the release of safranine (trace c).
Similar to synaptosomes, mitochondria become depolarized by micromolar concentrations of phytanic acid (20 and 50 μM; Figure 2, traces a and b). Compared with palmitic acid, phytanic acid depolarizes mitochondria much more (traces a and b compared with traces c and d). Furthermore, palmitic acid causes a small, approximately constant depolarization (traces c and d), whereas the phytanic acid-induced depolarization develops progressively (trace b). Despite the great difference in amplitude of depolarization induced by the two fatty acids, depolarization is almost completely reversed by the addition of BSA, which is well known to extract non-esterified fatty acids from mitochondria. This latter observation excludes that a severe damage of mitochondria by phytanic acid, e.g. lytic activity, underlies the strong depolarizing effect.
Figure 2. Effect of phytanic acid or palmitic acid on safranine accumulation by RBM (membrane potential).

Mitochondria (1 mg of protein/ml) suspended in buffer M were equilibrated with membrane potential probe safranine (10 μM). The concentration of phytanic acid (Phyt) added was 20 and 50 μM (traces a and b) and that of added palmitic acid was 50 and 100 μM (traces c and d). Incubation mixture was supplemented with BSA (0.5 mg of protein) to remove fatty acids from mitochondria (traces b and d). Inset: calibration of changes in the safranine fluorescence, as described in the Experimental section.
Effect of phytanic acid on the electron flow in the respiratory chain of isolated mitochondria
In principle, depolarization by phytanic acid could result from a combination of increase in the H+ conductance of the inner mitochondrial membrane (uncoupling) and a partial inhibition of the electron flow in the respiratory chain. To understand better the mechanism by which phytanic acid depolarized mitochondria, we next studied the effect of phytanic acid on the oxygen uptake. The oxygen uptake (respiration) was investigated in the resting state of mitochondria and in mitochondria stimulated by ADP (state 3 respiration) or by the protonophore FCCP (uncoupled respiration). In the resting state, the addition of low concentrations of phytanic acid increased the non-phosphorylating respiration (Figure 3), which indicates an increase in the H+ conductance across the inner membrane. State 3 respiration, in contrast, was decreased by phytanic acid (Figure 3). Interestingly, when the respiratory chain is stimulated by FCCP, low concentrations of phytanic acid (up to 10 nmol/mg of protein) did not reduce oxygen uptake.
Figure 3. Effect of phytanic acid on respiration of isolated RBM.

Mitochondria (1 mg of protein/ml) suspended in buffer M were exposed to increasing concentrations of phytanic acid (Phyt) in the resting state (•), in state 3 (▪) and after uncoupling (▴). ADP- and FCCP-stimulated respiration was adjusted by 2.5 mM ADP and 75 nM FCCP respectively. Incubation samples in the resting state were supplemented with oligomycin (5 μM). The results are mean values obtained with three preparations.
Stimulation of non-phosphorylating respiration by phytanic acid (20 nmol/mg of protein) was approx. 1.6-fold higher than that induced by the unbranched long-chain fatty acid palmitic acid (Figure 4A). In addition, non-phosphorylating respiration stimulated by phytanic acid is to a smaller degree recoupled by CAT (see Figure 4A, inset). Recoupling by CAT was found as the characteristic feature of protonophoric activity of straight-chain fatty acids [23]. This might indicate that in addition to the protonophoric activity of phytanic acid, this fatty acid increases the H+ conductance of the inner membrane by a further action, presumably by induction of H+ leakage.
Figure 4. Effect of phytanic acid or palmitic acid on the respiration and MTT reduction of isolated RBM.

