

Abstract
Equinatoxin II (Eqt-II) is a member of the actinoporins, a unique family of cytotoxins comprising 20 kDa pore-forming proteins isolated from sea anemones. Actinoporins bind preferentially to lipid membranes containing sphingomyelin, and create cation-selective pores by oligomerization of three to four monomers. Previous studies have shown that regions of Eqt-II crucial for its cytolytic mechanism are an exposed aromatic cluster and the N-terminal region containing an amphipathic α-helix. In the present study, we have investigated the transfer of the N-terminal α-helix into the lipid membrane by the use of three mutants containing an additional tryptophan residue in different positions within the amphipathic α-helix (Ile18→Trp, Val22→Trp and Ala25→Trp). The interaction of the mutants with different model systems, such as lipid monolayers, erythrocytes and ghost membranes, was extensively characterized. Intrinsic fluorescence measurements and the use of vesicles containing brominated phospholipids indicated a deep localization of the N-terminal amphipathic helix in the lipid bilayer, except for the case of Val22→Trp. This mutant is stabilized in a state immediately prior to final pore formation. The introduction of additional tryptophan residues in the sequence of Eqt-II has proved to be a suitable approach to monitor the new environments that surround defined regions of the molecule upon membrane interaction.
Keywords: actinoporin, amphipathic helix, membrane interaction, pore-forming toxin
Abbreviations: ANS, 8-anilinonaphthalene-1-sulphonic acid; ANTS, 8-aminonaphthalene-1,3,6-trisulphonic acid; DPX, p-xylene-bis-pyridinium bromide; Eqt-II, equinatoxin II; I18W, etc., a mutant protein of Eqt-II bearing an amino acid replacement of Ile18 with Trp, etc.; L/T, lipid/toxin molar ratio; LUV, large unilamellar vesicle; MLV, multilamellar vesicle; PC, phosphatidylcholine; PFT, pore-forming toxin; SM, sphingomyelin; St-II, sticholysin-II; RBC, red-blood cells; SUV, small unilamellar vesicle
INTRODUCTION
Pore-forming toxins (PFTs) are cytotoxic proteins, which have the ability to exist in two well-differentiated states: a soluble state and a membrane-associated state. In the soluble state, the regions of the PFTs that will insert into the lipid membrane remain hidden within the general toxin fold. Upon membrane interaction, conformational changes favour the insertion of those regions into the lipid membrane, thus permeabilizing the target cell [1]. Depending on the structural element that is inserted into the lipid membrane upon pore formation, PFTs have been divided into two groups: (a) those that insert β-hairpins forming a pore structured as a β-barrel, such as the Staphylococcus aureus α-toxin, cholesterol-dependent cytolysins from Gram-positive bacteria or anthrax protective antigen [2], and (b) those that make use of α-helices as structural elements for spanning the lipid membrane, such as colicins [3]. The latter are less well understood, in comparison with β-barrel PFTs, due to the inherent instability of the resulting pores.
An interesting group of PFTs is the actinoporin family consisting of toxins from sea anemones (Class: Anthozoa; Order: Actiniaria). It has been suggested that these cytolytic toxins, in combination with neurotoxins, may have a role in the predation of these organisms on small crustaceans and fishes, or in repelling predators (reviewed in [4]). They are lethal for crabs [5], fish [6] and mammals (the intravenous LD50 in mice is 35 μg/kg for Eqt-II (equinatoxin II)) [7], and have been shown to cause platelet aggregation [8], pulmonary oedema [9] and cardiac arrest [10]. A common feature contributing to this variety of pharmacological effects is an increase in permeability for Na+, K+, Ca2+ and other smaller solutes in targeted membranes. This is also the cause of actinoporin-induced lysis in erythrocytes, which is the result of colloidal osmotic shock. They are one of the most potent groups of haemolysins, effective at nanomolar concentrations. Such a potent lytic ability renders them suitable molecules for the design of immunotoxins, which could be used to fight against tumour cells or parasites [4].
Actinoporins are closely related proteins lacking cysteine residues, which form pores in lipid membranes in an SM (sphingomyelin)-dependent manner [4]. Eqt-II from Actinia equina and St-II (sticholysin-II) from Stichodactyla helianthus are the most studied representatives, for which high-resolution three-dimensional structures have recently been solved [11–13]. Both toxins are composed of a tightly folded β-sandwich flanked on two sides by α-helices (see Figure 1A). The first 30 N-terminal residues, encompassing the N-terminal α-helix, exhibit an amphipathic character, and this is the only region of the protein that can be separated from the body of the molecule without disrupting the β-sandwich.
Figure 1. The three-dimensional model of Eqt-II.

(A) The three-dimensional model of Eqt-II. The N-terminal amphipathic helix comprising residues 16–25 is shown in green. (B) The helical wheel analysis of the 16–25 region. The residues are coloured according to physical properties (black, hydrophobic; yellow, polar; red, negatively charged; blue, positively charged). (C) A scheme of Eqt-II pore-formation. The pore formation involves at least four different conformational states of the toxin: a soluble form, a membrane-bound form attached to the membrane with the aromatic cluster and phosphorylcholine site, a membrane-bound non-lytic form with the N-terminal helix lying parallel to the plane of the membrane, and an oligomeric lytic form with the N-terminal helix inserted in a perpendicular orientation to the plane of the membrane as a part of the conductive pathway. The lipid–water interface is shaded grey. Reproduced from [14] with permission. © (2002) The American Society for Biochemistry and Molecular Biology.
Pore formation by Eqt-II has been shown to be a multi-step process (Figure 1C) [14]. First, the toxin binds to the lipid bilayer. This step is governed by the aromatic amino acid cluster located on a broad loop at the tip of the β-sandwich and on the C-terminal α-helix [11,14,15]. Recent studies involving co-crystallization of St-II together with phosphocholine have revealed the existence of a phosphocholine-binding pocket that probably participates in the initial binding to the membrane [13]. In the next step, the N-terminal segment translocates to the lipid–water interface [14,16], and, finally, a transmembrane pore is formed by helices from three or four monomers [16,17]. Actinoporin pores are not resistant to SDS, and have not yet been directly visualized. The number of monomers in the pore was deduced from cross-linking and kinetic experiments [17,18]. Lipids themselves might have a role in constructing the pore walls together with the protein, by forming a so-called toroidal pore [19,20].
