

Abstract
Flavonoids and other polyphenolic compounds have been shown to inhibit human topoisomerase IB (topo I) through both inhibition of relaxation activity and through stabilization of the cleavable complex (poisoning). Some flavonoids have also been shown to intercalate DNA, and an association of topoisomerase inhibition with intercalation has been noted. We surveyed 34 polyphenolic compounds, primarily flavonoid glycones and aglycones, for their ability to inhibit topo I and to intercalate DNA using an in vitro gel electrophoresis method. We show that the most potent topo I poisons are the flavones and flavonols, and that these generally, but not always, are found to be DNA intercalators. There was no clear correlation, however, of topo-I-poisoning activity with the degree of DNA unwinding. Surprisingly, both DNA intercalation and topo I poisoning were shown to occur with some flavone glycones, including the C-glycosylflavone orientin. Inhibition of relaxation activity by flavonoids was found to be difficult to quantify and was most likely to be due to non-specific inhibition through flavonoid aggregation. As part of a structure–activity analysis, we also investigated the acid–base chemistry of flavonoids and determined that many flavonoids show acid–base activity with a pKa in the physiological pH region. For this reason, subtle pH changes can have significant effects on solution activity of flavonoids and their concomitant biological activity. In addition, these effects may be complicated by pH-dependent aggregation and oxidative degradation. Finally, we develop a simple model for the intercalation of flavonoids into DNA and discuss possible consequences of intercalation and topoisomerase inhibition on a number of cellular processes.
Keywords: aggregation, flavonoid, cleavage complex stabilization, DNA intercalation, topoisomerase I inhibition, structure–activity analysis
Abbreviations: λmax, wavelength of maximum absorbance; DTT, dithiothreitol; topo I, topoisomerase IB; topo II, topoisomerase II
INTRODUCTION
Flavonoids are a diverse group of naturally occurring polyphenolic compounds with wide-ranging biological properties (e.g. antiviral, anti-inflammatory, mutagenic, antimutagenic, antiproliferative and other effects; for reviews see [1–3]). The biochemical and molecular mechanisms by which flavonoids act are poorly understood, however. The potent antioxidant properties of many of these compounds and their ability to chelate redox-active metal ions have frequently been shown, or inferred, to play a role in their mechanisms of action in the cell [4]. Other proposed mechanisms include the specific inhibition of certain enzymes or signal transduction pathways [5], activation or stimulation of enzymes [6], hormone antagonism or inhibition [7], and oxidative damage [8].
Certain flavonoids can inhibit the DNA-maintenance enzymes topoisomerase IB (topo I) and topoisomerase II (topo II) [9,10]. These enzymes regulate the supercoiling of chromosomal DNA, and play pivotal roles in chromosome replication, transcription, recombination, segregation, condensation and repair [11]. They facilitate the relaxation of supercoiled DNA, essentially through a mechanism involving the breakage of a phosphodiester bond of either one strand (topo I) or both strands (topo II) of the duplex DNA. The inhibition of topoisomerases may involve ‘conventional’ inhibition where the activity of the enzyme is slowed or arrested by, for example, binding of the inhibitor to the active site or alteration of the binding behaviour of the enzyme with its substrate. This type of inhibition is generally referred to as inhibition of catalytic (relaxation) activity.
However, for the topoisomerases another type of inhibition may occur in which only the later stages of the enzymic relaxation process are inhibited. The relaxation process for topo I appears to involve the following simplified steps: (i) binding of the enzyme at the DNA-reaction site, (ii) strand scission, followed by transesterification in which an enzymic tyrosine residue covalently attaches to the 3′ end of the broken strand, and (iii) relaxation (unwinding) of the DNA strand followed by re-ligation of the broken strand and release of the enzyme for either another cycle of relaxation or detachment from the DNA. The conversion of the strand-cleaved into the intact form is easily reversible, and can be stabilized by inhibiting compounds that slow or prevent the re-ligation and detachment of the enzyme. This type of inhibition, referred to as ‘cleavable complex stabilization’ or ‘poisoning’, can be observed in a cell-free system as DNA-strand nicking (topo I) or breakage (topo II), or in cells as the appearance of various types of chromosomal damage [12].
Topoisomerase poisons may have long-term deleterious effects on the genomic stability of non-tumorous cells by increasing the incidence of homologous and non-homologous recombination events [13,14]. Such events are error-prone, resulting in chromosomal changes that may play a role in many poorly understood cellular processes, such as aging and carcinogenesis [3,15]. The ability of some flavonoids to act as topoisomerase poisons has been used to explain the results of mutagenicity testing in which it was shown that many flavonoids test positive for clastogenicity in mammalian cell systems [3]. It has also been proposed that the maternal consumption of flavonoids may play a role in the development of some childhood leukaemias [16]. And yet, numerous epidemiological studies suggest that flavonoid consumption is linked to a decrease in the incidence of certain cancers and, therefore, flavonoids may act as chemopreventive agents [3,5,17]. Our understanding of the health effects of consuming flavonoids and naturally occurring topoisomerase-inhibiting compounds is clouded by a number of problems, including limited knowledge of the identity and characteristics of these compounds, and a poor understanding of and lack of agreement regarding appropriate biological end-points.
While much attention has been placed on the study of topo II inhibition by polyphenolic compounds, comparatively less attention has centred on topo I inhibitors. Topo I inhibition by flavonoids has been assayed in vitro by several groups, but the results have not always been in agreement [9,18]. In the present study, we surveyed a broad spectrum of polyphenolic compounds, many of which have apparently never been tested, for their ability to inhibit human topo I. Because the ability to intercalate into DNA is often associated with the ability of compounds to stabilize the cleavable complex, and because we feel some methods used to assess topo I inhibition failed to distinguish between inhibition, poisoning and intercalation, we also evaluated the relative intercalative binding of these compounds. Using an assay developed in our laboratory [19], we were able to quantify intercalation and topo I poisoning, and develop a general explanation for these properties of flavonoids based on a structure–activity analysis.
Historically, widely varying experimental conditions and buffer solutions have been used to study flavonoids in aqueous solution. We carefully studied the behaviour of flavonoids in aqueous solution, and examined the effect of solution conditions on solubility and flavonoid interactions with proteins and DNA. We found that flavonoids in dilute aqueous solution can be significantly affected by a number of solution variables, including pH, salinity, and solvent strength, and that these variables appear to primarily influence self-aggregation, precipitation and oxidative degradation. This behaviour has significant effects on cell-free experiments, such as those reported here, but may also significantly affect any experimental study in which dilute aqueous solutions of flavonoids are used. Furthermore, because subtle structural features affect the pKa of each compound in unique ways, there may be significant differences in assay activity between very similar compounds. Those differences, however, may primarily represent solution effects, such as altered solubility or accelerated degradation, making structure–activity analyses prone to misinterpretation if solution behaviour of each compound is not taken into account.
MATERIALS AND METHODS
Reagents and solutions
pGEM®-9Zf(−) DNA plasmid (2.9 kb; Promega, Madison, WI, U.S.A.) was purchased at a concentration of 1 mg/ml in 10 mM Tris buffer (pH 7.5) containing 1 mM EDTA, and was stored frozen until use, at which time it was diluted in reaction buffer [consisting of 50 mM Tris/HCl (pH 7.5), 20 mM KCl, 1 mM EDTA, 0.3 mg/ml BSA and 1 mM DTT (dithiothreitol)]. Human topo I (TopoGEN, Columbus, OH, U.S.A.) in solution at 2 units/μl (1 unit is the amount of enzyme needed to relax 0.25 μg of plasmid in 30 min at 37 °C) was used without further purification. Proteinase K derived from Tritirachium album was obtained from USB (Cleveland, OH, U.S.A.). DNase-free BSA was purchased from Amersham Biosciences (Piscataway, NJ, U.S.A.). Agarose (molecular biology grade), the lactone form of (S)-(+)-camptothecin {1H-pyrano[3′,4′,6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione,4-ethyl-4-hydroxy-,(4S)-(9CI)} (90%), ethidium bromide (95%), DTT, Bromophenol Blue, boric acid, Triton X-100 and SDS were purchased from Sigma Chemical Corp. (St. Louis, MO, U.S.A.). DMSO (HPLC grade) was purchased from Aldrich Chemical Co. (St. Louis, MO, U.S.A.).
The polyphenols luteolin, gossypetin (98%), 5,7-dihydroxyflavone, 3′,4′-dihydroxyflavone (97%), 5,7,3′,4′-tetramethoxyflavone (97%), 5,6,7,3′,4′,5′-hexamethoxyflavone (98%), apigenin (98%), naringenin, hesperitin, genistein, prunetin, 7,3′,4′-trihydroxyisoflavone, tamarixetin and rhamnetin were purchased from Indofine Chemical Co. (Hillsborough, NJ, U.S.A.). Luteolin-4′-O-glucoside, luteolin-7-O-glucoside and orientin were purchased from Extrasynthese (Genay, France). Quercetin dihydrate (98%), morin, daidzein (98%), resveratrol (99%), phloretin (99%), phloridzin dihydrate (99%), myricetin (99%), diosmin (96.6%), rutin (95%), kaempferol (93%), (+)-catechin (98%), (−)-epicatechin, piceatannol (99.4%), silibinin (100%), fisetin (99%), quercitrin (89%), (−)-epigallocatechin gallate (95%) and (−)-epicatechin gallate were obtained from Sigma Chemical Co.
