

Summary
Astroglia play crucial neuroprotective roles by internalizing pathogenic aggregates and facilitating their degradation. Here, we show that α-SYN protofibril-induced organelle toxicities and reactive oxygen species (ROS) cause premature cellular senescence in astrocytes and astrocyte-derived cancer cells, resulting in a transient increase in the biogenesis of tunneling nanotubes (TNTs). TNT-biogenesis and TNT-mediated cell-to-cell transfer lead to clearance of α-SYN-induced organelle toxicities, reduction in cellular ROS levels, and reversal of cellular senescence. Enhanced cell proliferation is seen in the post-recovered cells after recovering from α-SYN-induced organelle toxicities. Further, we show that α-SYN-induced senescence promotes the transient localization of focal adhesion kinase (FAK) in the nucleus. FAK-mediated regulation of Rho-associated kinases plays a significant role in the biogenesis of TNTs and their subsequent proliferation. Our study emphasizes that TNT biogenesis has a potential role in the clearance of α-SYN-induced cellular toxicities, the consequences of which cause enhanced proliferation in the post-recovered astroglia cells.
Subject areas: Biological sciences, Molecular biology, Neuroscience, Cell biology
Graphical abstract
Highlights
-
•
α-SYN treated astroglia cells proliferate upon the transient biogenesis of TNTs
-
•
Transient TNT formation precedes clearance of α-SYN toxicities & reversal of senescence
-
•
Senescence results in nuclear FAK localization and ROCK-mediated TNT biogenesis
-
•
The rescued cells enhance proliferation via ROCK-mediated NF-κB signaling cascades
Biological sciences; Molecular biology; Neuroscience; Cell biology
Introduction
Parkinson’s disease (PD) is characterized by the loss of dopaminergic neurons in the substantia nigra (SN) due to the cytoplasmic accumulation of Lewy bodies or Lewy neurites, which mainly consist of α-synuclein (α-SYN) protein.1,2 Along with the accumulated cytoplasmic α-SYN, extracellular α-SYN also plays a significant role in neurodegeneration, progressive intercellular spreading of pathology, and neuroinflammation.3,4 Studies have shown that the elevation of neuronal activity and various stress conditions increase extracellular release of α-SYN.4,5 Pathogenic aggregates of α-SYN can be released from degenerating neurons, which can be taken up by surrounding neurons and glial cells. Glial cells normally express low levels of α-SYN, however, at the advanced stage of PD, α-SYN aggregates in astrocytes and glial synucleinopathies are often detected.6 Additionally, gliosis is a typical pathological feature of neurodegenerative diseases. Sustained activation and fibrous proliferation of glial cells, mainly astrocytes and microglia, are central features of dopaminergic neurodegeneration in PD.7 Neuronal activity modulates cellular crosstalk and intercellular communication between neurons and the surrounding neuroglial cells.8,9,10
Recent studies have shown that the mode of intercellular transfer of neurodegenerative proteins between glial cells facilitates the clearing of neurodegenerative aggregates, such as α-SYN and amyloid-β (Aβ).11,12,13 The spreading of neurodegenerative proteins through an intercellular mode of transfer and its role in pathology progression have been widely studied in several model systems.14,15 Exosomes, unconventional secretion, and direct cell-to-cell transfer via membrane nanotubes or tunneling nanotubes (TNTs) have been demonstrated by several studies as modes of intercellular transfer.14
The discoveries of TNTs have opened up the possibility of direct long-range cell-to-cell communication.16 TNTs have been shown to have open-ended, and thin (diameter around 50–700 nm) intercellular membrane-actin continuity (long up to 300 μm) between distant cells. TNTs are reported to be a conduit for the direct transfer of organelles,16 neurodegeneration-associated protein aggregates,15,17 viruses,18 and RNA.19 between cells. Neurodegenerative aggregate-induced endo-lysosomal toxicities and mitochondrial stress promote the biogenesis of TNTs.15,20 Propagation of pathogenic aggregates via TNTs has also been shown to aid in the progression of neurodegeneration.13,15,21 In this context, several studies have shown that α-SYN aggregates in lysosomes transfer from neuron to neuron via TNTs in PD.15,22,23 On the other hand, TNTs mediate crosstalk between astrocytes, and microglia to degrade toxic α-SYN through cell-to-cell transfer of aggregates and help to reduce ROS accumulation and oxidative stress.11,13 Oxidative stress and ROS promote the formation of TNTs and cell-to-cell transfer.24,25
ROS induced by the accumulation of neurotoxic proteins in neurons can induce cell senescence, which can aggravate neurodegeneration.26,27 Cellular senescence is a biologically homeostatic phenomenon that is defined by a persistent degree of cell cycle inhibition and cellular aging. Cell cycle arrest in senescence cells is believed to be irreversible, however, studies have shown that stress-induced premature senescence that is induced by the DNA damage-mediated amplification of the p53/p21 signaling pathway can be reversed by modulating the levels of p53 and p21 expressions.28,29
Senescent cells are more prevalent in brain tissues and exert roles in brain aging and neurodegeneration.30 Brain cells, including astrocytes, microglia, oligodendrocytes, and epithelial cells, undergo cellular senescence under oxidative stress, which aggravates neurodegeneration.31 On the contrary, glial cells, especially astrocytes, and microglia, play a critical role in protecting neurons by clearing toxic accumulations of neurodegenerative proteins from neurons and brains.11,13 α-SYN induced ROS generation can produce cellular senescence in response to DNA damage.27 Thus, the investigation into the mechanisms of TNT biogenesis and the role of TNTs in facilitating the clearance of toxic α-SYN protofibrils from brain to improve cell survival is needed.
In this study, we have shown that the transient localization of FAK/pFAK to the nucleus upon treatment with α-SYN protofibrils caused transient biogenesis of TNTs in the astroglia cells. Transient biogenesis of TNTs precedes the clearance of α-SYN-induced organelle toxicities and reversal of ROS-induced premature senescence, leading to enhanced cell proliferation in the post-recovered astroglia cells. This study emphasizes the potential role of TNTs in facilitating cellular clearance in pathological stress conditions, which maintains the survival of astroglia cells, by enhancing cell proliferation.
Results
α-synuclein protofibrils induce the biogenesis of transient tunneling nanotubes
The contributions of extracellular α-SYN protofibrils in the pathological spreading of PD in neuronal cell culture models are well-studied.32 Recent studies have shown that aggregates of α-SYN can modulate cellular crosstalk between neurons and the surrounding neuroglial cells,33 which plays an important role in getting rid of toxic loads of aggregates.11,13 However, the mechanism of the establishment of intercellular communications and their fate are yet to be understood. Therefore, we looked into the primary astrocytes and astrocyte-origin cancer cell lines (U-87 MG and U251) to understand the possible fate of the astroglia cells over time on α-SYN protofibril treatment.
To understand this, purified α-SYN was converted into protofibrils that were either unlabeled or fluorescently (TMR) labeled and characterized using transmission electron microscopy (Figure 1A). Interestingly, after treatment with 1μM α-SYN protofibrils at 3h, 6h, 12h, and 24h, we observed the formation of TNT-like membrane networks between primary astrocytes (Figure 1B), U-87 MG (Figures 1C and S1A) and U251 cells (Figure S1B) for a transient period at early hours. Time points were chosen to quantify the optimum biogenesis of membrane nanotubes after treatment with 1μM α-SYN protofibrils over 24 h (Figure S1C). Quantification of TNT-like structures was performed on confocal images of phalloidin-stained astrocytes (Figure 1B). DiD dye-stained membranes were imaged using a fluorescence microscope to detect the abundance of stained TNT-like membrane conduits in live cells (Figure 1C). DIC (Differential Interference Contrast) images of U-87 MG (Figure S1A) and U251 (Figure S1B) cells were also taken to detect TNT-like structures. Quantification showed an increase in several hovering thin TNT-like structures per cell compared to controls in the primary astrocytes at early time points (3h and 6h) (Figure 1D). DiD imaging of U-87 MG showed a transient increase in thin (diameters <1 μm) cell-to-cell membrane connections at early time points (3h and 6h) (Figures 1E and 1F). Similarly, the quantification of DIC images of U-87 MG cells (Figure S1D) and U251 (Figure S1E) showed enhanced transient biogenesis of TNTs at early time points compared to their respective controls and later time points.
Figure 1.

Characterization of α-SYN protofibrils and their effect on TNT formation in astroglia cells
(A) TEM (transmission electron microscope) images of α-SYN protofibrils unlabeled and TMR fluorescent dye labeled, respectively. Magnified structures of protofibrils are shown in the zoomed panel.
(B) Primary astrocytes were treated with 1μM α-SYN protofibrils for 3h, 6h, 12h, and 24h, stained for actin (red) and GFAP (green), white arrows indicate the formation of shorter and longer TNTs at 3h and 6h.
(C) DiD (membrane dye) stained U-87 MG cells showing a transient increase of TNT-like structures (red arrows) in the cells treated with α-SYN protofibrils (1 μM) for 3–6 h, compared to the respective controls and the cells treated for 12 h and 24 h. Blue arrows indicate relatively thicker tumor microtubes (TMs) like connections.
(D) The percentage of TNTs from primary astrocytes was quantified by counting the numbers normalized with the number of cells.
(E and F) Quantification of the percentage of TNT-like structures and pairs of cells connected by them upon α-SYN protofibril treatment in U-87 MG cells stained with DiD. Quantifications are done from 10 to 15 image frames of a set and each image frame has 10–20 cells. Scale bars are denoted on the images. Data are expressed as mean ± SD, ∗∗∗p ≤ 0.001. Statistics were analyzed using a two-way ANOVA. n = 3.
α-synuclein protofibrils induce open-ended tunneling nanotubes and cell-to-cell transfer
TNTs are actin-rich, nano-sized in diameter, and primarily defined as open-ended membrane tubes.34 TNTs are distinguished from filopodia and neurites by their distinct characteristic of hovering between two distant cells.35 Actin-binding phalloidin and GFAP-stained, 3D-reconstructed confocal images show actin-positive thin TNTs that hover between two cells in primary astrocytes (Figure 2A). Astrocyte-origin cancer cell lines (U-87 MG and U251) are known to form tumor microtubes (TMs), close-ended thicker membrane networks (diameters >1 μm) between cells and electrically coupled via gap junctions (GJ).36 To distinguish TNTs and TMs, confocal z stack images were obtained after immunocytochemical staining of cells using β-tubulin antibody and phalloidin dye. Thin actin-positive, hovering TNTs (Figure 2B) and thicker TMs (Figure 2C) are identified in U-87 MG cells. TMs are positive for both actin and β-tubulin. Studies have indicated that astrocytoma/glioblastoma cells express lower levels of neuronal marker β-tubulin and are stained faintly.37 Despite that, β-tubulin staining is distinctly detected in thicker TMs and absent in TNTs (Figures S2A and S2B). DIC images clearly showed the nano-sized diameters of TNTs and thicker TMs (Figures 2A–2C-upper panels). Reconstructed 3D volume images of confocal z-stacks show actin-positive, thin TNTs hover between two cells and are not attached to the substratum, whereas the tubulin-positive thicker TMs are present on the substratum (Figures 2A–2C-lower panels).
Figure 2.

Characterization of TNTs formed upon treatment with α-SYN protofibrils in astroglia cells
(A) The red arrow in the DIC image (a’) indicates TNT formed between two astrocytes on α-SYN protofibril treatment. The bottom panel shows a 3D volume view of Phalloidin and GFAP-positive astrocytes connected by both long (a’’) and short TNTs (b).
(B and C) DIC images of U-87 MG cells show TNTs (B) and TMs (C). The cells were stained with phalloidin and β-tubulin; both bottom panels show 3D volume views of TNT as an actin-positive hovering structure (B) and tubulin-positive TM at the surface (C). Red arrows indicate actin-positive TNTs and blue arrows indicate β-tubulin-positive tumor microtubes (TM), respectively.
(D and E) Characterization of GJ-negative (D) and -positive (E) TNTs based on Phalloidin and connexin43 staining at the tip of the TNT in U-87 MG cells. Pink arrows indicate close-ended TNT. The bottom panel shows 3D volume views of the same.
(F) Closed-ended TM characterized by the presence of connexin43 staining, indicated by pink arrows. The bottom panel shows 3D volume views of the TM at the surface.