In the present study, we have made use of three different Eqt-II N-terminal mutants [Ile18→Trp (I18W), V22W and A25W] to monitor the interaction of the N-terminal segment of the toxin with lipid membranes. These mutations did not affect binding to lipid membranes. The introduction of tryptophan residues at various positions along the N-terminal helix allowed us to explore the movement of this region to the core of the lipid bilayer by measuring the changes in intrinsic tryptophan fluorescence. The introduction of tryptophan at position 22 inhibited the N-terminal region from switching from the membrane non-lytic state to the transmembrane lytic state. In the case of I18W and A25W, the changes in the intrinsic fluorescence of the newly introduced tryptophan upon interaction with the membrane, and its quenching by SUV (small unilamellar vesicle)-containing brominated lipids, suggested that the N-terminal helix inserts deep into the lipid bilayer.
MATERIALS AND METHODS
Materials
Egg-yolk PC (phosphatidylcholine), bovine-brain SM, 1-palmitoyl-2-stearoyl-(6,7)-dibromo-sn-glycero-3-phosphocholine, 1-pal-mitoyl-2-stearoyl-(9,10)-dibromo-sn-glycero-3-phosphocholine and 1-palmitoyl-2-stearoyl-(11,12)-dibromo-sn-glycero-3-phosphocholine were from Avanti Polar Lipids (Alabaster, AL, U.S.A.). Triton X-100 was from Sigma (St Louis, MO, U.S.A.). ANTS (8-aminonaphthalene-1,3,6-trisulphonic acid), ANS (8-anilinonaphthalene-l-sulphonic acid) and DPX (p-xylene-bispyridinium bromide) were obtained from Molecular Probes (Eugene, OR, U.S.A.). Acrylamide was from Merck (Darmstadt, Germany), and horse RBCs (red-blood cells) were from Biomedics (Alcobendas, Spain).
Purification of native Eqt-II
Native Eqt-II was purified from the liquid obtained from freshly collected A. equina specimens in the Bay of Biscay, as described by Maček and Lebez [7]. The purified protein was concentrated to approx. 10 mg/ml with an Amicon 8050 (Danvers, MA, U.S.A.) ultrafiltration unit equipped with a regenerated nitrocellulose filter (Millipore, Bedford, MA, U.S.A.) with a molecular-mass cut-off of 10 kDa. Aliquots of the concentrated protein were stored at −24 °C, and once thawed they were not refrozen. Protein concentration was estimated spectrophotometrically using a molar absorption coefficient at 280 nm of 42800 M−1·cm−1, as estimated following the procedure of Perkins [21].
Cloning, expression and purification of Eqt-II mutants
I18W, V22W and A25W mutants were produced by substituting the corresponding wild-type amino acid residue with tryptophan, as described previously [22]. Mutants were expressed from a T7-based expression vector in Escherichia coli BL21(DE3) strain, and recombinant proteins were purified from bacterial supernatants as described previously [23]. Protein concentration was estimated spectrophotometrically using a molar absorption coefficient at 280 nm of 48500 M−1·cm−1 [21]. All mutants were purified to homogeneity, as observed on SDS/PAGE gels.
Binding to MLVs (multilamellar vesicles) of SM/PC (in a 1:1 ratio)
Bovine-brain SM and egg-yolk PC were mixed in organic solvent and evaporated thoroughly. MLVs were prepared by hydration of the lipid film with 10 mM Hepes buffer, pH 7.5, containing 200 mM NaCl and subsequent vortex-mixing. Each toxin at a final concentration of 1.67 μM was mixed with either MLVs at an L/T (lipid/toxin) ratio of 600 or the same volume of buffer. All mixtures were incubated overnight at 25 °C under constant stirring, and centrifuged at 30000 rev./min (56000 g) using a TLA-45 rotor in a Beckman Optima TLX centrifuge (Palo Alto, CA, U.S.A.) in order to separate MLV-bound from free toxin. Aliquots from each supernatant were loaded on an SDS/PAGE gel. The binding ability of each toxin to the membranes was estimated from comparison of the bands corresponding to incubations in the presence or the absence of MLVs.
Haemolytic activity
The haemolytic activity of the toxins against horse RBCs was determined by measuring the decrease in the turbidity of a cell suspension at 700 nm [24]. RBCs were washed five times with PBS [5 mM phosphate buffer (pH 8.0)/150 mM NaCl]. Washed RBCs were used to prepare a cell suspension with the desired initial attenuance (D700) value. Haemolysis was initiated after addition of the appropriate amount of toxin (15 nM final concentration) to 3 ml of RBC suspension stabilized at 25 °C. All measurements were performed in a UV–visible (UV–VIS) light spectrophotometer (Cary 3; Varian, Australia) with a thermostat-controlled cuvette holder and constant stirring.
Leakage of ANTS/DPX encapsulated in resealed ghost membranes
The vesicles formed by resealed RBC ghost membranes were obtained as described previously [25], but with slight modifications. Horse RBCs were washed five times with PBS. Aliquots (2 ml) of washed RBCs were added to 30 ml of ice-cold 5 mM phosphate buffer, pH 8.0, in order to achieve osmotic lysis. The resulting ghost membranes were washed four times with the same buffer by centrifugation at 16000 rev./min (24000 g) for 20 min at 4 °C in a Centrikon T-2080 centrifuge (Kontron Instruments, Basel, Switzerland). The membrane pellet obtained in the last washing step was resuspended in 2 ml of 10 mM Hepes buffer, pH 7.5, containing 50 mM NaCl, 25 mM ANTS and 90 mM DPX, and subjected to ten freeze–thaw cycles. In order to facilitate their resealing, the membranes were incubated overnight at 37 °C. Resealed ghosts were pelleted by centrifugation at 20000 rev./min (37000 g) for 20 min at 4 °C and resuspended in 2 ml of assay buffer, composed of 10 mM Hepes, pH 7.5/200 mM NaCl. Non-encapsulated dyes were separated further from the resealed ghosts by chromatography on a Sephadex G-75 column eluted with the assay buffer. Fractions corresponding to the resealed ghosts were collected, and the size of the vesicles was measured by quasi-elastic light scattering (QELS) in a Zetasizer-4 instrument (Malvern, Worcs., U.K.). In all the cases, the diameter of the resealed ghost vesicles ranged from 300–310 nm, with polydispersity values below 0.2.