Polyphenol solutions were prepared as stock solutions in DMSO and were either frozen at −20 °C or used immediately after dilution with purified water. Unless otherwise noted, topo-I-catalysed reactions were carried out in relaxation (reaction) buffer. Sample electrophoresis (solubilizing) buffer consisted of 2% (w/v) SDS, 14% (w/v) Ficoll 400 and 0.1% (w/v) Bromophenol Blue. Proteinase K was dissolved in a buffer solution consisting of 50 mM Tris/HCl (pH 7.5) and 1 mM CaCl2. Gel electrophoresis buffer (TBE) was prepared from 89 mM Tris, pH 8.3, 89 mM boric acid, 2 mM EDTA and 0.1% (w/v) SDS. Phosphate buffer solutions were prepared according to the method of Sorensen [20]. Basically, this buffer consisted of solutions of monopotassium phosphate and disodium phosphate mixed in proportions to give buffer solutions of specific pH. Modified Sorensen's phosphate buffer was prepared by molar substitution of monopotassium phosphate with monosodium phosphate. All samples assayed for topoisomerase activity and all unheated samples measured spectrophotometrically were equilibrated in the open air.
Spectrophotometry
Spectrophotometric analyses were carried out using a Hewlett Packard 8453 UV–visible spectrophotometer in the scanning mode using photodiode array detection. Scans were generally from 250 nm to 500 nm at room temperature (23–25 °C) or higher. Quartz flow cells of either 1 or 0.2 cm were used as appropriate, and all samples were read at room temperature, except for heated samples which were sampled and measured as quickly as possible to minimize cooling.
Combined DNA-unwinding and topo-I-poisoning assay
The assay for DNA unwinding and topo I poisoning has been described in detail elsewhere [19], but is described briefly here. Supercoiled pGEM DNA was diluted into relaxation buffer to a concentration of ∼4.4 ng/μl. Fully relaxed plasmid was prepared from an aliquot of this solution by the addition of the appropriate amount of topo I calculated to give full relaxation when incubated at 37 °C for 2 h. To 17 μl of either the relaxed or supercoiled plasmid dilution (75 ng of DNA in total) 2 μl of test compound solutions, control compound or blank solution was added. All reactions were carried out in DNase-free 0.5 ml microcentrifuge tubes. Immediately before use, test and control compounds were diluted in 33% (v/v) DMSO to maintain DMSO concentration at or below 3.3% of the final incubation volume. A stock solution of 8 mM camptothecin in DMSO was diluted, immediately before use, in 33% (v/v) DMSO. Samples were incubated for 20 min at room temperature, after which 1 μl of topo I solution (at 10 times the concentration necessary to give full plasmid relaxation) was added. This solution was then incubated further in the dark at 37 °C for 1 h with gentle shaking. The reaction was stopped by the addition of 2.5 μl of a 10% (w/v) SDS solution followed by 2.5 μl of proteinase K at 1 mg/ml. Protein digestion was carried out in total darkness for 1 h at 45 °C, after which 6.5 μl of electrophoresis loading buffer was added. The samples were analysed immediately by gel electrophoresis as described below or refrigerated for later analysis.
Inhibition of catalytic relaxation activity was determined using an additional dilution series of test compound containing supercoiled plasmid only (75 ng) and the amount of topo I added was 1–2 times the amount of enzyme necessary to fully relax the plasmid (instead of the 10-fold amount used above). From this point, the samples were treated as described above.
Samples were analysed by gel electrophoresis using 1% (w/v) agarose gels containing 0.1% (w/v) SDS. The presence of SDS during electrophoresis improves band resolution, allowing for more reliable measurement of topoisomer bands. Electrophoresis was carried out using TBE buffer in a horizontal electrophoresis apparatus (Bio-Rad Sub-cell GT) for, typically, 10 h at 1.25 V/cm. The gel was then stained in 1 μg/ml ethidium bromide/TBE solution for 1 h. Following staining, the gel was re-inserted in the electrophoresis apparatus and was electrophoresed again for 45–60 min at 3 V/cm in either TBE buffer or TBE buffer containing ethidium bromide at 1 μg/ml. This step ensures a clear separation of nicked, covalently closed relaxed and linearized plasmid bands. The gel was visualized under UV illumination using a ChemiImager 4000 Low-light Imaging System (Transilluminator Model TM-26; Alpha Innotech Corp., San Leandro, CA, U.S.A.) and photographed. Band fluorescence intensities (reported in arbitrary units) were measured by scanning the sample bands using AlphEase software (v. 3.3d). In no case was DNA detectable in gel sample wells following electrophoresis.
RESULTS
Explanation of topo-I-poisoning (cleavable complex stabilization) and DNA-intercalation assay
Using the electrophoresis assay described above, nicked plasmid (Figure 1A, uppermost band, labelled ‘Nik’) was separated from topoisomers that are relaxed to different degrees by topo I (Figure 1A, intermediate bands, labelled ‘Rn’). Increasingly supercoiled topoisomers extended downward to the fully supercoiled form (Figure 1A, lowermost band labelled ‘SC’). Linear plasmid (produced by strand breaks on opposite strands in close proximity to one another) migrated between the nicked and uppermost relaxed topoisomer, but was not detected in any of the experiments in the present study.
Figure 1. Intercalation and topo-I-induced cleavage by luteolin.

(A) Lanes 1–5, fully relaxed pGEM DNA+topo I (10×)+ethidium bromide at 0.1, 0.13, 0.15, 0.17 or 0.2 μg/ml respectively; lanes 6 and 7 (duplicates), fully relaxed pGEM DNA+camptothecin at 10 μM+topo I (10×); lane 8, fully relaxed pGEM DNA; lanes 9 and 10 (duplicates), fully relaxed pGEM DNA+topo I (10×); lane 11, supercoiled pGEM DNA; lane 12, supercoiled pGEM DNA+topo I (10×); lane 13, supercoiled pGEM DNA+luteolin (150 μM); lanes 14–18, fully relaxed pGEM DNA+topo I (10×)+luteolin at 10, 25, 75, 150 and 200 μM respectively. Sample treatments and gel electrophoresis conditions are as described in the Materials and methods section. Nik, nicked plasmid; Rn, relaxed plasmid forms. (B) Graphical representation of topo-I-induced cleavage by luteolin. The y-axis shows fluorescence band intensity of ethidium-bromide-stained upper band (single-strand nicked plasmid) band. Column numbers refer to gel lanes as shown in (A). Plasmid used was either supercoiled (SC) or topo I covalently closed relaxed (Rel). Gels run under identical conditions show identical trends; relative S.D. of nicked band intensity for replicate measurements of luteolin was 15%. Campto, camptothecin.
The effect of increasing concentrations of ethidium bromide (0.1–0.2 μg/ml) (Figure 1A, lanes 1–5) on the migration of the relaxed topoisomers produced by topo I provided a reference for the quantitative evaluation of intercalation by the test compounds (ordinarily only two concentrations of ethidium bromide are used). The test compound, in increasing concentration, was incubated with plasmid and then relaxed with topo I (Figure 1A, lanes 14–18). Extraction of the intercalated compound (after reaction termination with SDS or during electrophoresis) resulted in the re-supercoiling of the plasmid to a degree proportional to the amount of intercalated compound. Intercalation was quantified, for comparative purposes, by relating the degree of migration of the topoisomers to the migration of the ethidium bromide standards. By using an excess of enzyme, and including both relaxed and supercoiled plasmid samples in the same assay, errors in interpretation due to inhibition of catalytic relaxation activity were minimized [19].
Figure 1(A) also includes samples for the evaluation of cleavable complex stabilization. Lanes 6 and 7 are duplicate reference samples showing the effect of the potent topo I poison, camptothecin, on the generation of nicked plasmid in the presence of excess topoisomerase. The degree of cleavable complex stabilization induced by the test compound was determined by incubating relaxed or supercoiled plasmid DNA with the test compound and a 10× level of topoisomerase, trapping the covalently bound topoisomerase–DNA complex by the addition of SDS, digestion of the protein portion of the complex with proteinase K and, finally, separation of the plasmid forms by gel electrophoresis. The UV-induced fluorescence intensity of the ethidium-bromide-stained bands was measured to quantify the absolute amount of nicked form of plasmid in each lane. The degree of nicking increased with increasing concentrations of the test compound, luteolin (Figure 1A, lanes 14–18). For comparison, an arbitrarily chosen concentration of 100 μM test compound was used and then related to the amount of nicking induced by camptothecin at 10 μM (using the same sample materials and the same gel). In this way, a standardized measurement of topo I poisoning induced by the test compound could be determined. The measurement was corrected for background nicking produced by excess enzyme alone (Figure 1A, lanes 9 and 10); a control was also included for possible non-enzymic cleavage produced by a high concentration of the test compound independent of the enzyme (Figure 1A, lane 13).
