

Abstract
Disulfide-constrained peptides (DCPs) have gained increased attention as a drug modality due to their exceptional stability and combined advantages of large biologics and small molecules. Chemical synthesis, although widely used to produce DCPs, is associated with high cost, both economically and environmentally. To reduce the dependence on solid phase peptide synthesis and the negative environmental footprint associated with it, we present a highly versatile, low-cost, and environmentally friendly bioproduction platform to generate DCPs and their conjugates as well as chemically modified or isotope-labeled DCPs. Using the DCP against the E3 ubiquitin ligase Zinc and Ring Finger 3, MK1-3.6.10, as a model peptide, we have demonstrated the use of bacterial expression, combined with Ser ligation or transglutaminase-mediated XTEN ligation, to produce multivalent MK1-3.6.10 and MK1-3.6.10 with N-terminal functional groups. We have also developed a bioproduction method for the site-specific incorporation of unnatural amino acids into recombinant DCPs by the amber codon suppression system. Lastly, we produced 15N/13C-labeled MK1-3.6.10 with high yield and assessed the performance of a semiautomated resonance assignment workflow that could be used to accelerate binding studies and structural characterization of DCPs. This study provides a proof of concept to generate functionalized DCPs using bioproduction, providing a potential solution to alleviate the reliance on hazardous chemicals, reduce the cost, and expedite the timeline for DCP discovery.
Keywords: disulfide-constrained peptide, bioproduction, green chemistry, serine ligation, transglutaminase, amber codon, ZNRF3
Introduction
In the past decade, disulfide-constrained peptides (DCPs) containing up to around 40 amino acids and multiple disulfide bonds have gained increased attention as a drug modality due to their exceptional stability and combined advantages of large biologics and small molecules.1−3 Their increased surface area and 3D topology grant them the ability to bind and interrupt challenging protein–protein interaction interfaces better than small molecules or linear peptide drugs, while their reduced size compared to large molecule therapeutics allows greater tissue penetration for more effective treatment and smaller dosing volumes. The hypervariable loops between the disulfide bonds provide attractive templates for molecular evolution. Novel bioactive peptides have been developed through modification of the native sequences in loop regions of DCPs to target kinases, proteases, G-protein coupled receptors, growth factors, and protein–protein interactions through substituting the loop regions with known linear bioactive epitopes.4−10 Recently, we have developed a collection of DCP phage libraries by randomizing the loop regions of wild-type DCPs to identify potent binders against different protein targets successfully.11−13 However, challenges still exist in the hit discovery process using these libraries. For example, generation of lead peptides identified from the libraries often requires production of large number of DCPs (>50 per target) due to multiple rounds of affinity maturation. Chemical synthesis, although widely used to produce peptides, is associated with high cost both economically and environmentally. Solid phase peptide synthesis (SPPS)14,15 generates a large amount of hazardous waste including trifluoroacetic acid (TFA), N,N-dimethylformamide (DMF), piperidine, N-methylpyrrolidone (NMP), and dichloromethane (DCM).16,17 Regulatory agencies are progressively more stringent with the usage of these solvents due to their negative impact on the environment and health. For instance, the European Commission has recently adopted a regulation amending Annex XVII of REACH to restrict the use of DMF on the EU market. To address this issue, we and others have developed strategies to produce cyclic or linear DCPs recombinantly, from bacteria or yeast.18−20 These strategies provide environmental sustainability and also greatly reduce the economic burden of DCP production.
While DCPs with antibody-like affinity have been identified for a range of extracellular targets,11−13 some of these DCPs still display unfavorably fast off-rates compared to antibodies that contain greater surface area for binding. This can complicate the development of DCPs for difficult-to-drug targets, particularly for antagonistic molecules that compete with natural agonists that display more durable binding in vivo. Additionally, antibodies can engage multiple copies of the same target, which can also contribute to their activity. As a way to further increase the antibody-like behavior of smaller peptide-based ligands, we recently explored the use of multivalent DCPs to target membrane proteins.11−13 This strategy improves the residence time on the target and, in some cases, grants new functions to the monomeric DCPs. For example, in a study where we aimed to activate Wnt signaling through inhibition of the membrane bound E3 ubiquitin ligase Zinc and Ring Finger 3 (ZNRF3), we generated a series of multivalent DCPs (dimer, trimer, tetramer, and hexamer) using a nonfunctional monomeric DCP (MK1-3.6.10) targeting ZNRF3. The multimeric MK1-3.6.10 molecules not only demonstrated reduced off-rates compared to the monomeric MK1-3.6.10 when binding to ZNRF3 but also became potent ZNRF3 antagonists/Wnt signaling agonists.12 In another study, we developed DCPs that bound the third β-propeller domain of the Wnt coreceptor LRP6. These DCPs antagonize Wnt3a while sparing Wnt1 signaling. Following development of these DCPs into multivalent Wnt agonists, these multimeric DCPs showed improved affinity and could potentiate Wnt1 signaling through clustering of the LRP6 coreceptor.13 These studies highlight the utility of multivalent DCPs, and we believe that the benefits afforded to this strategy could be readily translatable to other challenging targets. Currently, the preferred method for DCP ligation is click chemistry, which is the 1,3-dipolar cycloaddition of an azide and alkyne to form 1,2,3-triazole.21,22 Briefly, monomeric azido- or alkynyl-DCPs are generated through SPPS and oxidative folding (to form intramolecular disulfide bonds), followed by click chemistry reaction using various lengths of PEG linkers with alkynyl- or azido-groups. While this strategy has found utility, it presents drawbacks such as low yield (due to challenges with folding of certain DCPs) and high cost of azido- or alkynyl-DCPs. Thus, alternative methods for production and ligation of DCPs that are chemoselective, inexpensive, rapid, safe, and more environmentally friendly would be beneficial.
Another challenge of DCP production lies in the incorporation of unnatural amino acids (UAAs) into the lead DCP molecules. In our efforts to generate DCPs with drug-like properties, we have often found it necessary to resort to using UAAs in order to improve affinity, stability, solubility, and pharmacokinetics. However, different from linear peptides or macrocycles, addition of UAAs sometimes complicates the oxidative folding process of DCPs and this, combined with the increased cost of UAAs during chemical synthesis, is often associated with higher cost than DCPs comprising only natural amino acids. Amber codon suppression-mediated site-specific incorporation of UAAs into proteins has proven to be a powerful approach that has witnessed increasing applications in the past decades.23 While the system has been widely adapted to therapeutic proteins,24 production of UAA-incorporated DCPs recombinantly has not been achieved. Such methodology would greatly benefit the discovery of DCP binders with desired drug-like properties.
Taking advantage of the unique property of multimeric MK1-3.6.10 (gain of functionality) as well as our previously reported bioproduction method, here, we address the aforementioned issues by employing different ligation strategies using recombinant MK1-3.6.10 to produce multivalent MK1-3.6.10 and MK1-3.6.10 with N-terminal functional groups, including Ser/Thr ligation (STL) and transglutaminase-mediated XTEN ligation. Importantly, we have developed a method for the site-specific incorporation of UAAs into recombinant DCPs by the amber codon system. Lastly, we produced 15N- and 15N/13C-labeled MK1-3.6.10 with high yield and assessed the performance of a semiautomated resonance assignment workflow that could be used to accelerate binding studies and structural characterization of DCPs. This study provides a proof of concept to generate various forms of DCPs as potential therapeutics or assay reagents using bioproduction.
Results and Discussion
Bioproduction of Active ZNRF3-DCPs
In order to develop a streamlined process for large-scale bioproduction of active DCPs, we first evaluated bioproduction of ZNRF3-DCPs using a previously established method.18 We selected several ZNRF3-DCPs with different affinities and identified from different scaffolds (MK1-2.A2.B9.1 and MK1-2.A1.B3 from λ-MK1a and MAU-1.2.4 from maurocalcin; KD ranging from 2500 to 5 nM).12 The His(6)-SUMO-DCP fusion proteins were overexpressed in Escherichia coli (1 L culture) and purified using the Ni-NTA resin in high yield (∼30 mg/L). Following digestion with TEV protease to release the peptide from the fusion protein, we assessed various purification strategies to separate the protein tag [His(6)-SUMO] and TEV protease from the DCPs. Since both His(6)-SUMO and TEV protease possess N-terminal His(6) tags, we began with His-tag pulldown using the Ni-NTA resin. Using this method, all three DCPs were separated from His(6)-SUMO and TEV protease, although with different efficiencies (Figure 1a). MK1-2.A2.B9.1 was successfully purified with a high yield and purity (3.6 mg/L, purity: 96%). However, for MK1-2.A1.B3 and MAU-1.2.4, the recovery rate was very low (1 and 0.3 mg/L, respectively). Further investigation revealed that for MK1-2.A1.B3 and MAU-1.2.4, the majority of the DCPs bound to the Ni-NTA resin, as incubation with high concentration of NaCl solution (1 M) could recover more DCPs after imidazole elution. For all three DCPs, His-tag pulldown failed to remove a high-molecular-weight impurity (apparent molecular weight: ∼50 kDa on SDS-PAGE; accurate mass: 68,985 Da on LC–MS; Figure 1a). Moreover, attempts using centrifugal ultrafiltration with membranes of different molecular weight cutoff (MWCO) failed to separate MK1-2.A1.B3 or MAU-1.2.4 from the rest of the mixture, as majority of the DCPs stuck on the membrane. We speculate that the “stickiness” of MK1-2.A1.B3 and MAU-1.2.4 might be due to nonspecific interaction between the hydrophobic patches on the DCPs with the membrane/resin.
