

Abstract
Purpose
The aim of this study was to investigate the efficacy and safety of orally administered branched-chain amino acids (BCAAs) on disease progression in patients with retinitis pigmentosa (RP).
Methods
A double-blind, randomized, placebo-controlled study was conducted at the Kyoto University Hospital. Seventy patients with RP aged 20 years or above were randomly assigned to the TK-98 (a combination of BCAAs in granule form) or placebo group. One packet (4.15 g) of the study drug was administered orally thrice daily for 78 weeks.
Results
There was no significant difference in the rate of change in the total point score, the primary endpoint, between the TK-98 (−52.4 ± 10.3 dB/year) and placebo (−42.9 ± 13.8 dB/year) groups. Ellipsoid zone length decreased by −76.5 ± 8.9 and −95.5 ± 12.2 µm/year in the TK-98 and placebo groups, respectively; although this difference was not significant, the TK-98 group showed slower degeneration. No serious adverse events were associated with the oral administration of TK-98 in patients with RP.
Conclusions
This study did not yield conclusive evidence supporting BCAA combination granules’ effectiveness in slowing visual field progression in patients with RP. An insignificant trend toward a slower reduction in ellipsoid zone length was found in morphological tests. Further studies are required to fully understand the potential benefits of BCAA supplementation in RP.
Translational Relevance
Our study demonstrates the safety of administering BCAAs to patients with RP. Accordingly, larger, more homogeneous clinical studies with longer durations may suggest their potential as therapeutic agents.
Keywords: neuroprotection, retinitis pigmentosa, BCAA
Introduction
Retinitis pigmentosa (RP) is a retinal disease primarily affecting photoreceptors and pigment epithelial cells. Photoreceptor cell death progresses gradually, typically resulting in night blindness, narrowing of the visual field, and loss of vision. It is caused by genetic variants, and more than 60 causative genes have been identified (https://sph.uth.edu/Retnet/). The disease affects one in every 4000 to 5000 people,1,2 with more than 1.5 million patients worldwide. Various treatment methods have been assessed, including gene therapy, regenerative medicine, and artificial retina.3 For example, in recent years, Luxturna, an adeno-associated virus vector-based gene therapy, has been approved worldwide and administered to patients with RP caused by retinal pigment epithelium-specific 65 kDa protein (RPE65) gene mutations.4,5 Clinical studies of gene therapy for RP have been reported for several other genes6,7 along with RPE65. In contrast, transplantation therapies using induced pluripotent stem (iPS) cells or embryonic stem (ES) cells,8 as well as treatments using artificial retina,9–12 currently tend to be used for patients with advanced stages of the disease, specifically those with poor visual function. In contrast, numerous studies have explored drug therapies that can stop or slow disease progression, regardless of the genetic variant. Based on the results of randomized controlled trials, oral administration of vitamin A13 or docosahexaenoic acid14,15 slows RP progression; however, recent research has raised questions regarding their efficacy.16 Oral administration of the antioxidant N-acetylcysteine in patients with RP improves visual acuity and suppresses the decline in retinal sensitivity after 12 weeks.17,18 Additionally, 9-cis-retinyl acetate, a synthetic 9-cis retinoid precursor, improves visual function when administered to patients with RPE65- or LRAT-related retinitis pigmentosa.19–21 However, no established treatment can stop or slow disease progression regardless of the genetic variant.
We focused on decreasing intracellular energy, specifically adenosine triphosphate (ATP) levels, during the processes of cell death and cellular degeneration induced by various stressors in different cultured cells, including neurons, to develop neuroprotective therapies that suppress the degeneration of photoreceptors and RPE cells in RP. We hypothesized that if we could inhibit the decrease in intracellular ATP levels, it may be possible to suppress cell death and cellular degeneration. One approach is to suppress cellular ATP consumption. Kyoto University Substances (KUSs) inhibit the ATPase activity of valosin-containing protein and suppress the decrease in intracellular ATP concentration and endoplasmic reticulum stress.22 This results in the suppression of cell death both in vitro and in vivo. KUSs slow down the disease progression in animal models of retinal diseases, including RP,22,23 and may exert a protective effect on retinal neurons, as evidenced in a clinical trial of central retinal artery occlusion.24
Another approach for maintaining ATP levels is to enhance intracellular ATP production. Branched-chain amino acids (BCAAs), including isoleucine, leucine, and valine, are essential for humans. We focused on using BCAAs for energy production by energy-demanding neurons and muscles. Using cultured cells (HeLa and 661W), we found that BCAAs increased intracellular glucose uptake and enhanced intracellular ATP production by recruiting glucose transporters to the plasma membrane.25 Additionally, we demonstrated that administration of BCAAs slows photoreceptor and retinal degeneration in animal models, including rd10 and rd12 mice.26 Thus BCAAs may provide a potential treatment strategy for RP, regardless of genetic variants.
TK-98 is the investigational drug identification code for LIVACT combination granules, containing three active ingredients (L-isoleucine, L-leucine, and L-valine) in the ratio of 1.0:2.0:1.2. LIVACT combination granules, which have been approved for improving hypoalbuminemia in patients with non-compensated liver cirrhosis who suffer from hypoalbuminemia despite adequate dietary intake, are currently being marketed in some countries including Japan (manufacturer and distributor: EA Pharma Co., Ltd., Tokyo, Japan).
We aim to develop TK-98 as a treatment for patients with RP, with the additional indication of “slowing visual function loss in RP.” We conducted a double-blind, randomized, placebo-controlled, comparative, investigator-initiated clinical trial to assess the inhibitory effect of orally administered BCAAs (TK-98) on disease progression in patients with RP.
Methods
Trial Design
In this exploratory, placebo-controlled, double-blind, comparative study, we assessed the efficacy and safety of TK-98 in patients with RP (https://jrct.niph.go.jp/en-latest-detail/jRCT2051180072).
Ethical Considerations
This clinical trial was conducted in strict adherence to ethical principles derived from the Declaration of Helsinki (1964) and its subsequent revisions, the “Law Concerning the Quality, Efficacy and Safety of Drugs, Medical Devices and Other Products,” the “Ministerial Ordinance on Standards for the Conduct of Clinical Trials of Drugs” (GCP Ordinance), as well as its related notifications and procedures, and the designed protocol for the clinical trial. This investigator-initiated clinical trial plan was developed based on the results of a face-to-face advisory meeting with the Pharmaceuticals and Medical Devices Agency of JAPAN. This study was approved by the Kyoto University Institutional Review Board (K051).
