

Abstract
Mitotic centromere-associated kinesin (MCAK) is a microtubule (MT)-destabilizing molecular motor. In the present study we show that the final 8 amino acids of the C-terminus of MCAK inhibit lattice-stimulated ATPase activity of the motor. Surprisingly, loss of this C-terminal ‘tail’ (MCAK-Q710) leads to more rapid depolymerization of MTs relative to full-length MCAK (wt-MCAK). Biochemical and microscopic assays revealed that MCAK-Q710 bound to the MT lattice with higher apparent affinity as compared with wt-MCAK. End-stimulated depolymerization was similar for both enzymes. These data suggest that lattice-bound MCAK can increase the rate of MT depolymerization, but at an energy cost. The function of the C-terminus of MCAK may be to selectively inhibit lattice-stimulated ATPase activity, resulting in limited interactions of the motor with the MT lattice. This increases the coupling between ATP hydrolysis and tubulin dimer release, but it also limits MT depolymerization.
Keywords: ATPase activity, Kin I, mitosis, microtubule depolymerization, mitotic centromere-associated kinesin (MCAK), Xenopus kinesin catastrophe modulator-1 (XKCM1)
Abbreviations: ADP·AlFx, ADP+aluminium+sodium fluoride; p[NH]ppA, adenosine 5′-[β,γ-imido]triphosphate (‘AMP-PNP’); CHO, Chinese-hamster ovary; EGFP, enhanced green fluorescent protein; GFP, green fluorescent protein; pp[CH2]pG, guanosine 5′-[α,β-methylene]triphosphate (‘GMP-CPP’); MCAK, mitotic centromere-associated kinesin; MCAK-Q710, truncated MCAK; MT, microtubule; wt-MCAK, full-length MCAK
INTRODUCTION
The kinesin superfamily consists of motor proteins that convert chemical energy, through ATP hydrolysis, into physical work [1–4]. These motors are involved in a wide range of cellular functions, such as vesicle transport along microtubule (MT) tracks, signal transduction and MT polymer dynamics [1,2,5]. MCAK (mitotic centromere-associated kinesin, the hamster orthologue of mouse Kif2C [6,7]) belongs to the Kin I (internal motor domain) or M-type [6] subfamily of kinesin-related proteins and shares a highly conserved ATP-hydrolysing motor (head) domain with other members of this subfamily [2,8]. Unlike many other kinesins, which transport cargo along the surface of MTs, MCAK and its homologues depolymerize them [9–12]. This activity is critical for proper cell division [10,13–15] and in modulating interphase MT dynamics [10,11].
In the presence of ATP, purified MCAK and its homologues are very efficient MT depolymerizers. However, exactly where on the MT and when MCAK hydrolyses ATP are still a matter of debate. Because MCAK localizes preferentially to the ends of stabilized MTs in the presence of a non-hydrolysable form of ATP, p[NH]ppA (adenosine 5′-[β,γ-imido]triphosphate, ‘AMP-PNP’) [9], one current model suggests that MCAK associates loosely with the lattice of MTs, perhaps through electrostatic interactions, and then translocates via one-dimensional diffusion to the MT ends where depolymerization takes place [9,16,17]. MT destabilization (in the form of protofilament peels) occurs even in the presence of p[NH]ppA when the stoichiometry of Kin I motor to tubulin in polymer is high [18]. These results led researchers to propose that the sole purpose of ATP hydrolysis is to trigger the dissociation of the terminal tubulin dimer [9,17].
In contrast with MCAK, conventional kinesin translocates along the MT lattice using a mechanism that is tightly coupled to ATP hydrolysis [19,20]. Conventional kinesin exists in two different conformations, compact/folded and unfolded [21]. Folding is promoted by an interaction between the C-terminus and the head/neck region. In the folded state, ATP hydrolysis is inhibited [22,23]. Irrespective of the striking functional differences, we show here that when the C-terminus of MCAK is truncated, MT-stimulated ATPase activity is increased. Interestingly, the MT depolymerization rate is increased both in vivo and in vitro in the absence of the C-terminus. Truncated MCAK (MCAK-Q710) binds with higher apparent affinity to the MT lattice as compared with the full-length protein (wt-MCAK). These results suggest that inhibition of lattice ATPase by the C-terminus may be a fundamental property of N-type and M-type kinesins.
We propose that the C-terminus of wt-MCAK, analogous to conventional kinesin, acts as a selective inhibitor of ATP hydrolysis that negatively influences the protein's interaction with the MT lattice. For MCAK, the tail may inhibit a process we call ‘lattice priming’, in which the motor becomes tightly coupled to the MT lattice during specific stages of the ATP hydrolysis cycle. As the MT ends approach, active motors present along the lattice can participate in depolymerization, thus affording the system more effective depolymerization as it mitigates the limitations of the simple diffusive end targeting of the wild-type motor.
EXPERIMENTAL
Constructs
MCAK constructs were made by ligating MCAK PCR products (from pOPRSVICAT-GFP-MCAK [15]) with TOPO NT-GFP fragments (Invitrogen, Carlsbad, CA, U.S.A.). Deletions were confirmed by DNA sequencing.
Cell culture, transfection and immunofluorescence
Cells were cultured, fixed and immunofluorescently labelled as described previously [11]. For cells cultured in the presence of paclitaxel, 15 μM paclitaxel was added 2 h into the transfection (using Lipofectamine, Invitrogen) and maintained for 20 h until fixation. Tubulin was labelled with mouse anti-tubulin DM1α (Sigma ImmunoChemicals, St. Louis, MO, U.S.A.) and Texas Red anti-mouse (Jackson ImmunoResearch, West Grove, PA, U.S.A.) antibodies. Analysis was done with a Nikon FX-A microscope equipped with a 60X/1.4 NA Plan Apo oil objective. Digital images were acquired with a Sensys cooled charge-coupled-device camera (Photometrics, Tucson, AZ, U.S.A.) controlled by QED camera software (QED Imaging, Tucson, AZ, U.S.A.).