In a typical leakage assay, the desired amount of toxin was added to the suspension containing resealed ghosts with encapsulated ANTS/DPX in 25 °C thermostat-controlled cuvettes with constant stirring. The changes in the fluorescence intensity were registered in an SLM-Aminco 8100 spectrofluorimeter (Spectronic Instruments, Rochester, NY, U.S.A.) with excitation and emission wavelengths of 350 and 510 nm respectively. Excitation and emission slits were set at 8 nm. An interference filter with a nominal cut-off value of 475 nm was placed in the emission light path to minimize the scattered-light contribution of the vesicles to the fluorescence signal. The percentage of leakage was calculated after the entire fluorescent probe was released by the addition of the non-ionic detergent Triton X-100 (0.1% final concentration, w/v), when 100% leakage was achieved. The detergent itself did not contribute towards or affect the fluorescence signal (results not shown).
ANS fluorescence measurements
The fluorescence quantum yield of ANS increases upon binding to hydrophobic regions exposed during the thermal denaturation of proteins [26]. The fluorescence intensity of ANS in the presence of Eqt-II and the different mutants was recorded in a PerkinElmer LS-50 spectrofluorimeter (Beaconsfield, Bucks., U.K.) equipped with a thermostat-controlled cell holder and a magnetic stirrer. Measurements were taken between 15 °C and 75 °C in 10 mM Hepes buffer, pH 7.5/200 mM NaCl. The heating rate was 1 °C/min. The actual temperature in the cuvette was measured with a thermocouple immersed in a second cuvette located in the same sample holder. The excitation wavelength was 370 nm, and emission was registered at 468 nm. Excitation and emission slits were set at 5 nm. Concentrations of ANS and Eqt-II were 6 μM and 15 μM respectively. The denaturation temperature was calculated as the mean value between the temperature at which fluorescence starts to increase and the temperature at which maximum fluorescence signal is reached.
Surface pressure measurements
Surface pressure measurements were performed with a Micro-Trough-S system from Kibron (Helsinki, Finland) at 25 °C with constant stirring. The aqueous subphase consisted of 1 ml of 10 mM Hepes, pH 7.5, containing 200 mM NaCl. An equimolar mixture of egg-PC and brain-SM, dissolved in chloroform/methanol (2:1, v/v), was gently spread over the subphase. The desired initial surface pressure was attained by changing the amount of lipid applied to the air–water interface. After 10 min (to allow for solvent evaporation) the protein was injected through a hole connected to the subphase. The final toxin concentration in the Langmuir trough was 1 μM. The increment in surface pressure with time was recorded until a stable signal was obtained.
Intrinsic fluorescence measurements
Changes in the intrinsic fluorescence of Eqt-II and the various mutants upon addition of LUVs (large unilamellar vesicles) made of SM/PC (1:1) were recorded in an SLM-Aminco 8100 spectrofluorimeter (Spectronic Instruments). LUVs were prepared by the extrusion method using polycarbonate filters with a pore size of 100 nm, as described previously [27]. The toxin concentration was 250 nM, and the L/T ratio was 600. Excitation wavelength was fixed at 295 nm in order to eliminate the contribution of the tyrosine residues and the emission spectra were recorded between 305 and 400 nm. Excitation and emission slits were set at 4 nm. The contribution of the light scattered by the vesicles to the fluorescence signal was minimized by using crossed polarizers (excitation polarizer was set at 90° and emission polarizer was set at 0°; [28]), and by subtraction of control spectra of the vesicles alone. All measurements were performed at 25 °C in a thermostat-controlled cuvette with constant stirring. The buffer used was 10 mM Hepes, pH 7.5/200 mM NaCl.
Fluorescence quenching measurements
We measured the quenching by acrylamide of the intrinsic fluorescence of Eqt-II in solution. The toxin concentration was 350 nM. Spectra were recorded in the absence or the presence of increasing acrylamide concentrations in an SLM-Aminco 8100 spectrofluorimeter (Spectronic Instruments). The excitation wavelength was fixed at 295 nm in order to eliminate the contribution of the tyrosine residues, and the emission spectra were recorded between 305 and 400 nm. Excitation and emission slits were set at 4 nm. The fraction of fluorophores accessible to the soluble quencher (fa) was calculated by using the Stern–Volmer equation modified for multiple emission centres [29,30]:
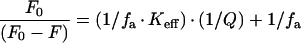 |
where F0 is the fluorescence intensity in the absence of acrylamide, F is the fluorescence intensity in the presence of acrylamide, fa is the fraction of accessible tryptophan residues, Keff is the effective quenching constant and Q is the acrylamide concentration.
We also measured the quenching of the intrinsic fluorescence upon addition of SM/brominated PC (1:1) SUVs, having bromine atoms covalently attached at different positions within the acyl chain. For preparing SUVs, ultrasounds were applied to an MLV suspension of the desired composition, followed by centrifugation at 14000 g for 10 min to eliminate the titanium particles released from the sonicator probe.
The toxin concentration used was 250 nM and the L/T ratio was 600. Fluorescence spectra were recorded under the same conditions described above in the presence of LUVs. The percentage of quenching (%Q) upon addition of brominated SUVs was calculated by applying the following equation:
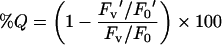 |
where F0 is the fluorescence intensity of the toxin in solution, Fv is the fluorescence intensity of the toxin upon addition of SM/PC SUVs without bromine, F0′ is the fluorescence intensity of the toxin in solution before the addition of the brominated vesicles, and Fv′ is the fluorescence intensity of the toxin upon addition of the brominated vesicles.