Participants
The main inclusion criteria were as follows: (1) patients diagnosed with typical RP, (2) patients aged 20 years or above, and (3) patients who provided written informed consent for participation in the study. The main exclusion criteria were as follows: (1) eyes in which the difference between the mean deviation (MD) values of two consecutive Humphrey visual field tests (10-2 Swedish interactive threshold algorithm standard program; HFA 10-2) in the last four or more tests (1–3 years before registration) was greater than 5.0 dB at least twice, (2) eyes with an MD value greater than −5.0 dB or less than −25.0 dB in the HFA10-2 before registration, (3) eyes with a history of internal ocular surgery other than cataract surgery, (4) eyes with cataracts equal to or greater than Nuclear Sclerosis grade 3 or with posterior subcapsular opacity progression within the previous year, (5) patients taking calcium blockers or sodium valproate, (6) patients taking medications or supplements containing dark-adjusted drugs, vitamins A and E, docosahexaenoic acid, taurine, lutein, or amino acids, and (7) patients who were using isopropyl unoprostone ophthalmic solution or brimonidine tartrate ophthalmic solution at that time.
The target number of patients was 70. This study was an exploratory phase II trial conducted within a limited budget involving a restricted number of patients. We recruited patients who had participated in natural history observation studies for one year or more for comparison with the progression of visual fields before treatment. Consequently, we could accumulate data from a maximum of 74 patients. We tested the difference in the mean change per year in the total point score (TPS, see below) on the HFA10-2 between the TK-98 and placebo groups using a mixed-effects linear model that considers the correlation between the left and right eyes. From a naturalistic observational study, the mean change per year in the primary endpoint “TPS of retinal sensitivity in the HFA 10-2” was −41.2 dB/year with a standard deviation of approximately 56.5 dB/year (unpublished data May 16, 2018). Based on clinical judgment, the effect we wanted to detect was set at 24.72 dB/year, or 60% of −41.2 dB/year, with a standard deviation of 56.5 dB/year. We assumed a correlation coefficient of 0.5, which should be considered when analyzing binocular data from the same patient. A two-tailed test using a mixed-effects linear model for the null hypothesis (i.e., mean of TK-98 group ≠ mean of placebo group).
The TK-98 and placebo groups were allocated in a 2:1 ratio. The rationale for this allocation was based on our previous animal experimental studies, which indicated that assigning more participants to the treatment arm would be more beneficial for patients. In reality, the trial encompassed 134 eyes (TK-98, 45 patients [43 with both eyes, two with one eye each]; Placebo, 25 patients [21 with both eyes, four with one eye each]), resulting in a power of 75.3% to detect significance at α = 20%.
Treatments
After enrollment, one packet (4.15 g) of the study drug was orally administered three times daily after meals. The study drug contained the following ingredients in one packet (4.15 g): 952 mg L-isoleucine, 1904 mg L-leucine, and 1144 mg L-valine (TK-98, the same ingredients as LIVACT combination granules). The placebo was a powdered formulation without BCAAs. The treatment and placebo packets were both packaged in aluminum foil for an identical appearance. Both the treatment and placebo packets contained similar white powdered granules, and their indistinguishability was confirmed by the trial's medication allocation supervisor. BCAAs are readily soluble in citric acid, slightly soluble in water, and almost insoluble in ethanol (95%). They dissolve readily in dilute hydrochloric acid. They are stable for three years when stored at room temperature. These granules are consumed directly with water.
The treatment duration was determined based on previous clinical studies on RP, which have investigated visual field progression over durations ranging from six months to four years. Specifically, previous studies, including our exploratory natural history observation study, suggested that cases with MD values ranging from −5.0 to −25.0 dB exhibited a faster rate of progression.27,28 The frequency of visual field tests was set to seven to assess the treatment effect over a shorter period. Therefore, to ensure effective capture of TK-98's “inhibition of visual field deterioration” effect, a treatment duration of one and a half years (78 weeks) was selected for this clinical trial, without interim evaluations. After the initiation of drug administration, visits were scheduled at four, 13, 26, 39, 52, 65, and 78 weeks. On each visit, participants were instructed to bring all remaining investigational drugs, and the coordinator calculated the number of doses taken during that period based on the prescribed amount and the remaining drug count. The medication compliance rate was calculated based on the total number of doses taken over the entire period. All patients were screened and followed up at Kyoto University Hospital with multiple visual field tests at the Morooka Eye Clinic (Kyoto, Japan).
Primary and Secondary Endpoints
The TPS of retinal sensitivity (the sum of the visual sensitivities for all 68 measured locations)29 by HFA10-2 using Carl Zeiss Meditec software (Carl Zeiss Meditec, Jena, Germany) was set as the primary endpoint to evaluate the efficacy of TK-98. Data were not considered reliable if the test result was ≥20% poor fixation, ≥15% false positive, or ≥33% false negative. If reliable data were not obtained at pre-enrollment testing and the end of the study drug administration, the test was repeated only once within two weeks. Adverse events and side effects were defined as secondary endpoints to investigate safety. For safety evaluation, hematology, urinalysis, and comprehensive ophthalmic examinations, including slit-lamp biomicroscopy, ophthalmoscopy, visual acuity, perimetry, and intraocular pressure measurements, were performed. All adverse events and side effects were recorded. Serious adverse events were defined as fatal or life-threatening, requiring hospitalization, causing permanent serious damage, or causing congenital anomalies. Other secondary endpoints included retinal sensitivity by HFA10-2 (mean deviation [MD] value), retinal sensitivity by HFA 30-2 (TPS), number of readable letters by the Early Treatment Diabetic Retinopathy Study (ETDRS) visual acuity test (under correction), and the logarithm of the minimum angle of resolution (logMAR) of the best-corrected visual acuity. High-resolution macular cross-sectional images were captured in follow-up mode using a Spectralis HRA+OCT system (Heidelberg Engineering, Heidelberg, Germany) to evaluate changes in retinal morphology. The ellipsoid zone length was assessed from these images. We measured the area of the relatively preserved retinal area in the macula through the analysis of fundus autofluorescence (FAF) images obtained using an Optos device (Nikon Corporation, Tokyo, Japan). In eyes displaying ring-shaped hyperautofluorescence, we quantified the area encompassed by this hyperautofluorescent ring. Conversely, in eyes without ring-shaped hyperautofluorescence, we measured the area of the macular region that exhibited no abnormal hypoautofluorescence.30 The ellipsoid zone length was determined as the average of the horizontal and vertical values.29 The frequency and timing of each test are summarized in Supplementary Table S1.