Figure 1(B) shows a graphical representation of the nicked form of plasmid intensity from the experiment shown in Figure 1(A) (excluding ethidium bromide lanes 1–5). Sample treatments had little or no effect on the amount of nicked plasmid present (i.e. Figure 1A, lanes 8–13), unless high levels of topo I were present with either luteolin (Figure 1A, lanes 14–18) or camptothecin (Figure 1A, lanes 6 and 7). The generation of relaxed plasmid from the stock supercoiled form did not increase the level of nicking [compare sample 11 (supercoiled) with sample 8 (relaxed) in Figure 1]. In the presence of a 10× level of topo I, a modest increase in the degree of nicking was seen [compare samples 12 (supercoiled) and 9 and 10 (relaxed) with sample 11 (supercoiled) in Figure 1], presumably representing the nicked plasmid produced by enzymically active topo I trapped on the plasmid at the moment of SDS addition. The addition of 150 μM luteolin to supercoiled plasmid alone (Figure 1, sample 13) also induced a slight amount of nicking, possibly by auto-oxidation of luteolin (none of the flavonoids used in the present study produced significant nicking of DNA plasmid independent of topo I, probably because of the presence of DTT and DMSO during the incubations). Incubation of relaxed plasmid in the presence of topo I (10×) and 10 μM camptothecin produced extensive nicking of the plasmid (Figure 1, samples 6 and 7). When luteolin was included in the incubation with relaxed plasmid and topo I (10×), a semi-linear dose–response curve was obtained (Figure 1, samples 14–18).
Flavon(ol)s are the most active intercalators and topo I poisons
Using the above procedure, we surveyed a wide range of polyphenolic compounds for their ability to stabilize the topo I cleavable complex and to intercalate DNA (Table 1). Of the 34 compounds studied, the most active compounds with regard to both poisoning and intercalation are the flavones and flavonols. Note that poisoning values reflect a direct relationship to potency and intercalation values reflect an inverse relationship (higher values represent decreasing degree of intercalation). Although intercalation, on first examination, appears to be associated with poisoning, a plot of the degree of intercalation against the degree of poisoning for those compounds that show both properties shows a poor correlation (R2=0.213).
Table 1. Topo I poisoning and intercalation of selected polyphenolic compounds.
 |
Topo-I-poisoning compounds tested in the present study generally showed a dose–response curve similar to that shown in Figure 1(B) (columns 14–18). However, for a number of compounds, the dose–response curve collapsed at the higher concentrations, showing a sudden drop in both the degree of nicking and intercalation. In addition, some test compounds gave variable topo-I-poisoning results. To better understand the basis for these results and to gain an understanding of the underlying mechanism of topoisomerase poisoning, the following experiments were performed.
BSA stabilizes flavonoids in solution and enhances their activity
In Figure 2(A), the effects of BSA and NaCl concentration on cleavable complex stabilization and intercalation are examined. Lanes 1–4 show a concentration gradient of quercetin (0, 30, 125 and 300 μM respectively) assayed under standard conditions, except that BSA was excluded from the reaction buffer. There is a progression of increasingly supercoiled topoisomers as quercetin concentration increases, as demonstrated by the increase in migration distance of the centroid of the topoisomer distribution in each sample. This progression is proportional to the concentration of quercetin, and represents the rewinding of plasmid after the intercalator is extracted from the plasmid. In Figure 2(A), lanes 5–8 are equivalent to lanes 1–4 except that samples were incubated in reaction buffer supplemented with NaCl (125 mM). The distribution of topoisomers is similar to those in lanes 1–4, except that, in the sample containing 300 μM quercetin (lane 8), the apparent intercalation has decreased dramatically to approximately the level of the 30 μM sample. Lanes 9–12 show samples treated exactly as the samples in lanes 1–4 except that reaction buffer was supplemented with 0.6 mg/ml BSA. Here, the pattern of topoisomer migration is again altered, with the most significant difference being in lane 12, where the topoisomers appear to migrate as the fully or nearly fully supercoiled plasmid, indicating a high degree of intercalation in this sample. This cannot be explained by inhibition of topoisomerase relaxation activity, since the plasmid was in the fully relaxed form before incubation with either topo I or quercetin.
Figure 2. Effect of NaCl and BSA on quercetin intercalation and quercetin-induced topo I DNA cleavage.

(A) All lanes represent 100 ng samples of fully relaxed pGEM DNA incubated in standard reaction buffer without BSA (except where supplemented) with a 10× concentration of topo I. Samples were treated according to the standard assay conditions, except where otherwise noted below. Lanes 1–4 show samples incubated with an increasing concentration gradient of quercetin (0, 30, 125 and 310 μM respectively). Lanes 5–8 again represent a quercetin concentration gradient of 0, 30, 125 and 310 μM respectively, supplemented with NaCl to a concentration of 125 mM. Lanes 9–12 similarly show a quercetin concentration of 0–310 μM for samples which were supplemented with BSA to a concentration of 0.62 mg/ml. Lanes 13 and 14 are duplicate samples containing 125 μM quercetin to which NaCl was added (to give 410 mM) immediately after a 1 h incubation with topo I and before SDS addition. Lanes 15 and 16 are duplicate samples in which topo I was incubated with 125 μM quercetin for 2 min at room temperature before the addition of pGEM DNA. These samples were then treated according to the standard procedure. Nik, nicked plasmid; Rn, relaxed plasmid forms; SC, supercoiled plasmid. (B) Quantification of nicked form pGEM DNA in the gel of (A) Band intensity is quantified as described in the Materials and methods section and is expressed in arbitrary units (AU). Lane numbers correspond to those in (A), except that sample 13 is the average of lanes 13 and 14, and sample 15 is the average of lanes 15 and 16. Gels run under identical conditions show identical trends; relative S.D. of nicked band intensity for replicate measurements of quercetin was 27%.
When the degree of topo-I-dependent plasmid nicking is considered, a general increase in the degree of plasmid nicking is seen as the concentration of quercetin increases (Figures 2A and 2B, lanes 1–4). At the highest quercetin concentration, however, the level of nicking decreases. This pattern is fairly consistent for the topo-I-poisoning flavonoids examined in the present study: poisoning is proportional to concentration of flavonoid until the concentration exceeds approx. 100–200 μM, at which point the level of poisoning decreases (the degree of intercalation generally decreases as well, although this is not always clearly evident). When NaCl is increased by 125 mM (Figure 2B, lanes 5–8) the degree of nicking diminishes. This is expected, since it is known that the binding efficiency of topo I to DNA decreases with increasing salt concentration [21]. As discussed below, however, changes in the activity of quercetin in solution resulting from the presence of NaCl probably also contribute to this effect.
In contrast with NaCl, the addition of BSA to the samples (Figure 2B, lanes 9–12) significantly increases the degree of DNA nicking at all concentrations and eliminates the drop in poisoning seen at the 300 μM concentration. Such apparent enhancement of enzyme activity in the presence of BSA is often attributed to non-specific binding effects [22]. In a simplified explanation of this phenomenon, non-specific binding of enzyme or substrate is disrupted by competitive binding with the more abundant BSA molecules. Topoisomerase could, for example, bind non-specifically to the surface of the sample vial, to DNA, or to itself, resulting in fewer functionally active enzyme molecules. In this model, the addition of a relatively high amount of BSA would out-compete topo I for these non-specific binding sites, thus freeing the enzyme and increasing its activity, presumably by increasing its accessibility to substrate. To test this possibility we increased the normal 10× level of topoisomerase present in the sample 2–3-fold. This increase in enzyme level, however, had either no significant effect on the level of poisoning by quercetin or decreased it somewhat (results not shown). Although not conclusive, this experiment does not support inactivation of topo I by non-specific binding as the explanation for the BSA-enhancement effect seen here.
Alternatively, a BSA-enhancement effect may occur if BSA disrupts the inhibition of topo I that occurs through specific or non-specific binding of quercetin to the enzyme. For example, many small planar compounds tend to self-aggregate in aqueous solution, causing non-specific inhibition of soluble enzymes (apparently by trapping the enzyme within or on the surface of the resulting micelle-like aggregates), and BSA can disrupt this process to some degree [22]. A decrease in the level of flavonoid aggregation in such a scenario would decrease non-specific enzyme inhibition and also increase the concentration of the specifically active non-aggregated form of the compound. In the topoisomerase assay, the result would be an apparent increase in intercalation activity by increasing the concentration of active flavonoid (assuming aggregated flavonoid is unable to intercalate or disaggregate efficiently) and, possibly, by alleviating topo I inhibition. Poisoning would be increased in a similar manner, since stabilization of the cleavable complex requires functional topoisomerase and, presumably, non-aggregated flavonoid.
When topo I was pre-incubated with 125 μM quercetin with no plasmid present for 2 min before pGEM addition (Figure 2B, sample 15), the result was a diminishing of poisoning-induced nicking (compare with samples 1 and 3 in Figure 2B). This is most easily interpreted as evidence that, under these conditions (no DNA, plasmid or BSA present), quercetin can inhibit topo-I-relaxation activity. Whether quercetin acts in a specific manner to inhibit topo I or in a non-specific manner, however, is not clear from these experiments. Our attempts to measure inhibition of topo I relaxation by quercetin at lowered topoisomerase levels gave inconsistent results, and we conclude that the most likely explanation for the enhancement effect of BSA on quercetin intercalation and topo I poisoning is both stabilization of the free form of quercetin and indirect enhancement of topo I activity. However, further close analysis of our data and the results of spectrophotometric experiments presented below lead us to believe that the primary cause of the BSA-enhancement effect is a stabilization of quercetin in its free monomeric form by BSA.
