

Abstract
β-Glycosidase from the hyperthermophilic archaeon Sulfolobus solfataricus is a homotetramer with a higher number of ion pairs compared with mesophilic glycoside hydrolases. The ion pairs are arranged in large networks located mainly at the tetrameric interface of the molecule. In the present study, the structure and thermal stability of the wild-type β-glycosidase and of three mutants in residues R488 and H489 involved in the C-terminal ionic network were examined by FTIR (Fourier-transform IR) spectroscopy. The FTIR data revealed small differences in the secondary structure of the proteins and showed a lower thermostability of the mutant proteins with respect to the wild-type. Generalized 2D-IR (two-dimensional IR correlation spectroscopy) at different temperatures showed different sequences of thermal unfolding events in the mutants with respect to the wild-type, indicating that punctual mutations affect the unfolding and aggregation process of the protein. A detailed 2D-IR analysis of synchronous maps of the proteins allowed us to identify the temperatures at which the ionic network that stabilizes the quaternary structure of the native and mutant enzymes at the C-terminal breaks down. This evidence gives support to the current theories on the mechanism of ion-pair stabilization in proteins from hyperthermophilic organisms.
Keywords: β-glycosidase, protein structure, quaternary structure, Sulfolobus solfataricus, thermal stability, two-dimensional infrared spectroscopy
Abbreviations: Amide I', amide I band in a 2H2O medium; 2D-IR, two-dimensional IR correlation spectroscopy; FTIR, Fourier-transform IR; GH1, glycoside hydrolase family 1; GST, glutathione S-transferase; Ssβ-gly, recombinant Sulfolobus solfataricus β-glycosidase; H489A, His489→Ala mutant of Ssβ-gly; R488A, Arg488→Ala mutant of Ssβ-gly; Δ1, Ssβ-gly mutant obtained by deletion of the C-terminal His489; Tm, melting temperature; Topt, optimal growth temperature
INTRODUCTION
Studies on enzymes and proteins from thermophilic and hyperthermophilic micro-organisms (growing at 50 °C<Topt<80 °C and 80 °C<Topt<120 °C respectively, where Topt is the optimal growth temperature) are of interest for a number of reasons. The characterization and the understanding of the high stability of these biomolecules at high temperatures and with common protein denaturants, such as organic solvents, detergents and extreme pHs, is fundamental for basic science, for biotechnological applications and for the design of new biocatalysts that are stable under extreme conditions. Thermophilic proteins and their mesophilic counterparts share 40–80% sequence similarity, their three-dimensional structures are superimposable and they have the same catalytic mechanism. Despite these similarities, thermophilic proteins are intrinsically more stable than their mesophilic counterparts and the adaptive changes, in terms of free-energy differences, correspond to the equivalent of a few weak intermolecular interactions induced by multiple factors [1].
Experimental evidence [2,3], supported by computational analysis of genomic data [4,5] has shown that ion-pair networks are dominant elements in proteins from hyperthermophiles. However, some hyperthermophilic proteins have fewer ion pairs than their mesophilic counterparts [6,7], and mutational analyses have given contradictory results [8,9]. Therefore the role of electrostatic interactions in the stabilization of thermostable proteins is still debated.
Among hyperstable enzymes, the β-glycosidase from the hyperthermophilic archaeon Sulfolobus solfataricus (Ssβ-gly), which grows at 80 °C and pH≤3.0, is one of the most studied. This enzyme shows maximal activity at temperatures above 90 °C, and extreme stability against heat and other protein denaturants [10]. Ssβ-gly is a member of a large family of enzymes, the glycoside hydrolase family 1 (GH1), widely conserved among animals, plants, archaea and bacteria. Its structure has been resolved at 2.6 Å (1 Å=0.1 nm) resolution [11] and the enzyme is a homotetramer of 226884 Da with 489 amino acid residues per subunit (Figure 1). The thermal denaturation of Ssβ-gly is an irreversible process and occurs through aggregation [10,12,13]. In order to clarify the basis of its thermostability and thermoactivity, this enzyme was studied under different conditions [10,13–17], and the inspection of its three-dimensional structure allowed us to address the studies at the molecular level. The most striking structural difference observed in Ssβ-gly when compared with mesophilic β-glycosidases from GH1 is its increased number of ion pairs, 60% of which occur as part of multiple ion-pair networks [11]. In particular, the largest ionic network is located at the tetrameric interface of Ssβ-gly, and it is formed by a 16-residue tail (residues 473–489) running along the interface of one of the two non-crystallographic symmetry-related dimers [16] (A–D or B–C, Figure 1). This is a specific characteristic of Ssβ-gly, even when compared with other hyperthermophilic glycoside hydrolases from GH1 [16]. The intersection of the C-terminal tails at the tetrameric interface of the molecule creates this ionic network that involves more than 16 ion pairs and hydrogen bonds between several residues from the four subunits. Inspection of the three-dimensional structure predicted that the interactions formed by Arg488 of one subunit with the amino acids present in other subunits have a stabilizing effect [11] (Figure 1). In particular, the carboxy group of the C-terminal His489 and the side chain of Glu345 of the same subunit are involved in two ion pairs with the guanidinium group of each Arg488 of the adjacent subunit (A–D and B–C). In addition, Arg488 forms hydrogen bonds with Pro486 of the opposite subunit (A–C and B–D) and with Arg346 of the same subunit. Conversely, His489 is hydrogen bonded to Ser348, Leu487 and Arg488 of the same subunit and their imidazole rings are at van der Waals distance between the crystallographic units A–B and C–D.
Figure 1. Three-dimensional structure of Ssβ-gly.

Upper panel: native tetrameric structure resolved at 2.6 Å resolution (Protein Data Bank code 1GOW). Lower panel: schematic representation of the C-terminal ion-pair network that stabilizes the quaternary structure at the A–D and C–B interfaces.
Ssβ-gly is active only in its tetrameric form [18], therefore mutations affecting the stability of the tetramer would increase the inactivation of the enzyme at high temperatures. By following this line of approach, we showed recently that the replacement of the C-terminal residues His489 and Arg488 with alanine or depletion of His489 (Δ1) led to faster enzyme inactivation, and the mechanism of inactivation was dependent on the mutation introduced [16]. However, this approach precluded the analysis of the effect of the mutations at the structural level.
The aim of the present work was to analyse the stabilizing role of the same residues in Ssβ-gly by studying the effect of temperature, and of the mutations on the secondary structure, thermal stability and quaternary structure of the enzyme. The study was performed using FTIR (Fourier-transform IR) spectroscopy and generalized 2D-IR (two-dimensional IR correlation spectroscopy) analysis was applied to IR spectra [19]. The combination of site-directed mutagenesis and 2D-IR analysis of IR spectra allowed us to detect different sequences of thermal unfolding events in the mutants with respect to the wild-type and a temperature-dependent destabilization of the quaternary structure of the enzyme, giving support to the current theories on the mechanism of ion-pair stabilization in proteins from hyperthermophiles.