(G) TNT-like structures (red arrows) are detectable in DIC images as focused structures at z = 4. At z = 0, filopodia-like extensions (magenta arrows) on the substratum are at focus.
(H and I) U87MG cells treated with 1μM α-SYN protofibrils for 3h and 6h, stained with lysotracker and MitoTracker, respectively. Red arrows indicate lysotracker and MitoTracker through TNTs at 3h and 6h. Movement of lysotracker (H) and MitoTracker (I) positive vesicles through TNTs were tracked using the TrackMate plug-in of Fiji.
(J) Quantification of the percentage of TNTs and (K) percentage of GJ-negative TNTs, GJ-positive TNTs, and TMs from the confocal z-stack images of U-87 MG cells. Quantification of length (L) and diameter (M) of TNTs formed by U-87 MG cells, U251 cells, and astrocytes. Quantifications are done from 10 to 15 image frames of a set and each image frame has 10–20 cells. Scale bars are denoted on the images. Data are expressed as mean ± SD, ∗∗∗p ≤ 0.001. Statistics were analyzed using a two-way ANOVA (2J and 2K) and one-way ANOVA (2L and 2M). n = 3.
Several studies have shown that TNTs are open-ended membrane channels, and can transfer organelles directly from one cell to another.16,23 Occasionally, these channels are electrically connected via GJ proteins. This may lead to the formation of closed membrane continuities that might not be involved in organelle transfer between cells.38,39 The altered distribution of Connexin43, a crucial GJ protein, may create selectively open ends. TNTs were immunostained with connexin43 to identify the presence of GJ-like structures at the connecting end of the cell body. Images captured populations of both connexin43 negative (Figure 2D) and positive (Figure 2E) at the endpoint of TNTs. Non-hovering thicker TMs are connexin43 positive structures (Figure 2F).
We further functionally characterized TNTs based on their ability to transfer organelles such as lysosomes and mitochondria. Time-lapse and z-stack images were taken using confocal microscopy (using both fluorescence channels and DIC channels) in live U-87 MG cells, stained with lysotracker and MitoTracker, post-treatment with 1μM α-SYN protofibrils (3h, 6h, 12h, and 24h). Time-lapse videos in DIC microscopy images revealed a transient increase in the biogenesis of thin cell-to-cell membrane connections and unidirectional movements of organelles via the membrane nanotubes at early time points (3h and 6h) (Videos S1 and S2). Time-lapse videos captured cell-to-cell movement of α-SYN-TMR-accumulated lysosomes and mitochondria through long (20 μm–120 μm) thin TNTs after 3h and 6 h of treatments, respectively (Videos S3 and S4). We validated from z-stack images, that the thin nano-sized membrane tubes, or TNTs, are hovering structures connecting two distant cells. The DIC images show thin membrane nanotubes (red arrows) between cells that are clearly visible at a higher plane of z = 4. On the surface, at z = 0, filopodia-like membrane extensions (violet arrows) are visible (Figure 2G) which are unlike the TNTs. We tracked the movements of lysotracker (Figure 2H) and MitoTracker-labelled (Figure 2I) organelles along the TNTs (identified from z-stacks) using the TrackMate plug-in available in FIJI and we observed that both organelles travel unidirectionally from one cell toward another, with an average speed of 0.062 ± 0.003 μm/s. Further, we captured time-lapse videos to detect direct cell-to-cell transfer of α-SYN-TMR accumulated lysotracker and mitochondria via open-ended TNTs between U-87 MG cells after 3h of treatments (Videos S5 and S6). Our results reveal the formation of open-ended functional TNTs and cell-to-cell transfer through them at 3h and 6h post-treatment with α-SYN.
Organelles were observed to move (indicated by white and black arrows) unidirectional inside the TNTs.
Organelles were observed to move unidirectionally (indicated by a white arrow) inside the TNTs.
The composite video shows unidirectional movements of vesicles colocalized (yellow) with α-SYN-TMR (red) and lysotracker (green) in long, thin TNTs (indicated by white arrow).
The composite video shows unidirectional movements of mitochondria (cyan) in long, thin TNTs (indicated by a white arrow).
Direct cell-to-cell transfer of α-SYN-TMR (red) accumulated lysotracker (green) positive vesicles (yellow vesicles; indicated by black arrow) via TNTs between neighboring U-87MG cells.
Direct cell-to-cell transfer of α-SYN-TMR (red) treated moto tracker positive vesicles (cyan vesicles; indicated by black arrow) via TNTs between neighboring U-87MG cells.
We observed a transient increase in the biogenesis of thin (diameters <1 μm), hovering TNTs upon 1μM α-SYN protofibrils treatment at early hours (3h and 6h) by quantifying from the 3D reconstruction of confocal z-stack images (Figure 2J). Though control cells contain a higher percentage of TMs, we observed a transient increase of both GJ-negative and GJ-positive TNTs, based on connexin43 staining after 3h and 6h post-treatment with α-SYN (Figure 2K). Length and diameters of TNTs were measured using the confocal z-stack images in all the cell types, primary astrocytes, U-87 MG, and U251 cells. The careful measurements of z-stack images detected that the diameter of TNTs ranges between 1 and 3 pixels, that is, around 220 nm–660 nm, and length varies between 20 μm and 100 μm across all the cell types (Figures 2L and 2M).
Transient lysosomal toxicities in astroglia cells upon treatment with α-synuclein protofibrils
Internalization of extracellularly applied α-SYN protofibrils into endo-lysosomal pathways induces transient accumulations and toxicities (3h and 6h) in primary astrocytes (Figure 3A) and U-87 MG cells (Figures 3B and 3C). α-SYN and LAMP1 were co-immunostained in primary astrocytes (Figure 3A) and U-87 MG cells treated with TMR-labelled α-SYN were stained with lysotracker (Figure 3B). Accumulations of α-SYN protofibrils in endo-lysosomes and resulting toxicities were studied following enlarged and distorted morphology of α-SYN-accumulated endo-lysosomal vesicles at 3h and 6h (Figures 3A and 3B). The extent of cathepsin leakage from LAMP2-positive vesicles was also observed to follow lysosomal toxicities (Figure 3C). α-SYN protofibrils were internalized in primary astrocytes and colocalized in substantial amounts to LAMP1-immunostained positive vesicles (analyzed using Coloc 2 plug-in in FIJI). Maximum colocalizations were observed at 3 h of treatment, which gradually decreased over time (Figure 3D). Internalization of protofibrils into the lysosomal pathway caused transient accumulation and lysosomal toxicities, resulting in larger-sized LAMP1 positive vesicles that were transiently detectable at 3h and 6h after α-SYN protofibrils treatment in astrocytes (Figure 3E). Total areas of red puncta of α-SYN-TMR oligomers per cell were quantified, measuring the size of each punctum. The results show that endo-lysosomal machinery in U-87 MG cells gradually clears up toxic protofibrils (Figure 3F). The half-life of the clearance of toxic protofibrils was determined to be 14.31 ± 5.24 h by fitting the one-phase decay equation to the α-SYN-TMR aggregates per cell data (Figure 3F). The half-life of extracellular α-SYN protofibrils (1μM) in the media of U-87 MG-treated cells was quantified using Western blot to follow the fate of the protofibrils (Figure S2C). The results show the half-life of elimination of the protofibrils is 12 h.
Figure 3.

Effect of α-SYN protofibrils on lysosomes in U-87 MG cells and astrocytes
(A) Mouse primary astrocytes treated with 1μM α-SYN protofibrils for 3h, 6h, 12h, and 24h. α-SYN protofibrils (green) in lamp1 positive vesicles (red). At 3h and 6h, larger sizes of lamp1 positive vesicles colocalized with α-SYN (yellow) are visible.
(B) U-87 MG cells treated with α-SYN-TMR protofibrils (red) in lysotracker-positive vesicles (green).
(C) U-87 MG cells treated with α-SYN protofibrils (1μM) were stained using Cathepsin D (red) and Lamp2 (green). At 3h larger sizes of lamp2 positive vesicles colocalized with cathepsin-D were observed, compared to the other time points (magnified images highlighted in upper-left corner).
(D) Quantification of the co-localization of α-SYN protofibrils in lamp1 positive lysosomes and (E) Size of α-SYN accumulated in lysosomes in astrocytes.
(F) Quantification of α-SYN accumulation per cell and (G) Lysosome size in U-87 MG cells. Quantifications are done by analyzing randomly selected 10–15 cells from different image frames of an experimental set. Scale bars are denoted on the images. Data are expressed as mean ± SD, ∗∗∗p ≤ 0.001. Statistics were analyzed using a two-way ANOVA. n = 3.
The analysis of cathepsin leakage in LAMP2-positive lysosomal vesicles indicated no significant increase of cathepsin-D to the cytosol (Figure 3C). However, we observed larger cathepsin-D and LAMP2-positive vesicles after 3h of protofibril treatments (Figure 3G). Morphology of LAMP2-positive lysosomes at 3h is elongated and rough in comparison to control, 6h, 12h, and 24h treated cells (magnified images highlighted in the upper-left corner of Figure 3C). The time of transient lysosomal toxicity coincides with the transient increase of TNT biogenesis (Figures 1B–1F). Altogether, the results suggest that the transfer of organelles via TNTs probably aids the cells in coping with protofibril-induced lysosomal toxicities.
Transient mitochondrial toxicities in astroglia cells upon treatment with α-synuclein protofibrils
Similar to transient lysosomal toxicities and biogenesis of TNTs, mitochondrial toxicities were observed through morphological changes in the early time points (3h and 6h) using MitoProbe JC-1 dye, which measures mitochondrial membrane potential (ΔΨM). Lipophilic, cationic JC-1 dye can enter healthy mitochondria and aggregate (emission @590 nm in the red channel) inside, whereas in unhealthy mitochondria with decreased membrane potential, the dye crosses in and out through the relatively open membrane pores and stays as monomers (emission @527 nm in the green channel). Adding to our previous observations, we noticed a decrease of ΔΨM transiently in the early time points (3h and 6h) in primary astrocytes, whereas at the later time points (12h and 24h), mitochondria looked healthier and were rescued from protofibril-induced mitochondrial toxicities (Figures 4A and 4B). The quantification shows JC-1 labeled red-colored healthier, larger mitochondria in the control and recovered healthier cells after 24h (Figure 4C). The toxic mitochondria in early time points (3h and 6h) were observed to possess shorter branch lengths compared to the control and the recovered cells at later times (12h and 24h) (Figure 4D). Mitochondrial toxicities were also observed in U-87 MG cells through morphological changes using MitoTracker staining (Figure 4E, upper panel) and mitochondrial membrane potential (ΔΨM) using MitoProbe JC-1 (Figure 4E, lower panel). Similar to our results in primary astrocytes, images of U-87 MG cells showed that at the early time points (3h and 6h), mitochondria were smaller and had fragmented morphology (Figure 4E, upper panel) and quantifications showed increased JC-1-stained greener mitochondria or mitochondria with decreased ΔΨM (Figure 4F). Thus, it is evident that primary astrocytes and U-87 MG cells adapt and are rescued from α-SYN protofibril-induced transient mitochondrial toxicities at later times.
Figure 4.

Effect of α-SYN protofibrils on mitochondria in astrocytes and U-87 MG cells
Primary astrocytes and U-87 MG were treated with 1μM α-SYN protofibrils for 3h, 6h, 12h, and 24h, and mitochondrial membrane potentials were detected using JC-1 staining (emission ratio of 527/590 nm).
(A) Primary astrocytes stained with JC-1.
(B) Quantification of green vs. red intensities, (C) Size of green/red mitochondria, and (D) mitochondria branch length by MiNA analysis were quantified from JC-1 astrocyte images.
(E) U87MG cells treated with 1μM α-SYN protofibrils for 3–24 h, were stained with MitoTracker (top panel -cyan) and JC-1 (bottom panel) to visualize mitochondrial morphology.
(F) Quantification of green vs. red intensities from JC-1 U-87 MG images. Quantification of green vs. red intensities. Quantifications are done from 10 to 15 image frames of a set, analyzing 10–20 cells from each image. Scale bars are denoted on the images. Data are expressed as mean ± SD, ∗∗∗p ≤ 0.001. Statistics were analyzed using a two-way ANOVA. n = 3.
