

Abstract
Discovery and development of small molecule therapeutics effective against Gram-negative pathogens is a highly challenging task. Most compounds active in biochemical settings fail to exhibit whole cell activity. The major reason for this lack of activity is the effectiveness of bacterial cell envelopes as permeability barriers. These barriers originate from the nutrient-selective outer membranes, which act synergistically with polyspecific efflux pumps. Guiding principles to enable rational optimization of small molecules for efficient penetration and intracellular accumulation in Gram-negative bacteria would have a transformative impact on the discovery and design of chemical probes and therapeutics. In this perspective, we draw on inspiration from traditional medicinal chemistry approaches for eukaryotic drug design to present a broader call for action in developing comparable approaches for Gram-negative bacteria.
Graphical Abstract

1. INTRODUCTION
Antibiotic resistance is a major threat to public health. In 2019, 1.27 million deaths were attributed to bacterial resistance, and multidrug-resistant (MDR) bacteria are predicted to give rise to 10 million deaths annually by 2050.1 Significant efforts are devoted to the discovery and optimization of new antibiotics and several new antibiotics and antibiotic/adjuvant combinations have recently been approved for clinical use.2–4 However, the emergence of resistance is an inevitable fate of each new antibiotic introduced into the clinic.5 Resistance emergence currently outpaces our ability to discover new chemotypes with whole cell activity, especially against Gram-negative bacteria (GNB) which represent the majority of the pathogens listed as public health threats by the Centers for Disease Control and Prevention (CDC).6–8 New strategies are needed to enable the rational design of molecules capable of infiltrating GNB. This is critical to maintaining and expanding the pipeline of clinical antibiotics in a more reliable fashion.9
Although the identification of new hits from biochemical or computational screening campaigns is trivial, identified molecules are typically not active against the most challenging Gram-negative pathogens, such as MDR Pseudomonas aeruginosa, Acinetobacter baumannii, and Enterobacteriaceae. The major contributor to the antibiotic resistance of these bacteria is the synergistic nature of the protective cell envelope components, comprised of chemically disparate outer (OM) and inner (IM) membranes, and promiscuous efflux pumps.10 In particular, the OM, composed of the asymmetric layers of lipopolysaccharides (LPS) and phospholipids embedded with porins, restricts penetration of large and hydrophobic compounds. Conversely, hydrophilic compounds are limited by the IM.11 The membranes are further fortified by efflux transporters acting across both the OM and IM.10 The synergistic interactions between these small molecule exclusion barriers make the bacterial cell essentially impermeable to most drug-like chemotypes.12 The challenge is further complicated in that evolutionary divergence between species and within strains leads to unparalleled complexity.13
Guiding principles and molecular design approaches have been instrumental in developing drug leads for intracellular eukaryotic targets. If similar guidelines could be developed for Gram-negative targeted molecules, the impact would revolutionize probe development and antibacterial drug discovery. Recent efforts to elucidate molecular architectures that affect efflux avoidance and OM permeation provide proof of concept that the development of guiding principles for GNB small molecule permeation and accumulation is feasible. Evolving research demonstrates that while some observations are chemotype- and/or species-specific, other features may be more broadly applicable across various molecular scaffolds and/or pathogens.
1.1. Current State and Challenges in Rational Antibacterial Design
Despite the growing threat to public health, most large pharmaceutical companies have abandoned antibacterial discovery efforts. Among many financial reasons for the exodus, the extended time of development, delayed return-on-investment, high attrition, and rapid resistance development to novel antibiotics were likely the main drivers. The complete DNA sequencing of Haemophilus influenzae in 1995, however, provided new hope and a list of potential unexploited targets, thus rekindling interest within several companies.14 One of which was GlaxoSmithKline (GSK), which evaluated more than 350 genes through allelic-replacement mutagenesis, eventually identifying ~100 genes as essential.15 Despite the prospect of the new gene targets, only 5 leads were generated from 70 biochemical high-throughput screening (HTS) campaigns, all of which turned out to exhibit non-specificity towards eukaryotic and bacterial cells, or lacked activity in a suitable spectrum of Gram-positive or Gram-negative species. Unfortunately, this outcome is a common theme when target-based approaches are applied to Gram-negatives, as exemplified by other failed large scale screening attempts (AstraZeneca: 2M compounds, Merck: 3M compounds).16 While phenotypic screening is often better at identifying antibacterial compounds with whole-cell activity, the ability to identify hits significantly suffers and mechanisms of action are often unknown.
Retaining whole cell activity during hit-lead optimization has proven to be the major driver of attrition in GNB drug discovery. The efficiency of the bacterial membrane and efflux pump exclusion significantly limits our ability to engineer biochemical hits into whole cell active compounds. Four approaches are typically taken to enhance the whole cell accumulation of molecules in GNB: 1) cotreatment with OM permeabilizing agents, 2) cotreatment with efflux pump inhibitors (EPIs), 3) attachment of siderophores, or 4) strategic structural modification. Cotreatment with common permeabilizing agents such as polymyxin B nonapeptide (PMBN) can enhance OM permeation but fails to address active efflux and is often hampered by dose-limiting issues.17 EPIs, on the other hand, block active drug transport out of the cell, resulting in improved accumulation but fail to address membrane permeation issues.18 The use of siderophore-antibiotic conjugates exhibits great promise but has largely been limited to compounds with periplasmic drug targets like β-lactams.19 The fourth approach, which centers on rational structural engineering to enhance whole cell accumulation represents the pinnacle for medicinal chemistry but requires large scale studies across diverse chemotypes and organisms to unravel the interplay of bacterial physiology and small molecule permeation. Excitingly, the antibacterial community has begun to unravel and define the features of small molecules that influence membrane permeation and efflux susceptibility, providing compelling evidence that rational molecular design to enhance accumulation is feasible.
In this perspective, we frame Gram-negative antibacterial discovery in the context of traditional medicinal chemistry rules/approaches and expose the gaps where more research is required to enable the development of guiding principles for GNB penetrance and accumulation. We first discuss physicochemical descriptors and highlight parallels between classical guidelines to antibacterial approaches. We then unpack the differences in the structure-activity relationship workflow between eukaryotic targeted therapies and antibacterials. Finally, we conclude with a discussion on the integration of artificial intelligence in antibacterial drug discovery and highlight the prospect of this union with medicinal chemistry.
2. PHYSICOCHEMICAL DESCRIPTORS
Central to the problem is a limited understanding of the physicochemical properties that influence small-molecule accumulation. Physicochemical rules correlating to enhanced absorption and oral bioavailability have been established for eukaryotic targeted molecules since the early 2000s. The Lipinski’s rule of 5 (Ro5), for example, represents the first systematic attempt to correlate compound physicochemical properties to their success as late-stage leads and has been adopted in drug discovery as foundational guidelines.20,21 Since the development of the Lipinski rules, other guidelines have been developed such as Veber’s rules,22 trends for blood-brain barrier (BBB) penetrance,23 beyond the “rule of five” (BRo5),24 and Abbott Physicochemical Tiering (APT),25 demonstrating the influence that descriptor rules can play in small molecule pharmacological efficiency. Comparable guidelines for small molecule accumulation in bacteria, however, represent a significant gap in knowledge and the lack of progress likely arises from the complexity of Gram-negative barriers. Recent effort in this space has begun to clarify influential physicochemical parameters for small molecule permeation, providing evidence that trends can be identified.
2.1. Insights from Retrospective Analyses
2.1.1. Achaogen Pharmaceuticals.
In 2008, O’Shea and Moser recognized the architectural differences between eukaryotic and bacterial cells and conducted a comparative physicochemical property assessment between antibacterial and non-antibacterial drugs.26 The analysis further categorized compounds into Gram-positive and Gram-negative whole cell actives. In the study, 147 active antibacterial compounds were selected (approved drugs and those under clinical investigation) and organized according to the physicochemical profiles of each (average molecular weight (MW), calculated LogP (cLogP), calculated LogD (cLogD7.4), polar surface area (PSA), Hydrogen-bond donors (HBD), and Hydrogen-bond acceptors (HBA)). A significant difference in the physicochemical profiles of Gram-positive and Gram-negative actives was revealed. A ~2-fold higher average MW was observed in Gram-positive actives (Avg. MW = 813) over Gram-negative actives (Avg. MW = 414), exceptions being azithromycin (MW = 719 Da) and polymyxin B1 (MW = 1203 Da). The lipophilicity of the Gram-positive actives (cLogP = 2.1) tracked with the non-antibacterial reference group (cLogP = 2.7), while a significant increase in polarity was noted among the Gram-negative actives (cLogP = −0.1). The increased polarity of Gram-negative actives was also captured in cLogD7.4 (Avg. = −2.8) and relative PSA percentage (Avg. = 42%). The report notes that the physicochemical space required to achieve both Gram-negative activity (higher polarity) and oral bioavailability (sufficient lipophilicity to allow lipid membrane permeability) rarely overlaps. One notable exception to this dichotomy is zwitterionic compounds (e.g., fluoroquinolones), which often exhibit absorption from the gut and exploit porin-mediated uptake.27,28
2.1.2. AstraZeneca.
In attempts to understand the discrepancy between typical HTS libraries and whole cell active antibacterials, scientists at AstraZeneca compared and profiled the physical properties of ~3,200 compounds identified in antibacterial projects to hits from traditional HTS.29 In summary, this retrospective analysis determined that the properties of hits exhibited by typical HTS libraries were significantly “disconnected” from the chemical space represented by whole cell active antibacterials. Specifically, HTS libraries were found to be significantly more hydrophobic (2–4 log units higher) than the antibacterial actives. The attempts to optimize HTS hits to retain on-target activity while reducing lipophilicity, often failed, leading to the abandonment of projects. Even in scenarios for which biochemical activity could be improved with a reduction in hydrophobicity, whole cell antibacterial activity was still often lacking. The authors note that a significantly higher percentage of whole cell GNB actives from ongoing antibacterial projects contained a formal positive or negative charge compared to typical HTS libraries, which primarily consist of neutral cores. In addition to OM permeation, the study also considered properties of molecules that influence efflux susceptibility. The analysis revealed low efflux to be associated with small polar molecules and large zwitterionic compounds. Small polar molecules are hypothesized to capitalize on an increased influx rate due to porin-mediated uptake, which compensates for slower efflux processes.30,31 For large molecules, the authors speculate that intracellular ion-trapping and/or yet to be determined modes of active transport may be responsible for accumulation.32,33
2.2. Insights from Chemotype-focused Prospective Assessments
2.2.1. Sulfamoyl Adenosines.
Early physicochemical studies were limited to few antibiotic classes and primarily focused on the influences of hydrophobicity on accumulation. For example, early investigations in Escherichia coli demonstrated that more hydrophobic β-lactams showed decreased rates of diffusion.34 Similarly, quinolones with higher levels of hydrophobicity were shown to accumulate at lower levels in E. coli and P. aeruginosa.35 In response to the limited physicochemical information, Davis and Tan developed an integrated platform for the quantitative analysis of small-molecule permeability in bacteria.36 Principal component analysis (PCA) was initially used to determine nonobvious correlations between 20 structural and physicochemical properties with accumulation levels of 10 sulfamoyl adenosine (AMS) probes in E. coli. The top three accumulators, benzoyl-AMS (8), 4-phenylbenzoyl-AMS (9), and decanoyl-AMS (10), shown in Figure (1A), clustered with hydrophobicity parameters such as LogP (ALogPs) and LogD. Consistent with this, the parameters found in the opposite quadrant of the biplot were related to polarity, such as topological polar surface area (tPSA), relative polar surface area (relPSA), HBA and HBD, and aqueous solubility. Pearson pairwise correlation analysis was then used to provide a quantitative assessment of the impact of each parameter individually. Consistent with the PCA results, hydrophobicity showed significant positive correlation with accumulation in E. coli while polarity was significantly negatively correlated. The analysis further revealed ring content (the number of ring systems and aryl rings) and size (MW and van der Waals surface area) to positively correlate with accumulation. Other properties found to negatively correlate with accumulation in E. coli were ring complexity, heteroatom count (O and N), and three-dimensional topology (number of stereocenters, stereochemical density, and fraction of sp3 hybridization (Fsp3)) (Figure 1B). Similar analyses were done in the presence of EPIs to determine whether certain structural or physicochemical properties sensitize the probes to efflux pump activity. No strong trends were observed, however, hydrophobicity and rotatable bonds seemed to positively correlate with sensitivity to phenylalanine-arginine β-naphthylamide (PAβN), a well-studied EPI, while polarity was shown to correlate negatively.