Randomization and Blinding
At the time of enrollment in the study, patients were randomly assigned to the “TK-98 group” or the “Placebo group.” A third party, independent of the patients, investigators, sub-investigators, and collaborators, randomly assigned the investigational drug to a set of six patients: four to the study drug, TK-98, and two to the placebo. The allocation adjustment factors were as follows. Stratification was performed in four strata: (1) age at the time of obtaining consent (less than 53 years old, 53 years old or older), (2) MD value before registration (less than −15.0 dB, equal or more than −15.0 dB). The mean values of both eyes were used when the study included both eyes. All patients, investigators, sub-investigators, and collaborators were blinded to the group until all results were confirmed.
Statistical Analysis
Patients who were enrolled in the study and received at least one dose of the study drug were defined as the largest analysis population (full analysis set [FAS]). Patients who received the study drug for at least 53 weeks had available measurements for the primary endpoint and did not seriously violate the study protocol (e.g., failure to comply with the enrollment criteria, failure to obtain consent, failure to enroll, or other serious violations of study procedures) or GCP violations were defined as the analysis population compliant with the study protocol (per protocol set [PPS]) and were subjected to efficacy evaluation. Any eye with cataract progression during the observation period, assessed by comparing slit-lamp biomicroscopy photographs taken at the beginning and end of the observation period, was excluded from the analysis of HFA and visual acuity in the PPS set. Patients who enrolled in the study and received any part of the study drug were defined as the safety analysis set (SAF) and used for safety evaluation.
The measured values for the primary and secondary endpoints were evaluated as the change in the amount per year, obtained by dividing the amount of change by the duration, as well as the change rate, calculated by dividing the amount of change by the baseline value and multiplying by 100. The measured values for HFA 10-2 were obtained from multiple valid tests (∼7 times), and the annual change amount and rate were calculated using the least-squares method. For the primary and secondary analyses, we calculated summary statistics. Group differences in mean values and their two-sided 95% confidence intervals (CI) were also calculated. A mixed-effects linear model that considered the correlation between the right and left eyes of the same patient was used to determine whether the TK-98 group was superior to the placebo group. We also performed a stratified analysis of the primary endpoint by TPS value of Humphrey visual field test 30-2 at the time of allocation (<400 dB), age at the time of inclusion (≥50 years), time from onset of first subjective symptoms (≥30 years), sex, and presumed causative gene (EYS). All significance tests were performed at α level = 0.05 (two-sided).
Results
Patient Characteristics
This investigator-initiated clinical trial was conducted at Kyoto University Hospital from March 2019 to December 2020 and involved 70 patients with RP. The average age was 51.7 ± 12.6 (range 21–77) years, and the average MD value of HFA 10-2 at inclusion was −14.79 ± 4.96 dB (Tables 1 and 2). The presumed causative gene was found in 42.9% of the patients (30 patients), whereas EYS was found in 24.3% (17 patients) (Table 1, Supplementary Table S2). Enrolled patients were randomly assigned; 45 (88 eyes) to the TK-98 group and 25 (46 eyes) to the placebo group. Patient characteristics are shown in the Figure, and the baseline characteristics are summarized in Tables 1 and 2 and Supplementary Tables S3 and S4. There were no significant differences between the two groups, except for the lens status (phakia or pseudophakia, P = 0.004), MD slope at the start (calculated by least square methods using visual field exam results within one to three years before inclusion, including the test at inclusion) (P = 0.081), ellipsoid zone length at the start (P = 0.049), and visual acuity at the start (P = 0.092 and 0.087 for readable letter counts and logMAR, respectively) (Table 2, Supplementary Table S4).
Table 1.
Baseline Characteristics of the Patients
| TK-98 (n = 45 Patients) | Placebo (n = 25 Patients) | Total (n = 70 Patients) | |||||
|---|---|---|---|---|---|---|---|
| Characteristic | Mean ± SD | Median (Range) | Mean ± SD | Median (Range) | Mean ± SD | Median (Range) | Intergroup Comparison P Value |
| Sex | |||||||
| Female | 24 (53.3%) | 14 (56.0%) | 38 (54.3%) | ||||
| Male | 21 (46.7%) | 11 (44.0%) | 32 (45.7%) | >0.999 | |||
| Age at time of inclusion (yr) | 51.7 ± 11.9 | 54.0 (21, 77) | 51.6 ± 14.2 | 50.0 (26, 75) | 51.7 ± 12.6 | 52.5 (21, 77) | 0.977 |
| MD value at the time of allocation (average for binocular cases), dB | −14.85 ± 4.93 | −14.74 (−23.14, −5.12) | −14.67 ± 5.12 | −13.08 (−23.96, −5.91) | −14.79 ± 4.96 | −14.25 (−23.96, −5.12) | 0.884 |
| Unilateral and binocular cases | 0.177 | ||||||
| Unilateral | 2 (4.4) | 4 (16.0) | 6 (8.6) | ||||
| Binocular | 43 (95.6) | 21 (84.0) | 64 (91.4) | ||||
| Genetic form, n patients | 0.444 | ||||||
| Autosomal dominant | 10 (22.2%) | 2 (8.0%) | 12 (17.1%) | ||||
| Autosomal recessive | 18 (40.0%) | 12 (48.0%) | 30 (42.9%) | ||||
| X-linked | 1 (2.2%) | 1 (4.0%) | 2 (2.9%) | ||||
| Sporadic | 16 (35.6%) | 10 (40.0%) | 26 (37.1%) | ||||
| Presumed causative gene, n patients | 0.316 | ||||||
| Confirmed cases of causative genes | 17 (37.8%) | 13 (52.0%) | 30 (42.9%) | ||||
| EYS | 8 (17.8%) | 9 (36.0%) | 17 (24.3%) | 0.144 | |||
| Time from onset of first subjective symptoms (yr)* | 29.3 ± 13.9 | 30.0 (3, 61) | 27.0 ± 12.3 | 25.0 (9, 67) | 28.5 ± 13.3 | 28.0 (3, 67) | 0.485 |
dB, decibel; MD, mean deviation; SD, standard deviation.
Baseline characteristics of patients in the full analysis set (FAS).
Time since onset of any of the following symptoms: night blindness, narrowing of the field of vision, loss of vision, or photophobia.
Table 2.