Figure 1.

Production of recombinant ZNRF3 DCPs in E. coli. (a) SDS-PAGE analysis of expression and purification of MK1-2.A2.B9.1, MK1-2.A1.B3, and MAU-1.2.4 in E. coli. The His(6)-SUMO-DCP fusion proteins were overexpressed in E. coli, purified using the Ni-NTA resin, and digested with TEV protease. DCPs were separated from His(6)-SUMO and TEV protease using the Ni-NTA resin with different efficiencies. MK1-2.A2.B9.1 can be fully recovered. The recovery rate was very low for MK1-2.A1.B3 and MAU-1.2.4, possibly due to nonspecific interaction between the hydrophobic patches on the DCPs with the resin. Incubation with a high concentration of NaCl solution can recover more DCPs. (b) LC–MS analysis of purified MK1-2.A2.B9.1 (by the Ni-NTA resin), MK1-2.A1.B3 (by the Ni-NTA resin and 1 M NaCl), and MAU-1.2.4 (by HPLC). HPLC purification produced DCPs with high yield and purity. (c) Recombinant ZNRF3 DCPs (MK1-2.A2.B9.1, MK1-2.A1.B3, MAU-1.2.4, and MK1-3.6.10) show similar affinity to their synthetic counterparts. The affinity values were measured by SPR. All values were averaged from three independent runs. (d) Representative SPR sensorgrams of different forms of MK1-2.A2.B9.1 and MK1-3.6.10 binding to human ZNRF3 (immobilized on the sensor). Black: raw data; red: fitted data. SUMO-DCP: His(6)-SUMO-DCP fusion protein; SUMO-DCP, digested: SUMO-His(6)-DCP fusion protein digested with TEV protease; Rec DCP: purified recombinant DCP; Syn DCP: chemically synthesized DCP.
To streamline the purification step, we then evaluated initial high-performance liquid chromatography (HPLC) purification, followed by TEV protease digestion. As shown in Figure 1b, MAU-1.2.4 was successfully purified from the mixture with good yield (∼2 mg/L for DCP and ∼30 mg/L for the fusion protein) using HPLC. This served as a general method for purification of all DCPs. We then measured the affinity of various forms of DCPs [His(6)-SUMO-DCP fusion, digested His(6)-SUMO-DCP/TEV protease mixture, and HPLC purified recombinant DCP] using surface plasmon resonance (SPR). All forms of DCPs showed KD values comparable to those of their synthetic counterparts (Figure 1c,d) when binding to ZNRF3, confirming the production of active DCPs from E. coli. The data indicate that it is possible to rank the affinity of various DCPs including the SUMO-tagged DCP fusion protein. Production of recombinant His(6)-SUMO-DCP requires one-step purification without any hazardous organic solvent. This provides a potential solution to alleviate the reliance on hazardous chemicals, reduce the cost, and expedite the timeline for DCP discovery.
In general, bioproduction demonstrates lower Complete Environmental Impact Factor (cEF) and is less costly compared to SPPS. To produce 10 mg of DCP, SPPS costs ∼$1500 and generates ∼5 L organic solvent, versus ∼$200 and <0.5 L organic solvent for bioproduction. Furthermore, scalability of the bioproduction method was demonstrated on a 70 mg target scale by expressing and isolating one DCP (Ktag-MK1-3.6.10, see the section below) from a 10 L culture volume. There is still significant opportunity for further improvement in yields with the use of commercial bioreactor systems. It is noteworthy that sometimes recombinant peptides/proteins are contaminated with bacterial endotoxin. Removal of endotoxin might be necessary to ensure the safety of recombinant DCP when used in animal studies.
Semibioproduction of Multivalent MK1-3.6.10 with Different Geometries
The monomeric MK1-3.6.10 binds to its target ZNRF3 but does not enhance the Wnt signaling in cells. Multivalent analogues of MK1-3.6.10, on the other hand, could potently activate Wnt signaling in both cells and human tissues.12 We took advantage of this unique property of MK1-3.6.10 to investigate bioproduction of dimeric DCPs with head-to-tail (connected from the C-terminus to the N-terminus25) or head-to-head (connected through the N-termini) geometries. First, we produced a recombinant MK1-3.6.10 monomer with the same binding affinity as its synthetic counterpart (Figure 1c,d). We then overexpressed two head-to-tail MK1-3.6.10 dimer constructs, di-MK1-3.6.10GS (two MK1-3.6.10 molecules connected by a GSGSGS linker) and di-MK1-3.6.10TEV (a control molecule with two MK1-3.6.10 molecules connected by the TEV protease digestion site, SENLYFQ) (Figure 2a). We were able to express the two SUMO-tagged dimeric DCPs successfully in good yield (3.7 mg/L for di-MK1-3.6.10GS and 6.3 mg/L for di-MK1-3.6.10TEV). Upon TEV digestion, di-MK1-3.6.10GS produced one major DCP species with six disulfide bonds; while di-MK1-3.6.10TEV produced two monomeric DCPs with three disulfide bonds in each (MK1-3.6.10-SENLYFQ and MK1-3.6.10), indicating the two DCPs likely fold independently without forming any intermolecular disulfide bonds (Figure 2b). SPR measurement with the SUMO-tagged DCP fusion protein and a mixture of the digested His(6)-SUMO, DCP, and TEV protease indicated that all of the recombinant monomeric and dimeric DCPs have comparable KD values to their synthetic counterparts for binding to ZNRF3 (Figure 2c). We then purified di-MK1-3.6.10GS using HPLC (purity >95%, yield ∼2 mg/L) and tested its cellular activity in HEK293 TOPbrite (TB) luciferase reporter cells. In contrast to the synthetic head-to-head dimer, the recombinant head-to-tail dimer di-MK1-3.6.10GS did not enhance the Wnt signaling in the reporter assay (Figure 2d). We reasoned that the inactivity of di-MK1-3.6.10GS is likely due to the fact that the head-to-tail orientation of the peptides is not optimal for generating a functional dimeric molecule. As a matter of fact, the crystal structure of MK1-3.6.10 in complex with ZNRF3 confirms the relative long distance between N- and C-termini of MK1-3.6.10 (∼23.5 Å, Figure S1a). This could dramatically alter the geometry between head-to-head and head-to-tail dimers. While di-MK1-3.6.10GS lacks bioactivity due to its suboptimal geometry for promoting Wnt signaling, its ability to bind with high affinity suggests that head-to-tail dimers that are readily made through bioproduction could be functional multivalent ligands for the right target.25
Figure 2.

Production of functional dimeric and multimeric MK1-3.6.10 using recombinant monomeric MK1-3.6.10. (a) Construct design for recombinant head-to-tail dimeric MK1-3.6.10. In the di-MK1-3.6.10GS construct, two DCPs were separated by a GSGSGS linker and TEV protease digestion would produce one major DCP species with six disulfide bonds. For the control construct, di-MK1-3.6.10TEV, two DCPs were separated by the TEV protease digestion site and TEV protease digestion would produce two monomeric DCPs (MK1-3.6.10-SENLYFQ and MK1-3.6.10) with three disulfide bonds in each if the two DCPs fold independently without forming any intermolecular disulfide bonds. (b) LC–MS and SDS PAGE analysis of di-MK1-3.6.10GS and di-MK1-3.6.10TEV expression. di-MK1-3.6.10GS produced one major species with six disulfide bonds, with a minor species (∼10%) also containing six disulfide bonds. di-MK1-3.6.10TEV produced two DCPs with three disulfide bonds in each. (c) Different forms of recombinant dimeric MK1-3.6.10 (head-to-tail) show similar affinity to their synthetic counterparts. The affinity values were measured by SPR. All values were averaged from three independent runs. (d) Recombinant head-to-tail MK1-3.6.10 dimers do not activate Wnt signaling in the TOPbrite reporter cells. Representative data from three independent runs are shown. (e) Generalized reaction scheme for STL. (f) Different forms of head-to-head dimeric MK1-3.6.10, produced through STL, show similar affinity to their synthetic counterparts. The affinity values were measured by SPR. All values were averaged from three independent runs. (g) Head-to-head dimeric and multimeric MK1-3.6.10 can activate Wnt signaling in the reporter assay. Representative data from three independent runs are shown.