The BSA-enhancing effect is not unique to quercetin. It can be seen that with 160 μM kaempferol, the level of topo-I-induced nicking more than doubles (above background levels) when 0.16 mg/ml BSA is added (Figure 3A). The degree of nicking then remains constant to 0.31 mg/ml BSA, but slowly decreases as BSA concentration is increased up to 1.24 mg/ml. This pattern is essentially repeated with respect to DNA intercalation: BSA at 0.16 mg/ml (Figure 3B, lane 2) dramatically increases intercalation relative to control levels (Figure 3B, lane 1), then remains fairly constant until BSA concentration exceeds 0.6 mg/ml (Figure 3B, lane 4), at which point it begins to decrease. It seems unlikely that, under the conditions used here (high levels of topoisomerase, pre-relaxed plasmid and a relatively long incubation time of 1 h), BSA would be capable of directly enhancing topo I activity sufficiently to increase intercalation to the degree seen here.
Figure 3. Effects of BSA concentration on kaempferol-induced cleavable complex stabilization.

The experiment was carried out essentially using the standard assay described in the Materials and methods section using 100 ng of fully relaxed pGEM plasmid, except that BSA was supplemented at the concentrations indicated. (A) Stippled bars, no topo I and no kaempferol; striped bars, no topo I and 160 μM kaempferol; closed bars, 10× topo I and no kaempferol; open bars, 10× topo I and 160 μM kaempferol. AU, arbitrary units. (B) Gel electrophoresis results from sample containing 160 μM kaempferol and 10× topo I. Lane 1, 0 mg/ml BSA; lane 2, 0.16 mg/ml BSA; lane 3, 0.3 mg/ml BSA; lane 4, 0.6 mg/ml BSA; lane 5, 1.2 mg/ml BSA. Gels run under identical conditions show identical trends; relative S.D. of nicked band intensity for replicate measurements of kaempferol was 11%. Nik, nicked plasmid; Rn, relaxed plasmid forms; SC, supercoiled plasmid.
Topo I poisoning by flavonoids is reversible by NaCl
Even though quercetin stabilizes the topo I–DNA cleavable complex, this stabilization is fairly labile and can be reversed by addition of high concentrations of salt, even after long-term incubation of the flavonoid with topo I and plasmid (Figure 2A, lanes 13 and 14; Figure 2B, sample 13). This effect has been reported by other investigators with camptothecin poisoning [21], and has been used as evidence to support a model of cleavable complex formation that involves an equilibrium between the stabilized complex, covalently bound topo-I–DNA complex, the non-covalently attached topo-I–DNA complex and the free forms of these three constituents. In this model, the addition of high concentrations of salt shifts the equilibrium far in the direction of the free forms, since high-salt concentrations appear to interfere with the binding of topo I to DNA. We found that nearly complete reversal of camptothecin (20 μM) and luteolin (200 μM) poisoning occurs in less than 1 min after addition of 350 mM NaCl (results not shown). This effect also occurs when NaCl is included in the relaxation buffer during the relaxation incubation step (Figure 4). At an NaCl concentration of 240 mM, the reversal of poisoning is nearly complete, affecting both intercalating flavonoid poisons, such as myricetin (Figure 4, samples 7–9), and non-intercalators, such as taxifolin (Figure 4, samples 10–12), as well as camptothecin (Figure 4, samples 4–6).
Figure 4. Effects of NaCl on cleavable complex stabilization by camptothecin, myricetin and taxifolin.

Topo I nicking was evaluated as described in the Materials and methods section, except that NaCl in the amounts of 40, 140 or 240 mM was included in the reaction buffer. Bars 1, 2 and 3, 75 ng of supercoiled pGEM plasmid with 10× topo I and 40, 140 or 240 mM NaCl respectively; bars 4, 5 and 6, 75 ng of supercoiled pGEM plasmid with 10× topo I, 10 μM camptothecin and 40, 140 or 240 mM NaCl respectively; bars 7, 8 and 9, 75 ng of supercoiled pGEM plasmid with 10× topo I, 100 μM myricetin and 40, 140 or 240 mM NaCl respectively; bars 10, 11 and 12, 75 ng of supercoiled pGEM plasmid with 10× topo I, 100 μM taxifolin and 40, 140 or 240 mM NaCl respectively. AU, arbitrary units.
Topo I poisoning and DNA intercalation by quercetin and luteolin is not the result of their artifactual degradation
Since some flavonoids are known to degrade in neutral aqueous solution, especially under aerobic conditions, the possibility that the above observed effects were the result of degradation products rather than the flavonoids themselves was investigated. Solutions of quercetin and luteolin (120 μM each) were prepared in standard reaction buffer and then pre-incubated at 37 °C. After 2 h at 37 °C, a portion of the sample was removed and incubated for 20 min with plasmid at room temperature. A 10× concentration of topo I was added, and the samples were treated as in the standard assay. After the 2 h pre-incubation, luteolin nicking was found to have increased by 1% (average of two experiments), while quercetin decreased by 3% relative to nicking induced by ‘fresh’ flavonoid. There was no detectable change in degree of intercalation for either compound, even after 3 h of pre-incubation. When DTT was excluded from the pre-incubation buffer (but supplemented in the final incubation buffer to ensure the standard 1 mM concentration during the topoisomerase incubation), a decrease was seen in the intercalation of quercetin after 2 h of pre-incubation. A slight decrease also occurred in the intercalation of luteolin. Under these conditions, quercetin-induced nicking decreased by 12.7% and luteolin-induced nicking decreased by 19.5%. In the case of quercetin and luteolin, then, it does not appear that artifactual degradation products produced during the experimental procedure can account for intercalation or poisoning, since both effects are stable or decrease with increasing time of incubation.
Solubility and stability of flavonoids in aqueous solution is highly dynamic and pH-dependent
We next sought to determine how various solution conditions affect the stability of the flavonoids themselves by using UV–visible spectrophotometry to monitor absorption changes (Figure 5). Over the pH range 3–9, naringenin exhibits a shift in the wavelength of maximum absorbance (λmax) from ∼290 nm to 325 nm with a clear isosbestic point, indicating that two distinct forms of this compound exist over this pH range (Figure 5a). This appears to represent the titration of the acidic hydroxy group at the C-7 position, indicating a pKa of slightly less than 7, which is in agreement with previous reports [23,24]. In very low-ionic-strength solution and 10% methanol (Figure 5a), the spectral data for naringenin describes an unambiguous titration curve. However, when naringenin was placed in isotonic Sorensen's phosphate buffer solutions (12 mM) covering the pH range 5–8, a cloudy solution developed in the lower pH range, and a precipitate slowly formed. The spectrophotometric result (Figure 5b) was a progressive loss of absorbance at 290 nm at the lower pHs, and, to a lesser extent, the loss of absorbance at 325 nm until the pH exceeded 7. This we interpreted as aggregation and eventual precipitation of the less soluble protonated form of naringenin in the pH region below the pKa. A slight loss of the 325 nm peak also occurs, in a time-dependent manner, as protonated naringenin is lost from solution, causing a progressive equilibrium shift toward the protonated form.
Figure 5. Effects of solution conditions on protonation state and solubility of naringenin.

(a) Titration of 370 μM naringenin in 10% methanol with pH adjusted by minimal addition of NaOH or HCl to produce solutions of pH 3, 4, 5, 6, 7, 8 and 9. (b) Titration of 340 μM naringenin in 10% methanol with pH adjusted to 5, 6, 7, 7.6 and 8 using Sorensen's phosphate buffer (12 mM). (c) Titration of 340 μM naringenin in 10% DMSO with pH adjusted to 5, 6, 7, 7.6, and 8 using Sorensen's phosphate buffer (12 mM). (d) Effect of NaCl: naringenin (370 μM) in pH 7.6 phosphate buffer (12 mM) and 10% methanol (curve 1) and with 4 M NaCl (curve 2). (e) Effect of methanol (MeOH) concentration: naringenin (370 μM) in pH 8 phosphate buffer (12 mM) and 10% methanol (curve 1) and the same conditions with methanol decreased to 1% (curve 2). (f) Effect of naringenin concentration on absorption spectrum. Shown are naringenin solutions at 18, 37, 74, 147, 220 and 370 μM in 1% methanol and 12 mM phosphate buffer at pH 8. AU, absorbance units.
Since the concentration of sodium and potassium varies with pH in this buffer system, the buffer was modified to contain only sodium salts of phosphate, and the titrations were repeated with no significant difference in the results (not shown). Apparently, phosphate buffer at 12 mM is sufficient to induce aggregation of naringenin in its fully protonated form under these conditions. Replacing methanol with an equal concentration of DMSO, however, restored the solubility of the protonated form (Figure 5c), demonstrating the influence of solvent type on the solubility of the non-ionic form of the flavonoid.
The equilibrium concentrations of the two flavonoid forms is also altered by solvent concentration and by the concentration of naringenin itself (Figures 5d–5f). In pH 7.6 phosphate buffer and 10% methanol, a solution of naringenin (370 μM) will be predominately deprotonated. Addition of NaCl causes a progressive shift to the protonated form, so that, at a concentration of 4 M NaCl, naringenin is predominantly in its protonated form (Figure 5d). Progressive lowering of the methanol concentration will also gradually shift the equilibrium in the direction of protonation, so that naringenin is almost fully protonated at a methanol concentration of slightly less than 1% (Figure 5e). Figure 5(f) shows the effect of concentration on the absorption spectrum of naringenin in 1% methanol at pH 8. At 18 μM, under these conditions, naringenin appears to be primarily deprotonated. As the concentration increases, however, the absorption ratio 290/325 increases, so that, at 370 μM, the proportion of protonated flavonoid has increased to at least four times the level of the deprotonated form.
