

Abstract
Background:
Previous epidemiological studies have repeatedly found per- and polyfluoroalkyl substances (PFAS) exposure associated with higher circulating cholesterol, one of the greatest risk factors for development of coronary artery disease. The main route of cholesterol catabolism is through its conversion to bile acids, which circulate between the liver and ileum via enterohepatic circulation. Patients with coronary artery disease have decreased bile acid excretion, indicating that PFAS-induced impacts on enterohepatic circulation may play a critical role in cardiovascular risk.
Objectives:
Using a mouse model with high levels of low-density and very low-density lipoprotein (LDL and VLDL, respectively) cholesterol and aortic lesion development similar to humans, the present study investigated mechanisms linking exposure to a PFAS mixture with increased cholesterol.
Methods:
Male and female mice were fed an atherogenic diet (Clinton/Cybulsky low fat, 0.15% cholesterol) and exposed to a mixture of 5 PFAS representing legacy, replacement, and emerging subtypes (i.e., PFOA, PFOS, PFHxS, PFNA, GenX), each at a concentration of , for 7 wk. Blood was collected longitudinally for cholesterol measurements, and mass spectrometry was used to measure circulating and fecal bile acids. Transcriptomic analysis of ileal samples was performed via RNA sequencing.
Results:
After 7 wk of PFAS exposure, average circulating PFAS levels were measured at 21.6, 20.1, 31.2, 23.5, and in PFAS-exposed females and 12.9, 9.7, 23, 14.3, and in PFAS-exposed males for PFOA, PFOS, PFHxS, PFNA, and GenX, respectively. Total circulating cholesterol levels were higher in PFAS-exposed mice after 7 wk ( vs. in female mice and vs. in male mice exposed to vehicle or PFAS, respectively). Total circulating bile acid levels were higher in PFAS-exposed mice ( vs. in female mice and vs. in male mice exposed to vehicle or PFAS, respectively). In addition, total fecal bile acid levels were lower in PFAS-exposed mice ( vs. in females and vs. in males exposed to vehicle or PFAS, respectively). In the ileum, expression levels of the apical sodium-dependent bile acid transporter (ASBT) were higher in PFAS-exposed mice.
Discussion:
Mice exposed to a PFAS mixture displayed higher circulating cholesterol and bile acids perhaps due to impacts on enterohepatic circulation. This study implicates PFAS-mediated effects at the site of the ileum as a possible critical mediator of increased cardiovascular risk following PFAS exposure. https://doi.org/10.1289/EHP14339
Introduction
Per- and polyfluoroalkyl substances (PFAS) are a large family of synthetically manufactured chemicals used for their surfactant properties in industrial and consumer products, including nonstick cookware and food storage, clothing, carpets, and aqueous film-forming foams (AFFF) used in firefighting.1–5 PFAS have become global environmental contaminants, found in water and food sources around the world.6–8 Consumption of PFAS through these contaminated sources have led to the accumulation of PFAS in humans, resulting in 98% of American adults having detectable PFAS levels in their blood.9 PFAS, especially long-chain or legacy PFAS, have long half-lives and are resistant to degradation, leading to their inclusion in a group of chemicals termed forever chemicals.10,11 Although production of legacy long-chain PFAS has been phased out in the United States, emerging PFAS of unknown toxicity, with shorter chain lengths or fluoroether alternatives, have been used as replacements.12–14 Research into the effects of emerging PFAS, such as hexafluoropropylene oxide dimer acid (GenX), is a growing field. Although GenX has a much shorter biological half-life compared with legacy PFAS, animal studies have demonstrated that GenX exposure can lead to adverse health effects similar to other PFAS.15–17
Research into the health effects of PFAS has revealed that exposure is associated with numerous adverse outcomes in humans.18 Perfluorooctanoic acid (PFOA) exposure in humans has been associated with kidney and testicular cancers,19–21 as well as negative reproductive and developmental effects, including impaired fertility22,23 and decreased birth weight.24 PFAS exposure has also been shown to have hepatotoxic effects and is associated with biomarkers of liver function and metabolic dysfunction-associated steatotic liver disease (MASLD; formerly known as nonalcoholic fatty liver disease) in humans.25–27 Recent studies have also highlighted the association of PFAS exposure with markers of cardiovascular risk,28,29 although a consistent association between increasing PFAS levels in humans and cardiovascular disease were not found in recent meta-analyses.30,31 Cardiovascular diseases are the leading cause of premature mortality worldwide.32,33 Despite effective interventions for historical risk factors [e.g., low-density lipoprotein (LDL) cholesterol, blood pressure, smoking], considerable residual risk remains for atherosclerotic heart disease, implicating a role for other environmental stressors, such as PFAS.34,35
High levels of cholesterol are an important factor in the formation and progression of atherosclerotic plaques, leading to blood clots and related events, such as heart attacks, strokes, or aneurisms.36–38 Previous epidemiological studies have repeatedly determined PFAS exposure to be associated with elevated cholesterol (i.e., hypercholesterolemia).31,39–42 Most PFAS, especially PFOA and perfluorooctanesulfonic acid (PFOS), were found associated with increased total and LDL cholesterol, whereas certain PFAS, such as perfluorohexane sulfonic acid (PFHxS) and perfluorodecanoic acid (PFDA), demonstrated a positive association with high-density lipoprotein (HDL) cholesterol.43 Even at lower “background” PFAS levels within the general US population, analysis of National Health and Nutrition Examination Survey (NHANES) data (2003–2004) reported positive associations between PFOS, PFOA, perfluorononanoic acid (PFNA) and both total and non-HDL cholesterol.40
The primary route of cholesterol catabolism is through its conversion to bile acids, accounting for of its daily turnover.44 Bile acids are synthesized in the liver from cholesterol and then move into the bile, through the intestines, and then can be either excreted or reabsorbed at the distal small intestine (ileum) and sent back to the liver, a cycle that is known as the enterohepatic circulation.45 Previous studies have shown that PFAS exposure has been significantly associated with alterations of bile acid profiles.46–49 Furthermore, previous studies have shown that having lower levels of bile acid excretion in patients was associated with coronary artery disease and its risk factors.50,51
To be able to circulate within the enterohepatic circulation, bile acids bind to active membrane transporters.52 The ileum is the major site of bile acid intestinal absorption, where the apical sodium-dependent bile acid transporter (ASBT) is responsible for most of the active absorption of bile acids, facilitating their transport from the lumen of the ileum across the apical membrane of ileal enterocytes.52–55 It has previously been shown that PFAS are able to directly bind several bile acid transporters, including ASBT,56 which is critical for PFAS transport through the enterohepatic circulation.57 Taken together, these studies support the idea that alterations in bile acid metabolism and transport within the ileum may play an important role in the development of coronary artery disease. In the present study, we aimed to investigate mechanisms linking exposure to a PFAS mixture with impacts on cholesterol and bile acid metabolism. To do this, we used LDL receptor–deficient mice (), which have high levels of LDL/very LDL (VLDL) cholesterol and aortic lesion development similar to humans when fed the Clinton/Cybulsky diet,58 that were exposed to water containing a mixture of five environmentally relevant PFAS (PFOA, PFOS, PFHxS, PFNA, and GenX) for 7 wk.
