

Abstract
In the present paper, we describe cloning and expression of two outer membrane proteins, BpsOmp38 (from Burkholderia pseudomallei) and BthOmp38 (from Burkholderia thailandensis) lacking signal peptide sequences, using the pET23d(+) expression vector and Escherichia coli host strain Origami(DE3). The 38 kDa proteins, expressed as insoluble inclusion bodies, were purified, solubilized in 8 M urea, and then subjected to refolding experiments. As seen on SDS/PAGE, the 38 kDa band completely migrated to ∼110 kDa when the purified monomeric proteins were refolded in a buffer system containing 10% (w/v) Zwittergent® 3-14, together with a subsequent heating to 95 °C for 5 min. CD spectroscopy revealed that the 110 kDa proteins contained a predominant β-sheet structure, which corresponded completely to the structure of the Omp38 proteins isolated from B. pseudomallei and B. thailandensis. Immunoblot analysis using anti-BpsOmp38 polyclonal antibodies and peptide mass analysis by MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight) MS confirmed that the expressed proteins were BpsOmp38 and BthOmp38. The anti-BpsOmp38 antibodies considerably exhibited the inhibitory effects on the permeation of small sugars through the Omp38-reconstituted liposomes. A linear relation between relative permeability rates and Mr of neutral sugars and charged antibiotics suggested strongly that the in vitro re-assembled Omp38 functioned fully as a diffusion porin.
Keywords: Burkholderia, cloning, diffusion pore, expression, outer membrane protein, refolding
Abbreviations: BCA, bicinchoninic acid; CSA, non-hygroscopic ammonium (+)-10-camphorsulphonate; FTIR, Fourier transform IR; GlcNAc, N-acetylglucosamine; IPTG, isopropyl β-D-thiogalactoside; LB, Luria–Bertani; LD50, 50% lethal dose; LDAO, N,N-dimethyldodecylamine N-oxide; LPS, lipopolysaccharide; MALDI–TOF, matrix-assisted laser-desorption ionization–time-of-flight; MOMP, major outer membrane protein; MRE, mean residue molar ellipticity; β-OG, n-octyl β-D-glucoside; PEG, poly(ethylene glycol)
INTRODUCTION
Burkholderia pseudomallei is a Gram-negative bacterium that causes melioidosis, a potentially fatal disease in humans and other animals, including dolphins, sheep, pigs and goats [1–3]. In Thailand, melioidosis is endemic and is most widespread in the north-east region, where, for instance, in an hospital it was responsible for 19% of community-acquired sepsis and 40% of deaths from community-acquired septicaemia. The clinical spectrum of melioidosis consists of four major forms, including acute fulminant septicaemia, sub-acute illness, chronic infection and subclinical disease. Syndromes may be localized or disseminated and affect different organs, depending on whether the disease is acute or chronic. In humans, the incubation period generally takes approx. 2–3 days. However, the disease can be developed 6–26 years after exposure [4]. By comparing the 16 S rRNA gene sequences of bacterial species in the genus Burkholderia, B. pseudomallei was shown to be phylogenetically closely related to Burkholderia thailandensis, with only 15 nucleotide dissimilarities [5]. The main characteristic for separation of the two species is the difference in their ability to utilize L-arabinose. Almost all clinical isolates of B. pseudomallei are unable to utilize L-arabinose as a single substrate (Ara−), while B. thailandensis can utilize L-arabinose (Ara+) [6,7]. Moreover, these two bacteria have a distinct difference in their relative virulence. The LD50 (50% lethal dose) for B. pseudomallei in the Syrian hamster model of acute melioidosis is less than ten organisms, whereas the LD50 for B. thailandensis is approx. 106 organisms [8]. Successful treatment of melioidosis has been difficult due to the lack of effective drugs and the inherent resistance of B. pseudomallei to various groups of antibiotics, including β-lactams, aminoglycosides, macrolides and polymyxins [9,10]. It has been suggested that the resistance may be associated with low permeability of antibiotics through porin channels located at the outer membrane of the bacterium [11].
Bacterial porins have been classified into non-specific (general diffusion) and specific porins. Non-specific porins allow the passage of hydrophilic solutes up to an exclusion size of Mr∼600 and show a linear relation between the permeation rate and solute concentration gradient [12,13]. On the other hand, specific porins exhibit Michaelis–Menten kinetics for the transport of specific solutes [14]. A number of bacterial porins with related structures has been characterized, for instance OmpF, PhoE, Omp32 and LamB [15–17]. Most porins, if not all, are stable in trimeric form [18]. This rigid structure generally is detergent-resistant, but heat-sensitive [19]. The entire polypeptide of a porin subunit typically comprises 300–420 amino acids, which fold into a 16- or 18-stranded antiparallel β-barrel. X-ray crystallography [15,20] and neutron crystallography [21] have demonstrated that the hydrophobic part of the β-barrel is inserted into the core of the outer membrane to form the transmembrane pore. The pore is, in turn, constricted by an internal loop or eyelet that folds inwardly and is attached to the inner side of the barrel wall. The β-strands are connected on the periplasmic side by short loops or turns and on the extracellular side by long irregular loops [18]. These loops display high sequence variation among homologous porins, which give rise to the structural variability at the cell surface that may help the bacteria to escape from cellular recognition, e.g. specific antibodies, invading phages or certain proteases [18].
We reported recently the isolation of BpsOmp38 and Bth-Omp38 from B. pseudomallei type strain ATCC23343 and B. thailandensis type strain ATCC700388 respectively [22]. Like most other porins, Omp38 exhibited heat-modifiable, SDS-stable behaviour and functioned as a trimer (Mr 110000). The trimer was dissociated into three identical monomeric subunits (Mr 38000) when heated to 95 °C. We also described the isolation of the genes that encode full-length BpsOmp38 and BthOmp38 from genomes of B. pseudomallei and B. thailandensis. Analysis of the putative amino acid sequences revealed that both proteins were almost identical. Liposome-swelling assays revealed that Omp38 could form a general diffusion pore, allowing hydrophilic sugars (Mr<650) to permeate.
In the present paper, we describe cloning and expression of two 38 kDa proteins corresponding to the processed BpsOmp38 and BthOmp38 using the pET23d(+) vector and Escherichia coli host strain Origami(DE3) system. Both proteins were expressed as insoluble inclusion bodies, and were then subsequently subjected to denaturing–refolding experiments. Immunoblotting using anti-BpsOmp38 polyclonal antibodies and MALDI-TOF (matrix-assisted laser-desorption ionization–time-of-flight) identified the expressed proteins to be Omp38. CD spectral data confirmed that the trimeric proteins obtained from a refolding system in the presence of Zwittergent® 3-14 were properly folded. Function of the refolded Omp38 as a general diffusion pore has been verified by liposome-swelling assays.
EXPERIMENTAL
Chemicals
Bacteria culture media and bacteriological agar were purchased from Scharlau Chemie (Barcelona, Spain). DTT (dithiothreitol), iodoacetamide, ammonium hydrogen carbonate, D-stachyose, D-melezitose, D-sucrose, L-arabinose, D-glucose and D-mannose were from Acros Organics (Morris Plains, NJ, U.S.A.). Proteinase K and trypsin (sequencing grade) were from Promega. Sephacryl S-200® HR resin and dextran T-40 were from Amersham Biosciences. Zwittergent® 3-14 (n-tetradecyl-N,N-dimethyl-3-ammonio-1-propanesulphonate) was from Sigma–Aldrich. Other detergents used for protein preparation were purchased from Carlo Erba Reagenti (Milan, Italy). PEG [poly(ethylene glycol)] 20000 was from Fluka. Pfu DNA polymerase and T4 ligase were purchased from Promega. Oligonucleotides for PCR amplification were synthesized by Proligo Singapore (Singapore Science Park II, Singapore). PCR purification and plasmid preparation kits were purchased from Qiagen. All restriction endonucleases were from New England Bio-labs. All other reagents for general laboratory use were from Sigma–Aldrich and Carlo Erba Reagenti.