Quantitation of MT depolymerization in vivo
GFP (green fluorescent protein)–MCAK fusion constructs were transfected into CHO (Chinese-hamster ovary) cells and cultured in the presence of 15 μM paclitaxel for 20 h. Cells were then fixed and labelled with DM1α antibody. Digital images were acquired using a cooled charge-coupled-device camera. Images of transfected cells were collected for GFP and MT fluorescence using the same exposure times. Images were saved as 8-bit TIFF files with a 256 grey-scale range. Quantification was performed using NIH Image 1.62 and Microsoft Excel. Interphase cells chosen for quantification displayed similar levels of cytoplasmic GFP expression. The average pixel intensity (mean grey value) of GFP in the cytoplasm was measured for both constructs to control for variations in the extent of nuclear sequestration. Using a grey-scale image of MTs, where MTs appear in intensity between white and light grey (grey-scale value between 0 and 120) on a nearly black (grey-scale value near 256) background, we recorded the pixels present within the grey-scale range of 0 and 120. This number was divided by the total pixels in the cell. The resulting number gave us a normalized value representing the amount of tubulin polymer per cell.
Expression and purification of recombinant MCAK
MCAK-Q710 was constructed by ligating a PCR fragment containing the truncated C-terminus into pVL1393-MCAK digested with NotI and NcoI. EGFP (enhanced GFP)-labelled MCAK constructs were made by digesting EGFP–MCAK from pYOY152 (from Y. Ovechkina, Department of Physiology and Biophysics, University of Washington, Seattle, WA, U.S.A.) and ligating it into the Baculovirus expression vector, pVL1393 (Pharmingen, San Diego, CA, U.S.A.). Baculovirus expression of recombinant His6-tagged MCAK in Sf9 cells (Pharmingen) and its purification on Ni-NTA (Ni2+-nitrilotriacetate)–agarose columns (Qiagen) were performed as described previously [15]. Peak fractions in elution buffer [15] were stored at −70 °C. The number of active nucleotide-binding sites was measured radiometrically as described previously [23]. Because MCAK is a dimer [15], the concentrations are always expressed as the number of active heads.
In vitro MT assembly
Bovine brain tubulin was acquired from Cytoskeleton (Denver, CO, U.S.A.). For in vitro pelleting assays, 2.2 μM of tubulin was added to BRB80 (80 mM Pipes, pH 6.8, 1 mM EGTA and 1 mM MgCl2) with 6 mM MgCl2, 1.5 mM GTP and 0.5 M DMSO, and incubated for 30 min at 37 °C. Following the incubation, MTs were diluted 1:20 BRB80 at 37 °C plus 10 μM paclitaxel. Variable MT lengths were achieved by passing MTs through a 30-gauge syringe of either 1.27 cm in length (Becton Dickinson, Franklin Lakes, NJ, U.S.A.) or 5.08 cm in length (Hamilton, Reno, NV, U.S.A.). Resulting lengths (5 μm and 15 μm respectively) were determined via immunofluorescent microscopy. Unsheared MTs can extend to approx. 40 μm in length, but may retain bundled MT polymer and have a less consistent length distribution as compared with the sheared polymer. For ATPase assays, 11 μM tubulin was added to BRB80 with 8 mM MgCl2, 2 mM GTP and 0.7 M DMSO, and grown at 37 °C as described above. MTs were then diluted 1:2 with BRB80 plus paclitaxel to give a final paclitaxel concentration of 7.5 μM.
pp[CH2]pG (guanosine 5′-[α,β-methylene]triphosphate, ‘GMP-CPP’) rhodamine-labelled MTs were grown by combining 4 μg of unlabelled bovine brain tubulin, 1 μg of rhodamine-labelled bovine brain tubulin, 6 mM MgCl2, 1.5 mM pp[CH2]pG and 0.5 M DMSO, and incubated for 30 min at 37 °C. Following the incubation, MTs were diluted 1:12.5 in 37 °C BRB80. MTs were then spun in an airfuge at 149000 g for 10 min in order to separate out any unincorporated tubulin. All MTs were used within 24 h of being polymerized.
In vitro MT depolymerization assays
Elution buffer containing 50 nM active motor protein was mixed with 0.8 mM DTT, 1.2 mM MgATP, 75 mM KCl and 2.2 μM paclitaxel-stabilized MT polymer. Reactions were incubated at room temperature (25±1 °C) for 16 min and centrifuged in an airfuge at 149000 g for 10 min (50 μM MgATP and a 3-min incubation were used to generate the binding curve for the motor). Supernatants and pellets were assayed for the presence of tubulin and motor on Coomassie-stained SDS polyacrylamide gels (Invitrogen). Bands were quantified using NIH Image 1.62. The extent of depolymerization with the two constructs was assayed by comparing the total amount of tubulin in the supernatant minus the supernatant of a no-motor control. Motor affinity was assayed by comparing the percentage of motor in the pellet over a range of motor-to-tubulin stoichiometries. Asymptotic values were obtained using an exponential association curve (see below).
In vitro motor/rhodamine-MT ‘squashes’
Rhodamine-labelled MTs (1.8 mM) were combined with 30 nM of active EGFP-labelled motor. MT/motor mixture was incubated in BRB80 containing 3 mM p[NH]ppA or ADP (or no nucleotide), 75 mM KCl, 1 mg/ml BSA and an anti-fade (40 mM D-glucose, 200 μg/ml glucose oxidase, 35 μg/ml catalase, 1 mM DTT and 0.5% β-mercaptoethanol). After 5 min, 2 μl of this solution was squashed on to a microscope slide under a glass coverslip. ADP·Pi mimic experiments (ADP·AlFx, ADP+aluminium+sodium fluoride) were performed as described previously [24] with the minor substitution of using NaF as opposed to KF.