Figure 1.

Proof-of-concept quantitative analysis of small-molecule permeability in E. coli. (A) AMS core scaffold and the three analogs with the greatest predicted accumulation in the multivariate PCA analysis. The hydrophobicity parameter ALogPs clustered with the accumulation control. (B) Pearson pairwise correlations between bacterial accumulation and structural/physicochemical properties in E. coli. Positive correlations are in green, negative correlations are in red.
2.2.2. Globularity and Rotatable bonds.
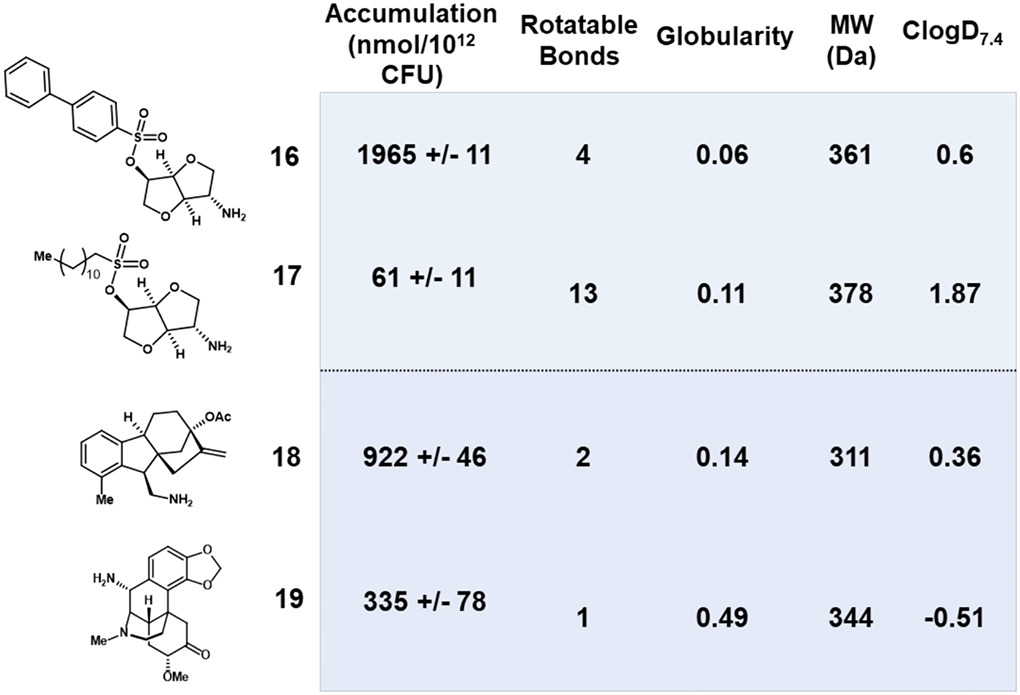
In a seminal study, the Hergenrother lab conducted a systematic analysis of the accumulation of an unbiased and structurally diverse library in E. coli.37 Compound accumulation was assessed in a whole cell context to ensure a more reliable measure of efficacy. Charge was found to be the primary factor dictating accumulation in E. coli. Specifically, inclusion of a positively charged primary amine elicited a significant improvement in accumulation over more sterically hindered amines or neutral and negatively charged molecules. However, accumulators were identified in the collection devoid of a primary amine, so an additional 297 molecular descriptors were profiled for each compound. Shape, best described as globularity (T), and flexibility, captured as the number of rotatable bonds (R), were determined to be important physicochemical factors. This was exemplified in a case study wherein amine 16 with four rotatable bonds accumulated well, whereas amine 17, with similar MW and globularity but significantly more rotatable bonds, exhibited >30-fold lower accumulation (Table 1). In another case study, the authors demonstrated that compound 18 accumulated ~3-fold better than the high globularity comparator (19), despite similar functional groups, MW, and rotatable bonds (Table 1). Together, these physicochemical properties that improve accumulation in E. coli were termed the eNTRy rules: unhindered primary amine (N), low three-dimensionality (T ≤0.25), and R ≤5. The eNTRy rules have now been applied to numerous scaffolds in various structural contexts, however, few present accumulation data so it is unknown whether increases in potency are due to enhanced permeability or binding affinity.38,39
Table 1.
Examples of globularity and rotatable bond influence on accumulation.
|
2.3. Perspective
Drug design for eukaryotic targets has benefited greatly from guiding principles centered on physicochemical properties. As mentioned, Lipinski’s rules of 5 (MW, HBD, HBA, logP) for oral bioavailability and good absorption, later expanded by Veber to include rotatable bonds and polar surface area, are widely applied. Similar guidelines exist for CNS active molecules which mimic Lipinski/Veber guidelines but require more stringency in each of the descriptors. One could imagine, more tailored guidelines for various organs or desired areas of distribution (e.g., eye, lungs, gut). While the desired descriptor ranges are framed as guidelines for bioavailability, implicit in this property is efficient permeation through eukaryotic cell membranes. A key limitation exists for antibacterials active against Gram-negative pathogens, as these molecules ultimately face an additional barrier prior to target engagement, the chemically and biologically complex bacterial cell envelope. However, the medicinal chemistry community can certainly use existing paradigms in eukaryotic drug discovery as inspiration to define cell permeation and efflux avoidance properties, in hopes of elucidating new Gram-negative centric guidelines. Several points of consideration for further pursuit of defining physicochemical descriptors for GNB permeation and accumulation follow.
2.3.1. Context matters.
The retrospective studies by Achoagen26 and AstraZeneca29 have illuminated the core problems with traditional hit-to-lead strategies and provided the first steps towards identifying physicochemical properties exhibited by Gram-negative actives as a whole. Observations made from these retrospective studies, however, are skewed toward existing antibiotic classes, potentially limiting insight to existing chemical space and thus trends may not be applicable to novel chemotypes. While physicochemical trends for Gram-negative actives were uncovered, there are many approved antibiotics that are overt exceptions to the observed trends. Of course, many of these exceptions result directly from disparate pharmacology (e.g., target location, mechanism of action, and uptake mechanism). When conducting physicochemical analyses, the mechanism of action, location of the target, chemical family, and mode of uptake (if known) should be properly classified as part of the assessment. For example, if the target is located in the periplasm, then parameters identified for actives would be indicative of OM permeation but would not include information regarding the ability of compounds to permeate both membranes. Differentiating the contributing factors of each barrier (i.e., OM, IM, and efflux) is critical to obtaining a more accurate picture of influencing properties. A significant limiting factor, however, is a current lack of subcellular accumulation assays. Development of methods to determine compound location within bacterial cells (e.g., periplasm, cytoplasm, membrane) would provide even more resolution in mapping compound properties for targets in specific cell compartments.
2.3.2. Bacterial diversity will likely limit broadly applicable trends.
Evidence is accumulating that different bacterial species, and even strains within a species, can vary drastically in the chemical composition and biological efficiency of their membrane, uptake, and efflux systems.13 For example, P. aeruginosa exhibits only one-eighth the permeability of E. coli.10 This is defined, in large part, by the repertoire of Resistance-Nodulation-Division (RND) efflux pumps and lack of general porins in P. aeruginosa. A glimpse into a third pathogen, A. baumannii, reveals the OM to play a minor role in small molecule permeation, while the overexpression of efflux pumps precludes most small molecules from accumulating.40,41 As such, physicochemical trends observed for permeation in one organism are not likely to be applicable to evolutionarily diverged organisms. For this reason, there is a significant need for species-specific analyses. Defining species-specific trends and cross-validating these trends over various chemotypes will more accurately define organism barrier characteristics, a feature not possible when grouping Gram-negatives together as a whole. With a growing interest in narrow-spectrum therapeutics, continued research in this area to define species-specific barriers in the context of small molecule permeation would be highly impactful to the field in general. Eventually, this level of analysis will enable cross-chemotype and -organism comparisons, revealing similarities and disparities between pathogens.
2.3.3. Property guidelines may find most use in hit prioritization rather than ligand optimization.
Ideally, a medicinal chemist could optimize the pharmacophore for target engagement and then focus on auxophore manipulation to install desired properties for accumulation. Scenarios that require pharmacophore alteration to manipulate whole molecule properties often lead to suboptimal on target-activity or other complications. As a rule, physicochemical descriptors are a characteristic of a molecule as a whole and thus it may be difficult to significantly improve certain physicochemical properties without sacrificing target interactions. Likewise, it has been shown that the OM and efflux pumps of an organism are influenced by distinct physicochemical properties. In fact, descriptors that improve efflux avoidance often correlate with significant liabilities for OM exclusion.42–44 This highlights the synergy of the OM and efflux, wherein optimizing a physicochemical profile for one barrier strengthens the effect of the other barrier. Another challenge with engineering a specific physicochemical profile into a scaffold of interest is that many descriptors are abstract or not readily applied from a design perspective. For example, lowering logD7.4 is a relatively straightforward objective that can likely be accomplished through incorporation of a single motif. Lowering the globularity of a compound, however, without interfering with established active site interactions is more challenging. The underlying need for practical and accessible physicochemical guidelines is rarely prioritized in physicochemical analysis and should be considered. This is not to say that properties like globularity are not useful, they certainly are. It is merely to suggest that properties with broader actionability should be prioritized. As such, while a tendency may be to include all possible descriptors in an assessment, one should consider the capability of chemists to act on a descriptor, with actionable properties being of highest utility. All told, physicochemical guidelines may find the most utility in prioritizing scaffolds post-screening for hit-to-lead optimization, with hits exhibiting optimal physicochemical profiles being prioritize higher, as the starting point is more desirable.
3. STRUCTURE UPTAKE RELATIONSHIPS
Motif driven approaches for molecular optimization remain the main strategies employed by medicinal chemists in eukaryotic targeted molecules. Well-established tools like the Topliss tree essentially provide medicinal chemists a roadmap for molecular optimization, in this case for a hit molecule possessing a phenyl group. Bioisosteric replacement, the most employed strategy in molecular design, provides a means for chemists to rationally substitute one functionality for another to enhance biological activity and ADME properties. In screening initiatives, much work has gone into characterizing pan-assay interference (PAINS) compounds and structural alerts to aid in hit prioritization. Reimagining these motif-centric strategies to provide comparable tools for Gram-negative probe and therapeutic optimization would be transformational. Recent work towards understanding how structural features/motifs influence OM penetration and/or efflux recognition has begun to point towards the feasibility of this goal.