To generate a functional dimeric MK1-3.6.10 similar to our previously made synthetic dimer, we sought to produce a head-to-head dimer using STL.26−28 STL enables the conjugation of unprotected peptides to other peptides or functional groups that contain a salicylaldehyde (SAL) ester, forming an amide bond at the ligation site. We first expressed a variant of MK1-3.6.10 that produced an N-terminal serine residue after TEV cleavage (MK1-3.6.10S), synthesized the SAL-PEG5-SAL diester linker, and then performed STL with the linker and peptide in a 1:1 pyridine/AcOH buffer, followed by treatment with TFA (Figures 2e and S1b). The reaction was monitored by liquid chromatography–mass spectrometry (LC–MS) and used a slight excess of peptide to ensure complete conversion to the dimer product. In SPR experiments, both the recombinant MK1-3.6.10S and the head-to-head dimer di-MK1-3.6.10S showed affinity comparable to that of their synthetic counterparts for binding to ZNRF3 (Figure 2f). Importantly, the head-to-head dimer di-MK1-3.6.10S was able to induce Wnt signaling activation to a similar extent to the synthetic MK1-3.6.10 dimer in the reporter assay (Figure 2g), confirming that the head-to-head geometry is the optimal configuration for generating ZNRF3 antagonists within this family of peptide. Furthermore, trimeric and tetrameric (head-to-head) MK1-3.6.10S made using the same STL strategy showed trends for activating Wnt signaling in cells similar to those of the fully synthetic higher order multimers (Figure 2g). In summary, STL can readily produce multivalent ligands with head-to-head geometries using bioproduced DCPs.
Production of N-Terminal Biotinylated DCP Using Recombinant MK1-3.6.10S
In addition to making multivalent ligands, we envision that STL can be a useful way to functionalize the N-terminus of bioproduced DCPs for a range of applications. We demonstrate one simple proof of concept by generating biotinylated DCPs that can be broadly used in different types of assays, including for immobilizing DCPs on surfaces for SPR or ELISA, or for use as detection tools in immunofluorescence and fluorescence-activated cell sorting. Since linker length can influence the efficiency of such applications, we synthesized biotin-PEG2-SAL and biotin-PEG12-SAL esters and conjugated them to the N-terminus of the recombinant MK1-3.6.10S via STL. In the presence of a slight excess (2 equiv) of the corresponding biotin-SAL esters, we observed complete biotinylation of the MK1-3.6.10S, as determined by analytical HPLC. We also obtained good recovery after purification (∼50–60%). We then immobilized either biotin-PEG2- or biotin-PEG12-MK1-3.6.10S on an SPR sensor coated with streptavidin and tested the binding affinity of ZNRF3. To emphasize, this approach differs from our previous SPR protocols that subject immobilized protein to untagged DCP. Gratifyingly, both biotinylated MK1-3.6.10S molecules demonstrated similar binding to ZNRF3 (KD = 149 ± 8 nM and KD = 118 ± 3.2 nM for biotin-PEG2- and biotin-PEG12-MK1-3.6.10S, respectively) (Figures 3a and S2a). Furthermore, the immobilized peptides did not bind to the functionally related ligase Ring Finger Protein 43 (RNF43), confirming that this approach retained the desired DCP specificity (Figure 3a). The slightly reduced affinity of biotinylated MK1-3.6.10S toward free ZNRF3 compared to recombinant MK1-3.6.10S binding to immobilized protein (KD = 58 ± 5 nM) is likely due to the sensor surface spatially hindering ZNRF3′s access to the binding site of the immobilized DCPs. This hypothesis is supported by the fact that the shorter biotin-PEG2-MK1-3.6.10S linker, which would create more crowding effects around the sensor chip, showed a more pronounced decrease in binding affinity. Nevertheless, the data here confirm that biotinylation via STL can be a useful tool to immobilize bioproduced DCPs on a surface to probe binding interactions.
Figure 3.

Production and application of biotinylated MK1-3.6.10S (through STL) and octameric MK1-3.6.10 (through TG). (a) Biotinylated MK1-3.6.10S can be used as a binding assay reagent. Representative SPR sensorgram of biotin-MK1-3.6.10-PEG12 (immobilized on the sensor) binding to human ZNRF3 (black: raw data; red: fitted data) or RNF43 (purple). Biotin-MK1-3.6.10-PEG12 was generated through STL using recombinant MK1-3.6.10. (b) Biotin-MK1-3.6.10-PEG12 can be used to report cellular distribution of ZNRF3 in HEK293 cells transiently expressing HA-tagged ZNRF3. Biotin-MK1-3.6.10-PEG12 and the anti-HA antibody detected overexpressed ZNRF3 in the same cells, with a similar cellular distribution pattern. Scale bar: 10 μm. (c) Representative SPR sensorgram of Ktag-MK1-3.6.10 binding to human ZNRF3 (immobilized on the sensor). Black: raw data; red: fitted data. (d) Graphical representation of the enzymatic ligation of Ktag-MK1-3.6.10 to a Q-tag containing an XTEN polypeptide scaffold by transglutaminase. (e) Representative SPR sensorgram of octameric MK1-3.6.10 (MK1-3.6.10-XTEN80-8) binding to human ZNRF3 (immobilized on the sensor). Black: raw data; red: fitted data. MK1-3.6.10-XTEN generated through transglutaminase-mediated XTEN ligation demonstrated high affinity and extremely slow Koff (e) but failed to activate Wnt signaling in TOPbrite cells (f). Representative data from three independent runs are shown for all the assays.
We also explored the application of biotinylated MK1-3.6.10S as an immunofluorescence detection reagent for ZNRF3 expression in and within cells. In HEK293T cells transiently transfected with HA-tagged full length ZNRF3, biotinylated MK1-3.6.10S and the anti-HA antibody detected overexpressed ZNRF3 in the same cells, with a similar cellular distribution pattern (Figures 3b and S2b). In cells with lower expression levels of ZNRF3, it seems that ZNRF3 can be detected throughout the cell, with a more pronounced distribution at the perinuclear region and plasma membrane (Figure 3b). The data demonstrate that biotinylated MK1-3.6.10S can be a successful immunofluorescence reagent to report on ZNRF3 expression and localization. Taken together, these studies show that bioproduced DCPs can be readily functionalized by STL for different applications.
Transglutaminase-Mediated Bioproduction of Multivalent MK1-3.6.10
As another orthogonal and environmentally friendly conjugation method using only standard amino acids, we investigated the enzymatic ligation of a recombinant monomeric DCP using microbial transglutaminase (TG) to generate multivalent ligands displayed on an XTEN scaffold. As an alternative to chemical ligation, enzymatic ligation is more sustainable and economically viable for large-scale manufacturing of therapeutic peptides.29 TGs catalyze the acyl-transfer reaction between glutamine and lysine to form N6-(5-glutamyl)-lysine isopeptide cross-links and have been widely used to attach payloads to proteins or antibodies,30,31 but its application to DCP conjugation has not been previously explored. XTEN, a family of unstructured hydrophilic protein polymers, were originally designed to prolong the in vivo half-life of protein or peptide therapeutics.32 XTEN can be recombinantly overexpressed as a fusion with proteins and peptides, but this approach limits the total number of ligands that can be fused to the scaffold as well as limiting fusion location to the termini. Ligands can also be conjugated to XTEN post expression, for the sake of gaining better control over the positioning of the payloads along the length of XTEN.32 We generated MK1-3.6.10 with an N-terminal Ktag (GRSKL) followed by a GSGSGS linker (Ktag-MK1-3.6.10) and showed that the addition of this short peptide sequence had minimal impact on its affinity (KD = 79 ± 2.8 nM, Figure 3c). For our test case, we used an 80 kDa XTEN scaffold containing eight spaced out Q-tags (VLQSP) within exposed loops (XTEN80-8Q). We then used TG to ligate bioproduced Ktag-MK1-3.6.10 to form an octameric display of the peptide ligand (MK1-3.6.10-XTEN80-8, Figure 3d). The more arms within a multivalent scaffold, the more difficult it is to react all of the functional groups. To avoid a complicated purification process to separate the fully reacted product from incomplete intermediates, we used an excess of peptide to drive the reaction forward. The ligation was left overnight, and LC–MS confirmed complete conversion to the desired 8-mer product (Figure S2c).
Using SPR, we demonstrated that MK1-3.6.10-XTEN80-8 showed high affinity (subpicomolar range) against ZNRF3, in large part due to the extremely slow Koff (Figure 3e). This high affinity likely confirms that MK1-3.6.10-XTEN80-8 can simultaneously bind multiple copies of ZNRF3 at the same time; however, this did not lead to enhanced Wnt signaling in the reporter assay (Figure 3f). As observed with our previous head-to-tail recombinant dimer that also did not have activity, we speculate that the geometry and orientation of the ligands are very important for promoting Wnt signaling and that the linear arrangement of the DCP ligand on the XTEN scaffold is not optimal for this specific function (Figure 3f). Nonetheless, these data show that TG can be readily used to attach bioproduced DCPs to polypeptide scaffolds with the potential to achieve multivalency or increased half-life. This system relies on recombinant expression and enzymatic ligation only and therefore further reduces the use of an organic solvent and lowers the cEF.
Site-Specific Incorporation of Unnatural Amino Acids into Recombinant MK1-3.6.10
UAA incorporation by amber codon suppression is a powerful and environmentally friendly tool to modify the properties of antibodies or proteins at will. Here, we explored its application to the DCP bioproduction platform. We prepared a pBAD33-based plasmid (pAzF) that encodes a Methanococcus jannaschii tyrosyl-tRNA synthetase (Mj TyrRS) mutant evolved for the specific aminoacylation of an orthogonal tRNACUA, also encoded in the same plasmid, with the noncanonical amino acid 4-azidophenylalanine (AzF).33 To measure the compatibility and efficiency of AzF incorporation in our bioproduction system, we expressed amber stop codon mutants of the superfolder green fluorescent protein (sfGFP) at K2 or Y151 through the same pST239-SUMO plasmid used for DCPs. We observed robust fluorescence when these sfGFP mutants are coexpressed with pAzF and when AzF is supplemented in the culture media at the optimal concentration of 5 mM (Figure S3a). Notably, even in the presence of the pAzF plasmid, no GFP signal was observed when AzF was absent. This suggests the high fidelity of this amber codon suppression system.