ATPase assays
Assays were performed as described previously [17].
‘On’ and ‘off’ rates for ATPase experiments were evaluated with an exponential association curve fit over a 60-min time interval:
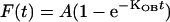 |
where A is the asymptotic value; t, time; kOB, observed association rate expressed in inverse time; kOB, kON[MCAK]+kOFF.
Constants were determined with a non-linear least-squares fit. Regression analysis and curves were generated using MATLAB (The MathWorks) and Microsoft Excel.
RESULTS
C-terminal truncations of MCAK exhibit increased MT depolymerization in vivo
CHO cells transfected with full-length MCAK (wt-MCAK) display a complete loss of MT polymer by 20 h post-transfection (Figures 1A and 1B). However, when wt-MCAK-transfected cells are cultured for the same time duration in the presence of 15 μM paclitaxel, they exhibit only a partial loss of MT polymer (Figures 1C and 1D). Partial abrogation of MCAK activity with paclitaxel allowed us to differentiate between MCAK constructs with slightly dissimilar rates of MT depolymerization.
Figure 1. C-terminal truncations increase the depolymerization activity of MCAK.

The MT depolymerization in CHO cells transfected with wt-MCAK and MCAK-Q710 is shown. (A, C, E) show MTs, with antibody against α-tubulin. (B, D, F) show fluorescent expression of GFP-tagged motor protein. (A) and (B) show a cell transfected with GFP-tagged wt-MCAK. (C) and (D) show a cell transfected with GFP-tagged wt-MCAK, cultured in the presence of 15 μM paclitaxel. (E) and (F) show a cell transfected with GFP-tagged MCAK-Q710 in the presence of 15 μM paclitaxel. (G) shows the C-terminal truncations of GFP-tagged MCAK in conjunction with their amino acid truncation sites. Each construct was transiently transfected into CHO cells cultured in the presence of 15 μM paclitaxel. Cells were fixed 16 h post transfection and antibody labelled against α-tubulin. The extent of MT depolymerization was scored for each of the constructs. + was the maximum observed depolymerization represented by the shortest bundles; ++++ was the minimum represented by longer MT bundles. AA, amino acid.
Maney et al. [11] observed that wt-MCAK exhibited slightly lower MT depolymerization activity than a construct of MCAK lacking the entire C-terminus (the region immediately following the motor domain). In order to precisely map this inhibitory domain, we prepared successively smaller C-terminus deletions of MCAK and transfected these constructs into CHO cells cultured in the presence of 15 μM paclitaxel. By inspection, cells transfected with C-terminus deletions between Ser583 and Gln710 exhibited equally enhanced MT depolymerization when compared with wt-MCAK (Figure 1G). We determined that it is the terminal 8 amino acids that are responsible for the partial auto-inhibition of MT depolymerization activity of full-length MCAK.
Quantitation of the C-terminal inhibition of MT depolymerization in vivo
MCAK-Q710 was chosen for quantitative comparison with wt-MCAK, as it was the most intact truncation construct with maximally enhanced MT depolymerization. Figures 1(E) and 1(F) show a cell transfected with MCAK-Q710, cultured in the presence of paclitaxel. MCAK-Q710 is able to depolymerize bundled MTs to a significantly greater extent than wt-MCAK.
In order to quantitate the extent of depolymerization for both MCAK constructs, we devised a method using digital images of transfected cells (described in the Experimental section). The relative amount of tubulin polymer remaining in a population of transfected cells is shown in Figure 2(A). wt-MCAK and MCAK-Q710 transfected cells exhibited a significant difference in the extent of polymer loss according to a paired t test with 95% confidence. In addition, we measured the relative levels of cytoplasmic MCAK expression and found that these two populations of cells exhibited statistically indistinguishable levels of cytoplasmic protein expression (Figure 2B). This suggests that the difference in depolymerization is not due to sequestration of MCAK-Q710 away from cytoplasmic MTs.
Figure 2. MCAK-Q710 shows enhanced MT depolymerization in vivo.

(A) shows the difference in tubulin polymer levels for the same populations of cells. (B) is a comparison of the cytoplasmic MCAK expression levels between wt-MCAK and MCAK-Q710. Fluorescent intensity levels are measured over 256 grey-scale values. Error bars are shown for each (S.E.M., n=44).
In vitro MT depolymerization is enhanced by the loss of MCAK's C-terminus
To confirm that the difference in MT depolymerization seen in vivo was intrinsic to the motor and not the result of interactions with other cellular proteins, we performed a series of in vitro depolymerization assays. wt-MCAK and MCAK-Q710 purified from Sf9 cells (see the Experimental section) were incubated with paclitaxel-stabilized MTs (about 15 μm in length, on average) and MgATP and then pelleted. The amount of tubulin released into the supernatant was quantified on an SDS gel as an indication of the extent of MT depolymerization. Any motor found in the supernatant is either unbound or associated with free tubulin. Motor protein observed with the pellet is presumably bound to the MT polymer. Quantitation of these gels revealed that MCAK-Q710 was approx. 25% more potent as a MT depolymerizer than wt-MCAK in vitro (Figure 3A). In addition, more MCAK-Q710 was found in the MT pellet as compared with wt-MCAK, suggesting that the deletion construct may have a higher apparent affinity for the MT polymer. Pelleting assays performed over a range of motor-to-tubulin stoichiometries confirmed that MCAK-Q710 does, in fact, have a higher affinity for the MT (Figure 3B). Saturating experiments revealed that in the presence of 50 μM MgATP and 44 μM MT polymer, MCAK-Q710 saturates the MT polymer at 527 μM whereas wt-MCAK saturates at 453 μM, a 16% increase (the saturating equation is described in the Experimental section).
Figure 3. MCAK-Q710 depolymerizes paclitaxel-stabilized MTs faster than wt-MCAK in vitro and exhibits increased apparent affinity for the MT.