3.1. Motif-enhanced Uptake
3.1.1. Ionizable Amine.
In the original eNTRy rule study, 6-deoxynybomycin (6DNM) was identified as a Gram-positive only antibacterial exhibiting good globularity (≤0.25) and ≤5 rotatable bonds.37 Installation of a primary amine resulted in a drastic shift in MIC from >32 μg/ml (6DNM) to 0.5–16 μg/ml in E.coli (6DNM-NH3) (Figure 2). In another example, primary amine installation was successfully utilized to re-design Debio-1452, a Gram-positive-only antibiotic into Debio-1452-NH3, a compound with activity against WT E. coli (MIC=4 μg/ml), E. cloacae (MIC=8 μg/ml), K. pneumoniae (MIC=8 μg/ml), and A. baumannii (MIC=4 μg/ml) (Figure 2).45 The enhancement in activity using this strategy is believed to be through porin-mediated uptake, provided by the complementarity of the positively charged amino group with the topology of general porins. The Hergenrother lab has continued to expand upon the application of the eNTRy rules, leading to the design of a flavin mononucleotide (FMN) riboswitch inhibitor with activity against WT E. coli (BW25113) (MIC=4 μg/ml), E. cloacae (ATCC BAA-2341) (MIC=4 μg/ml), and K. pneumoniae (ATCC 27736) (MIC=4 μg/ml)46 and have begun to pursue alternative positively charged functionalities such as guanidiniums and pyridiniums to enhance small molecule accumulation in E. coli.47
Figure 2.

Examples of increased activity with the addition of an ionizable amine.
Others have now also leveraged this approach, illustrating it promise as a cross-chemotype effect. Researchers at King’s College London implemented this strategy on 1,5-diphenyl-pyrroles, which were identified as a novel antibacterial class, represented by compound 2 (Figure 2).48 The original compounds were unable to permeate the OM without the assistance of PMBN (MIC>128 μg/ml). The addition of a protonatable amine (8w) or guanidine moiety (8x) showed improved activity against WT E. coli (NCTC 12923) (MIC=8 μg/ml), K. pneumoniae (M6) (MIC=16–32 μg/ml), and A. baumannii (MIC=8–32 μg/ml), (ATCC 17978) (Figure 2). Likewise, Achaogen began with a well-characterized biotin carboxylase (BC) inhibitor discovered in a HTS against an efflux pump-deficient strain of E. coli (tolC, imp).49 Aiming to increase the polarity of compound 1 (cLogD = 3.3), an ionizable amine was installed (14a), resulting in a 32-fold improvement in potency against WT E. coli (MIC=1 μg/ml), 8-fold against WT K. pneumoniae (MIC=8 μg/ml), and 16-fold against an efflux-compromised strain of P. aeruginosa (MIC=1 μg/ml) (Figure 2). Similarly, in an SAR study on 2,3,4,9-tetrahydro-1H-carbazoles, we determined that the presence of a primary amine is essential for efficient cell permeation, with IU-1 exhibiting superior accumulation and activity (EC50 = 24.8 μM) in comparison to the des-amino comparator IU-3 (EC50 >160 μM) (Figure 2).50 Notably, a derivative of this anti-virulence chemotype was recently reported to elicit in vivo efficacy in clearing uropathogenic E. coli in a murine model of infection.51
3.1.2. Oxazolidinone Structure-uptake relationships.
Due to the advancement in microbiological and analytical methods, critical information regarding permeation and accumulation can still be gained from well-interrogated compound families. One example is the oxazolidinone family that were rationally developed in the 1990s, producing multiple FDA approved compounds, most notably linezolid. Linezolid, however, exhibits only modest activity against fastidious Gram-negative respiratory tract pathogens, such as H. influenzae and Moraxella catarrhalis, and is primarily limited to the treatment of Gram-positive infections. The high conservation between Gram-positive and Gram-negative ribosomes, however, implies that if oxazolidinone analogs could accumulate and avoid efflux, this family would exhibit broad spectrum activity. We have recently leveraged the well-established oxazolidinone SAR trends to identify motif-driven enhancement in Gram-negative permeation and accumulation.52 Using a reagent clustering and selection approach, a chemically diverse library of oxazolidinones was synthesized and assessed for activity against three ESKAPE pathogens: E. coli, P. aeruginosa, and A. baumannii with varying levels of barrier compromise (i.e., efflux deletion and/or OM hyperporination). Orientation, location, and composition of functional groups were all determinants of chemotype accumulation. Broad spectrum activity was observed with two of the substituted pyridyl analogs 8d and 8o along with a substituted phenyl analog 3e (Figure 3). Notably, two of these compounds 8o and 3e include a primary ionizable amine.
Figure 3.
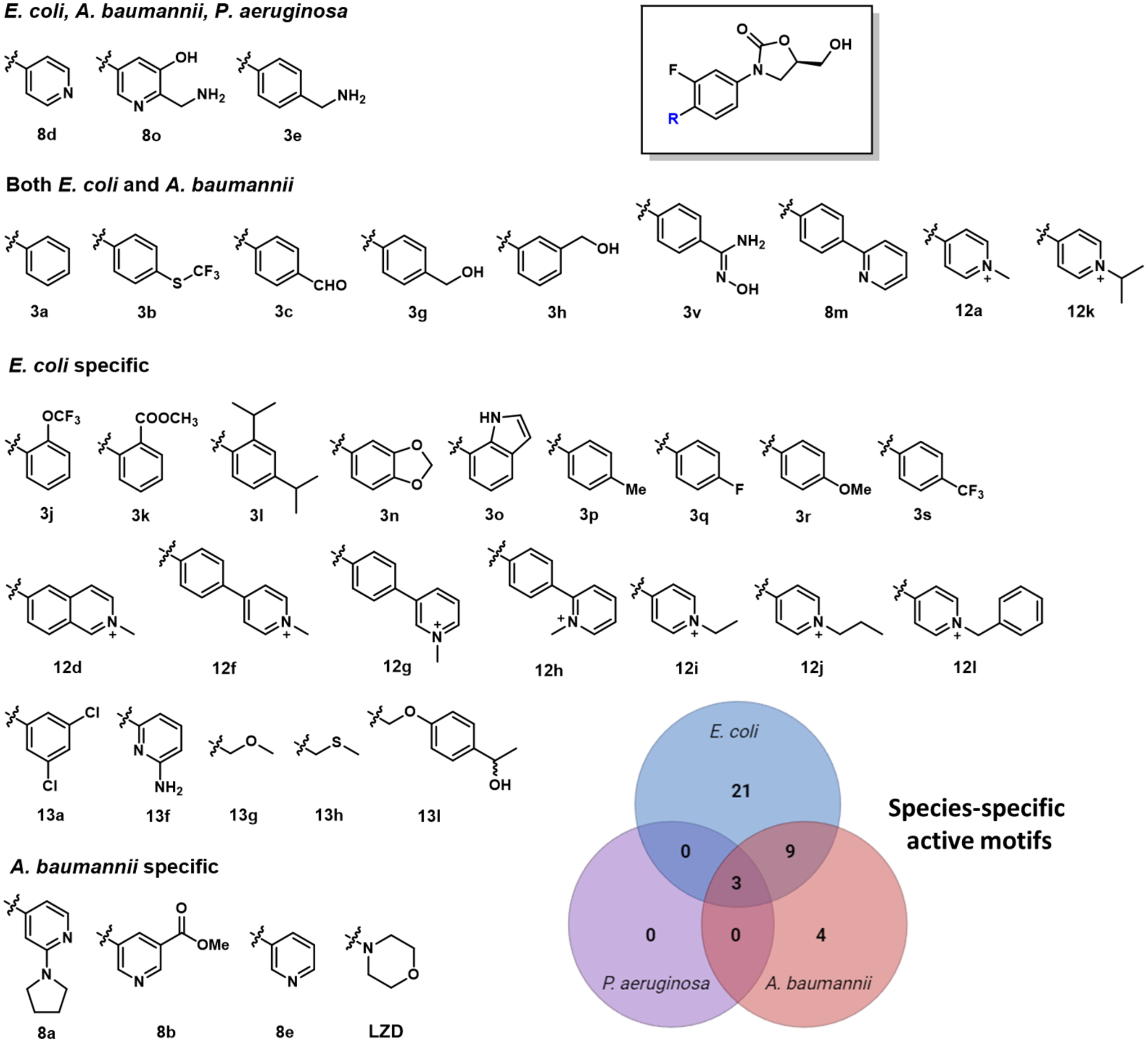
Motifs conferring WT activity classified by organism.
Overall, more motif effects were observed in WT E. coli than WT A. baumannii and WT P. aeruginosa. Specifically, 9 substituted phenyls (3j, 3k, 3l, 3n, 3o, 3p, 3q, 3r, 3s; IC50 = 23.0–99.1 μM), 7 substituted pyridiniums (12d, 12f, 12g, 12h, 12i, 12j, 12l; IC50 = 15.4–98.4 μM) and 5 substituted alkynyls (13a 13f, 13g, 13h,13l; IC50 = 59.0–94.9 μM) produced enhanced permeation in E. coli. Three other substituted pyridyls (8a, 8b, 8e; IC50 = 19.0–83.2 μM) exhibited improved permeation in WT A. baumannii. No permeation enhancing motifs were identified specifically for WT P. aeruginosa. Additional phenyl, pyridinium, and pyridyl analogs displayed enhanced permeation profiles for both WT E. coli and A. baumanii, (3a, 3b, 3v, 3c, 3h, 3g, 12a, 12k, 8m; IC50 = 13.5–89.5 μM) (Figure 3).
3.2. Accumulation Hindering Entities (ACHEs)
In addition to identifying structures that increase accumulation in GNB, there is value in identifying problematic motifs associated with efflux susceptibility and poor OM permeation. The elucidation of accumulation hindering entities (ACHEs), which present OM permeation liabilities and/or efflux susceptibility, would provide a list of “structural alerts” that could be useful in hit-to-lead optimization. In our oxazolidinone study, all compounds exhibiting a measurable IC50 in at least one double compromised strain were analyzed for OM permeation and efflux liabilities. A broad spectrum (i.e., observed in WT E. coli, P. aeruginosa, and A. baumannii) increase in efflux susceptibility was observed for 6 different motifs: 3c, 3n, 3p, 3r, 3s, and 3u (Figure 4A). On the contrary, no specific motifs were identified that associated with poor OM permeability across all three species. Select motifs were observed to increase efflux susceptibility or OM permeation in E. coli or A. baumannii alone, however, many of the motifs were classified as P. aeruginosa-specific ACHEs. Additionally, against P. aeruginosa, seven of the motifs identified as problematic for OM permeation also presented as efflux liabilities: 3b, 3t, 8j, 8o, 12a, 12h and Linezolid. (Figure 4B). It will be interesting to see if these motifs exhibit similar OM and efflux liabilities across other chemotypes.
Figure 4.
