

Summary
Microglia play a crucial role in a range of neuropathologies through exacerbated activation. Microglial inflammatory responses can be influenced by prior exposures to noxious stimuli, like increased levels of extracellular adenosine and ATP. These are characteristic of brain insults like epileptic seizures and could potentially shape subsequent responses through epigenetic regulation. We investigated DNA methylation and expression changes in human microglia-like cells differentiated from monocytes following ATP-mediated preconditioning. We demonstrate that microglia-like cells display homeostatic microglial features, shown by surface markers, transcriptome, and DNA methylome. After exposure to ATP, TLR-mediated activation leads to an exacerbated pro-inflammatory response. These changes are accompanied by methylation and transcriptional reprogramming associated with enhanced immune-related functions. The reprogramming associated with ATP-mediated preconditioning leads to profiles found in microglial subsets linked to epilepsy. Purine-driven microglia immune preconditioning drives epigenetic and transcriptional changes that could contribute to altered functions of microglia during seizure development and progression.
Subject areas: Microglia, Purines, Epilepsy, Epigenetics, Transcriptomics
Graphical abstract
Highlights
-
•
Microglia-like cells replicate microglial transcriptome and DNA methylome
-
•
Purinergic preconditioning promotes enhanced pro-inflammatory response
-
•
Methylation and transcription of inflammatory genes are altered by ATP pre-exposure
-
•
ATP-related expression changes mimic microglial population expanded in epilepsy
Microglia; Purines; Epilepsy; Epigenetics; Transcriptomics
Introduction
Microglia, as the brain’s resident macrophage population, represent the first line of immune defense within the central nervous system (CNS). Despite their low abundance in relation to other cell types (around 10%),1,2 consistent evidence has demonstrated an essential role of microglia in maintaining overall brain function.3,4,5 Furthermore, mutations of microglia-specific genes have been associated with different neurodegenerative diseases.6,7 The involvement of microglia in neuropathology is often attributed to its aberrant activation. Recently, exacerbated and uncontrolled pro-inflammatory activation has been proposed to be due to the existence of innate immune memory, in a process tightly regulated by epigenetic reprogramming. In the literature, there has not been unanimous consensus on the nomenclature used to describe innate immune memory. According to the model proposed by Neher and Cunningham (2019),8 microglia manifest a “primed” state in neuropathological settings, reflected by an activated state or a state of elevated permissiveness to activation. This has been supported by an exacerbated pro-inflammatory response to lipopolysaccharide (LPS) in mice models of neurodegenerative diseases.9,10,11 The concept of immune priming is now defined by a more precise nomenclature. Innate immune memory represents a conditioned response to a secondary stimulus induced by prior exposure to initial stimuli. This response can be expressed in two directions: “immune training” (formerly priming) reflects an exacerbated pro-inflammatory response, while “immune tolerance” is characterized by a desensitized response.12
The study of purinergic signaling may represent a promising pathway to better understand the role of microglial conditioning in epileptogenesis. Adenosine and ATP have been proposed to be essential mediators of epilepsy.13 However, there is no consensus on the precise underlying mechanisms that are involved. Generally, adenosine is primarily considered under the light of its anticonvulsant effects through activation of adenosine A1 receptors,14 and may also exert excitatory and neurodegenerative effects associated with adenosine A2A receptor activation.15,16 Like adenosine, ATP is a ubiquitous endogenous molecule with multiple implications on the CNS through modulation of cell survival, proliferation and differentiation, axonal growth and maturation, excitability and glial activation.17 The impact of ATP in epileptogenesis has mainly been studied in the context of the ionotropic P2X7 receptor (P2RX7), the activation of which is predominantly associated with microglia activation and pro-inflammation. P2RX7 is generally increased in epilepsy with anticonvulsive effects attributed to its antagonistic treatment.13 Purine metabolism is widely pleiotropic, with the respective receptors being expressed throughout all the major cell types in the CNS, and therefore exerting potentially contrary effects depending on the cell type, the receptor, the brain region, the used model of epilepsy, and genetic heterogeneity. Understanding in depth the intricacies of the impact of purinergic imbalance in epileptogenesis may represent an overwhelming task. What appears to be more widely accepted is that both adenosine and ATP extracellular levels increase significantly upon brain insult, including after the occurrence of seizures. It is also important to take into consideration that ATP has a short half-life and is quickly converted into adenosine by ectonucleotidases. Therefore, the increased release of ATP also contributes to elevated levels of adenosine.18,19,20,21
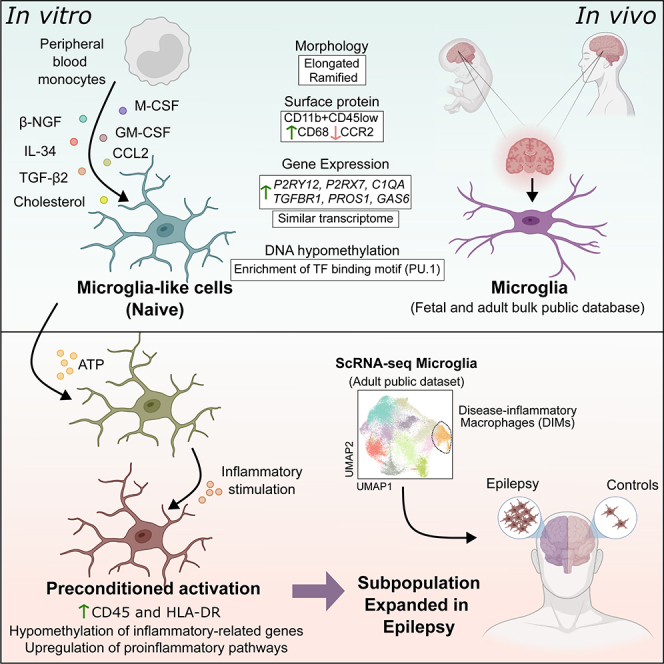
Human microglia research has been significantly hampered by the low replicability of in vivo traits in cell culture models. The use of fetal or adult brain biopsies for human primary microglia cultures is limited by ethical and logistic reasons, together with relatively low yields of isolation.22 In addition, replicating the homeostatic microglia phenotype is particularly challenging due to the substantial transcriptomic and epigenetic changes that cells undergo in culture,23 and may be influenced by postmortem conditions.24 To overcome these limitations in experimental scalability of human microglia cultures, alternative protocols have been developed, including the differentiation of peripheral blood monocytes into microglia-like cells. In this study, we aimed to investigate how ATP-driven preconditioning could influence posterior inflammatory activation in microglia by using monocyte-derived microglia-like cells. We have characterized the DNA methylation and transcriptional changes associated with inflammatory preconditioning, the associated features and its potential relationship with the phenotype of microglia in epileptogenesis.
Results
Monocyte-derived microglia-like cells show features of homeostatic microglia
We first differentiated microglia-like cells from peripheral blood monocytes using an adapted version of a previously described procedure.25 Monocytes were differentiated in serum-free culture medium supplemented with M-CSF, GM-CSF, β-NGF, IL-34, and CCL2. Moreover, we added TGF-β2 and cholesterol solution, which have been shown to increase viability of mice microglia in vitro26 (Figure 1A). In parallel, we differentiated monocytes to pro-inflammatory macrophages as a reference to validate the homeostatic nature of microglia-like cells. Microglia-like cells acquired a stable phenotype at seven days. After three days, we observed that cells acquired an elongated and ramified morphology resembling homeostatic microglia in vivo (Figure 1B). Gene expression evaluation by quantitative PCR demonstrated upregulation of the canonical homeostatic microglia marker P2RY12 in microglia-like cells in comparison to monocytes (Figure 1C). We also performed flow cytometry evaluation of a panel of markers consistently used to describe and distinguish microglia-like from monocytes and macrophages. Our gating strategy aimed to delimitate the CD11b+ macrophage lineage population in the microglia-like and monocyte-derived macrophage samples (Figure S1A). We observed a decrease in CD14 and CCR2 in microglia-like cells in relation to monocytes and macrophages, as previously described.27 We also determined an increase in CD68, a standard microglia marker in brain tissue that is also expressed by other macrophages, in relation to monocytes. Finally, we measured a decrease in CD45 in relation to macrophages (Figures 1D and S1B). It is important to note that the dual CD11b/CD45 signal is the most predominantly used strategy to distinguish homeostatic microglia (CD11b+/CD45low) from macrophages (CD11b+/CD45high), and this model successfully replicated these conditions.
Figure 1.

Monocyte-derived microglia-like cells show features of homeostatic microglia
(A) Schematic representation of the in vitro differentiation protocol from freshly isolated monocytes to monocyte-derived microglia-like cells and monocyte-derived macrophages. For microglia-like differentiation, monocytes were plated in serum-free conditions with medium supplemented with M-CSF, GM-CSF, β-NGF, IL-34, CCL2, TGF-β2, and cholesterol. Macrophages were obtained by culturing monocytes in medium supplemented with 10% FBS and M-CSF; plus, LPS treatment one day prior to collection.
(B) Representative optical microscopy images of microglia-like differentiation at days three and six of culture with 20× magnification.
(C) Barplot representation of the gene expression of P2RY12 in microglia-like cells in comparison to monocytes obtained by RT-qPCR. The significance was calculated using a paired t test (∗p < 0.05).
(D) Boxplot representation of the median fluorescence intensity (MFI) values obtained by flow cytometry for CD14, CCR2, CD68, CD11b, and CD45 in monocytes, macrophages, and microglia-like cells. The significance was calculated using a paired t test (∗p < 0.05, ∗∗p < 0.01).
(E) Boxplot representation of RNA-seq normalized counts for P2RY12, P2RX7, C1QA, PROS1, TGFBR1, and GAS6 in monocytes and microglia-like cells. All six genes were significantly upregulated in microglia-like cells in the regression model.
(F) Principal-component analysis (PCA), using variance stabilizing transformation (VST) values considering all transcriptome of the RNA-seq data generated in this study for monocytes and microglia-like, together with public data of CD14+/CD16-and CD14-/CD16+ monocytes, dendritic cells, monocyte-derived macrophages, monocyte-derived microglia-like, iPSC-derived microglia, microglial cell lines (C20 and HMC3), and primary adult and fetal microglia. See also Figures S1 and S2, and Table S1.