RESULTS AND DISCUSSION
Several strategies have been applied to determine the regions of PFTs that are involved in spanning the membrane in order to create transmembrane pores. One of the most employed strategies consists of introducing cysteines along the sequence, and, by labelling newly introduced thiol groups with a fluorescent probe, evaluating their localization after membrane interaction using membrane-embedded quenchers [16,22,31]. We have studied the localization of the N-terminal amphipathic α-helix of Eqt-II after interaction with the membrane. In the present study we have used tryptophan residues as ‘natural probes’ instead of cysteines, because residue labelling with fluorescent probes can sometimes introduce diverse disturbances in the toxin–membrane interaction [32]. Such an approach was used in topological studies of some other membrane-interacting protein toxins, such as the poreforming domain of bacterial toxin colicin E1 and the transmembrane domain of diphtheria toxin [33–35]. We have designed three mutants, I18W, V22W and A25W, each of them containing an additional tryptophan residue within the N-terminal amphipathic α-helix of Eqt-II (Figures 1A and 1B). I18, V22 and A25 were chosen in order to cover the whole length of the α-helix (Figure 1A). As these residues are located in the apolar side of the helix (Figure 1B), it is expected that they face the acyl chains of the phospholipids after helix insertion, thus exhibiting different fluorescent properties from the soluble state. The changes in the fluorescent properties associated with the newly introduced tryptophan, either in solution or in the presence of different lipid systems, may allow us to monitor the interaction of this region of the toxin with lipid membranes and its localization after interaction with the membrane.
Characterization of the mutants
Initial characterization of the mutants in solution was achieved by fluorescence spectroscopy, calculation of the fraction of tryptophans accessible to quenching by acrylamide (fa), and the determination of the thermal denaturation temperature (Tm). Fluorescence spectra of mutants in solution showed higher intensities (Figure 2, continuous lines, and Table 1), and emission maxima that were red-shifted 1–2 nm (Table 1) in comparison with native Eqt-II, indicating a solvent-exposed location of the newly introduced tryptophan. These results correlate well with the acrylamide-quenching results shown in Figure 3(A). By using the modified Stern–Volmer equation for multiple emission centres ([29,30]; see the Materials and methods section), we obtained fa values of 58%, 77%, 78% and 68% for native Eqt-II, I18W, V22W and A25W proteins respectively. Eqt-II contains five tryptophans in its sequence. Three of them, W149, W112 and W116, are exposed to the solvent, whereas W117 and W45 are oriented towards the hydrophobic core of the toxin [11,12]. This is in agreement with the fa of 58% that we obtained for the native Eqt-II. The incorporation of a new tryptophan residue in a solvent-accessible location results in increased fa values for the mutants.
Figure 2. Intrinsic fluorescence spectra.

Emission fluorescence spectra of Eqt-II, I18W, V22W and A25W in solution (continuous lines) and in the presence of SM/PC (1:1) LUVs (broken lines). The toxin concentration was 250 nM, and the L/T ratio was 600. Excitation wavelength was set at 295 nm and emission spectra were recorded from 305–400 nm. Excitation and emission slits were fixed at 4 nm. Buffer used was 10 mM Hepes, pH 7.5/200 mM NaCl. Measurements were carried out at 25 °C with constant stirring. Spectra shown are typical of three independent experiments. Averages of fluorescence parameters in the absence and presence of LUVs are shown in Table 1.
Table 1. Changes in the intrinsic fluorescence parameters of native Eqt-II and the I18W, V22W and A25W mutants in solution, and after addition of SM/PC (1:1) LUVs.
λmax, emission maximum wavelength in solution; λ′max, emission maximum wavelength in the presence of LUVs; −Δλmax, blue-shift of emission maximum wavelength after LUV addition; If333, fluorescence intensity at 333 nm of toxins in solution; If333nat, fluorescence intensity at 333 nm of native Eqt-II in solution; ΔIf, increase in fluorescence intensity upon LUV addition. Results are the means for three independent experiments±S.D. Means for ΔIf were compared using the Student's paired t test. *P<0.05.
| Toxin | λmax (nm) | λ′max (nm) | −Δλmax (nm) | If333/If333nat | ΔIf (%) |
|---|---|---|---|---|---|
| Eqt-II | 333 | 331 | 2 | 1±0 | 1.7±0.9 |
| I18W | 334 | 330 | 4 | 1.2±0.1 | 16.7±2.1* |
| V22W | 335 | 333 | 2 | 1.4±0.2 | 4.1±1.5 |
| A25W | 335 | 331 | 4 | 1.3±0.1 | 13.9±1.5* |
Figure 3. Characterization of the I18W, V22W and A25W mutants.

(A) Stern–Volmer plots for the quenching of wild-type Eqt-II (circles) and mutants I18W (squares), V22W (triangles) and A25W (diamonds) with acrylamide. Excitation wavelength was 295 nm. F0 is the fluorescence intensity in the absence of acrylamide and F is the fluorescence intensity in the presence of different concentrations of acrylamide. The toxin concentration was 350 nM. All measurements were carried out at 25 °C with constant stirring. The buffer used was 10 mM Hepes, pH 7.5/200 mM NaCl. (B) ANS fluorescence as a function of temperature in the presence of Eqt-II (continuous line), I18W (dashed line), V22W (dotted line) and A25W (dashed–dotted line). Toxin concentrations were 6 μM; the ANS concentration was 15 mM. Buffer used was 10 mM Hepes, pH 7.5/200 mM NaCl. The temperature was increased by 1 °C/min. ANS fluorescence was excited at 370 nm, and emission was registered at 468 nm. In both cases, duplicate experiments were performed, producing nearly identical values.
The stability of the mutants was tested with the ANS-binding assay. Eqt-II structure is composed of a tightly packed β-sandwich, which encloses a highly hydrophobic core (Figure 1) [11,12]. Amino acid substitutions within the N-terminal amphipathic α-helix are not expected to induce major changes in the thermal stability of the mutants. The denaturation temperature for each mutant was calculated by measuring the changes in the fluorescence of ANS in the presence of protein. From the thermograms shown in Figure 3(B), we calculated the Tm for each toxin: 58.5 °C, 53 °C, 58 °C and 56.5 °C for native Eqt-II, I18W, V22W and A25W proteins respectively. Tm values for V22W and A25W were almost identical with that of native Eqt-II. Only I18W showed a slightly lower Tm value. These results indicate that the mutations do not significantly alter the correct packing of the molecule and, therefore, its stability.