MATERIALS AND METHODS
Materials
2H2O (99.9%) was purchased from Aldrich. All other reagents and solvents were commercial samples of the highest purity.
Purification of wild-type and mutant Ssβ-gly
The preparation of the Ssβ-gly mutants R488A (Arg488→Ala), H489A (His489→Ala) and Δ1 is described elsewhere [16]. The wild-type and mutant enzymes were expressed in Escherichia coli as a fusion with GST (glutathione S-transferase) as described previously [16]. This procedure was adopted to avoid the heating steps currently used to remove the E. coli thermolabile proteins [20], since incubation at high temperature could inactivate mutants impaired in their stability. As an effect of this procedure, the wild-type and mutant Ssβ-gly show a short tail of four extra amino acids at their N-terminus [16]; in the present paper, Ssβ-gly containing this tail is considered to be the wild-type.
The purification of the H489A and Δ1 mutants was performed as described previously [16]. The purification procedure of the wild-type and R488A proteins used in the present study was slightly modified as follows: the GST binding was performed by adding to the E. coli cellular extract 3 ml of the glutathione–Sepharose 4B matrix (Amersham Biosciences), equilibrated in 50 mM sodium phosphate buffer, pH 7.4, 150 mM NaCl, 1% (v/v) Triton X-100 (PBS–Triton buffer), and incubating overnight at 4 °C. After binding, the matrix was packed, and after 30 column volumes washing with PBS buffer (without Triton), the matrix was resuspended in 1 vol. of PBS buffer and incubated overnight at 4 °C with 60 units of thrombin solution. The efficiency of thrombin cleavage was assayed by subjecting to SDS/PAGE an amount of the matrix slurry before and after the thrombin treatment. Thereafter, the soluble and GST-free β-glycosidase protein was recovered by five column washes with PBS buffer. Washes containing the β-glycosidase protein were pooled and dialysed against 20 mM sodium phosphate buffer, pH 7.0. After this treatment, the wild-type and the R488A mutant were >95% pure by SDS/PAGE, and were used for all the subsequent characterizations. The purification procedure yielded approx. 3 and 20 mg of pure protein for wild-type and R488A mutant Ssβ-gly respectively, from approx. 30 g of wet cell pellet. The samples stored at 4 °C were stable for several months.
Preparation of samples for IR measurements
Typically, 1–1.5 mg of protein dissolved in the buffer used for its purification were centrifuged in a 30K Centricon micro-concentrator (Amicon) at 3000 g and 4 °C, and concentrated to a volume of approx. 40 μl. Then, 300 μl of 50 mM phosphate buffer (prepared in 2H2O), p2H 6.5, were added and the sample concentrated again; the p2H value corresponds to the pH-meter reading +0.4 [21]. This procedure was repeated several times in order to completely replace the original buffer with the 50 mM phosphate buffer, p2H 6.5. The washings took 24 h, which is the time of contact of the protein with 2H2O before FTIR analysis. In the last washing, the protein sample was concentrated to a final volume of approx. 35 μl and used for the IR measurements.
IR spectra
The concentrated protein samples were placed in a thermostatically controlled Graseby Specac 20500 cell (Graseby-Specac, Orpington, Kent, U.K.) fitted with CaF2 windows and 25 μm Teflon spacers. FTIR spectra were recorded by means of a PerkinElmer 1760-X FTIR spectrometer using a deuterated triglycine sulphate detector and a normal Beer-Norton apodization function. At least 24 h earlier, and during data acquisition, the spectrometer was continuously purged with dry air at a dew point of −40 °C. Spectra of buffers and samples were acquired at 2 cm−1 resolution under the same scanning and temperature conditions. In the thermal denaturation experiments, the temperature was raised in 5 °C steps from 20 °C to 95 °C. The actual temperature in the cell was controlled by a thermocouple placed directly on to the CaF2 window. Spectra were collected and processed using the Spectrum software from PerkinElmer. The deconvoluted parameters for the amide I band were set with a gamma value of 2.5 and a smoothing length of 60. Second-derivative spectra were calculated over a nine-data-point range (9 cm−1).
The melting temperature (Tm) in thermal denaturation curves was calculated by fitting the curves with a sigmoid function as described in [22].
Generalized 2D-IR analysis
Generalized 2D-IR analysis of IR band intensities of deconvoluted spectra was performed according to the method of Noda [19]. To generate synchronous and asynchronous plots, 2Dcos toolbox software and Matlab 6.0 software (The MathWorks, Natick, MA, U.S.A.) were used. The 2Dcos toolbox software was developed in Professor Y. Ozaki's laboratory (Kwansei Gakuin University, Sanda, Japan).
RESULTS
Secondary structure of Ssβ-gly
Figure 2 shows the absorbance and resolution-enhanced IR spectra of wild-type Ssβ-gly in the 1750–1500 cm−1 region. These spectra are similar to those obtained from a previous IR analysis of the enzyme at p2H 7.5 and 10.0 [13–15]. The bands displayed in the 1700–1620 cm−1 region (amide I band) can be attributed to secondary-structural elements [23] as β-sheet (1636.9), α-helix (1654.6), turns (1666.1), unordered structures and/or loops (1643.2). The band at 1625.3 has been assigned to β-strand(s) particularly exposed to solvent, while the origin of the 1683.5 band is less certain, because both turns and coupled high-frequency vibrations of β-segments [24] can contribute in the spectral region between 1670 and 1690 cm−1. The peak close to 1550 cm−1 represents the residual amide II band, i.e. the amide II band (1600–1500 cm−1 range) after 1H/2H exchange of the amide hydrogens of the polypeptide chain. Indeed, this band is particularly sensitive to the exchange of amide hydrogen with deuterium. In experiments performed in 1H2O medium, the intensity of the amide II band is approx. two thirds that of amide I band, while in 2H2O medium it decreases significantly [13,14,25]. The greater the intensity decrease, the greater the 1H/2H exchange. Moreover, a big 1H/2H exchange indicates that the protein structure is very accessible to the solvent (2H2O). The fact that the IR spectrum of the wild-type Ssβ-gly displays a residual amide II band, indicates that at 20 °C, the protein segments were not completely accessible to the solvent. The other peaks shown in Figure 2 are due to amino acid side chain absorption [26,27].
Figure 2. IR spectra of wild-type Ssβ-gly.

Original absorbance (A), deconvoluted (B), and second derivative (C) spectra in 2H2O medium at 20 °C.