SDS/PAGE gel shows the difference in localization and depolymerization activity between wt-MCAK and MCAK-Q710. Supernatant lanes (S) show the amount of free tubulin dimer and unbound protein left in solution after the reaction was centrifuged. Pellet lanes (P) show the amount of larger polymer and any bound motor protein after centrifugation. The upper bands in the gel are MCAK motor and the lower bands are tubulin. (A) In each experiment, 50 nM of active motor was added to 2200 nM paclitaxel-stabilized MTs and 1 mM MgATP. MTs were sheared to a length of 15 μm on average. The depolymerization reactions were carried out for 16 min at room temperature (25±1 °C). (B) shows the binding of motor to the MT over a range of motor concentrations. Saturating experiments demonstrate that in the presence of 50 μM MgATP and 8.8 μM MT polymer, MCAK-Q710 saturates the MT polymer at 527 μM, whereas wt-MCAK saturates at 453 μM, a 16% increase. Curves were fitted over data points from 3 independent experiments.
C-terminal truncation of MCAK increases MT lattice-stimulated ATPase activity
MCAK exhibits both free tubulin-stimulated and MT-stimulated ATP hydrolysis [17]. One possible model suggests that ATP hydrolysis or Pi release may serve to dissociate MCAK from the removed terminal tubulin dimer [6,9,17,18]. Some researchers believe that this happens before the tubulin–MCAK complex dissociates from the MT protofilament, resulting in processive depolymerization [17]. Others believe that it occurs afterwards [6,18]. In either case, we wanted to determine if MCAK-Q710 had a higher depolymerization rate, because it was able to promote more quickly the dissociation of the motor from the terminal tubulin dimer. To test this, we measured the tubulin-stimulated ATPase activity for both wt-MCAK and MCAK-Q710 (Figures 4A and 4B). MCAK-Q710 and wt-MCAK had nearly identical tubulin-stimulated Pi release rates, suggesting that the enhanced MT depolymerization of MCAK-Q710 is not due to an enhanced rate of tubulin dissociation from the motor.
Figure 4. Longer MTs increase MT-stimulated ATPase activity of MCAK.

Active motor (50 nM) was incubated in the presence of 11 μM tubulin, 250 μM [γ-32P]ATP (6000 Ci/mmol) and 250 μM unlabelled MgATP. All Figures display Pi release (μM) over time. Linear curve fits (A, B) were generated by Microsoft Excel. All others (C–E) were generated with MATLAB (The Mathworks). The curves fitted to the data in (C–E) are exponential association curves, fitted via non-linear least-squares regression. (A) shows free tubulin-stimulated ATPase with wt-MCAK and (B) is with MCAK-Q710. (C) shows ATPase stimulation in the presence of unsheared MTs. (D) shows ATPase stimulation in the presence of paclitaxel-stabilized MTs, 5 μm in length. (E) shows ATPase stimulation in the presence of MTs, 15 μm in length. Curves were fitted over data points from 4 independent experiments.
We measured the MT-stimulated ATPase activity using paclitaxel-stabilized MTs. MCAK-Q710 exhibited a higher rate of ATPase activity in the presence of MTs, as compared with wt-MCAK (Figures 4C–4E). This was most significantly pronounced with longer MTs, which have a higher ratio of lattice to ends.
We fit our ATPase data (the Pi released in the experiments) to an exponential association curve (described in Experimental section). We interpreted the ATPase activity in these experiments as an indicator of an ATP-stimulating tight-coupling reaction between MCAK and the MT. This particular measurement does not discriminate between end and lattice binding. ‘On’ and ‘off’ rates were determined by varying MCAK concentrations (results not shown).
We chose 15 μm MTs because their length is more consistent with those found in cells. ‘On’ and ‘off’ rates are listed in Table 1(A). The MT on rate for wt-MCAK, as indicated by the hydrolysis of ATP, is approx. 2.3-fold less than that of MCAK-Q710, suggesting that MCAK-Q710 finds high-affinity, ATPase-stimulating binding sites more quickly than wt-MCAK. In contrast, the off rates for both were comparable for the two proteins (Table 1A).
Table 1. Kinetics of wt-MCAK and MCAK-Q710 as measured by ATP hydrolysis.
(A) kon association constant of the motor to the MT as measured by ATPase activity, and dissociation constant of the motor from the MT as measured by ATPase activity. Experimental conditions: 11 μM tubulin, 15 μm MTs and 12.5–50 nM active MCAK heads. (B) Experimental conditions: 11 μM tubulin and 50 nM active MCAK heads.
| (A) | ||||
|---|---|---|---|---|
| kon (μM−1·s−1) | koff (s−1) | |||
| wt-MCAK | 7.5±1.76 | 0.123±0.084 | ||
| MCAK-Q710 | 16.4±1.51 | 0.183±0.037 | ||
| (B) | ||||
| Initial ATPase activity (s−1) | ||||
| MTs… | 5 μm | 15 μm | Unsheared | |
| wt-MCAK | 0.102 | 0.106 | 0.26 | |
| MCAK-Q710 | 0.302 | 0.318 | 0.676 | |
We evaluated the initial velocities of ATPase activities for both wt-MCAK and MCAK-Q710 in order to compare the initial motor–MT interactions (Table 1B). It can be seen for each MT length that MCAK-Q710 has an approx. 3-fold increase in ATPase activity at the initial rates.
To compare ATPase activity with MT depolymerization, we plotted the tubulin released from comparable pelleting assays at a number of time points, alongside the ATPase curve (Figure 5). It can be seen that wt-MCAK displays an ATPase activity that more closely tends toward a 1:1 stoichiometry with tubulin dimer release in comparison with MCAK-Q710. The ATPase activity of MCAK-Q710 quickly diverges from the rate of tubulin release.
Figure 5. wt-MCAK exhibits tighter 1:1 coupling between MT depolymerization and ATP hydrolysis in comparison with MCAK-Q710.