Bacterial strains and plasmids
B. pseudomallei type strain ATCC23343, E. coli type strain ATCC25922, Staphylococcus aureus type strain ATCC25923 and Pseudomonas aeruginosa type strain ATCC27853 were gifts from Ms Worada Samosornsuk (Department of Microbiology, Faculty of Allied Health Science, Thammasat University, Thailand). B. thailandensis type strain ATCC700388 was kindly provided by Dr Richard H. Ashley (Department of Biomedical Sciences, University of Edinburgh, U.K.). E. coli strains DH5α and Origami-(DE3) were obtained from Novagen. pGEM®-T vector was obtained from Promega, and pET23d(+) expression vector was from Novagen.
Purification of native Omp38 from B. pseudomallei and B. thailandensis
BpsOmp38 and BthOmp38 were prepared from B. pseudomallei and B. thailandensis respectively following the protocols described previously [22,23]. Briefly, the bacteria were grown in LB (Luria–Bertani) broth at 37 °C with vigorous shaking. Cells from 2 litres of late-exponential culture were suspended in 10 ml of 10 mM Tris/HCl, pH 8.0, containing 1 mM PMSF and 2 mg of hen's-egg lysozyme. The cell suspension was sonicated using a Sonopuls Ultrasonic homogenizer with a 6-mm-diameter probe (50% duty cycle; amplitude setting, 20%; total time, 5 min), and large cellular debris and unbroken cells were removed by centrifugation at 10000 g for 30 min at room temperature (25 °C). Cell membranes were recovered from the cell lysates by microcentrifugation at 12000 g for 1 h, suspended in 10 mM Tris/HCl, pH 8.0, containing 0.5% (w/v) SDS, and incubated at 30 °C for 1 h. A complex of non-solubilized peptidoglycan sheets was separated from solubilized cytoplasmic and membrane components by microcentrifugation at 12000 g for 1 h. The crude peptidoglycan pellet was then solubilized in 4 ml of 10 mM Tris/HCl, pH 8.0, containing 2% (w/v) SDS and 0.5 M NaCl, and was incubated at 37 °C for 1 h. Solubilized peptidoglycan-associated proteins, separated from insoluble peptidoglycan by microcentrifugation at 12000 g for 1 h, were applied twice on to a Sephacryl S-200® HR column (1.5 cm×95 cm) equilibrated previously with 10 mM Tris/HCl, pH 8.0, containing 1% (w/v) SDS and 0.5 M NaCl. The chromatography was carried out at a flow rate of 1 ml/min and fractions of 2 ml were collected. Concentrations of eluted proteins were determined by measuring the A280, and the protein profile was analysed further using SDS/12% PAGE. Fractions containing 38 kDa proteins were pooled and precipitated with 50% (v/v) ethanol at −30 °C overnight [24]. The precipitated protein was collected by centrifugation at 20000 g for 30 min, then dissolved in 1 ml of 10 mM Tris/HCl, pH 8.0, containing 2% (w/v) SDS and 0.5 M NaCl. The protein solution was dialysed against a large volume of 10 mM Tris/HCl, pH 8.0, for 48 h at room temperature with four changes of the same buffer. A final concentration of Omp38 was determined using the BCA (bicinchoninic acid) assay.
Determination of protein concentration
Protein concentrations were estimated using the BCA assay kit (Pierce, Rockford, IL, U.S.A.) according to the manufacturer's instructions. A protein sample (12.5 μl) was mixed with 100 μl of the BCA working reagent. After the reaction mixture was incubated at 37 °C for 30 min, absorbance at 540 nm was measured with a microplate reader spectrophotometer (Labsystem, Finland). BSA at varied concentrations ranging from 0.025–2.0 mg/ml was used to construct a standard calibration curve and to determine protein concentrations of unknown samples.
Cloning of the DNA encoding BpsOmp38 and BthOmp38
The genes encoding full-length BpsOmp38 and BthOmp38 were isolated from genomic DNA of B. pseudomallei and B. thailandensis and cloned into the pGEM-T cloning vector using PCR-based method as described previously [22]. The recombinant plasmids, designated pGEM-T-BpsOmp38 and pGEM-T-BthOmp38, were used as templates for PCR amplification of the Omp38 fragments. The forward primer included the initiation codon ATG following an NcoI restriction site and the nucleotides that encode Omp38 lacking a signal peptide fragment. The reverse primer included an XhoI restriction site following the nucleotides that encode the C-terminal end of Omp38. The primer sequences are: NcoI (sequence underlined) forward primer, 5′-CATGCC-ATGGCTCAAAGCAGCGTCACGC-3′; XhoI (sequence underlined) reverse primer, 5′-CCGCTCGAGTTAGAAGCGGTGACGCAGACC-3′.
PCRs were carried out with Pfu DNA polymerase in a Gene-Amp™ PCR System 9700 thermocycler (PE Applied Biosystems, Foster City, CA, U.S.A.). The PCR products of expected size (1.1 kb) were purified and concentrated using the Qiagen PCR purification kit, then digested with the corresponding restriction enzymes to generate cohesive ends. The 1.1 kb DNA fragments were re-extracted from an 1% agarose gel using the Qiagen gel extraction kit. The purified DNA fragments were ligated to the plasmid pET-23d(+) previously digested with the same restriction endonucleases and transformed into E. coli host strain DH5α, according to the standard protocol. The recombinant plasmids, designated pET-23d(+)-BpsOmp38 and pET-23d(+)-BthOmp38, were isolated from the transformed E. coli DH5α cells using the Qiagen plasmid miniprep kit, and were analysed on a 1% agarose gel. The success of cloning of the Omp38 DNA fragments was verified by PCR amplification using the same set of primers as described above.
Expression of Omp38 in E. coli and preparation of inclusion bodies
Approx. 100 ng of pET23d(+)-Omp38 DNA was transformed into E. coli Origami(DE3), and single colonies were transferred to 50 ml of LB medium containing ampicillin (100 μg/ml) and 1% glucose [25]. After incubation for 8 h at 37 °C, the culture was transferred to four 1-litre flasks, each containing 500 ml of LB medium with ampicillin (100 μg/ml). Incubation was carried out at 37 °C in a shaking incubator until a D600 of approx. 0.6 was reached. At this point, IPTG (isopropyl β-D-thiogalactoside) was added to the culture medium to a final concentration of 0.4 mM. The incubation was continued for an additional 90 min, then cells were harvested by centrifugation at 10000 g for 30 min. The cell pellet was resuspended in 50 mM Tris/HCl, pH 8.0, containing 1 mM EDTA and 100 mM NaCl (TEN buffer), using the ratio of 3 ml of buffer per g (wet weight) of cells [26]. PMSF (1 mM) and lysozyme (2 mg/ml) were subsequently added to the cell suspension, then sonicated using a Sonopuls Ultrasonic homogenizer with a 6-mm-diameter probe (50% duty cycle; amplitude setting, 20%; total time, 5 min). Unbroken cells were removed by centrifugation, and proteins from a whole-cell lysate were pelleted at 20000 g at 4 °C. The insoluble pellet was washed once with 10 ml of 2 M urea and 0.05% (v/v) Tween 20 in 10 mM Tris/HCl, pH 8.0, then centrifuged further at 20000 g at room temperature for 30 min [27]. At this stage, the white pellet obtained mainly contained partially purified inclusion bodies of the expressed 38 kDa protein.