Interaction with lipid membranes
Next, we studied the interaction of mutants with RBCs, resealed ghost membranes and lipid monolayers composed of SM/PC (1:1).
The pore-forming activity of mutants I18W and A25W (monitored as RBC haemolysis or the release of ANTS/DPX encapsulated in resealed ghost membranes) was practically identical with that of native Eqt-II (Figures 4A and 4B). This result indicates that, in these mutants, the functional role of the N-terminal α-helix in the cytolytic mechanism is not being affected by the newly introduced tryptophan. In the case of V22W, the haemolytic and permeabilizing activities were markedly reduced (Figures 4A and 4B). The substitution of V22 for tryptophan hinders the formation of the pore. The reduced pore-forming ability of this mutant could be the result of: (i) reduced binding; (ii) inability of the N-terminal α-helix to insert into the membrane; or (iii) limited oligomerization ability.
Figure 4. Pore-forming activity of the I18W, V22W and A25W mutants.

(A) Attenuance (D700) decrease after addition of native Eqt-II, I18W, V22W and A25W to a 3 ml suspension of horse RBCs with an initial attenuance of 0.5 is shown. The upper arrow indicates the addition of each toxin (final concentration 15 nM). Experiments were performed at 25 °C with constant stirring in PBS buffer. (B) Leakage of ANTS/DPX from resealed RBC ghost membranes after addition of Eqt-II, I18W, V22W and A25W. The first arrow indicates the addition of each toxin (final concentration 0.67 μM). The second arrow indicates the addition of the detergent Triton X-100 (0.1% final concentration, w/v) in order to achieve 100% of the leakage. All measurements were carried out at 25 °C with constant stirring. The buffer used was 10 mM Hepes, pH 7.5/200 mM NaCl. In both cases, a representative result from two independent experiments that yielded similar results is shown.
The binding ability of the different mutants to MLVs composed of SM/PC (1:1) was then measured. The absence of protein bands in supernatants after centrifugation in the presence of MLVs demonstrates that all the toxins, including V22W, are able to bind to the membranes under these conditions (Figure 5). Therefore the reduced pore-forming activity of V22W must be due to either the inability of the N-terminal α-helix to insert into the membrane or limitations in the oligomerization process, or both, since insertion and oligomerization are most likely to be concerted processes.
Figure 5. Binding of Eqt-II and the I18W, V22W, A25W mutants to SM/PC (1:1) MLVs.

The toxins (concentration 1.67 μM) were incubated overnight at 25 °C in the absence or presence of SM/PC (1:1) MLVs (L/T ratio 600:1). The incubation buffer was 10 mM Hepes, pH 7.5/200 mM NaCl. The SDS/PAGE gel shows the bands corresponding to the supernatants obtained after centrifugation of the samples. Lanes 3, 5, 7 and 9 correspond to the toxins incubated in the presence of MLVs, whereas lanes 2, 4, 6 and 8 correspond to the toxins incubated in the absence of MLV. Lane 1 corresponds to the molecular-mass markers. The same result was obtained in a duplicate experiment.
We also investigated the interaction of each mutant with a lipid monolayer made of SM/PC (1:1). In Figure 6 we show the increase in surface pressure generated after injection of each toxin into the subphase of monolayers of SM/PC (1:1) at different initial pressures. From the intersection of the plots with the abscissa, we can calculate the value of the critical pressure, i.e. the initial pressure at which the toxin cannot insert into the monolayer. This parameter gives information about the avidity of each toxin for the monolayer. The critical pressures calculated from the plots shown in Figure 6 were 36.2, 37.1, 36.8 and 38.7 mN/m for native Eqt-II, I18W, V22W and A25W respectively. All mutants show slightly higher critical pressures than the native toxin. The presence of one additional tryptophan residue, whose affinity for interfaces has been well documented [36], favours the transfer of the N-terminal region to the lipid–water interface, even at slightly higher initial pressures. The differential behaviour of the V22W mutant is no longer detected in the monolayer system. This result suggests that the V22W mutant would be able to transfer its N-terminal α-helix with a parallel orientation with respect to the plane of the membrane (Figure 1C), a preferred orientation in this system due to the lack of half of the bilayer [37]. Thus its decreased rate of haemolysis and permeabilization activity would arise from the inability to insert the α-helix in the perpendicular orientation that would give rise to the oligomeric pores in bilayers (Figure 1C).
Figure 6. Changes in surface pressure after insertion of Eqt-II and the I18W, V22W and A25W mutants into lipid monolayers of SM/PC (1:1).

Increment of surface pressure after addition of Eqt-II (circles), I18W (squares), V22W (triangles) and A25W (diamonds) into the subphase as a function of the initial surface pressure of the monolayer is shown. Each point is the average of two independent experiments. The subphase buffer was 10 mM Hepes, pH 7.5/200 mM NaCl. The experiment was performed at 25 °C with constant stirring.
Fluorescence measurements
The five tryptophans present in the sequence of Eqt-II are responsible for its intrinsic fluorescence properties [38]. Upon interaction with membranes, a blue-shift in the maximum emission wavelength and an increase in the fluorescent intensity are observed, caused by the interaction of W112 and W116 with the lipid membrane [14,15,38,39]. These two tryptophans are implicated in the initial shallow binding of the toxin to the membranes [14]. In the case of I18W, V22W and A25W, the environment of the newly introduced tryptophan residue should alter their fluorescent properties, both in solution and in the presence of vesicles. These changes would allow us to conclude the localization of the N-terminal α-helix in the membrane-bound state of the toxin.