Most other natural flavonoid compounds also have a C-7 hydroxy group due to constraints in biosynthesis and, provided this hydroxy group has not been substituted in some way, or affected by inductive or resonance effects of other A-ring substitutions, these compounds will display solution behaviour similar to that of naringenin. The flavonols, quercetin, rutin, fisetin, kaempferol and myricetin, and the flavones, luteolin and apigenin, examined in the present study, all showed pH-dependent titration curves in the pH region 6.5–8.5, although most are difficult to measure due to poor solubility in the lower pH region and rapid degradation in the higher pH regions. While similar in some respects, these compounds are nevertheless unique with regard to their detailed solution behaviour due to differences in solubility and acid–base characteristics.
Flavonoid stability in reaction buffer is a function of pKa and interactions with buffer constituents
In the topo I assay, the flavonoids were generally diluted in reaction buffer consisting of 50 mM Tris (pH 7.5), 20 mM KCl, 3.3% DMSO, 0.3 mg/ml BSA and 1 mM DTT. These solutions were incubated at room temperature for 20 min in the presence of pGEM DNA (generally, 3.75–6.25 μg/ml), topo I was added and the mixture was then incubated at 37 °C for 1 h before the reaction was stopped by the addition of SDS. Except during the 37 °C incubation, when the samples were sealed, all samples and buffer solutions were equilibrated in open air. By monitoring changes in spectral properties, we next studied changes in flavonoid stability using conditions designed to simulate the conditions of the topoisomerase assay. It can be seen that in a reaction buffer containing 0.1 mg/ml BSA, quercetin (100 μM, 37 °C) degrades quite rapidly (26% loss after 2 h) (Figure 6a), but when 1 mM DTT is included in the buffer (Figure 6b), degradation is slowed considerably (5% loss after 2 h). Assuming that DTT is acting as a reductant, we conclude that degradation of quercetin under these conditions is primarily oxidative. In contrast, 100 μM luteolin is quite stable under assay conditions, even when BSA or DTT are not present (Figure 6c), but lowering the pH from 7.5 to 7.0 dramatically destabilizes the solution (Figure 6d). Apigenin (Figures 6e and 6f) is also quite unstable in assay buffer lacking BSA, but is significantly stabilized by 5 mg/ml BSA. The stability of morin was not affected by either BSA, DTT or pH changes of ±0.5 units from pH 7.5 under the conditions of the topo I assay (results not shown). This, we believe, is due to its very low pKa of 3.5 (fully ionized at pH 7.5) and inherent resistance to oxidation (lack of a catechol-containing B-ring).
Figure 6. Stability of selected flavon(ol)s under conditions similar to the topoisomerase assay and the effect of some solution modifications.

Stock solutions of flavonoids were dissolved in DMSO and diluted into reaction buffer to give a concentration of 100 μM and 3.3% DMSO. Reaction buffer consisted of 50 mM Tris buffer at pH 7.5, 20 mM KCl and 1 mM EDTA. (a and b) Effect of DTT on quercetin. (a) Quercetin at 100 μM dissolved as above in reaction buffer supplemented with 0.1 mg/ml BSA. UV–visible absorbance spectra were obtained after the following treatments: 1, immediately after addition of quercetin to reaction buffer (uppermost curve at 380 nm); 2, after 2 h at 37 °C; 3, after 4 h at 37 °C; and 4, after 6 h at 37 °C. Each successive treatment results in a progressive decrease in absorbance at 380 nm. (b) Sample treated as in (a) supplemented with 1 mM DTT. (c and d) Effect of pH on the stability of luteolin. (c) Luteolin (100 μM) was dissolved in topoisomerase reaction buffer (pH 7.5) containing no BSA or DTT, or (d) in reaction buffer adjusted to pH 7.0 again containing no BSA or DTT. Three absorbance readings were taken for each solution: 1, immediately after dissolving; 2, after 20 min at room temperature; and 3, after an additional 60 min at 37 °C. (e and f) Effect of BSA on apigenin stability. (e) Apigenin (100 μM) in reaction buffer containing no BSA immediately after dissolving (1), 20 min at room temperature (2), an additional 60 min at 37 °C (3), an additional 3 h at room temperature (4), and an additional 12 h at room temperature (5). (f) Same as (e) with reaction buffer supplemented with 5 mg/ml BSA. AU, absorbance units.
The effects of BSA and DTT on the solution stability of quercetin and luteolin were examined in more detail by monitoring the change in the absorbance (λmax) for each compound under varying solution conditions (Figure 7). As evidenced by the rapid loss in absorbance, quercetin was least stable at pH 7 and was stabilized somewhat by BSA or BSA in combination with DTT, but not by DTT alone (Figure 7a). At pH 7.5, quercetin was relatively stable in the presence of BSA, and the presence of DTT alone contributed to stabilization (Figure 7b). At pH 8, DTT alone was quite effective at stabilizing quercetin, but BSA no longer had a significant effect (Figure 7c). For luteolin (Figures 7d–7f), solution stability was poorest in the lower pH range, and this was mitigated by BSA or BSA plus DTT, but not by DTT alone. At higher pH, luteolin is relatively stable even after 20 h in solution, and neither BSA nor DTT affects solution stability.
Figure 7. Effects of BSA and DTT on quercetin and luteolin in solution at different pH.

(a–c) Quercetin (100 μM) in topoisomerase reaction buffer (50 mM Tris, 20 mM KCl, 1 mM EDTA and 3.3% DMSO) at pH 7.0 (a), 7.5 (b) and 8.0 (c). Samples were incubated with 0.3 mg/ml BSA and 1 mM DTT (■), 1 mM DTT only (◆), 0.3 mg/ml BSA only (×), or no amendment (▲) for the times indicated at 24 °C. (d–f) Repeat of (a–c) with luteolin.
DISCUSSION
Solution behaviour of polyphenols
Using naringenin as an example (pKa≈7; Figure 5), we propose the following model to describe the dynamic and unstable nature of flavonoids (and presumably other polyphenols) in aqueous, aerobic solution:
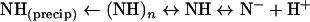 |
The deprotonated anionic form of naringenin (N−) absorbs at 325 nm, and, owing to its charged state, is highly soluble in water and is not prone to aggregation. The fully protonated form of naringenin (NH) absorbing at 290 nm is much less soluble and tends to aggregate [(NH)n] in units of n. This aggregated form remains in solution and appears to absorb at or near the absorbance of the protonated, unaggregated (monomeric) form. In low-ionic-strength solution at a pH near the pKa, naringenin in the predominately deprotonated form can be shifted to the apparent protonated form (absorbance at 290 nm) by increasing the ionic strength (and polarity of the solution), since this will favour the formation of aggregates, causing a shift in the equilibrium to the left in the above equation (see Figure 5d). Solvents such as methanol or DMSO will have the opposite effect, since increasing solvent concentration will lower solution polarity, tending to make aggregation less favourable and shifting the above equilibrium to the right (Figure 5e). Increasing concentration of naringenin will also tend to increase the potential for aggregation (when solvent strength is low), resulting in a shift in the equilibrium to the left (Figure 5f). Under certain conditions, such as high ionic strength, low solvent strength, high concentrations of solute, low pH, or an appropriate combination of these, the soluble aggregate will tend to precipitate out of solution, resulting in an extreme equilibrium shift to the left.
Most of the flavonoids examined in the present study were also titratable in the pH range 6.5–8.5, and show solution effects similar to those of naringenin. In addition, some flavonoids, particularly the flavonols, appear to be susceptible to degradation in aqueous solution, presumably by auto-oxidation under aerobic conditions. When the stability of the flavonol, quercetin, and the flavone, luteolin, are compared at several different pHs (Figure 7), it can be seen that pH affects the rate of loss of both quercetin and luteolin from solution. As the pH approaches 7, the rate of loss of quercetin (pKa≈7) from solution increases, but is lowered by the presence of BSA (Figure 7a). At higher pH, quercetin is still lost from solution, but only DTT, and not BSA, can slow this loss. Again, we interpret this as showing that, at lower pH, the loss of quercetin in solution is primarily due to aggregation/precipitation and that this process is counteracted by BSA. As the pH increases, quercetin becomes increasingly ionized, and loss appears to occur primarily through oxidation, since the reductant DTT can prevent the apparent loss, but BSA has no effect. Luteolin (pKa≈7.5) loss from solution also increases as the pH drops and aggregation increases, unless BSA is present. At higher pH, however, luteolin is quite stable in the absence of either DTT or BSA, indicating minimal oxidation under these conditions.