We first tested whether AzF can be incorporated at the N-terminus of MK1-3.6.10. Since the first glycine residue of the DCP is required in the TEV cleavage step during purification, we inserted the amber codon at the second position. However, the expression of this construct was inefficient and led to truncation products (Figure 4a). We hypothesized that this poor ribosomal readthrough could be rescued by inserting a flexible GS linker between the AzF incorporation site and the DCP. Indeed, we were able to obtain nAzF-GS-MK1-3.6.10 with reasonable yields (0.22–1.35 mg/L for DCPs) (Figure 4a,b). Having successfully incorporated an azide handle into a bioproduced DCP, we envisioned that this would enable various types of modifications using commercially available terminal or strained alkyne reagents. We then prepared dimers of this DCP via copper(I)-assisted azido-alkyne cycloaddition (CuAAC) with a bis-propargyl-PEG5 linker. The nAzF-GS-MK1-3.6.10 dimer showed similar affinity in SPR measurements (Figures 4b and S3b) and similar activity in Wnt activation assays compared to the synthetic MK1-3.6.10 PEG5 dimer counterpart (Figure 4c). As AzF is a minimal, genetically encoded tag that can be readily functionalized with diverse moieties (e.g., biotin, fluorophores, PEG linkers, etc.), we propose that UAA incorporation can be a versatile technique for altering the structure, properties, and utility of our bioproduced DCPs.
Figure 4.

Incorporation of unnatural amino acids into DCPs via amber codon suppression. (a) SDS-PAGE analysis of the Ni-NTA-purified His(6)-SUMO-MK1-3.6.10 fusion constructs with incorporated AzF at different positions. Green asterisks indicate the correct mass of desired full-length product. Red asterisks indicate observed truncation products. (b) Summary table of purified yield and measured affinities (via SPR) of azidophenylalanine-containing MK1-3.6.10 peptides compared to wild-type. n.b.: no binding; n.a.: not available. (c) Dimeric MK1-3.6.10 linked at the N-terminal AzF activates Wnt signaling in TOPbrite cells with a similar efficiency as a fully synthetic version dimerized at an N-terminal azidolysine group. (d) Crystal structure of ZNRF3-MK1-3.6.10 (PDB code: 8G4Y) complex showing a putative available surface for derivatization of F30. (e) Models of the extension of F30 aromatic moiety with either the azido group or the aminomethyl-triazole group.
In the crystal structure of MK1-3.6.10 bound to the ZNRF3 extracellular domain, MK1-3.6.10 contacts the outer face of the ZNRF3 β sheet, including the extended beta “flap” (residues 66–76).12 The ZNRF3/DCP interface comprises a range of hydrophobic, polar, and aromatic ring interactions involving multiple regions of the DCP. We noted that the side chain of F30 of MK1-3.6.10 provides minimal interaction despite being in close proximity to a potential binding pocket (Figure 4d); hence, we hypothesized that this residue would be amenable to medicinal chemistry substitutions to modulate MK1-3.6.10 binding affinity. We tested this hypothesis by incorporating AzF at the F30. Again, we were able to bioproduce MK1-3.6.10 AzF30 with good yield (20.5 mg/L for the fusion protein and 1.35 mg/L for DCP) (Figure 4b). After purification of this intermediate, we functionalized it further with propargylamine via CuAAC. Using SPR, we measured a 9-fold reduction in affinity when F30 (KD = 38.5 ± 4.5 nM) is substituted with AzF (KD = 326 ± 19 nM). Upon extension with propargylamine, the affinity was improved relative to the AzF variant (KD = 55 ± 6.5 nM), similar to the original, wild-type MK1-3.6.10 DCP (Figures 4b and S3b). To rationalize these observations, we modeled these structural modifications onto the crystal structure (Figure 4e) and found that AzF30 may introduce a potential clash with ZNRF3 Y118 that could push the DCP away from the interface, while extension with propargylamine may gain additional H-bonding interactions with T152 and R178. For comparison, we also performed substitutions similar to those for MK1-3.6.10 residue Y32. Both AzF and propargylamine-clicked AzF substitutions abolished binding. This is consistent with the crystal structure showing that Y32 points directly into the ZNRF3 interface with minimal space available for amino acid extensions (Figure S3c). Taken altogether, these experiments demonstrate the utility of AzF incorporation for late-stage chemical modification of bioproduced DCPs to improve their drug-like properties.
Bioproduction of Isotope Labeled MK1-3.6.10 for NMR Studies
The ability to produce recombinant DCPs in E. coli enables isotopic labeling of these peptides for heteronuclear solution NMR studies. Taking advantage of a previously reported protocol to produce naturally occurring disulfide-rich venom peptides,19 as well as our bioproduction method, we sought to generate isotopic labeled DCPs identified from our phage DCP libraries with high yield.
Production of functional DCPs in E. coli requires the formation of disulfide bonds. This is achieved through targeting the constructs to the periplasmic space for oxidative folding, taking advantage of the alkaline phosphatase (phoA) promoter combined with the heat-stable enterotoxin II (STII) leader sequence.18 For Fab production, the PhoA promoter showed higher secretion ability than the T7 promoter, and the signal peptide STII demonstrated better extracellular secretion efficiency than pelB.34,35 It was speculated that this is because the T7lac system, although a stronger expression system, could overwhelm the native type II secretion system STII employs.34,35 Here, we confirmed that the yield of His(6)-SUMO-MK1-3.6.10 indeed was much lower with the T7 promoter (1–5 mg/mL) compared with the PhoA promoter (∼30 mg/mL). We reasoned that since the use of minimal media usually leads to a reduced protein yield, the T7lac-STII expression system could be advantageous in this regard. Furthermore, lactose-induced T7lac expression system is cost-effective, unlike the autoinduction PhoA promoter system, which necessitates the use of costly 13C-labeled lactose.19 We expressed and purified 15N labeled and 15N/13C labeled His(6)-SUMO-MK1-3.6.10 with the T7lac-STII expression system in BL21 Star (DE3) cells. We obtained a relatively high yield (13 mg/mL) for isotope-labeled His(6)-SUMO-MK1-3.6.10. After TEV digestion and HPLC purification, we harvested 2 mg of 15N/13C labeled MK1-3.6.10 from 3 L of culture, with labeling efficiency of 99.3 and 97.1% for 15N labeled and 15N/13C labeled MK1-3.6.10, respectively (Figure 5a).
Figure 5.

Production of [15N,13C]-labeled MK1-3.6.10 for NMR studies. (a) LC–MS analysis of 15N and 15N/13C labeled recombinant MK1-3.6.10. Both labeled MK1-3.6.10 can be expressed and purified from E. coli with good yield (∼0.67 mg/L) and labeling efficiency. (b) Correctly assigned resonances obtained from [15N,13C]-labeled MK1-3.6.10 as determined by the FLYA routine of CYANA. Correct assignments were determined by comparing the resulting high-confidence assignment list from FLYA to a manually assigned reference. The accuracy of assignments was preserved when reducing the number of data sets, allowing for shorter data collection times. (c) Graphical representation of FLYA results. Backbone amides’ assignments matching the manual reference are colored in green, whereas low-confidence assignments are colored in orange. Residues without backbone NH signals or no expected signal in the [1H–15N] HSQC spectrum are colored in gray; black lines indicate disulfide connectivity. (d) [1H–15N] HSQC spectrum of MK1-3.6.10. Assigned amino acids are labeled according to their position in the peptide sequence. Peaks arising from minor conformations are labeled with blue asterisks.
Bioproduced 15N/13C labeled MK1-3.6.10 provides the necessary starting conditions for implementing a semiautomated resonance assignment workflow that could accelerate structural characterization of DCPs and binding site mapping studies through [1H–15N]-HSQC chemical shift fingerprinting. The application of NMR to characterize the structure of DCPs and their interaction with a protein target requires, as a first step, the resonance assignment of most of the nuclei of interest (proton, carbon, and nitrogen). This process can be time-consuming if carried out in a manual fashion, so we sought to test how an automated assignment routine like FLYA, which has been designed for the resonance assignment of proteins, would perform when applied to constrained peptides such as DCPs.
To assess the performance of FLYA, we tested how the use of various data set combinations affects the accuracy of FLYA-derived resonance assignments when compared to the manually determined assignments. Using a standard set of triple-resonance data sets (HNCA, HNcoCA, HNCO, HncaCO, CBCANH, and CBCAcoNH), FLYA correctly assigned 31 of the 31 manually assigned backbone amide NH resonances (Figure 5b,c). Note in Figure 5d that the [1H–15N]-HSQC displays more peaks than expected from the sequence; however, FLYA is able to accurately determine most assignments while only erroneously assigning minor conformation peaks to residues for which no resonances were assigned (these are low confidence assignments, which are not consistent in all data sets).