Denaturing and refolding of Omp38 expressed in E. coli
The insoluble inclusion bodies obtained from the previous step were resuspended in 10 ml of a freshly prepared denaturing buffer (8 M urea in 25 mM sodium acetate buffer, pH 5.0), and centrifuged at 20000 g at room temperature for 2 h. The clear supernatant (5 ml) was applied on to a prepacked SP Fast Flow HiTrap™ column (1.5 cm×3 cm) (Amersham Biosciences), followed by a prepacked DEAE Fast Flow HiTrap™ column (1.5 cm×3 cm) (Amersham Biosciences). For both chromatographic steps, proteins were fractionated with a 0–0.3 M step gradient of NaCl in 25 mM sodium acetate buffer, pH 5.0, and an applied flow rate of 1 ml/min. Eluted fractions of 2 ml were collected and the A280 was measured for every fraction. Protein fractions were analysed further on SDS/12% PAGE, and the fractions containing the 38 kDa protein were pooled and precipitated by 50% (v/v) ethanol at −20 °C overnight [24]. The protein pellet containing monomeric Omp38 was collected by centrifugation at 20000 g for 20 min and dissolved with 2 ml of 8 M urea in 20 mM Tris/HCl, pH 8.0. Concentration of the purified protein was determined with the BCA kit, and the final concentration of the protein was adjusted to 5 mg/ml with the Tris/HCl/urea buffer.
For refolding of Omp38, the protein solution was diluted 1:1 with a refolding solution [20 mM Tris/HCl containing 10% (w/v) Zwittergent® 3-14, 200 mM NaCl, 20 mM CaCl2, 10 mM EDTA and 0.02% NaN3] [25,26], and heated at 95 °C for 5 min. The protein solution was incubated overnight at room temperature, then purified using a Sephacryl S-200® HR column (1.5 cm×95 cm) previously equilibrated with 20 mM Tris/HCl, pH 7.0, containing 0.05% Zwittergent® 3-14. A flow rate of 0.5 ml/min was applied, and fractions of 2 ml were collected. The A280 was measured and the 110 kDa proteins, as observed using SDS/PAGE under non-heated conditions, were combined and concentrated to 1 ml in a dialysis bag wrapped with PEG 20000 powder. The concentrated solutions were dialysed further against 20 mM Tris/HCl, pH 7.0, containing 0.05% Zwittergent® 3-14 at room temperature for 2 days, with frequent changes of the dialysis buffer. A final concentration of the protein was determined by the BCA assay, and the protein solution was stored at −30 °C until use.
Secondary-structure determination by CD spectroscopy
Secondary-structure composition indicating unfolded and folded states of the expressed proteins was determined with a Jasco J-715 spectropolarimeter (Japan Spectroscopic Co., Tokyo, Japan). The purified unfolded (38 kDa) and refolded (110 kDa) proteins (0.5 mg/ml) were solubilized in the appropriate buffers and subjected to CD measurements by comparing with the native Omp38. CD spectral data were acquired at both far UV (180–250 nm) and near UV (250–320 nm) regions. CD measurements were performed at 25 °C with a scan speed of 20 nm/min, 2 nm bandwidth, 100 mdeg sensitivity, an average response time of 2 s and an optical path length of 0.2 mm. A minimum of three consecutive scans was accumulated, and the average spectra were stored. The baseline buffer for the native proteins was 10 mM Tris/HCl, pH 8.0. The baseline buffer for the unfolded proteins was 8 M urea in 20 mM Tris/HCl, pH 7.0. The baseline buffer for the refolded proteins was 20 mM Tris/HCl, pH 7.0, containing 0.05% Zwittergent® 3-14. Each protein spectrum was standardized with its corresponding buffer spectrum. The raw data were transformed to MRE (mean residue molar ellipticity) using the following equation:
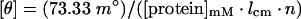 |
where [θ] is the MRE in deg·cm2/dmol, n is the number of amino acids in the polypeptide chain, m° is the measured ellipticity and lcm is the path length in centimetres.
The intensity of standard CSA (non-hygroscopic ammonium (+)-10-camphorsulphonate) at wavelength 290 nm was approx. 45 units, giving the calculated conversion factor from 3300/CSA intensity using the above equation to be 73.33. From its corresponding nucleotide sequence, the number of amino acid residues per Omp38 monomer was predicted to be 354.
SDS/PAGE following immunoblotting
Production of anti-BpsOmp38 polyclonal antibodies was carried out as described in our previous work [22]. To confirm Omp38 expression, whole-cell lysates prepared from E. coli Origami-(DE3) cells harbouring the recombinant pET23d(+)-Omp38 plasmids with and without IPTG induction were electrophoresed on a 12% polyacrylamide gel. Immunoblotting was performed using a 1:10000 dilution of anti-BpsOmp38 antiserum in PBS, pH 7.4, containing 0.2% (v/v) Tween 20 and 5% (w/v) non-fat dried milk and detected with the ECL® (enhanced chemiluminescence) reagent (Amersham Biosciences), according to the manufacturer's instructions. The purified native BpsOmp38 and BthOmp38 (5 μg) were used as positive controls.
Peptide mass analysis by MALDI–TOF MS
The 38 kDa proteins expressed from E. coli Origami(DE3) cells were denatured in the Tris/HCl/urea buffer and purified as described above. Protein samples were electrophoresed on a 12% polyacrylamide gel, and the 38 kDa band was excised and subjected to trypsin digestion according to the method of Shevchenko et al. [28]. After overnight digestion at 37 °C, peptides were extracted and dried in a vacuum centrifuge. A small fraction of these tryptic peptides was analysed by MALDI–TOF MS (Voyager-DE Pro in reflective mode) in an α-cyano-4-hydroxycinnamic acid matrix. To confirm that the obtained peptides were Omp38 peptides, databank searching was performed with MS-Fit (http://prospector.ucsf.edu/) for MALDI–TOF mass fingerprint data.
Liposome-swelling assays
Preparation of proteoliposomes and reconstitution of Omp38 (50 μg) into the liposomes were described previously [22]. Permeation rates of small sugars through the refolded Omp38 were determined by the liposome-swelling method. The tested sugars included L-arabinose (Mr 150), D-glucose (Mr 180), D-mannose (Mr 180), D-galactose (Mr 180), GlcNAc (N-acetylglucosamine; Mr 221), D-sucrose (Mr 342), D-melezitose (Mr 522) and D-stachyose (Mr 667). Permeability of antibiotics that are potentially used for melioidosis treatment, including amikacin (Mr 782), gentamicin (Mr 709), ceftazidime (Mr 637), cefepime (Mr 572), clindamycin (Mr 505), cefotaxime (Mr 477) and meropenem (Mr 383), were also tested using the same procedure. Changes in the swelling rate of the Omp38-reconstitued liposomes upon addition of the solutes were observed spectrometrically at 400 nm for 60 s. The relative permeabilities of the pore-forming proteins were assumed to be proportional to the initial swelling rates [29].