(A) Non-phosphorylating respiration. Mitochondria (1 mg of protein/ml) suspended in buffer M (supplemented with 5 μM oligomycin). Respiration was stimulated by the addition of 20 μM phytanic or palmitic acid. Resting state respiration was 10 nmol of O2·min−1·(mg of protein)−1. The concentration of added CAT was 2 μM. Results shown are the means±S.D. obtained from four preparations. Inset: recoupling is given by the percentage of CAT-sensitive oxygen uptake, referred to as the total oxygen uptake observed with phytanic acid or palmitic acid. (B) MTT reduction. Mitochondria (0.25 mg of protein) suspended in 1 ml of buffer M (plus 0.1 mg of MTT) were treated with 20 and 50 μM phytanic or palmitic acid. Activity of the control incubation (without additions) was ΔA595=0.44±0.05·(5 min)−1·(mg of protein)−1 (100%). Results are means±S.D. from four preparations.
To estimate directly the effect of phytanic acid on the electron flow in the respiratory chain, reduction of the tetrazolium dye MTT by the electron-transport chain was measured in the presence of phytanic acid. Exposure of mitochondria to 20 or 50 μM phytanic acid significantly slowed down the electron flow in the respiratory chain (Figure 4B), whereas palmitic acid (20 μM) decreased MTT reduction to only approx. one-third.
Inhibition of the AAC
Reduction of the oxygen uptake in state 3 respiration by low concentrations of phytanic acid, as seen in Figure 3, indicates that phytanic acid could partly inhibit the exchange of adenine nucleotides across the inner membrane by the AAC. Therefore in the first series of experiments, we compared the inhibition of the AAC activity by CAT with inhibition of state 3 respiration by CAT. AAC activity was determined as uptake of [14C]ADP in brain mitochondria, which were pretreated with various amounts of CAT. Figure 5 shows that the rate of [14C]ADP uptake decreases steadily with the amount of CAT applied (Figure 5A), and, that already the addition of small amounts of CAT decreased state 3 respiration (Figure 5B). For analysis, data of state 3 respiration were plotted against the corresponding data of the AAC activity obtained at the same concentrations of CAT. The resulting plot, which is given in Figure 5(C), shows clearly that state 3 respiration is tuned by the reduction of AAC activity. Thus state 3 respiration decreased by approx. 10%, when the inhibition of the AAC was between 0 and 20%. With a reduction of the AAC activity by more than 30%, state 3 respiration decreased more severely. This finding fits nicely with the estimation of CAAC (see the Experimental section) from the initial slope of the ‘titration curve’, which is shown in Figure 5(B). The estimated flux control coefficient of 0.23 indicates that the AAC has a significant share of the control exerted on state 3 respiration. The value of 0.23 corresponds to 23% of the total control, which is kept by all the protein components of the oxidative phosphorylation and by the proton leak.
Figure 5. Inhibition of state 3 respiration and AAC activity by CAT.

(A) AAC activity. Uptake of [14C]ADP by mitochondria was measured as described in the Experimental section. Activity of the uninhibited AAC was 588±88 pmol·min−1·(mg of protein)−1 (n=4). The results are mean values obtained with four preparations. (B) State 3 respiration. Mitochondria (1 mg of mitochondrial protein) were incubated in 2 ml of buffer M. Respiration was measured after stepwise addition of aliquots of CAT solution (10 μM), and was expressed as percentage of uninhibited state 3 respiration (control). Results are mean values obtained from three preparations. (C) Dependence of state 3 respiration on AAC activity in brain mitochondria. The data points from (A) were plotted against those from (B) obtained after CAT-induced inhibition.
In the next series of experiments, we studied the influence of phytanic acid on AAC activity. Phytanic acid inhibits the adenine nucleotide exchange across the inner membrane, but, in contrast with CAT, inhibition reaches only approx. 50% of maximal activity (Figure 6A). In addition, we made an estimate of the control, which the AAC has on state 3 respiration in the presence of phytanic acid. Therefore state 3 respiration was determined with increasing amounts of CAT in the presence of 20 μM phytanic acid (Figure 6B). The initial slope of this ‘titration curve’ gives the flux control coefficient in the presence of phytanic acid. The flux control coefficient is shifted by phytanic acid from 0.23 in control conditions up to an average value of 0.46 (Figure 6B, data in inset).
Figure 6. Inhibition of the AAC activity by phytanic acid.