Secondary structure of R488A, H489A and Δ1 mutants
Comparison of the spectrum of the wild-type with those of R488A, H489A and Δ1 mutant Ssβ-gly allows detection secondary-structural changes induced by the mutations (Figure 3, spectra A–C). A better view of spectral differences is obtained by calculating difference spectra (Figure 3, spectra D–F). In these spectra, negative and positive bands reflect a lower and higher intensity, respectively, of a particular peak also present in the spectrum of the wild-type. A particular case is related to the presence of negative and positive adjacent bands of similar intensity, which reflects band-shift of a particular band [28,29]. A more complicated difference spectrum is obtained when a particular peak simultaneously undergoes band-shift and changes in its intensity [30]. Figure 3 shows that the deconvoluted spectra of Δ1 and H489A differ slightly from that of the wild-type, since the bands related to α-helix (1654.1) and to β-sheet (1636.9) are lower and higher in intensity respectively than in the wild-type. Difference spectra also show negative bands close to 1625 cm−1 (β-strands). These data indicate that the secondary structure of Δ1 and H489A mutants is slightly different from that of the wild-type. Other differences are shown by difference spectra below 1620 cm−1, indicating changes in the amino acid side chain absorption with respect to the wild-type. The positive broad band centred at 1545 cm−1 (difference spectra) indicates that, in the Δ1 and H489A mutants, the extent of 1H/2H exchange is slightly higher than in the wild-type, indicating a larger accessibility of the solvent (2H2O) to the protein. The different 1H/2H exchange could be due to the changes in the secondary structure and/or to different dynamics of the structure induced by the mutations. In the case of the R488A mutant, very tiny differences can be detected only in the difference spectrum, indicating very small or no differences in the secondary structure and in the 1H/2H exchange ability of the protein.
Figure 3. Comparison of IR spectra of wild-type and Ssβ-gly mutants.

Upper set of spectra: comparison of deconvoluted spectra of wild-type, Δ1, R488A and H489A. Spectra were obtained in 2H2O medium at 20 °C. Dotted lines refer to the spectra of the wild-type. Continuous lines refer to spectra of R488A (A), Δ1 (B) and H489A (C) mutant proteins. Lower set of spectra: difference spectra between two deconvoluted spectra. The difference spectra were obtained by subtracting the R488A (D), Δ1 (E) or H489A (F) spectrum from that of the wild-type. For a better view, the difference spectra were multiplied by a factor of 3.
Thermal denaturation
Figure 4 shows the deconvoluted and difference spectra of wild-type Ssβ-gly and mutants in the 20–99.5 °C range. With the increase in temperature the amide I' (amide I band in 2H2O) band (1700–1620 cm−1) intensity decreases and the position shifts. Moreover, at high temperatures, the residual amide II band (1600–1500 cm−1) intensity decreases, as a consequence of further 1H/2H exchange due to relaxation of tertiary/quaternary structure and to thermal denaturation. Two new bands at 1616 and 1686 cm−1 also appear at high temperatures as a consequence of protein unfolding and aggregation. Comparison of Figures 4(A)–4(D) or Figures 4(E)–4(H) shows that the above mentioned phenomena, and in particular the temperature-dependent changes in the amide I' or 1616 cm−1 band intensities, are different in the mutants with respect to the wild-type, suggesting different thermal stability of the proteins. Indeed, thermal denaturation curves obtained from the amide I' band intensity decrease (Figure 5A) show small, but significant, differences in the Tm that were 92.5, 91.3, 90.8 and 89.5 for Ssβ-gly, R488A, H489A and Δ1 respectively. Moreover, the curves related to mutant proteins shown in Figures 5(A) and 5(B) have a steeper gradient than the curves related to the wild-type, further indicating a lower thermal stability and a different aggregation ability of the mutants with respect to the wild-type.
Figure 4. Deconvoluted and difference spectra of the wild-type and Ssβ-gly mutants in the 20–99.5 °C temperature range.

Deconvoluted spectra (A–D) were collected in the 20–95 °C temperature range every 5 °C. Then, three other spectra were collected at 98, 99 and 99.5 °C. Difference spectra (E–H) were generated from the deconvoluted spectra by the 2Dcos toolbox software using the spectrum obtained at 20 °C as reference [19]. (A, E), (B, F), (C, G) and (D, H) refer to the wild-type, R488A, H489A and Δ1 Ssβ-gly respectively.
Figure 5. Temperature-induced changes in the spectra of the wild-type and mutants.

Thermal denaturation and protein aggregation were followed by monitoring the amide I' band intensity (A) and the 1616 cm−1 band intensity (B) as a function of the temperature.
Generalized 2D-IR analysis
In order to gain more information on the thermal denaturation pattern of the proteins, we performed generalized 2D-IR analysis of the IR spectra [19]. The method allows us to obtain synchronous and asynchronous spectra of dynamic spectral intensity variations induced by the increase in temperature. The former represents the simultaneous or coincidental changes of spectral intensities measured at two discrete and independent wavenumbers ν1 and ν2 (on x- and y-axes respectively). The latter represents sequential, or unsynchronized, changes in spectral intensities measured at ν1 and ν2. Autopeaks are present only on the diagonal of a synchronous map. Cross-peaks are present in either synchronous or asynchronous maps at the off-diagonal positions, and they can be positive or negative. The sign of synchronous cross-peaks becomes positive if the spectral intensities at corresponding wavenumbers either increase or decrease together as a function of temperature. Negative synchronous cross-peaks indicate that one of the spectral intensities is increasing while the other is decreasing. The sign of asynchronous cross-peaks becomes positive if the intensity change at ν1 occurs predominantly before ν2. On the other hand, it becomes negative if the change at ν1 occurs after ν2. This rule is reversed if the synchronous cross-peak at ν1 and ν2 is negative [19,31]. Multiple lines reflect the intensity of the peaks.