Cell-to-cell transfer of mitochondria in α-synuclein protofibril-treated astroglia cells
The transfer of mitochondria between α-SYN protofibril-treated U-87 MG cells was further examined by co-culturing these cells (Figure 5A). MitoDsRed (red) and EGFP-lifeact (green) transfected U-87 MG cells were co-cultured and treated with α-SYN protofibrils for 3h, 6h, 12h, and 24h. Results demonstrate mitoDsRed-labeled red mitochondria in the green EGFP lifeact-stained cells upon treatment with α-SYN protofibrils (Figure 5A). The transferred red mitochondria in the green cells were observed at their maximum at early time points (3h and 6h) when cells show enhanced mitochondrial toxicities and increased numbers of TNTs (Figure 5B). When conditional media from the mitoDsRed transfected cells were given to the green cells transfected with EGFP- lifeact, we did not observe any significant transfer of mitochondria (Figure S3A). The zoomed images of (Figure 5A) the transferred mitochondria in the acceptor cells after 3h and 6h treatment with α-SYN are shown in Figure S3B. The result excludes the probability of the transfer of mitochondria through exosomes. The morphology of the transferred mitochondria in the green cells mostly appears round in shape rather than elongated and healthy. Quantification shows that transferred mitochondria are smaller in size and round-shaped compared to the mitochondria of the control cells (Figure 5C). Therefore, we further studied the fate of cell-to-cell transfer of toxic mitochondria, following ROS levels in the cells upon protofibril treatment at different time intervals. We also observed increased total cellular ROS at an early time point (3h) and a gradual decrease in ROS levels at later time points (6h, 12h, and 24h) in U-87 MG cells (Figures 5D and 5E). The fluorescence intensity of DCFDA was analyzed using a flow cytometer to quantify the cellular ROS levels (Figure 5E). The quantification of DCFDA flow cytometer data shows similar results in primary astrocytes, with a significant increase in cellular ROS levels at 3h compared to the control cells, which then decreased at later time points (6h, 12h, and 24h) (Figures 5F and 5G).
Figure 5.

Cell-cell transfer of mitochondria and α-SYN-induced ROS-mediated cellular senescence
(A) Transfer of mitochondria was detected in a co-culture experiment. U87MG cells were co-cultured using transiently transfected with Mito-ds-Red (red) and EGFP-lifeact (green) respectively and treated with 1μM α-SYN protofibrils for 3h–24h. Confocal microscopy images show the transfer of Mito-ds-Red labeled mitochondria to EGFP-lifeact labeled green cell population.
(B) Quantification of the percentage of acceptor cells with transferred mitochondria.
(C) quantification of the size of the transferred mitochondria vs. the control mitochondria. Quantifications are done from 5 image frames of a set and each image frame has 10–15 cells.
(D and F) Estimation of total cellular ROS by DCFDA assay in the cells treated with 1μM α-SYN protofibrils for 3h–24h by flow cytometry in U-87 MG and astrocytes, respectively.
(E and G) Quantification of the percentage of the gating population of the DCFDA-positive cells was obtained from the same.
(H) Western blot analysis to estimate the levels of senescence marker p21 in the cells treated with 1μM α-SYN protofibrils. Full-length western blots of four repeats are represented in Figure S8.
(I) In astrocyte images, the white arrows indicate DAPI-stained fragmented nuclei, while the red arrows indicate the formation of TNTs from those cells.
(J) Quantification of the percentage of fragmented nucleus on α-SYN protofibril treatment. Quantifications are done from around 10 image frames of a set and each image frame has 10–20 cells. Scale bars are denoted on the images. Data are expressed as mean ± SD, ∗∗∗p ≤ 0.001. Statistics were analyzed using a two-way ANOVA except graph C, which was analyzed by Student’s t test. n = 3.
α-synuclein protofibril-induced reactive oxygen species-mediated cellular senescence and its correlation with the transient biogenesis of tunneling nanotubes
Increased levels of cellular ROS play a significant role in inducing cellular senescence and reduction of ROS accumulation can reverse p-21-mediated cellular senescence.29 Studies have also shown the critical role of α-SYN-induced ROS in cellular senescence-associated neurodegeneration.40,41 The increased ROS levels and its elimination in correlation to TNT biogenesis motivated us to look into the senescence states in astroglia cells.29 We have observed a transient increase in p-21 levels with α-SYN protofibrils treatment in western blot analysis at an earlier time point (3h) in U-87 MG, then p-21 levels decrease gradually over time (6h, 12h, and 24h) (Figure 5H). Further, we have observed irregular nuclei, invaginations, and fragmented nuclei during the transient time window 3h and 6h, which corresponds to the α-SYN protofibril-induced transient organelle toxicities, ROS production, and the biogenesis of TNTs in primary astrocytes (Figures 5I and 5J) and U251 cells (Figures S4A and S4B). We established that cells undergo transient cellular senescence at an early time point (3h) by measuring β-galactosidase activity as the senescence marker (Figure S4C). It is known that DNA damage-related nuclear size irregularities are often associated with the hyperproduction of cellular ROS and mediated senescence. The transient cellular senescence corresponds to the transient biogenesis of TNTs at early hours in α-SYN-treated cells. Eventually, the reduction of ROS accumulation caused a reversal of p-21-dependent cellular senescence at later times.
Clearance of α-synuclein induced organelle toxicities and reactive oxygen species in cell survival and proliferation
It is a well-established fact that extracellularly applied α-SYN protofibrils are toxic to neurons and cause gradual neuronal death.42 Conversely, we observed astrocytes and astrocyte-derived cancer cells survive by alleviating α-SYN protofibril-induced organelle toxicities and cellular ROS levels. We also observed that the biogenesis of TNTs and cell-to-cell transfer precedes the clearance of α-SYN protofibril-induced organelle toxicities and cellular ROS levels. Recent studies have shown that cell-to-cell transfer of mitochondria in astrocytes via TNTs facilitates cell survival.43 Therefore, we investigated cell viability using α-SYN protofibrils in primary astrocytes, U-87 MG, and U251 astroglia cells. MTT assay measures cell metabolic activity and is used as an indicator for cell viability. The results pertaining to this assay show a concentration-dependent increase of cell viability upon the treatment of toxic α-SYN protofibrils at later time points (12h and 24h) in astrocytes (Figure 6A), compared to the respective controls and early time points (3h and 6h). α-SYN protofibril-induced organelle toxicities at earlier times also resulted in decreased metabolic activity (Figure 6A). α-SYN protofibril-treated U-87 MG (Figure 6B) and U251 (Figure 6D) cells show proliferation at later times (12h and 24h) compared to their respective controls. U-87 MG cells also show concentration-dependent proliferation at later times (24h) (Figure 6C). Further, we manually counted the cells to reconfirm that the increasing concentrations of toxic protofibrils caused an increase in cell numbers or cell proliferation after 24 and 48 h of treatments in astroglia cells (Figures S5A and S5B). Overall, the results showed that, instead of developing progressive toxicities or cell death overtime α-SYN protofibril treatment, the astroglia cells adapt to overcome the stress and proliferate during post-recovery time. We have verified the toxic effects of the protofibrils in the neurons derived from the differentiation of neuroblastoma (N2a) cells. The results show time- and concentration-dependent cell death in the treated cells (Figures S5C and S5D).
Figure 6.

TNT biogenesis pathways in cell proliferation
(A) Absorbance measured at 570 nm after MTT assay performed in astrocytes treated with varying concentrations (0.5μM, 1μM, 2μM, and 3μM) of α-SYN protofibrils at 3h, 6h, 12h and 24h.
(B) Cell number quantification of U-87 MG cells treated with 1μM α-SYN protofibrils after 3h, 6h, 12h, 18h and 24h.
(C) MTT absorbance estimated at 24h on treatment with varying concentrations (0.5μM, 1μM, 2μM, and 3μM) of α-SYN protofibrils in U-87 MG cells.
(D) Estimation of MTT absorbance at 12h and 24h after treatment with 1μM α-SYN protofibrils in U251 cells.
(E) Fluorescence images of astrocytes and U87MG cells treated with 1μM α-SYN protofibrils for 3h–24h were stained with proliferation marker Ki67 along with nuclear stain.
(F and G) Quantification of the intensity of Ki67 per cell in astrocytes and U-87 MG respectively. Quantifications are done from 15 image frames of a set and each image frame has 20–25 cells.
(H) Flow chart depicting the mode of action of actin inhibitors.
(I) DiD (membrane dye) stained, control and 1μM α-SYN protofibrils treated U-87 MG cells pre-treated (before 30min) with 3 μM IPA3, 0.5 μM cytochalasin D, 50 μM CK-666 (Arp2/3 inhibitor), 75 μM blebbistatin and 5 μM Y-27632 (ROCK inhibitor). Red arrows indicate the formation of TNT-like structures observed at 3 h and (K) shows the quantification of TNT numbers. (J) representative images showing cell numbers with the above-mentioned treatment with inhibitors and α-SYN protofibrils at 24 h.
(L and M) quantification of MTT absorbance and cell numbers of the same, respectively. Quantifications are done from 5 image frames of a set and each image frame has 15–20 cells. Scale bars are denoted on the images. Data are expressed as mean ± SD, ∗∗∗p ≤ 0.001. Statistics were analyzed using a two-way ANOVA, and only graph 6B was analyzed using one-way ANOVA. n = 3.
Since we have observed a significant increase in cell numbers and cell viability upon α-SYN protofibrils treatment, we checked for cell proliferation using Ki67 as the marker. Astrocytes and U-87 MG cells immunostained with Ki67 antibody and nuclear stain DAPI (Figure 6E) show a significant increase in Ki67 overexpression in the nucleus at 12h and 24h in comparison to control and early time points (3h and 6h) (Figures 6F and 6G). Overall, the results indicate toxic α-SYN-induced initial stress (till 6 h) promotes the biogenesis of TNTs and cell-to-cell transfer of organelles, consequences of which aid in rescuing the cells from organelle toxicities or ROS-induced cellular stresses. Further, to deal with recovery, cells probably facilitate proliferation.
Tunneling nanotube biogenesis pathways in astroglia proliferation
TNTs are structurally open-ended membrane actin conduits. Thereby, it is obvious that the modulation of membrane and cytoskeleton will play a major role in their biogenesis. However, the exact mechanism of TNT biogenesis is not known. Studies on the screening of inhibitors in the actin signaling pathways could unfold molecular events behind the α-SYN toxicity-induced biogenesis of TNTs. Therefore, we studied different actin inhibitors (Figure 6H) to see their effects on the biogenesis of TNTs at an early time (3h) (Figure 6I) and cell proliferation at a later time (24h) in U-87 MG cells (Figure 6J). We observed that cytochalasin D (inhibits actin polymerization and interaction of G-actin-cofilin) and IPA-3 (PAK1/2 inhibitor) inhibited α-SYN protofibril-induced TNTs formation, whereas, CK-666 (Arp2/3 inhibitor), blebbistatin (myosin-II-specific ATPase inhibitor), and Y-27632 (ROCK inhibitor) promote the biogenesis of TNTs (Figure 6K). Similar to the reports of earlier studies,44 we have also observed that Arp2/3 inhibitor CK-666 caused the formation of significantly longer TNTs (Figures 6I and S6).
To understand the role of TNTs in cell proliferation, cell viability was assayed using MTT (Figure 6L), and cell numbers were determined by manual counting (Figure 6M). TNT numbers (Figure 6K) and cell proliferation (Figures 6L and 6M) data showed that the actin inhibitors, cytochalasin D and IPA-3, which prevent the biogenesis of TNTs, inhibit α-SYN protofibril-induced cell proliferation as well (Figures 6L and 6M). The actin inhibitors,CK-666 and blebbistatin, which facilitate the biogenesis of TNTs, did not alter α-SYN protofibril-induced proliferation. However, the inhibitors alone (CK-666 and blebbistatin) did not show a significant effect on cell proliferation. We observed that the ROCK inhibitor Y-27632 in the presence and absence of α-SYN protofibrils significantly increased proliferation (Figures 6L and 6M).