(A) Inhibition of [14C]ADP uptake. Mitochondria were preincubated with various concentrations of phytanic acid (Phyt) for 5 min. Uptake of [14C]ADP by mitochondria was measured as described in the Experimental section. The results are mean values obtained with four preparations. (B) Effect of phytanic acid on the control of state 3 respiration by AAC. State 3 respiration of mitochondria in the presence of 20 nmol of phytanic acid/mg of mitochondrial protein was inhibited successively by adding CAT. CAAC values were calculated from ‘titration curves’ (Figures 5A and 6B), as described in the Experimental section and shown in the inset. Results are the mean values obtained from three preparations.
Triggering of permeability transition by phytanic acid
Depolarization of the inner mitochondrial membrane is well known to facilitate the opening of the PTP of mitochondria when they are pretreated with low amounts of Ca2+ (see [11] for a review). Therefore we measured the potency of phytanic acid to induce PT (permeability transition) in brain mitochondria. For that, mitochondria were preloaded with 100 nmol of Ca2+ per mg of mitochondrial protein and were exposed to various concentrations of phytanic acid. Then the collapse of Δψm, which is obvious by the rapid release of safranine, was determined. The sudden increase in safranine fluorescence resulting from release of safranine indicates opening of PTP. Figure 7 shows that the addition of a low concentration of phytanic acid, such as 5 nmol/mg of mitochondrial protein, causes the collapse of Δψm after a lag phase of approx. 3 min. An increase in phytanic acid concentration to 10 nmol/mg of mitochondrial protein considerably shortened the lag phase. With a concentration of phytanic acid corresponding to 20 nmol/mg of mitochondrial protein, Δψm collapsed almost immediately after addition of phytanic acid. We analysed the kinetics of the fluorescence traces obtained with various concentrations of phytanic acid to get a measure for the susceptibility to undergo PT. The time period required for reaching a 50% increase in the safranine fluorescence (ΔtPT50, time of half-maximal permeability transition) was used as a measure. A decrease in ΔtPT50 values reflects an increase in the tendency of mitochondria to undergo PT. When cyclosporin A was added before phytanic acid, this collapse of Δψm was prevented completely, which is a clear proof that phytanic acid induced PT. In addition, we found the following correlation. When FCCP was added at concentrations of 25, 50 and 100 nM, the time course of depolarization was similar to the curves, which are presented in Figure 7 for phytanic acid.
Figure 7. Permeability transition of RBM induced by phytanic acid.

Mitochondria (1 mg of protein) were suspended in 1 ml EDTA-free buffer M supplemented with 10 μM safranine plus 100 μM Ca2+. Phytanic acid was added to final concentrations of 5, 10 or 20 μM. Cyclosporin A concentration was 1 μM. The sudden increase in safranine fluorescence resulting from the release of safranine indicates opening of the PTP. ΔtPT50, the time required for half-maximal fluorescence increase.
To elucidate the role of depolarization as a trigger of PT, in an additional series of experiments, mitochondria were suspended in medium without the addition of Ca2+. Then, Δψm values were measured using the [3H]tetraphenylphosphonium cation as a probe for quantitative analysis of Δψm. We determined Δψm values after adding either 5, 10 and 20 μM phytanic acid or 25, 50 and 100 nM FCCP. Changes in Δψm values account for partial steady-state depolarization due to the protonophoric activity of either phytanic acid or FCCP. In the analysis in Figure 8, ΔtPT50 values were plotted against the corresponding changes in ΔΔψm for either phytanic acid or FCCP. Interestingly, the dependence between ΔtPT50 and depolarization (ΔΔψm) was different for phytanic acid and FCCP. The curves in Figure 8 show that the tendency to undergo PT at a given depolarization is higher in the presence of phytanic acid when compared with that of FCCP. This suggests that induction of PT by FCCP requires a stronger depolarization of the inner membrane than the PT induced by phytanic acid. Therefore we conclude that besides the protonophoric activity of phytanic acid most probably an additional deleterious effect of phytanic acid contributes to the induction of PT. This is probably the interaction of phytanic acid with the AAC, which locks the AAC. This would be, in analogy to the influence of CAT, the cytosolic conformation [38,39].