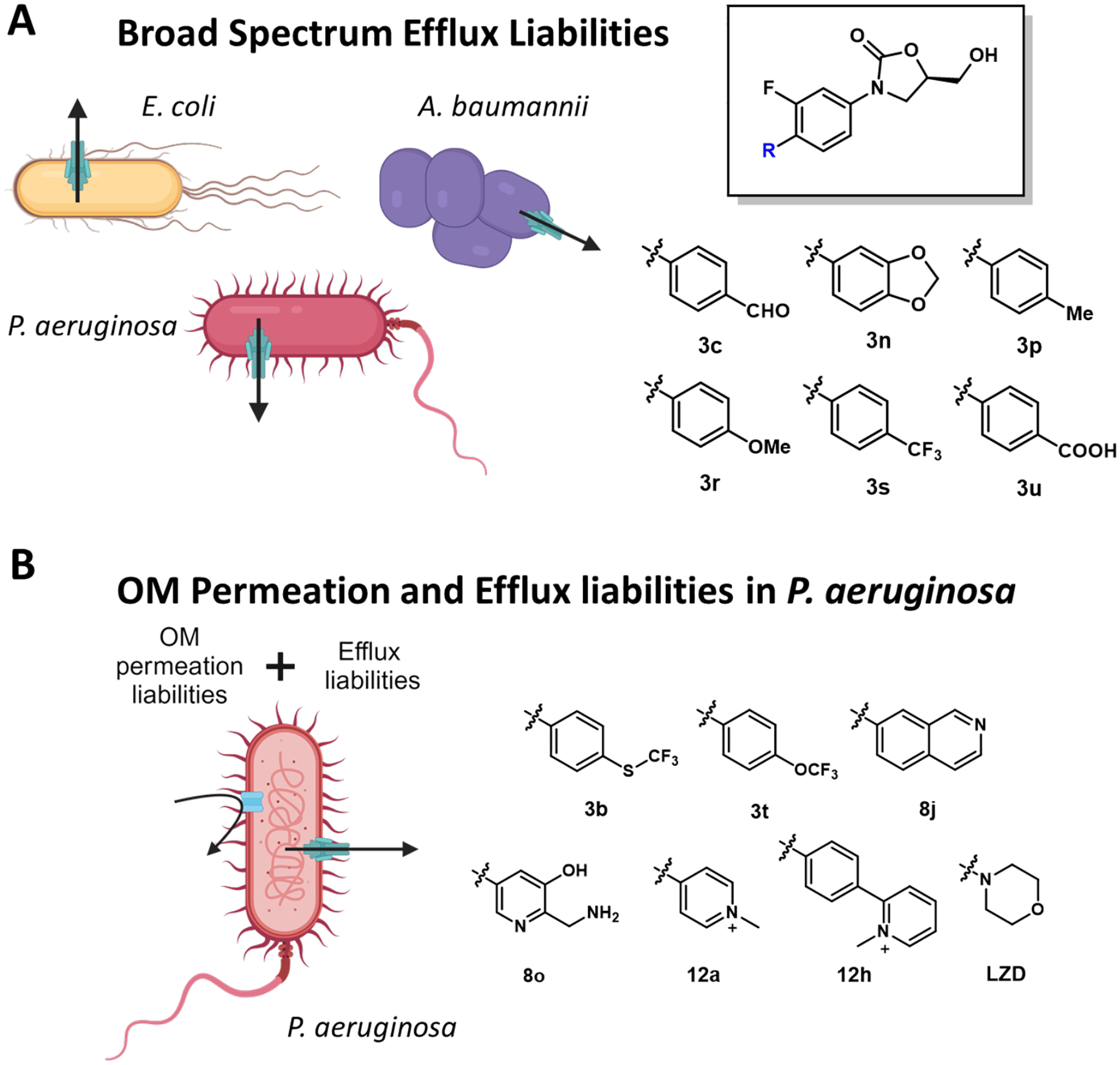
Chemical motifs that create accumulation liabilities in Gram-negative bacteria. (A) Six motifs that correlated to an increased efflux liability in E. coli, A. baumannii, and P. aeruginosa. (B) Seven motifs that contributed to both OM permeation and efflux liabilities in P. aeruginosa. LZD = Linezolid.
3.3. Perspective
Examples described in this section clearly indicate that motifs alone can influence permeation and accumulation in GNB. These motif driven effects can arise from the installation of specific functional groups (e.g., ionizable amine), motif substitution, or even location. However, outside of the incorporation of an ionizable amine, it remains to be determined which motif driven effects are cross-chemotype compatible. Many motif effects seem to be biased toward specific organisms; however, evidence suggests that broader spectrum trends can be identified, at least for select pathogens. This is exemplified by the enhanced accumulation seen in E. coli, K. pneumoniae, and A. baumannii for various chemotypes upon incorporation of an ionizable amine.37,45 With this fundamental knowledge, one can begin to draw corollaries on traditional medicinal chemistry approaches for eukaryotic targets, to provide inspiration for strategies that can be leveraged to rationally improve Gram-negative permeation and accumulation. Points of consideration for interrogating and leveraging motif driven trends in the context of Gram-negative bacteria follow.
3.3.1. Motif driven strategies are actionable.
As described previously, it would be beneficial to medicinal chemists to provide options for manipulating a single location on a molecule to influence OM permeation and/or efflux susceptibility. This can often be challenging when considering alteration of whole molecule descriptors but would be more attainable for motifs. Take for example a scenario in which a molecule binds to the target of interest with high-affinity and contains a solvent exposed region. One could envision installing a permeation enhancing motif at this location, similarly to the approach taken for the installation of alkynes and azides for pull-down experiments or other functionalities to influence ADME properties. This would leave the pharmacophore intact while addressing OM and/or efflux liabilities.
3.3.2. Utility of motif driven studies to identify structural alert ACHEs.
In traditional medicinal chemistry, the term structural alert is typically reserved for PAINs or functionalities known to have metabolic liabilities. For example, core scaffolds such as isothiazolones, quinones/catechols, and phenol-sulphonamides are flagged in medicinal chemistry campaigns in order to avoid undesired covalent modification, redox recycling, and nonspecific interactions.53 As demonstrated in several of the presented studies, this approach should be broadened in the antibacterial space to include accumulation hindering entities (ACHEs) that are subject to OM exclusion and efflux recognition. In this context, classification of motifs as ACHEs would provide further assistance in hit filtering/prioritization following HTS screens. If for example, one was deciding between two hit molecules for an SAR campaign, the molecule devoid of ACHEs could be prioritized. On the contrary, if the binding mode of a hit is known, and an identified ACHE is solvent exposed, one could proceed with the hit, expecting to substitute the problematic motif to gain Gram-negative activity.
3.3.3. Motif influence sets the stage for rational replacement strategies.
In drug discovery, several approaches have proven influential in activity optimization, such as bioisosteric replacement, scaffold hopping, and the Topliss assessment. While these strategies are primarily focused on enhancing target engagement and manipulating ADME properties, one could envision similar approaches to enhancing Gram-negative penetrance. As more motifs are identified that influence permeation and accumulation (both positively and negatively), replacement strategies for addressing liabilities are expected to emerge. For this to occur, however, more comprehensive studies need to be conducted to further define trends that are dependent or independent on chemotype and organism.
4. COMPUTATIONAL APPROACHES
In recent years computational power has increased exponentially along with the development of new algorithms capable of handling more complex challenges. As such, artificial intelligence (AI) and machine learning (ML) have gained significant traction in drug discovery, especially in the context of antibacterials. Corwin Hansch’s seminal work, using quantitative structure-activity relationships (QSAR) in the late 1960s, set the foundation for what has now not only led to efforts to address the complexity of GNB permeation and accumulation but to even uncover novel antibacterials.54 The power of ML lies in the assimilation of diverse data sets in order to provide predicted outputs of desired outcomes. Specifically, it positions the field to decipher the effects of physicochemical properties and molecular architecture on Gram-negative permeation and accumulation in a simultaneous and comprehensive fashion. Experimental validation of several models has now begun to appear in literature, painting a very promising picture for AI in antibacterial discovery and optimization.
4.1. Computational Determination of Physicochemical Accumulation Descriptors
4.1.1. Physicochemical scoring functions for porin-mediated entry.
The majority of antibiotics in clinical use cross the Gram-negative OM through polar water-filled channels provided by outer membrane porins (OMP).55 The transport of β-lactams through the general porins of E. coli has been used as a model system to study porin-mediated entry.56,57 The Omp family of porins exhibits high sequence homology consisting of 16-stranded β-barrel trimers encompassing a narrow centralized constriction region instilled by extracellular loop L3, resulting in an hourglass shape.58,59 It has been shown that small changes in the narrow constriction region of OmpF directly affect small molecule permeation.60,61 During translocation, positively and negatively charged amino acids are segregated on opposite sides of the porin creating a transverse electric field.62,63 Based on the crystal structures of OmpF and OmpC orthologues of clinically relevant Enterobacter cloacae, Klebsiella aerogenes, and Klebsiella pneumoniae, Acosta-Gutierrez and Ceccarelli created a scoring function to predict small-molecule permeation.63 An experimental permeability data set of nine clinically relevant antibiotics that exploit eight distinct channels was curated, resulting in 72 independent measurements. The structural and experimental data was leveraged to create a scoring function driven by molecular dynamics simulations to predict the permeability of small molecules through select porins (OmpF, OmpC, Omp35, Omp36, OmpE35, OmpE36, OmpK35, OmpK36).
Both β-lactam and non-β-lactam scaffolds were analyzed, and the scoring function was shown to accurately predict changes in permeability based on structural disparities between molecules. Cefepime (zwitterionic) was predicted to permeate better in a larger number of OM porins than Ceftazidime (anionic). The prediction was validated through comparative time-kill results across a number of porin engineered E. coli (K-12 W3110) cell lines. Faster bactericidal effects were seen in cells expressing OmpF-like porins (~0.01% survival at t=150 min) compared to cells expressing OmpC-like porins (~1–10% survival at t=150 min) when treated with Ceftazidime. In contrast, potent killing kinetics observed across all cell lines treated with Cefepime (~0.001–0.1% survival at t=150 min) was attributed to the zwitterionic nature of the compound. Increased permeability occurred upon inclusion of a positive charge and was linked to a change in dipole moment, which increased favorable interactions within the pore. The accuracy of the scoring function was further tested on published non β-lactam structures from Richter and Hergenrother’s initial eNTRy rule study.37 Changes in membrane permeability due to altered charge, size, shape, and dipole moment were correctly predicted. In agreement with the experimental data, molecules with a net positive charge were predicted to have higher accumulation than corresponding neutral analogs.
4.1.2. Kinetic modeling of small molecule uptake.
In 2017, a kinetic model was developed by Westfall and Rybenkov to accurately describe permeation properties influencing drug uptake through the Gram-negative cell envelope.64 The synergism of the outer and inner membranes and active efflux by multidrug transporters was analyzed. Two kinetic parameters, efflux (KE) and barrier (B) constants, were introduced to replace traditional Michaelis Menten kinetics. KE defines a measure by which the intracellular concentration of the drug is reduced compared to the thermodynamic equilibrium at which the transporter operates below saturation. B describes the effects of high drug concentrations and discriminates between efficient and semi-efficient efflux. Thus, these parameters relate the efficiencies of active efflux and passive diffusion at high and low drug concentrations. The model was experimentally validated by measuring the uptake of bis-benzimide (Hoechst 33342), a topoisomerase inhibitor,65 in E. coli (BW25113). Linear concentration dependence of the initial rate should have been observed if the system operated under Fick’s law of diffusion or Michaelis-Menten behavior, however, hyperbolic concentration dependence was observed, agreeing with the model.
4.1.3. Models for efflux pump recognition.
Clinical isolates of MDR P. aeruginosa are resistant to nearly all available antibiotics and have been identified as a serious threat by the CDC.66 Their cell envelopes are extremely effective in protecting cells from antibiotics due to the synergy between active efflux pumps and the OM.12 Recently, new models have been developed by Mehla and Zgurskaya that identify scaffolds associated with the avoidance and/or inhibition of active efflux in P. aeruginosa.43 A library of 260 active peptidomimetics were analyzed in four P. aeruginosa strains: WT PAO1, Δ6 efflux pump deficient (ΔmexAB-oprM, ΔmexCD-oprJ, ΔmexXY, ΔmexJKL, ΔmexEF-oprN, and ΔtriABC), and hyperporinated variants (PAO1-Pore and PΔ6-Pore). The compounds were categorized and placed into groups based on whether they contribute to permeation through the OM or active efflux. The IC50 ratios of PAO1/PΔ6 and PAO1-Pore/PΔ6-Pore defined the contribution of efflux. The experimental measurements were used to train a linear predictive model to determine the relative importance of different chemical descriptors on efflux avoidance. The best performing and generalizable descriptors were subject to binomial regression models to further categorize them as efflux avoiders (IC50 PΔ6-Pore/IC50 PAO1-Pore of ≥0.25) or efflux inhibitors (both growth-dependent and growth-independent IC50 ratios >0.5). It was determined that the largest contributing descriptors for efflux inhibition were molecular shape, presented as average values: acylindricity (~2.00), amphiphilicity, represented by anisotropic polarizability (~180 au), number of aromatic rings (2–3), contacts with active site residues like Pro668 and Leu674, LogD (~−3), and number of HBA (4–5). These properties were then designed into a small library of compounds resulting in increased efflux pump inhibition. For example, many of the predicted properties were included in the design of OU-109 (Acylindricity = 1.7, Aromatic rings = 2, HBA= 5, Contacts with residues = 3) which contained no innate activity against P. aeruginosa but successfully enhanced the activity of co-administered antibiotics through efficient efflux inhibition (Figure 5).