Due to the small size of the peptide and relatively good signal dispersion observed in the [1H–15N]-HSQC spectrum, we assessed a minimal data set combination required to confidently assign amide NH signals. Figure 5b highlights the accuracy in resonance assignment for various combinations of triple-resonance data sets. Reducing the data sets to those containing both the intra- and adjacent-carbon resonances (Set 2) was sufficient; further, for well-dispersed spectra, the CBCANH and CBCAcoNH (Set 3) were sufficient to match 98.5% of the backbone atoms assigned manually (excluding carbonyl carbons), which reduced the acquisition time by a factor of 2. When more comprehensive atom assignments are desired, the addition of through-bond side-chain experiments such as HNHA, 13C- and 15N-resolved TOCSY experiments can be utilized with FLYA as well, in our case achieving 95% backbone, 78% side chain, and 85% of all atoms assigned manually (Figure S4a,b).
To further assess what would be possible with isotopically labeled DCPs, data corresponding to only the 15N-labeled peptide were used as input for assignment with FLYA. Initially, results were poor, even with the addition of 15N-NOESY-derived data, with very few NOESY peaks being assigned with high confidence (Figure S4b,c). To improve this process, we used an AlfaFold2 model of MK1-3.6.10 as input along with the 15N-NOESY-derived peak lists and noticed substantial improvement as shown in Figure S4c. Combining a structural model with experimental data not only produced very similar results to the backbone assignments generated when using a full suite of triple resonance data but it is also very close to matching assignments obtained for the side chain atoms in the 15N-edited TOCSY data set in the context of a full suite of [15N,13C]-labeled data set (Figure S4d).
NMR heteronuclear methods are broadly applied to characterize structure and dynamics of disulfide constrained peptides.36,37 In the context of disulfide-constrained peptides’ discovery and optimization, [1H–15N]-HSQC fingerprinting provides a useful and quick method to assess peptide binding to its target and to gain information on the binding epitope.38,39 In order to perform these types of NMR studies, it is essential to obtain the resonance assignment of the peptide backbone HN nuclei, a process that can take several days due to data collection time and manual data analysis. In order to accelerate the resonance assignment workflow, we tested the application of FLYA (an automated assignment routine) in combination with fast data collection methods using non-uniform sampling (NUS). In the case of 15N/13C-labeled MK1-3.6.10, we found that the minimal set of data required to obtain most of the backbone assignment consists of three experiments: [1H–15N]-HSQC, CBCANH, and CBCAcoNH. These experiments can be run in less than 8 h when using the NUS data acquisition approach. We also found that a 15N-labeling scheme can be sufficient, in combination with an Alphafold model, to achieve the same coverage of the backbone amide assignment obtained with the more costly 15N/13C-labeled peptide.
The use of NUS methods to shorten data collection time and the application of an automated assignment routine like FLYA to obtain a reliable assignment of backbone atoms should expedite the resonance assignment process and enable more focused structure–activity relationship studies.
Conclusions
In this study, we present a versatile and highly efficient bioproduction platform to generate various forms of DCPs as an environmentally sustainable alternative to SPPS. We first demonstrated a streamlined process for DCP hit generation and affinity ranking using bioproduction (Figure 6). We then demonstrated the ability to functionalize the bioproduced DCPs through different methods using MK1-3.6.10 as a test case (Figure 6). Previous efforts only describe the bioproduction of monomeric DCPs.18−20 Our current platform demonstrates the ability to generate completely recombinant head-to-tail dimers, head-to-head dimeric/multimeric ligands through tandem bioproduction and STL, and also multivalent ligands through enzymatic ligation to macromolecular scaffolds, in which all components used in this case (DCP, XTEN, and TG) were produced recombinantly and required only aqueous solutions for functionalization. While all of the multivalent DCPs produced in this study have improved affinity toward the target ZNRF3, only some of these ligands have improved functional activity, highlighting the need for diverse chemistries and scaffolds that can be applied to bioproduced DCPs in order to generate the desired function in multivalent ligands. We also demonstrated that STL could add useful functional groups to the N-termini of DCPs, exemplified by the production of biotinylated MK1-3.6.10S and its utility in both surface binding experiments (e.g. SPR) and immunofluorescence studies. These functionalized ZNRF3-DCPs specifically bind to ZNRF3 but not RNF43 (unlike their natural ligand R-spondin) and therefore can serve as powerful tools for probing the distinct roles between ZNRF3 and RNF43 in Wnt biology. Finally, we demonstrated that we could use this platform to introduce other specialty functionalities into a DCP, including UAA incorporation and isotopic labels. The installation of UAAs into recombinantly expressed peptides and proteins is still a challenge and we demonstrated that we could accomplish this with a DCP using amber codon suppression, while labeling a DCP with 15N/13C can enable sophisticated NMR studies. Collectively, these efforts demonstrate that our bioproduction platform can incorporate diverse functionalities into DCPs that previously have only been done using completely synthetic methods and, therefore, serves as a potential solution to alleviate the reliance on hazardous chemicals for DCP production.
Figure 6.
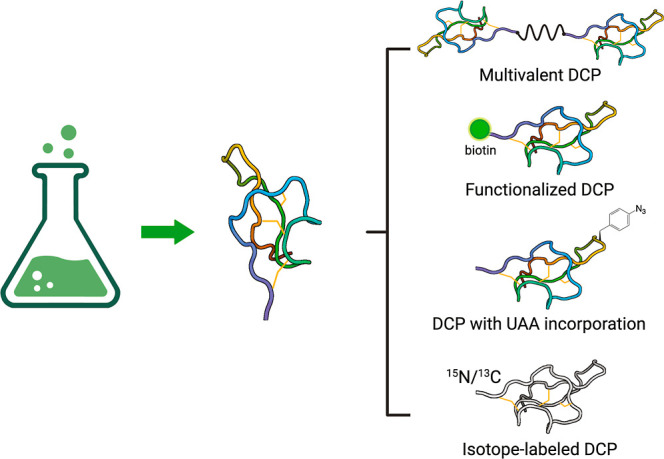
A bioproduction platform to generate various forms of DCPs as an environmentally sustainable alternative to SPPS. The platform can be used to generate: (1) multivalent DCPs, including completely recombinant head-to-tail dimers, head-to-head dimeric/multimeric ligands using tandem bioproduction and STL, and also through enzymatic ligation onto multivalent polypeptide scaffolds; (2) DCPs with functional chemical groups (such as biotin); (3) DCPs with UAAs through amber codon suppression; (4) 15N/13C labeled DCPs for heteronuclear solution NMR studies. This figure was created using Biorender (http://biorender.com).
Materials and Methods
Expression and Purification of DCPs
Fusion DCP protein was generated as previously described.18 Briefly, plasmid (pST239) containing N-terminal His(6) and SUMO followed by a TEV protease cleavage site was transformed into the 44H9 strain (Genentech).18 A 20 mL overnight culture was grown at 30 °C in Luria broth (LB) medium with 50 μg/mL carbenicillin. For overexpression, 1 L of culture of soy C.R.A.P. phosphate limiting media [3.57 g of (NH4)2SO4, 0.71 g of Na citrate–2xH2O, 1.07 g of KCl, 5.36 g of yeast extract (certified), 5.36 g of HycaseSF-Sheffield, pH adjusted with KOH to 7.3, volume adjusted to 872 mL with deionized water and supplemented with 110 mL of 1 M MOPS, pH 7.3, 11 mL of 50% glucose, 7 mL of 1 M MgSO4] with 50 μg/mL carbenicillin was inoculated with 20 mL of the overnight culture and grown for additional 24 h at 30 °C. Cells were then harvested by centrifugation at 6000 rpm for 15 min and stored as a pellet at −80 °C until purification. For purification of the fusion protein, the cell pellet was thawed on ice and resuspended in 5 times its volume of lysis buffer consisting of 50 mM Tris, pH 8.0, 300 mM NaCl, 5 mM imidazole, 5% glycerol, 0.1% Triton X-100 and cOmplete, EDTA-free protease inhibitor cocktail (Roche, 1 tablet/50 mL buffer). The resuspended pellet was lyzed by a microfluidizer (SC Hydraulic Engineering Corp) for four cycles on ice. The lysed pellet was centrifuged at 18,000 rpm for 30 min to remove all debris. The resulting supernatant was then purified using a HiTrap FF column (Cytiva) on an AKTA Pure system (Cytiva) with a linear gradient of 20 to 500 mM imidazole in 50 mM Tris, pH 8.0, and 300 mM NaCl. Purified protein was quantitated using a Nanodrop spectrophotometer (Thermo Fisher) and analyzed with SDS-PAGE. For TEV protease digestion, the purified protein was incubated with the TEV protease at a ratio of 50:1 (w/w) overnight at room temperature in the presence of 0.5 mM EDTA. The products were then analyzed using an analytical LC–MS (Agilent 1200 series) equipped with a PLRP-S column (Agilent, PL1912-1802), 6224 time-of-flight mass spectrometer (TOF-MS), diode array detection (DAD) detector, with a flow rate of 0.5 mL/min and a linear gradient of 15–60% aqueous acetonitrile containing 0.05% TFA over 6 min. The final HPLC purification was performed by preparative RP-HPLC (Agilent 1200 series), equipped with a Kinetex EVO C18 column (Phenomenex, 00D-4633-P0-AX), with a flow rate of 20 mL/min and a linear gradient of 15–60% aqueous acetonitrile containing 0.1% trifluoroacetic acid (TFA) over 35 min.