To further assess the acquisition of the resting microglia phenotype at the transcriptomic level, we performed RNA-seq analysis of monocytes and microglia-like cells. The differentiation of monocyte to microglia-like cells resulted in the upregulation of 6692 genes and downregulation of 6189 genes (adjusted p value [FDR] < 0.05) (Figure S2A and Table S1), with the involvement of a wide range of transcription factors (TFs) (Figure S2B). In this process, we observed upregulation of P2RY12, P2RX7, C1QA, PROS1, TGFBR1, and GAS6 (Figure 1E and Table S1), microglia markers previously shown to be increased in microglia-like cells.25 We integrated our RNA-seq data with public datasets corresponding to microglia and other myeloid populations (Figure 1F). These were described by Rai et al. (2020),28 and included CD14+/CD16- (classical) and CD14-/CD16+ (non-classical) monocytes, conventional dendritic cells, two sets of induced pluripotent stem cell (iPSC)-derived microglia, monocyte-derived macrophages, the monocyte-derived microglia-like cells generated in that study, microglia cell lines (C20 and HMC3) and primary human adult and fetal microglia. We observed that our monocytes clustered together with those from the other datasets, and that the microglia-like cells generated with our method clustered closer to adult and fetal microglia. These results support the validity of microglia-like cells to replicate microglial transcriptomic features.
DNA methylation changes during differentiation associate with the acquisition of microglial features
We next determined the DNA methylation changes associated with the differentiation to microglia-like cells. In comparison to monocytes, homeostatic microglia-like cells had 2879 differentially methylated positions (DMPs) (adjusted p value [FDR] < 0.05 and absolute mean Beta difference >0.2) (Table S2). Most DMPs (2799) corresponded to CpG sites that are hypomethylated in microglia-like cells in comparison to monocytes (Figure 2A), which demonstrated prevalence in intergenic regions and were enriched in enhancer regions (Figures S2C and S2D). Gene ontology (GO) analysis of hypomethylated DMPs revealed enrichment of terms associated with inflammation and leukocyte differentiation (Figure 2B). Moreover, TF binding motif enrichment analysis yielded multiple TFs associated with the myeloid lineage (Figure 2C). Within those, we were able to identify several TFs previously associated with microglia-specific epigenetic modeling in human brain tissue.23,29 Notably, PU.1 emerged as the leading TF in this list (Figure 2D). To further validate the microglial nature of our microglia-like cells, we integrated our DNA methylation profiles with public data from microglia isolated from 56 human brain tissue samples (GSE191200). We observed that microglia-like cells clustered closer to primary microglia samples than to monocytes (Figure 2E). Integration of DNA methylation and RNA-seq showed that the genes associated with hypomethylated DMPs were upregulated in microglia-like cells vs. monocytes (Figure 2F). Selected examples of genes displaying both hypomethylation near the transcription start sites (TSS) and upregulation during microglia-like differentiation include CD68 and CSF1R, which are implicated in differentiation within the macrophage lineage and are essential for microglia maintenance and survival in vivo30 (Figure 2G). We also compared the DNA methylation profile of the microglia-like cells generated in vitro with macrophages, also differentiated in vitro from monocytes31 (similar differentiation protocol; see STAR method details). For such dataset, we applied the same preprocessing and DMP calculation pipeline and observed a majority pattern of hypomethylation (2806 DMPs) (Figure S2E). We next overlapped the lists of hyper and hypomethylated DMPs for macrophages vs. monocytes with the ones determined for microglia-like cells vs. monocytes (Figure S2F). As expected, there was a substantial intersection between both sets of DMPs, considering the macrophagic nature of both cell types and their similar monocyte-derived in vitro differentiation. In addition, both microglia-like cells and macrophages present DMPs that are exclusive to each differentiation (Figure S2F). GO analysis of the hypomethylated DMPs exclusive to macrophages showed a higher enrichment of pro-inflammatory and activation-related terms such as ones associated with wound healing, inflammatory response, and myeloid cell activation; while microglia-like exclusive methylation changes were enriched for cytokine synthesis and negative regulation of immune system (Figure S2G). These results support the acquisition of a homeostatic phenotype by microglia-like cells in relation to other models of monocyte-derived macrophages.
Figure 2.

DNA methylation changes during differentiation associate with the acquisition of microglial features
(A) Heatmap representation of the DNA methylation of the differentially methylated positions (DMPs) obtained for the microglia-like vs. monocytes comparison. DNA methylation is represented as the Z score of the beta values. The significance cutoff was of adjusted p value (FDR) < 0.05 and difference in mean beta values >0.2.
(B) Selected list of significantly enriched gene ontology (GO) terms for the hypomethylated DMPs in microglia-like. The significance of enrichment is represented by the negative of the log of the adjusted p value, the enrichment fold change and the number of gene hits.
(C) Motif enrichment of the most significant TFs (p value < 1E-21) from the list of hypomethylated DMPs. The TFs are annotated by family and the enrichment is represented by the negative of the log of the p value and the percentage of sequences matching the motif.
(D) Barplot showing the TF motif enrichment of a selected group of TFs previously show to be associated with microglia-specific chromatin accessibility. All show a p value lower than 0.01. The enrichment is represented by the negative of the log of the p value.
(E) Principal-component analysis (PCA) considering the beta values corresponding to all pairwise DMPs between monocytes, microglia-like cells and primary microglia from postmortem brain (GSE191200).
(F) Gene set enrichment analysis (GSEA) of the list of genes associated with hypomethylated DMPs in the differential expression comparison between microglia-like and monocytes. Genes associated with hypomethylated DMPs are upregulated in microglia-like cells. The enrichment is represented by the positive Normalized Enrichment Score (NES) and the statistical significance by the adjusted p value (FDR).
(G) Graphical representation of the beta values of the CpGs located nearby the CD68 and CSF1R genes for monocytes and microglia-like cells (left panels). The genes and the individual probes are represented in relation to the annotated genes in the UCSC Ref Seq. The CpGs highlight in red demonstrate a statistically significant hypomethylation in microglia-like vs. monocytes. Boxplot representation of RNA-seq normalized counts for CD68 and CSF1R in monocytes and microglia-like cells (right panels). Both genes were significantly upregulated in microglia-like cells in the regression model. See also Figure S2 and Table S2.
Exposure to ATP in microglia-like cells leads to epigenetic and transcriptomic preconditioning
To interrogate whether an initial ATP stimulus influences microglia responses to secondary inflammatory activation, we stimulated microglia-like cells with ATP and then subjected them to TLR4-mediated activation with LPS (Figure 3A). To this end, after a 6-day microglia differentiation, we treated a set of samples with ATP one day before harvesting (+ATP), whereas in parallel we left a second set of samples without ATP treatment (Naive). In addition, we performed microglia activation using LPS for both microglia sample sets and collected the cells after two days (+ATP+LPS and +LPS, respectively). These experiments were conducted with four biological replicates. Firstly, we observed upregulation of the pro-inflammatory gene IL1B, and a similar trend for IL6, in the +ATP+LPS condition in comparison to +LPS (Figure 3B). The levels of IL-1β protein in the supernatant of cells treated with +ATP+LPS also showed a non-significant increasing trend (Figure 3C). However, we acknowledge the lack of strength in demonstrating a higher activation at the functional level and cannot exclude the possibility that IL-1β might be released from non-inflammatory cell death. Therefore, additional functional assays would be required. The exacerbated pro-inflammatory profile of +ATP+LPS microglia-like cells was corroborated by flow cytometry. They showed higher levels of CD14, CD45, and HLA-DR in comparison to +LPS (Figures 3D and S3A). Of note, the increase in HLA-DR appears to result from additive effect of the ATP and LPS stimuli, as ATP alone significantly enhances its levels.
Figure 3.

ATP-driven preconditioning promotes a posterior exacerbated pro-inflammatory response to LPS, coupled with bidirectional DNA methylation modifications
(A) Schematic representation of the preconditioning and inflammatory stimulation strategy in microglia-like cells. The first set of conditions were collected at day seven of culture and were either preconditioned (+ATP) or non-preconditioned (Naive) with 100 μM ATP for 24 h. The preconditioned and non-preconditioned microglia-like were stimulated with 100 ng/mL LPS two days before collection and collected at day ten of culture (+ATP+LPS and +LPS, respectively).
(B) Barplot representation of the gene expression of IL1B and IL6 in Naive, +LPS, +ATP+ and +ATP+LPS microglia-like obtained by RT-qPCR.
(C) Boxplot representation of protein levels of IL-1B in the supernatant of +ATP+LPS vs. +LPS.
(D) Boxplot representation the median fluorescence intensity (MFI) values obtained by flow cytometry for CD14, CD68, CD45, and HLA-DR in Naive, +LPS, +ATP and +ATP+LPS. The statistical significance for panels (B), (C), and (D) was calculated using a paired t test (∗p < 0.05, ∗∗p < 0.01).
(E) Heatmap representation of the DNA methylation of the differentially methylated positions (DMPs) obtained for the pairwise comparison between all microglia-like conditions. Unsupervised clustering originated eight modules of DMPs (M1-M8). DNA methylation is represented as the Z score of the beta values. The significance cutoff was of p value <0.05 and absolute difference in mean beta values >0.1.
(F) Line plots representing the progression of DNA methylation from monocytes (MO) to non-activated microglia-like cells (No LPS) and to activated microglia-like cells (LPS) separated by the presence (+ATP and +ATP+LPS) or absence (Naive, +LPS) of the ATP stimulus. DNA methylation is represented as the mean of the Z score for all DMPs in each module for each group of cells.
(G) Selected list of significantly enriched gene ontology (GO) terms for each of the modules of DMPs. The significance of enrichment is represented by the negative of the log of the p value, the enrichment fold change and the number of gene hits.
(H) Transcription factor (TF) motif enrichment of a selected list of TFs (p value <0.01) for each module of DMPs. The TFs are annotated by family and the enrichment is represented by the negative of the log of the p value and the percentage of sequences matching the motif. See also Figure S3 and Tables S3 and S4.