Depolymerization and ATP hydrolysis were compared for MTs 15 μm in length. (A) is wt-MCAK and (B) is MCAK-Q710. The stoichiometry of ATP hydrolysis and MT depolymerization quickly diverges with MCAK-710. However, this activity is suppressed with wt-MCAK.
MCAK-Q710 binds lattice more readily in the zero ATP state
In order to determine microscopically where MCAK was interacting with the MT during ATP hydrolysis, we constructed and purified EGFP-fusion constructs of both motors. We incubated EGFP-fusion motors with MTs in the presence of p[NH]ppA (to mimic the ATP-bound state), ADP·AlFx (which mimics the ADP·Pi state), ADP and no nucleotide. The results are shown in Figures 6(A) and 6(B) (ADP·AlFx results not shown). Both EGFP–wt-MCAK and EGFP–MCAK-Q710 accumulate at MT ends in p[NH]ppA. In the ADP·Pi state, there was very little detectable binding of wt-MCAK to the MT within the first 15 min of the incubation (results not shown). After a 45-min incubation, there was a slight increase in binding (from approx. 1–10%) solely at the microtubule ends. We were unable to detect any association of MCAK-Q710 in this ADP·Pi mimicked state with MTs within 45 min. This is likely to reflect a difference in the limits of detection of MCAK-Q710 (due to its tendency to aggregate less, as seen in Figures 6A and 6B, motor only), rather than a difference in end binding between the two proteins. In the presence of ADP, there is no detectable binding of either motor to the MT on the lattice or the MT end.
Figure 6. wt-MCAK shows more significant aggregation than MCAK-Q710 and differential MT binding in the absence of nucleotide.

Images of EGFP–wt-MCAK (A) and EGFP–MCAK-Q710 (B) bound to pp[CH2]pG-stabilized MTs with varying nucleotide conditions are presented. Motor only-labelled rows show the interactions of the motor alone in the absence of MTs. wt-MCAK displays more significant aggregation than MCAK-Q710. p[NH]ppA-labelled (AMP-PNP) rows illustrate the motor in the presence of MTs and p[NH]ppA. Both motors bind the ends of MTs in the presence of p[NH]ppA. Again, wt-MCAK displays larger aggregate on the MT ends in comparison with MCAK-Q710. ADP-labelled rows show the motor with MTs in the presence of ADP. Neither motor appears to bind the MT in the presence of ADP. No nucleotide-labelled rows show the motors with MTs, but in the absence of nucleotide. Both motors bundle MTs and decorate the MT lattice. However, wt-MCAK decorates the lattice in a more punctate fashion, whereas MCAK-Q710 appears to more uniformly decorate the MT lattice with increased apparent-affinity. All binding experiments were done in 75 mM KCl. Incubations were for 5 min at room temperature (25±1 °C).
In the absence of nucleotide, both MCAK constructs significantly bundle MTs. However, there is a distinct difference in the lattice decoration of EGFP–wt-MCAK in comparison with EGFP–MCAK-Q710. wt-MCAK appears to decorate the lattice sparsely in a punctate fashion, whereas MCAK-Q710 appears to bind more uniformly throughout the lattice.
We tested the hypothesis that ATP is required to free MCAK from free tubulin dimers by determining whether incubation with free tubulin could inhibit the MT bundling observed in the nucleotide free state. At a 1:1 stoichiometry of free-to-polymerized tubulin, MT bundling was reduced approx. 20%. At an 8:1 stoichimoetry, bundling was reduced approx. 90%. Results were similar for both motors. These results suggest that tubulin can sequester MCAK away from the MT lattice in the absence of nucleotide.
DISCUSSION
MCAK has a high affinity for MT ends which are the substrate for its depolymerizing activity [17]. In the present study we show that lattice-bound MCAK can also significantly increase the efficacy of MT depolymerization, although at an energy cost (uncoupling). The interaction of MCAK with the lattice is inhibited by 8 amino acids at the C-terminus of the protein. Therefore, MCAK has a mechanism to suppress lattice interactions in preference for end binding, which is the productive substrate for MT depolymerization [17]. What is surprising, however, is that lattice-bound MCAK increases the velocity with which MTs are depolymerized, rather than serving as a sink for unproductive interactions.
Extreme C-terminus of MCAK is an auto-inhibitor of ATPase activity along the MT lattice
wt-MCAK and MCAK-Q710 exhibit a significant difference in MT-stimulated ATPase activity. This disparity increased with the length of the MTs (and by extension, with longer exposure to the MT lattice), presumably because MCAK-Q710 can interact productively (in a manner that stimulates ATP hydrolysis) with more sites along the MT lattice in comparison with wt-MCAK.
A comparison of the on rates revealed that MCAK-Q710 finds an ATPase-stimulating binding site along the MT at a rate 2.3 times faster than wt-MCAK. Furthermore, by comparing ATPase activity with MT depolymerization, we saw that wt-MCAK tends toward a 1:1 stoichiometry, whereas MCAK-Q710 quickly diverges from this trend. This suggests that with paclitaxel-stabilized MTs, ATP hydrolysis is more strongly coupled to MT depolymerization for wt-MCAK. Our results suggested that when the ratio of lattice-to-ends increases, both motors tend to exhibit non-end stimulated ATP hydrolysis. Hunter et al. [17] also observed that wt-MCAK exhibited low lattice-stimulated ATPase activity in particular, when the concentration of motor exceeded the concentration of exposed MT ends. However, wt-MCAK significantly suppresses this activity in comparison with MCAK-Q710. Again, this suggests that the role of the C-terminus may be to suppress lattice-stimulated ATPase activity. Relief of inhibition by the C-terminus (via protein interactions or ‘clipping’) might be one method that organisms could employ to disassemble long cellular MTs rapidly.