Surface Plasmon Resonance
SPR measurements were carried out on a Biacore S200 instrument (GE Healthcare) with PBS-T (PBS with 0.05% Tween 20, pH 7.4) as the assay buffer as described previously.11,40 For kinetic measurements, biotinylated ZNRF3 was captured on three sensor channels of a Series S SA Sensor chip (GE Healthcare). The sensor was then blocked with 0.1 mg/mL of biotin. 3× serial dilutions of the DCP were injected to the chip. Data were double-referenced by subtracting the signal from a control sensor channel (coated with biotin only) and the signal from a buffer injection. Binding data were fitted using the manufacturer’s software.
Wnt Reporter Assay
The assay was performed as previously described.12 Briefly, HEK293 cells with stably integrated firefly luciferase-based Wnt reporter (TOPbrite) and pRL-SV40 Renilla luciferase (Promega) were used for the Wnt reporter assays. 20,000 cells/well were seeded in each well of a clear-bottom white polystyrene 96-well plate (Corning, cat no. 3903) and incubated for 24 h. The next day, cells were stimulated with 50 μg/mL recombinant human Wnt3a protein (R&D systems, 5036-WN) in the absence or presence of DMSO-based DCPs. After 6 h of incubation, cells were subjected to the Dual-Luciferase Reporter Assay (Promega, no. E2920) according to the manufacturer’s instructions. Luciferase signals were measured on a PerkinElmer EnVision multilabel reader and the ratio of firefly luminescence to Renilla luminescence was calculated and normalization to appropriate controls (DMSO or Wnt3A + DMSO) was followed, if necessary. Graphs of normalized RLU were plotted with GraphPad Prism 9.
Serine/Threonine Ligation
The SAL linker synthesis and STL methods were based on previously established protocols.26,28 To synthesize the SAL linker, 1 equiv of acid linker (as either mono, di-, tri-, or tetra-acid) was added to a vial. 1.1–2 equiv of DCC and DMAP per number of acid groups was added to the vial and the reagents were dissolved in DMF. After a few minutes of stirring, 1.1–2 equiv of salicylaldehyde per number of acid groups was then added to the reaction. The Starting linker concentration was between 50 and 150 mM. Reactions were monitored by LC–MS until completion (typically within a few hours) and then diluted with water/acetonitrile and purified by preparative RP-HPLC. The purity of the collected fractions was assessed by LC–MS, with the pure fractions combined and lyophilized.
For multimerization, 1 equiv of SAL linker (as either di-, tri-, or tetra-ester) and 1–1.25 equiv of MK1-3.6.10S per salicylaldehyde ester functional group were added to a 1.5 mL centrifuge tube. For biotinylation, 2 equiv of biotin-PEG2 or biotin-PEG12 SAL ester were added per equiv of MK1-3.6.10S.
A solution of 1:1 pyridine/acetic acid was added to make the final concentration of MK1-3.6.10S 5 mM. The tube was vortexed for 30 s, briefly spun down, and then placed on a shaker at room temperature. The reactions were monitored by LC–MS and typically completed within 6 h. The solution was dried by a stream of nitrogen gas and then dissolved in 250 μL of 95:5 TFA/water. The acidolysis reaction went on for ∼15 min, was subsequently diluted with water/acetonitrile, and purified by preparative RP-HPLC. The purity of the collected fractions was assessed by LC–MS, with the pure fractions combined and lyophilized. Typical yields for STL after HPLC purification were 30–60%.
Cell Culture
HEK293 cells were grown in high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and 2 mM GlutaMAX (Gibco). HEK293 luciferase reporter cells were maintained in DMEM with a nutrient mixture F12 (50:50), 10% (v/v) FBS, 2 mM GlutaMAX (Gibco), and 40 μg/mL hygromycin (cellgro). All cells were incubated in a 5% CO2 humidified incubator at 37 °C for 24 h before experiments.
Immunofluorescence
HEK293 cells (transiently expressing HA-tagged ZNRF3) were fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature, washed 3 times with PBS, and permeabilized with 1% Triton X-100 in PBS for 5 min at room temperature. Cells were washed again and incubated with 20 μg/mL biotinylated MK1-3.6.10S or an anti-HA antibody (Cell Signaling, 2367S) for 1 h at room temperature, as indicated. Cells were then washed and incubated with Streptavidin-Alexa Fluor 488 (Thermo Fisher, S11223) or Alexa Fluor 647 secondary antibody (Thermo Fisher, A21235) for 30 min at room temperature. To stain the nuclei, cells were incubated with Hoechst 33342 (5 μg/mL in PBS) for 10 min, washed 3 times with PBS, and then stored in 100 μL of PBS in the dark until image acquisition. Fluorescence images were captured on a high throughput ImageXpress Confocal HT.ai imaging system (Molecular Devices) and images were analyzed by MetaXpress 6.7.41,42
Transglutaminase Ligation to XTEN
The XTEN 80 kDa scaffold containing 8X Q-tags (XTEN80-8Q) in evenly spaced loop regions was produced by E. coli expression. The E. coli cell pellet was resuspended in a buffer containing 50 mM citrate, pH 4.0 (30% w/v), and incubated at 4 °C overnight. After heat lysis, the sample was centrifuged at 23,000 RCF for 30 min. The supernatant was collected and clarified by the addition of 0.1% w/v Celite and incubation at 4 °C with gentle mixing for 1 h. The supernatant was then filtered through a 0.2 μm filter and purified on a Ni-NTA column. The purified protein was digested by trypsin, and XTENs were further purified by using a Capto-Q Impres anion exchange column (GE Healthcare). To perform TG-medicated XTEN-DCP ligation, the reagents were mixed in a 1.5 mL centrifuge tube to achieve the following final concentrations: [XTEN80-8Q] = 24 μM; [Ktag-MK1-3.6.10] = 2000 μM; [TG] = 0.1 μM; 100 mM Tris at pH 8.0. The ligation reaction mixture was placed on a heated shaker overnight set at 30 °C and 500 rpm. The reaction mixture was assessed by LC–MS (negative mode) and then purified by size exclusion chromatography (Superdex 200 10/300 GL, GE Healthcare). Individual fractions were analyzed by LC–MS. The most pure fractions were combined and then concentrated using an Amicon Ultra 30k MWCO (Millipore).
Evaluation of AzF Incorporation Efficiency (on GFP) in 44H9 Cells
Plasmid pAzF was constructed via NEBuilder HiFi DNA assembly (NEB) of the synthetic gene encoding the orthogonal tRNA and tRNA synthetase33 into the pBAD33 plasmid (ATCC). Wild-type GFP was similarly cloned into the pST239 vector by using NEBuilder. Point mutations on GFP were introduced via Q5 site-directed mutagenesis (NEB). 44H9 cells were made chemically competent, transformed with pAzF, and grown in 12.5 μg/mL chloramphenicol media. The resulting strain (44H9-pAzF) was again made chemically competent, transformed with either wild-type or mutant GFP pST239 plasmids, and selected over plates containing 25 μg/mL carbenicillin and 12.5 μg/mL chloramphenicol. Single colonies were picked and grown in LB with 25 μg/mL carbenicillin and 12.5 μg/mL chloramphenicol overnight at 37 °C. A 50 μL portion of the saturated overnight culture was transferred into 500 μL of soy C.R.A.P. medium with 12.5 μg/mL chloramphenicol, 25 μg/mL carbenicillin, 0.1% arabinose, and the indicated amount of p-azidophenylalanine. The cultures were then grown at 20 °C for 24 h. GFP yield was determined by measuring fluorescence (excitation 488 nm/emission 507 nm) and normalizing to cell density (OD 600).
Expression and Purification of Azidophenylalanine-Containing DCPs
Amber codon mutants were prepared via Q5 site-directed mutagenesis (NEB) of the MK1-3.6.10 expression plasmid. Expression and purification procedures are similar to the protocols described in the Materials and Methods section, with the addition of 0.1% arabinose and 5 mM AzF in the final expression media. Exposure to light was minimized to prevent potential photodegradation of the AzF amino acid.
Click Chemistry Functionalization of AzF-Containing DCPs
A freshly prepared mixture of copper sulfate pentahydrate (2 equiv) and tris-hydroxypropyltriazolylmethylamine (THPTA) (4 equiv) was added to a solution of propargylamine (1.2 equiv) or bis-PEG5-propargyl linker (0.45 equiv). Sodium ascorbate (10 equiv) was added to this reagent cocktail, followed by the addition of the purified azido-MK1-3.6.10 monomer (1 mM final peptide concentration, 1 equiv) in 20 mM phosphate, pH 7.5. The reaction was monitored for completion by analytical LC–MS after 30–60 min. The reaction was diluted in water before being subjected to preparative RP-HPLC. Fractions containing the desired product were pooled and lyophilized.