Biogenesis of α-synuclein induced tunneling nanotubes through the modulation of ROCK pathway, resulting in increased cell survival and proliferation
A recent study has shown that the ROCK inhibitor (Y-27632) is an intriguing compound that boosts the biogenesis of TNTs via Myosin II-mediated actin modulation.11 However, it is not yet known how α-SYN dominates the modulation of ROCK inhibition signaling mediated TNT biogenesis, over the cofilin-G-actin interaction (cytochalasin-D inhibited pathway) mediated actin remodulation. To unfold the mechanism, we followed the internalization of α-SYN-TMR protofibrils in the presence of cytochalasin-D and ROCK inhibitor (Y-27632) using flow cytometry quantification (Figure 7A). We observed that cytochalasin-D inhibited the internalization of α-SYN protofibrils, however, Y-27632 did not show any effect (Figure 7B).
Figure 7.

FAK translocation, ROCK remodulation, and activation of proliferation pathway
(A) Estimation of uptake of α-SYN-TMR protofibrils by U251 cells treated with 1μM α-SYN protofibrils and pre-treated (before 30min) with 0.5 μM cytochalasin D and 5 μM Y-27632 (ROCK inhibitor) by flow cytometry.
(B) Quantification of percentage gated fluorescence intensity of α-SYN-TMR protofibrils in the above experiment.
(C) Fluorescence images of maximum intensity projected confocal z-stacks in astrocytes stained with Phalloidin (green) and FAK (red). The upper panel has nucleus stained with DAPI and the lower panel is without DAPI. White arrows indicate the nuclear colocalization of FAK, red arrows indicate TNTs and pink arrows indicate FAK at focal adhesion.
(D) Quantification of FAK intensity in the nucleus of the cells with and without TNTs.
(E and G) Fluorescence images of maximum intensity projected confocal z-stacks in U-87 MG cells stained with FAK (red) and pFAK (red) with DAPI, respectively.
(F and H) Quantification of FAK and pFAK fluorescence intensity per cell in the nucleus. Quantifications are done from 15 image frames in a set, and each image frame has 15–20 cells.
(I) Western blot images showing the change in ROCK1 and ROCK2 expressions with time on 1μM α-SYN protofibril treatment in U-87 MG cells.
(J) Quantification of ROCK1 and ROCK2 western blots. Full-length western blots of four repeats are represented in Figure S9.
(K) Western blot images showing increased intensity of pERK1/pERK2, NF-κB, and Cdk1 with time on 1μM α-SYN protofibril treatment in U-87 MG cells. Full-length western blots of three repeats for pERK1/pERK2 and NF-κB and five repeats for Cdk1 were represented in Figure S10.
(L) Quantification of pERK1/pERK2, NF-κB, and Cdk1 western blots. Scale bars are denoted on the images. Data are expressed as mean ± SD, ∗∗∗p ≤ 0.001. Statistics were analyzed using a two-way ANOVA. n = 3.
α-synuclein-induced cellular senescence results in the translocation of focal adhesion kinase in the nucleus
Our results showed α-SYN protofibrils induced nuclear deformity and cellular senescence in astroglia cells at early time points (3h and 6h) (Figures 5H–5J). Several studies have shown that focal adhesion kinase (FAK) inhibition could induce DNA damage and nucleus deformity that accompany cellular senescence.45,46 Loss of integrin-dependent cell adhesion in cellular stress modulates FAK and facilitates its translocation from the plasma membrane to the nucleus.47,48,49 We observed de-adhesion of astroglia cells (U-87 MG) with α-SYN protofibril treatments at early time points (3h and 6h) when cells show lysosomal-mitochondrial toxicities and transient biogenesis of TNTs, compared to the control and cells at later times (12h and 24h) (Figure S7A). Further, we noticed treatments with α-SYN protofibrils cause nuclear translocation of FAK in the astrocytes that are connected by TNTs at early time points (3h and 6h) (Figures 7C and 7D). The presence of nuclear FAK was verified by 3D images at xz and yz planes (Figure S7B). We also observed treatments with α-SYN protofibrils cause nuclear translocation of FAK to the nucleus in the U-87 MG (Figures 7E and 7F) and U251 (Figures S7C and S7D) cells at early time points (3h and 6h), whereas, at later timepoints in the post-recovered cells, FAK relocates back to PM and cytosol. We have also observed the nuclear translocation of activated phospho-FAK (Tyr 397) transiently at early time points (3h and 6h) in astrocytes (Figures 7G and 7H). These results clearly demonstrate that there is a transient translocation of FAK in the nucleus at the early and late time points on α-SYN treatment.
Nuclear translocation of focal adhesion kinase modulates the ROCK pathway to induce tunneling nanotube biogenesis and cell proliferation
Inhibition of integrin-mediated FAK activation prevents ROCK activation and inhibits cell adhesion. Moreover, when active/inactive FAK is displaced from cell-adhesion sites in non-adherent cells, Rho-mediated activation of ROCK kinases maintains cytoskeleton tension by regulating actin re-modulation.50 To understand the FAK-mediated regulation of ROCK kinases, we have performed western blots and quantified expressions of ROCK1 and ROCK2 in the α-SYN treated cells (Figures 7I and 7J). Results show ROCK2 inhibition was observed in the early hour (3 h) after α-SYN treatment, later expressions increased gradually (Figures 7I and 7J). However, ROCK1 expression with α-SYN treatment showed gradual inhibition over time.
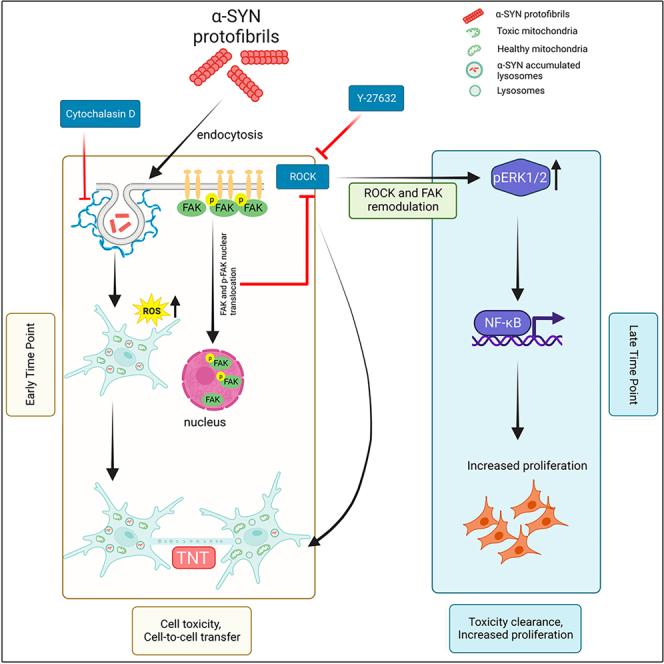
ROCK activation pathways are involved in cell proliferation as well.50 Our western blots showed increased levels of ROCK-mediated cell proliferation markers ERK1/2, NF-κB, and Cdk1 (Figures 7K and 7L). We observed higher ROCK2 expression in the rescued cells (Figures 7I and 7J). Probably, ROCK2 activation at later hours results in increased cell proliferation through ERK1/2 and NF-κB signaling cascades. Overall, the results delineate that α-SYN protofibril treatment caused transient localization of FAK/pFAK to the nucleus of astroglia cells, which could modulate ROCK inhibitory pathways to promote the biogenesis of TNTs in the astroglia cells for a transient time in early hours after the treatment. The rescued cells, post-α-SYN treatment and transient TNT biogenesis, eventually may re-activate ROCK signaling to restore cytoskeleton tension and promote enhanced cell proliferation (graphical abstract).
Discussion
PD pathology progresses with the spreading of misfolded α-SYN aggregates in the brain.2 Even though cytoplasmic inclusion of α-SYN in the neurons is the central hallmark of neuropathology development in PD, evidence suggests the release of α-SYN aggregates from degenerated neurons in the extracellular brain plays a significant role in PD pathology progression. The role of extracellular α-SYN in cell-to-cell transfer and PD pathology progression has been widely studied in several model systems.14 Recent studies have shown that extracellular α-SYN aggregates, taken up by astrocytes and microglia, promote the biogenesis of TNTs. The TNT-mediated glial crosstalk and cell-to-cell transfer facilitate the degradation of the aggregates and clearance of toxic organelles.11,13 Previously, the study by Loria et al. showed that astrocytes facilitate the degradation of α-SYN protofibrils by promoting TNT-mediated transfer.51 The crosstalk between astrocytes and microglia via TNTs facilitates the degradation of α-SYN protofibrils even faster.12 A recent study has reported an on-demand formation of TNTs to clear toxic α-SYN protofibrils and alleviate ROS levels in microglia.11 The study showed the rescue of the toxic burden of α-SYN via TNT-mediated borrowing of healthier mitochondria from the healthier neighbors and the transfer of toxic aggregates to them.11 Recently, a study demonstrated that neuronal cells transfer toxic mitochondria to microglia via TNTs, whereas microglia protect neurodegeneration by transferring healthier mitochondria to neurons.33 However, the molecular players involved in the biogenesis of transient TNTs, the fates of TNT-mediated cell-to-cell transfer, and their dynamic interplay in the clearance of toxic neurodegenerative aggregates are not well explored.
Our study has shown, for the first time, that the transient translocation of FAK/pFAK to the nucleus upon α-SYN protofibril treatment causes transient biogenesis of TNTs, which eventually contributes to enhanced cell proliferation in the astroglia cells. Our study has also established a strong correlation between the transient biogenesis of TNTs and α-SYN-induced toxic burden at early hours (3h and 6h). Cell-to-cell transfer through transient TNTs corresponds to cellular toxicities, such as lysosomal, and mitochondrial toxicities, and increased levels of cellular ROS and associated cellular senescence. However, the astroglia cells recover from these cellular toxicities at later hours (12h and 24 h). The post-recovered astroglia cells proliferate with increasing concentrations of α-SYN protofibril treatments. Understanding the molecular players involved in the proliferation of astrocytes in response to α-SYN aggregates is highly important since astrogliosis and microgliosis act as essential mediators in maintaining cellular homeostasis in the PD-degenerative brain.7
Several studies have reported cell-to-cell transfer of mitochondria through TNTs in several pathophysiological conditions, where the transfer of healthy mitochondria results in the rescue of cells from apoptosis or toxicities.52,53,54 However, it is not the case that TNTs transfer only healthy mitochondria to neighboring cells. Microglia and astrocytes probably facilitate the dilution of its toxic burden by transferring toxic α-SYN aggregates and toxic mitochondria by sharing with their neighbors through TNTs.11,51 We have observed that α-SYN protofibrils induce increased ROS production at early time points (3h and 6h), causing reduced mitochondrial membrane potential, resulting in the generation of fragmented mitochondria and the disintegration of the mitochondrial network. In the co-culture experiment, we observed only the unidirectional transfer of toxic fragmented mitochondria to their neighboring cells. Several studies have shown that accumulated ROS from transferred toxic mitochondria could facilitate cell proliferation.55,56 The observations suggest that astrocytes and astroglia cells share their toxic organelles (lysosomes and mitochondria) with surrounding neighbors to dilute the toxic burden, and the transferred toxic mitochondria may contribute to facilitating proliferation.
We have observed that α-SYN protofibrils induced increased ROS levels directly affect mitochondrial dysfunction, and DNA damage, which play key roles in inducing cellular senescence in reactive astrocytes.41,57,58 We have observed α-SYN protofibrils induced mitochondrial abnormality, fragmented irregular nuclei, increased β-galactosidase activity, and p21-pathway-dependent premature cellular senescence for a transitory period. Astroglia cells recover from senescence-related toxicities by alleviating ROS levels after the transient biogenesis of TNTs and TNT-mediated cell-to-cell transfer. ROS-induced DNA damage and mitochondrial toxicity do not always lead to apoptosis.59 Cells activate repair mechanism/s in cases of delayed apoptosis. Moreover, a reduction in ROS accumulation can reverse p21-mediated stress-induced premature senescence or vice versa.29
Cell-to-cell transfer of neurodegenerative aggregates drives the spreading of aggregates, which act as seeds for further propagation in neurons,60 whereas, astrocytes and astroglia cells rescue cellular toxicities.13,51,60 The role of the inherent conventional clearance efficiency of astrocytes cannot completely be ruled out. Unlike neurons, astrocytes lack endogenous α-SYN, and the capacity of continuous seeding to form higher-order toxic aggregates is limited. This is to emphasize here that TNTs are predominantly observed in cell types that possess inherent anti-apoptotic properties, such as primary neurons, neuronal cells, neuroglial cells, and cancer cells.16,61 Neurons possess inherent anti-apoptotic properties such as other brain cells; however, unlike glial cells, they are mitotically incompetent.62,63 We observed that α-SYN-induced TNT-mediated cell-to-cell transfer eventually leads to cell death in mitotically incompetent neurons, whereas astrocytes and astroglia cells clear toxic burdens and enhance proliferation and cell survival. TNT-mediated cellular clearance and proliferation in astrocytes could play a significant role in PD.