Fluorescence spectra of the mutants in solution and in the presence of SM/PC (1:1) LUVs are shown in Figure 2 and summarized in Table 1. The L/T ratio was kept at 600, a value at which almost all the toxin appears to be bound to the membrane [39]. Upon interaction with LUVs, native Eqt-II showed a 2 nm blue shift of the emission maximum and a 1.7% increase in fluorescence intensity due to the interaction of tryptophan residues W112 and W116 with the lipid membrane [14,15,38,39]. Addition of LUVs to mutants I18W and A25W resulted in a bigger blue shift and an increase in the intensity, which is significantly higher than that observed with the native toxin (Table 1). These results indicate that the additional tryptophan occupies a more hydrophobic environment after interaction with the LUVs, which is due to an insertion of the N-terminal α-helix into the bilayer. For the V22W mutant, the blue-shift and the increase in intensity are almost identical with those of the native Eqt-II (Table 1). It seems that in this mutant the α-helix does not proceed deeper into the bilayer. This is in agreement with the reduced haemolytic and permeabilizing activities observed in Figures 4(A) and 4(B). Apparently, the substitution of V22 with tryptophan stabilizes this mutant in a non-lytic state, a conformation in which the N-terminal α-helix lies parallel to the plane of the membrane (Figure 1C).
In order to gain more information on the localization of the N-terminal α-helix of Eqt-II in the membrane-bound state, we measured the quenching of the fluorescence after interaction with SUVs composed of SM and brominated PC (1:1). We used PCs which were brominated in positions 6/7, 9/10 and 11/12 of one of their two acyl chains. SUV were used in order to minimize the light-scattering contribution of the vesicles to the fluorescence. In addition, these results could be compared with a previous study [14], which confirmed a shallow interaction of W112 and W116 of Eqt-II with the lipid membrane using the same model system. Bromine is a short-range quencher, having a Förster distance (distance at which 50% of the quenching occurs) of 0.925 nm [40]. This means that tryptophan residues need to come into close proximity with the bromine in order to be quenched. Quenching efficiencies for each toxin are detailed in Table 2. Native Eqt-II shows low percentages of quenching, reaching a maximum of 6.3% with the bromine at the position 9/10. The ‘hook-like’ exposed orientation of W112 [11,12] and the big separation between polar heads of phospholipids in SUVs would favour the interaction of W112 with the bromine at this position, and give rise to the observed quenching in the case of the native Eqt-II. V22W shows quenching values close to those of the native toxin. This result indicates that the newly introduced tryptophan does not insert deep into the bilayer, an observation which is fully consistent with the changes observed in its intrinsic fluorescence upon LUV interaction (Table 1).
Table 2. Quenching by brominated SUVs.
Percentages of quenching for Eqt-II, I18W, V22W and A25W after addition of SM/brominated PC (1:1) SUVs. Positions of bromine atoms within the PC acyl chain are indicated in the Table. Results are the means for three independent experiments±S.D. Means were compared using the Student's paired t test.*P<0.05; †P≤0.01.
| Quenching (%) | |||
|---|---|---|---|
| Toxin | Br 6/7 | Br 9/10 | Br 11/12 |
| Eqt-II | 2.2±3.2 | 6.3±0.3 | 0.7±2.1 |
| I18W | 8±4.5 | 14.1±1.6† | 6.7±1.3† |
| V22W | 5.1±2.6 | 8.7±2.1 | 4.1±1.0 |
| A25W | 6.4±1.4 | 13.5±1.6* | 6.1±1.1* |
The N-terminal helix lies parallel to the membrane when interacting with lipid monolayers, a system in which toxins interact with only one hemilayer [14,16,37]. This means that V22W has no difficulties in transferring the N-terminal α-helix in an orientation parallel to the membrane (Figure 6). However, its haemolytic and permeabilizing activities are markedly reduced (Figure 4). The newly introduced tryptophan residue could be establishing interactions with the interface of the membrane that stabilize the α-helix in the parallel orientation, thus inhibiting its transition to a perpendicular orientation and the formation, together with other monomers, of functional pores. This is supported by the fact that the residue at position 22 is more conserved among homologues within the actinoporin family than are residues 18 and 25 [16], and therefore its substitution generates larger disturbances in the activity of the toxin. V22W allowed us to distinguish between two crucial steps in the mechanism of pore formation of the toxin. It represents an interesting variant of Eqt-II, which is stabilized in a state immediately preceding the final lytic state, and therefore it could be of great value in future studies on these last steps of the actinoporin pore-formation mechanism.
I18W and A25W exhibit higher quenching values at all positions. The maximum quenching (close to 14%) is observed when the bromine atoms are located at positions 9/10 (Table 2). Therefore the newly introduced tryptophan is transferred to positions near to the carbons 9/10 of acyl chains where its fluorescence is largely quenched, suggesting a deep interaction with the membrane of the N-terminal helix in the case of these mutants.
Concluding remarks
In recent studies many clues concerning the pore-forming mechanism of Eqt-II have been unveiled. The α-helix located at the N-terminal region has been postulated to be transferred to the membrane in two steps (Figure 1C) [14,16]. In the first one, (the non-lytic state), the amphipathic helix lies parallel to the plane of the membrane. At L/T ratios below 1000, a fraction of the toxin will oligomerize and proceed to a second (lytic) state, where the N-terminal amphipathic α-helix elongates and, together with helices from other monomers, adopts a perpendicular orientation and creates the pore. Characterization of the tryptophan mutants used in this study confirms these two states. The pore-forming ability of I18W and A25W remained intact, whereas the fluorescence changes resulting from the presence of an additional tryptophan residue within the N-terminal α-helix suggest a membrane-bound topology, where the α-helix adopts a perpendicular orientation across the membrane. In contrast, the pore-forming activity of V22W is markedly reduced. Its new tryptophan is not transferred to a more non-polar environment; nor does it seem to be quenched by membrane-embedded bromine, thus suggesting a membrane-bound topology where the α-helix lies parallel to the plane of the membrane in a non-lytic state, even at L/T ratios a long way below 1000. This mutant could be used in future studies on the actinoporin pore-formation mechanism in order to investigate the last steps of helix insertion, and their correlation with the oligomerization process.