DNA methylation analysis of the four studied microglia conditions showed some differences (Figure 3E and Table S3). We performed a pairwise comparison between all studied microglia-like groups. Significant DMPs were considered for a p value <0.05 and absolute mean Beta difference >0.1. A total of 2440 DMPs were grouped by unsupervised clustering in eight modules (Figure 3E and Table S4). The progression of each module throughout the differentiation from monocytes to homeostatic microglia (no LPS) to activated microglia (LPS) was evaluated separately for microglia-like cells untreated (Naive and +LPS) and treated with ATP (+ATP and +ATP+LPS) (Figure 3F). Some modules do not show visible differences caused by ATP treatment, such as M3 (573 DMPs) and M4 (347 DMPs) in which methylation increases with LPS stimulation independently of the ATP treatment; and M5 (592 DMPs) which shows an opposite behavior, meaning decreased methylation post-LPS invariably in ATP-treated and untreated cells. The impact of ATP is observed in M1 (148 DMPs), which exhibits the same trend of increased methylation in response to LPS as untreated cells, but at reduced levels. The opposite tendency is observed for M2 (167 DMPs), whose methylation also increased with LPS, but at higher levels in ATP treated cells. M7 (237 DMPs) shows an ATP-mediated decrease in methylation and the trend post-LPS is toward demethylation. Moreover, M6 (185 DMPs) is characterized by a spike in methylation after ATP treatment, but with no impact in the response to LPS. Lastly, in M8 (191 DMPs) the LPS-induced demethylation is impeded in ATP treated cells. Moreover, we evaluated the distribution of these modules in relation to gene location and CpG islands, as well as the association with chromatin states (Figures S3B and S3C). We highlight M6 and M8 which demonstrated higher prevalence near TSS. GO analysis showed that the modules of DMPs not influenced by ATP preconditioning present a striking enrichment of terms associated with pro-inflammatory activation, like glucocorticoid receptor binding and positive regulation of transforming growth factor β production (M3), negative regulation of chronic inflammatory response and microglial cell activation involved in immune response (M4), and monocyte chemotaxis and positive regulation of T cell differentiation (M5), thus representing general LPS-mediated patterns. ATP treatment hampers the methylation levels of DMPs enriched for pathways involved in response to DNA damage and hypoxia, and inflammation (e.g., inflammatory response to antigen stimulus; interleukin-4 secretion) (M1); and mitochondrial membrane fission, lipid modification, excitatory signaling (kainite selective glutamate receptor complex) and interleukine-7-mediated signaling pathway (M7). Plus, ATP treatment demonstrated the opposite effect (increased methylation) in DMPs involved in regulation of response to reactive oxygen species and phagocytic vesicle membrane (M2). Module M6, which experience a spike in methylation solely as response to ATP, shows enrichment for synapse-related processes and purine phosphorylation. Finally, DMPs in M8 were associated with mechanisms involved in synaptic function (excitatory synapse and synapse organization), purine metabolism (dADP metabolic process), and neurotransmission (alpha-2B adrenergic receptor binding) (Figure 3G). TF motif enrichment analysis also generated distinct profiles between methylation modules (Figure 3H). M2, M3, and M4 showed analogous TF enrichment; namely, Etv2 and ERG. In addition, both M1 and M5 demonstrated enrichment of multiple NFKB iterations and members of the AP-1 family (e.g., Jun, Fos, and ATF), which are known to regulate the surveillant microglia phenotype, but also immune activation.32 Examining the modules that showed a more striking influence by ATP-treatment, we observed enrichment in M6 (ATP-driven methylation) of E2F3, a factor associated with proliferation and migration in glioma cells.33 M8, which shows the clearest preconditioning-related effect, uniquely showed significant enrichment for Rfx1, mostly known as a transactivator of hepatitis B virus enhancer I, and also described to regulate MHC class II genes.34 In the context of microglia, increased levels of Rfx1 have been shown to downmodulate APOE expression, and, consequently, impair Aβ uptake.35 Furthermore, Rfx1 presents site-specific and DNA methylation-dependent binding activity.36
ATP-preconditioning resulted in significant changes in gene expression. We detected 378 differentially expressed genes (DEGs) (205 upregulated and 173 downregulated) in +ATP vs. Naive microglia-like cells; and 1223 DEGs (667 upregulated and 556 downregulated) in +ATP+LPS vs. +LPS (Figure 4A). Of note, a significant proportion of the upregulated and downregulated DEGs were coincident in both comparisons (Figure 4B), suggesting that exposure to ATP by itself promotes gene expression changes that are maintained after activation. Moreover, we performed unsupervised clustering of all DEGs from all pairwise comparisons (Table S5) between the four microglia-like study conditions, consisting in a total of 7362 genes. Unsupervised clustering of all DEGs from pairwise comparisons separated them into twelve modules (E1-E12) (Figures 4C and 4D, and Table S6). Similar to the described in the methylation modules, a large portion of the identified DEGs were unaffected by the ATP stimulus, behaving similarly in response to LPS, namely the ones belonging to expression modules E1 (1330 genes), E2 (924 genes), E8 (450 genes), E9 (371 genes), E11 (819 genes), and E12 (1166). The remaining modules demonstrated ATP-mediated patterns, including E3 (227 genes) in which the expression increases in both ATP-treated conditions (+ATP, +ATP+LPS), and E7 (339 genes) in which ATP and LPS present an additive effect by progressively causing downregulation. In E5 (471 genes) and E10 (403) gene expression upregulated in response to LPS, but exponentiated in +ATP+LPS. E4 genes (424) demonstrated a hampered upregulation following LPS in +ATP+LPS cells. In the opposite direction, the downregulation of E6 genes (438) post-LPS is also impeded by ATP. In this regard, the behaviors observed in E4, E5, E6, and E10 are interpreted as a potential preconditioning effect of ATP (Figure 4D). From the GO analysis of the expression modules, the most prominent results include the enrichment of inflammation-related terms in the aforementioned preconditioning modules (E5, cytokine activity; E10, regulation of leukocyte mediated cytotoxicity and phagocytic vesicle). In addition, immunologic terms were not observable for E4, which was enriched for pathways involved in vesicle transport; however, E6 demonstrated high enrichment of pro-inflammatory terms such as immune response-activating signaling pathway and phagocytosis (Figure 4E). Analysis of regulon enrichment based on gene expression of TF targets demonstrated a higher activation of inflammation-related TFs caused by ATP pre-exposure, such as a member of the interferon-regulatory factor (IRF1), NF-kB subunits (RELA and NFKB1), and STAT proteins (STAT1 and STAT5B) in the +ATP+LPS vs. +LPS comparison (Figure 4F). To integrate the transcriptomics and DNA methylation data, we calculated the enrichment of the overlap between the genes from each gene expression module (E1-E12) and the genes associated with the methylation modules (M1-M8). (Figure S3D). Of note, no significant enrichment was observed for the main modules that we deemed previously as related to preconditioning.
Figure 4.

The pro-inflammatory response caused by ATP preconditioning is observed at the transcriptional level
(A) Volcano plots depicting the differential expression between +ATP and Naive, and between +ATP+LPS and +LPS microglia-like cells. Differentially expressed genes (DEGs) are considered for adjusted p value (FDR) < 0.05. Upregulated DEGs (log2(fold change) > 0) are highlighted in red, and downregulated DEGs (log2(fold change) < 0) in green.
(B) Venn diagram of the overlap between the list of genes up and downregulated in the two comparisons.
(C) Heatmap representation of the DEGss obtained for the pairwise comparison between all microglia-like conditions. Gene expression is represented as the Z score of the normalized counts. The significance cutoff was of adjusted p value (FDR) < 0.05. Unsupervised clustering divided the DEGs in twelve modules (E1-E12). DEGs associated with differentially methylated positions (DMPs) are highlighted (black lines).
(D) Violin plot representation of the mean Z score of the normalized counts of each DEG module in monocytes and in all microglia-like cell groups.
(E) Selected list of significantly enriched gene ontology (GO) terms for each of modules of DEGs. The significance of enrichment is represented by the negative of the log of the adjusted p value, the enrichment fold change and the number of gene hits.
(F) Transcription factor (TF) enrichment of all significant TF (adjusted p value (FDR) < 0.05) in both differential expression comparisons (+ATP vs. Naive and +ATP+LPS vs. +LPS). Significance is represented by the NES (Normalized Enrichment Score) and the negative of log of the adjusted p value (FDR). See also Figure S3 and Tables S5 and S6.
ATP-mediated preconditioning induces a molecular signature in microglia-like cells that correspond to subsets expanded in epilepsy
Finally, we examined the transcriptomic changes observed in ATP preconditioned microglia-like cells in the context of human microglia subpopulations in patients with epilepsy and healthy individuals. To this end, we used an integrated single-cell (sc)RNA-seq object consisting of 36,927 microglia cells from both patients with epilepsy and healthy controls without any diagnosed neuropathology.37,38,39,40,41 The integrated object consisted of twelve clusters of microglial cells (Figure 5A), expressing high levels of canonical markers, P2RY12 and CX3CR1, and were obtained after exclusion of cells expressing markers for macrophages, T cells and other CNS cell types (see STAR Methods). Each subpopulation showed variable expression of known homeostatic genes (P2RY12 and CX3CR1) and pathology-related microglia markers (SPP1 and APOE). In addition, we investigated the average expression of gene signatures characterizing the phenotypes of “disease-associate microglia” (DAM) and “disease-inflammatory macrophages” (DIMs), described elsewhere,42 and the module score expression of the genes in the expression modules described in our study. We observed a high expression of the E5 and E10 modules, described as upregulated in ATP pre-exposed cells, in cluster 7, which also demonstrated a specific upregulation of the DIM signature (Figure 5B). We then subsetted the scRNA-seq object, considering only cluster 7, which consisted of 1569 and 622 cells from epilepsy patients and controls, respectively. Reclustering of these cells resulted in five clusters, one of which (cluster 0) was expanded in epilepsy in comparison to controls (Figure 5D). To further explore the association of both the DNA methylation and the gene expression modules obtained in our microglia-like experiment, we investigated the module score expression of the genes within each methylation and expression module in the DIM subpopulations, with special attention to DIM cluster 0 (Figure 5E). The expression modules E5 and E10 showed high expression in DIM cluster 0. We acknowledge the lack of association between the DNA methylation modules and the gene expression analysis, and the scRNA-seq validation. Even when inspecting lists of genes shared between E5 and E10 and the methylation module 8 (inhibition of demethylation by ATP pre-exposure), no clear pattern of high expression in DIM cluster 0 was observed (Figure 5E). Of note, genes from M8 were considered here based on the abovementioned closer location to TSS and potential influence in transcriptomic regulation. However, at the transcriptomic level we were able to observe the replication of the expression patterns observed in our microglia-like experiment and the scRNA-seq validation. In particular, we represent four genes belonging to E5 (P2RX4, LILRB4) and E10 (OTUD1, SIGLEC10) that have been described in microglia activation and immune regulation and demonstrated high levels of expression in the epilepsy-associated DIM cluster 0 and were significantly upregulated in +ATP+LPS vs. +LPS (Figures 5E and 5F). Therefore, these data demonstrate that an exacerbated inflammatory activation of microglia-like cells promoted by ATP preconditioning is replicated in a population of inflammatory microglia-like cells that infiltrate the brain during injury and are expanded in epilepsy patients.