Figure 8. Dependence of permeability transition on mitochondrial depolarization induced by either phytanic acid or FCCP.

ΔtPT50 values were determined from the increase in safranine fluorescence initiated by 5, 10 and 20 μM phytanic acid (Phyt; Figure 7) or 25, 50 and 100 nM of FCCP. An increasing tendency of mitochondria to undergo PT is reflected by decreasing ΔtPT50 values. The results are mean values obtained with three mitochondrial preparations. Depolarization (ΔΔψm=ΔψmPhyt/FCCP–ΔψmControl) of the inner mitochondrial membrane induced by phytanic acid or FCCP was measured by treating RBM suspended in buffer M. Mitochondria used for measuring depolarization were not preloaded with Ca2+. ΔψmControl corresponds to the membrane potential generated in the resting state without additions. Results of depolarization were obtained with four preparations.
DISCUSSION
The enormous tissue enrichment of a long-chain, saturated fatty acid due to the metabolic situation in Refsum disease is unique. In other pathological situations, such as in type II diabetes mellitus [40], chronically increased fatty acid levels do not reach comparable levels. The proposed toxicity of a supraphysiological concentration of phytanic acid has been attributed to changes in membrane functions, which occur as a consequence of an incorporation of the esterified, branched-chain phytanic acid into membrane lipids. Thus distortions of the phospholipid arrangement were suggested to be responsible for de-myelination of peripheral and central nervous system or heart and bone abnormalities, which are seen in patients suffering from Refsum disease [9]. Indeed, fungal membranes (Neurospora crassa) enriched with phytanic acid-containing phospholipids show alterations in the physicochemical properties [10]. In addition, a considerable amount of phytanic acid is accumulated in the non-esterified form, most probably bound to membranes and proteins [13,41]. Studies with isolated cells indicate that non-esterified phytanic acid is toxic for cells [42–44].
In the present study, we demonstrate that low micromolar concentrations of phytanic acid caused significant de-energization of RBM, which were investigated in situ in synaptosomes and as isolated mitochondria under in vitro conditions (Figures 1 and 2). De-energization by phytanic acid is attributed to interactions of phytanic acid with various membrane constituents. It is obvious that phytanic acid increases the H+ cycling across the inner mitochondrial membrane, as indicated by the stimulation of non-phosphorylating respiration (Figure 3) and, in addition, the decrease in this respiration caused by addition of CAT (Figure 4A). Secondly, there is presumably a H+ leakage induced in the inner membrane. The latter suggestion is based on the finding that de-energization by phytanic acid develops in a progressive manner (Figure 2, trace c). Distortion of the arrangement of membrane lipids by incorporation of bulky phytanic acid is most probably the cause of this supposed H+ leakage. The lack of progressive de-energization (depolarization) with addition of the unbranched palmitic acid (Figure 2, trace d) supports this conclusion. In addition, severe de-energization by phytanic acid was also found with rat liver mitochondria, suggesting that phytanic acid acts on various mitochondria [39].
The depression of state 3 respiration and FCCP-uncoupled respiration (Figure 3) and the decrease in MTT reduction (Figure 4B) by phytanic acid clearly indicate that phytanic acid can slow down the electron flow between the complexes of the respiratory chain. In analogy to findings obtained with unbranched fatty acids, complexes I and III are presumable sites for this inhibitory interaction [45]. Concerning the increase in H+ conductance of the inner membrane and the inhibition of electron flow of the respiratory chain, phytanic acid acts more severely than palmitic acid, one of the most active among the saturated, unbranched fatty acids, which exerts deleterious activities on mitochondrial energy transduction [45].