Acknowledgments
This work was supported by grant 042.310-13552/2001 from the University of the Basque Country and BIO2003-BI09056 from the Basque Government. I.G.-A. was the recipient of a predoctoral fellowship from the Basque Government. A.B. was the recipient of a postdoctoral fellowship from the Basque Government. Slovenian authors acknowledge financial support from the Slovenian Ministry of Education, Science and Sport.
References
- 1.Gouaux E. Channel-forming toxins: tales of transformation. Curr. Opin. Struct. Biol. 1997;7:566–573. doi: 10.1016/s0959-440x(97)80123-6. [DOI] [PubMed] [Google Scholar]
- 2.Heuck A. P., Tweten R. K., Johnson A. E. Beta-barrel pore-forming toxins: intriguing dimorphic proteins. Biochemistry. 2001;40:9065–9073. doi: 10.1021/bi0155394. [DOI] [PubMed] [Google Scholar]
- 3.Lakey J. H., Slatin S. L. Pore-forming colicins and their relatives. Curr. Top. Microbiol. Immunol. 2001;257:131–161. doi: 10.1007/978-3-642-56508-3_7. [DOI] [PubMed] [Google Scholar]
- 4.Anderluh G., Maček P. Cytolytic peptide and protein toxins from sea anemones (Anthozoa: Actiniaria) Toxicon. 2002;40:111–124. doi: 10.1016/s0041-0101(01)00191-x. [DOI] [PubMed] [Google Scholar]
- 5.Giese C., Mebs D., Werding B. Resistance and vulnerability of crustaceans to cytolytic sea anemone toxins. Toxicon. 1996;34:955–958. doi: 10.1016/0041-0101(96)00051-7. [DOI] [PubMed] [Google Scholar]
- 6.Mebs D. Anemonefish symbiosis: vulnerability and resistance of fish to the toxin of the sea anemone. Toxicon. 1994;32:1059–1068. doi: 10.1016/0041-0101(94)90390-5. [DOI] [PubMed] [Google Scholar]
- 7.Maček P., Lebez D. Isolation and characterization of three lethal and hemolytic toxins from the sea anemone Actinia equina L. Toxicon. 1988;26:441–451. doi: 10.1016/0041-0101(88)90183-3. [DOI] [PubMed] [Google Scholar]
- 8.Teng C. M., Lee L. G., Lee C. Y., Ferlan I. Platelet aggregation induced by equinatoxin. Thromb. Res. 1988;52:401–411. doi: 10.1016/0049-3848(88)90024-2. [DOI] [PubMed] [Google Scholar]
- 9.Lafranconi W. M., Ferlan I., Russell F. E., Huxtable R. J. The action of equinatoxin, a peptide from the venom of the sea anemone, Actinia equina, on isolated lung. Toxicon. 1984;22:347–352. doi: 10.1016/0041-0101(84)90078-3. [DOI] [PubMed] [Google Scholar]
- 10.Bunc M., Drevenšek G., Budihna M., Šuput D. Effects of equinatoxin II from Actinia equina (L.) on isolated rat heart: the role of direct cardiotoxic effects in equinatoxin II lethality. Toxicon. 1999;37:109–123. doi: 10.1016/s0041-0101(98)00168-8. [DOI] [PubMed] [Google Scholar]
- 11.Athanasiadis A., Anderluh G., Maček P., Turk D. Crystal structure of the soluble form of equinatoxin II, a pore-forming toxin from the sea anemone Actinia equina. Structure (Cambridge) 2001;9:341–346. doi: 10.1016/s0969-2126(01)00592-5. [DOI] [PubMed] [Google Scholar]
- 12.Hinds M. G., Zhang W., Anderluh G., Hansen P. E., Norton R. S. Solution structure of the eukaryotic pore-forming cytolysin equinatoxin II: implications for pore formation. J. Mol. Biol. 2002;315:1219–1229. doi: 10.1006/jmbi.2001.5321. [DOI] [PubMed] [Google Scholar]
- 13.Mancheño J. M., Martín-Benito J., Martínez-Ripoll M., Gavilanes J. G., Hermoso J. A. Crystal and electron microscopy structures of sticholysin II actinoporin reveal insights into the mechanism of membrane pore formation. Structure (Cambridge) 2003;11:1319–1328. doi: 10.1016/j.str.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 14.Hong Q., Gutiérrez-Aguirre I., Barlič A., Malovrh P., Kristan K., Podlesek Z., Maček P., Turk D., González-Mañas J. M., Lakey J. H., Anderluh G. Two-step membrane binding by Equinatoxin II, a pore-forming toxin from the sea anemone, involves an exposed aromatic cluster and a flexible helix. J. Biol. Chem. 2002;277:41916–41924. doi: 10.1074/jbc.M204625200. [DOI] [PubMed] [Google Scholar]
- 15.Malovrh P., Barlič A., Podlesek Z., Maček P., Menestrina G., Anderluh G. Structure-function studies of tryptophan mutants of equinatoxin II, a sea anemone pore-forming protein. Biochem. J. 2000;346:223–232. [PMC free article] [PubMed] [Google Scholar]
- 16.Malovrh P., Viero G., Dalla Serra M., Podlesek Z., Lakey J. H., Maček P., Menestrina G., Anderluh G. A novel mechanism of pore formation: membrane penetration by the N-terminal amphipathic region of equinatoxin. J. Biol. Chem. 2003;278:22678–22685. doi: 10.1074/jbc.M300622200. [DOI] [PubMed] [Google Scholar]
- 17.Belmonte G., Pederzolli C., Maček P., Menestrina G. Pore formation by the sea anemone cytolysin equinatoxin II in red blood cells and model lipid membranes. J. Membr. Biol. 1993;131:11–22. doi: 10.1007/BF02258530. [DOI] [PubMed] [Google Scholar]
- 18.Tejuca M., Dalla Serra M., Ferreras M., Lanio M. E., Menestrina G. Mechanism of membrane permeabilization by sticholysin I, a cytolysin isolated from the venom of the sea anemone Stichodactyla helianthus. Biochemistry. 1996;35:14947–14957. doi: 10.1021/bi960787z. [DOI] [PubMed] [Google Scholar]