Figure 5.

ATP-mediated preconditioning induces a molecular signature in microglia-like cells that correspond to subsets expanded in epilepsy
(A) UMAP representation of the integrated single-cell (sc)RNA-seq object composed of 36,927 microglia cells from epilepsy patients and healthy individuals, distributed across twelve clusters.
(B) Dot plot representation of the expression of genes associated with homeostatic (P2RY12 and CX3CR1) and pathological (SPP1 and APOE) microglial phenotypes, and the module score expression of gene signatures characteristic of “disease-associated microglia” (DAM) and “disease inflammatory-macrophages” (DIMs), and the gene expression modules from our analysis.
(C) UMAP representation, split by pathological conditions (Epilepsy and Controls), of only the cells from cluster 7 which represent the DIMs. Reclustering of the DIM population resulted in five subclusters (DIM cluster 0–4). The object accounts for 1569 cells from epilepsy patients and 622 cells from controls.
(D) Differential composition analysis of each DIM subpopulation in epilepsy patients vs. controls using sccomp. Population expansion (right-shift) or depletion (left-shift) are represented by the credible interval of the slope (95% confidence). The dashed lines represent the default threshold for consideration of significance (−0.2–0.2). Statistical significance is considered for an adjusted p value (FDR) < 0.05 and is highlighted in red. Boxplot representation of the proportion distribution of the five DIM subpopulations in each individual sample from the epilepsy patients and controls groups. Cluster 0 is significantly expanded in epilepsy in comparison to controls. The blue boxes represent the posterior predictive check, which consists of a simulation from the fitted model. The overlap of the stimulated proportions with the real data validates the adequacy of the model. The red triangles represent predicted outliers.
(E) Dot plot representation of the expression of the module score of the genes from the DNA methylation and the gene expression modules, the overlapping genes between M8 and E5 (7 genes), E10 (7 genes) and E4 (10 genes), and a selected list of immune-related genes from E5 (P2RX4, LILRB4) and E10 (OTUD1, SIGLEC10).
(F) Boxplot representation of the normalized counts (RNA-seq) of four DEGs from the +ATP+LPS vs. +LPS comparison in the four studied microglia-like conditions (Naive, +LPS, +ATP and +ATP+LPS), and belonging to E5 (P2RX4 and LILRB4) and E10 (OTUD1 and SIGLEC10). All four genes are significantly upregulated in +ATP+LPS vs. +LPS (adjusted p value (FDR) < 0.05).
Discussion
In this study, we have shown that ATP-mediated preconditioning of microglia-like cells is accompanied by epigenetic and transcriptional reprogramming that is associated with their subsequent inflammatory responses. The occurrence of innate immune memory and the participation of epigenetic changes in this context provides new potential mechanisms to explain some of the roles of microglia in neuropathology. In this case, in the context of epileptogenesis. Secondary immune responses following a first insult can exhibit different outcomes depending on the nature of the stimuli. In the case of trained immunity, the response is more pronounced or exacerbated, whereas in the case of tolerance, the response is diminished.8 Evidence supporting the existence of memory in microglia remains limited. Nevertheless, studies have shown that sequential treatment of microglia with LPS can influence the production of cytokines and reactive oxygen species, phagocytic activity, and persistent alterations in gene expression profiles.43,44 Processes leading to the acquisition of innate immune memory are thought to be tightly regulated by epigenetic modifications. Two studies have conducted RNA-seq, ATAC-seq, and ChIP-seq of multiple histone marks on microglia, revealing epigenetically dependent transcriptomic changes associated with immune preconditioning.45,46 The participation of DNA methylation was not explored in these studies, despite its relevance in myeloid differentiation. DNA methylation is the most stable and one of the best studied epigenetic modifications. It consists of the addition of a methyl group to cytosine nucleotides in the DNA and contributes and/or reflects the transcriptional competence of chromosomal regions47,48
As mentioned previously, the use of human microglia models is necessary to study the molecular mechanisms involved in pathogenesis. A common concern regarding the use of microglia-like models is their distinct ontogeny compared to in vivo microglia, which differentiate from precursor cells that migrate into the brain in early embryogenesis and are independent from peripheral monocytes.49,50 A common in vitro approach to study human microglia is iPSC-derived microglia, which aims at emulating the microglial ontogeny.51 Using microglia-like cells derived from monocytes offers the advantage of a simpler and quicker experimental protocol in comparison to iPSC-derived microglia, which consists of a longer multi-step procedure.52 Furthermore, the use of the differentiation of microglia-like cells from monocytes has gained momentum due to the recent discovery of a novel myeloid population within the cerebral microglial population. Using single-cell transcriptomics, Silvin and colleagues (2022), described a subset of cells known as “disease-inflammatory macrophages” (DIMs). Although DIMs share features with microglia, what made them be annotated under the microglia spectrum, they are actually derived from infiltrating monocytes. These DIMs present a general pro-inflammatory profile with neurotoxic implications in neurodegeneration and aging.42
In vitro models of microglia-like cells have been shown to mimic in vivo microglia features, including morphology, specific surface protein expression, expression of specific canonical homeostatic markers, and overall transcriptomic profile.25,27,28,52,53,54 In this study, we have performed a multi-level characterization of microglia-like cells differentiated from monocytes demonstrating the acquisition of the elongated and ramified structure, and the acquisition of standard microglia surface markers. At the transcriptomic level, our microglia-like cells showed high resemblance with primary human microglia, outperforming those generated by previous protocols. Furthermore, we obtained the DNA methylation profiles of microglia-like cells, which also support its cellular identity, given their cell type specificity.55 In this analysis, we have demonstrated a successful replication of microglial DNA methylation patterns. The DNA methylation profiles of microglia-like cells cluster closer to in vivo primary microglia than to primary monocytes and are enriched for TF binding motifs associated not only with myeloid cells but also with microglia. In an encompassing study, Gosselin et al. performed transcriptomic and epigenomic characterization of mice and human microglia, including ATAC-seq (defines open chromatin), and ChIP-seq for H3K4me2 (dimethylation of histone H3 lysine 4, annotates regulatory regions like enhancers) and H3K27ac (acetylation of histone H3 lysine 27, annotates regions with high transcriptional activity). ATAC-seq peaks associated with H3K4me2 and H3K27ac in microglia were predominantly enriched for PU.1, denoted as the main driver of microglial identity, but also for IRF, MEF2, SMAD, and MAF factors.23 Our results indicate that these same factors, with prominence for PU.1, are associated with the DNA methylation changes in microglia-like differentiation.
Our model of microglia preconditioning aligns with the pathogenesis landscape observed in various neurodegenerative diseases, characterized by the presence of widespread primed/activated/trained microglia states reported in these conditions.8 In addition, we recognize the specific relevance of integrating purinergic signaling within this framework, particularly when considering its implications in the context of epileptogenesis. Mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE-HS) is particularly relevant considering the high incidence of reported initial precipitating injuries in early childhood, namely trauma, hypoxia, intracranial infection, and febrile seizures.56,57,58,59 It could be postulated that purine-driven microglia preconditioning caused by the initial insult may influence posterior microglia responses in ways that could influence the structural and molecular changes associated with the latent phase of epilepsy development. Furthermore, considering the progressive nature of MTLE, seizure activity may precondition microglia activation through the release of extracellular ATP and adenosine, with impact on the continuous aggravation of neuroinflammation and prognosis worsening. An important aspect that deserves attention in this discussion is the previously described adenosine kinase (ADK) hypothesis of epileptogenesis, which proposes that, in epileptogenic conditions, the levels of ADK, an enzyme responsible for converting endogenous adenosine into AMP, increase. This elevation of ADK results in the extracellular depletion of adenosine.60 This mechanism is directly associated with DNA methylation. The conversion of S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH) serves as a major source of methyl groups. Subsequently, SAH is hydrolyzed to produce homocysteine and adenosine. Hence, any disturbance in the intracellular and extracellular balance of adenosine levels can directly impact DNA methylation by altering the thermodynamics of the SAM metabolism. In the early stages, this process was proposed to be unidirectional leading to overall DNA hypermethylation due to increased adenosine consumption by ADK in the context of epileptogenesis.61,62 However, studies have demonstrated that in complex and heterogeneous pathologies such as epilepsy, DNA methylation alterations occur in a region-specific manner, with some methylation sites showing hypermethylation and others hypomethylation. In a recent study conducted by our group, we observed that DNA methylation of multiple sites associated with neuroinflammation correlate with disease duration, suggesting the implication of microglia in epilepsy progression.63
In the present study, we have observed that ATP-driven preconditioning is associated with the acquisition of a more pro-inflammatory phenotype. This shift in the phenotype is accompanied by bidirectional changes in both transcriptomic and DNA methylation profiles, with a tendency toward DNA hypomethylation in pathways associated with inflammation. Furthermore, we were able to replicate the changes caused by ATP preconditioning in vitro in scRNA-seq data from epilepsy patients and controls. These results provide support for the replicability of these mechanisms described in the context of epileptogenesis. Additionally, we have determined that genes upregulated in activated and preconditioned microglia-like cells, as well as the genes associated with immune training, are elevated in subpopulations of DIMs that are expanded in epilepsy. P2RX4 codes a purinergic receptor, expected to be directly modulated by ATP that has been shown to be increased in a reactive microglia phenotype with implications in neuropathic pain.64 In addition, LILRB4 and SIGLEC10 code two immune checkpoints,65,66 and OUTD1, a regulator of inflammatory responses,67 close the list of candidate genes that depict an ATP-preconditioned pro-inflammatory enhancement in microglia-like cells and a potential implication in epileptogenesis. In any case, we acknowledge the minimal relationship between DNA methylation and transcriptomic changes. This could be due to the short time interval between the two stimuli, which may not allow for the slower DNA methylation changes to occur.
Our in vitro model utilizing microglia-like cells has demonstrated that exposure to ATP, which mimics the conditions observed during brain insults such as epileptic seizures, leads to a concurrent increase in inflammatory activation and alterations in transcriptional patterns. Various studies have used similar models in patients to study pathological mechanisms related to microglia.25,68,69,70,71,72 By comparing our ATP-mediated preconditioning model with microglia subsets from scRNA-seq data derived from epilepsy patients and healthy donors, we provide evidence for the involvement of purinergic conditioning in the acquisition of disease associated microglial states. Activated preconditioned microglia-like cells resemble subpopulations of DIMs that are expanded in the brains of epilepsy patients, potentially contributing to the heightened neuroinflammatory profile associated with epileptogenesis. Further epigenetic and functional analysis of microglia cells from patients with MTLE-HS compared to controls will shed light on the distinct responsiveness of microglia in epilepsy.