The generalized 2D-IR analysis of deconvoluted spectra shown in Figures 4(A)–4(D) generates the difference spectra (Figures 4E–4H) from which synchronous and asynchronous maps are obtained [19]. Figure 6 shows the synchronous and asynchronous maps of the wild-type. The maps were calculated considering the 40–99.5 °C (Figures 6A and 6B) or 20–99.5 °C (Figures 6C and 6D) temperature interval and the 1700–1500 cm−1 (Figures 6A and 6B) or the 1700–1600 cm−1 (Figures 6C and 6D) spectral range. This separation of IR spectra in different temperature sets and/or in different spectral ranges was done because it allows a more detailed description of the spectral events [32]. Further separations were carried out to describe in full the spectral changes (results not shown). The synchronous spectra show main autopeaks at 1685 (Figure 6C), 1654 (Figures 6A and 6C), 1617 (Figures 6A and 6C) and 1545 (Figure 6A) cm−1. Minor autopeaks were detected at 1666, 1643, 1636 and 1625 cm−1 in an expanded scale of the synchronous map (results not shown). The schematic representation of the synchronous spectrum, showing all autopeaks and cross-peaks, is reported in Table 1, which summarizes the peaks shown in the upper part of the synchronous spectra of wild-type Ssβ-gly (Figure 6). The negative cross-peak (1687↓–1675↑) represents a shift of the 1687 cm−1 β-sheet band to 1675 cm−1. This phenomenon was checked by analysing the difference spectra which revealed a significant decrease in the 1687 cm−1 band and a corresponding increase in the 1675 cm−1 band intensity in the 45–70 °C interval (results not shown). It is worth noting that, in difference spectra, the position of negative and positive peaks may not correspond exactly to those found in the second derivative or deconvoluted spectra, especially in the case of peaks generated by band-shifts [28,29]. Hence the 1687 cm−1 peak corresponds to the 1683.5 cm−1 band (turn/β-sheet) shown in Figure 2. The shift is caused by further 1H/2H exchange [25].
Figure 6. Generalized 2D-IR analysis of Ssβ-gly IR spectra.

The generalized 2D-IR analysis was performed on deconvoluted spectra of the wild-type considering different sets of spectral and/or temperature ranges. The Figure shows the synchronous (A, C) and asynchronous (B, D) maps calculated considering the 40–99.5 °C (A, B) or the 20–99.5 °C (C, D) temperature interval and the 1700–1500 cm−1 (A, B) or the 1700–1600 cm−1 (C, D) spectral range. Negative peaks are shown in grey. Multiple lines represent intense peaks. The same procedure of analysis was followed for the spectra of R488A, H489A and Δ1 mutants (not shown).
Table 1. Schematic representation of the synchronous spectrum of wild-type Ssβ-gly.
The synchronous spectrum of wild-type Ssβ-gly was obtained analysing the 1700–1500 cm−1 spectral range considering the 40–99.5 °C temperature interval, and analysing the 1700–1600 cm−1 spectral range considering the 20–99.5 °C temperature interval. Autopeaks are represented by the letter A. Positive and negative cross-peaks are represented by + and − respectively, and they are described by ν1 (last row) and ν2 (first column). ↑ and ↓ indicate the increase and decrease of peak intensity respectively. The peaks marked * were observed in an expanded scale (1630–1680 cm−1) in the 40–99.5 °C temperature interval of the synchronous map. ** indicates peaks observed analysing the 1700–1600 cm−1 spectral range in the 20–65 °C temperature interval. The negative peak (1687↓–1675↑) represents a shift of the 1687 cm−1 β-sheet band to 1675 cm−1.
| 1518↓ | − | ||||||||||
| 1545↓ | + | − | A | ||||||||
| 1617↑ | + | + | − | − | − | − | A | ||||
| 1625↓ | +** | −** | A** | ||||||||
| 1636↓ | − | +* | + | + | A | ||||||
| 1643↓ | − | + | + | A | |||||||
| 1654↓ | − | − | + | A | |||||||
| 1666↓ | − | A* | |||||||||
| 1675↑ | −** | ||||||||||
| 1685↑ | A | ||||||||||
| 1687↓ | A** | ||||||||||
| cm−1 | 1687↓ | 1685↑ | 1675↑ | 1666↓ | 1654↓ | 1643↓ | 1636↓ | 1625↓ | 1617↑ | 1545↓ | 1518↓ |
The schematic representation of the asynchronous spectrum of the wild-type in Figure 6 is shown in Table 2. The analysis of both synchronous and asynchronous maps allowed us to describe the sequence of events that leads to protein unfolding and aggregation. Table 3 reports these events for all proteins studied. The data show that for all proteins the 1687→1675 cm−1 shift is concomitant with the decrease in the 1550 cm−1 band intensity, i.e. with further 1H/2H exchange. Differences can be observed at the level of the 1635 cm−1 band (β-sheet) that decreases in intensity in the case of wild-type and H489A concomitantly with the formation of protein aggregates, while in the case of R488A and Δ1 mutants, the decrease in intensity of the 1635 cm−1 band occurs before and after protein aggregation respectively. The decrease in intensity of the other peaks, in all cases, occurs before protein aggregation.
Table 2. Schematic representation of the asynchronous spectrum of Ssβ-gly.
The asynchronous spectrum of Ssβ-gly was obtained analysing the 1700–1500 cm−1 spectral range considering the 40–99.5 °C temperature interval, and analysing the 1700–1600 cm−1 spectral range considering the 20–99.5 °C temperature interval. Each peak is described by ν1 (last row) and ν2 (first column). + and − represent positive and negative peaks respectively. The letters a and b mean that the intensity change at ν1 occurs predominantly after and before ν2 respectively. ↑ and ↓ indicate the increase and decrease of peak intensity respectively.
| 1518↓ | +a | −a | +a | |||||||
| 1550↓ | +a | −a | −a | −a | −a | +a | ||||
| 1611↑ | +a | +a | −a | |||||||
| 1618↑ | +b | −b | −b | −b | +a | |||||
| 1635↓ | +b | |||||||||
| 1645↓ | +a | +b | +b | |||||||
| 1654↓ | +a | −b | +b | |||||||
| 1666↓ | +a | |||||||||
| 1675↑ | ||||||||||
| 1685↑ | ||||||||||
| cm−1 | 1685↑ | 1675↑ | 1666↓ | 1654↓ | 1645↓ | 1635↓ | 1618↑ | 1611↑ | 1550↓ | 1518↓ |
Table 3. Sequence of unfolding events for the wild-type and Ssβ-gly mutants.