Effects of anti-BpsOmp38 polyclonal antibodies on sugar uptake of Omp38
Varied dilutions of anti-BpsOmp38 polyclonal antibodies, 1:10, 1:100, 1:1000, 1:10000 and 1:100000, were mixed with 50 μg of the refolded and native Omp38, then incubated at 37 °C for 2 h. The protein mixtures were subsequently reconstituted into proteoliposomes and the liposome swelling assays were carried out with the same set of sugars as mentioned above. The proteoliposome mixtures containing various dilutions of the antibodies in the presence of BSA or in the absence of protein were used as negative controls. An additional control was conducted using refolded BpsOmp38 and BthOmp38 mixed with anti-(rabbit β-glucosidase) antibodies raised against an E. coli-expressed Dalbergia nigrescens Kurz β-glucosidase antigen. Concentrations of the antibodies were the same as described for anti-BpsOmp38 antiserum. Permeability of D-glucose through the Omp38 pores was determined using the above permeation assay.
RESULTS
Expression of Omp38 in E. coli
The recombinant plasmids, pET23d(+)-BpsOmp38 and pET23d(+)-BthOmp38, were purified from E. coli DH5α cells and transformed into E. coli Origami(DE3) cells for a large-scale production of monomeric Omp38. When the cells were grown to exponential phase (D600∼0.6), Omp38 expression was induced by adding IPTG (0.4 mM) with an incubation period of 1.5 h at 37 °C. Figure 1(A) represents the SDS/PAGE of the whole cell lysates of E. coli expressing BpsOmp38 (Figure 1A, lane 2) and BthOmp38 (Figure 1A, lane 4).
Figure 1. Expression of Omp38 in E. coli Origami(DE3).

The whole-cell lysate with IPTG induction or without IPTG induction of E. coli carrying the pET23d(+)-Omp38 plasmids were subjected to (A) SDS/PAGE following Coomassie Blue stain and (B) immunoblot analysis using anti-BpsOmp38 antiserum. Lanes: Std, standard proteins; 1, E. coli Origami(DE3) carrying pET23d(+)-BpsOmp38 without IPTG induction; 2, IPTG-induced E. coli Origami(DE3) carrying pET23d(+)-BpsOmp38; 3, E. coli Origami(DE3) carrying pET23d(+)-BthOmp38 without IPTG; 4, IPTG-induced E. coli Origami(DE3) carrying pET23d(+)-BthOmp38; 5, native BpsOmp38; 6, native BthOmp38. Sizes indicated in kDa to the left of the gel.
Western blot analysis using anti-BpsOmp38 polyclonal antibodies reacted with a major 38 kDa band of the whole-cell lysate prepared from the cells with IPTG induction (Figure 1B, lanes 2 and 4), while no signal was detected with the whole-cell lysate obtained from the cells without IPTG induction (Figure 1B, lanes 1 and 3). The antibodies strongly detected native BpsOmp38 and BthOmp38 (Figure 1B, lanes 5 and 6 respectively). This indicated that the expressed 38 kDa proteins were BpsOmp38 and BthOmp38.
Purification and refolding of Omp38 expressed from E. coli
As the 38 kDa proteins expressed from E. coli Origami(DE3) cells were insoluble, these proteins were readily separated from other components in the whole-cell lysate by centrifugation. The insoluble pellets containing aggregated monomeric Omp38 were then subjected to inclusion-body preparation, yielding white pellets, which were commonly recognized as protein inclusion bodies. The insoluble pellets were completely solubilized with 8 M urea in Tris/HCl, pH 8.0, buffer, then purified further using strong cation-exchange (SP Fast Flow HiTrap™), followed by weak anion-exchange (DEAE Fast Fflow HiTrap™) columns. The purified Omp38 monomers were subsequently used in refolding experiments. With all the detergents tested, the monomeric subunits were found to re-associate into a trimer when an equal volume of a refolding solution [20 mM Tris/HCl, pH 7.0, containing 10% (w/v) Zwittergent™ 3-14, 200 mM NaCl, 20 mM CaCl2, 10 mM EDTA and 0.02% NaN3] was added to the protein solution.
When the refolding process had taken place slowly overnight at room temperature, trimer (Mr∼110000) was mainly detected (Figure 2A). However, a pale band corresponding to the dimer (Mr∼76000) of BpsOmp38 (Figure 2A, lane 2) and BthOmp38 (Figure 2A, lane 5) was still observed. The monomers were completely re-associated into the trimer when the protein mixture was subsequently heated at 95 °C for 5 min (Figure 2A, lanes 3 and 6).
Figure 2. SDS/PAGE analysis of refolded Omp38 and the Mr determination.

(A) SDS/PAGE analysis of refolded Omp38 expressed in E. coli. The purified monomeric Omp38 solubilized in the Tris/HCl/urea buffer was subjected to refolding using 20 mM Tris/HCl, pH 7.0, containing 10% (w/v) Zwittergent® 3-14, 200 mM NaCl, 20 mM CaCl2, 10 mM EDTA and 0.02% NaN3. Lanes: 1, unfolded BpsOmp38; 2, incompletely refolded BpsOmp38; 3, completely refolded BpsOmp38; 4, unfolded BthOmp38; 5, incompletely refolded BthOmp38; 6, completely refolded BthOmp38. (B) Determination of the Mr of Omp38. The native, refolded or unfolded Omp38 was applied on a Sephacryl S-200® HR (1.5 cm×95 cm) column. A flow rate of 1.0 ml/min was applied, and fractions of 2 ml were collected. Protein concentrations of eluted fractions were determined by measuring the A280. The inset logarithmic plot displays a calibration curve determining the Mr of Omp38. ◆, Native Omp38; ◇, refolded Omp38; ●, unfolded Omp38.
The trimeric proteins remained stable, even though the concentration of Zwittergent® 3-14 was reduced from 10% to 0.05% with all salts removed by a Sephacryl S-200® HR filtration column. The Mr of the refolded Omp38 was determined by the same gel-filtration column to be approx. 100000 (Figure 2B). This value corresponded to the Mr of the native Omp38 determined under the same conditions.
Confirmation of Omp38 expression by MALDI–TOF MS
The purified monomeric BpsOmp38 and BthOmp38 in 20 mM Tris/HCl, pH 7.0, containing 8 M urea were resolved on a 12% polyacrylamide gel, and the 38 kDa band was excised and subjected to in-gel digestion using trypsin. MALDI–TOF MS identified at least seven out of the 18 theoretical tryptic peptides in the putative amino acid sequence of Omp38 that was reported previously [22].
Positions of the identified peptides, designated P1–P7, in the Omp38 sequence are given in Figure 3. These results re-confirmed that the 38 kDa proteins expressed in E. coli Origami(DE3) were BpsOmp38 and BthOmp38.
Figure 3. Identification of tryptic digests of the expressed proteins by MALDI–TOF MS.

The seven identified peptides (P1–P7) of the 38 kDa proteins expressed from E. coli Origami(DE3) that gave a complete match with the putative peptides of Omp38 are presented in bold and underlined. The start of the peptide P6 is indicated as an asterisk.