The basis for this BSA effect is not known. BSA may act by binding flavonoids and preventing aggregation and/or precipitation, but, if this is the case, the binding mechanism is unclear. The literature indicates (somewhat unconvincingly) that BSA binds the anionic form of flavonoids only [25], and, if this is true, BSA may affect the apparent stability of flavonoids by binding the anionic form, shifting the acid–base equilibrium towards deprotonation, thereby decreasing the tendency to aggregate. Alternatively, flavonoids may simply bind BSA weakly, creating an equilibrium pool of both bound and free forms of quercetin, thus lowering the concentration of free flavonoids and slowing aggregation. BSA may also act as an antioxidant in this system, but, if so, the effect is minor, since BSA has almost no effect on quercetin at pH 8, where oxidation is maximal (Figure 7c). BSA at concentrations of 0.8 and 5 mg/ml increased the pH of topoisomerase reaction buffer by 0.03 and 0.2 units respectively, and these changes might have some effect on flavonoid stability, but only for flavonoids with a pKa very close to pH 7.5.
The stability of quercetin in dilute aqueous solution was also studied by Mocek and Richardson [26] who noticed phenomena similar to those reported here. Using 50% ethanol solutions and acetate or phosphate buffers covering the pH range 4–7, they suggested that their observations could be explained by ionization at the 3 position hydroxy group [26]. This would allow oxygen to react at the 2′ position, leading to the formation of a quinone which polymerizes and eventually precipitates. This mechanism explains the increased susceptibility of quercetin to oxidation in the higher pH range, but fails to provide an explanation for precipitation in the low pH range. Also, if the titration curve of quercetin over this pH range is due to ionization of the 3 position hydroxy group, it would follow that the 3-OH-substituted rutinoside of quercetin, rutin, would show no titration behaviour over this pH range, yet, acid–base titration of rutin produces a curve quite similar to that of quercetin (results not shown). In addition, the titration of the 7-methoxy derivative of quercetin (rhamnetin) shows no pH-induced absorbance changes over the pH range 5–9 (results not shown). We propose that the behaviour of quercetin observed in the present study is best explained by our above proposed model, where addition of BSA primarily inhibits aggregation/precipitation, and DTT primarily inhibits oxidation.
These results indicate that the stability of flavonoids in solution is affected by solution conditions in ways that are unique to the individual compounds. Careful attention to flavonoid solution behaviour is therefore necessary to interpret the results of in vitro and in vivo studies properly. The results of the topoisomerase assay, discussed below, were evaluated with these considerations in mind.
Relationship between intercalation activity and topo I inhibition/poisoning
The ability of low-molecular-mass compounds to stabilize the topo-I–DNA cleavable complex is often, but not always, associated with the ability of these compounds to intercalate DNA, but intercalating compounds are frequently incapable of inhibiting the topoisomerases. Camptothecin, for example, one of the most potent known topo I poisons, has never been demonstrated to intercalate DNA, while such strong intercalative binders as ethidium bromide and chloroquine are completely ineffective at either poisoning or inhibiting catalytic activity [27]. Our results indicate that, for the flavonoids examined in the present study, DNA intercalation is not necessary, but may be sufficient for stabilization of the topo-I–DNA cleavable complex. In the present study, all of the flavonoids shown to be capable of intercalative binding to DNA were also efficient poisons of topo I (Table 1). It is clear that some non-intercalative flavonoids are also capable of poisoning topo I, however, and there is a very poor correlation (R2=0.213) between intercalation, as measured by the degree of DNA unwinding, and poisoning potency for the compounds surveyed in the present study. A lack of correlation has also been reported for a series of intercalating indolocarbazole compounds which are also capable of topo I poisoning [27].
Structure–activity relationship: intercalation and poisoning
Only flavones and flavonols which have a double bond between the 2 and 3 carbons of the C-ring are capable of intercalative binding with DNA (Table 1). This double bond ensures a sufficiently planar structure to allow placement between adjacent bases of the DNA duplex [28]. The flavanonol analogue of quercetin, taxifolin, lacks this double bond and loses all ability to intercalate in our assay. The absence of intercalating ability by morin, even though it has the necessary planar structure, is best explained by its unusually low pKa of 3.5, making it anionic at neutral pH and unlikely to intercalate DNA or bind to the topo-I–DNA complex.
A closer analysis of the flavone/flavonol compounds also shows a fairly consistent requirement for at least one free hydroxy group on the B-ring at the 4′ position for intercalation to occur. Chrysin, for example, has a B-ring which is completely free of hydroxy groups and is unable to intercalate or poison topo I. This suggests stabilization of binding by hydrogen bonding between flavonoid and DNA; the loss of intercalative ability by tamarixetin, in which the 4′ hydroxy group is replaced by a methoxy group, supports this model. The fact that luteolin-4′-O-glucoside is still capable of weak intercalation can be rationalized if the necessary stabilization is provided by hydrogen bonding of the conjugated glucose to the bound DNA.
In this survey, the simplest flavonoid structure still capable of intercalation and poisoning is 3′,4′-dihydroxyflavone. This would indicate that the 3′ hydroxy group is important in some manner for stabilizing intercalative binding (or enabling sufficient solubility in aqueous solution). Although the flavonol analogue of apigenin, kaempferol, quite effectively intercalates despite the absence of the 3′ hydroxy group, it is conceivable that the 3 position hydroxy group of the flavonols is sufficient to stabilize intercalation, at least enough to compensate for loss of the 3′ hydroxy group. However, as this model would predict, the ability to intercalate is enhanced further when the 3′ hydroxy group is added to the kaempferol structure, forming quercetin, although poisoning ability changes little.
The results indicate that hydroxy groups at the 5 and 7 positions play a minor role in stabilizing the intercalation of flavonoids, or possibly a slightly destabilizing role. Apigenin, which restores the 5 and 7 position hydroxy groups, but loses the 3′ hydroxy group (compared with 3′,4′-dihydroxyflavone), intercalates very poorly (not measurable by this assay), but is a reasonably potent topo I poison. Fisetin, which lacks the 5 position hydroxy group, is a potent poison and intercalator, again demonstrating the relative unimportance of the 5 position hydroxy group.
The present paper reports, we believe for the first time, evidence that the C-8 glycosylated flavone, orientin, is capable of intercalating DNA and acting as a topo I poison. Of the 34 compounds that we tested, orientin was the most potent topo I poison and a moderately effective intercalator. The 7-O-glucoside of luteolin also demonstrated activity, being the most effective DNA-unwinding compound and a moderately potent poison of topo I. These findings were unexpected, since previous reports regarding glycosylated flavon(ol)s indicated that conjugation with various saccharides resulted in loss of topo I and II poisoning and intercalation activity [1,16,29]. Our findings may help explain the reported clastogenicity of glycosylated flavones which are widely distributed in foods [30].
The essentially equivalent behaviour of luteolin and its flavonol analogue quercetin with regard to DNA unwinding and topo I poisoning indicates that the 3 position hydroxy group plays a relatively minor role in either process. Therefore the loss of intercalative capacity by flavonols which have been glycosylated at the 3 position hydroxy group (e.g. rutin and quercitrin) suggests that steric constraints on intercalation play a more important role than isomerization, ionization or hydrogen bonding at the 3 position hydroxy group. If intercalation of the active compounds involves entry of the fused A- and C-rings along the edge defined by the 4 and 5 position carbons, a bulky moiety attached at the 3, 4 or 5 carbon would significantly hinder this process. This provides an explanation for the inability of genistein to intercalate despite its planar structure, and explains how luteolin 7-O-glucoside and orientin are still able to intercalate despite the attachment of bulky glucose molecules. This also raises the possibility that intercalation of flavon(ol)s is only partial, in that the 7 and 8 position carbons may protrude from the stacked DNA bases even when the intercalating compound is fully bound. In this way, bulky moieties attached at the 7 and 8, and possibly the 6, position carbons do not interfere with intercalation.
The potent poisoning behaviour of luteolin 7-O-glucoside and orientin also suggests that chemical groups attached to the 7 or 8 position of the A-ring may enhance the poisoning effect. Since glucose itself would not be expected to be chemically reactive under these conditions, this effect is again best explained by reduced topo I efficiency due to steric interference. It has been pointed out in other studies that the potency of non-flavonoid-intercalating topo I poisons is enhanced by the presence of functional groups on the poisoning compound which lie in the minor groove and presumably hinder the functioning of the enzyme [31]. Thus the above model of flavonoid–DNA binding may explain why sugars attached at the 7 and 8 positions enhance topo I poisoning.
Other models have also been described. For example, it has been proposed that flavonoids containing catechol or pyrogallol B-rings convert into quinone/semiquinone forms, and it is these forms which are responsible for topo II poisoning [28]. Although it is possible that such a mechanism is specific for topo II, the ability of kaempferol and apigenin, two non-catechol-containing flavonoids, to poison topo I in the present study would indicate that quinone/semiquinone formation cannot adequately explain topo I poisoning by flavonoids. There also does not appear to be any clear correlation of topo-I-poisoning activity in the present study with the antioxidant or pro-oxidant properties of the poisoning flavonoids [4,32].
It is important to note that for the flavonoids studied, the ability to intercalate is very weak. For example, the molar ratio of luteolin/ethidium bromide at which luteolin induces DNA unwinding equivalent to ethidium-bromide-induced unwinding is approx. 150:1. Flavonoids, being weakly electrophilic planar compounds with weak binding properties, are less likely to be mutagenic or genotoxic than intercalators with cationic substituents or adduct-forming reactive electrophilic substituents [33].