Baseline Characteristics of the Eyes
| TK-98 (n = 88 Eyes) | Placebo (n = 46 Eyes) | Total (n = 134 Eyes) | |||||
|---|---|---|---|---|---|---|---|
| Characteristic | Mean ± SD | Median (Range) | Mean ± SD | Median (Range) | Mean ± SD | Median (Range) | Intergroup Comparison, P Value |
| Lens, n eyes | 0.004 | ||||||
| Phakia | 76 (86.4%) | 29 (63.0%) | 105 (78.4%) | ||||
| Pseudophakia | 12 (13.6%) | 17 (37.0%) | 29 (21.6%) | ||||
| Humphrey visual field test 10-2 | |||||||
| TPS (dB) | 1223.0 ± 332.8 | 1212.0 (615, 1901) | 1245.3 ± 363.7 | 1284.0 (586, 1836) | 1230.7 ± 342.5 | 1217.0 (586, 1901) | 0.591 |
| MD value (dB) | −14.9 ± 4.9 | −14.6 (−23.7, −5.0) | −14.4 ± 5.2 | −13.9 (−24.0, −5.6) | −14.7 ± 5.0 | −14.3 (−24.0, −5.0) | 0.496 |
| MD slope (dB/y)* | −0.12 ± 0.92 | −0.31 (−1.98, 3.44) | −0.52 ± 1.05 | −0.45 (−4.27, 1.21) | −0.26 ± 0.98 | −0.38 (−4.27, 3.44) | 0.081 |
| Humphrey visual field test 30-2 | |||||||
| TPS (dB) | 478.7 ± 350.3 | 357.0 (80, 1628) | 458.2 ± 306.8 | 343.0 (101, 1223) | 471.8 ± 335.2 | 356.0 (80, 1628) | 0.884 |
| Visual acuity (ETDRS) | |||||||
| Readable letter count | 65.2 ± 17.6 | 71.0 (17, 86) | 69.1 ± 15.1 | 73.0 (9, 87) | 66.5 ± 16.8 | 72.0 (9, 87) | 0.092 |
| LogMAR | 0.35 ± 0.32 | 0.26 (−0.12, 1.18) | 0.28 ± 0.27 | 0.22 (−0.06, 1.38) | 0.33 ± 0.30 | 0.24 (−0.12, 1.38) | 0.087 |
| Ellipsoid zone length (µm) | 1541.8 ± 1362.4 | 1414.8 (0.0, 5042.5) | 2028.0 ± 1308.2 | 1913.5 (0.0, 5861.0) | 1708.7 ± 1359.0 | 1620.3 (0.0, 5861.0) | 0.049 |
| Preserved retinal area (mm2) | 18.7 ± 14.9 | 15.2 (1.6, 58.1) | 17.8 ± 12.6 | 14.5 (2.3, 41.9) | 18.4 ± 14.1 | 14.9 (1.6, 58.1) | 0.730 |
Baseline characteristics of the eyes in the FAS.
Calculated using MD values observed up to three years before the time of observation at the start of the study (including the time of observation at the start of the pretreatment period).
Figure.
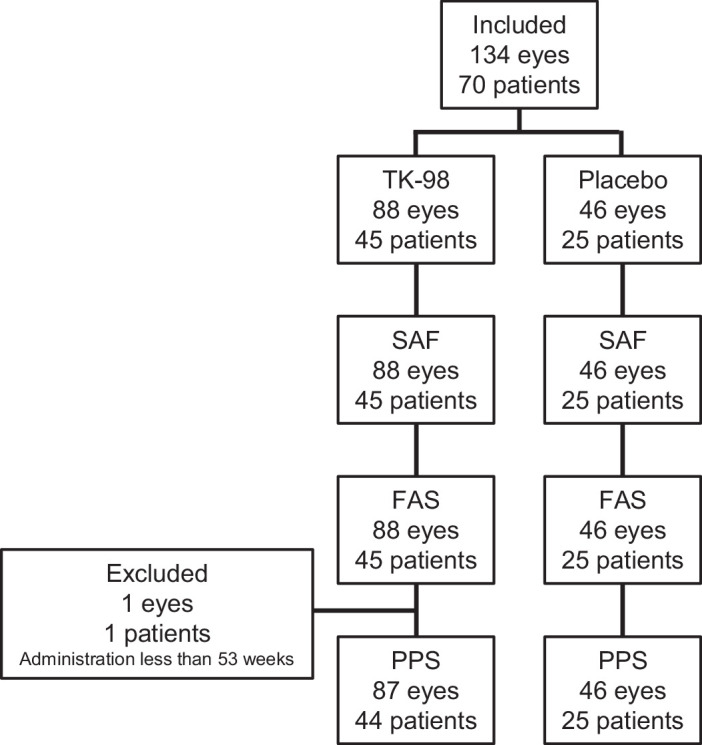
Patient disposition.
Treatment Compliance
Calculated compliance was 90% or higher for 88.9% of the patients in the TK-98 group and 96% of the patients in the placebo group (Table 3). Two patients in the TK-98 group discontinued the trial due to personal reasons. One patient (one eye) was excluded from the PPS analysis because the duration of the study drug administration was less than 53 weeks (Figure).
Table 3.
Compliance Rate
| TK-98 (n = 45 Patients) | Placebo (n = 25 Patients) | |
|---|---|---|
| ≥95% | 30 (66.7) | 20 (80.0) |
| ≥90% | 40 (88.9) | 24 (96.0) |
| ≥70%, <90% | 4 (8.9) | 1 (4.0) |
| <70% | 1 (2.2) | 0 (0.0) |
Adherence to the study drug in the SAF population was calculated.
Treatment Efficacy
The results of the HFA 10-2, used as the primary endpoint, were valid six times or more in more than 90% of the eyes (Supplementary Table S5). The one-year equivalent change in the TPS for retinal sensitivity of the HFA10-2 test performed for the FAS population is shown in Table 4. The analysis using the linear mixed-effects model, which considers the correlation between the right and left eyes in the same subject, suggested that the difference between the groups in the least squares mean was not statistically significant (−52.4 ± 10.3 vs. −42.9 ± 13.8 dB/y [least-squares mean ± standard error], TK-98 vs. placebo group, respectively, P = 0.582). The results of the secondary endpoints, including the MD on the HFA 10-2 test, TPS on the HFA 30-2 test, BCVA, ellipsoid zone length on the OCT test, and relatively preserved retinal area on the FAF images, are shown in Table 5 (FAS population) and Supplementary Table S6 (PPS population). The difference between the two groups in any of the endpoints was not statistically significant. However, the TK group tended to have less deterioration in the morphological examinations (e.g., ellipsoid zone length: −76.4 ± 66.4 vs. −95.6 ± 78.3 µm/y, P = 0.215 [FAS population]). Two eyes in the placebo group showed cataract progression; therefore they were excluded from the visual field and visual acuity analyses in the PPS population.