Secondary-structure determination using CD spectroscopy
The folding states of the expressed Omp38 from E. coli were investigated by means of CD spectroscopy. The CD spectra of the refolded proteins were essentially compared with the spectra of the native proteins. Identification of the CD spectral patterns has been assessed based on the previously reported data of E. coli OmpA [30]. The absorption spectra illustrating secondary-structure composition of native and refolded Omp38 are demonstrated in Figure 4. It can be seen that the spectra of the refolded proteins were almost identical with those of the native ones, indicating that the expressed proteins were folded into their native conformations. The full spectra gave a minus peak near 215 nm, suggesting a predominant β-sheet structure of the Omp38 proteins. The spectra of the urea-denatured Omp38 were also examined (see Figure 4). The unfolded form of Omp38 was identified to be random coils as the corresponding spectra gave a minus peak below 205 nm, which were very similar to those obtained for the urea-denatured OmpA porin from E. coli [30].
Figure 4. CD spectra of Omp38.

The native Omp38 was prepared by SDS extractions following Sephacryl S-200® HR filtration, then dialysed extensively to remove SDS. The unfolded protein was solubilized in Tris/HCl/urea buffer and subjected to refolding using 10% (w/v) Zwittergent® 3-14 in 20 mM Tris/HCl, pH 7.0. The protein was subsequently dialysed to reduce the concentration of Zwittergent® 3-14 to 0.05%. Spectra of Omp38 were obtained with a Jasco J-715 spectropolarimeter. A solution of 20 mM Tris/HCl, pH 7.0, containing 0.05% Zwittergent® 3-14 or 8 M urea was used for background subtraction. —, Unfolded BthOmp38; - - -, unfolded BpsOmp38; ◇, native BpsOmp38; ◆, refolded BpsOmp38; □, native BthOmp38; ■, refolded BthOmp38.
Pore-forming activity of the refolded Omp38
The trimeric BpsOmp38 and BthOmp38 refolded with Zwittergent™ 3-14 were reconstituted into proteoliposomes and tested for pore-forming activity. Diffusion of solutes through the protein pores caused the liposomes to swell, which lowers their absorbance at a wavelength of 400 nm. As expected, L-arabinose (Mr 150), which is the smallest sugar tested in this experiment, gave the highest rate of diffusion through the Omp38 pores (Figure 5A), followed by the diffusion rates of D-glucose, D-mannose and D-galactose (Mr 180), GlcNAc (Mr 221), D-sucrose (Mr 342) and D-melezitose (Mr 522) respectively. On the other hand, D-stachyose (Mr 667) showed the slowest diffusion rate, as it has the highest Mr compared with the other sugars. It was noticeable that the diffusion rates of D-glucose, D-mannose and D-galactose were only slightly different, since their Mr values are the same. A reconstitution of the native proteins into proteoliposomes and an exposure of the formed pores to the individual sugars led to the same results (Figure 5B).
Figure 5. Liposome-swelling assays of refolded Omp38.

Diffusion rates for neutral saccharides were determined by liposome-swelling assays using proteoliposomes reconstituted with the native (A) and refolded (B) BpsOmp38. The following symbols represent L-arabinose (■); D-glucose (▲); D-mannose (▼); D-galactose (◆); GlcNAc (●); D-sucrose (□); D-melezitose (△) and D-stachyose (▽). Each experiment was repeated three times, and liposomes with BSA and without any protein were used as negative controls. (C) Relative permeation rates of sugars through liposomes reconstituted with native (△) or refolded (▲) BpsOmp38. The values are normalized to the permeation rate of L-arabinose and plotted on a logarithmic scale. The broken and solid lines are a regression fit to all the data for native and refolded Omp38 respectively.
Figure 5(C) represents the diffusion rates of the selected sugars through BpsOmp38 pores in relation to the rate of L-arabinose, which was set to 100%. The relative rates of diffusion through the native and refolded proteins were almost identical, and decreased with increasing size of the sugars. The size-exclusion limit of the Omp38 pores was estimated to be Mr∼650.
Penetration of seven antibiotics through the refolded and native Omp38 was also determined using the liposome-swelling method. The permeability rates of the antibiotics through the native and refolded Omp38 pores were normalized to the diffusion rate of L-arabinose. The results in Table 1 clearly demonstrated that the permeability decreased with increasing size of the antibiotics. The highest rate was observed for the smallest antibiotic, meropenem (Mr 383), followed by ciprofloxacin (Mr 421), clindamycin (Mr 505), cefepime (Mr 572) and ceftazidime (Mr 637) respectively. On the other hand, amikacin (Mr 782) and gentamicin (Mr 709) both could not pass through the pores, because their Mr apparently exceeds the size-exclusion limit of the Omp38 pores. Figure 6 shows a linear relationship between the relative rates of diffusion with the Mr of the antibiotics. It can be seen that the corresponding curve fit was almost indistinguishable from that of the neutral sugars, which indicated that only the molecular size determined the ability of the antibiotics to penetrate through the Omp38 pores. No significant difference was observed in the transport of the antibiotics through the BpsOmp38 and BthOmp38 pores, and no diffusion of them was observed with the BSA-reconstituted liposomes.
Table 1. Relative permeability rates of antibiotics through Omp38 porins.
Permeability rates of antibiotics were calculated relative to the rate of L-arabinose (Mr 150), which was set to 100%.Values given as mean values±S.D. are obtained from three independent assays, and liposomes with BSA and liposomes without any protein were used as negative controls.
| Permeability rate (%) | |||
|---|---|---|---|
| Antibiotic | Mr | Native BpsOmp38 | Refolded BpsOmp38 |
| Amikacin | 782 | 0 | 0 |
| Gentamicin | 709 | 0 | 0 |
| Ceftazidime | 637 | <1 | <1 |
| Cefepime | 572 | 4±1.6 | 5±0.5 |
| Clindamycin | 505 | 8±0.9 | 8±0.5 |
| Ciprofloxacin | 421 | 15±0.9 | 15±0.9 |
| Meropenem | 383 | 20±0.5 | 20±0.5 |
Figure 6. Relative permeation of neutral and charged solutes through Omp38.

Purified native BpsOmp38 was reconstituted into liposome vesicles. The permeabilities of various sugars and antibiotics through BpsOmp38 were determined as described in the text, and the permeability of the channel was assumed to be proportional to the swelling rate. The size-dependent permeation rates for sugars (□) and antibiotics (■) were plotted on a logarithmic scale, and the values are normalized to the permeation rate of L-arabinose. The broken and solid lines are a regression fit to all the data for sugars and antibiotics respectively.
Effects of anti-BpsOmp38 antibodies on Omp38 pore activity
Proteoliposome-swelling assays were also used to determine the effects of anti-BpsOmp38 antibodies on the activity of Omp38 pores. The refolded BpsOmp38 and BthOmp38 were incubated at 37 °C for 2 h with various dilutions of anti-BpsOmp38 polyclonal antibodies before mixing with proteoliposomes.
Figure 7 reveals the inhibitory effects of the antibodies on the pore-forming activity of BpsOmp38 and BthOmp38. Lower diffusion rates of the chosen sugars through the Omp38 pores were typically observed as concentrations of added antibodies increased. For all the sugars, the control reactions using BSA, BSA–antibodies or antibodies alone did not give detectable swelling of the liposomes. In addition, the permeation behaviour of glucose was not at all altered by anti-β-glucosidase polyclonal antibodies. This suggested that the non-related anti-rabbit antibodies did not influence the Omp38 activity in the permeation assay.
Figure 7. Effects of anti-BpsOmp38 polyclonal antibodies on Omp38 activity.