Therefore, the important question that needs to be understood is how the biogenesis of TNTs and clearance of cellular toxicities are associated with proliferation in astrocytes. Several studies have indicated that the biogenesis of TNTs may induce cell proliferation, to protect the cells from cell death under pathogenic conditions and chemo- or radio-therapy-related stress.36,54,64,65 The studies show mostly limited or indirect correlation between TNTs and cell proliferation. Our results on small molecule actin modulators indicate ROCK inhibitory pathway mediated biogenesis mechanism/s of TNTs and its correlation with enhanced proliferation. Small molecules actin modulators (cytochalasin D, and IPA-3) inhibit the biogenesis of TNTs and prevent α-SYN protofibril-induced cell proliferation. On the other hand, the actin modulators CK-666 and blebbistatin, which facilitate the biogenesis of TNTs, did not show a significant effect on the enhancement of cell proliferation. Even though these actin inhibitors (CK-666 and blebbistatin) show better cell survival compared to the molecules that inhibit TNTs. Only ROCK inhibitor Y-27632 mediated biogenesis of TNTs significantly promotes proliferation, similar to proliferation induced by α-SYN protofibrils. ROCK inhibitor Y-27632 inhibits LIMK-dependent cofilin-G-actin interaction and inhibits actin polymerization whereas, IPA-3 and cytochalasin-D could also inhibit actin polymerization through the LIMK pathway.11 However, we observed ROCK inhibition promotes the biogenesis of TNTs, whereas, IPA-3 and cytochalasin-D inhibit TNTs. Our result suggests, that the inhibition of ROCK kinase pathways may modulate TNT biogenesis by regulating actin cytoskeleton through myosin-II-specific downstream signaling molecules, which dominates over cofilin-mediated actin modulation. Similarly, a recent study11 has shown that α-SYN protofibrils promote the biogenesis of TNTs in microglia cells via regulating ROCK inhibitory pathway by the phosphorylation of myosin light chain phosphatase (MLCP). Thus, we tried to unfold how α-SYN protofibrils induced actin-remodulation may modulate ROCK inhibitory signaling cascades to induce TNT biogenesis.
We have found that α-SYN-induced transient senescence-related toxicities caused nuclear localization of FAK and pFAK for the transitory period, which corresponds to TNT biogenesis at early time points (3h and 6h). Organization of FAK at focal adhesion sites of PM regulates the activation of Rho-mediated ROCK signaling, and its de-localization from focal adhesion inhibits the ROCK pathway.66 On the other hand, in low adhesion conditions displacement of inactive/active FAK/pFAK from focal adhesion sites leads to the Rho kinase-mediated activation of ROCK signaling, probably to restore the cytoskeletal tension.50 Then, the restoration of cytoskeleton tension may cause the re-translocation of FAK/pFAK to PM, which could reinstate ROCK activation. In this line, we have detected α-SYN toxicities induced FAK translocation dynamics, which could modulate the transient inhibition of ROCK2 signaling to induce the biogenesis of TNTs at early times and later may activate to restore cytoskeleton tension. We have also seen the proliferation of astroglia cells via the activation of ROCK signaling mediated ERK1/2 and NF-κB proteins, in the post-recovered astroglia cells. Thus, our results suggest that the biogenesis of TNTs is not the root cause of cell proliferation, rather modulation of α-SYN protofibrils induced ROCK-mediated actin-regulatory signaling pathway related to the biogenesis of TNTs triggers cell proliferation.
In conclusion, our study reveals that α-SYN protofibrils induce the biogenesis of TNTs, which aids in enhancing the clearance of toxic burden as a cellular survival strategy to rescue astroglia cells from ROS-induced cell death/cellular senescence. α-SYN protofibrils regulate the FAK-mediated modulation of ROCK signaling cascades to rescue cells from toxic burden by promoting TNT biogenesis. The rescued cells eventually re-activate ROCK2 signaling, probably to restore cytoskeleton tension and enhance cell proliferation via ERK1/2 and NF-κB signaling. α-SYN inclusions in the astroglia cells and glial inclusion or fibrous gliosis in the areas of neurodegeneration are widespread in PD.7 Thus, the study is important for understanding the relevance of TNT-mediated crosstalk in the clearance of α-SYN protofibrils by astrocytes and its implication in astroglia cell proliferation. Thus, the study will open up new strategies to design therapeutics targeting astrocytes in the PD brain.
Limitations of the study
The study does not reveal the consequences of lysosome and mitochondria transfer. The fate of transferred toxic lysosomes and mitochondria is closely related to both the cell-to-cell spreading of toxic organelles and the clearance of toxic burdens to sustain cells' survival, making it a complex subject that requires further detailed studies, which we have not explored in this study. Also, the in vivo presence of TNTs upon α-SYN treatment was not checked in this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GAPDH Mouse | Cloud clone | CAB932Hu22 |
| ROCK1 | CST | 4035T |
| ROCK2 | CST | 8236S |
| NF-kB p65/RelA | Abclonal | A10609 |
| pERK1-T202 + pERK2- T18 | Abclonal | AP0485 |
| CDK1 | Abclonal | A0220 |
| p21 Waf1/Cip1 | CST | 2947 |
| Goat anti-mouse | Invitrogen | 32430 |
| Goat anti-Rabbit | Invitrogen | 32460 |
| Ki67 | Millipore | B9260 |
| FAK | CST | 3285T |
| phospho-FAK (Tyr397) | Invitrogen | 44-624G |
| Cathepsin D | GeneTex | 42368 |
| LAMP-1 | BD Biosciences | 611042 |
| LAMP-2 | Invitrogen | MA1-205 |
| βIII-tubulin | Cloud-Clone | PAE711Hu01 |
| Connexin43 | CST | 3512S |
| SNCa | Cloud-Clone | PAB222Hu01 |
| GFAP | Cloud-Clone | PAA068Mu01 |
| Phalloidin | Abcam | 176756 |
| Alexa- 488 anti-rabbit | Invitrogen | A-11070 |
| Alexa- 555 anti-mouse | Invitrogen | A-1413312 |
| Alexa- 488 anti-mouse | Invitrogen | A-11059 |
| Alexa-555 anti-rabbit | Invitrogen | A-21428 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Gibco | 2120395 |
| FBS | Gibco (US Origin) | 1600004 |
| PSN | Thermo Fisher Scientific | 15640055 |
| Glutamax | Gibco | 35050 |
| Retinoic acid | Sigma-Aldrich | R2625 |
| trypsin-EDTA | Gibco | 25200-072 |
| Minimum Essential Media | Gibco | 61100-087 |
| D-glucose | Sigma Aldrich | G7021 |
| TCEP | Sigma Aldrich | C4706 |
| TMR-maleimide | Sigma Aldrich | 94506 |
| PD-10 columns | Himedia | TKC287 |
| 4-Hydroxynonenal | Sigma-Aldrich | 393204 |
| MTT reagent | Himedia | TC191-1G |
| DMSO | Himedia | TC185 |
| ProLong Gold | Invitrogen | P36941 |
| ECL solution | Invitrogen | 34094 |
| lysotracker | Invitrogen | L12492 |
| mitotracker | Invitrogen | M7514 |
| DiD®Vybrant | Invitrogen | V22887 |
| JC-1 | BD Biosciences | 551302 |
| Lipofectamine 3000 | Invitrogen | 44386 |
| IPA3 | Merck | 506106-5MG |
| Cytochalasin D | Thermo Fisher Scientific | PHZ1063 |
| CK-666 | Sigma-Aldrich | SML0006 |
| Blebbistatin | Merck | 203390-5MGCN |
| Y-27632 | Sigma-Aldrich | Y0503 |
| HEPES | Sigma Aldrich | H0887 |
| Critical commercial assays | ||
| Mycoplasma detection kit (MycoFlour) | Sigma-Aldrich | M7006 |
| β-galactosidase staining kit | Cell Biolabs Inc. | AKR- 100 |
| Experimental models: Cell lines | ||
| U-87 MG | ECACC | #89081402 |
| U251 | ECACC | #09063001 |
| N2a | NCCS, Pune, India, Cell repository | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6J (Strain#:000664) | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Recombinant DNA | ||
| α-SYN wild-type | Addgene | 36046 |
| α-SYN (wt)-141C | Addgene | 108866 |
| pLV-mitoDsRed | Addgene | 44386 |
| mEGFP-lifeact-7 | Addgene | 58470 |
| Software and algorithms | ||
| Fiji | NIH | https://imagej.net/software/fiji/ |
| Endnote | Clarivate | https://endnote.com/ |
| Graphpad Prism v8 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| FCS Express | De Novo Software | https://denovosoftware.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sangeeta Nath: sangeeta.nath@manipal.edu.
Materials availability
No unique reagents were generated in this project.
Data and code availability
-
•
Data and images that support the findings of this study are available on request from the lead contact.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data in this paper is available from the lead contact upon request.
Experimental model and study participant details
Mice
Primary astrocyte culture was conducted using five-week-old mice (C57BL/6J) of either sex. The original source of C57BL/6J mice strain (RRID:IMSR_JAX:000664) was procured from The Jackson Laboratory. All experimental protocols have been approved and conducted in accordance with the national policies of the use of animals under the regulation of the Institutional Animal Ethics Committee, Indian Institute of Science, CAF/Ethics/869/2021. The mice were kept on a standard 12/12-hour light-dark cycle. They were housed in same-sex groups or sustained breeding pairs whenever possible and provided with fresh bedding, nesting material, food and water. Both male and female mice were used in the study without any bias.
Primary astrocytes culture
The five-week-old mice (C57BL/6J) of either sex were sacrificed using CO2, and the cerebral cortices were dissected from the mice's brains and observed using an Olympus SZ51 stereomicroscope. Pieces of cortical tissue were trypsinized using 0.25% trypsin-EDTA solution (Gibco Canada origin #25200-072) for 5 min. The tissue was washed twice in warm HBSS and transferred to Minimum Essential Media (Gibco 61100-087) with 10% FBS (Gibco #1600004, US Origin) containing HEPES (Sigma Aldrich H0887) and D-glucose (Sigma Aldrich G7021). The tissue was triturated using a fire-polished pipette and counted using Trypan Blue in an automated cell counter. Dissociated cells were plated in MEM with 10% FBS on Corning® 100mm TC-Treated Culture Dish at a density of 10 million cells per dish. The media was replaced with fresh MEM with 10% FBS on the next day and maintained the same for 1 week. After 1 week, cells were trypsinized and re-plated for experiments.
Cell lines and culture maintenance
U-87 MG and U251 cell lines (astrocytoma-glioblastoma origin cancer cell lines) were kind gifts from Prof. Kumaravel Somasundaram of the Indian Institute of Science, Bangalore, India (Original source; European Collection of Authenticated Cell Cultures, ECACC). Both cell lines were tested for mycoplasma contamination using Mycoplasma detection kit (Sigma-Aldrich #M7006). Cells were cultured and maintained in DMEM (Gibco #2120395) media supplemented with 10% FBS (fetal bovine serum; Gibco #1600004, US Origin), along with 1% PSN (Penicillin-Streptomycin-Neomycin Mixture; Thermo Fisher Scientific #15640055) incubated at 37°C, 5% CO2.
Neuro 2a (N2a) neuroblastoma cell line was procured from NCCS, India. These cells were also tested for mycoplasma contamination. The cells were cultured and maintained in DMEM media with 10% FBS and 1X Glutamax (Gibco #35050). N2a cells were differentiated with 10 μM Retinoic acid (RA; Sigma-Aldrich #R2625) in the presence of 2% FBS-supplemented media for 2-3 days. Differentiation was established from the neurites like morphology.