Our experiments indicate an on rate for wt-MCAK that is approx. 8-fold less than that found by Hunter et al. [17] However, two important differences in experimental design should be noted. The first is that different MT-stabilizing mechanisms were used: paclitaxel verses pp[CH2]pG. It has not yet been determined if the differences in these stabilizations affect the method or efficiency in which MCAK depolymerizes MTs. It can, however, be noted that the extreme disparity that Hunter et al. [17] observed between the tubulin-stimulated ATPase activity and that stimulated by MTs is not present in our present experiments. Our measurements are more consistent with Moores et al. [25] in which they saw comparable maximum ATPase activity with free tubulin and paclitaxel-stabilized polymer. The second difference is that there is a considerable difference in our MT lengths. MTs in the study by Hunter et al. [17] were on the order of 2.2 μm, whereas ours are approx. 15 μm. If we consider the notion that wt-MCAK primarily employs Brownian motion to diffuse to MT ends, and MT ends are the primary site of ATP hydrolysis for wt-MCAK, then the difference in diffusion times may account for the difference in the on rates.
A model: enhanced depolymerization of MCAK-Q710 may result from additional motors being distributed along the MT lattice
We propose that MCAK, analogous to kinesin and myosin II [22,26], may exist in two conformations: folded (inactive, limited ATPase activity) and unfolded (active ATPase activity). The C-terminus of MCAK may contribute to keeping the motor in the folded conformation when not at the highest-affinity binding site (MT ends) to inhibit excess ATP hydrolysis along the lattice (Figure 7). This may serve as an important regulatory mechanism, particularly when the cell is not actively remodelling its MT array. Once MCAK encounters a protofilament end, high-affinity binding to the conformationally distinct terminal tubulin [27,28] may out compete the C-terminus for the binding of the motor and releases the ATPase inhibition. Alternatively, the MCAK C-terminus may possess a binding domain for the end of the terminal tubulin dimer, comparable with that of Ncd [29] (Figure 7A). Selective binding to the terminal tubulin dimer may also provide some insight as to why MCAK, unlike most other kinesins, has free tubulin-stimulated ATPase activity.
Figure 7. Proposed model of the difference between wt-MCAK and MCAK-Q710.

(A) is a detailed illustration of the proposed ATP hydrolysis cycle of wt-MCAK based on the EGFP–wt-MCAK binding assays. The model proposes that the binding of MCAK to the MT lattice promotes the dissociation of ADP from the motor and the subsequent binding of ATP. MCAK oscillates along the lattice in the ATP bound state until it finds a MT end. Association with the high-affinity-end binding sites triggers ATP hydrolysis and terminal tubulin dimer dissociation. Pi release then liberates the motor from the cleaved tubulin dimer. This model does not differentiate between processivity and non-processivity. Thus both scenarios are illustrated. (B) illustrates how wt-MCAK oscillates back and forth along the MT in a weak binding state until it finds an end, at which time the C-terminal inhibition is relieved. ATPase activity is uninhibited by this interaction and the ATP-binding state then allows the motor to bind tightly to the high-affinity binding site on the terminal tubulin dimer. (C) illustrates how MCAK-Q710 may interact with the MT. While it may primarily oscillate in the loose binding state, similar to wt-MCAK, it may also be able to hydrolyse ATP sporadically along the MT lattice. This ATP hydrolysis along the MT may allow the motor to bind tightly to the lattice. As the MT end approaches these tightly bound motors, they may be able to facilitate more efficient MT depolymerization. This may happen as a result of more efficient end targeting via a power stroke (in comparison with the diffusion-based targeting of wt-MCAK) or it may result from a cooperative interaction between the lattice-bound motors and those bound to the MT end.
Figure 7(A) illustrates our proposed ATP hydrolysis cycle of wt-MCAK. The model is similar to others that have been put forth [6,17] in that it suggests that the binding of MCAK to the MT lattice promotes the release of ADP, which results in the binding of ATP. We suggest that the motor oscillates in the ATP-bound state until it finds a MT end. This is supported by our observation that MCAK binds with significantly lower affinity to MT ends in the ADP·Pi state in comparison with the ATP-bound state. ATP hydrolysis may promote the active release of the terminal tubulin dimer from the MT ends. Subsequent release of Pi might then trigger the dissociation of the released dimer from the MCAK motor, leaving the motor in the ADP state and ready for another cycle. Our assays do not resolve whether or not MCAK functions as a processive motor (both cases are illustrated in Figure 7A). Our model for the activity of MCAK-Q710 accommodates either case.
We have identified the C-terminus as an inhibitor of high-affinity ATPase-stimulating lattice interactions. We have also demonstrated that MCAK-Q710 exhibits increased depolymerization activity relative to wt-MCAK. Lattice binding is presumed to be a sink for end-stimulated depolymerization. How do we reconcile these observations? We put forth two plausible explanations.
The first possibility is compatible with the observation that MCAK may be processive [17]. Biased diffusional motility of MCAK, which involves intermittent ATP hydrolysis, can potentially explain the enhanced activity of MCAK-Q710. If the C-terminus tail inhibits lattice interactions that would lead to ATP hydrolysis, then the absence of the tail (MCAK-Q710) may result in ATP-dependent biased diffusion of the motor along the lattice, as is seen for Kif1A monomers [30]. Considering the time it would take for a motor to reach a MT ends via simple Brownian motion along the lattice, it stands to reason that directed movement via a processive mechanism may result in faster end targeting. This precise mechanism has been elegantly established for Kif1A [30].
Because we have never observed such ATP-dependent lattice motility and because the processivity measured for MCAK is relatively weak [17], we also considered the formal possibility that MCAK is not processive. Our biochemical and microscopic assays confirm that the higher apparent affinity of MCAK-Q710 is not due to enhanced accumulation of motor at the MT end, but rather on the lattice. Therefore, we suggest a model in which MCAK's mechanism of MT depolymerization includes a feature that we call ‘lattice priming’. While directed movement along the lattice and lattice priming are not mutually exclusive mechanisms, it is important to note that lattice priming has the potential to mimic processivity, possibly resulting from the tendency of wt-MCAK to aggregate.