Table 4.
Main Outcome Measurement
| Intergroup Comparison | |||||||
|---|---|---|---|---|---|---|---|
| Case | Eye | Mean ± SD | Median (Range) | Least-Squares Mean ± SE | Difference in Least-Squares Mean (95% CI) | P Value | |
| TK-98 | 45 | 88 | −52.8 ± 75.0 | −40.9 (−480.6, 117.5) | −52.4 ± 10.3 | −9.5 (−44.0, 24.9) | 0.582 |
| Placebo | 25 | 46 | −38.5 ± 63.8 | −41.4 (−192.7, 113.4) | −42.9 ± 13.8 | ||
Changes in the total point score per year on the Humphrey visual field test (10-2) were analyzed in the FAS population. The coefficients of change per year were obtained using a linear simple regression analysis based on the least squares method, with the evaluation value from the start to 78 weeks (or at the time of discontinuation for the discontinued eye) as the objective variable and the time point as the explanatory variable. The least-squares mean is the mean value for each group estimated using a linear mixed-effects model that considers the correlation between the two eyes. The P values represent the results of a two-tailed test of the null hypothesis “the difference between the least squares means of the two groups is zero” at a significance level of α = 0.05.
Table 5.
Secondary Outcome Measurements
| TK-98 | Placebo | Intergroup comparison | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Eye | Mean ± SD | Median (Range) | Least-Squares Mean ± SE | Case | Eye | Mean ± SD | Median (Range) | Least-Squares Mean ± SE | Difference in Least-Squares Mean (95% CI) | P Value | |
| Humphrey visual field test 10-2 | ||||||||||||
| TPS, dB | ||||||||||||
| CR | 45 | 88 | −5.0 ± 8.9 | −4.0 (−70.4, 9.9) | −4.9 ± 1.2 | 25 | 46 | −4.0 ± 6.7 | −3.5 (−29.8, 7.3) | −5.0 ± 1.6 | 0.1 (−3.9, 4.0) | 0.969 |
| MD value, dB | ||||||||||||
| CA | 45 | 88 | −0.76 ± 1.28 | −0.62 (−9.10, 1.90) | −0.74 ± 0.17 | 25 | 46 | −0.57 ± 0.99 | −0.64 (−3.18, 1.72) | −0.68 ± 0.23 | −0.05 (−0.62, 0.52) | 0.851 |
| CR | 45 | 88 | −5.6 ± 8.6 | −4.3 (−42.2, 13.0) | −5.6 ± 1.3 | 25 | 46 | −4.1 ± 9.5 | −4.9 (−28.1, 27.7) | −4.1 ± 1.7 | −1.5 (−5.7, 2.8) | 0.490 |
| Humphrey visual field test 30-2 | ||||||||||||
| TPS, dB | ||||||||||||
| CA | 44 | 85 | −37.8 ± 57.6 | −21.7 (−241.9, 47.4) | −38.0 ± 7.4 | 25 | 45 | −21.8 ± 56.6 | −19.7 (−237.9, 145.6) | −20.2 ± 10.1 | −17.7 (−42.7, 7.3) | 0.161 |
| CR | 44 | 85 | −6.4 ± 10.8 | −5.4 (−30.9, 29.5) | −6.3 ± 1.4 | 25 | 45 | −6.5 ± 12.7 | −5.4 (−40.3, 20.6) | −6.3 ± 1.9 | 0.0 (−4.8, 4.7) | 0.987 |
| Visual acuity (ETDRS) | ||||||||||||
| Readable letter count | ||||||||||||
| CA | 44 | 87 | −1.1 ± 2.9 | −0.7 (−13.9, 4.0) | −1.1 ± 0.3 | 25 | 46 | −0.7 ± 2.8 | −0.7 (−8.5, 6.6) | −0.8 ± 0.4 | −0.2 (−1.3, 0.9) | 0.699 |
| CR | 44 | 87 | −2.3 ± 7.3 | −1.3 (−38.5, 15.5) | −2.2 ± 0.7 | 25 | 46 | −1.3 ± 5.5 | −1.1 (−22.3, 12.7) | −1.4 ± 1.0 | −0.9 (−3.3, 1.6) | 0.477 |
| LogMAR | ||||||||||||
| CA | 44 | 87 | 0.026 ± 0.072 | 0.013 (−0.079, 0.396) | 0.026 ± 0.008 | 25 | 46 | 0.020 ± 0.061 | 0.013 (−0.132, 0.198) | 0.022 ± 0.011 | 0.004 (−0.023, 0.031) | 0.757 |
| CR | 44 | 87 | 16.6 ± 57.1 | 4.0 (−39.6, 330.2) | 12.0 ± 4.3 | 25 | 46 | 19.9 ± 55.3 | 8.3 (−31.5, 337.6) | 17.5 ± 5.8 | −5.5 (−19.9, 8.9) | 0.448 |
| Ellipsoid zone length, µm | ||||||||||||
| CA | 45 | 88 | −76.4 ± 66.4 | −69.7 (−371.2, 0.0) | −76.5 ± 8.9 | 25 | 46 | −95.6 ± 78.3 | −80.4 (−344.1, 0.0) | −95.5 ± 12.2 | 18.9 (−11.2, 49.1) | 0.215 |
| CR | 42 | 75 | −8.7 ± 9.2 | −5.0 (−46.4, −0.6) | −9.3 ± 1.3 | 25 | 44 | −6.9 ± 7.7 | −4.0 (−33.2, −0.1) | −7.4 ± 1.7 | −1.9 (−6.1, 2.3) | 0.371 |
| Preserved retinal area, mm2 | ||||||||||||
| CA | 44 | 84 | −0.92 ± 0.88 | −0.64 (−3.57, −0.01) | −0.93 ± 0.14 | 25 | 46 | −0.96 ± 1.19 | −0.36 (−5.29, −0.01) | −0.97 ± 0.18 | 0.05 (−0.41, 0.51) | 0.843 |
| CR | 44 | 84 | −5.5 ± 4.4 | −4.1 (−22.0, −0.1) | −5.5 ± 0.5 | 25 | 46 | −4.9 ± 3.7 | −3.7 (−13.8, −0.3) | −4.9 ± 0.7 | −0.6 (−2.4, 1.1) | 0.483 |
CA, change amount; CR, change rate.