Limitations of the study
We acknowledge the limitations of this model, particularly regarding the use of LPS to induce pro-inflammatory activation. This method may not fully replicate the in vivo epileptogenic process, as neuroinflammation does not imply the presence of infectious agents. Nevertheless, exposure to LPS is intended here to induce a general pro-inflammatory activation and is commonly used in the literature as a secondary stimulus to study innate immune memory in microglia.43,44 We must also recognize that the demonstration of the ATP-driven pro-inflammatory profile in microglia-like cells would benefit from functional experiments, like phagocytosis assays and quantification of the secretion of activated cytokines.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-human CD14 | Biolegend | Cat# 367116; RRID: AB_2571929 |
| PE anti-human CX3CR1 | Biolegend | Cat# 341604; RRID: AB_1595456 |
| PE-Cy5 anti-human CD11b | Biolegend | Cat# 301308; RRID: AB_314160 |
| PE-Cy7 anti-human HLA-DR | Biolegend | Cat# 307616; RRID: AB_493588 |
| APC anti-human CD192 (CCR2) | Biolegend | Cat# 357208; RRID: AB_2562239 |
| APC-Cy7 anti-human CD68 | Biolegend | Cat# 333822; RRID: AB_2571965 |
| PE anti-human P2RY12 | Biolegend | Cat# 392104; RRID: AB_2716007 |
| APC anti-human MERTK | Biolegend | Cat# 367612; RRID: AB_2687289 |
| KO anti-human CD45 | BeckmanCoulter Life Sciences | Cat# B36294 |
| Biological samples | ||
| Peripheral blood from healthy donors | Catalan Blood and Tissue Bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI Medium 1640+ GlutaMAXTM | Gibco, Thermo Fisher | Cat# 61870036 |
| α-minimal essential medium (α-MEM) | Invitrogen | Cat# 32561-029 |
| Penicillin – Streptomycin | Labclinics, S.A. | L0022-100 |
| Human M-CSF | Peprotech | Cat# 300-25 |
| Human GM-CSF | Peprotech | Cat# 300-03 |
| Human beta-NGF | Peprotech | Cat# 450-01 |
| Human MCP-1/MCAF (CCL2) | Peprotech | Cat# 300-04 |
| Human IL-34 | Peprotech | Cat# 200-34 |
| Human TGF-beta 2 (Mammalian) | Peprotech | Cat# 100-35B |
| Cholesterol | Merck | Cat# C3045-5G |
| ATP (adenosine 5′-triphosphate disodium salt) | InvivoGen | Cat# tlrl-atpl |
| LPS (lipopolysaccharide) | Merk | Cat# C3045-5G |
| FBS (Fetal Bovine Serum) | ThemoFisher Scientific | Cat# 10270106 |
| PBS | ThemoFisher Scientific | Cat# 20012019 |
| Paraformaldehyde | Aname, SL | Cat# 15710-S |
| Versene | ThemoFisher Scientific | Cat# 15040033 |
| Critical commercial assays | ||
| CD14+ MicroBeads | Miltenyi Biotec | Cat# 130-050-201 |
| Maxwell RSC simplyRNA Cells Kit | Promega | Cat# AS1390 |
| Transcriptor First Strand cDNA Synthesis Kit | Roche | Cat# 4897030001 |
| LightCycler® 480 SYBR Green Mix | Roche | Cat# 4887352001 |
| IL-1 beta Human ELISA Kit | Invitrogen | Cat# KHC0011 |
| LIVE/DEADTM Fixable Violet | ThemoFisher Scientific | Cat# L34964 |
| Deposited data | ||
| DNA methylation and gene expression | This paper | GSE235104 |
| Oligonucleotides | ||
| qRT-PCR primers, see STAR method details | This paper | N/A |
| Software and algorithms | ||
| Primer3 | Koressaar et al.73 | https://primer3.ut.ee/ |
| shinyÉpico | Morante-Palacios et al.74 | https://github.com/omorante/shinyepico |
| Minfi | Aryee et al.75 | https://bioconductor.org/packages/release/bioc/html/minfi.html |
| Limma | Ritchie et al.76 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Sva | Leek et al.77 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| HISAT2 | Kim et al.78 | https://daehwankimlab.github.io/hisat2/ |
| featureCounts | Liao et al.79 | https://subread.sourceforge.net/featureCounts.html |
| DESeq2 | Love et al.80 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Seurat | Hao et al. (2021 and 2023)81,82 | https://satijalab.org/seurat/ |
| DoubletFinder | McGinnis et al.83 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Sccomp | Mangiola et al.84 | https://github.com/MangiolaLaboratory/sccomp |
| Gplots | Warnes et al.85 | https://cran.r-project.org/web/packages/gplots/index.html |
| ComplexHeatmap | Gu et al.86 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Gviz | Hahne et al.87 | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| VennDiagram | Chen et al.88 | https://cran.r-project.org/web/packages/VennDiagram/index.html |
| HOMER | Heinz et al.89 | http://homer.ucsd.edu/homer/motif/ |
| GREAT | McLean et al.90 | http://great.stanford.edu/public/html |
| DoRothEA | Garcia-Alonso et al.91 | https://saezlab.github.io/dorothea/articles/dorothea.html |
| clusterProfiler | Wu et al.92 | https://guangchuangyu.github.io/software/clusterProfiler/ |
| GeneOverlap | Shen et al.93 | https://github.com/shenlab-sinai/GeneOverlap |
| Fgsea | Korotkevich et al.94 | https://www.bioconductor.org/packages/release/bioc/html/fgsea.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Esteban Ballestar (eballestar@carrerasresearch.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All DNA methylation and expression datasets for this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through a GEO accession numbers GSE235104 and GSE272030 respectively.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Monocyte purification and in vitro differentiation to microglia-like cells
Peripheral blood was collected under the form of buffy coats obtained from anonymous adult male donors through the Catalan Blood and Tissue Bank. All donors signed an informed consent and sample collections were performed as per the guidelines of the World Medical Association (WMA) Declaration of Helsinki.
Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque gradient centrifugation. Monocytes were then purified using positive selection with CD14+ MicroBeads (Miltenyi Biotec). For microglia-like differentiation, monocytes were cultured in RPMI Medium 1640+ GlutaMAX (Gibco, Thermo Fisher) containing 100 units/mL penicillin, 100 μg/mL streptomycin, and supplemented with 10 ng/mL human M-CSF, 10 ng/mL human GM-CSF, 10 ng/mL human β-NGF, 100 ng/mL human IL-34, 100 ng/mL human CCL2, 2 ng/mL human TGF-β2 (PeproTech) and 1.5 μg/mL cholesterol solution dissolved in ethanol. Microglia-like plating density was of 1.5 M cells per well (6-well plates). Homeostatic (Naive) microglia-like cells were obtained after seven days of culture without media changes. For monocyte-derived macrophage differentiation, monocytes were attached to plates by incubation with serum-free medium and posteriorly cultured in α-minimal essential medium (α-MEM; Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin and supplemented with 25 ng/mL human M-CSF. Plating density for monocyte-derived macrophages was 3 M cells per well (6-well plates). No media changes were performed for the macrophage culture, and cells were activated with 10 ng/mL of LPS at day six of culture and collected 24h after.
Activation and preconditioning of microglia-like cells
The design of the preconditioning analysis consisted in the development of the following four microglia-like activation states: microglia-like without any stimuli and collected at day seven (Naive); microglia-like stimulated with 100 μM ATP at day six and collected 24h after (+ATP); microglia-like stimulated with 100 ng/mL LPS at day eight and collected 48h after (+LPS); Microglia-like stimulated with 100 μM ATP at day six and with 100 ng/mL LPS at day eight, and collected at day ten of culture (+ATP+LPS).
Method details
Real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR)
Total RNA was isolated using the Maxwell RSC simplyRNA Cells Kit (Promega) and reverse-transcribed to cDNA with Transcriptor First Strand cDNA Synthesis Kit (Roche) per the manufacturer’s instructions. qRT-PCR primers were designed with Primer3 software73 (P2RY12: forward – CCACTCTGCAGGTTGCAATA; reverse – GGCTTGCATTTCTTGTTGGT; CX3CR1: forward – CACAAAGGAGCAGGCATGGAAG; reverse – CAGGTTCTCTGTAGACACAAGGC; RPL38 (housekeeping): forward – TGGGTGAGAAAGGTCCTGGTC; reverse - CGTCGGGCTGTGAGCAGGAA). Technical triplicates were run for each sample using LightCycler 480 SYBR Green Mix (Roche) and analyzed with a LightCycler instrument (Roche).
RNA-seq
RNA-seq libraries of monocytes and microglia-like cells were generated and sequenced by Novogene (Cambridge), in 150-bp paired-end, with the Illumina NovaSeq 6000 platform, using four biological replicates for each group. An average of more than 65 million reads were obtained for all samples.
DNA methylation profiling
Genomic DNA was isolated from cell lysates in Proteinase K using an in-house salting out protocol. We then performed bisulfite conversion with the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA) and DNA methylation profiling using Infinium MethylationEPIC BeadChips. These arrays cover 850,000 single-nucleotide positions, accounting for 99% of the annotated RefSeq genes.
Quantification of supernatant cytokines
Supernatants from cell cultures were collected and stored at - 80°C. Before use, frozen samples were thawed at room temperature. Enzyme-linked immunosorbent assays (ELISA) were performed to detect IL-1β (Invitrogen, Thermo Fisher Scientific).