Figure 5.
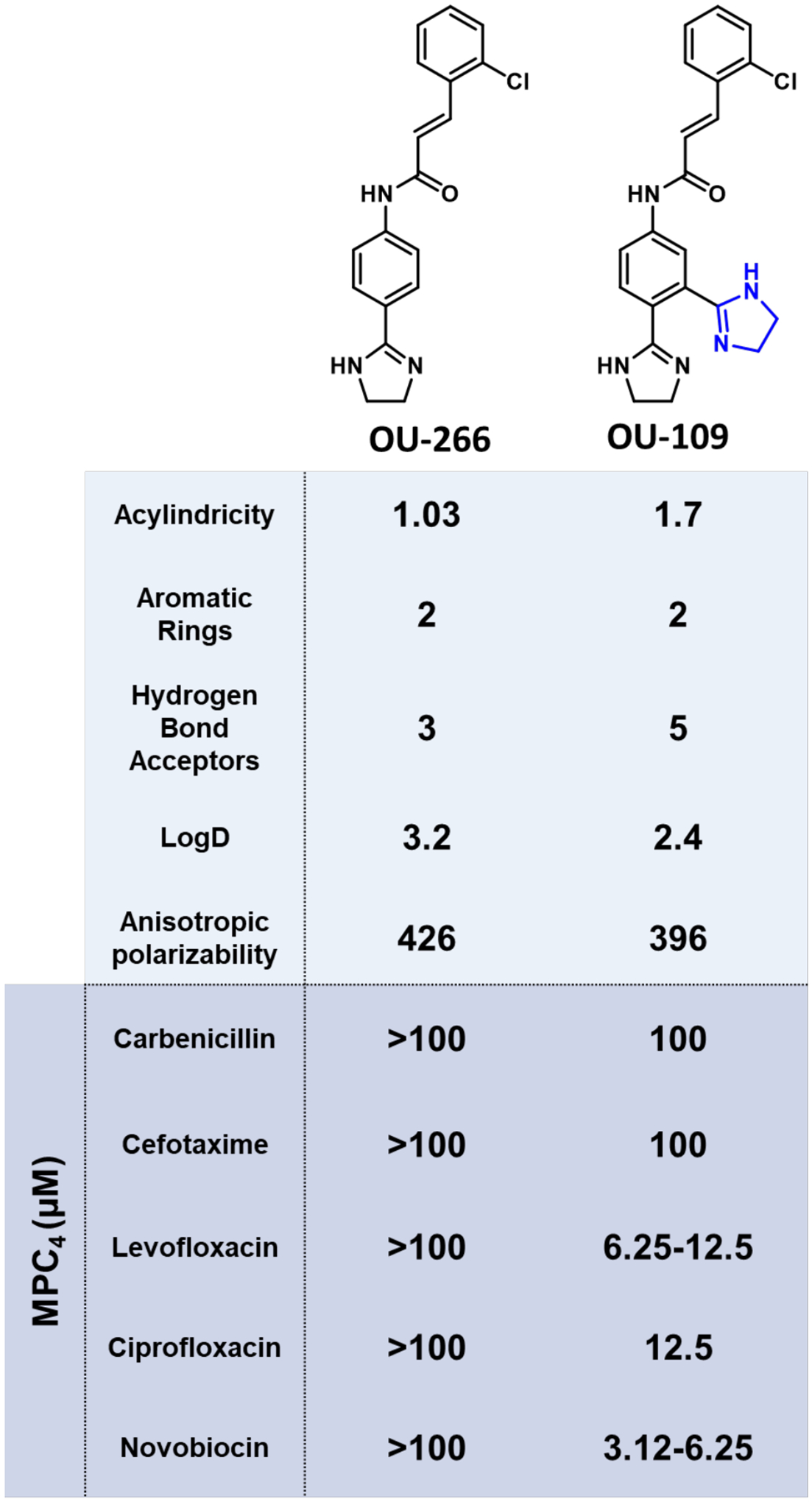
Example application of the identified efflux inhibition rules on OU-266 derivatives. Derivatives were tested for EPI activity against PΔ6-Pore (MexAB-OprM) in combination with the displayed antibiotics. MPC4 = Minimal potentiating concentration able to reduce 4-fold the antibiotic minimal inhibitory concentration (MIC).
4.2. Computational Approaches to Identify Motifs Important for Accumulation
4.2.1. Topological analysis by artificial neural networks.
Topological methods represent the structure of a molecule as indices, a series of numbers that contain information on the number and connectivity between comprising atoms. QSAR studies are the most widely used topological method and employ theoretical molecular descriptors to predict physicochemical, pharmacological, and toxicological properties of molecules based on a training set.67 Historically, QSAR is limited by the use of linear discriminant analysis (LDA) but more recently, artificial neural networks (ANNs) have been incorporated to create QSAR methods capable of recognizing nonlinear relationships in small-molecule accumulation.68 In one example, an ANN model was used in parallel with LDA to discriminate and select new antibacterial agents from a chemical library.69 The models were trained with 70% of the selected 217 active and 216 non-active compounds resulting in 62 topological indices (e.g., atom number and type, bond number and type, conjugated double bonds, and distance between atoms). The remaining 30% of the compounds were subject to the defined indices using both LDA and ANN as discrimination methods to obtain a discriminant function. The discriminant function was used to search for active antibacterial compounds in a large chemical database (Available Chemical Directory). The compounds classified as actives were tested against both Gram-positive (Enterococcus faecalis ATCC29212 and Staphylococcus aureus ATCC6538P) and Gram-negative (E. coli ATCC10536 and P. aeruginosa ATCC27853) strains to confirm the prediction. The overall accuracy for the ANN (98.69%) surpassed that of the LDA (77.78%).
4.2.2. Data driven derivation of bioisosteric replacements.
A recent study from the Zachariae group used a data set of 1887 compounds with experimentally determined MICs in both E. coli and S. aureus to develop a data-driven model using matched molecular pair analysis (MMPA) and ML.70 The results revealed that neither molecular weight nor LogP served as the primary predictor of permeability across the OM. Fifteen key substructural descriptors were predicted to significantly enhance Gram-negative activity including the addition of a thiophene or aryl chloride and the removal of esters or lactone groups. Experimental E. coli MIC measurements from the ChEMBL database were standardized into pMIC to observe structural patterns linked to Gram-negative activity. For example, replacing a carbonyl with a thiophene resulted in a 0.74 ΔpMIC while replacing a nitrile with a thiophene had much less effect on Gram-negative activity with a −0.04 ΔpMIC (Figure 6).
Figure 6.

Examples of predicted motif replacements and correlating pMIC in diverse scaffolds. The replacement of the red functionality by the blue motif increases Gram-negative activity.
4.3. Identification of New Antibacterials and Targets
Recently, deep learning approaches have also been successfully applied to small molecule antibacterial discovery. Stokes and Collins trained a deep neural network to predict molecules with antibacterial activity against E. coli.71 The deep learning model was first trained on hits from a virtual screen of 2,335 chemically diverse compounds to predict growth inhibition of E. coli. This library consisted of FDA-approved drugs and various natural products and was screened at 50 μM against E. coli using 80% growth inhibition as the cut off. The model was then applied against the Drug Repurposing Hub containing over 100 million compounds to identify potential leads. Through this method halicin, a structurally unique antibiotic with activity against E. coli (MIC=2 μg/mL), a Carbapenem-resistant Enterobacteriaceae (CRE) panel (MIC=1–10 μg/mL), and an A. baumannii MDR panel (MIC=1–10 μg/mL), was identified as a novel antibacterial. A subsequent study by the Stokes group screened 7,684 compounds for inhibition against A. baumannii to be used to train a message-passing neural network for antibiotic discovery.72 The model was then applied to the Drug Repurposing Hub (6,680 molecules). The lead compound abaucin (MIC = ~2 μg/ml against A. baumannii ATCC 17978) was discovered and exhibited a narrow spectrum of activity through the perturbation of lipoprotein trafficking facilitated by LolE. In an A. baumannii wound infection mouse model, abaucin-treated mice (4% wt/vol with Glaxal Base Moisturizing Cream) carried nearly identical bacterial load to the pretreated infection control mice (~4.0×107 CFU/g) compared to the vehicle-treated mice (~6.9×108 CFU/g).
4.4. Perspective
The discovery of novel antibiotics is becoming increasingly difficult and the current strategies that are relied upon (e.g., screening, derivatization of known chemotypes, human experience) are becoming less productive. Given the multidimensional nature of the permeation and accumulation challenge, computational models are optimally positioned to provide an extra level of understanding, enabling medicinal chemistry and drug discovery efforts for Gram-negative pathogens. ML approaches have the potential to substantially decrease the cost and increase the rate of success of antibacterial drug discovery campaigns, providing a win-win for the future. Considerations for the future of AI, ML, and cheminformatics in Gram-negative discovery follow.
4.4.1. Models provide unparalleled multivariable assimilation.
Significant efforts have been made to understand fundamental properties associated with permeation across the Gram-negative barriers. Until recently, a lack of quantitative models capable of matching the complexity of the Gram-negative barriers limited progress toward understanding and predicting Gram-negative penetrance to human intuition or retrospective analyses. Integration of cheminformatics allowed multivariable assimilation to set a foundation for which more complex machine learning models could be built. New models now have the capability to parameterize physicochemical profiles, structural connectivity, and experimental data, dramatically elevating SAR understanding beyond human capabilities, leading to new knowledge. For example, Westfall and Rybenkov’s analysis revealed novel physical principles in the behavior of the cellular envelope, providing a new approach to analyze structure-activity relationships of novel antibiotics. While this model was built in the context of E. coli, it is reasonable to assume that a similar approach can be taken for other pathogens of interest. Acosta-Gutierrez and Ceccarelli’s approach offers yet another strategy for predicting permeability by assessing molecular compensation for the inherent entropic barrier of outer membrane porins, instead of searching for an energy minimum in docking studies. This scoring function, which was developed in the context of E. cloacae, K. aerogenes, and K. pneumoniae has the potential to be extrapolated to other organisms and used in virtual screening to improve the rate of discovering efficient accumulators. Models focusing on structural features are also proving to be insightful. For example, Gurvic and Zachariae’s model now provides suggestions for structural replacements of a disparate motifs for increased activity in E. coli, giving rise to the first evidence that a bioisosteric replacement approach for Gram-negative accumulation may be feasible. These results, however, have not been demonstrated experimentally in a prospective fashion, especially in a cross-chemotype context. While the identified structural replacements are likely to be limited to E. coli, similar models are undoubtedly possible for other organisms.
4.4.2. Experimental validation is essential.
Machine learning is not perfect and there are many pieces that must be considered to ensure a successful model. These approaches must be coupled and built with sufficient experimental data and then (re)confirmed experimentally, followed by iterative model refinement. In order to design a quality model, the representative biological assay of choice must be considered and the desired biological outcome following compound treatment should be incorporated into the design. For example, Stokes and Collins implemented growth inhibition as the defining metric and base for training the model to identify both broad- and narrow-spectrum Gram-negative actives. Extrapolating this approach to anti-virulence agents, however, will require alternative experimental readouts, as these agents fail to produce anti-growth effects. Another consideration is the composition of the training data itself. The output of a machine learning campaign is only as good as the training set. Attributes that make a good training set include sufficient chemical diversity and comparable data sets for active and inactive compounds. Additionally, the active molecules should be structurally diverse, or the model will likely be limited to narrow chemical space and thus not generalize well to new scaffolds. Ideally, the accuracy of the model will lead to high prediction scores translating to experimental validation and novel leads. However, it is worth considering that synthetic tractability of outputs, structural uniqueness, and molecular stability are equally important.