Various dilutions of the antibodies were incubated with 50 μg of the refolded BpsOmp38 and BthOmp38 in a final volume of 200 μl. The reaction mixtures were subsequently mixed with liposomes, and the permeation rates of the tested sugars were determined. Each experiment was repeated three times using BSA, BSA–antibodies and anti-BpsOmp38 antibodies without any protein as control tests. Permeation of glucose through the refolded Omp38 mixed with anti-(rabbit β-glucosidase) antibodies at various concentrations was measured as an additional control. ABs, antibodies; ara, L-arabinose; gl, D-glucose; man, D-mannose; gal, D-galactose; glcNAc, N-acteylglucosamine; suc, D-sucrose; mel, D-melezitose; rabbit, anti-(rabbit β-glucosidase) polyclonal antibodies.
DISCUSSION
Although several porins have been identified and characterized from Gram-negative bacteria, previous reports have demonstrated that expression of porins using conventional plasmid systems turned out to be difficult, since high expression of intact heterologous gene products was usually lethal to E. coli host cells [31–35]. However, a few studies have reported expression of porins lacking signal peptide sequences as aggregated inclusion bodies. Examples are the expression of Hib (Haemophilus influenzae type b) in Bacillus subtilis using pKTH288 [36], Neisseria species class 3 porin in E. coli BL21(DE3)ΔompA using pET17b [26], Rhodopseudomonas blastica porin in E. coli BL21(DE3)pLysS using pET3a [37], Burkholderia cepacia OpcP in E. coli JM109 using pTrc99A [38], Orientia tsutsugamushi r56 in E. coli BL21 using pET11a [39] and Mycobacterium smegmatis MspA in E. coli BL21(DE3) using pET24(+) [40]. In the present study, BpsOmp38 and BthOmp38 lacking signal peptide sequences were expressed successfully as insoluble inclusion bodies in E. coli Origami(DE3) using the pET23d(+) vector system. Generally, E. coli Origami(DE3) cells are used to express highly soluble proteins. However, this host strain was chosen to express Omp38, based on our findings that anti-Bps-Omp38 antibodies did not react with all proteins from the whole-cell lysate of E. coli Origami(DE3) (Figure 1B, lanes 1 and 3). In contrast, other E. coli host strains, including HMS174(DE3), NovaBlue(DE3) and BL21(DE3)pLysS, appeared to express endogenous outer membrane proteins, which crossed-reacted with the BpsOmp38 antiserum. In addition, these protein bands gave overlapping Mr (∼35000–40000) with the Mr of monomeric Omp38. Thus these E. coli strains were not applicable for Omp38 expression. Anti-BpsOmp38 polyclonal antibodies strongly recognized the expressed 38 kDa protein, and seven tryptic peptides identified by MALDI–TOF MS were found to be identical with those of the native proteins, giving strong evidence that the expressed 38 kDa proteins were Omp38.
Amphiphilic molecules, for instance phospholipids and detergents, were reported to facilitate refolding of bacterial porins [21,37,41]. In the present study, we have tried several refolding systems using different amphiphilic molecules to functionally refold the expressed Omp38. To examine the influence of the type of detergents and phospholipids on the refolding capability of Omp38, a combination of different phospholipids and detergents was added to the urea-solubilized 38 kDa proteins. These compounds included SDS, CHAPS, LDAO (N,N-dimethyldodecylamine N-oxide), MEGA-9 (n-nonanoyl-N-hydroxyethylglucamide), β-OG (n-octyl β-D-glucoside), Tween 20, Triton X-100, octyl-POE (octyl-polyoxyethylene), phosphatidylcholine (lecithin), and a short-chain phospholipid of diheptanoyl phosphatidylcholine (C7). It has been found that 1% SDS in combination with phosphatidylcholine helped the proteins to refold. However, phosphatidylcholine has been reported to be inappropriate for refolding proteins for crystallization purposes, because of its relatively large amphiphilic structure, thus generating poor X-ray diffraction patterns [41]. In addition, SDS seriously interfered with functional assays and disrupted protein crystallization due to the fact that SDS strongly binds to biological macromolecules in such a way that it is difficult to be removed or exchanged. A mixture of LDAO and phospholipid (C7), and a mixture of β-OG and phospholipid (C7), also partially promoted refolding of Omp38, however, with the monomers (∼30%) and dimers (∼5%) remaining in the protein mixture. When other detergents were tried, the proteins tended to aggregate instead of refolding. Moreover, using a detergent or phospholipid on its own failed to provide well-solubilized or refolded Omp38.
Since Zwittergent® 3-14 has been suggested to promote refolding of several bacterial porins [25,26,42], various concentrations of Zwittergent® 3-14 have ultimately been tried for refolding of our Omp38. It was found that the refolding buffer system (200 mM NaCl, 0 mM EDTA and 0.02% NaN3 in 20 mM Tris/HCl, pH 7.0) containing 10% Zwittergent® 3-14 and 20 mM CaCl2 was best to help the monomeric Omp38 to re-associate into the trimeric conformation. Under such conditions, the protein could form the trimer almost completely, leaving only trace amounts of monomer (<3%) and dimer (∼5%) (see Figure 2A, lanes 2 and 5). Complete folding was observed when the protein mixture was subsequently heated at 95 °C for 5 min and incubated overnight at room temperature. Apparently, the heat treatment following the prolonged incubation at the lower temperature assisted the misfolded protein to undergo conformational rearrangements, eventually leading to refolding of the protein to its trimeric form. When the concentration of the Zwittergent® 3-14 was reduced to 0.05%, the protein solution still remained clear. This indicated a relatively stable conformation of the re-associated proteins. Identical secondary-structure composition of the refolded and native proteins, as revealed by CD spectroscopy (Figure 4), verified the proper folding of the trimeric Omp38. The adsorption spectra obtained reflected a predominant β-sheet structure of the proteins, which corresponded well with our FTIR (Fourier-transform IR) spectroscopy measurements [22]. It was noticeable that the spectrum of native BpsOmp38 did not overlap precisely with other protein spectra, especially at the lower wavelengths. This could be an effect of LPS (lipopolysaccharide) that was tightly bound to the protein prepared from B. pseudomallei. The LPS was also found to alter the FTIR spectral shape of BpsOmp38 in the ester carbonyl stretching regions as discussed in our previous report [22]. It is important to note that a dramatic decrease in yield of the trimer was observed when concentrations of the proteins in 8 M urea were in excess of 10 mg/ml before dilution with Zwittergent® 3-14. Instead of refolding, the proteins re-aggregated and formed a turbid solution. A similar problem had also been observed in a previous report describing attempts to refold Neisseria porins expressed in E. coli [26]. Moreover, refolding of the expressed Omp38 could not be achieved when the 38 kDa proteins were solubilized with denaturing systems other than 6 M urea (i.e. 2% SDS or 6 M guanidinium chloride) before they were subjected to the refolding step using Zwittergent® 3-14 detergent.