In addition, phytanic acid partly impairs the AAC-mediated exchange of adenine nucleotides across the inner membrane (Figure 6A), even at concentrations as low as 2.5 μM (corresponding to 2.5 nmol/mg of mitochondrial protein). This inhibition increased the control of state 3 respiration by the AAC. Thus, in the presence of phytanic acid, the control coefficient increases from 0.23 to 0.46 (Figure 6B, inset). Even when there was only a slight inhibition of the AAC activity (Figure 5A) already a clear decrease in state 3 respiration was seen (Figure 5B). Thus, in our experiments, for the decrease in state 3 respiration and, consequently, for decrease in oxidative ATP generation there is practically no threshold, which the decrease in AAC transport capacity has to exceed to be effective. In contrast, a clear threshold has been found previously when enzymic activities of the respiratory chain complexes were inhibited. In isolated RBM, the activities of the respiratory chain complexes could be considerably reduced by the respective inhibitors (rotenone, antimycin A) without causing a major change in state 3 respiration [46,47]. Lastly, phytanic acid acts as a powerful inducer of permeability transition (Figure 7). This is probably due to the depolarization of the inner membrane and, in addition, the locking of the AAC in the c-conformation.
In summary, uncoupling of mitochondria, inhibition of the electron flow in the respiratory chain and inhibition of the adenine nucleotide exchange across the inner mitochondrial membrane are harmful effects exerted by non-esterified phytanic acid, which finally disturbs the ATP supply. On top of that, the inner mitochondrial membrane becomes depolarized by uncoupling and inhibition of the H+ ejection by complexes of the respiratory chain. Thus phytanic acid sensitizes brain mitochondria for permeability transition. Hence, we suggest that in neural tissue enriched with phytanic acid, the long-lasting reduction in the ATP supply and the facilitation of the opening of the PTP initiate neurodegeneration. This damaging mechanism seems also to be involved in phytanic acid-induced disturbance of Ca2+ homoeostasis and mitochondrial potential in rat hippocampal astrocytes, thus reducing cell viability [48]. In contrast with the rapid response of the mitochondrial physiology to an exposure with phytanic acid, the manifestation of clinical features of Refsum disease is a long-lasting process. For understanding this discrepancy one has to take into account the following facts: (i) the accumulation of non-esterified phytanic acid in the body to toxic levels is an ongoing process; (ii) a moderate reduction in the oxidative ATP generation is compensated by an up-regulation in the glycolytic ATP generation (see e.g. [49]); (iii) several observations indicate that the clinical manifestation of diseases based on defective mitochondria occurs only when a threshold level is reached [50]. In analogy, delayed tissue response underlies chronic exposure of animals to the complex I inhibitor rotenone, which causes Parkinson-like symptoms [51].
Besides these direct impairments of mitochondrial functions, phytanic acid might also induce other processes by which cell death could be accelerated, such as stimulation of the secretion of tumour necrosis factor α and the induction of inducible nitric oxide synthase [44].
Acknowledgments
We thank Mrs H. Goldammer for her skilful technical assistance in performing the experiments. This work was supported by grants from BMBF (01ZZ0107), Medizinische Fakultät (‘Neuroverbund’), Land Sachsen-Anhalt (2923A) and Fonds der chemischen Industrie (G. R.).