- 19.Valcarcel C. A., Dalla Serra M., Potrich C., Bernhart I., Tejuca M., Martinez D., Pazos F., Lanio M. E., Menestrina G. Effects of lipid composition on membrane permeabilization by sticholysin I and II, two cytolysins of the sea anemone Stichodactyla helianthus. Biophys. J. 2001;80:2761–2774. doi: 10.1016/S0006-3495(01)76244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderluh G., Dalla Serra M., Viero G., Guella G., Maček P., Menestrina G. Pore formation by equinatoxin II, a eukaryotic protein toxin, occurs by induction of nonlamellar lipid structures. J. Biol. Chem. 2003;278:45216–45223. doi: 10.1074/jbc.M305916200. [DOI] [PubMed] [Google Scholar]
- 21.Perkins S. J. Protein volumes and hydration effects. Eur. J. Biochem. 1986;157:169–180. doi: 10.1111/j.1432-1033.1986.tb09653.x. [DOI] [PubMed] [Google Scholar]
- 22.Anderluh G., Barlič A., Podlesek Z., Maček P., Pungerčar J., Gubenšek F., Zecchini M. L., Dalla Serra M., Menestrina G. Cysteine-scanning mutagenesis of an eukaryotic pore-forming toxin from sea anemone. Eur. J. Biochem. 1999;263:128–136. doi: 10.1046/j.1432-1327.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- 23.Anderluh G., Pungerčar J., Štrukelj B., Maček P., Gubenšek F. Cloning, sequencing, and expression of equinatoxin II. Biochem. Biophys. Res. Commun. 1996;220:437–442. doi: 10.1006/bbrc.1996.0391. [DOI] [PubMed] [Google Scholar]
- 24.Louw A., Visser L. Kinetics of erythrocyte lysis by snake cardiotoxins. Biochim. Biophys. Acta. 1977;498:143–153. doi: 10.1016/0304-4165(77)90095-2. [DOI] [PubMed] [Google Scholar]
- 25.Steck T. L., Kant J. A. Preparation of impermeable ghosts and inside-out vesicles from human erythrocyte membranes. Methods Enzymol. 1974;31:172–180. doi: 10.1016/0076-6879(74)31019-1. [DOI] [PubMed] [Google Scholar]
- 26.Stryer L. The interaction of a naphthalene sulfonate dye with apomyoglobin and apohemoglobin. A fluorescent probe for nonpolar sites. J. Mol. Biol. 1965;13:482–495. doi: 10.1016/s0022-2836(65)80111-5. [DOI] [PubMed] [Google Scholar]
- 27.Mayer L. D., Hope M. J., Cullis P. R. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta. 1986;858:161–168. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- 28.Ladokhin A. S., Jayasinghe S., White S. H. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal. Biochem. 2000;285:235–245. doi: 10.1006/abio.2000.4773. [DOI] [PubMed] [Google Scholar]
- 29.Lehrer S. S. Solute perturbation of protein fluorescence. Quenching of tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry. 1971;10:3254–3263. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 30.Eftink M. R., Ghiron C. A. Exposure of tryptophanyl residues in proteins. Quantitative determination by quenching studies. Biochemistry. 1976;15:672–682. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- 31.Shepard L. A., Heuck A. P., Hamman B. D., Rossjohn J., Parker M. W., Ryan K. R., Johnson A. E., Tweten R. K. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens Perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry. 1998;37:14563–14574. doi: 10.1021/bi981452f. [DOI] [PubMed] [Google Scholar]
- 32.Heuck A. P., Jonson A. E. Pore-forming protein structure analysis in membranes using multiple independent fluorescence techniques. Cell. Biochem. Biophys. 2002;36:89–101. doi: 10.1385/CBB:36:1:89. [DOI] [PubMed] [Google Scholar]
- 33.Merrill A. R., Palmer L. R., Szabo A. G. Acrylamide quenching of the intrinsic fluorescence of tryptophan residues genetically engineered into the soluble colicin E1 channel peptide. Structural characterization of the insertion-competent state. Biochemistry. 1993;32:6974–6981. doi: 10.1021/bi00078a023. [DOI] [PubMed] [Google Scholar]
- 34.Malenbaum S. E., Collier R. J., London E. Membrane topography of the T domain of diphtheria toxin probed with single tryptophan mutants. Biochemistry. 1998;37:17915–17922. doi: 10.1021/bi981230h. [DOI] [PubMed] [Google Scholar]
- 35.Tory M. C., Merrill A. R. Adventures in membrane protein topology. A study of the membrane-bound state of colicin E1. J. Biol. Chem. 1999;274:24539–24549. doi: 10.1074/jbc.274.35.24539. [DOI] [PubMed] [Google Scholar]
- 36.White S. H., Wimley W. C. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 37.Escrive C., Laguerre M. Molecular dynamics simulations of the insertion of two ideally amphipathic lytic peptides LK15 and LK9 in a 1,2-dimyristoyl-phosphatidylcholine monolayer. Biochim. Biophys. Acta. 2001;1513:63–74. doi: 10.1016/s0005-2736(01)00343-1. [DOI] [PubMed] [Google Scholar]
- 38.Maček P., Zecchini M., Pederzolli C., Dalla Serra M., Menestrina G. Intrinsic tryptophan fluorescence of equinatoxin II, a pore-forming polypeptide from the sea anemone Actinia equina L. monitors its interaction with lipid membranes. Eur. J. Biochem. 1995;234:329–335. doi: 10.1111/j.1432-1033.1995.329_c.x. [DOI] [PubMed] [Google Scholar]
- 39.Caaveiro J. M. M., Echabe I., Gutiérrez-Aguirre I., Nieva J. L., Arrondo J. L. R., González-Mañas J. M. Differential interaction of Equinatoxin II with model membranes in response to lipid composition. Biophys. J. 2001;80:1343–1353. doi: 10.1016/S0006-3495(01)76107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolen E. J., Holloway P. W. Quenching of tryptophan fluorescence by brominated phospholipid. Biochemistry. 1990;29:9638–9643. doi: 10.1021/bi00493a019. [DOI] [PubMed] [Google Scholar]