Main second outcome measurements in the FAS Population. All changes (CA) were changes per year. The CR is the amount of change divided by the absolute value of the initial measurement and multiplied by 100. The least squares mean is the mean of each group estimated in a mixed-effects model that accounts for binocular correlation. The P values represent the results of a two-tailed test of the null hypothesis “the difference between the least squares means of the two groups is zero” at a significance level of α = 0.05.
The results of the stratified analysis of the PPS population for the primary endpoints are shown in Table 6. Although several strata (TPS value of Humphrey visual field test 30-2 at the time of allocation <400 dB, age ≥50 years, time from onset of first subjective symptoms ≥30 years, male, causative gene of EYS) showed greater least squares means in the TK treatment group, which means that the TK group tended to have less deterioration, none of the differences were significant.
Table 6.
Stratified Analysis
| TK-98 | Placebo | Intergroup Comparison | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Eye | Mean ± SD | Median (Range) | Least-Squares Mean ± SE | Case | Eye | Mean ± SD | Median (Range) | Least-Squares Mean ± SE | Difference in Least-Squares Mean (95% CI) | P Value | |
| TPS value of Humphrey visual field test 30-2 at the time of allocation <400 dB | 27 | 50 | −43.6 ± 60.0 | −39.1 (−183.6, 117.5) | −43.6 ± 10.2 | 15 | 23 | −55.2 ± 58.9 | −66.1 (−192.7, 74.1) | −53.5 ± 14.2 | 9.9 (−25.5, 45.3) | 0.576 |
| Age at time of inclusion ≥50 y | 26 | 51 | −39.5 ± 61.8 | −28.3 (−163.6, 117.5) | −40.5 ± 11.5 | 13 | 24 | −44.4 ± 67.9 | −54.9 (−192.7, 113.4) | −45.0 ± 16.4 | 4.5 (−36.0, 45.0) | 0.824 |
| Time from onset of first subjective symptoms ≥30 y | 22 | 43 | −42.2 ± 56.3 | −36.2 (−181.9, 57.7) | −41.5 ± 10.1 | 7 | 13 | −65.3 ± 52.6 | −59.0 (−192.7, 21.7) | −65.9 ± 18.2 | 24.4 (−18.4, 67.2) | 0.252 |
| Male sex | 21 | 42 | −42.0 ± 43.2 | −37.4 (−129.6, 32.5) | −42.4 ± 8.5 | 11 | 18 | −52.7 ± 46.2 | −61.1 (−127.5, 27.1) | −56.5 ± 12.2 | 14.2 (−16.2, 44.6) | 0.349 |
| Presumed causative gene, EYS | 8 | 16 | −46.1 ± 61.4 | −34.7 (−183.6, 29.6) | −49.4 ± 17.9 | 9 | 16 | −59.4 ± 59.2 | −57.6 (−192.7, 27.1) | −57.3 ± 17.4 | 7.9 (−45.3, 61.2) | 0.755 |
CA, change amount; CR, change rate.
Stratified analysis of the PPS population for the primary endpoint of change per year in TPS by the Humphrey visual field test (10-2) (dB/y). The P values represent the results of a two-tailed test of the null hypothesis “the difference between the least squares means of the two groups is zero” at a significance level of α = 0.05.
Safety of the Treatment
Adverse events were observed in 44 patients (97.8%) in the TK-98 group and 25 patients (100.0%) in the placebo group. Ophthalmological adverse events were observed in 39 patients (86.7%) in the TK-98 group and 22 patients (88.0%) in the placebo group (Supplementary Table S7). Cataract progression was observed in only three eyes of three patients in the placebo group (P = 0.042). The most common adverse event was visual field deterioration (negative MD slope on the HFA 10-2 tests during the study period). Serious adverse events that required hospitalization were observed in four patients (8.9%, hemorrhoid, contusion, leiomyoma of the uterus, and brain infarction) in the TK-98 group and two patients (8.0%, cataract and colorectal polyps) in the placebo group, which were ruled out to be associated with drug administration. Possible side effects related to the investigational drug, all gastrointestinal symptoms which were not severe, were observed in three patients (6.7%) in the TK-98 group and two patients (8.0%) in the placebo group (Table 7).
Table 7.
Side Effects
| TK-98 (n = 45 Patients) | Placebo (n = 25 Patients) | |||||||
|---|---|---|---|---|---|---|---|---|
| Event | Mild | Moderate | Severe | Total | Mild | Moderate | Severe | Total |
| Abdominal distension | 2 | 2 (4.4%) | 1 | 1 (4.0%) | ||||
| Constipation | 1 | 1 (2.2%) | ||||||
| Gastroesophageal reflux disease | 1 | 1 (4.0%) | ||||||
| Total | 3 (6.7%) | 2 (8.0%) | ||||||
Side effects of the investigational drugs in the SAF population.
Discussion
In this investigator-initiated clinical trial involving 70 patients with RP, we evaluated the efficacy and safety of TK-98 compared with a placebo. There was no statistically significant difference in the one-year equivalent change in the TPS for retinal sensitivity on HFA 10-2, the primary endpoint, between the TK-98 and placebo groups (Table 4). Similarly, the secondary endpoints were not different between the two groups, including MD on HFA 10-2, TPS on HFA 30-2, BCVA, ellipsoid zone length on OCT, and preserved retinal area on FAF images. However, there was a trend toward less deterioration in the morphological examinations in the TK group (Table 5). Safety analyses revealed that serious adverse events were infrequent and unrelated to the drug (Table 7).
In this clinical trial, we used age and baseline MD values for allocation, which was performed appropriately. However, there was a difference between the two groups in that the TK-98 group had a higher population of phakia, shorter ellipsoid zone length at baseline, slightly worse visual acuity, and a slower rate of decline in MD just before allocation, which was calculated based on examinations within one to three years, including the baseline examination (Table 2). In addition, because cataracts progress gradually, cataract progression can occur in phakic eyes even if it is not readily apparent on slit-lamp examination, although cases of clinically obvious cataract progression that were excluded from subjective tests, such as visual field testing, were only present in the placebo group. The higher proportion of phakic eyes in the TK-98 group at baseline than in the placebo group may have affected the evaluation of efficacy using visual fields. These factors may have influenced efficacy evaluation using the visual field.