5. CONCLUDING REMARKS
The challenge of translating biochemical hits into whole cell actives against Gram-negative bacteria remains the single biggest challenge in medicinal chemistry. Rightfully so, preliminary efforts to provide insight into this challenge focused on retrospective analyses of physicochemical descriptors of Gram-negative active molecules in comparison to eukaryotic and Gram-positive drugs. Conceptually, this was a worthy endeavor that elucidated the chemical space of molecules likely to accumulate in bacteria. The limitations of these studies, however, are that 1) Gram-negative bacteria were treated as a whole and a limited number of antibacterial classes were represented, 2) mechanism of action and target location were rarely considered, and 3) uptake mechanism was ignored. We now know that all these attributes play an influential role in small molecule penetrance. Because of evolutionary divergence, it should not be surprising that little evidence of broad consensus regarding the key physicochemical determinants has been identified for compound accumulation.
We anticipate that the most productive angle will be to interrogate species-specific requirements for small molecule permeation and accumulation. Historically, broad spectrum activity was essential for antibacterial development. However, given the improving diagnostic capabilities, dire need of new antibacterials, and the complicating issues of microfloral disturbance caused by broad-spectrum agents, narrow spectrum antibacterials are finally garnering the consideration they deserve.
The acceptance of narrow spectrum efforts, especially in the context of basic research, drastically improves the feasibility of developing guiding principles for priority pathogens. This allows researchers to focus on an organism of choice, to understand the molecular requirements of small molecule penetrance and accumulation more rigorously. Hergenrother’s eNTRy rules perform very well for E. coli, and continued efforts from the group and others have now shown the trends to translate, in some fashion, to other organisms. Exceptions to these rules, however, provide compelling evidence that more work towards porin-independent uptake is warranted. Nonetheless, this advancement is exciting and provides proof-of-concept that Lipinski-esque guidelines can be generated. The call to action is to extend these types of studies to other pathogens of interest, especially those evolutionarily divergent from E. coli.
In our opinion, motif driven approaches are likely to lead to the most tractable strategies for enhancing permeation and accumulation in Gram-negative bacteria. Increasing efforts in microbiology to understand active uptake mechanisms and recognition features of efflux pumps are likely to lead to privileged motifs. Those identified to be detrimental to penetrance/efflux should be classified as Accumulation Hindering Entities (ACHEs), while those enhancing accumulation (e.g., ionizable amines) could be added to the medicinal chemist’s toolbox for rational design. With more progress, a picture will begin to clarify for specific organisms that provide re-imagined versions of commonly used medicinal chemistry approaches for accumulation enhancement.
A considerable effort has been made to develop whole cell mutants of various species to enable the deconvolution of individual barrier contributors.73 However, recent large compound accumulation screens have relied on low throughput assays.36,37,74 Future analysis of large libraries will require more high throughput assays capable of incorporating compound incubation, bacterial lysis, lysate extraction, and sample analysis. One promising example uses an engineered filter plate to separate cells from unbound compound paired with solid-phase extraction (SPE) technology and MS analysis.75 An alternative promising method is described by Zhou and Miller, which leverages LCMS to conveniently measure accumulation by observing the change in extracellular drug concentration.76 A comparison of the levels of accumulation of linezolid in WT (0.3 pmol in 2×109 cells) and ΔtolC E. coli (3.0 pmol in 2×109 cells) was used as a proof-of-concept. Nonetheless, even with new tools and methods enabling broader participation in this initiative, assay standardization is critical to allow cross-study comparison.
Studies are still in their infancy. Progress will take time but will be informative. The field is young and there is plenty of opportunity for contribution. Resources for antibacterial drug development are becoming more accessible through web applications (e.g., eNTRyway), which allow chemists to analyze molecules of interest for specific desired properties. Resources like the Chemistry Center for Combating Antibiotic Resistant Bacteria (CC4CARB) at RTI International are establishing Gram-negative focused libraries for public dissemination, which will further enable independent labs to contribute to the understanding of their organism of interest.
The success of guiding principles and computational methods in eukaryotic drug discovery provides a compelling reason to pursue comparable tools for Gram-negative drug discovery. The stage is set, as examples are now evolving for physicochemical descriptors and motif effects on permeation and accumulation of small molecules in Gram-negative pathogens. Machine learning provides an additional level of analysis, capable of identifying trends beyond what is capable from human interpretation. The challenge is complex, and will require teams with microbiology, medicinal chemistry, and computational expertise to provide the most meaningful results. A long road ahead remains, but progress made thus far indicates a bright future.
SIGNIFICANCE.
Retrospective and prospective studies have provided proof of concept that the identification of molecular architectures that improve Gram-negative permeation and accumulation is feasible.
Existing assays and resources now provide foundational tools to define chemotype-dependent and -independent trends.
Pathogen-specific and -general trends for permeation and accumulation will enable novel chemical probe and therapeutic development, impacting both basic and translational research.
ACKNOWLEDGEMENTS
The table of contents graphic, Figure 1, and Figure 4 were created with BioRender. Alexis Stoorza is supported by the National Institute of General Medical Sciences of the National Institutes of Health (T32GM132029). The authors are grateful for funding by the National Institute of Allergy and Infectious Disease of the National Institutes of Health (R01AI136795). The content included in this manuscript does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred.
ABBREVIATIONS USED
- ACHEs
Accumulation hindering entities
- AI
Artificial Intelligence
- ALogPs
LogP
- ALogpS
LogS
- AMS
Sulfamoyl adenosine
- ANN
Artificial neural network
- APT
Abbott Physicochemical Tiering
- Avg
Average
- B
Barrier constant
- BC
Biotin carboxylase
- Bro5
Beyond the “rule of five”
- CC4CARB
Chemistry Center for Combating Antibiotic Resistant Bacteria
- cLogD7.4
Calculated LogD
- EPI
Efflux pump inhibitors
- FMN
Flavin mononucleotide
- Fsp3
Fraction sp3 hybridization
- GNB
Gram-negative bacteria
- GSK
GlaxoSmithKline
- IM
Inner membrane
- KE
efflux constant
- LDA
Linear discriminant analysis
- ML
Machine learning
- MMPA
Matched molecular pair analysis
- N
Nitrogen
- N
Unhindered primary amine
- nStereo
Stereocenters
- nStMW
stereochemical density (nStereo/MW)
- O
Oxygen
- OM
Outer membrane
- OMP
Outer membrane porins
- PAβN
Phenylalanine-arginine β-naphthylamide
- PAINS
Pan-assay interference compounds
- PMBN
Polymyxin B nonapeptide
- R or RotB
Rotatable bonds
- relPSA
relative polar surface area
- RND
Resistance-Nodulation-Division
- RngAr
Aryl rings
- RngSys
Ring systems
- RRSys
Rings per ring system
- RTI
Research Triangle Institute
- SPE
Solid-phase extraction
- T
Globularity
- tPSA
topological polar surface area
Biographies
Alexis M. Stoorza received her B.S. in Chemistry with an emphasis in Biochemistry from Northwest Nazarene University in 2021. She is currently an NIH Chemical Biology Training Grant fellow in the Duerfeldt Lab at the University of Minnesota. Alexis Stoorza’s research focuses on investigating anti-virulence strategies and understanding molecular features correlating with small-molecule accumulation in Gram-negative bacteria.
Adam S. Duerfeldt received his B.A. in Chemistry from Central College and his Ph.D. in medicinal chemistry from the University of Kansas. Dr. Duerfeldt then conducted his postodoctoral studies at The Scripps Research Institute. Dr. Duerfeldt began his independent career at the University of Oklahoma in the Department of Chemistry before moving to the Department of Medicinal Chemistry at the University of Minnesota, where he is currently an Associate Professor. The research foci of the Duerfeldt lab are on the development of chemical tools to enhance our understanding of disease mechanisms and the development of probes and therapeutic leads for infectious and retinal diseases.