Method details
Purification and Labelling of α-SYN protein
The human α-SYN wild-type (Addgene ID #36046) and α-SYN (wt)-141C (Addgene ID #108866) with a cysteine residue at C-terminus, constructs were purchased and overexpressed in E. coli. Then purification was done from periplasmic fraction as described in.67,68 Samples were aliquoted and flash frozen in liquid nitrogen and stored immediately at -80°C.
The purified α-SYN (wt)-141C protein was thawed and pre-incubated with reducing agent TCEP (tris(2-carboxyethyl) phosphine-hydrochloride); Sigma Aldrich-C4706; 20mM) to reduce/dissociate any pre-existing cysteine bonds. Then the protein was labelled adding two-fold molar excess of TetramethylRhodamine-5-maleimide (TMR-maleimide; Sigma Aldrich #94506) in 20 mM HEPES buffer (pH 7.4). The protein solution was vortexed and the mixture was incubated for 1h at 20°C. The excess label was removed by using PD-10 columns (Himedia #TKC287).69
Preparation and characterisation of α-SYN protofibrils
Protofibrils of α-SYN (labelled and unlabelled) was prepared with 1 μM of unlabelled α-SYN wild-type and labelled α-SYN-141C by incubating with 0.65% 4-Hydroxynonenal (10mg/ml stock) (Sigma-Aldrich #393204-1MG) at 37°C for 7 days with moderate shaking.70
The α-SYN protofibrils formed at the end of 7 days were then characterized by transmission electron microscopy (TEM). The unlabelled and labelled α-SYN protofibrils were resuspended in 1X PBS, coated on a carbon grid with uranyl acetate and dried in a desiccator. The grids were visualized using FEI Tecnai T20 at 200 KV. The α-SYN protofibrils were then lyophilized and stored at -20°C and resuspended in 1X TBS (Tris-buffered saline) buffer before experiments.
The toxicity of the protofibrils was assessed by treating the differentiated neuronal cells seeded at a density of 30,000 cells / well in 24 well plates. Cell viability assay was performed using different concentrations of α-SYN protofibrils (0.5μM, 1μM, 2μM, 3μM) over different periods by MTT assay.
Treatment conditions of astroglia cells by α-SYN protofibrils
All the experiments were carried out by treatment of primary astrocytes, U-87 MG, and U251 cells with 1μM α-SYN protofibrils- unlabelled or TMR-labelled for 3h, 6h, 12h, and 24h at 37°C, 5% CO2 unless mentioned otherwise. The cells were seeded at the same time for all the treated time points and controls. Once cells were adhered properly and appeared healthy after 24h of seeding, then treatments were done by adding (α-SYN 1μM) protofibrils in a reverse order of the time points. For example, 24h time points were treated first and then sequentially 12 h, 6 h, and 3 h points. Control cells did not receive any treatment. Then, experiments were performed for all the time points and controls at a time. Therefore, the condition of the untreated control cells corresponds to all the treatment points.
MTT assay and cell numbers counting
MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide) assay is a colorimetric assay used to measure cellular metabolic activity. Astrocytes, U-87 MG, and U251 cells were seeded at a density of 4000 cells per well in a 96-well plate and incubated overnight at 37°C, 5% CO2. Different concentrations of α-SYN protofibrils (0.5μM, 1μM, 2μM, 3μM) were added to the wells over different time periods (3h, 6h, 12h, 24h, and 48h) and treated with MTT reagent (Himedia # TC191-1G) for 2h. The insoluble formazan crystals were dissolved by adding dimethyl sulfoxide (DMSO; Himedia #TC185) which was later quantified by measuring absorbance at 570 nm using the PerkinElmer-Multimode plate reader spectrophotometer. Cell numbers were counted from the phase contrast microscopy images or DIC images using the multi-point option in Fiji (a Java-based program developed at the National Institutes of Health, USA).
MTT assay and cell number counting were performed similarly with actin polymerization pathway-modulating inhibitors. The U-87 MG cells were pre-treated (before 30min) with 3 μM IPA3 (Merck, 506106-5MG), 0.5 μM cytochalasin D (Thermo Fisher Scientific, PHZ1063), 50 μM CK-666 (Arp2/3 inhibitor, Sigma-Aldrich, SML0006), 75 μM Blebbistatin (Merck, 203390-5MGCN) and 5 μM Y-27632 (ROCK inhibitor, Sigma-Aldrich, Y0503). First, we have selected the range of concentrations based on the literature,11,21 then we screened to validate the effect on U87-MG cells and finalized the effective working concentration.
Immunocytochemistry (ICC)
After treatment with the above-mentioned concentrations and time points adding α-SYN protofibrils, the cells were washed with 1X PBS and fixed with 4% PFA. The cells were then incubated with incubation buffer (1mg/ml Saponin and 5%FBS) for 20 min at RT. Respective primary antibodies (as mentioned below) were added and incubated overnight at 4°C, the cells were then washed 3 times with 1X PBS. All the secondary antibody (1:700 dilution) incubations were carried out in the dark for 2 hrs at RT, and 1X PBS washes were repeated. The coverslips were mounted on glass slides with ProLong Gold antifade reagent with DAPI, (Invitrogen P36941). Alternately, glass bottom 35mm dishes (Cellvis, D35-14-1.5-N) were used in a few experiments.
Primary antibodies Ki67 anti-rabbit (Millipore B9260) as cell proliferation marker; FAK Rabbit (CST 3285T) and phospho-FAK ((Tyr397) pAb; Invitrogen 44-624G) as total and active FAK marker; Cathepsin D anti-rabbit (GeneTex #42368), LAMP-1 anti-mouse (BD Biosciences # 611042), LAMP-2 anti-mouse (Invitrogen #MA1-205) as lysosomal markers; βIII-tubulin anti-rabbit (Cloud-Clone #PAE711Hu01), Connexin43 Rabbit Ab (CST #3512S), SNCa rabbit pAb (Cloud-Clone #PAB222Hu01), GFAP rabbit pAb (Cloud-Clone #PAA068Mu01) and Phalloidin conjugated with iFlour555 (Abcam #176756) were used to stain TNTs and TMs. Secondary antibodies Alexa- 488 goat anti-rabbit (#A-11070; Invitrogen), Alexa- 555 anti-mouse (#A-1413312; Invitrogen), and Alexa- 488 anti-mouse IgG (H+L) (#A-11059; Invitrogen) were used respectively following the above-mentioned protocol. All primary antibodies were used at a dilution of 1:300, secondary antibodies at 1:500, and phalloidin at 1:700. ICC-stained cells were imaged with a confocal microscope (Zeiss LSM880, Carl Zeiss, Germany) or fluorescence microscope (IX73-Olympus).
Western Blot
U-87 MG cells were seeded at a density of 1 million per well in a 6-well plate and were treated with 1μM α-SYN protofibrils from 3h-24h. Post-treatment media was removed and the cells were washed with 1X PBS. RIPA buffer was added and the cells were scrapped and collected in 1.5 ml tubes. The tubes were incubated on ice with intermittent vortexing. Later the suspension was spun at 12,000 rpm for 10 min and the supernatant was collected and stored at -200C. After normalizing the protein concentration, western blot assay was performed with primary antibodies GAPDH Mouse (Cloud-Clone # CAB932Hu22 dilution 1:1000), ROCK1 (CST # 4035T dilution 1:1000), ROCK2 (CST # 8236S dilution 1:1000), NF-kB p65/RelA Mouse mAb (Abclonal # A10609 dilution 1:1000), phospho ERK1-T202 + ERK2- T18 (Abclonal # AP0485 dilution 1:1000), Cdk1 Rabbit pAb (Abclonal #A0220 dilution 1:1000), p21 Waf1/Cip1 (12D1) Rabbit mAb (CST #2947 dilution 1:1000) and secondary antibodies, Goat anti-mouse (H+L) (Invitrogen #32430 dilution 1:1500) and Goat Anti-Rabbit (H+L) (Invitrogen #32460 1:1500). The blot was developed with ECL solution (SuperSignal West Femto Trial kit Invitrogen #34094) and were quantified by densitometry using gel analyzer plugin of Fiji software.
α-SYN internalization
U-87 MG cells were seeded in a 24-well plate at a density of 0.1million/well, pre-treated with 0.5 μM cytochalasin D and 5 μM Y-27632 (ROCK inhibitor), then treated (before 30min) with TMR labelled 1μM α-SYN protofibrils. Post-treatment U-87 MG cells were trypsinized (0.25% trypsin-EDTA solution; Gibco Canada origin #25200-072). The pellet was collected and resuspended in 1X PBS and twice washed with 1X PBS. α-SYN-TMR levels were quantified by measuring the fluorescence intensity of TMR at excitation/emission 555/585nm by using a flow cytometer (BD LSR II) and analysis was done using FCS express software.
Live cell imaging using mitotracker and lysotracker
U-87 MG cells (80,000 cells/well) were seeded on a 35 mm glass bottom dish (Cellvis, D35-14-1.5-N) and treated with 1μM TMR labelled α-SYN for 0h, 3h, 6h, 12h, and 24 h. The 0h cells were immediately washed before staining with lysotracker and mitotracker. Cells were stained with mitotracker green (Invitrogen #M7514) and lysotracker deep red (Invitrogen #L12492) and were incubated at 37°C for 15 min. Post incubation, the cells were washed with 10% DMEM and time-lapse and z-stack images were taken using confocal microscopy (Zeiss LSM880, Carl Zeiss, Germany). This was done to comprehend the localization of α-SYN and the transfer of organelles through TNTs.
Live Cell imaging using DiD®vybrant membrane dye
U-87 MG cells (80,000 cells/well) were seeded on a 35 mm dish and treated with 1μM α-SYN as mentioned above. Cells were stained with cell-labelling dye DiD®Vybrant (Invitrogen #V22887) to label cell membranes. The dye was diluted with 10% DMEM (phenol red free) in the ratio 1:200 and incubated at 37°C for 20 min. Post incubation, the cells were washed with DMEM for 10 min, and images of live cells were taken to visualise the number of thin membrane tubes like TNTs using a fluorescence microscope (IX73-Olympus).
Estimation of cytosolic ROS
2′-7′-Dichlorodifluorescein diacetate (DCFDA) is used to detect ROS production in the cell. U-87 MG cells were seeded in a 24-well plate at a density of 0.1 million/well and treated with 1μM α-SYN protofibrils as mentioned above. Post-treatment U-87 MG cells were trypsinized (0.25% trypsin-EDTA solution; Gibco Canada origin #25200-072). The pellet was collected and resuspended in DMEM with 10% FBS and 20μM DCFDA. Cells were incubated for 30 min at 37°C then washed with 1X PBS to remove the extra dye. ROS levels were quantified by measuring the fluorescence of DCFDA stained cells at excitation/emission 488/520nm by using a flow cytometer (BD LSR II) and analysis was done using FCS express software.
Mitochondrial membrane potential using JC-1 dye
JC-1 (5,5,6,6’-tetrachloro-1,1’,3,3’ tetraethylbenzimi-dazolylcarbocyanine iodide) is the indicator of mitochondrial membrane potential. JC-1 is a cationic, lipophilic dye. Normal healthy cells show negative mitochondrial membrane potential where JC-1 dye can enter into the mitochondria and form J-aggregates. The aggregates exhibit excitation/emission maxima at 485/590 nm (red spectrum). Unhealthy cells show lower mitochondrial membrane potential as it loses the balance of electrochemical potential. At this condition, a lesser amount of JC-1 dye enters into the mitochondria and retains its monomeric form. These monomers exhibit excitation/emission maxima at 514/529 nm (green spectrum). To know the effect of α-synuclein aggregates on mitochondrial membrane potential, JC-1 assay (BD Biosciences #551302) was done. Astrocytes and U-87 MG cells were seeded at a density of 15,000 cells / well in the glass bottom area of the 35 mm imaging dishes (Cellvis, D35-14-1.5-N). Cells were treated with 1μM α-SYN for 3h-24h. Confocal images were also taken at an excitation of 490 nm and emission at 527 nm (green) and 590 nm (red). The ratio of green fluorescence versus red fluorescence per cell was analysed using Fiji image analysis software to quantify mitochondrial membrane potential.