1550, 1H/2H exchange; 1675, β-sheet/turns; shift from 1687 to 1675 cm−1; 1687, β-sheet/turns; shift to 1675 cm−1; 1625, β-sheet; 1666, turns; 1654, α-helices; 1644, unordered/bends; 1618, protein aggregation; 1685, protein aggregation; 1635, β-sheet. ↑ and ↓ indicate the increase and decrease of peak intensity respectively.
| Sequence of events in… | ||||
|---|---|---|---|---|
| Temperature (°C) | Wild-type | R488A | Δ1 | H489A |
| 20 | 1550↓, 1675↑, 1687↓, 1625↓ | 1550↓, 1675↑, 1687↓, 1625↓ | 1550↓, 1675↑, 1687↓, 1625↓ | 1550↓, 1675↑, 1687↓, 1625↓ |
| ↓ | 1666↓ | 1666↓, | 1666↓, 1654↓, 1644↓ | 1666↓, 1654↓, 1644↓ |
| ↓ | 1654↓ | 1654↓, 1644↓, 1635↓ | ||
| ↓ | 1644↓ | |||
| ↓ | 1618↑, 1685↑, 1635↓ | 1618↑, 1685↑ | 1618↑, 1685↑ | 1618↑, 1685↑, 1635↓ |
| 99.5 | 1635↓ | |||
Generalized 2D-IR analysis is a powerful tool that allows the detection of small changes in a particular peak. In an attempt to detect differences in the spectral characteristics of the amino acids involved in the ionic network at the C-terminal of Ssβ-gly, we applied the analysis in the 1620–1500 cm−1 range, where amino acid side chain absorption typically occurs [27]. Figure 7 shows the synchronous spectra of Ssβ-gly wild-type and mutants. The maps are displayed in the 1620–1500 cm−1 interval and in different temperature sets. Considering the wild-type map in the 20–60 °C temperature range (first column/first row) we can observe that, besides other peaks, a 1584/1550 cm−1 negative cross-peak is present (arrow). This peak represents the simultaneous or coincidental changes in spectral intensities measured at 1584 and 1550 cm−1 (on x- and y-axes respectively). In particular, the absorption at 1584 cm−1 is due to arginine and/or to ionized aspartate residues [27], while the 1550 cm−1 peak is due to residual amide II band. The peak is negative, meaning that one band increases and the other decreases in intensity. Since the 1550 cm−1 band intensity decreases with the increase in temperature (due to further 1H/2H exchange), the 1584 cm−1 band intensity increases. If we consider the 20–65 °C temperature interval (first column/second row), we can see that the 1584/1550 cm−1 peak increases in intensity. The peak intensity then remains constant up to the 20–75 °C interval (first column, fourth row); in the 20–80 °C range it decreases in intensity, while it is absent in the 20–85 °C and 20–90 °C temperature ranges. One possible explanation for these results is that the increase in temperature weakens the ionic interactions, allowing the arginine and/or aspartate residue(s) to become more free; consequently, it modifies the arginine and/or aspartate absorption. Furthermore, since a cross-peak in a synchronous spectrum appears only if two events are associated, i.e. if they are in phase (occurring concomitantly), the changes in arginine and/or aspartate absorption can be seen until the 20–80 °C interval, because up to these temperatures, they are associated (in phase) with the changes in the residual amide II band intensity. In fact, with the increase in temperature, the ionic interactions of arginine and/or aspartate residue(s) will weaken and this will increase the flexibility of the protein. As a consequence, a deeper contact of the solvent (2H2O) with the polypeptide chain will take place [27], and more hydrogens will be exchanged with deuterium. The absence of changes in arginine and/or aspartate absorption or the absence of the ‘in-phase’ changes in intensity of the amide II band will result in the absence of the 1584/1550 cm−1 peak in the synchronous map. This situation will occur when a particular temperature breaks the ionic interactions completely. In the case of wild-type Ssβ-gly, the temperature would be 85 °C. A particular case is that the disappearance of the 1584/1550 cm−1 peak could be due to the breaking of the ionic interaction of Arg488 with Glu345 and His489. This would increase the flexibility of the dimer–dimer assembly, allowing a closer contact of the solvent (2H2O) with the C-terminal of the protein. At this point one can say that 32 aspartate and 32 arginine residues are present in each Ssβ-gly monomer, and thus it is not possible to ascribe the 1584/1550 cm−1 peak only to Arg488.
Figure 7. Synchronous spectra of the wild-type and Ssβ-gly mutants.

The columns, from the left to the right, relate to wild-type, R488A, H489A and Δ1 Ssβ-gly respectively. Each column displays seven synchronous maps that represent synchronous spectra originated considering different temperature intervals. The rows, from the top to the bottom, display maps originated from deconvoluted spectra obtained in the 20–60, 20–65, 20–70, 20–75, 20–80, 20–85 and 20–90 °C temperature ranges respectively. Each map is displayed in the 1620–1500 cm−1, as shown by the bottom map in the first column. Negative peaks are shown in grey. Multiple lines reflect intense peak. The arrow in the map of first column/first row shows the 1584/1550 cm−1 negative cross-peak due to Arg488 and residual amide II band respectively. The arrow in the map of the first column/second row shows the 1600/1550 cm−1 negative cross-peak due to His489 and residual amide II band respectively.
However, if we consider the synchronous spectra (Figure 7, second column) of the R488A mutant (that lacks Arg488) we cannot see any peak at 1584/1500 cm−1, suggesting that no changes in the absorption band of arginine and/or aspartate residues occur at any temperature intervals, or that the changes in the 1584 and 1550 cm−1 band intensities are not in phase. Nevertheless, it is very probable that, as in the case of the wild-type, the increase in temperature leads to weakening/breaking of ionic interactions between arginine and/or aspartate residues and other amino acids. Hence, the lack of the 1584/1550 cm−1 peak in R488A is ascribable to the absence of in-phase changes in absorption of the arginine and/or glutamate residues present in the protein and the 1H/2H exchange process, i.e. with the decrease in the 1550 cm−1 band intensity. This means that the 1584/1550 cm−1 negative cross-peak in the wild-type Ssβ-gly synchronous maps is due to correlated changes in the absorption of Arg488 and the amide II band. Hence the temperature at which the 1584/1550 cm−1 negative cross-peak disappears corresponds to a free Arg488, i.e. to the breaking of the ionic bridges that contribute to keeping together the crystallographic dimer–dimer assembly at the C-terminal (Figure 1, A–B and C–D).
Similar results to those of Ssβ-gly were obtained with H489A and Δ1 mutants (Figure 7, third and fourth columns respectively). The latter mutation maintains the ion pair between Arg488 and Glu345, but abolishes the original ion pair between Arg488 and the carboxy group of the C-terminal His489, weakening the interactions between the two non-crystallographic dimers (A–D and C–B). In H489A, the interactions of the histidine imidazole ring are lost, but the original ion pair between the carboxy group of the C-terminal His489 and Arg488 is substituted with the ion pair between Arg488 and the terminal carboxy group of Ala489 (Figure 1). The synchronous spectra of the two mutants display the 1584/1550 cm−1 cross-peak, but it disappears at lower temperature (80 °C) with respect to Ssβ-gly, which is consistent with the alteration/deletion of the salt bridge at the A–D and C–B tetrameric interface of the mutants. Table 4 summarizes the results related to the 1584/1550 cm−1 cross-peak for all proteins.