It is equally important that the stabilization of the cleavable complex, even for the potent poison camptothecin, has been shown to be reversible. A model for camptothecin poisoning has been proposed in which the ‘poisoned’ cleavable complex is in equilibrium with the various intermediate forms of the complex [21]. We have shown in the present paper that the cleavable complex formed by both intercalating and non-intercalating flavonoids is reversible with high NaCl concentrations in a manner similar to that of camptothecin, and that this reversibility occurs relatively quickly. It is therefore likely that the stabilized complex formed in vivo by flavonoids is transient and would need to be transformed into a more permanent lesion before serious damage or genotoxicity could occur.
Intercalating compounds may be topoisomerase poisons or they may also inhibit the catalytic activity of the enzyme, in theory, precluding the possibility of poisoning the enzyme. We have attempted to control for inhibition of catalytic activity in our assay and to quantify it where it can be shown to occur [19]. Low levels of catalytic inhibition were detected for some flavonoids in the present study, but, with the exception of luteolin and morin, we were unable to quantify catalytic inhibition due to irreproducible results. We believe the source of this poor reproducibility lies in the nature of the inhibition itself, which is probably non-specific in nature and due primarily to aggregation of the test compounds. However, other factors, such as the dilute enzymes solutions needed for this measurement, may have also contributed to poor reproducibility.
It should be noted that there is significant overlap in the flavonoids shown in the present study to be topo I poisons and those known to be topo II poisons [10,16,18,34]. The promiscuous nature of flavonoid topoisomerase poisons has implications for in vivo experiments in which flavonoid-induced genotoxic endpoints are attributed to topo II poisoning alone, and raises the possibility that flavonoids may be active against the topoisomerase IA family of enzymes, which have recently been shown to be important for efficient, error-free homologous recombination [35].
Implications for in vivo effects of flavonoids
Numerous reports have shown that various flavonoids are clastogenic in mammalian cell culture [28,30,36] and some have attributed this to damage induced by topoisomerase cleavable complex stabilization [28]. Yet flavonoids have never been conclusively shown to be carcinogenic. In fact, some epidemiological studies of flavonoid consumption have shown correlations of lower cancer incidence with increased flavonoid consumption [5,17] and flavonoids have inhibited mutagenesis and carcinogenesis in chemically induced and transgenic models of these processes [37–39]. There are also many reports of flavonoids acting as antiproliferative agents in a number of different cell systems [40,41].
Evidence exists throughout the literature that flavonoids and other genotoxic polyphenolic compounds can show a biphasic dose–response curve, with negative effects at high concentrations and positive effects at low concentrations [1,6,46]. This type of response, termed hormesis, has been noted for other carcinogens [42]. If such an effect occurs with the topoisomerase poisons it could explain their apparently contradictory biological effects. In cell-culture experiments, where cells are typically exposed to abnormally high levels of flavonoids for relatively short periods of time, deleterious effects on genomic stability and cell viability would be observed. However, at the whole-organism level, where exposure is through diet, intracellular concentrations of flavonoids are generally maintained at very low levels through a number of mechanisms [43]. At these low levels, flavonoid effects on long-term genomic stability may turn out to be counter to those seen in most cell-culture experiments.
If such a low-level positive effect is real for the flavonoids, we can only speculate on its mechanism. Cells are reported to be exquisitely sensitive to the types of damage likely to be caused by topoisomerase poisons: blocked replication forks, stalled transcription complexes and double-strand breaks [44]. Thus it is likely that, even at very low levels, poisoning flavonoids could elicit a DNA-damage cell response. The DNA-damage detection and repair systems in cells are complicated and are still poorly understood, but it is possible that induction of repair systems at low levels of genome damage might have the effect of promoting more general, stabilizing effects on the genome. It has recently been proposed that early detection of DNA double-strand breaks may actually be mediated through changes in supercoiling topology caused by breaks [45]. Theoretically, poisoned topo I could mimic this effect, since it can act as a swivel point for release of supercoiling tension without causing complete breakage of the chromosome. It may be that the ease of reversibility of the cleavable complex produced by flavonoids, the relative ease of repair of nicked sites, or the S-phase-specificity of topo I poison genotoxic effects play important roles. But topo I is also known to be involved directly in DNA repair [11], and has been shown to bind the tumour suppressor protein p53 and the Werner syndrome helicase WRN, both known to be crucial for different aspects of DNA repair [46,47]. Clearly, much more needs to be learned about the role of topoisomerases in genome maintenance and repair before we can understand the effects of low levels of flavonoids on these systems.
The behaviour of flavonoids in solution as characterized in the present study has a number of in vivo implications. Flavonoids with a pKa near physiological pH will display complex solution behaviour, since there will be an equilibrium pool of both the protonated and deprotonated forms of the compound. Selective binding of both or either form to proteins, or selective absorption of the protonated form by lipids/membranes will affect this equilibrium in unpredictable ways, as will oxidative reactions which appear to involve primarily, the deprotonated form. The aggregation and precipitation of the protonated forms will tend to ensure that high concentrations of these compounds in solution at a pH below their pKa will not be stable. All of these effects may be yet another means by which intracellular flavonoid concentrations are maintained at very low levels. It may be possible that flavonoids only ‘survive’ and enter cells when they are bound to macromolecules; in this case, the active concentration of flavonoids would be determined more by their binding properties than their actual total concentration. Finally, the ability of flavonoids and other polyphenolic compounds to interact with topo I and DNA may provide another mechanism by which these compounds affect many aspects of cell functioning, especially at the very low concentrations normally attained in cells by dietary means.
Acknowledgments
This work was supported by a fellowship from the National Institute of Environmental Health Sciences (National Institutes of Health grant 5T32E507059), the American Vineyard Foundation and California Competitive Grant Program for Research in Viticulture and Enology, the Wine Spectator and a Jastro Shields scholarship. We also thank Nicole Rabaud for her generous support in preparation of the manuscript, Dr David Mills for use of his laboratory facilities and his technical advice, and Andrew Peckham and Crystal Viegelman for their technical assistance.
References
- 1.Formica J. V., Regelson W. Review of the biology of quercetin and related bioflavonoids. Food Chem. Toxicol. 1995;33:1061–1080. doi: 10.1016/0278-6915(95)00077-1. [DOI] [PubMed] [Google Scholar]
- 2.Nijveldt R. J., van Nood E., van Hoorn D. E., Boelens P. G., van Norren K., van Leeuwen P. A. Flavonoids: a review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001;74:418–425. doi: 10.1093/ajcn/74.4.418. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson L. R. Role of plant polyphenols in genomic stability. Mutat. Res. 2001;475:89–111. doi: 10.1016/s0027-5107(01)00073-2. [DOI] [PubMed] [Google Scholar]
- 4.van Acker S. A., van den Berg D. J., Tromp M. N., Griffioen D. H., van Bennekom W. P., van der Vijgh W. J., Bast A. Structural aspects of antioxidant activity of flavonoids. Free Radical Biol. Med. 1996;20:331–342. doi: 10.1016/0891-5849(95)02047-0. [DOI] [PubMed] [Google Scholar]
- 5.Le Marchand L. Cancer preventive effects of flavonoids – a review. Biomed. Pharmacother. 2002;56:296–301. doi: 10.1016/s0753-3322(02)00186-5. [DOI] [PubMed] [Google Scholar]
- 6.Howitz K. T., Bitterman K. J., Cohen H. Y., Lamming D. W., Lavu S., Wood J. G., Zipkin R. E., Chung P., Kisielewski A., Zhang L. L., et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature (London) 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 7.Manach C., Regerat F., Texier O., Agullo G., Demigne C., Remesy C. Bioavailability, metabolism, and physiological impact of 4-oxo-flavonoids. Nutr. Res. 1996;16:517–544. [Google Scholar]