Co-culture model and mitochondria transfer
One population of U-87 MG cells was transiently transfected with pLV-mitoDsRed (Addgene #44386; was a gift from Dr. Pantelis Tsoulfas's lab) plasmid and another population with mEGFP-lifeact-7 (Addgene #58470; was a gift from Michael Davidson) plasmid using lipofectamine 3000 (Invitrogen #44386) transfection reagent. An equal number of transfected cells from both populations were seeded together at a total density of 60,000 cells per glass bottom 35mm dish (Cellvis, #D35-14-1.5-N) and treated with α-SYN protofibrils. After treatment, the cells were fixed with 4 % PFA and images were taken using confocal microscopy (Zeiss LSM880).
β- galactosidase activity assay
U-87 MG cells were seeded at the density of 10,000 cells per well in a 24-well plate. The cells were treated with 1μM α-SYN for 3h and 24h, these time points were chosen according to the data from other experiments. Post-treatment the β-galactosidase activity was measured by using the β-galactosidase staining kit (AKR- 100 Cell Biolabs Inc). Brightfield images of the stained cells were taken using a colour camera.
Microscopy
Fluorescence images were taken using Zeiss LSM880 confocal laser scanning microscope (Carl Zeiss, Germany) or fluorescence microscope (IX73-Olympus). The confocal images were taken using objectives Plan-Apochromat 40x/1.40 or 63x/1.40 Oil Dic M27, with the fluorescence filter sets DAPI, FITC, and TRITC (Carl Zeiss, Germany). Sequential images of the different fluorescence channels were taken with 405 nm, 488 nm, and 561 nm lasers. The images were captured with a pixel dwell of 1.02 μs and each xy-pixel of 220 nm2. For all the experiments at least 5-10 images per condition were taken from randomly selected areas. DIC (differential interference contrast) images were captured along with fluorescence channels to understand the morphology and cell boundary for both fixed and live imaging experiments. Time-lapse and z-stacks (6 – 12 stacks of z-scaling ∼ 415 nm) were captured from the bottom to the top of the cells using a confocal microscope to identify TNTs and TMs. Trafficking of organelles through TNTs and TMs was tracked from time-lapse images. Wide-field fluorescence microscope (IX73-Olympus) was used to capture images for a few experiments using 20X/0.4 NA, and 40X/1.3 NA plan-apochromatic objectives.
Image analysis
TNT characterization
Confocal images were analysed using Fiji a Java-based image processing software developed at the National Institutes of Health (NIH) and the Laboratory for Optical and Computational Instrumentation (LOCI). TNTs were characterized by their unique nature to hover and not attach to the substratum, images were taken with z-stacks using confocal microscopy as mentioned above. TNTs (less than 1μM in diameter) were found in the middle z-stacks while TMs are found in the initial z-stacks since they are attached to the surface. Images of z-stacks were reconstructed using the 3D volume view plugin of Fiji as described earlier (Dilna et al., 2021). TNTs were manually counted and plotted as the ratio of the number of TNTs to the number of cells per field.35
Tracking of organelles
Movements of lysotracker and mitotracker positive vesicles through TNTs and TMs were tracked using the Trackmate (manual) plugin in Fiji. Analysis was done from time-lapse videos taken for a minimum of 30 frames at an interval of 36 seconds for 18-20 mins. By setting each organelle as an object we used the semi-automatic tracking method of the trackmate plugin. The speed of the organelles was determined from the average displacement between each consecutive image.
Size analysis
The area of protofibrils, size of aggregates, and size of lysosomes and mitochondria were analysed per cell from the randomly taken images of 200-300 cells per condition. The analysis was performed by setting a threshold to separate the particles as individual points and then by using the ‘Analyze particle’ option of Fiji.
Intensity analysis
Similarly, the expression of Ki67, FAK, pFAK, α-SYN aggregates, lysosomes, and the ratio of green versus red fluorescence of the JC-1 experiment was analysed by quantifying the intensities of the labelled proteins per cell from the images of 200-300 cells for each condition. Intensities were analysed by drawing and selecting regions of interest using the ROI-plugin in Fiji.
Mitochondria morphology analysis
The quantification of mitochondria branching and branch length were determined using the MiNA plugin in the Fiji software. We downloaded the MiNA analysis plug-in from the Git Hub repository of Stuart Lab. We cropped each cell stained with mitochondria and ran the MiNA plugin to determine the mean branch length of mitochondria.
Quantification and statistical analysis
To validate the significance of the analysed data, one-way and two-way ANOVA tests were performed as per the experimental parameters as mentioned in the figure legends. For Figure 5C, we performed a student t-test since we had only two conditions. The statistics were calculated from the three biological repeats.
Acknowledgments
We thank Mrs. Suma (JNCASR) for the confocal microscopy and Mrs. Usha (JNCASR) for TEM imaging. We thank Dr. Anujith Kumar (MIRM-MAHE) for sharing his resources and Ms. Smitha Bhaskar (MIRM-MAHE) for helping with transfection using the electroporation technique. We thank Prof. Dipankar Nandi, Indian Institute of Science, for the DCFDA reagent. We thank Dr Lakshmi Balasubramanian (C-CAMP, Bangalore) and Mr. Vedam Pruthvi (MIRM-MAHE) for giving suggestions to develop an image analysis flow diagram. Figure 6H, Graphical Abstract and SI S3 A were created with Biorender.com.
Funding A.R, and S.C thank Manipal Academy of Higher Education for Dr. TMA Pai fellowship; A.R thanks ICMR SRF- Direct fellowship, R.K. thanks the Indian Council of Medical Research of India (#5/4-5/Ad-hoc/Neuro/216/2020-NCD-I) for her JRF fellowship; S.N thanks the Science and Engineering Research Board of India for the SERB-SRG (#SRG/2021/001315) grant; the Indian Council of Medical Research of India (#5/4-5/Ad-hoc/Neuro/216/2020-NCD-I and #IIRP-2023-0084) and the Intramural fund of Manipal Academy of Higher Education, Manipal, India (#MAHE/CDS/PHD/MIFR/2019) for financial support; and DST FIST grant #SR/FST/LS-I/2018/121. The financial support from the DBT-RA program in Biotechnology and Life Sciences and DST-SERB NPDF to SP (JNCASR) is gratefully acknowledged; The financial support from Department of Biotechnology (DBT) grant in Life Science Research, Education and Training at JNCASR (BT/INF/22/SP27679/2018), S. Ramachandran-National Bioscience Award for Career Development (NBACD)-2020-21 (SAN No. 102/IFD/SAN/990/2021-22) and JNCASR intramural funds to RM is acknowledged. S.P (NIMHANS) thanks the Science and Engineering Research Board, Government of India for the funding support (ECR/2018/002219).
Author contributions
S.N conceived and conducted the research; A.R, R.K, S.P (JNCASR), R.M (IISc), R.M (JNCASR), S.P (NIMHANS), and S.N designed and interpreted data; A.R, R.K, S.P (JNCASR), S.J, S.C, A.A, M.N.D, and M.G performed experiments; A.R, R.K, S.P (JNCASR), S.C, and A.A analyzed the data; A.R, R.M (JNCASR), and S.N wrote the article taking valuable inputs from all the authors.
Declaration of interests
Authors declare no competing interest.
Published: July 23, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110565.
Supplemental information
References
- 1.Forno L.S. Neuropathology of Parkinson's disease. J. Neuropathol. Exp. Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Kalia L.V., Lang A.E. Parkinson's disease. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 3.Lee H.J., Bae E.J., Lee S.J. Extracellular alpha--synuclein-a novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 2014;10:92–98. doi: 10.1038/nrneurol.2013.275. [DOI] [PubMed] [Google Scholar]
- 4.Yamada K., Iwatsubo T. Extracellular alpha-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 2018;13:9. doi: 10.1186/s13024-018-0241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fortin D.L., Nemani V.M., Voglmaier S.M., Anthony M.D., Ryan T.A., Edwards R.H. Neural activity controls the synaptic accumulation of alpha-synuclein. J. Neurosci. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krejciova Z., Carlson G.A., Giles K., Prusiner S.B. Replication of multiple system atrophy prions in primary astrocyte cultures from transgenic mice expressing human alpha-synuclein. Acta Neuropathol. Commun. 2019;7:81. doi: 10.1186/s40478-019-0703-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacMahon Copas A.N., McComish S.F., Fletcher J.M., Caldwell M.A. The Pathogenesis of Parkinson's Disease: A Complex Interplay Between Astrocytes, Microglia, and T Lymphocytes? Front. Neurol. 2021;12 doi: 10.3389/fneur.2021.666737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gibson E.M., Purger D., Mount C.W., Goldstein A.K., Lin G.L., Wood L.S., Inema I., Miller S.E., Bieri G., Zuchero J.B., et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 2014;344 doi: 10.1126/science.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venkatesh H.S., Johung T.B., Caretti V., Noll A., Tang Y., Nagaraja S., Gibson E.M., Mount C.W., Polepalli J., Mitra S.S., et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161:803–816. doi: 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venkatesh H.S., Morishita W., Geraghty A.C., Silverbush D., Gillespie S.M., Arzt M., Tam L.T., Espenel C., Ponnuswami A., Ni L., et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573:539–545. doi: 10.1038/s41586-019-1563-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheiblich H., Dansokho C., Mercan D., Schmidt S.V., Bousset L., Wischhof L., Eikens F., Odainic A., Spitzer J., Griep A., et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell. 2021;184:5089–5106.e21. doi: 10.1016/j.cell.2021.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rostami J., Mothes T., Kolahdouzan M., Eriksson O., Moslem M., Bergström J., Ingelsson M., O'Callaghan P., Healy L.M., Falk A., Erlandsson A. Crosstalk between astrocytes and microglia results in increased degradation of alpha-synuclein and amyloid-beta aggregates. J. Neuroinflammation. 2021;18:124. doi: 10.1186/s12974-021-02158-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rostami J., Holmqvist S., Lindström V., Sigvardson J., Westermark G.T., Ingelsson M., Bergström J., Roybon L., Erlandsson A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 2017;37:11835–11853. doi: 10.1523/JNEUROSCI.0983-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neupane S., De Cecco E., Aguzzi A. The Hidden Cell-to-Cell Trail of alpha-Synuclein Aggregates. J. Mol. Biol. 2023;435 doi: 10.1016/j.jmb.2022.167930. [DOI] [PubMed] [Google Scholar]
- 15.Victoria G.S., Zurzolo C. The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J. Cell Biol. 2017;216:2633–2644. doi: 10.1083/jcb.201701047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rustom A., Saffrich R., Markovic I., Walther P., Gerdes H.H. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–1010. doi: 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- 17.Ramírez-Jarquín U.N., Sharma M., Shahani N., Li Y., Boregowda S., Subramaniam S. Rhes protein transits from neuron to neuron and facilitates mutant huntingtin spreading in the brain. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jansens R.J.J., Tishchenko A., Favoreel H.W. Bridging the Gap: Virus Long-Distance Spread via Tunneling Nanotubes. J. Virol. 2020;94 doi: 10.1128/JVI.02120-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haimovich G., Ecker C.M., Dunagin M.C., Eggan E., Raj A., Gerst J.E., Singer R.H. Intercellular mRNA trafficking via membrane nanotube-like extensions in mammalian cells. Proc. Natl. Acad. Sci. USA. 2017;114:E9873–E9882. doi: 10.1073/pnas.1706365114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raghavan A., Rao P., Neuzil J., Pountney D.L., Nath S. Oxidative stress and Rho GTPases in the biogenesis of tunnelling nanotubes: implications in disease and therapy. Cell. Mol. Life Sci. 2021;79:36. doi: 10.1007/s00018-021-04040-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dilna A., Deepak K.V., Damodaran N., Kielkopf C.S., Kagedal K., Ollinger K., Nath S. Amyloid-beta induced membrane damage instigates tunneling nanotube-like conduits by p21-activated kinase dependent actin remodulation. Biochim. Biophys. Acta, Mol. Basis Dis. 2021;1867 doi: 10.1016/j.bbadis.2021.166246. [DOI] [PubMed] [Google Scholar]