Table 4. Temperature of disappearance of the 1584/1550 cm−1 and 1600/1550 cm−1 cross-peaks in the synchronous spectra of the wild-type and mutant Ssβ-gly.
| Ssβ-gly | 1584/1550 cm−1 peak | 1600/1550 cm−1 peak |
|---|---|---|
| Wild-type | 85 °C | 80 °C |
| Δ1 | 80 °C | Absent |
| H489A | 80 °C | Absent |
| R488A | Absent | Absent |
Further support for the interpretation that the presence of the 1584/1550 cm−1 cross-peaks is due to changes in intensity of Arg488 and to the in-phase changes of the amide II band intensity, is obtained by looking at the presence of the 1600/1550 cm−1 peak in the synchronous map of the wild-type enzyme (Figure 7, first column/second row, see arrow). The peak, whose absorption at 1600 cm−1 is due to histidine residue(s) [27], is visible up to the 20–75 °C map (Figure 7, first column/fourth row), while it is missing in all maps of the mutants. According to the above explanation, the appearance of the 1600/1550 cm−1 peak should be due to concomitant changes in absorption of histidine/residual amide II band in the 20–65 °C, 20–70 °C and 20–75 °C intervals, i.e. to the relaxation/breaking of ionic bridges between His489 and Arg488. At temperatures above 75 °C, His489 should be free from ionic interactions. Similarly to the discussion concerning Arg488, each Ssβ-gly monomer contains 14 histidine residues and thus the peak could be due to all of them. However, the peak is absent in the maps of R488A, which is not able to form an ionic bridge with His489, and in the maps of H489A and Δ1 that both lack His489. Hence we conclude that the 1600/1550 cm−1 negative cross-peak is due to correlated changes; i.e. in-phase changes, in the absorption of His489 and amide II band absorption.
DISCUSSION
The combination of site-directed mutagenesis, FTIR spectroscopy and generalized 2D-IR analysis of IR spectra has allowed us to describe in detail the effects of punctual mutation on the secondary structure, on the sequence of thermal unfolding events, and on the thermal stability of the secondary and quaternary structure of Ssβ-gly. Our data indicate that single mutations affect the structural properties of the protein, including its propensity to aggregate, as observed also in a recent study concerning the structural and thermal stability analysis of E. coli and Alicyclobacillus acidocaldarius thioredoxin [29].
The sequence of unfolding events resulted differently in the mutants with respect to Ssβ-gly. Indeed, the 2D-IR analysis revealed that for all proteins, the first event that occurs with the increase in temperature is a further 1H/2H exchange that causes a shift in the 1687 cm−1 band (turns/β-sheet) to 1675 cm−1. This event occurs before any unfolding process, indicating that the protein structure or part of it starts undergoing relaxation at relatively low temperatures. Subsequently, and for all proteins, the unfolding process takes place at the level of the β-structures belonging to the 1625 cm−1 band. From this point, the sequence of events starts differing in the mutant proteins with respect to the wild-type. The temperature-induced unfolding process continues with the loss in turns (1666 cm−1 band) that, in the case of Δ1 and H489A mutants, is accompanied by a decrease in the number of α-helices and bends. Instead, in the case of the wild-type, the decrease in band intensity of α-helices and bends occurs after the decrease in intensity of the 1666 cm−1 band. Hence the data indicate that the Δ1 and H489A mutations destabilize the α-helices and bends, suggesting a specific role of His489 in the stabilization of these secondary-structural elements. In addition, a destabilization of β-sheet is suggested to occur in the R488A mutant, since the decrease in the 1635 cm−1 band intensity starts before the onset of the aggregation process. Instead, the Δ1 mutation seems to stabilize the β-sheets related to the 1635 cm−1 band, since the decrease in the band intensity starts after the onset of protein aggregation.
Summarizing, the sequence of unfolding events indicates that in all proteins there are secondary-structural elements that are not affected by the punctual mutations and that they are particularly sensitive to temperature (1687 and 1625 cm−1 bands). On the other hand, the data indicate that the mutations differently influence the aggregation process and the stability of other secondary-structural elements that, in turn, start undergoing unfolding at different temperatures. Hence it is possible that the different ability of the proteins to undergo intermolecular interactions (aggregation) at high temperatures, i.e. after partial or total unfolding, is driven by the different sequence of exposure to the solvent of single amino acids or of a particular sequence of amino acids. In fact, if, in the native protein, the amino acid(s) responsible for protein aggregation are present in different secondary-structural elements, they will interact after unfolding of these secondary structures and aggregation will take place. However, if the sequence of unfolding events changes, the exposure of amino acid(s) responsible for protein aggregation will change. Hence the possibility exists that the pattern of amino acid(s) interactions will also change with the consequence of alterations in the susceptibility of the protein to undergo aggregation.
More interestingly, and in agreement with our previous studies [16], we show in the present study that the mutated residues play a major role in the quaternary structure stabilization of Ssβ-gly. We showed previously that the stability of the Δ1, H489A and R488A mutants, measured by experiments of thermal inactivation, was reduced if compared with the wild-type [16]. In H489A, the most evident effect of the mutation was the formation of a polar cavity, facing the four subunits, filled with several water molecules and occupied by the hydrophobic methyl chains of the new alanine residue [16]. In this case, the unfavourable location of alanine was suggested to be the key factor of reduced protein stability. In Δ1, the mutation also induced the formation of a hydrophilic cavity, larger than that in H489A. In this case, the free α-carboxy group of Arg488 faces the cavity created by the mutation and does not interact with any residue. The reduced stability of the protein is ascribable to the abolition of the original ion pair between Arg488 and His489. In the case of the R488A mutant, the formation of a cavity facing the four subunits was not detected, and the observed reduced protein stability is ascribable to the abolition of the original ionic network Arg488/Glu345/His489 connecting the four subunits. The results presented here are consistent with previous data and experimentally validate the proposed models [16]. In fact, it is worth noting that the IR spectra show a reduced thermal stability of the mutant proteins and an increased 1H/2H exchange ability in the Δ1 and H489A, but not in R488A, at 20 °C. Moreover, the generalized 2D-IR analysis demonstrated that relaxation of protein structure starts before any unfolding process and that the ion-pair interactions that stabilize the A–D and C–B interfaces (Figure 1) start to weaken at relatively low temperatures, and that they are lost at 85 °C for the wild-type, and at 80 °C for the Δ1 and H489A mutants. Mutant R488A is also destabilized if compared with the wild-type [16]. However, in this case, it was not possible to identify the temperature of quaternary structure destabilization, since the method of analysis is based on the absorption of Arg488. In conclusion, our study shows direct experimental evidence that the residues involved in the major ionic network at the tetrameric interface stabilize the quaternary structure of Ssβ-gly.