Flow cytometry
Levels of cell surface protein markers were evaluated by flow cytometry using a BD LSR Fortessa cytometer. Cells were washed once with PBS, after which Versene, a non-enzymatic dissociation buffer (ThermoFisher), was added for detachment. After the addition of RPMI Medium 1640 + GlutaMAXTM (Gibco, Thermo Fisher) containing 100 units/mL penicillin, 100 μg/mL streptomycin and 10% FBS to the wells, cells were gently scrapped and collected. After collection, cells were resuspended in the staining buffer (PBS with 4% FBS and 2 mM EDTA). Cells were incubated at 4°C with the viability dye LIVE/DEAD Fixable Violet (ThermoFisher), following the manufacturer’s protocol. Cell staining followed a protocol developed in-house consisting of two separate panels of antibodies [Panel 1: CD14 (FITC) (#367116 BioLegend), CX3CR1 (PE) (#341604 BioLegend), CD11b (PE-Cy5) (#301308 BioLegend), HLA-DR (PE-Cy7) (#307616 BioLegend), CD192 or CCR2 (APC) (#357208 BioLegend), CD68 (APC-Cy7) (#333822 BioLegend), CD45 (KO) (#B36294 BeckmanCoulter Life Sciences); Panel 2: CD14 (FITC), P2RY12 (PE) (#392104 BioLegend), CD11b (PE-Cy5), HLA-DR (PE-Cy7), MERTK (APC) (#367612 BioLegend), CD45 (KO)]. After staining, cells were fixed in PBS +4% paraformaldehyde and analyzed within the following 24 h. A compensation matrix was developed from single-positive experiments with beads. Median fluorescence intensity (MFI) values were calculated by subtracting the value from the unstained control to the stained sample.
Quantification and statistical analysis
Processing and analysis of DNA methylation data
Methylation data were preprocessed and analyzed using R language with the shinyÉPICo web interface74 base on the minfi75 and limma76 packages. Beta values, comprised between 0 and 1 (0 and 100% ratio of methylated probe intensity/sum of methylated and unmethylated probe intensities), were used for visualization purposes. M values consist of the log2 ratio of the intensities of the methylated and unmethylated probes and were used for statistical purposes, since beta values are heteroscedastic for highly methylated and unmethylated sites.95 Normalization was performed by the Noob and Quantile methods from minfi. Probes with a detection significance of p < 0.01 were removed. CpHs positions were kept in the dataset. Since all subjects were male, methylation sites from X and Y chromosomes were kept, whereas all positions located at single nucleotide polymorphisms (SNP) loci were removed (minimum allele frequency (MAFs) = 0). Differentially methylated positions (DMPs) were calculated using an eBayes-moderated t test from limma. Donor was used as covariate. The same preprocessing and DMP calculation pipeline was applied for the analysis of the DNA methylation data on monocyte-derived macrophages vs. monocytes obtained from GSE131177.31 Public DNA methylation data of primary microglia (n = 56) isolated from postmortem brain samples (GSE191200) was integrated in the in-house generated data, pre-processed, and normalized all together and batch corrected with the ComBat function from the sva package.77
Processing and analysis of RNA-seq data
Fastq files were align to the hg19 transcriptome using HISAT278 with default settings, and read counts by gene were assigned with featureCounts.79 All the posterior analysis was performed using R language. Differentially expressed genes (DEGs) were calculated using DESeq2.80 Only genes with more than ten raw counts in at least three samples were included. Donor was used as a covariate in the model. Significant DEGs were considered for adjusted p value (FDR) < 0.05. Variance Stabilizing Transformation (VST) and normalized count values were used for representation.
For the integration with public RNA-seq, raw count values were merged and adjusted for dataset using the ComBat_seq function of the sva package.77 Raw count data for primary adult and fetal human microglia, C20 and HMC3 cell lines, CD14 and CD16 monocytes, dendritic cells, monocyte-derived macrophages, monocyte-derived microglia-like cells, and iPSC-derived microglia was collected from Rai et al. (2020).28 This study included data from other public datasets, namely GSE89189 and GSE117829.
Pre-processing, integration, and analysis of single-nucleus (sn) and single-cell (sc)RNA-seq data
We used public snRNA-seq37,38,39,40 and single-cell (sc)RNA-seq41 data from brain tissue samples of surgically treated epilepsy patients and autopsied individuals. The data were obtained in the count matrix format, at the raw or preprocessed stages. Demultiplexing had already been done and the sequences had been aligned to the human reference genome (GRCh38), accounting for intronic and exonic regions. The individual samples in each dataset were concatenated into a single count matrix, and the subsequent processing was performed using the Seurat (v4.0.2 and v5) R package.81,82 Genes were only considered if detected in at least three nuclei, and nuclei were excluded if presented unique genes inferior to 200 or superior to 5,000, total UMI counts less than 500 and over 20,000, mitochondrial RNA content superior to 20% and ribosomal RNA content superior to 5%. The latter was only applied to snRNA-seq datasets. In addition, a list of 105 genes shown to be influenced by postmortem interval in cerebral cortex were filtered out.96 Potential doublets were estimated using the doubletFinder_v3 function83 for each individual subject and removed. The individual Seurat objects for each dataset were normalized using SCTransform normalization97 with default 3000 variable genes. Dimensionality reduction was performed with Principal Component Analysis (PCA) and Uniform Manifold Approximation and Projection (UMAP) accounting for the 30 main principal components (PCs). The annotation for the main cell types (neurons, oligodendrocytes, OPCs, astrocytes, endothelial cells, and immune cells) in the CNS was performed for each dataset using the FindNeighbors (30 PCs) and FindClusters (res = 0.05) functions, after which known canonical gene markers were identified within the lists of cluster markers obtained using FindAllMarkers (min.pct = 0.1 and logfc.threshold = 0.25, test.use = Wilcox). The immune cell clusters were annotated based on high expression of CD74, DOCK8, APBB1IP, HLA-DRA, PTPRC, P2RY12, C1QB, CX3CR1, C3, CSF1R and AIF1. Individual subjects with unproportionally low nuclei number in relation to the other subjects in each dataset were removed before integration. The immune cell clusters from all datasets were integrated using the Seurat v5 pipeline82 adapted for SCTransform normalization. Of note, no major bias was observed between snRNA-seq and scRNA-seq samples. The main 20 PCs and the 3000 most variable features across all datasets were considered for the FindIntegrationAnchors and IntegrateData functions. Dimensionality reduction was performed using PCA and UMAP (50 PCs). Clustering was performed using FindNeighbors (50 PCs) and FindClusters (res = 0.5). Clusters corresponding to macrophages (based on MRC1 expression), T cells (based on CD247 expression) and potential doublets (based on SLC1A2, PLP1 and VCAN expression) were removed, and the final integrated object with 36,927 microglia nuclei and cells was reclustered using the aforementioned settings, resulting in twelve populations. For the visualization of gene expression, we used the normalized counts of the “RNA” assay. The average expression of the lists of genes was obtained with AddModuleScore. Reclustering of the DIM subset was once again done with the abovementioned settings. To infer on the significance of the differential distribution of DIM subpopulations in epilepsy patients vs. controls we used the sccomp package.84 The sccomp_glm function calculates the differential composition using a Bayesian method based on sum-constrained independent Beta-binomial distributions. The results are presented by of a credible interval of the slope (95% confidence) and FDR. A credible interval of the slope (95% confidence) higher or lower than zero represents expansion or depletion, respectively, of a cluster for a determined category. A significant differential composition is considered for FDR <0.05. The “contrasts” option was used to calculate the differential composition of epilepsy patients in relation to the control group.
Statistics, data analysis and representation
The R 4.2.0. software was used for statistical analyses. Group means were compared using the paired t-test for numeric variables. Fisher’s exact test was used to calculate the significance of non-random association between two categorical variables. Heatmaps were developed with heatmap.2 function of the gplots package,85 for Z score estimation, followed by the Heatmap function of the ComplexHeatmap package.86 The representation of DNA methylation in relation to genomic coordinates was obtained using the Gviz package.87 Overlaps were obtained using either the VennDiagram package.88 Transcription factor (TF) motif enrichment for DMPs was obtained using the findMotifsGenome.pl function of HOMER (Hypergeometric Optimization of Motif EnRichment),89 considering a window of ±250 bp. Gene ontology (GO) enrichment for DMPs and determination of CpG-gene pairs were performed using the GREAT online tool (http://great.stanford.edu/public/html),90 with default settings. All EPIC array coordinates were used as background for motif enrichment and GO analyses of methylation data. Functional enrichment of differential expression data from the RNA-seq was performed using the Discriminant Regulon Expression Analysis (DoRothEA)91 for TF activity, and the enrichGO function from the clusterProfiler package,92 for GO. DNA methylation and RNA-seq results were integrated by evaluating the enrichment of the overlap between DEGs and DMP-related genes using a Fisher’s exact test within GeneOverlap93 and Gene Set Enrichment Analysis (GSEA) using fgsea.94 The association of the lists of DMPs with chromatin states was evaluated by enrichment analysis using ChromHMM categories of monocytes (Roadmap Epigenomics Project). Fisher’s exact tests were calculated using all the positions annotated in the EPIC array as background.
Acknowledgments
We thank the CERCA Program/Generalitat de Catalunya, the Josep Carreras Foundation and ICBAS-UP for institutional support. We acknowledge all members of the Epigenetics and Immune Disease Group at the Josep Carreras Leukemia Research Institute, the Immunogenetics Laboratory of the Molecular Pathology and Immunology Department of the ICBAS-UP, and the Neurophysiology and Neurology Departments of Centro Hospitalar Universitário de Santo António (CHUdSA). E.B. is funded by the Spanish Ministry of Science and Innovation (MICINN) [PID2020117212RB-I00; AEI/10.13039/501100011033]. R.M-F. is funded by an FCT (Fundação para a Ciência e Tecnologia) fellowship (SFRH/BD/137900/2018). UMIB is funded by FCT Portugal (UIDB/00215/2020 and UIDP/00215/2020), and ITR (LA/P/006/2020).
Author contributions
E.B., R.M-F., and B.L. conceived the study. E.B. and R.M-F. designed the experiments. R.M-F., J.C-S., and L.C. performed the experiments. R.M-F. and J.C-S. performed bioinformatic analysis. R.M-F. and E.B. wrote the manuscript. All authors revised and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the author(s) used ChatGPT in order to check grammar. After using this tool/service, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the content of the publication.
Published: July 20, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110546.