Like with native Omp38, the diffusion rates of small neutral sugars through the refolded BpsOmp38 and BthOmp3 were Mr-dependent, with the size-exclusion limit of the protein pores estimated to be ∼Mr 650. Interesting findings were obtained with respect to the determination of the diffusion rates of a set of different antibiotics. Gentamicin (Mr 782), for example, could not entirely pass through the Omp38 channels (Table 1), although B. pseudomallei is known to be sensitive to this antibiotic. On the contrary, clindamicin (Mr 505) was able to pass through, even though the bacterium exhibits high resistance to such an antibiotic. Although positive charges on the amikacin and gentamicin molecules could possibly have an effect on penetration rates, it was obvious that both antibiotics could not pass through due to the fact that their molecular sizes were just too large to enter the constrictive size of the Omp38 pores. A linear relation between the relative permeation rates and the Mr of other small antibiotics (see Figure 6) clearly indicated that the uptake of such compounds occurred via a general diffusion mechanism. The nearly indistinguishable curve fit is a suggestion that the transport process of the charged antibiotics through the channels was taking place in an identical manner with the uncharged sugars, which was entirely dependent on the Mr with no interference from ionic charges on the solutes. These findings are in contrast with a previous report on OmpF, OmpC and PhoE channels [29], where negatively charged solutes dramatically exhibited a retarding effect on penetration rates through the OmpF and OmpC channels, but an accelerating effect on the PhoE channel. From our observations, it has to be concluded that a susceptibility of B. pseudomallei to certain antibiotics is not associated directly with the permeation of the antibiotics through Omp38 pores. It is likely that the antibiotic resistance of the bacterium occurs via a more complex molecular mechanism than just the hindrance of diffusion into the cell interior through the size-selectivity of porin channels. A relevant report has demonstrated that AmrAB-OprA, a multi-drug efflux system, plays a more specific role in transportation of aminoglycoside and macrolide antibiotics [9]. From their findings, two generated transposon mutants of the AmrAB-OprA proteins had led to hypersusceptibilities of B. pseudomallei to several antibiotics, including streptomycin, gentamicin, neomycin, tobramycin, kanamycin, spectinomycin and clindamycin.
Immunodepletion experiments revealed that anti-BpsOmp38 polyclonal antibodies had inhibitory effects on pore-forming activity of BpsOmp38 and BthOmp38 (Figure 6). This is in an agreement with a study from Wyllie et al. [43], who reported that the MOMP (major outer membrane protein) of Chlamydia psittaci-specific monoclonal antibody could modify the opening and closing states of MOMP channels that were inserted in planar lipid bilayer membranes. After addition of anti-MOMP monoclonal antibody, the opening states of MOMP were reduced to much lower amplitude. Based on the facts that the diffusion behaviour of the selected solutes were identical for the refolded and native Omp38 with the permeability rates dependent on the molecular sizes of the solutes, and that the passage of the solutes through the two proteins was inhibited indiscriminately by anti-BpsOmp38 polyclonal antibodies, we concluded that the refolded Omp38 functioned fully as a general diffusion pore.
In conclusion, we present the expression of BpsOmp38 and BthOmp38 using the pET23d(+) vector and E. coli Origami(DE3) system. We also report the successful in vitro re-assembly of the monomeric subunits into their trimeric forms. The refolded proteins have been proved to be structurally and functionally identical with their native counterparts. Currently, three-dimensional structure determination using the protein crystallization technique is being attempted to reveal the molecular structure of the Omp38 proteins. Future work will be focused on single channel measurements using the reconstitution of the proteins into planar lipid bilayers to elucidate the basic function of Omp38 as a porin channel.
Acknowledgments
This work was supported financially by a Suranaree University of Technology Grant (grant number: SUT-106-46-36-02) to W. S. and a Shell Studentship to J. S. We thank Miss Phimonphan Chuankhayan, School of Biochemistry, Institute of Science, Suranaree University of Technology, Thailand, for kindly providing the anti-β-glucosidase polyclonal antibodies, and Dr Albert Schulte, Department of Analytical Chemistry, Ruhr University of Bochum, Germany, for critical readings of the manuscript.
References
- 1.Vendrous N. A., Chow D., Liong E. Experimental vaccine against Pseudomonas pseudomallei infections in captive cetaceans. Dis. Aquat. Org. 1988;5:157–161. [Google Scholar]
- 2.Dance D. A. B. Melioidosis: the tip of the iceberg. Clin. Microbiol. Rev. 1991;4:52–60. doi: 10.1128/cmr.4.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Currie B., Smith-Vaughan H., Golledge C., Buller N., Sriprakash K. S., Kemp D. J. Pseudomonas pseudomallei isolates collected over 25 years from a non-tropical endemic focus show clonality on the basis of ribotyping. Epidemiol. Infect. 1994;113:307–312. doi: 10.1017/s0950268800051736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arakawa M. Infection with Pseudomonas pseudomallei. Rinsho Byori. 1990;38:1226–1231. [PubMed] [Google Scholar]
- 5.Brett P. J., DeShazer D., Woods D. E. Burkholderia thailandensis sp. nov., a Burkholderia pseudomallei-like species. Int. J. Syst. Bacteriol. 1998;48:317–320. doi: 10.1099/00207713-48-1-317. [DOI] [PubMed] [Google Scholar]
- 6.Smith M. D., Angus B., Wuthiekanun V., White N. J. Arabinose assimilation defines a non-virulent biotype of Burkholderia pseudomallei. Infect. Immun. 1997;65:4319–4321. doi: 10.1128/iai.65.10.4319-4321.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dharakul T., Tassaneetrithep B., Trakulsomboon S., Songsivilai S. Phylogenetic analysis of Ara+ and Ara− Burkhoderia pseudomallei isolates and development of a multiplex PCR procedure for rapid discrimination between the two biotypes. J. Clin. Microbiol. 1999;37:1906–1912. doi: 10.1128/jcm.37.6.1906-1912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reckseidler S., Deshazer D., Sokol P. A., Woods D. E. Detection of bacterial virulence genes by subtractive hybridization: identification of capsular polysaccharide of Burkholderia pseudomallei as a major virulence determinant. Infect. Immun. 2001;69:34–44. doi: 10.1128/IAI.69.1.34-44.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore R. A., Deshazer D., Reckseidler S., Weissman A., Woods D. E. Efflux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei. Antimicrob. Agents Chemother. 1999;43:465–470. doi: 10.1128/aac.43.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White N. J. Melioidosis. Lancet. 2003;361:1715–1722. doi: 10.1016/s0140-6736(03)13374-0. [DOI] [PubMed] [Google Scholar]
- 11.Bianco N., Neshat S., Poole K. Conservation of the multidrug resistance efflux gene oprM in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1997;41:853–856. doi: 10.1128/aac.41.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jap B. K., Walian P. J. Structure and functional mechanism of porins. Physiol. Rev. 1996;76:1073–1088. doi: 10.1152/physrev.1996.76.4.1073. [DOI] [PubMed] [Google Scholar]