Visual field testing, a subjective method, is used to evaluate RP progression in many clinical studies.15,31–37 However, drugs that have been definitively proven to effectively suppress visual field progression in RP, are lacking. In this study, our primary purpose was to use morphological indicators as the principal criteria for evaluating progression. However, because there was no precedent for accepting morphological examination as the primary endpoint by the Japanese regulatory agency Pharmaceuticals and Medical Devices Agency, we chose to primarily use the visual field test results. In previous clinical studies, static perimetry using the HFA 30-2 program was used.32,33 The 30-2 program comprises 76 test points, whereas the 10-2 program comprises 68 test points, indicating that the 10-2 program can capture changes in central vision more sensitively. Therefore in this study the total point score of the HFA 10-2 was chosen as the primary outcome measure, whereas HFA 30-2 was used as a secondary outcome measure. In the present study, to bolster the reliability of our analysis, we excluded patients showing substantial variability in visual field test results at baseline, filtered out unreliable test outcomes, and used multiple test results to calculate both the annual change in visual field tests and the rate of change in this parameter using the least-squares method. However, the correlation coefficient of linear regression for the visual field tests during the study period was lower than anticipated, with less than 25% of eyes in both the TK-98 and placebo groups showing a correlation coefficient of 0.5 or higher, suggesting that it is difficult to accurately determine efficacy using subjective testing of visual fields, especially within a relatively short period of 1.5 years to detect the progression of visual field in this study. For instance, several other clinical studies have assessed visual field progression over a period of four years.15,32 The standard deviation was large in both groups, particularly in the TK-98 group, indicating that it is challenging to compare the extent of progression between groups based on subjective testing.
Objective assessment of visual function may involve the use of electroretinography (ERG), with reports suggesting that changes in the amplitude of Flicker ERG serve as a good indicator.16 In this study, ERG was not adopted as an evaluation criterion since, at the initiation of the trial, it was nonrecordable for both rod and cone responses in 50 participants.
In contrast, the morphometric tests of ellipsoid zone length by OCT and the area of relatively preserved retina in the macula by fundus photography showed smaller deterioration in the TK-98 group than in the placebo group, suggesting the superiority of TK-98, although not statistically significant.
In a stratified analysis limited to more advanced cases, visual field testing was better in the TK-98 group compared with the placebo group (Table 5). In addition, the trend toward the superiority of TK-98 in subjects with EYS gene abnormalities, which is the most common genetic mutation in Japanese patients,38,39 suggests that TK-98 may be superior in a comparative study in a relatively homogeneous patient population.
The severity of the adverse events was mild or moderate, and no severe adverse events related to the medication were observed. All adverse reactions to the study medications were mild gastrointestinal symptoms, as stated in the attached TK-98 document. The only ophthalmologic adverse event with a statistically significant difference in incidence between groups was worsening of cataracts, which was higher in the placebo group (12.0% in placebo, 0% in TK-98, P = 0.042). Worsening visual field defect was the most frequently observed adverse event.
This study has some limitations that warrant further consideration, including remarkable variability in subjective visual function testing, stemming from factors, such as the diverse range of patient conditions, including genetic mutations and varying progression rates, as well as the relatively short duration of the follow-up period. The study's exploratory nature also contributes to the lack of comprehensive assessment. In the context of studies with limited numbers of patients, while a crossover trial could be considered, we opted for a parallel trial herein to avoid prolonged trial duration.
In conclusion, we did not find significant evidence to support the suppression of visual field progression in patients with RP upon the oral administration of one packet (4.15 g) of TK-98 after meals three times a day. Morphological evaluation showed an insignificant trend toward a slower reduction in ellipsoid zone length with TK-98 administration. Stratified analysis suggested that TK-98 may be superior in a relatively homogeneous patient population. In contrast, TK-98 administration to patients with RP was safe. Further research is warranted to provide clearer insights into the potential efficacy of TK-98 in RP treatment, requiring larger sample sizes and more homogeneous patient cohorts over longer durations.
Supplementary Material
Acknowledgments
The authors thank all the patients who participated in the study and the ophthalmologists who introduced them to the Kyoto University Hospital. We thank the members of the Efficacy and Safety Evaluation Committee for this clinical trial; Akira Murakami of Juntendo University, who chaired the committee; Mineo Kondo of Mie University; and Chie Sotozono of Kyoto Prefectural University of Medicine. We thank Satoshi Morooka of Morooka eye clinic for performing visual field tests. We are grateful to EA Pharma for providing us with information on TK-98. Although the bulk formulation of TK-98 was ordered from EA Pharma, the company was not involved in this study.
Supported by a research grant from the Practical Application Research Project for Intractable Diseases of the Japan Agency for Medical Research and Development (AMED) and a donation from the Ophthalmology Research Grant. Prior to conducting the clinical trial, review and approval by the conflict-of-interest review committee were obtained in accordance with the conflict-of-interest management policy of Kyoto University.