Expression and Purification of Isotope-Labeled DCPs
One Shot BL21 Star (DE3) strain (Thermo Fisher, C601003) was transformed with 100 ng of the plasmid encoding His(6)-SUMO-tagged DCP. 25 mL overnight culture in M9 medium was grown at 37 °C. Next day, 1 L of M9 medium [1× M9 salt (Genentech), 2 mM MgSO4, 1 mL of 100× BME vitamin (Sigma B6891), 1 g of 15NH4Cl (Cambridge Isotope Laboratories, NLM-467-25), 3 g of d-glucose (for 15N labeling, Sigma G7528) or d-glucose, 13C6 (for 15N/13C labeling, Cambridge Isotope Laboratories, CLM-1396-25), 1 g of Celtone base 15N (Cambridge Isotope Laboratories, CGM-1030P-N-1), 1 μM of CaCl2 and 50 μg/mL carbenicillin] was inoculated with 25 mL of the overnight culture and grown at 37 °C until OD reached between 0.6 and 0.8. Then, 0.3 mM IPTG (final concentration) was added to the culture to induce the protein expression. The culture was allowed to grow at 16 °C overnight and was harvested by centrifugation. Purification and tag cleavage were followed, as described above.
NMR Analysis with 15N/13C Labeled MK1-3.6.10
15N/13C-labeled MK1-3.6.10 was reconstituted at 250 μM (as quantified by UV absorbance) in HEPES buffer, pH 7.2. NMR experiments were performed on either a 500 MHz Bruker Avance-III spectrometer equipped with a QNP cryoprobe (all experiments from the standard TopSpin 3.6.4 library) or a 600 MHz Avance-III spectrometer equipped with a TCI HCN cryoprobe (TopSpin 3.6.0). NMR experiments were performed at 298 K. For assignment of backbone atoms, HNCO, HNCA, HNcaCO, HNcoCA, HNCACB, and CBCAcoNH spectra were performed on the 600 MHz instrument with NUS compression to 25% (schedule generated using TopSpin), for a cumulative experiment time of 17 h. Additionally, in order to assign side chains, an HCCH-TOCSY and an HcccoNNH-TOCSY were run on the 600 MHz instrument, with the latter using NUS compression of 50%. As potential supplements for assignment purposes, a 13C-NOESY-HSQC was acquired on the 600 MHz instrument, and HNHA, 15N-DIPSI2, and 15N-NOESY-HSQC were acquired on the 500 MHz spectrometer. Spectra were analyzed with NMRViewJ.43 Peak lists were obtained by using automated functions in NMRViewJ and edited manually to remove noise signals and duplicate peaks. Manual backbone assignments were performed using the RunAbout tool within NMRViewJ. Side-chain assignments were completed manually. Minor states were assigned using 15N-NOESY and 15N-DIPSI2. Automated resonance assignment was achieved with the FLYA module of CYANA 3.98.1.44 Tolerances for aligning signals were 0.03 ppm in 1H and 0.4 ppm for both 13C and 15N. The FLYA routine was run within CYANA using the manually assigned assignment list as a chemical shift reference to determine accuracy of various data set combinations. A total of 20 FLYA runs were performed with CYANA to consolidate the cumulative results to the final assignment. The results were then compared to the manually determined assignments to evaluate FLYA ability to correctly identify backbone and side chain resonances. When noted, an AlphaFold245 structural model of MK1-3.6.10 was used to generate expected peaks during the FLYA routine.
Acknowledgments
We thank Nataliya Popovych for providing technical support for bioproduction of isotope-labeled DCPs.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.4c00026.
Generation of the head-to-head dimer di-MK1-3.6.10S, characterization and application of biotinylated MK1-3.6.10S and MK1-3.6.10-XTEN80-8, incorporation of unnatural amino acids into DCPs via amber codon suppression, and comparison of chemical shift assignments for [13C,15N]- and [15N]-labeled MK1-3.6.10 and LC–MS analysis for all the peptides generated in the manuscript (PDF)
Author Contributions
∥ S.H. and A.T.B. contributed equally to this work. CRediT: Sunhee Hwang data curation, formal analysis, investigation, methodology, validation, visualization, writing-review & editing; Aaron T Balana data curation, formal analysis, investigation, methodology, validation, visualization, writing-review & editing; Bryan Martin formal analysis, investigation, methodology, writing-review & editing; Michael Clarkson formal analysis, investigation, methodology, writing-review & editing; Paola Di Lello formal analysis, investigation, methodology, writing-review & editing; Hao Wu formal analysis, investigation, methodology, writing-review & editing; Yanjie Li methodology, writing-review & editing; Jakob Fuhrmann methodology, writing-review & editing; Yavuz Dagdas formal analysis, investigation, methodology, writing-review & editing; Patrick Holder formal analysis, investigation, methodology, writing-review & editing; Christina I Schroeder formal analysis, methodology, visualization, writing-review & editing; Stephen E Miller data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing-review & editing; Xinxin Gao conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing-original draft, writing-review & editing.
The authors declare no competing financial interest.
Notes
The TOC figure was created using Biorender (http://biorender.com).
Supplementary Material
References
- Ho T. N. T.; Turner A.; Pham S. H.; Nguyen H. T.; Nguyen L. T. T.; Nguyen L. T.; Dang T. T. Cysteine-rich peptides: From bioactivity to bioinsecticide applications. Toxicon 2023, 230, 107173. 10.1016/j.toxicon.2023.107173. [DOI] [PubMed] [Google Scholar]
- Tammineni R.; Gulati P.; Kumar S.; Mohanty A. An overview of acyclotides: Past, present and future. Phytochemistry 2020, 170, 112215. 10.1016/j.phytochem.2019.112215. [DOI] [PubMed] [Google Scholar]
- Tyler T. J.; Durek T.; Craik D. J. Native and Engineered Cyclic Disulfide-Rich Peptides as Drug Leads. Molecules 2023, 28 (7), 3189. 10.3390/molecules28073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza C.; Henriques S. T.; Wang C. K.; Cheneval O.; Chan L. Y.; Bokil N. J.; Sweet M. J.; Craik D. J. Using the MCoTI-II Cyclotide Scaffold To Design a Stable Cyclic Peptide Antagonist of SET, a Protein Overexpressed in Human Cancer. Biochemistry 2016, 55 (2), 396–405. 10.1021/acs.biochem.5b00529. [DOI] [PubMed] [Google Scholar]
- Hilpert K.; Wessner H.; Schneider-Mergener J.; Welfle K.; Misselwitz R.; Welfle H.; Hocke A. C.; Hippenstiel S.; Höhne W. Design and characterization of a hybrid miniprotein that specifically inhibits porcine pancreatic elastase. J. Biol. Chem. 2003, 278 (27), 24986–24993. 10.1074/jbc.M212152200. [DOI] [PubMed] [Google Scholar]
- Kimura R. H.; Jones D. S.; Jiang L.; Miao Z.; Cheng Z.; Cochran J. R. Functional mutation of multiple solvent exposed loops in the Ecballium elaterium trypsin inhibitor-II cystine knot miniprotein. PLoS One 2011, 6 (2), e16112 10.1371/journal.pone.0016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R. H.; Levin A. M.; Cochran F. V.; Cochran J. R. Engineered cystine knot peptides that bind αvβ3, αvβ5, and α5β1 integrins with low-nanomolar affinity. Proteins 2009, 77 (2), 359–369. 10.1002/prot.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause S.; Schmoldt H.-U.; Wentzel A.; Ballmaier M.; Friedrich K.; Kolmar H. Grafting of thrombopoietin-mimetic peptides into cystine knot miniproteins yields high-affinity thrombopoietin antagonists and agonists. FEBS J. 2007, 274 (1), 86–95. 10.1111/j.1742-4658.2006.05567.x. [DOI] [PubMed] [Google Scholar]
- Garcia A. E.; Camarero J. A. Biological activities of natural and engineered cyclotides, a novel molecular scaffold for peptide-based therapeutics. Curr. Mol. Pharmacol. 2010, 3 (3), 153–163. 10.2174/1874-470211003030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeur E.; Gueret S. M.; Adihou H.; Gopalakrishnan R.; Lemurell M.; Waldmann H.; Grossmann T. N.; Plowright A. T. New Modalities for Challenging Targets in Drug Discovery. Angew. Chem., Int. Ed. Engl. 2017, 56 (35), 10294–10323. 10.1002/anie.201611914. [DOI] [PubMed] [Google Scholar]
- Hansen S.; Zhang Y.; Hwang S.; Nabhan A.; Li W.; Fuhrmann J.; Kschonsak Y.; Zhou L.; Nile A. H.; Gao X.; et al. Directed evolution identifies high-affinity cystine-knot peptide agonists and antagonists of Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. U.S.A. 2022, 119 (46), e2207327119 10.1073/pnas.2207327119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kschonsak Y. T.; Gao X.; Miller S. E.; Hwang S.; Marei H.; Wu P.; Li Y.; Ruiz K.; Dorighi K.; Holokai L.; et al. Potent and selective binders of the E3 ubiquitin ligase ZNRF3 stimulate Wnt signaling and intestinal organoid growth. Cell Chem. Biol. 2024, 31, 1176–1187.e10. 10.1016/j.chembiol.2023.11.006. [DOI] [PubMed] [Google Scholar]