In our previous study, the analysis of the residual enzymic activity at different temperatures, allowed us to calculate the reaction orders of thermal inactivation, which resulted in an integer being close to 1 for the wild-type and the H489A mutant, and higher than 2 for R488A and Δ1 mutants [16]. For the latter two mutants, a mechanism of thermal inactivation that includes active intermediates was postulated:
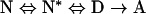 |
where N, N*, D and A stand for native and active intermediates, and denatured and aggregated enzyme respectively. Most likely, the active intermediates are dimers, since they were suggested to form in Ssβ-gly at pH 10 and in the presence of SDS [13], and were demonstrated recently to exist in the presence of SDS as active forms [17]. In an attempt to explain these observations at the molecular level, we analysed the three-dimensional structure of Ssβ-gly. The destabilization of the tetrameric structure of the wild-type and mutant proteins could lead to the formation of monomers or two possible dimers (A–C and A–D; Figure 1). However, as described recently [16,17], the interfaces A–C and D–B are stabilized by a large ion-pair network comprising 16 ion-pair interactions and several hydrogen bonds, while the other non-crystallographic interface (A–D and B–C) is stabilized only by a few interactions as depicted in Figure 1 (lower panel). Hence the most probable dimers formed by the destabilization of the quaternary structure are A–C and D–B. The weakening of the A–C and D–B interfaces is supported further by the observation that the weakening and/or the alterations of the ionic network that stabilize them allows a closer contact with the solvent (2H2O) as revealed by the analysis of the extent of 1H/2H exchange. It is worthy of note that the temperatures at which the quaternary structure of wild-type Ssβ-gly and mutants is destabilized are lower with respect to the corresponding Tms, indicating that the formation of possible dimers precedes the marked thermal denaturation of the proteins. The results of the present study thus indicate directly that high temperatures destabilize the quaternary structure of the wild-type, and Δ1 and H489A Ssβ-gly, while the formation of active intermediates was postulated to occur for R488A and Δ1 [16]. The discrepancy could be explained by the fact that in this previous study, thermal inactivation was followed by the analysis of residual enzymic activity, while in the present study, it was followed by the analysis of thermal denaturation.
Conclusions
In the present paper, we have demonstrated the importance of key residues located at the C-terminal of Ssβ-gly monomers in the stabilization of the quaternary structure of the enzyme. Ssβ-gly mutant proteins, with modifications at the monomer's C-terminal, were prepared by site-directed mutagenesis, and their thermal stability and structure were analysed by FTIR spectroscopy. Generalized 2D-IR analysis of IR spectra detected differences in the sequence of thermal unfolding events of the mutant proteins with respect to the control and identified the temperature at which the ionic network, which stabilizes the quaternary structure of the native and mutant enzymes at the C-terminal, breaks up. The quaternary structure of the mutant proteins was less stable, giving support to the current theories on the mechanism of ion-pair stabilization in proteins from hyperthermophilic organisms.
Acknowledgments
This work was supported by grants from the Ministero dell'Istruzione, dell'Università e della Ricerca Scientifica (MIUR) [Progetti di Ricerca di Interesse Nazionale (PRIN 2001 and PRIN 2003)] (F.T. and E.B.); by the Agenzia Spaziale Italiana project: “Extremophilic Archaea as model systems to study origin and evolution of early organisms: molecular mech-anisms of adaptation to extreme physical-chemical conditions” contract no. I/R/365/02 (B.C.-P., M.R. and M.M.), and by MIUR project “Folding di proteine: l'altra metà del codice genetico” (M.R and M.M.).
References
- 1.Petsko G. A. Structural basis of thermostability in hyperthermophilic proteins, or “there's more than one way to skin a cat”. Methods Enzymol. 2001;334:469–478. doi: 10.1016/s0076-6879(01)34486-5. [DOI] [PubMed] [Google Scholar]
- 2.Danson M. J., Hough D. W. Structure, function and stability of enzymes from the Archaea. Trends Microbiol. 1998;6:307–314. doi: 10.1016/s0966-842x(98)01316-x. [DOI] [PubMed] [Google Scholar]
- 3.Karshikoff A., Ladenstein R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: a ‘traffic rule’ for hot roads. Trends Biochem. Sci. 2001;26:550–556. doi: 10.1016/s0968-0004(01)01918-1. [DOI] [PubMed] [Google Scholar]
- 4.Chakravarty S., Varadarajan R. Elucidation of factors responsible for enhanced thermal stability of proteins: a structural genomics study. Biochemistry. 2002;41:8152–8161. doi: 10.1021/bi025523t. [DOI] [PubMed] [Google Scholar]
- 5.Suhre K., Clavery J. Genomic correlates of hyperthermostability, an update. J. Biol. Chem. 2003;278:17198–17202. doi: 10.1074/jbc.M301327200. [DOI] [PubMed] [Google Scholar]
- 6.Usher K. C., Cruz A. F., Dahlquist F. W., Swanson R. V., Simon M. I., Remington S. H. Crystal structures of CheY from Thermotoga maritima do not support conventional explanations for the structural basis of enhanced thermostability. Protein Sci. 1998;7:403–412. doi: 10.1002/pro.5560070221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao L., Honig B. Electrostatic contributions to the stability of hyperthermophilic proteins. J. Mol. Biol. 1999;289:1435–1444. doi: 10.1006/jmbi.1999.2810. [DOI] [PubMed] [Google Scholar]
- 8.Horovitz A., Serrano L., Avron B., Bycroft M., Fersht R. A. Strength and co-operativity of contributions of surface salt bridges to protein stability. J. Mol. Biol. 1990;216:1031–1044. doi: 10.1016/S0022-2836(99)80018-7. [DOI] [PubMed] [Google Scholar]
- 9.Vetriani C., Maeder D. L., Tolliday N., Yip K. S., Stillman T. J., Britton K. L., Rice D. W., Klump H. H., Robb F. T. Protein thermostability above 100 °C: a key role for ionic interactions. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12300–12305. doi: 10.1073/pnas.95.21.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pouwels J., Moracci M., Cobucci-Ponzano B., Perugino G., van der Oost J., Kaper T., Lebbink J. H. G., De Vos W. M., Ciaramella M., Rossi M. Activity and stability of hyperthermophilic enzymes: a comparative study on two archaeal β-glycosidases. Extremophiles. 2000;4:157–164. doi: 10.1007/s007920070030. [DOI] [PubMed] [Google Scholar]