REFERENCES
- (1).Murray CJL; Ikuta KS; Sharara F; Swetschinski L; Aguilar GR; Gray A; Han C; Bisignano C; Rao P; Wool E; Johnson SC; Browne AJ; Chipeta MG; Fell F; Hackett S; Haines-Woodhouse G; Hamadani BHK; Kumaran EAP; McManigal B; Achalapong S; Agarwal R; Akech S; Albertson S; Amuasi J; Andrews J; Aravkin A; Ashley E; Babin F-X; Bailey F; Baker S; Basnyat B; Bekker A; Bender R; Berkley JA; Bethou A; Bielicki J; Boonkasidecha S; Bukosia J; Carvalheiro C; Castañeda-Orjuela C; Chansamouth V; Chaurasia S; Chiurchiù S; Chowdhury F; Donatien RC; Cook AJ; Cooper B; Cressey TR; Criollo-Mora E; Cunningham M; Darboe S; Day NPJ; Luca MD; Dokova K; Dramowski A; Dunachie SJ; Bich TD; Eckmanns T; Eibach D; Emami A; Feasey N; Fisher-Pearson N; Forrest K; Garcia C; Garrett D; Gastmeier P; Giref AZ; Greer RC; Gupta V; Haller S; Haselbeck A; Hay SI; Holm M; Hopkins S; Hsia Y; Iregbu KC; Jacobs J; Jarovsky D; Javanmardi F; Jenney AWJ; Khorana M; Khusuwan S; Kissoon N; Kobeissi E; Kostyanev T; Krapp F; Krumkamp R; Kumar A; Kyu HH; Lim C; Lim K; Limmathurotsakul D; Loftus MJ; Lunn M; Ma J; Manoharan A; Marks F; May J; Mayxay M; Mturi N; Munera-Huertas T; Musicha P; Musila LA; Mussi-Pinhata MM; Naidu RN; Nakamura T; Nanavati R; Nangia S; Newton P; Ngoun C; Novotney A; Nwakanma D; Obiero CW; Ochoa TJ; Olivas-Martinez A; Olliaro P; Ooko E; Ortiz-Brizuela E; Ounchanum P; Pak GD; Paredes JL; Peleg AY; Perrone C; Phe T; Phommasone K; Plakkal N; Ponce-de-Leon A; Raad M; Ramdin T; Rattanavong S; Riddell A; Roberts T; Robotham JV; Roca A; Rosenthal VD; Rudd KE; Russell N; Sader HS; Saengchan W; Schnall J; Scott JAG; Seekaew S; Sharland M; Shivamallappa M; Sifuentes-Osornio J; Simpson AJ; Steenkeste N; Stewardson AJ; Stoeva T; Tasak N; Thaiprakong A; Thwaites G; Tigoi C; Turner C; Turner P; van Doorn HR; Velaphi S; Vongpradith A; Vongsouvath M; Vu H; Walsh T; Walson JL; Waner S; Wangrangsimakul T; Wannapinij P; Wozniak T; Sharma TEMWY; Yu KC; Zheng P; Sartorius B; Lopez AD; Stergachis A; Moore C; Dolecek C; Naghavi M Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. The Lancet 2022, 399 (10325), 629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keam SJ Sulbactam/Durlobactam: First Approval. Drugs 2023, 83 (13), 1245–1252. 10.1007/s40265-023-01920-6. [DOI] [PubMed] [Google Scholar]
- 3.Do M; Shin B; Lioudis E; Munn E Novel Drugs Approved in 2021–2022. Transform. Med. T-Med 2022, 1 (3), 72–77. 10.54299/tmed/zcno1399. [DOI] [Google Scholar]
- 4.Andrei S; Droc G; Stefan G FDA Approved Antibacterial Drugs: 2018–2019. Discoveries 7 (4), e102. 10.15190/d.2019.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silver LL Challenges of Antibacterial Discovery. Clin. Microbiol. Rev 2011, 24 (1), 71–109. 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucher HW; Talbot GH; Bradley JS; Edwards JE; Gilbert D; Rice LB; Scheld M; Spellberg B; Bartlett J Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am 2009, 48 (1), 1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 7.Tommasi R; Brown DG; Walkup GK; Manchester JI; Miller AA ESKAPEing the Labyrinth of Antibacterial Discovery. Nat. Rev. Drug Discov 2015, 14 (8), 529–542. 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- 8.CDC. The biggest antibiotic-resistant threats in the U.S Centers for Disease Control and Prevention. https://www.cdc.gov/drugresistance/biggest-threats.html (accessed 2022–03-30). [Google Scholar]
- 9.Laxminarayan R; Duse A; Wattal C; Zaidi AKM; Wertheim HFL; Sumpradit N; Vlieghe E; Hara GL; Gould IM; Goossens H; Greko C; So AD; Bigdeli M; Tomson G; Woodhouse W; Ombaka E; Peralta AQ; Qamar FN; Mir F; Kariuki S; Bhutta ZA; Coates A; Bergstrom R; Wright GD; Brown ED; Cars O Antibiotic Resistance—the Need for Global Solutions. Lancet Infect. Dis 2013, 13 (12), 1057–1098. 10.1016/S1473-3099(13)70318-9. [DOI] [PubMed] [Google Scholar]
- 10.Zgurskaya HI; López CA; Gnanakaran S Permeability Barrier of Gram-Negative Cell Envelopes and Approaches To Bypass It. ACS Infect. Dis 2015, 1 (11), 512–522. 10.1021/acsinfecdis.5b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lugtenberg B; Van Alphen L Molecular Architecture and Functioning of the Outer Membrane of Escherichia Coli and Other Gram-Negative Bacteria. Biochim. Biophys. Acta BBA - Rev. Biomembr 1983, 737 (1), 51–115. 10.1016/0304-4157(83)90014-X. [DOI] [PubMed] [Google Scholar]
- 12.Krishnamoorthy G; Leus IV; Weeks JW; Wolloscheck D; Rybenkov VV; Zgurskaya HI Synergy between Active Efflux and Outer Membrane Diffusion Defines Rules of Antibiotic Permeation into Gram-Negative Bacteria. mBio 2017, 8 (5), 10.1128/mbio.01172-17. 10.1128/mbio.01172-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sohlenkamp C; Geiger O Bacterial Membrane Lipids: Diversity in Structures and Pathways. FEMS Microbiol. Rev 2016, 40 (1), 133–159. 10.1093/femsre/fuv008. [DOI] [PubMed] [Google Scholar]
- 14.Fleischmann RD; Adams MD; White O; Clayton RA; Kirkness EF; Kerlavage AR; Bult CJ; Tomb J-F; Dougherty BA; Merrick JM; McKenney K; Sutton G; FitzHugh W; Fields C; Gocayne JD; Scott J; Shirley R; Liu L; Glodek A; Kelley JM; Weidman JF; Phillips CA; Spriggs T; Hedblom E; Cotton MD; Utterback TR; Hanna MC; Nguyen DT; Saudek DM; Brandon RC; Fine LD; Fritchman JL; Fuhrmann JL; Geoghagen NSM; Gnehm CL; McDonald LA; Small KV; Fraser CM; Smith HO; Venter JC Whole-Genome Random Sequencing and Assembly of Haemophilus Influenzae Rd. Science 1995, 269 (5223), 496–512. 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- (15).Payne DJ; Gwynn MN; Holmes DJ; Pompliano DL Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat. Rev. Drug Discov 2007, 6 (1), 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 16.manager C Converting Gram-positive-only compounds into broad-spectrum antibiotics REVIVE. https://revive.gardp.org/converting-gram-positive-only-compounds-into-broad-spectrum-antibiotics/ (accessed 2023–09-28). [DOI] [PMC free article] [PubMed]
- 17.Vaara M Polymyxin Derivatives That Sensitize Gram-Negative Bacteria to Other Antibiotics. Molecules 2019, 24 (2), 249. 10.3390/molecules24020249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhardwaj AK; Mohanty P Bacterial Efflux Pumps Involved in Multidrug Resistance and Their Inhibitors: Rejuvinating the Antimicrobial Chemotherapy. Recent Patents Anti-Infect. Drug Disc 7 (1), 73–89. [DOI] [PubMed] [Google Scholar]
- 19.Negash KH; Norris JKS; Hodgkinson JT Siderophore–Antibiotic Conjugate Design: New Drugs for Bad Bugs? Molecules 2019, 24 (18), 3314. 10.3390/molecules24183314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development settings1PII of Original Article: S0169–409X(96)00423–1. The Article Was Originally Published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1. Adv. Drug Deliv. Rev 2001, 46 (1), 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 21.Keller TH; Pichota A; Yin Z A Practical View of ‘Druggability.’ Curr. Opin. Chem. Biol 2006, 10 (4), 357–361. 10.1016/j.cbpa.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 22.Veber DF; Johnson SR; Cheng H-Y; Smith BR; Ward KW; Kopple KD Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem 2002, 45 (12), 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 23.Mikitsh JL; Chacko A-M Pathways for Small Molecule Delivery to the Central Nervous System across the Blood-Brain Barrier. Perspect. Med. Chem 2014, 6, PMC.S13384. 10.4137/PMC.S13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang M-Q; Wilkinson B Drug Discovery beyond the ‘Rule-of-Five.’ Curr. Opin. Biotechnol 2007, 18 (6), 478–488. 10.1016/j.copbio.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Cox PB; Gregg RJ; Vasudevan A Abbott Physicochemical Tiering (APT)--a Unified Approach to HTS Triage. Bioorg. Med. Chem 2012, 20 (14), 4564–4573. 10.1016/j.bmc.2012.05.047. [DOI] [PubMed] [Google Scholar]
- 26.O’Shea R; Moser HE Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem 2008, 51 (10), 2871–2878. 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 27.Koepsell H; Endou H The SLC22 Drug Transporter Family. Pflüg. Arch 2004, 447 (5), 666–676. 10.1007/s00424-003-1089-9. [DOI] [PubMed] [Google Scholar]
- 28.Nestorovich EM; Danelon C; Winterhalter M; Bezrukov SM Designed to Penetrate: Time-Resolved Interaction of Single Antibiotic Molecules with Bacterial Pores. Proc. Natl. Acad. Sci 2002, 99 (15), 9789–9794. 10.1073/pnas.152206799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown DG; May-Dracka TL; Gagnon MM; Tommasi R Trends and Exceptions of Physical Properties on Antibacterial Activity for Gram-Positive and Gram-Negative Pathogens. J. Med. Chem 2014, 57 (23), 10144–10161. 10.1021/jm501552x. [DOI] [PubMed] [Google Scholar]
- 30.Nikaido H; Rosenberg EY Effect on Solute Size on Diffusion Rates through the Transmembrane Pores of the Outer Membrane of Escherichia Coli. J. Gen. Physiol 1981, 77 (2), 121–135. 10.1085/jgp.77.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manchester JI; Buurman ET; Bisacchi GS; McLaughlin RE Molecular Determinants of AcrB-Mediated Bacterial Efflux Implications for Drug Discovery. J. Med. Chem 2012, 55 (6), 2532–2537. 10.1021/jm201275d. [DOI] [PubMed] [Google Scholar]
- 32.Kazmi F; Hensley T; Pope C; Funk RS; Loewen GJ; Buckley DB; Parkinson A Lysosomal Sequestration (Trapping) of Lipophilic Amine (Cationic Amphiphilic) Drugs in Immortalized Human Hepatocytes (Fa2N-4 Cells). Drug Metab. Dispos. Biol. Fate Chem 2013, 41 (4), 897–905. 10.1124/dmd.112.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlier M-B; Zenebergh A; Tulkens PM Cellular Uptake and Subcellular Distribution of Roxithromycin and Erythromycin in Phagocytic Cells. J. Antimicrob. Chemother 1987, 20 (suppl_B), 47–56. 10.1093/jac/20.suppl_B.47. [DOI] [PubMed] [Google Scholar]
- 34.Zimmermann W; Rosselet A Function of the Outer Membrane of Escherichia Coli as a Permeability Barrier to Beta-Lactam Antibiotics. Antimicrob. Agents Chemother 1977, 12 (3), 368–372. 10.1128/aac.12.3.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCaffrey C; Bertasso A; Pace J; Georgopapadakou NH Quinolone Accumulation in Escherichia Coli, Pseudomonas Aeruginosa, and Staphylococcus Aureus. Antimicrob. Agents Chemother 1992, 36 (8), 1601–1605. 10.1128/aac.36.8.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis TD; Gerry CJ; Tan DS General Platform for Systematic Quantitative Evaluation of Small-Molecule Permeability in Bacteria. ACS Chem. Biol 2014, 9 (11), 2535–2544. 10.1021/cb5003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ Predictive Compound Accumulation Rules Yield a Broad-Spectrum Antibiotic. Nature 2017, 545 (7654), 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Y; Shi H; Zhou M; Ren Q; Zhu W; Zhang W; Zhang Z; Zhou C; Liu Y; Ding X; Shen HC; Yan SF; Dey F; Wu W; Zhai G; Zhou Z; Xu Z; Ji Y; Lv H; Jiang T; Wang W; Xu Y; Vercruysse M; Yao X; Mao Y; Yu X; Bradley K; Tan X Discovery of Pyrido[2,3-b]Indole Derivatives with Gram-Negative Activity Targeting Both DNA Gyrase and Topoisomerase IV. J. Med. Chem 2020, 63 (17), 9623–9649. 10.1021/acs.jmedchem.0c00768. [DOI] [PubMed] [Google Scholar]