The lattice-priming model accommodates the situation in which a slightly higher apparent affinity of the motor for the MT lattice promotes a slightly enhanced level of depolymerization. The active conformation of MCAK-Q710 may allow it to exist sparsely along the MT lattice, at a higher apparent affinity at specific times during the ATP hydrolysis cycle. This activity will not directly translate to depolymerization, because the tubulin dimers are locked within the lattice. However, as the MT ends approach, active motors tightly bound along the lattice will participate in depolymerization, thus affording the system more efficient depolymerization, as it will be less limited by the simple diffusive property of the full-length motor. These models are illustrated in Figures 7(B) and 7(C).
Although we propose that MCAK-Q710 may bind with higher affinity along the MT lattice during certain stages of the ATP hydrolysis cycle, it is important to remember that this mutant, analogous to wt-MCAK, depends on loose tethering to the MT lattice through much of the cross-bridge cycle in order to maintain uninterrupted contact with the MT lattice. This is consistent with the results of our pelleting assays where salt concentrations were markedly increased. In these instances, both wt-MCAK and MCAK-Q710 were diminished in their ability to depolymerize MTs (results not shown).
Increasing the apparent affinity of MCAK for MTs can enhance its MT depolymerizing activity by promoting lattice priming. In the cell, simple diffusion along long cellular MTs may not be an efficient method for targeting MT ends, particularly when rapid re-ordering of the MT array is required. This represents an excellent mechanism of priming for MCAK-dependent depolymerization activity in situations in which MT ends are blocked by assembly-promoting factors (such as CLIP-170 and Eb1 [31,32]). It is worth noting that GFP–MCAK is often seen transiently associated with the MT lattice in vivo (results not shown). It stands to reason that the C-terminus of MCAK serves as an inhibitor of fruitless ATP hydrolysis along the MT lattice for MCAK that is free in solution or when active MT depolymerization is not occurring. Priming of MCAK could then be promoted by any factors that interact with the C-terminus of MCAK, thus relieving lattice inhibition. Furthermore, these results suggest that any factor (such as the Kin I activator ICIS [33]) that potentially increases the apparent affinity of MCAK for the MT lattice has the potential to stimulate or enhance MT depolymerizing activity of MCAK in a regulated fashion in response to the need for rapid MT remodelling.
Acknowledgments
We thank Michael Wagenbach for the unlabelled wt-MCAK protein and for the rhodamine-labelled tubulin. We thank Jeremy Cooper, Will Hancock (Department of Bioengineering, Pennsylvania State University, PA, U.S.A.), Marla Feinstein, Yulia Ovechkina and Kathleen Rankin for many discussions and comments on the manuscript. We also thank Steve Carlson and Mark Bothwell for many useful discussions on experimental techniques. This study was supported by National Institutes of Health Grant (GM53654A) and Department of Defense Grant (DAMD 17-01-1-0450 to L.W.) and National Institutes of Health Predoctoral Fellowship 1 F31 (GM65061 to A.M.).
References
- 1.Goldstein L. S., Philp A. V. The road less traveled: emerging principles of kinesin motor utilization. Annu. Rev. Cell Dev. Biol. 1999;15:141–183. doi: 10.1146/annurev.cellbio.15.1.141. [DOI] [PubMed] [Google Scholar]
- 2.Vale R. D., Fletterick R. J. The design plan of kinesin motors. Annu. Rev. Cell Dev. Biol. 1997;13:745–777. doi: 10.1146/annurev.cellbio.13.1.745. [DOI] [PubMed] [Google Scholar]
- 3.Vale R. D., Reese T. S., Sheetz M. P. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brady S. T. A novel brain ATPase with properties expected for the fast axonal transport motor. Nature (London) 1985;317:73–75. doi: 10.1038/317073a0. [DOI] [PubMed] [Google Scholar]
- 5.Vale R. D., Milligan R. A. The way things move: looking under the hood of molecular motor proteins. Science. 2000;288:88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- 6.Ogawa T., Nitta R., Okada Y., Hirokawa N. A common mechanism for microtubule destabilizers – M type kinesins stabilize curling of the protofilament using the class-specific neck and loops. Cell. 2004;116:591–602. doi: 10.1016/s0092-8674(04)00129-1. [DOI] [PubMed] [Google Scholar]
- 7.Miki H., Setou M., Kaneshiro K., Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc. Natl. Acad. Sci. U.S.A. 2001;98:7004–7011. doi: 10.1073/pnas.111145398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wordeman L., Mitchison T. J. Identification and partial characterization of mitotic centromere-associated kinesin, a kinesin-related protein that associates with centromeres during mitosis. J. Cell Biol. 1995;128:95–104. doi: 10.1083/jcb.128.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai A., Verma S., Mitchison T. J., Walczak C. E. Kin I kinesins are microtubule-destabilizing enzymes. Cell. 1999;96:69–78. doi: 10.1016/s0092-8674(00)80960-5. [DOI] [PubMed] [Google Scholar]
- 10.Walczak C. E., Mitchison T. J., Desai A. XKCM1: a Xenopus kinesin-related protein that regulates microtubule dynamics during mitotic spindle assembly. Cell. 1996;84:37–47. doi: 10.1016/s0092-8674(00)80991-5. [DOI] [PubMed] [Google Scholar]