- 22.Dilsizoglu Senol A., Samarani M., Syan S., Guardia C.M., Nonaka T., Liv N., Latour-Lambert P., Hasegawa M., Klumperman J., Bonifacino J.S., Zurzolo C. alpha-Synuclein fibrils subvert lysosome structure and function for the propagation of protein misfolding between cells through tunneling nanotubes. PLoS Biol. 2021;19 doi: 10.1371/journal.pbio.3001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abounit S., Bousset L., Loria F., Zhu S., de Chaumont F., Pieri L., Olivo-Marin J.C., Melki R., Zurzolo C. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016;35:2120–2138. doi: 10.15252/embj.201593411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y., Cui J., Sun X., Zhang Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011;18:732–742. doi: 10.1038/cdd.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desir S., Dickson E.L., Vogel R.I., Thayanithy V., Wong P., Teoh D., Geller M.A., Steer C.J., Subramanian S., Lou E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget. 2016;7:43150–43161. doi: 10.18632/oncotarget.9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Si Z., Sun L., Wang X. Evidence and perspectives of cell senescence in neurodegenerative diseases. Biomed. Pharmacother. 2021;137 doi: 10.1016/j.biopha.2021.111327. [DOI] [PubMed] [Google Scholar]
- 27.Yoon Y.S., You J.S., Kim T.K., Ahn W.J., Kim M.J., Son K.H., Ricarte D., Ortiz D., Lee S.J., Lee H.J. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp. Mol. Med. 2022;54:115–128. doi: 10.1038/s12276-022-00727-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beauséjour C.M., Krtolica A., Galimi F., Narita M., Lowe S.W., Yaswen P., Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macip S., Igarashi M., Fang L., Chen A., Pan Z.Q., Lee S.W., Aaronson S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002;21:2180–2188. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martínez-Cué C., Rueda N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020;14:16. doi: 10.3389/fncel.2020.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schousboe A., Bak L.K., Waagepetersen H.S. Astrocytic Control of Biosynthesis and Turnover of the Neurotransmitters Glutamate and GABA. Front. Endocrinol. 2013;4:102. doi: 10.3389/fendo.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dieriks B.V., Park T.I.H., Fourie C., Faull R.L.M., Dragunow M., Curtis M.A. alpha-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson's disease patients. Sci. Rep. 2017;7 doi: 10.1038/srep42984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chakraborty R., Nonaka T., Hasegawa M., Zurzolo C. Tunnelling nanotubes between neuronal and microglial cells allow bi-directional transfer of alpha-Synuclein and mitochondria. Cell Death Dis. 2023;14:329. doi: 10.1038/s41419-023-05835-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abounit S., Delage E., Zurzolo C. Identification and Characterization of Tunneling Nanotubes for Intercellular Trafficking. Curr. Protoc. Cell Biol. 2015;67:12.10.1–12.10.21. doi: 10.1002/0471143030.cb1210s67. [DOI] [PubMed] [Google Scholar]
- 35.Valappil D.K., Raghavan A., Nath S. Detection and Quantification of Tunneling Nanotubes Using 3D Volume View Images. J. Vis. Exp. 2022;186:e63992. doi: 10.3791/63992. [DOI] [PubMed] [Google Scholar]
- 36.Osswald M., Jung E., Sahm F., Solecki G., Venkataramani V., Blaes J., Weil S., Horstmann H., Wiestler B., Syed M., et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528:93–98. doi: 10.1038/nature16071. [DOI] [PubMed] [Google Scholar]
- 37.Abbassi R.H., Recasens A., Indurthi D.C., Johns T.G., Stringer B.W., Day B.W., Munoz L. Lower Tubulin Expression in Glioblastoma Stem Cells Attenuates Efficacy of Microtubule-Targeting Agents. ACS Pharmacol. Transl. Sci. 2019;2:402–413. doi: 10.1021/acsptsci.9b00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X., Veruki M.L., Bukoreshtliev N.V., Hartveit E., Gerdes H.H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. USA. 2010;107:17194–17199. doi: 10.1073/pnas.1006785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sowinski S., Jolly C., Berninghausen O., Purbhoo M.A., Chauveau A., Köhler K., Oddos S., Eissmann P., Brodsky F.M., Hopkins C., et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008;10:211–219. doi: 10.1038/ncb1682. [DOI] [PubMed] [Google Scholar]
- 40.Miller S.J., Campbell C.E., Jimenez-Corea H.A., Wu G.H., Logan R. Neuroglial Senescence, alpha-Synucleinopathy, and the Therapeutic Potential of Senolytics in Parkinson's Disease. Front. Neurosci. 2022;16 doi: 10.3389/fnins.2022.824191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verma D.K., Seo B.A., Ghosh A., Ma S.X., Hernandez-Quijada K., Andersen J.K., Ko H.S., Kim Y.H. Alpha-Synuclein Preformed Fibrils Induce Cellular Senescence in Parkinson's Disease Models. Cells. 2021;10 doi: 10.3390/cells10071694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y., Yuan Y., Li Y., Han D., Liu T., Yang N., Mi X., Hong J., Liu K., Song Y., et al. Inhibition of alpha-Synuclein Accumulation Improves Neuronal Apoptosis and Delayed Postoperative Cognitive Recovery in Aged Mice. Oxid. Med. Cell. Longev. 2021;2021 doi: 10.1155/2021/5572899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valdebenito S., Malik S., Luu R., Loudig O., Mitchell M., Okafo G., Bhat K., Prideaux B., Eugenin E.A. Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-93775-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henderson J.M., Ljubojevic N., Belian S., Chaze T., Castaneda D., Battistella A., Giai Gianetto Q., Matondo M., Descroix S., Bassereau P., Zurzolo C. Tunnelling nanotube formation is driven by Eps8/IRSp53-dependent linear actin polymerization. EMBO J. 2023;42 doi: 10.15252/embj.2023113761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chuang H.H., Wang P.H., Niu S.W., Zhen Y.Y., Huang M.S., Hsiao M., Yang C.J. Inhibition of FAK Signaling Elicits Lamin A/C-Associated Nuclear Deformity and Cellular Senescence. Front. Oncol. 2019;9:22. doi: 10.3389/fonc.2019.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou J., Yi Q., Tang L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: a focused review. J. Exp. Clin. Cancer Res. 2019;38:250. doi: 10.1186/s13046-019-1265-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lietha D., Cai X., Ceccarelli D.F.J., Li Y., Schaller M.D., Eck M.J. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim S.T., Miller N.L.G., Chen X.L., Tancioni I., Walsh C.T., Lawson C., Uryu S., Weis S.M., Cheresh D.A., Schlaepfer D.D. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J. Cell Biol. 2012;197:907–919. doi: 10.1083/jcb.201109067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lim S.T., Chen X.L., Lim Y., Hanson D.A., Vo T.T., Howerton K., Larocque N., Fisher S.J., Schlaepfer D.D., Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pirone D.M., Liu W.F., Ruiz S.A., Gao L., Raghavan S., Lemmon C.A., Romer L.H., Chen C.S. An inhibitory role for FAK in regulating proliferation: a link between limited adhesion and RhoA-ROCK signaling. J. Cell Biol. 2006;174:277–288. doi: 10.1083/jcb.200510062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loria F., Vargas J.Y., Bousset L., Syan S., Salles A., Melki R., Zurzolo C. alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017;134:789–808. doi: 10.1007/s00401-017-1746-2. [DOI] [PubMed] [Google Scholar]
- 52.Spees J.L., Olson S.D., Whitney M.J., Prockop D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA. 2006;103:1283–1288. doi: 10.1073/pnas.0510511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lou E., Fujisawa S., Morozov A., Barlas A., Romin Y., Dogan Y., Gholami S., Moreira A.L., Manova-Todorova K., Moore M.A.S. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han X., Wang X. Opportunities and Challenges in Tunneling Nanotubes Research: How Far from Clinical Application? Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22052306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heinke L. Mitochondrial ROS drive cell cycle progression. Nat. Rev. Mol. Cell Biol. 2022;23:581. doi: 10.1038/s41580-022-00523-5. [DOI] [PubMed] [Google Scholar]
- 56.Kidwell C.U., Casalini J.R., Pradeep S., Scherer S.D., Greiner D., Bayik D., Watson D.C., Olson G.S., Lathia J.D., Johnson J.S., et al. Transferred mitochondria accumulate reactive oxygen species, promoting proliferation. Elife. 2023;12 doi: 10.7554/eLife.85494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davalli P., Mitic T., Caporali A., Lauriola A., D'Arca D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016;2016 doi: 10.1155/2016/3565127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gallage S., Gil J. Mitochondrial Dysfunction Meets Senescence. Trends Biochem. Sci. 2016;41:207–209. doi: 10.1016/j.tibs.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 59.Borges H.L., Linden R., Wang J.Y.J. DNA damage-induced cell death: lessons from the central nervous system. Cell Res. 2008;18:17–26. doi: 10.1038/cr.2007.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nath S., Agholme L., Kurudenkandy F.R., Granseth B., Marcusson J., Hallbeck M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J. Neurosci. 2012;32:8767–8777. doi: 10.1523/JNEUROSCI.0615-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gousset K., Schiff E., Langevin C., Marijanovic Z., Caputo A., Browman D.T., Chenouard N., de Chaumont F., Martino A., Enninga J., et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009;11:328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 62.Tardivel M., Bégard S., Bousset L., Dujardin S., Coens A., Melki R., Buée L., Colin M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016;4:117. doi: 10.1186/s40478-016-0386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sharma M., Subramaniam S. Rhes travels from cell to cell and transports Huntington disease protein via TNT-like protrusion. J. Cell Biol. 2019;218:1972–1993. doi: 10.1083/jcb.201807068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saha T., Dash C., Jayabalan R., Khiste S., Kulkarni A., Kurmi K., Mondal J., Majumder P.K., Bardia A., Jang H.L., Sengupta S. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 2022;17:98–106. doi: 10.1038/s41565-021-01000-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang F., Chen X., Cheng H., Song L., Liu J., Caplan S., Zhu L., Wu J.Y. MICAL2PV suppresses the formation of tunneling nanotubes and modulates mitochondrial trafficking. EMBO Rep. 2021;22 doi: 10.15252/embr.202052006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schober M., Raghavan S., Nikolova M., Polak L., Pasolli H.A., Beggs H.E., Reichardt L.F., Fuchs E. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J. Cell Biol. 2007;176:667–680. doi: 10.1083/jcb.200608010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang C., Ren G., Zhou H., Wang C.C. A new method for purification of recombinant human alpha-synuclein in Escherichia coli. Protein Expr. Purif. 2005;42:173–177. doi: 10.1016/j.pep.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 68.Jos S., Gogoi H., Prasad T.K., Hurakadli M.A., Kamariah N., Padmanabhan B., Padavattan S. Molecular insights into alpha-synuclein interaction with individual human core histones, linker histone, and dsDNA. Protein Sci. 2021;30:2121–2131. doi: 10.1002/pro.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nath S., Meuvis J., Hendrix J., Carl S.A., Engelborghs Y. Early aggregation steps in alpha-synuclein as measured by FCS and FRET: evidence for a contagious conformational change. Biophys. J. 2010;98:1302–1311. doi: 10.1016/j.bpj.2009.12.4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sackmann V., Sinha M.S., Sackmann C., Civitelli L., Bergström J., Ansell-Schultz A., Hallbeck M. Inhibition of nSMase2 Reduces the Transfer of Oligomeric alpha-Synuclein Irrespective of Hypoxia. Front. Mol. Neurosci. 2019;12:200. doi: 10.3389/fnmol.2019.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Organelles were observed to move (indicated by white and black arrows) unidirectional inside the TNTs.
Organelles were observed to move unidirectionally (indicated by a white arrow) inside the TNTs.
The composite video shows unidirectional movements of vesicles colocalized (yellow) with α-SYN-TMR (red) and lysotracker (green) in long, thin TNTs (indicated by white arrow).
The composite video shows unidirectional movements of mitochondria (cyan) in long, thin TNTs (indicated by a white arrow).
Direct cell-to-cell transfer of α-SYN-TMR (red) accumulated lysotracker (green) positive vesicles (yellow vesicles; indicated by black arrow) via TNTs between neighboring U-87MG cells.
Direct cell-to-cell transfer of α-SYN-TMR (red) treated moto tracker positive vesicles (cyan vesicles; indicated by black arrow) via TNTs between neighboring U-87MG cells.
Data Availability Statement
-
•
Data and images that support the findings of this study are available on request from the lead contact.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data in this paper is available from the lead contact upon request.