References
- 1.Wanders R. J., Jansen G. A., Lloyd M. D. Phytanic acid alpha-oxidation, new insights into an old problem: a review. Biochim. Biophys. Acta. 2003;1631:119–135. doi: 10.1016/s1388-1981(03)00003-9. [DOI] [PubMed] [Google Scholar]
- 2.Wanders R. J. A., Jakobs C., Skjeldal O. H. Refsum disease. In: Scriver C. R., Beaudet A. L., Sly W. S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edn. New York: McGraw-Hill; 2001. pp. 3303–3321. [Google Scholar]
- 3.Wierzbicki A. S., Lloyd M. D., Schofield C. J., Feher M. D., Gibberd F. B. Refsum's disease: a peroxisomal disorder affecting phytanic acid alpha-oxidation. J. Neurochem. 2002;80:727–735. doi: 10.1046/j.0022-3042.2002.00766.x. [DOI] [PubMed] [Google Scholar]
- 4.Ferdinandusse S., Denis S., Ijist L., Dacremont G., Waterham H. R., Wanders R. J. Subcellular localization and physiological role of alpha-methylacyl-CoA racemase. J. Lipid Res. 2000;41:1890–1896. [PubMed] [Google Scholar]
- 5.Ferdinandusse S., Rusch H., van Lint A. E., Dacremont G., Wanders R. J., Vreken P. Stereochemistry of the peroxisomal branched-chain fatty acid alpha- and beta-oxidation systems in patients suffering from different peroxisomal disorders. J. Lipid Res. 2002;43:438–444. [PubMed] [Google Scholar]
- 6.Verhoeven N. M., Wanders R. J., Poll-The B. T., Saudubray J. M., Jakobs C. The metabolism of phytanic acid and pristanic acid in man: a review. J. Inherit. Metab. Dis. 1998;21:697–728. doi: 10.1023/a:1005476631419. [DOI] [PubMed] [Google Scholar]
- 7.Ferdinandusse S., Denis S., Clayton P. T., Graham A., Rees J. E., Allen J. T., McLean B. N., Brown A. Y., Vreken P., Waterham H. R., et al. Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Nat. Genet. 2000;24:188–191. doi: 10.1038/72861. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien J. S. Cell membranes – composition: structure: function. J. Theor. Biol. 1967;15:307–324. doi: 10.1016/0022-5193(67)90140-3. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg D. Refsum disease. In: Scriver C. R., Beaudet A. L., Sly W. S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. 7th edn. New York: McGraw-Hill; 1995. pp. 2351–2369. [Google Scholar]
- 10.Friedman K. J., Glick D. Role of lipids in the Neurospora crassa membrane: IV. Biochemical and electrophysiological changes caused by growth on phytanic acid. J. Membr. Biol. 1982;64:1–9. doi: 10.1007/BF01870763. [DOI] [PubMed] [Google Scholar]
- 11.Bernardi P., Penzo D., Wojtczak L. Mitochondrial energy dissipation by fatty acids. Mechanisms and implications for cell death. Vitam. Horm. 2002;65:97–126. doi: 10.1016/s0083-6729(02)65061-7. [DOI] [PubMed] [Google Scholar]
- 12.Wojtczak L., Schönfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim. Biophys. Acta. 1993;1183:41–57. doi: 10.1016/0005-2728(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 13.Atshaves B. P., Storey S. M., Petrescu A., Greenberg C. C., Lyuksyutova O. I., Smith R., III, Schroeder F. Expression of fatty acid binding proteins inhibits lipid accumulation and alters toxicity in L cell fibroblasts. Am. J. Physiol. Cell Physiol. 2002;283:C688–C703. doi: 10.1152/ajpcell.00586.2001. [DOI] [PubMed] [Google Scholar]
- 14.Ellinghaus P., Wolfrum C., Assmann G., Spener F., Seedorf U. Phytanic acid activates the peroxisome proliferator-activated receptor alpha(PPARalpha) in sterol carrier protein 2-/sterol carrier protein x-deficient mice. J. Biol. Chem. 1999;274:2766–2772. doi: 10.1074/jbc.274.5.2766. [DOI] [PubMed] [Google Scholar]
- 15.Lemotte P. K., Keidel S., Apfel C. M. Phytanic acid is a retinoid X receptor ligand. Eur. J. Biochem. 1996;236:328–333. doi: 10.1111/j.1432-1033.1996.00328.x. [DOI] [PubMed] [Google Scholar]