- 11.Maney T., Wagenbach M., Wordeman L. Molecular dissection of the microtubule depolymerizing activity of mitotic centromere-associated kinesin. J. Biol. Chem. 2001;276:34753–34758. doi: 10.1074/jbc.M106626200. [DOI] [PubMed] [Google Scholar]
- 12.Homma N., Takei Y., Tanaka Y., Nakata T., Terada S., Kikkawa M., Noda Y., Hirokawa N. Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell. 2003;114:229–239. doi: 10.1016/s0092-8674(03)00522-1. [DOI] [PubMed] [Google Scholar]
- 13.Tournebize R., Popov A., Kinoshita K., Ashford A. J., Rybina S., Pozniakovsky A., Mayer T. U., Walczak C. E., Karsenti E., Hyman A. A. Control of microtubule dynamics by the antagonistic activities of XMAP215 and XKCM1 in Xenopus egg extracts. Nat. Cell Biol. 2000;2:13–19. doi: 10.1038/71330. [DOI] [PubMed] [Google Scholar]
- 14.Rogers G. C., Rogers S. L., Schwimmer T. A., Ems-McClung S. C., Walczak C. E., Vale R. D., Scholey J. M., Sharp D. J. Two mitotic kinesins cooperate to drive sister chromatid separation during anaphase. Nature (London) 2004;427:364–370. doi: 10.1038/nature02256. [DOI] [PubMed] [Google Scholar]
- 15.Maney T., Hunter A. W., Wagenbach M., Wordeman L. Mitotic centromere-associated kinesin is important for anaphase chromosome segregation. J. Cell Biol. 1998;142:787–801. doi: 10.1083/jcb.142.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ovechkina Y., Wagenbach M., Wordeman L. K-loop insertion restores microtubule depolymerizing activity of a ‘neckless’ MCAK mutant. J. Cell Biol. 2002;159:557–562. doi: 10.1083/jcb.200205089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunter A. W., Caplow M., Coy D. L., Hancock W. O., Diez S., Wordeman L., Howard J. The kinesin-related protein MCAK is a microtubule depolymerase that forms an ATP-hydrolyzing complex at microtubule ends. Mol. Cell. 2003;11:445–457. doi: 10.1016/s1097-2765(03)00049-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moores C. A., Yu M., Guo J., Beraud C., Sakowicz R., Milligan R. A. A mechanism for microtubule depolymerization by KinI kinesins. Mol. Cell. 2002;9:903–909. doi: 10.1016/s1097-2765(02)00503-8. [DOI] [PubMed] [Google Scholar]
- 19.Hua W., Young E. C., Fleming M. L., Gelles J. Coupling of kinesin steps to ATP hydrolysis. Nature (London) 1997;388:390–393. doi: 10.1038/41118. [DOI] [PubMed] [Google Scholar]
- 20.Coy D. L., Wagenbach M., Howard J. Kinesin takes one 8-nm step for each ATP that it hydrolyzes. J. Biol. Chem. 1999;274:3667–3671. doi: 10.1074/jbc.274.6.3667. [DOI] [PubMed] [Google Scholar]
- 21.Hackney D. D., Levitt J. D., Suhan J. Kinesin undergoes a 9 S to 6 S conformational transition. J. Biol. Chem. 1992;267:8696–8701. [PubMed] [Google Scholar]
- 22.Hackney D. D., Stock M. F. Kinesin's IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat. Cell Biol. 2000;2:257–260. doi: 10.1038/35010525. [DOI] [PubMed] [Google Scholar]
- 23.Coy D. L., Hancock W. O., Wagenbach M., Howard J. Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat. Cell Biol. 1999;1:288–292. doi: 10.1038/13001. [DOI] [PubMed] [Google Scholar]
- 24.Rice S., Lin A. W., Safer D., Hart C. L., Naber N., Carragher B. O., Cain S. M., Pechatnikova E., Wilson-Kubalek E. M., Whittaker M., et al. A structural change in the kinesin motor protein that drives motility. Nature (London) 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- 25.Moores C. A., Hekmat-Nejad M., Sakowicz R., Milligan R. A. Regulation of KinI kinesin ATPase activity by binding to the microtubule lattice. J. Cell Biol. 2003;163:963–971. doi: 10.1083/jcb.200304034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. New York: Garland Science; 2002. Molecular Biology of the Cell. [Google Scholar]
- 27.Hyman A. A., Chretien D., Arnal I., Wade R. H. Structural changes accompanying GTP hydrolysis in microtubules: information from a slowly hydrolyzable analogue guanylyl-(α,β)-methylene-diphosphonate. J. Cell Biol. 1995;128:117–125. doi: 10.1083/jcb.128.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Severin F. F., Sorger P. K., Hyman A. A. Kinetochores distinguish GTP from GDP forms of the microtubule lattice. Nature (London) 1997;388:888–891. doi: 10.1038/42270. [DOI] [PubMed] [Google Scholar]
- 29.Wendt T., Karabay A., Krebs A., Gross H., Walker R., Hoenger A. A structural analysis of the interaction between ncd tail and tubulin protofilaments. J. Mol. Biol. 2003;333:541–552. doi: 10.1016/j.jmb.2003.08.051. [DOI] [PubMed] [Google Scholar]
- 30.Okada Y., Higuchi H., Hirokawa N. Processivity of the single-headed kinesin KIF1A through biased binding to tubulin. Nature (London) 2003;424:574–577. doi: 10.1038/nature01804. [DOI] [PubMed] [Google Scholar]
- 31.Ligon L. A., Shelly S. S., Tokito M., Holzbaur E. L. The microtubule plus-end proteins EB1 and dynactin have differential effects on microtubule polymerization. Mol. Biol. Cell. 2003;14:1405–1417. doi: 10.1091/mbc.E02-03-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez F., Diamantopoulos G. S., Stalder R., Kreis T. E. CLIP-170 highlights growing microtubule ends in vivo. Cell. 1999;96:517–527. doi: 10.1016/s0092-8674(00)80656-x. [DOI] [PubMed] [Google Scholar]
- 33.Ohi R., Coughlin M. L., Lane W. S., Mitchison T. J. An inner centromere protein that stimulates the microtubule depolymerizing activity of a KinI kinesin. Dev. Cell. 2003;5:309–321. doi: 10.1016/s1534-5807(03)00229-6. [DOI] [PubMed] [Google Scholar]