- 11.Aguilar C. F., Sanderson I., Moracci M., Ciaramella M., Nucci R., Rossi M., Pearl L. H. Crystal structure of the β-glycosidase from the hyperthermophilic archaeon Sulfolobus solfataricus: resilience as a key factor in thermostability. J. Mol. Biol. 1997;271:789–802. doi: 10.1006/jmbi.1997.1215. [DOI] [PubMed] [Google Scholar]
- 12.D'Auria S., Rossi M., Barone G., Catanzano F., Del Vecchio P., Graziano G., Nucci R. Temperature-induced denaturation of β-glycosidase from the archaeon Sulfolobus solfataricus. J. Biochem. (Tokyo) 1996;120:292–300. doi: 10.1093/oxfordjournals.jbchem.a021412. [DOI] [PubMed] [Google Scholar]
- 13.D'Auria S., Barone R., Rossi M., Nucci R., Barone G., Fessas D., Bertoli E., Tanfani F. Effects of temperature and SDS on the structure of β-glycosidase from the thermophilic archaeon Sulfolobus solfataricus. Biochem. J. 1997;323:833–840. doi: 10.1042/bj3230833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Auria S., Moracci M., Febbraio F., Tanfani F., Nucci R., Rossi M. Structure–function studies on β-glycosidase from Sulfolobus solfataricus: molecular bases of thermostability. Biochimie. 1998;80:949–957. doi: 10.1016/s0300-9084(00)88892-6. [DOI] [PubMed] [Google Scholar]
- 15.D'Auria S., Nucci R., Rossi M., Bertoli E., Tanfani F., Gryczynski I., Malak H., Lakowicz J. R. β-Glycosidase from the hyperthermophilic Archaeon Sulfolobus solfataricus: structure and activity in the presence of alcohols. J. Biochem. (Tokyo) 1999;126:545–552. doi: 10.1093/oxfordjournals.jbchem.a022484. [DOI] [PubMed] [Google Scholar]
- 16.Cobucci-Ponzano B., Moracci M., Di Lauro B., Ciaramella M., D'Avino R., Rossi M. Ionic network at the C-terminus of the β-glycosidase from the hyperthermophilic archaeon Sulfolobus solfataricus: functional role in the quaternary structure thermal stabilization. Proteins. 2002;48:98–106. doi: 10.1002/prot.10128. [DOI] [PubMed] [Google Scholar]
- 17.Gentile F., Amodeo P., Febbraio F., Picaro F., Motta A., Formisano S., Nucci R. SDS-resistant active and thermostable dimers are obtained from the dissociation of homotetrameric β-glycosidase from hyperthermophilic Sulfolobus solfataricus in SDS. J. Biol. Chem. 2002;277:44050–44060. doi: 10.1074/jbc.M206761200. [DOI] [PubMed] [Google Scholar]
- 18.Catanzano F., Graziano G., De Paola B., Barone G., D'Auria S., Rossi M., Nucci R. Guanidine-induced denaturation of β-glycosidase from Sulfolobus solfataricus expressed in Escherichia coli. Biochemistry. 1998;37:14484–14490. doi: 10.1021/bi980490w. [DOI] [PubMed] [Google Scholar]
- 19.Noda I. Generalized two-dimensional correlation method applicable to infrared, raman, and other types of spectroscopy. Appl. Spectrosc. 1993;47:1329–1336. [Google Scholar]
- 20.Moracci M., Ciaramella M., Rossi M. β-Glycosidase from Sulfolobus solfataricus. Methods Enzymol. 2001;330:201–215. doi: 10.1016/s0076-6879(01)30376-2. [DOI] [PubMed] [Google Scholar]
- 21.Salomaa P., Schaleger L. L., Long F. A. Solvent deuterium isotope effects on acid–base equilibria. J. Am. Chem. Soc. 1964;86:1–7. [Google Scholar]
- 22.Meersman F., Smeller L., Heremans K. Comparative Fourier transform infrared spectroscopy study of cold-, pressure-, and heat-induced unfolding and aggregation of myoglobin. Biophys. J. 2002;82:2635–2644. doi: 10.1016/S0006-3495(02)75605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arrondo J. L. R., Muga A., Castresana J., Goni F. M. Quantitative studies of the structure of proteins in solutions by Fourier-transform infrared spectroscopy. Prog. Biophys. Mol. Biol. 1993;59:23–56. doi: 10.1016/0079-6107(93)90006-6. [DOI] [PubMed] [Google Scholar]
- 24.Krimm S., Bandekar J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986;38:181–364. doi: 10.1016/s0065-3233(08)60528-8. [DOI] [PubMed] [Google Scholar]
- 25.Osborne H. B., Nabedryk-Viala E. Infrared measurements of peptide hydrogen exchange in rhodopsin. Methods Enzymol. 1982;88:676–680. [Google Scholar]
- 26.Chirgadze Y. N., Fedorow O. W., Trushina N. P. Estimation of amino acid residue side-chain absorption in the infrared spectra of protein solutions in heavy water. Biopolymers. 1975;14:679–694. doi: 10.1002/bip.1975.360140402. [DOI] [PubMed] [Google Scholar]
- 27.Barth A., Zsherp C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002;35:369–430. doi: 10.1017/s0033583502003815. [DOI] [PubMed] [Google Scholar]
- 28.Paolini S., Tanfani F., Fini C., Bertoli E., Pelosi P. Porcine odorant-binding protein: structural stability and ligand affinities measure by Fourier-transform infrared spectroscopy and fluorescence spectroscopy. Biochim. Biophys. Acta. 1999;1431:179–188. doi: 10.1016/s0167-4838(99)00037-0. [DOI] [PubMed] [Google Scholar]
- 29.Pedone E., Bartolucci S., Rossi M., Pierfederici F. M., Scirè A., Cacciamani T., Tanfani F. Structural and thermal stability analysis of Escherichia coli and Alicyclobacillus acidocaldarius thioredoxin revealed a molten globule-like state in thermal denaturation pathway of the proteins: an infrared spectroscopic study. Biochem. J. 2003;373:875–883. doi: 10.1042/BJ20021747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Umemura J., Cameron D. G., Mantsch H. H. A Fourier transform infrared spectroscopic study of the molecular interaction of cholesterol with 1,2-dipalmitoyl-sn-glycero-3-phosphocholine. Biochim. Biophys. Acta. 1980;602:32–44. doi: 10.1016/0005-2736(80)90287-4. [DOI] [PubMed] [Google Scholar]
- 31.Filosa A., Wang Y., Ismail A. A., English A. M. Two-dimensional infrared correlation spectroscopy as a probe of sequential events in the thermal unfolding of cytochromes c. Biochemistry. 2001;40:8256–8263. doi: 10.1021/bi002710n. [DOI] [PubMed] [Google Scholar]
- 32.Fabian H., Mantsch H. H., Schultz C. P. Two-dimensional IR correlation spectroscopy: sequential events in the unfolding process of the λ Cro-V55C repressor protein. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13153–13158. doi: 10.1073/pnas.96.23.13153. [DOI] [PMC free article] [PubMed] [Google Scholar]