Supplemental information
References
- 1.Kettenmann H., Kirchhoff F., Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77:10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 2.Mittelbronn M., Dietz K., Schluesener H.J., Meyermann R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001;101:249–255. doi: 10.1007/s004010000284. [DOI] [PubMed] [Google Scholar]
- 3.Sominsky L., De Luca S., Spencer S.J. Microglia: Key players in neurodevelopment and neuronal plasticity. Int. J. Biochem. Cell Biol. 2018;94:56–60. doi: 10.1016/j.biocel.2017.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Erblich B., Zhu L., Etgen A.M., Dobrenis K., Pollard J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. 2011;6 doi: 10.1371/journal.pone.0026317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhan Y., Paolicelli R.C., Sforazzini F., Weinhard L., Bolasco G., Pagani F., Vyssotski A.L., Bifone A., Gozzi A., Ragozzino D., Gross C.T. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 2014;17:400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- 6.Bianchin M.M., Martin K.C., de Souza A.C., de Oliveira M.A., Rieder C.R. de M. Nasu-Hakola disease and primary microglial dysfunction. Nat. Rev. Neurol. 2010;6:2. doi: 10.1038/nrneurol.2010.17-c1. [DOI] [PubMed] [Google Scholar]
- 7.Sims R., van der Lee S.J., Naj A.C., Bellenguez C., Badarinarayan N., Jakobsdottir J., Kunkle B.W., Boland A., Raybould R., Bis J.C., et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017;49:1373–1384. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neher J.J., Cunningham C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019;40:358–374. doi: 10.1016/j.it.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Ramaglia V., Hughes T.R., Donev R.M., Ruseva M.M., Wu X., Huitinga I., Baas F., Neal J.W., Morgan B.P. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2012;109:965–970. doi: 10.1073/pnas.1111924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pott Godoy M.C., Tarelli R., Ferrari C.C., Sarchi M.I., Pitossi F.J. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain. 2008;131:1880–1894. doi: 10.1093/brain/awn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin Z., Raj D., Saiepour N., Van Dam D., Brouwer N., Holtman I.R., Eggen B.J.L., Möller T., Tamm J.A., Abdourahman A., et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging. 2017;55:115–122. doi: 10.1016/j.neurobiolaging.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 12.Martins-Ferreira R., Leal B., Costa P.P., Ballestar E. Microglial Innate Memory and Epigenetic Reprogramming in Neurological Disorders. Prog. Neurobiol. 2021;200 doi: 10.1016/j.pneurobio.2020.101971. [DOI] [PubMed] [Google Scholar]
- 13.Beamer E., Kuchukulla M., Boison D., Engel T. ATP and adenosine-Two players in the control of seizures and epilepsy development. Prog. Neurobiol. 2021;204 doi: 10.1016/j.pneurobio.2021.102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amorim B.O., Hamani C., Ferreira E., Miranda M.F., Fernandes M.J.S., Rodrigues A.M., de Almeida A.-C.G., Covolan L. Effects of A1 receptor agonist/antagonist on spontaneous seizures in pilocarpine-induced epileptic rats. Epilepsy Behav. 2016;61:168–173. doi: 10.1016/j.yebeh.2016.05.036. [DOI] [PubMed] [Google Scholar]
- 15.Stockwell J., Jakova E., Cayabyab F.S. Adenosine A1 and A2A Receptors in the Brain: Current Research and Their Role in Neurodegeneration. Molecules. 2017;22 doi: 10.3390/molecules22040676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuda M., Suzuki Y., Hino H., Kuzume K., Morimoto T., Ishii E. Adenosine A1 receptor blockage mediates theophylline-associated seizures. Epilepsia. 2010;51:483–487. doi: 10.1111/j.1528-1167.2009.02382.x. [DOI] [PubMed] [Google Scholar]
- 17.Del Puerto A., Wandosell F., Garrido J.J. Neuronal and glial purinergic receptors functions in neuron development and brain disease. Front. Cell. Neurosci. 2013;7:197. doi: 10.3389/fncel.2013.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.During M.J., Spencer D.D. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann. Neurol. 1992;32:618–624. doi: 10.1002/ana.410320504. [DOI] [PubMed] [Google Scholar]
- 19.Dossi E., Blauwblomme T., Moulard J., Chever O., Vasile F., Guinard E., Le Bert M., Couillin I., Pallud J., Capelle L., et al. Pannexin-1 channels contribute to seizure generation in human epileptic brain tissue and in a mouse model of epilepsy. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aar3796. [DOI] [PubMed] [Google Scholar]
- 20.Lietsche J., Imran I., Klein J. Extracellular levels of ATP and acetylcholine during lithium-pilocarpine induced status epilepticus in rats. Neurosci. Lett. 2016;611:69–73. doi: 10.1016/j.neulet.2015.11.028. [DOI] [PubMed] [Google Scholar]
- 21.Dale N., Frenguelli B.G. Release of adenosine and ATP during ischemia and epilepsy. Curr. Neuropharmacol. 2009;7:160–179. doi: 10.2174/157015909789152146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olah M., Raj D., Brouwer N., De Haas A.H., Eggen B.J.L., Den Dunnen W.F.A., Biber K.P.H., Boddeke H.W.G.M. An optimized protocol for the acute isolation of human microglia from autopsy brain samples. Glia. 2012;60:96–111. doi: 10.1002/glia.21251. [DOI] [PubMed] [Google Scholar]
- 23.Gosselin D., Skola D., Coufal N.G., Holtman I.R., Schlachetzki J.C.M., Sajti E., Jaeger B.N., O’Connor C., Fitzpatrick C., Pasillas M.P., et al. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017;356 doi: 10.1126/science.aal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizee M.R., Miedema S.S.M., van der Poel M., Heutinck K.M., Adelia, Schuurman K.G., Schuurman K.G., van Strien M.E., Melief J., Smolders J., Hendrickx D.A., et al. Isolation of primary microglia from the human post-mortem brain: effects of ante- and post-mortem variables. Acta Neuropathol. Commun. 2017;5:16. doi: 10.1186/s40478-017-0418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryan K.J., White C.C., Patel K., Xu J., Olah M., Replogle J.M., Frangieh M., Cimpean M., Winn P., McHenry A., et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aai7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butovsky O., Jedrychowski M.P., Moore C.S., Cialic R., Lanser A.J., Gabriely G., Koeglsperger T., Dake B., Wu P.M., Doykan C.E., et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Etemad S., Zamin R.M., Ruitenberg M.J., Filgueira L. A novel in vitro human microglia model: characterization of human monocyte-derived microglia. J. Neurosci. Methods. 2012;209:79–89. doi: 10.1016/j.jneumeth.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 28.Rai M.A., Hammonds J., Pujato M., Mayhew C., Roskin K., Spearman P. Comparative analysis of human microglial models for studies of HIV replication and pathogenesis. Retrovirology. 2020;17:35. doi: 10.1186/s12977-020-00544-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeh H., Ikezu T. Transcriptional and Epigenetic Regulation of Microglia in Health and Disease. Trends Mol. Med. 2019;25:96–111. doi: 10.1016/j.molmed.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elmore M.R.P., Najafi A.R., Koike M.A., Dagher N.N., Spangenberg E.E., Rice R.A., Kitazawa M., Matusow B., Nguyen H., West B.L., Green K.N. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li T., Garcia-Gomez A., Morante-Palacios O., Ciudad L., Özkaramehmet S., Van Dijck E., Rodríguez-Ubreva J., Vaquero A., Ballestar E. SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Res. 2020;48:665–681. doi: 10.1093/nar/gkz1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holtman I.R., Skola D., Glass C.K. Transcriptional control of microglia phenotypes in health and disease. J. Clin. Invest. 2017;127:3220–3229. doi: 10.1172/JCI90604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen Z.-G., Liu X.-Z., Chen C.-X., Lu J.-M. Knockdown of E2F3 Inhibits Proliferation, Migration, and Invasion and Increases Apoptosis in Glioma Cells. Oncol. Res. 2017;25:1555–1566. doi: 10.3727/096504017X14897158009178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pugliatti L., Derré J., Berger R., Ucla C., Reith W., Mach B. The genes for MHC class II regulatory factors RFX1 and RFX2 are located on the short arm of chromosome 19. Genomics. 1992;13:1307–1310. doi: 10.1016/0888-7543(92)90052-t. [DOI] [PubMed] [Google Scholar]
- 35.Feng C.-Z., Yin J.-B., Yang J.-J., Cao L. Regulatory factor X1 depresses ApoE-dependent Aβ uptake by miRNA-124 in microglial response to oxidative stress. Neuroscience. 2017;344:217–228. doi: 10.1016/j.neuroscience.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 36.Reith W., Ucla C., Barras E., Gaud A., Durand B., Herrero-Sanchez C., Kobr M., Mach B. RFX1, a transactivator of hepatitis B virus enhancer I, belongs to a novel family of homodimeric and heterodimeric DNA-binding proteins. Mol. Cell Biol. 1994;14:1230–1244. doi: 10.1128/mcb.14.2.1230-1244.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran M.N., Maynard K.R., Spangler A., Huuki L.A., Montgomery K.D., Sadashivaiah V., Tippani M., Barry B.K., Hancock D.B., Hicks S.C., et al. Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron. 2021;109:3088–3103.e5. doi: 10.1016/j.neuron.2021.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pappalardo J.L., Zhang L., Pecsok M.K., Perlman K., Zografou C., Raddassi K., Abulaban A., Krishnaswamy S., Antel J., van Dijk D., Hafler D.A. Transcriptomic and clonal characterization of T cells in the human central nervous system. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.abb8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franjic D., Skarica M., Ma S., Arellano J.I., Tebbenkamp A.T.N., Choi J., Xu C., Li Q., Morozov Y.M., Andrijevic D., et al. Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron. 2022;110:452–469.e14. doi: 10.1016/j.neuron.2021.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thrupp N., Sala Frigerio C., Wolfs L., Skene N.G., Fattorelli N., Poovathingal S., Fourne Y., Matthews P.M., Theys T., Mancuso R., et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.108189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mancuso R., Van Den Daele J., Fattorelli N., Wolfs L., Balusu S., Burton O., Liston A., Sierksma A., Fourne Y., Poovathingal S., et al. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 2019;22:2111–2116. doi: 10.1038/s41593-019-0525-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silvin A., Uderhardt S., Piot C., Da Mesquita S., Yang K., Geirsdottir L., Mulder K., Eyal D., Liu Z., Bridlance C., et al. Dual ontogeny of disease-associated microglia and disease inflammatory macrophages in aging and neurodegeneration. Immunity. 