- 16.Aoyama T., Peters J. M., Iritani N., Nakajima T., Furihata K., Hashimoto T., Gonzalez F. J. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor α (PPARα) J. Biol. Chem. 1998;273:5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 17.Schluter A., Barbera M. J., Iglesias R., Giralt M., Villarroya F. Phytanic acid, a novel activator of uncoupling protein-1 gene transcription and brown adipocyte differentiation. Biochem. J. 2002;362:61–69. doi: 10.1042/0264-6021:3620061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schapira A. H. Primary and secondary defects of the mitochondrial respiratory chain. J. Inherit. Metab. Dis. 2002;25:207–214. doi: 10.1023/a:1015629912477. [DOI] [PubMed] [Google Scholar]
- 19.Beal M. F. Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Ann. N. Y. Acad. Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- 20.Swerdlow R. H., Kish S. J. Mitochondria in Alzheimer's disease. Int. Rev. Neurobiol. 2002;53:341–385. doi: 10.1016/s0074-7742(02)53013-0. [DOI] [PubMed] [Google Scholar]
- 21.Groen A. K., Wanders R. J., Westerhoff H. V., van der Meer R., Tager J. M. Quantification of the contribution of various steps to the control of mitochondrial respiration. J. Biol. Chem. 1982;257:2754–2757. [PubMed] [Google Scholar]
- 22.Kholodenko B., Zilinskiene V., Borutaite V., Ivanoviene L., Toleikis A., Praskevicius A. The role of adenine nucleotide translocators in regulation of oxidative phosphorylation in heart mitochondria. FEBS Lett. 1987;223:247–250. doi: 10.1016/0014-5793(87)80298-3. [DOI] [PubMed] [Google Scholar]
- 23.Skulachev V. P. Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett. 1991;294:158–162. doi: 10.1016/0014-5793(91)80658-p. [DOI] [PubMed] [Google Scholar]
- 24.Schönfeld P. Does the function of adenine nucleotide translocase in fatty acid uncoupling depend on the type of mitochondria? FEBS Lett. 1990;264:246–248. doi: 10.1016/0014-5793(90)80259-l. [DOI] [PubMed] [Google Scholar]
- 25.Halestrap A. P., Davidson A. M. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis–trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brustovetsky N., Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- 27.Kokoszka J. E., Waymire K. G., Levy S. E., Sligh J. E., Cai J., Jones D. P., MacGregor G. R., Wallace D. C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature (London) 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai J. C., Clark J. B. Preparation of synaptic and nonsynaptic mitochondria from mammalian brain. Methods Enzymol. 1979;55:51–60. doi: 10.1016/0076-6879(79)55008-3. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y., Fiskum G., Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 30.Kauppinen R. A., Nicholls D. G. Synaptosomal bioenergetics. The role of glycolysis, pyruvate oxidation and responses to hypoglycaemia. Eur. J. Biochem. 1986;158:159–165. doi: 10.1111/j.1432-1033.1986.tb09733.x. [DOI] [PubMed] [Google Scholar]
- 31.Akerman K. E., Wikstrom M. K. Safranine as a probe of the mitochondrial membrane potential. FEBS Lett. 1976;68:191–197. doi: 10.1016/0014-5793(76)80434-6. [DOI] [PubMed] [Google Scholar]
- 32.Joyce O. J., Farmer M. K., Tipton K. F., Porter R. K. Oxidative phosphorylation by in situ synaptosomal mitochondria from whole brain of young and old rats. J. Neurochem. 2003;86:1032–1041. doi: 10.1046/j.1471-4159.2003.01915.x. [DOI] [PubMed] [Google Scholar]
- 33.Halestrap A. P. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochim. Biophys. Acta. 1989;973:355–382. doi: 10.1016/s0005-2728(89)80378-0. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y., Peterson D. A., Kimura H., Schubert D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J. Neurochem. 1997;69:581–593. doi: 10.1046/j.1471-4159.1997.69020581.x. [DOI] [PubMed] [Google Scholar]
- 35.Barbour R. L., Chan S. H. Characterization of the kinetics and mechanism of the mitochondrial ADP-ATP carrier. J. Biol. Chem. 1981;256:1940–1948. [PubMed] [Google Scholar]