- 39.Renslo AR; Jaishankar P; Venkatachalam R; Hackbarth C; Lopez S; Patel DV; Gordeev MF Conformational Constraint in Oxazolidinone Antibacterials. Synthesis and Structure−Activity Studies of (Azabicyclo[3.1.0]Hexylphenyl)Oxazolidinones. J. Med. Chem 2005, 48 (15), 5009–5024. 10.1021/jm058204j. [DOI] [PubMed] [Google Scholar]
- 40.Fournier P-E; Vallenet D; Barbe V; Audic S; Ogata H; Poirel L; Richet H; Robert C; Mangenot S; Abergel C; Nordmann P; Weissenbach J; Raoult D; Claverie J-M Comparative Genomics of Multidrug Resistance in Acinetobacter Baumannii. PLOS Genet 2006, 2 (1), e7. 10.1371/journal.pgen.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon E-J; Courvalin P; Grillot-Courvalin C RND-Type Efflux Pumps in Multidrug-Resistant Clinical Isolates of Acinetobacter Baumannii: Major Role for AdeABC Overexpression and AdeRS Mutations. Antimicrob. Agents Chemother 2013, 57 (7), 2989–2995. 10.1128/aac.02556-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leus IV; Weeks JW; Bonifay V; Shen Y; Yang L; Cooper CJ; Nath D; Duerfeldt AS; Smith JC; Parks JM; Rybenkov VV; Zgurskaya HI Property Space Mapping of Pseudomonas Aeruginosa Permeability to Small Molecules. Sci. Rep 2022, 12 (1), 8220. 10.1038/s41598-022-12376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehla J; Malloci G; Mansbach R; López CA; Tsivkovski R; Haynes K; Leus IV; Grindstaff SB; Cascella RH; D’Cunha N; Herndon L; Hengartner NW; Margiotta E; Atzori A; Vargiu AV; Manrique PD; Walker JK; Lomovskaya O; Ruggerone P; Gnanakaran S; Rybenkov VV; Zgurskaya HI Predictive Rules of Efflux Inhibition and Avoidance in Pseudomonas Aeruginosa. mBio 2021, 12 (1), 10.1128/mbio.02785-20. 10.1128/mbio.02785-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooper CJ; Krishnamoorthy G; Wolloscheck D; Walker JK; Rybenkov VV; Parks JM; Zgurskaya HI Molecular Properties That Define the Activities of Antibiotics in Escherichia Coli and Pseudomonas Aeruginosa. ACS Infect. Dis 2018, 4 (8). 10.1021/acsinfecdis.8b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker EN; Drown BS; Geddes EJ; Lee HY; Ismail N; Lau GW; Hergenrother PJ Implementation of Permeation Rules Leads to a FabI Inhibitor with Activity against Gram-Negative Pathogens. Nat. Microbiol 2020, 5 (1), 67–75. 10.1038/s41564-019-0604-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Motika SE; Ulrich RJ; Geddes EJ; Lee HY; Lau GW; Hergenrother PJ Gram-Negative Antibiotic Active Through Inhibition of an Essential Riboswitch. J. Am. Chem. Soc 2020, 142 (24), 10856–10862. 10.1021/jacs.0c04427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perlmutter SJ; Geddes EJ; Drown BS; Motika SE; Lee MR; Hergenrother PJ Compound Uptake into E. Coli Can Be Facilitated by N-Alkyl Guanidiniums and Pyridiniums. ACS Infect. Dis 2021, 7 (1), 162–173. 10.1021/acsinfecdis.0c00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masci D; Hind C; Islam MK; Toscani A; Clifford M; Coluccia A; Conforti I; Touitou M; Memdouh S; Wei X; La Regina G; Silvestri R; Sutton JM; Castagnolo D Switching on the Activity of 1,5-Diaryl-Pyrrole Derivatives against Drug-Resistant ESKAPE Bacteria: Structure-Activity Relationships and Mode of Action Studies. Eur. J. Med. Chem 2019, 178, 500–514. 10.1016/j.ejmech.2019.05.087. [DOI] [PubMed] [Google Scholar]
- 49.Andrews LD; Kane TR; Dozzo P; Haglund CM; Hilderbrandt DJ; Linsell MS; Machajewski T; McEnroe G; Serio AW; Wlasichuk KB; Neau DB; Pakhomova S; Waldrop GL; Sharp M; Pogliano J; Cirz RT; Cohen F Optimization and Mechanistic Characterization of Pyridopyrimidine Inhibitors of Bacterial Biotin Carboxylase. J. Med. Chem 2019, 62 (16), 7489–7505. 10.1021/acs.jmedchem.9b00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y; Gardner JJ; Fortney KR; Leus IV; Bonifay V; Zgurskaya HI; Pletnev AA; Zhang S; Zhang Z-Y; Gribble GW; Spinola SM; Duerfeldt AS First-Generation Structure-Activity Relationship Studies of 2,3,4,9-Tetrahydro-1H-Carbazol-1-Amines as CpxA Phosphatase Inhibitors. Bioorg. Med. Chem. Lett 2019, 29 (14), 1836–1841. 10.1016/j.bmcl.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fortney KR; Smith SN; van Rensburg JJ; Brothwell JA; Gardner JJ; Katz BP; Ahsan N; Duerfeldt AS; Mobley HLT; Spinola SM CpxA Phosphatase Inhibitor Activates CpxRA and Is a Potential Treatment for Uropathogenic Escherichia Coli in a Murine Model of Infection. Microbiol. Spectr 2022, 10 (2), e02430–21. 10.1128/spectrum.02430-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu Z; Leus IV; Chandar B; Sherborne BS; Avila QP; Rybenkov VV; Zgurskaya HI; Duerfeldt AS Structure–Uptake Relationship Studies of Oxazolidinones in Gram-Negative ESKAPE Pathogens. J. Med. Chem 2022, 65 (20), 14144–14179. 10.1021/acs.jmedchem.2c01349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baell J; Walters MA Chemistry: Chemical Con Artists Foil Drug Discovery. Nature 2014, 513 (7519), 481–483. 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 54.Hansch C Quantitative Approach to Biochemical Structure-Activity Relationships. Acc. Chem. Res 1969, 2 (8), 232–239. 10.1021/ar50020a002. [DOI] [Google Scholar]
- 55.Masi M; Réfregiers M; Pos KM; Pagès J-M Mechanisms of Envelope Permeability and Antibiotic Influx and Efflux in Gram-Negative Bacteria. Nat. Microbiol 2017, 2 (3), 1–7. 10.1038/nmicrobiol.2017.1. [DOI] [PubMed] [Google Scholar]
- 56.Nikaido H; Rosenberg EY; Foulds J Porin Channels in Escherichia Coli: Studies with Beta-Lactams in Intact Cells. J. Bacteriol 1983, 153 (1), 232–240. 10.1128/jb.153.1.232-240.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kojima S; Nikaido H Permeation Rates of Penicillins Indicate That Escherichia Coli Porins Function Principally as Nonspecific Channels. Proc. Natl. Acad. Sci 2013, 110 (28), E2629–E2634. 10.1073/pnas.1310333110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cowan S; Garavito R; Jansonius J; Jenkins J; Karlsson R; König N; Pai E; Pauptit R; Rizkallah P; Rosenbusch J; Rummel G; Schirmer T The Structure of OmpF Porin in a Tetragonal Crystal Form. Structure 1995, 3 (10), 1041–1050. 10.1016/S0969-2126(01)00240-4. [DOI] [PubMed] [Google Scholar]
- 59.Baslé A; Rummel G; Storici P; Rosenbusch JP; Schirmer T Crystal Structure of Osmoporin OmpC from E. Coli at 2.0 Å. J. Mol. Biol 2006, 362 (5), 933–942. 10.1016/j.jmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 60.Simonet V; Malléa M; Pagès J-M Substitutions in the Eyelet Region Disrupt Cefepime Diffusion through the Escherichia Coli OmpF Channel. Antimicrob. Agents Chemother 2000, 44 (2), 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vidal S; Bredin J; Pagès J-M; Barbe J β-Lactam Screening by Specific Residues of the OmpF Eyelet. J. Med. Chem 2005, 48 (5), 1395–1400. 10.1021/jm049652e. [DOI] [PubMed] [Google Scholar]
- (62).Acosta-Gutierrez S; Scorciapino MA; Bodrenko I; Ceccarelli M Filtering with Electric Field: The Case of E. Coli Porins. J. Phys. Chem. Lett 2015, 6 (10), 1807–1812. 10.1021/acs.jpclett.5b00612. [DOI] [PubMed] [Google Scholar]
- 63.Bajaj H; Acosta Gutierrez S; Bodrenko I; Malloci G; Scorciapino MA; Winterhalter M; Ceccarelli M Bacterial Outer Membrane Porins as Electrostatic Nanosieves: Exploring Transport Rules of Small Polar Molecules. ACS Nano 2017, 11 (6), 5465–5473. 10.1021/acsnano.6b08613. [DOI] [PubMed] [Google Scholar]
- 64.Westfall DA; Krishnamoorthy G; Wolloscheck D; Sarkar R; Zgurskaya HI; Rybenkov VV Bifurcation Kinetics of Drug Uptake by Gram-Negative Bacteria. PLOS ONE 2017, 12 (9), e0184671. 10.1371/journal.pone.0184671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen AY; Yu C; Bodley A; Peng LF; Liu LF A New Mammalian DNA Topoisomerase I Poison Hoechst 33342: Cytotoxicity and Drug Resistance in Human Cell Cultures. Cancer Res 1993, 53 (6), 1332–1337. [PubMed] [Google Scholar]
- 66.2019-Ar-Threats-Report-508.Pdf https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed 2023–09-04).
- 67.Shahlaei M Descriptor Selection Methods in Quantitative Structure–Activity Relationship Studies: A Review Study. Chem. Rev 2013, 113 (10), 8093–8103. 10.1021/cr3004339. [DOI] [PubMed] [Google Scholar]
- 68.Kövesdi I; Dominguez-Rodriguez MF; Orfi L; Náray-Szabó G; Varró A; Papp JG; Mátyus P Application of Neural Networks in Structure-Activity Relationships. Med. Res. Rev 1999, 19 (3), 249–269. . [DOI] [PubMed] [Google Scholar]
- 69.Murcia-Soler M; Pérez-Giménez F; García-March FJ; Salabert-Salvador MT; Díaz-Villanueva W; Castro-Bleda MJ; Villanueva-Pareja A Artificial Neural Networks and Linear Discriminant Analysis: A Valuable Combination in the Selection of New Antibacterial Compounds. J. Chem. Inf. Comput. Sci 2004, 44 (3), 1031–1041. 10.1021/ci030340e. [DOI] [PubMed] [Google Scholar]
- 70.Gurvic D; Leach AG; Zachariae U Data-Driven Derivation of Molecular Substructures That Enhance Drug Activity in Gram-Negative Bacteria. J. Med. Chem 2022, 65 (8), 6088–6099. 10.1021/acs.jmedchem.1c01984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stokes JM; Yang K; Swanson K; Jin W; Cubillos-Ruiz A; Donghia NM; MacNair CR; French S; Carfrae LA; Bloom-Ackermann Z; Tran VM; Chiappino-Pepe A; Badran AH; Andrews IW; Chory EJ; Church GM; Brown ED; Jaakkola TS; Barzilay R; Collins JJ A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180 (4), 688–702.e13. 10.1016/j.cell.2020.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu G; Catacutan DB; Rathod K; Swanson K; Jin W; Mohammed JC; Chiappino-Pepe A; Syed SA; Fragis M; Rachwalski K; Magolan J; Surette MG; Coombes BK; Jaakkola T; Barzilay R; Collins JJ; Stokes JM Deep Learning-Guided Discovery of an Antibiotic Targeting Acinetobacter Baumannii. Nat. Chem. Biol 2023, 1–9. 10.1038/s41589-023-01349-8. [DOI] [PubMed] [Google Scholar]
- 73.Prajapati JD; Kleinekathöfer U; Winterhalter M How to Enter a Bacterium: Bacterial Porins and the Permeation of Antibiotics. Chem. Rev 2021, 121 (9), 5158–5192. 10.1021/acs.chemrev.0c01213. [DOI] [PubMed] [Google Scholar]
- 74.Iyer R; Ye Z; Ferrari A; Duncan L; Tanudra MA; Tsao H; Wang T; Gao H; Brummel CL; Erwin AL Evaluating LC–MS/MS To Measure Accumulation of Compounds within Bacteria. ACS Infect. Dis 2018, 4 (9), 1336–1345. 10.1021/acsinfecdis.8b00083. [DOI] [PubMed] [Google Scholar]
- 75.Widya M; Pasutti WD; Sachdeva M; Simmons RL; Tamrakar P; Krucker T; Six DA Development and Optimization of a Higher-Throughput Bacterial Compound Accumulation Assay. ACS Infect. Dis 2019, 5 (3), 394–405. 10.1021/acsinfecdis.8b00299. [DOI] [PubMed] [Google Scholar]
- 76.Zhou Y; Joubran C; Miller-Vedam L; Isabella V; Nayar A; Tentarelli S; Miller A Thinking Outside the “Bug”: A Unique Assay To Measure Intracellular Drug Penetration in Gram-Negative Bacteria. Anal. Chem 2015, 87 (7), 3579–3584. 10.1021/ac504880r. [DOI] [PubMed] [Google Scholar]