- 8.Galati G., Sabzevari O., Wilson J. X., O'Brien P. J. Prooxidant activity and cellular effects of the phenoxyl radicals of dietary flavonoids and other polyphenolics. Toxicology. 2002;177:91–104. doi: 10.1016/s0300-483x(02)00198-1. [DOI] [PubMed] [Google Scholar]
- 9.Boege F., Straub T., Kehr A., Boesenberg C., Christiansen K., Andersen A., Jakob F., Kohrle J. Selected novel flavones inhibit the DNA binding or the DNA religation step of eukaryotic topoisomerase I. J. Biol. Chem. 1996;271:2262–2270. doi: 10.1074/jbc.271.4.2262. [DOI] [PubMed] [Google Scholar]
- 10.Yamashita Y., Kawada S. Z., Nakano H. Induction of mammalian topoisomerase II dependent DNA cleavage by nonintercalative flavonoids, genistein and orobol. Biochem. Pharmacol. 1990;39:737–744. doi: 10.1016/0006-2952(90)90153-c. [DOI] [PubMed] [Google Scholar]
- 11.Wang J. C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 12.Trask D. K., Muller M. T. Stabilization of type I topoisomerase–DNA covalent complexes by actinomycin D. Proc. Natl. Acad. Sci. U.S.A. 1988;85:1417–1421. doi: 10.1073/pnas.85.5.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehmann M., Franco A., de Souza Prudente Vilar K., Lukza Reguly M., de Andrade H. H. Doxorubicin and two of its analogues are preferential inducers of homologous recombination compared with mutational events in somatic cells of Drosophila melanogaster. Mutat. Res. 2003;539:167–175. doi: 10.1016/s1383-5718(03)00162-1. [DOI] [PubMed] [Google Scholar]
- 14.Sabourin M., Nitiss J. L., Nitiss K. C., Tatebayashi K., Ikeda H., Osheroff N. Yeast recombination pathways triggered by topoisomerase II-mediated DNA breaks. Nucleic Acids Res. 2003;31:4373–5384. doi: 10.1093/nar/gkg497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L., Hickson I. D. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature (London) 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 16.Strick R., Strissel P. L., Borgers S., Smith S. L., Rowley J. D. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc. Natl. Acad. Sci. U.S.A. 2000;97:4790–4795. doi: 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterson J., Lagiou P., Samoli E., Lagiou A., Katsouyanni K., La Vecchia C., Dwyer J., Trichopoulos D. Flavonoid intake and breast cancer risk: a case-control study in Greece. Br. J. Cancer. 2003;89:1255–1259. doi: 10.1038/sj.bjc.6601271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constantinou A., Mehta R., Runyan C., Rao K., Vaughan A., Moon R. Flavonoids as DNA topoisomerase antogonists and poisons – structure–activity-relationships. J. Nat. Prod. 1995;58:217–225. doi: 10.1021/np50116a009. [DOI] [PubMed] [Google Scholar]
- 19.Webb M. R., Ebeler S. E. A gel electrophoresis assay for the simultaneous determination of topoisomerase I inhibition and DNA intercalation. Anal. Biochem. 2003;321:22–30. doi: 10.1016/s0003-2697(03)00459-7. [DOI] [PubMed] [Google Scholar]
- 20.Sober H. A. Handbook of Biochemistry: Selected Data for Molecular Biology. Cleveland, OH: The Chemical Rubber Co.; 1968. Buffer solutions. [Google Scholar]
- 21.Hertzberg R. P., Caranfa M. J., Hecht S. M. On the mechanism of topoisomerase I inhibition by camptothecin: evidence for binding to an enzyme–DNA complex. Biochemistry. 1989;28:4629–4638. doi: 10.1021/bi00437a018. [DOI] [PubMed] [Google Scholar]
- 22.McGovern S. L., Caselli E., Grigorieff N., Shoichet B. K. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 23.Jovanovic S. V., Steenken S., Tosic M., Marjanovic B., Simic M. G. Flavonoids as antioxidants. J. Am. Chem. Soc. 1994;116:4846–4851. [Google Scholar]
- 24.Wagner H., Chari V. M., Sonnenbichler J. 13C-NMR-spektren natuerlich vorkommender flavonoide. Tetrahedron Lett. 1976;21:1799–1802. [Google Scholar]
- 25.Dangles O., Dufour C., Manach C., Morand C., Remesy C. Binding of flavonoids to plasma proteins. Methods Enzymol. 2001;335:319–333. doi: 10.1016/s0076-6879(01)35254-0. [DOI] [PubMed] [Google Scholar]
- 26.Mocek M., Richardson P. J. Kinetics and mechanism of quercetin oxidation. J. Inst. Brew. 1972;78:459–465. [Google Scholar]
- 27.Bailly C., Dassonneville L., Colson P., Houssier C., Fukasawa K., Nishimura S., Yoshinari T. Intercalation into DNA is not required for inhibition of topoisomerase I by indolocarbazole antitumor agents. Cancer Res. 1999;59:2853–2860. [PubMed] [Google Scholar]
- 28.Snyder R. D., Gillies P. J. Evaluation of the clastogenic, DNA intercalative and topoisomerase II-interactive properties of bioflavonoids in Chinese hamster V79 cells. Environ. Mol. Mutagen. 2002;40:266–276. doi: 10.1002/em.10121. [DOI] [PubMed] [Google Scholar]
- 29.Rueff J., Laires A., Borba H., Chaveca T., Gomes M. I., Halpern M. Genetic toxicology of flavonoids: the role of metabolic conditions in the induction of reverse mutation, SOS functions and sister-chromatid exchanges. Mutagenesis. 1986;1:179–183. doi: 10.1093/mutage/1.3.179. [DOI] [PubMed] [Google Scholar]
- 30.Popp R., Schimmer O. Induction of sister-chromatid exchanges (SCE), polyploidy, and micronuclei by plant flavonoids in human lymphocyte cultures: a comparative study of 19 flavonoids. Mutat. Res. 1991;246:205–213. doi: 10.1016/0027-5107(91)90123-6. [DOI] [PubMed] [Google Scholar]
- 31.Pilch D. S., Yu C., Makhey D., LaVoie E. J., Srinivasan A. R., Olson W. K., Sauers R. R., Breslauer K. J., Geacintov N. E., Liu L. F. Minor groove-directed and intercalative ligand–DNA interactions in the poisoning of human DNA topoisomerase I by protoberberine analogs. Biochemistry. 1997;36:12542–12553. doi: 10.1021/bi971272q. [DOI] [PubMed] [Google Scholar]
- 32.Burda S., Oleszek W. Antioxidant and antiradical activities of flavonoids. J. Agric. Food Chem. 2001;49:2774–1779. doi: 10.1021/jf001413m. [DOI] [PubMed] [Google Scholar]
- 33.Wilson W. D. DNA intercalators. In: Kool E. T., editor. Comprehensive Natural Products Chemistry, vol. 7. Amsterdam: Elsevier; 1999. pp. 427–454. [Google Scholar]
- 34.Austin C. A., Patel S., Ono K., Nakane H., Fisher L. M. Site-specific DNA cleavage by mammalian DNA topoisomerase II induced by novel flavone and catechin derivatives. Biochem. J. 1992;282:883–889. doi: 10.1042/bj2820883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harmon F. G., DiGate R. J., Kowalczykowski S. C. RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: a conserved mechanism for control of DNA recombination. Mol. Cell. 1999;3:611–620. doi: 10.1016/s1097-2765(00)80354-8. [DOI] [PubMed] [Google Scholar]
- 36.Silva I. D., Gaspar J., da Costa G. G., Rodrigues A. S., Laires A., Rueff J. Chemical features of flavonols affecting their genotoxicity: potential implications in their use as therapeutical agents. Chem. Biol. Interact. 2000;124:29–51. doi: 10.1016/s0009-2797(99)00139-8. [DOI] [PubMed] [Google Scholar]
- 37.Uhl M., Ecker S., Kassie F., Lhoste E., Chakraborty A., Mohn G., Knasmuller S. Effect of chrysin, a flavonoid compound, on the mutagenic activity of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and benzo(a)pyrene (B(a)P) in bacterial and human hepatoma (HepG2) cells. Arch. Toxicol. 2003;77:477–484. doi: 10.1007/s00204-003-0469-4. [DOI] [PubMed] [Google Scholar]
- 38.Edenharder R., Krieg H., Kottgen V., Platt K. L. Inhibition of clastogenicity of benzo[a]pyrene and of its trans-7,8-dihydrodiol in mice in vivo by fruits, vegetables, and flavonoids. Mutat. Res. 2003;537:169–181. doi: 10.1016/s1383-5718(03)00078-0. [DOI] [PubMed] [Google Scholar]
- 39.Ebeler S. E., Brenneman C. A., Kim G. S., Jewell W. T., Webb M. R., Chacon-Rodriguez L., MacDonald E. A., Cramer A. C., Levi A., Ebeler J. D., et al. Dietary catechin delays tumor onset in a transgenic mouse model. Am. J. Clin. Nutr. 2002;76:865–872. doi: 10.1093/ajcn/76.4.865. [DOI] [PubMed] [Google Scholar]
- 40.Kuntz S., Wenzel U., Daniel H. Comparative analysis of the effects of flavonoids on proliferation, cytotoxicity, and apoptosis in human colon cancer cell lines. Eur. J. Nutr. 1999;38:133–142. doi: 10.1007/s003940050054. [DOI] [PubMed] [Google Scholar]
- 41.Wang I. K., Lin-Shiau S. Y., Lin J. K. Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukemia HL-60 cells. Eur. J. Cancer. 1999;35:1517–1525. [PubMed] [Google Scholar]
- 42.Calabrese E. J., Baldwin L. A. Can the concept of hormesis be generalized to carcinogenesis? Regul. Toxicol. Pharmacol. 1998;28:230–241. doi: 10.1006/rtph.1998.1267. [DOI] [PubMed] [Google Scholar]
- 43.Scalbert A., Williamson G. Dietary intake and bioavailability of polyphenols. J. Nutr. 2000;130:2073S–2085S. doi: 10.1093/jn/130.8.2073S. [DOI] [PubMed] [Google Scholar]
- 44.Svejstrup J. Q. Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Cell Biol. 2002;3:21–29. doi: 10.1038/nrm703. [DOI] [PubMed] [Google Scholar]
- 45.Bakkenist C. J., Kastan M. B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature (London) 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 46.Soe K., Grosse F. p53 stimulates human topoisomerase I activity by modulating its DNA binding. Nucleic Acids Res. 2003;31:6585–6592. doi: 10.1093/nar/gkg846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laine J. P., Opresko P. L., Indig F. E., Harrigan J. A., von Kobbe C., Bohr V. A. Werner protein stimulates topoisomerase I DNA relaxation activity. Cancer Res. 2003;63:7136–7146. [PubMed] [Google Scholar]