- 13.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003;267:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikaido H. Porins and specific channels of bacterial outer membranes. Mol. Microbiol. 1992;6:435–442. doi: 10.1111/j.1365-2958.1992.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 15.Cowan S. W., Schirmer T., Rummel G., Steiert M., Ghosh R., Pauptit R. A., Jansonius J. N., Rosenbusch J. P. Crystal structures explain functional properties of two E. coli porins. Nature (London) 1992;358:727–733. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- 16.Zeth K., Diederichs K., Welte W., Engelhardt H. Crystal structure of Omp32, the anion-selective porin from Comamonas acidovorans, in complex with a periplasmic peptide at 2.1 Å resolution. Structure Fold Des. 2000;8:981–992. doi: 10.1016/s0969-2126(00)00189-1. [DOI] [PubMed] [Google Scholar]
- 17.Charbit A. Maltodextrin transport through LamB. Front. Biosci. 2003;8:265–274. doi: 10.2741/1046. [DOI] [PubMed] [Google Scholar]
- 18.Schirmer T. General and specific porins from bacterial outer membranes. J. Struct. Biol. 1998;121:101–109. doi: 10.1006/jsbi.1997.3946. [DOI] [PubMed] [Google Scholar]
- 19.Nitzan Y., Orlovsky K., Pechatnikov I. Characterization of porins isolated from the outer membrane of Serratia liquefaciens. Curr. Microbiol. 1999;8:71–79. doi: 10.1007/s002849900406. [DOI] [PubMed] [Google Scholar]
- 20.Weiss M. S., Schulz G. E. Structure of porin refined at 1.8 Å resolution. J. Mol. Biol. 1992;277:493–509. doi: 10.1016/0022-2836(92)90903-w. [DOI] [PubMed] [Google Scholar]
- 21.Pebay-Peyroula E., Garavito R. M., Rosenbusch J. P., Zulauf M., Timmins P. A. Detergent structure in tetragonal crystals of OmpF porin. Structure. 1995;3:1051–1059. doi: 10.1016/s0969-2126(01)00241-6. [DOI] [PubMed] [Google Scholar]
- 22.Siritapetawee J., Prinz H., Samosornsuk W., Ashley R. H., Suginta W. Functional reconstitution, gene isolation and topology modelling of porins from Burkholderia pseudomallei and Burkholderia thailandensis. Biochem. J. 2004;377:579–587. doi: 10.1042/BJ20031118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gotoh N., White N. J., Chaowagul W., Woods D. Isolation and characterization of the outer-membrane proteins of Burkholderia (Pseudomonas) pseudomallei. Microbiology. 1994;140:797–805. doi: 10.1099/00221287-140-4-797. [DOI] [PubMed] [Google Scholar]
- 24.Garavito R. M., Rosenbusch J. P. Isolation and crystallization of bacterial porin. Methods Enzymol. 1986;125:309–328. doi: 10.1016/s0076-6879(86)25027-2. [DOI] [PubMed] [Google Scholar]
- 25.Pullen J. K., Liang S. M., Blake M. S., Mates S., Tai J. Y. Production of Haemophilus influenzae type-b porin in Escherichia coli and its folding into the trimeric form. Gene. 1995;152:85–88. doi: 10.1016/0378-1119(94)00706-x. [DOI] [PubMed] [Google Scholar]
- 26.Qi H. L., Tai J. Y., Blake M. S. Expression of large amounts of Neisseria porin proteins in Escherichia coli and refolding of the proteins into native trimers. Infect. Immun. 1994;62:2432–2439. doi: 10.1128/iai.62.6.2432-2439.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conlan S., Zhang Y., Cheley S., Bayley H. Biochemical and biophysical characterization of OmpG: a monomeric porin. Biochemistry. 2000;39:11845–11854. doi: 10.1021/bi001065h. [DOI] [PubMed] [Google Scholar]
- 28.Shevchenko A., Wilm M., Vorm O., Mann M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 29.Nikaido H., Rosenberg E. Y. Porin channels in Escherichia coli: studies with liposomes reconstituted from purified proteins. J. Bacteriol. 1983;153:241–252. doi: 10.1128/jb.153.1.241-252.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulieris P. V., Behrens S., Holst O., Kleinschmidt J. H. Folding and insertion of the outer membrane protein OmpA is assisted by the chaperone Skp and by lipopolysaccharide. J. Biol. Chem. 2003;278:9092–9099. doi: 10.1074/jbc.M211177200. [DOI] [PubMed] [Google Scholar]
- 31.Carbonetti N. H., Sparling P. F. Molecular cloning and characterization of the structural gene for protein I, the major outer membrane protein of Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U.S.A. 1987;86:2172–2175. doi: 10.1073/pnas.84.24.9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gotschlich E. C., Seiff M. E., Blake M. S., Koomey M. Porin protein of Neisseria gonorrhoeae: cloning and gene structure. Proc. Natl. Acad. Sci. U.S.A. 1987;84:8135–8139. doi: 10.1073/pnas.84.22.8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barlow A. K., Heckels J. E., Clarke I. N. Molecular cloning and expression of Neisseria meningitidis Class 1 outer membrane protein in Escherichia coli K12. Infect. Immun. 1987;55:2734–2740. doi: 10.1128/iai.55.11.2734-2740.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carbonetti N. H., Simnad V. I., Seifert H. S., So M., Sparling P. F. Genetics of protein I of Neisseria gonorrhoeae, construction of hybrid porins. Proc. Natl. Acad. Sci. U.S.A. 1988;85:6841–6845. doi: 10.1073/pnas.85.18.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolstad A., Jensen H. B. Complete sequence of Omp1, the structural gene encoding the 40-kDa outer membrane proteins of Fusobacterium nucleatum strain Fev1. Gene. 1993;132:107–112. doi: 10.1016/0378-1119(93)90521-4. [DOI] [PubMed] [Google Scholar]
- 36.Dahan D., Srikumar R., Laprade R., Coulton J. W. Purification and refolding or recombinant Haemophilus influenzae type b porin produced in Bacillus subtilis. FEBS Lett. 1996;392:304–308. doi: 10.1016/0014-5793(96)00841-1. [DOI] [PubMed] [Google Scholar]
- 37.Schmid B., Kromer M., Schulz G. E. Expression of porin from Rhodopseudomonas blastica in Escherichia coli inclusion bodies and folding into exact native structure. FEBS Lett. 1996;381:111–114. doi: 10.1016/0014-5793(96)00080-4. [DOI] [PubMed] [Google Scholar]
- 38.Tsujimoto H., Gotoh N., Yamagishi J., Oyamada Y., Nishino T. Cloning and expression of the major porin protein gene opcP of Burkholderia (formerly Pseudomonas) cepacia in Escherichia coli. Gene. 1997;186:113–118. doi: 10.1016/s0378-1119(96)00692-0. [DOI] [PubMed] [Google Scholar]
- 39.Ching W. M., Wang H., Eamsila C., Kelly D. J., Dasch G. A. Expression and refolding of truncated recombinant major outer membrane protein antigen (r56) of Orientia tsutsugamushi and its use in enzyme-linked immunosorbent assays. Clin. Diagn. Lab. Immunol. 1998;5:519–526. doi: 10.1128/cdli.5.4.519-526.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinz C., Karosi S., Niederweis M. High-level expression of the mycobacterial porin MspA in Escherichia coli and purification of the recombinant protein. J. Chromatogr. 2003;790:337–348. doi: 10.1016/s1570-0232(03)00130-2. [DOI] [PubMed] [Google Scholar]
- 41.Eisele J. L., Rosenbusch J. P. In vitro folding and oligomerization of a membrane protein: transition of bacterial porin from random coil to native conformation. J. Biol. Chem. 1990;265:10217–10220. [PubMed] [Google Scholar]
- 42.Ulmer J. B., Burke C. J., Shi C., Friedman A., Donnelly J. J., Liu M. A. Pore formation and mitogenicity in blood cells by the class 2 protein of Nesseria meningitidis. J. Biol. Chem. 1992;267:19266–19271. [PubMed] [Google Scholar]
- 43.Wyllie S., Ashley R. H., Longbottom D., Herring A. J. The major outer membrane protein of Chlamydia psittaci functions as a porin-like ion channel. Infect. Immun. 1998;66:5202–5207. doi: 10.1128/iai.66.11.5202-5207.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]