2022;55:1448–1465.e6. doi: 10.1016/j.immuni.2022.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Lajqi T., Lang G.-P., Haas F., Williams D.L., Hudalla H., Bauer M., Groth M., Wetzker R., Bauer R. Memory-Like Inflammatory Responses of Microglia to Rising Doses of LPS: Key Role of PI3Kγ. Front. Immunol. 2019;10:2492. doi: 10.3389/fimmu.2019.02492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaafsma W., Zhang X., van Zomeren K.C., Jacobs S., Georgieva P.B., Wolf S.A., Kettenmann H., Janova H., Saiepour N., Hanisch U.-K., et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav. Immun. 2015;48:205–221. doi: 10.1016/j.bbi.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 45.Wendeln A.-C., Degenhardt K., Kaurani L., Gertig M., Ulas T., Jain G., Wagner J., Häsler L.M., Wild K., Skodras A., et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–338. doi: 10.1038/s41586-018-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X., Kracht L., Lerario A.M., Dubbelaar M.L., Brouwer N., Wesseling E.M., Boddeke E.W.G.M., Eggen B.J.L., Kooistra S.M. Epigenetic regulation of innate immune memory in microglia. J. Neuroinflammation. 2022;19:111. doi: 10.1186/s12974-022-02463-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldberg A.D., Allis C.D., Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 49.Ginhoux F., Greter M., Leboeuf M., Nandi S., See P., Gokhan S., Mehler M.F., Conway S.J., Ng L.G., Stanley E.R., et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ginhoux F., Lim S., Hoeffel G., Low D., Huber T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasselmann J., Blurton-Jones M. Human iPSC-derived microglia: A growing toolset to study the brain’s innate immune cells. Glia. 2020;68:721–739. doi: 10.1002/glia.23781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sargeant T.J., Fourrier C. Human monocyte-derived microglia-like cell models: A review of the benefits, limitations and recommendations. Brain Behav. Immun. 2023;107:98–109. doi: 10.1016/j.bbi.2022.09.015. [DOI] [PubMed] [Google Scholar]
- 53.Leone C., Le Pavec G., Même W., Porcheray F., Samah B., Dormont D., Gras G. Characterization of human monocyte-derived microglia-like cells. Glia. 2006;54:183–192. doi: 10.1002/glia.20372. [DOI] [PubMed] [Google Scholar]
- 54.Banerjee A., Lu Y., Do K., Mize T., Wu X., Chen X., Chen J. Validation of Induced Microglia-Like Cells (iMG Cells) for Future Studies of Brain Diseases. Front. Cell. Neurosci. 2021;15 doi: 10.3389/fncel.2021.629279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dor Y., Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018;392:777–786. doi: 10.1016/S0140-6736(18)31268-6. [DOI] [PubMed] [Google Scholar]
- 56.Wieser H.-G. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia. 2004;45:695–714. doi: 10.1111/j.0013-9580.2004.09004.x. [DOI] [PubMed] [Google Scholar]
- 57.Pittau F., Bisulli F., Mai R., Fares J.E., Vignatelli L., Labate A., Naldi I., Avoni P., Parmeggiani A., Santucci M., et al. Prognostic factors in patients with mesial temporal lobe epilepsy. Epilepsia. 2009;50:41–44. doi: 10.1111/j.1528-1167.2008.01969.x. [DOI] [PubMed] [Google Scholar]
- 58.Abou-Khalil B., Andermann E., Andermann F., Olivier A., Quesney L.F. Temporal lobe epilepsy after prolonged febrile convulsions: excellent outcome after surgical treatment. Epilepsia. 1993;34:878–883. doi: 10.1111/j.1528-1157.1993.tb02105.x. [DOI] [PubMed] [Google Scholar]
- 59.O’Dell C.M., Das A., Wallace G., 4th, Ray S.K., Banik N.L. Understanding the basic mechanisms underlying seizures in mesial temporal lobe epilepsy and possible therapeutic targets: a review. J. Neurosci. Res. 2012;90:913–924. doi: 10.1002/jnr.22829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boison D. The adenosine kinase hypothesis of epileptogenesis. Prog. Neurobiol. 2008;84:249–262. doi: 10.1016/j.pneurobio.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobow K., Blümcke I. The emerging role of DNA methylation in epileptogenesis. Epilepsia. 2012;53:11–20. doi: 10.1111/epi.12031. [DOI] [PubMed] [Google Scholar]
- 62.Murugan M., Fedele D., Millner D., Alharfoush E., Vegunta G., Boison D. Adenosine kinase: An epigenetic modulator in development and disease. Neurochem. Int. 2021;147 doi: 10.1016/j.neuint.2021.105054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martins-Ferreira R., Leal B., Chaves J., Li T., Ciudad L., Rangel R., Santos A., Martins da Silva A., Pinho Costa P., Ballestar E. Epilepsy progression is associated with cumulative DNA methylation changes in inflammatory genes. Prog. Neurobiol. 2022;209 doi: 10.1016/j.pneurobio.2021.102207. [DOI] [PubMed] [Google Scholar]
- 64.Masuda T., Iwamoto S., Yoshinaga R., Tozaki-Saitoh H., Nishiyama A., Mak T.W., Tamura T., Tsuda M., Inoue K. Transcription factor IRF5 drives P2X4R+-reactive microglia gating neuropathic pain. Nat. Commun. 2014;5:3771. doi: 10.1038/ncomms4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Su M.-T., Inui M., Wong Y.L., Takahashi M., Sugahara-Tobinai A., Ono K., Miyamoto S., Murakami K., Itoh-Nakadai A., Kezuka D., et al. Blockade of checkpoint ILT3/LILRB4/gp49B binding to fibronectin ameliorates autoimmune disease in BXSB/Yaa mice. Int. Immunol. 2021;33:447–458. doi: 10.1093/intimm/dxab028. [DOI] [PubMed] [Google Scholar]
- 66.Zhang C., Zhang J., Liang F., Guo H., Gao S., Yang F., Guo H., Wang G., Wang W., Zhou G. Innate immune checkpoint Siglec10 in cancers: mining of comprehensive omics data and validation in patient samples. Front. Med. 2022;16:596–609. doi: 10.1007/s11684-021-0868-z. [DOI] [PubMed] [Google Scholar]
- 67.Oikawa D., Gi M., Kosako H., Shimizu K., Takahashi H., Shiota M., Hosomi S., Komakura K., Wanibuchi H., Tsuruta D., et al. OTUD1 deubiquitinase regulates NF-κB- and KEAP1-mediated inflammatory responses and reactive oxygen species-associated cell death pathways. Cell Death Dis. 2022;13:694. doi: 10.1038/s41419-022-05145-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ohgidani M., Kato T.A., Setoyama D., Sagata N., Hashimoto R., Shigenobu K., Yoshida T., Hayakawa K., Shimokawa N., Miura D., et al. Direct induction of ramified microglia-like cells from human monocytes: dynamic microglial dysfunction in Nasu-Hakola disease. Sci. Rep. 2014;4:4957. doi: 10.1038/srep04957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sellgren C.M., Sheridan S.D., Gracias J., Xuan D., Fu T., Perlis R.H. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol. Psychiatry. 2017;22:170–177. doi: 10.1038/mp.2016.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ohgidani M., Kato T.A., Hosoi M., Tsuda M., Hayakawa K., Hayaki C., Iwaki R., Sagata N., Hashimoto R., Inoue K., et al. Fibromyalgia and microglial TNF-α: Translational research using human blood induced microglia-like cells. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-11506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sellgren C.M., Gracias J., Watmuff B., Biag J.D., Thanos J.M., Whittredge P.B., Fu T., Worringer K., Brown H.E., Wang J., et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019;22:374–385. doi: 10.1038/s41593-018-0334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ormel P.R., Böttcher C., Gigase F.A.J., Missall R.D., van Zuiden W., Fernández Zapata M.C., Ilhan D., de Goeij M., Udine E., Sommer I.E.C., et al. A characterization of the molecular phenotype and inflammatory response of schizophrenia patient-derived microglia-like cells. Brain Behav. Immun. 2020;90:196–207. doi: 10.1016/j.bbi.2020.08.012. [DOI] [PubMed] [Google Scholar]
- 73.Koressaar T., Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 74.Morante-Palacios O., Ballestar E. shinyÉPICo: A graphical pipeline to analyze Illumina DNA methylation arrays. Bioinformatics. 2021;37:257–259. doi: 10.1093/bioinformatics/btaa1095. [DOI] [PubMed] [Google Scholar]
- 75.Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leek, J.T., Johnson, W.E., Parker, H.S., Fertig, E.J., Jaffe, A.E., Zhang, Y., Storey, J.D., and Torres, L.C. (2022). sva: Surrogate Variable Analysis. R package version 3.44.0.
- 78.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 80.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hao Y., Stuart T., Kowalski M.H., Choudhary S., Hoffman P., Hartman A., Srivastava A., Molla G., Madad S., Fernandez-Granda C., Satija R. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2024;42:293–304. doi: 10.1038/s41587-023-01767-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mangiola S., Roth-Schulze A.J., Trussart M., Zozaya-Valdés E., Ma M., Gao Z., Rubin A.F., Speed T.P., Shim H., Papenfuss A.T. sccomp: Robust differential composition and variability analysis for single-cell data. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2203828120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Warnes G.R., Bolker B., Bonebakker L., Gentleman R., Huber W., Liaw A., Lumley T., Maechler M., Magnusson A., Moeller S., et al. gplots: Various R Programming Tools for Plotting Data. R package version 3.1.3. 2022. https://cran.r-project.org/package=gplots
- 86.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 87.Hahne F., Ivanek R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016;1418:335–351. doi: 10.1007/978-1-4939-3578-9_16. [DOI] [PubMed] [Google Scholar]
- 88.Chen H. _VennDiagram: Generate High-Resolution Venn and Euler Plots_. R package version 1.7.3. 2022. https://cran.r-project.org/package=VennDiagram
- 89.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garcia-Alonso L., Iorio F., Matchan A., Fonseca N., Jaaks P., Peat G., Pignatelli M., Falcone F., Benes C.H., Dunham I., et al. Transcription Factor Activities Enhance Markers of Drug Sensitivity in Cancer. Cancer Res. 2018;78:769–780. doi: 10.1158/0008-5472.CAN-17-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shen, L., and IS.M.M Sinai (2021). GeneOverlap: Test and visualize gene overlaps. Preprint.
- 94.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 95.Du P., Zhang X., Huang C.-C., Jafari N., Kibbe W.A., Hou L., Lin S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinf. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu Y., Wang L., Yin Y., Yang E. Systematic analysis of gene expression patterns associated with postmortem interval in human tissues. Sci. Rep. 2017;7:5435. doi: 10.1038/s41598-017-05882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All DNA methylation and expression datasets for this publication have been deposited in the NCBI Gene Expression Omnibus and are accessible through a GEO accession numbers GSE235104 and GSE272030 respectively.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.