- 36.Heinrich R., Rapoport T. A. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 1974;42:89–95. doi: 10.1111/j.1432-1033.1974.tb03318.x. [DOI] [PubMed] [Google Scholar]
- 37.Kacser H., Burns J. A. The control of flux. Symp. Soc. Exp. Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- 38.Chavez E., Zazueta C., Garcia N. Carboxyatractyloside increases the effect of oleate on mitochondrial permeability transition. FEBS Lett. 1999;445:189–191. doi: 10.1016/s0014-5793(99)00128-3. [DOI] [PubMed] [Google Scholar]
- 39.Schönfeld P., Bohnensack R. Fatty acid-promoted mitochondrial permeability transition by membrane depolarization and binding to the ADP/ATP carrier. FEBS Lett. 1997;420:167–170. doi: 10.1016/s0014-5793(97)01511-1. [DOI] [PubMed] [Google Scholar]
- 40.McGarry J. D., Dobbins R. L. Fatty acids, lipotoxicity and insulin secretion. Diabetologia. 1999;42:128–138. doi: 10.1007/s001250051130. [DOI] [PubMed] [Google Scholar]
- 41.Schönfeld P., Struy H. Refsum disease diagnostic marker phytanic acid alters the physical state of membrane proteins of liver mitochondria. FEBS Lett. 1999;457:179–183. doi: 10.1016/s0014-5793(99)01009-1. [DOI] [PubMed] [Google Scholar]
- 42.Powers J. M., Kenjarski T. P., Moser A. B., Moser H. W. Cerebellar atrophy in chronic rhizomelic chondrodysplasia punctata: a potential role for phytanic acid and calcium in the death of its Purkinje cells. Acta Neuropathol. 1999;98:129–134. doi: 10.1007/s004010051060. [DOI] [PubMed] [Google Scholar]
- 43.Dubois-Dalcq M., Menu R., Buyse M. Influence of fatty acids on fine structure of cultured neurons. An experimental approach to Refsum's disease. J. Neuropathol. Exp. Neurol. 1972;31:645–667. doi: 10.1097/00005072-197210000-00008. [DOI] [PubMed] [Google Scholar]
- 44.Idel S., Ellinghaus P., Wolfrum C., Nofer J. R., Gloerich J., Assmann G., Spener F., Seedorf U. Branched chain fatty acids induce nitric oxide-dependent apoptosis in vascular smooth muscle cells. J. Biol. Chem. 2002;277:49319–49325. doi: 10.1074/jbc.M204639200. [DOI] [PubMed] [Google Scholar]
- 45.Takeuchi Y., Morii H., Tamura M., Hayaishi O., Watanabe Y. A possible mechanism of mitochondrial dysfunction during cerebral ischemia: inhibition of mitochondrial respiration activity by arachidonic acid. Arch. Biochem. Biophys. 1991;289:33–38. doi: 10.1016/0003-9861(91)90438-o. [DOI] [PubMed] [Google Scholar]
- 46.Davey G. P., Clark J. B. Threshold effects and control of oxidative phosphorylation in nonsynaptic rat brain mitochondria. J. Neurochem. 1996;66:1617–1624. doi: 10.1046/j.1471-4159.1996.66041617.x. [DOI] [PubMed] [Google Scholar]
- 47.Davey G. P., Peuchen S., Clark J. B. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J. Biol. Chem. 1998;273:12753–12757. doi: 10.1074/jbc.273.21.12753. [DOI] [PubMed] [Google Scholar]
- 48.Kahlert S., Schönfeld P., Reiser G. The Refsum disease marker phytanic acid, a branched chain fatty acid, affects Ca2+ homeostasis and mitochondria, and reduces cell viability in rat hippocampal astrocytes. Neurobiol. Dis. 2004 doi: 10.1016/j.nbd.2004.08.010. in the press. [DOI] [PubMed] [Google Scholar]
- 49.Kahlert S., Reiser G. Requirement of glycolytic and mitochondrial energy supply for loading of Ca2+ stores and InsP3-mediated Ca2+ signaling in rat hippocampus astrocytes. J. Neurosci. Res. 2000;61:409–420. doi: 10.1002/1097-4547(20000815)61:4<409::AID-JNR7>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 50.Rossignol R., Faustin B., Rocher C., Malgat M., Mazat J. P., Letellier T. Mitochondrial threshold effects. Biochem. J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sherer T. B., Betarbet R., Testa C. M., Seo B. B., Richardson J. R., Kim J. H., Miller G. W., Yagi T., Matsuno-Yagi A., Greenamyre J. T. Mechanism of toxicity in rotenone models of Parkinson's disease. J. Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]