Disclosure: H.O. Ikeda, a patent related to this study (6764233, Japan); T. Hasegawa, a patent related to this study (6764233, Japan); H. Abe, None; Y. Amino, None; T. Nakagawa, None; H. Tada, None; M. Miyata, None; A. Oishi, None; S. Morita, None; A. Tsujikawa, None
References
- 1. Hartong DT, Berson EL, Dryja TP.. Retinitis pigmentosa. Lancet . 2006; 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 2. O'Neal TB, Luther EE.. Retinitis pigmentosa. StatPearls. Treasure Island, FL: StatPearls Publishing. 2023. [PubMed] [Google Scholar]
- 3. Cross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L.. Current and future treatment of retinitis pigmentosa. Clin Ophthalmol. 2022; 16: 2909–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Russell S, Bennett J, Wellman JA, et al.. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet . 2017; 390: 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maguire AM, Russell S, Wellman JA, et al.. Efficacy, safety, and durability of Voretigene Neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: results of phase 1 and 3 trials. Ophthalmology . 2019; 126: 1273–1285. [DOI] [PubMed] [Google Scholar]
- 6. Ghazi NG, Abboud EB, Nowilaty SR, et al.. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. 2016; 135: 327–343. [DOI] [PubMed] [Google Scholar]
- 7. Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de la Camara C, et al.. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med . 2020; 26: 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mandai M. Pluripotent stem cell-derived retinal organoid/cells for retinal regeneration therapies: a review. Regen Ther. 2023; 22: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ho AC, Humayun MS, Dorn JD, et al.. Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology . 2015; 122: 1547–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaffrath K, Schellhase H, Walter P, et al.. One-year safety and performance assessment of the argus ii retinal prosthesis: a postapproval study. JAMA Ophthalmol. 2019; 137: 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Edwards TL, Cottriall CL, Xue K, et al.. Assessment of the electronic retinal implant alpha ams in restoring vision to blind patients with end-stage retinitis pigmentosa. Ophthalmology 2018; 125: 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stingl K, Bartz-Schmidt KU, Besch D, et al.. Subretinal visual implant alpha IMS—clinical trial interim report. Vision Res. 2015; 111: 149–160. [DOI] [PubMed] [Google Scholar]
- 13. Berson EL, Rosner B, Sandberg MA, et al.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993; 111: 761–772. [DOI] [PubMed] [Google Scholar]
- 14. Hoffman DR, Locke KG, Wheaton DH, Fish GE, Spencer R, Birch DG.. A randomized, placebo-controlled clinical trial of docosahexaenoic acid supplementation for X-linked retinitis pigmentosa. Am J Ophthalmol. 2004; 137: 704–718. [DOI] [PubMed] [Google Scholar]
- 15. Hoffman DR, Hughbanks-Wheaton DK, Spencer R, et al.. Docosahexaenoic acid slows visual field progression in x-linked retinitis pigmentosa: ancillary outcomes of the DHAX trial. Invest Ophthalmol Vis Sci. 2015; 56: 6646–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Comander J, Weigel DiFranco C, Sanderson K, et al.. Natural history of retinitis pigmentosa based on genotype, vitamin A/E supplementation, and an electroretinogram biomarker. JCI Insight . 2023; 8: e167546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kong X, Hafiz G, Wehling D, Akhlaq A, Campochiaro PA.. Locus-level changes in macular sensitivity in patients with retinitis pigmentosa treated with oral N-acetylcysteine. Am J Ophthalmol . 2021; 221: 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Campochiaro PA, Iftikhar M, Hafiz G, et al.. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J Clin Invest . 2020; 130: 1527–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rotenstreich Y, Belkin M, Sadetzki S, et al.. Treatment with 9-cis β-carotene-rich powder in patients with retinitis pigmentosa: a randomized crossover trial. JAMA Ophthalmol . 2013; 131: 985–992. [DOI] [PubMed] [Google Scholar]
- 20. Koenekoop RK, Sui R, Sallum J, et al.. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1b trial. Lancet . 2014; 384: 1513–1520. [DOI] [PubMed] [Google Scholar]
- 21. Scholl HP, Moore AT, Koenekoop RK, et al.. Safety and proof-of-concept study of oral QLT091001 in retinitis pigmentosa due to inherited deficiencies of retinal pigment epithelial 65 Protein (RPE65) or Lecithin:retinol acyltransferase (LRAT). PLoS One. 2015; 10: e0143846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ikeda HO, Sasaoka N, Koike M, et al.. Novel VCP modulators mitigate major pathologies of rd10, a mouse model of retinitis pigmentosa. Sci Rep. 2014; 4: 5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hasegawa T, Muraoka Y, Ikeda HO, et al.. Neuoroprotective efficacies by KUS121, a VCP modulator, on animal models of retinal degeneration. Sci Rep . 2016; 6: 31184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ikeda HO, Muraoka Y, Hata M, et al.. Safety and effectiveness of a novel neuroprotectant, KUS121, in patients with non-arteritic central retinal artery occlusion: an open-label, non-randomized, first-in-humans, phase 1/2 trial. PLoS One . 2020; 15: e0229068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iwai S, Hasegawa T, Ikeda HO, Tsujikawa A.. Branched chain amino acids promote ATP production via translocation of glucose transporters. Invest Ophthalmol Vis Sci. 2022; 63: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasegawa T, Ikeda HO, Iwai S, et al.. Branched chain amino acids attenuate major pathologies in mouse models of retinal degeneration and glaucoma. Heliyon . 2018; 4: e00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ito N, Miura G, Shiko Y, Kawasaki Y, Baba T, Yamamoto S.. Progression rate of visual function and affecting factors at different stages of retinitis pigmentosa. Biomed Res Int . 2022; 2022: 7204954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iga Y, Hasegawa T, Ikeda HO, et al.. Progression of retinitis pigmentosa on static perimetry, optical coherence tomography, and fundus autofluorescence. Sci Rep. 2023; 13: 22040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hasegawa T, Oishi A, Ikeda HO, et al.. Detection sensitivity of retinitis pigmentosa progression using static perimetry and optical coherence tomography. Transl Vis Sci Technol. 2021; 10: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oishi M, Oishi A, Ogino K, et al.. Wide-field fundus autofluorescence abnormalities and visual function in patients with cone and cone-rod dystrophies. Invest Ophthalmol Vis Sci. 2014; 55: 3572–3577. [DOI] [PubMed] [Google Scholar]
- 31. Pasantes-Morales H, Quiroz H, Quesada O.. Treatment with taurine, diltiazem, and vitamin E retards the progressive visual field reduction in retinitis pigmentosa: a 3-year follow-up study. Metab Brain Dis . 2002; 17: 183–197. [DOI] [PubMed] [Google Scholar]
- 32. Berson EL, Rosner B, Sandberg MA, et al.. Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Arch Ophthalmol. 2004; 122: 1297–1305. [DOI] [PubMed] [Google Scholar]
- 33. Berson EL, Rosner B, Sandberg MA, et al.. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol . 2010; 128: 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakazawa M, Ohguro H, Takeuchi K, Miyagawa Y, Ito T, Metoki T.. Effect of nilvadipine on central visual field in retinitis pigmentosa: a 30-month clinical trial. Ophthalmologica 2011; 225: 120–126. [DOI] [PubMed] [Google Scholar]
- 35. Birch DG, Weleber RG, Duncan JL, Jaffe GJ, Tao W, Ciliary Neurotrophic Factor Retinitis Pigmentosa Study Groups. Randomized trial of ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for retinitis pigmentosa. Am J Ophthalmol. 2013; 156: 283–292.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Birch DG, Bernstein PS, Iannacone A, et al.. Effect of oral valproic acid vs placebo for vision loss in patients with autosomal dominant retinitis pigmentosa: a randomized phase 2 multicenter placebo-controlled clinical trial. JAMA Ophthalmol. 2018; 136: 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Akiyama M, Ikeda Y, Yoshida N, et al.. Therapeutic efficacy of topical unoprostone isopropyl in retinitis pigmentosa. Acta Ophthalmol . 2014; 92: e229–234. [DOI] [PubMed] [Google Scholar]
- 38. Numa S, Oishi A, Higasa K, et al.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020; 10: 20770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oishi M, Oishi A, Gotoh N, et al.. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Invest Ophthalmol Vis Sci . 2014; 55: 7369–7375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.