- Thakur A. K.; Miller S. E.; Liau N. P. D.; Hwang S.; Hansen S.; de Sousa E. M. F.; Sudhamsu J.; Hannoush R. N. Synthetic Multivalent Disulfide-Constrained Peptide Agonists Potentiate Wnt1/beta-Catenin Signaling via LRP6 Coreceptor Clustering. ACS Chem. Biol. 2023, 18 (4), 772–784. 10.1021/acschembio.2c00753. [DOI] [PubMed] [Google Scholar]
- Amblard M.; Fehrentz J. A.; Martinez J.; Subra G. Methods and protocols of modern solid phase Peptide synthesis. Mol. Biotechnol. 2006, 33 (3), 239–254. 10.1385/MB:33:3:239. [DOI] [PubMed] [Google Scholar]
- Behrendt R.; White P.; Offer J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22 (1), 4–27. 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidro-Llobet A.; Kenworthy M. N.; Mukherjee S.; Kopach M. E.; Wegner K.; Gallou F.; Smith A. G.; Roschangar F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84 (8), 4615–4628. 10.1021/acs.joc.8b03001. [DOI] [PubMed] [Google Scholar]
- Martin V.; Egelund P. H. G.; Johansson H.; Le Quement S. T.; Wojcik F.; Pedersen D. S. Greening the synthesis of peptide therapeutics: an industrial perspective. RSC Adv. 2020, 10 (69), 42457–42492. 10.1039/d0ra07204d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Kaluarachchi H.; Zhang Y.; Hwang S.; Hannoush R. N. A phage-displayed disulfide constrained peptide discovery platform yields novel human plasma protein binders. PLoS One 2024, 19 (3), e0299804 10.1371/journal.pone.0299804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klint J. K.; Senff S.; Saez N. J.; Seshadri R.; Lau H. Y.; Bende N. S.; Undheim E. A.; Rash L. D.; Mobli M.; King G. F. Production of recombinant disulfide-rich venom peptides for structural and functional analysis via expression in the periplasm of E. coli. PLoS One 2013, 8 (5), e63865 10.1371/journal.pone.0063865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap K.; Du J. Q.; Looi F. Y.; Tang S. R.; de Veer S. J.; Bony A. R.; Rehm F. B. H.; Xie J.; Chan L. Y.; Wang C. K.; et al. An environmentally sustainable biomimetic production of cyclic disulfide-rich peptides. Green Chem. 2020, 22 (15), 5002–5016. 10.1039/D0GC01366H. [DOI] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem., Int. Ed. 2002, 41 (14), 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Wang Q.; Chan T. R.; Hilgraf R.; Fokin V. V.; Sharpless K. B.; Finn M. G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125 (11), 3192–3193. 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- Shandell M. A.; Tan Z.; Cornish V. W. Genetic Code Expansion: A Brief History and Perspective. Biochemistry 2021, 60 (46), 3455–3469. 10.1021/acs.biochem.1c00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wals K.; Ovaa H. Unnatural amino acid incorporation in E. coli: current and future applications in the design of therapeutic proteins. Front. Chem. 2014, 2, 15. 10.3389/fchem.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassagnon I. R.; McCarthy C. A.; Chin Y. K. Y.; Pineda S. S.; Keramidas A.; Mobli M.; Pham V.; De Silva T. M.; Lynch J. W.; Widdop R. E.; et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. U.S.A. 2017, 114 (14), 3750–3755. 10.1073/pnas.1614728114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine P. M.; Craven T. W.; Li X.; Balana A. T.; Bird G. H.; Godes M.; Salveson P. J.; Erickson P. W.; Lamb M.; Ahlrichs M.; et al. Generation of Potent and Stable GLP-1 Analogues Via “Serine Ligation”. ACS Chem. Biol. 2022, 17 (4), 804–809. 10.1021/acschembio.2c00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. C.; Lam H. Y.; Zhang Y. F.; Chan C. K. Salicylaldehyde Ester-Induced Chemoselective Peptide Ligations: Enabling Generation of Natural Peptidic Linkages at the Serine/Threonine Sites. Org. Lett. 2010, 12 (8), 1724–1727. 10.1021/ol1003109. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Xu C.; Lam H. Y.; Lee C. L.; Li X. Protein chemical synthesis by serine and threonine ligation. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (17), 6657–6662. 10.1073/pnas.1221012110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlas J.; Nuijens T.; Persson J.; Svensson T.; Schmidt M.; Toplak A.; Nilsson M.; Rasmussen J. H. Sustainable, cost-efficient manufacturing of therapeutic peptides using chemo-enzymatic peptide synthesis (CEPS). Green Chem. 2019, 21 (23), 6451–6467. 10.1039/C9GC03600H. [DOI] [Google Scholar]
- Schneider H.; Deweid L.; Avrutina O.; Kolmar H. Recent progress in transglutaminase-mediated assembly of antibody-drug conjugates. Anal. Biochem. 2020, 595, 113615. 10.1016/j.ab.2020.113615. [DOI] [PubMed] [Google Scholar]
- Walsh S. J.; Bargh J. D.; Dannheim F. M.; Hanby A. R.; Seki H.; Counsell A. J.; Ou X. X.; Fowler E.; Ashman N.; Takada Y.; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev. 2021, 50 (2), 1305–1353. 10.1039/D0CS00310G. [DOI] [PubMed] [Google Scholar]
- Ding S.; Song M.; Sim B. C.; Gu C.; Podust V. N.; Wang C. W.; McLaughlin B.; Shah T. P.; Lax R.; Gast R.; et al. Multivalent Antiviral XTEN-Peptide Conjugates with Long in Vivo Half-Life and Enhanced Solubility. Bioconjugate Chem. 2014, 25 (7), 1351–1359. 10.1021/bc500215m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J. W.; Santoro S. W.; Martin A. B.; King D. S.; Wang L.; Schultz P. G. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 2002, 124 (31), 9026–9027. 10.1021/ja027007w. [DOI] [PubMed] [Google Scholar]
- Gundinger T.; Kittler S.; Kubicek S.; Kopp J.; Spadiut O. Recombinant Protein Production in E. coli Using the phoA Expression System. Fermentation 2022, 8 (4), 181. 10.3390/fermentation8040181. [DOI] [Google Scholar]
- Luo M.; Zhao M.; Cagliero C.; Jiang H.; Xie Y.; Zhu J.; Yang H.; Zhang M.; Zheng Y.; Yuan Y.; et al. A general platform for efficient extracellular expression and purification of Fab from Escherichia coli. Appl. Microbiol. Biotechnol. 2019, 103 (8), 3341–3353. 10.1007/s00253-019-09745-8. [DOI] [PubMed] [Google Scholar]
- Frederick R. O.; Haruta M.; Tonelli M.; Lee W.; Cornilescu G.; Cornilescu C. C.; Sussman M. R.; Markley J. L. Function and solution structure of the Arabidopsis thaliana RALF8 peptide. Protein Sci. 2019, 28 (6), 1115–1126. 10.1002/pro.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher I.; Chang S. C.; Li Y.; Chhabra S.; Palmer A. G.; Norton R. S.; Chill J. H. Conformational flexibility in the binding surface of the potassium channel blocker ShK. Chembiochem 2014, 15 (16), 2402–2410. 10.1002/cbic.201402295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C. H. Y.; King G. F.; Mobli M. Molecular basis of the interaction between gating modifier spider toxins and the voltage sensor of voltage-gated ion channels. Sci. Rep. 2016, 6, 34333. 10.1038/srep34333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A. H.; Edwards I. A.; Mishra B. P.; Sharma G.; Healy M. D.; Elliott A.; Blaskovich M. A. T.; Cooper M. A.; Collins B. M.; Jia X. Y.; Mobli M. Elucidating the Lipid Binding Properties of Membrane-Active Peptides Using Cyclised Nanodiscs. Front. Chem. 2019, 7, 238. 10.3389/fchem.2019.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Maziere A.; Beard R.; Klumperman J.; Hannoush R. N. Fatty acylation enhances the cellular internalization and cytosolic distribution of a cystine-knot peptide. iScience 2021, 24 (11), 103220. 10.1016/j.isci.2021.103220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; De Maziere A.; Iaea D. B.; Arthur C. P.; Klumperman J.; Ciferri C.; Hannoush R. N. Visualizing the cellular route of entry of a cystine-knot peptide with Xfect transfection reagent by electron microscopy. Sci. Rep. 2019, 9, 6907. 10.1038/s41598-019-43285-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Stanger K.; Kaluarachchi H.; Maurer T.; Ciepla P.; Chalouni C.; Franke Y.; Hannoush R. N. Cellular uptake of a cystine-knot peptide and modulation of its intracellular trafficking. Sci. Rep. 2016, 6, 35179. 10.1038/srep35179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. A. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004, 278, 313–352. 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- Schmidt E.; Guntert P. A new algorithm for reliable and general NMR resonance assignment. J. Am. Chem. Soc. 2012, 134 (30), 12817–12829. 10.1021/ja305091n. [DOI] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Zidek A.; Potapenko A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.