Materials and Methods
Animal Experiments
Male and female B6.129S7-/J mice (strain #002207) on a C57Bl/6J genetic background were purchased from Jackson Laboratories at 7 wk of age and were allowed to acclimate for 1 wk upon arrival. The mice were housed with five mice per cage in a 12-h light/dark cycle under controlled temperature (21–23°C) and humidity (30%–70%). The mice were then randomly divided into the following four experimental groups: a) (), b) (), c) (), and d) (). Subsequent to acclimation, the mice were fed an atherogenic diet (Clinton/Cybulsky low-fat diet, 0.15% cholesterol; Research Diets; product number D01061401C) (Table S1) for 1 wk prior to the beginning of PFAS exposure and then continued on the atherogenic diet for the remainder of the study.
The mice were exposed for 7 wk via their drinking water supply to either control water or water containing a PFAS mixture. Double-distilled water from Wayne State University was used to produce the control and PFAS water. The PFAS water contained a mixture of five PFAS: a) PFOA (Sigma-Aldrich; CAS No: 335-67-1; 95%), b) PFOS (Sigma-Aldrich; CAS No: 2795-39-3; ), c) PFHxS (J&K Scientific; CAS No: 3871-99-6; ), d) PFNA (Sigma-Aldrich; CAS No: 375-95-1; 97%), and e) GenX (Synquest Laboratories; CAS No: 62037-80-3; 95%). PFAS water was made from a concentrated stock (containing each PFAS at ) used throughout the experiment. Each of the five PFAS were present in the PFAS mixture water at a concentration of and the mice were allowed to drink the water ad libitum. The estrous cycle was not controlled for in the female mice throughout the study. Control groups were provided control water containing no PFAS. Water bottles were changed weekly. The animal protocol was approved by the Wayne State University Institutional Animal Care and Use Committee.
Sample Collection
The body weight per mouse and the food and water intake per cage (i.e., per five mice) were recorded weekly. Blood was collected longitudinally throughout the study via the submandibular vein at the start of weeks 3 (unfasted) and 5 (unfasted), as well as at study completion (fasted). Fasting was conducted overnight for 16 h prior to euthanasia. Mice were euthanized via carbon dioxide. Plasma was separated from blood via centrifugation at for 10 min at 4°C. Body composition was measured via an EchoMRI Body Composition Analyzer (Echo Medical System) for five mice of each group at weeks 2 and 5, as well as at study completion. Tissues and samples collected at study completion include liver, aortas, ileum, kidney, gonadal fat, and cecum contents. Sections of the liver’s large right lobe were either collected in Scigen Tissue-Plus O.C.T. Compound (Thermo Fisher Scientific, #4585), fixed in 10% neutral-buffered formalin for histology or homogenized in TRI Reagent Solution (Thermo Fisher Scientific, #AM9738). The remaining liver tissue was frozen at . The ileal section of the intestines was identified as distance from the cecum. Ileal tissue was homogenized in TRI reagent or frozen at . Ileal contents were not removed prior to RNA extraction. Cecum contents were emptied from the cecum and frozen at . Tissues for hematoxylin and eosin (H&E) staining were fixed in 10% neutral-buffered formalin (48 h) and embedded in paraffin (Sakura Tissue Tek VIP E300 Tissue Processor). Sections were cut at a thickness of (Sakura Tissue Tek TEC 5 Tissue Embedding Console System) and stained with H&E. The activity of alanine aminotransferase (ALT) was measured in plasma by using the Alanine Aminotransferase Activity Assay Kit (Sigma-Aldrich, #MAK052).
Urine and feces samples were collected from six mice per treatment group on week 6 of PFAS exposure. The mice were individually housed in metabolic cages (Tecniplast USA, Inc.) for 48 h. The mice were allowed 24 h to acclimate to the cages; then urine and feces samples were collected, measured for volume or weight, respectively, and frozen at until subsequent analysis. For urine and feces amounts collected, the fold change was calculated by dividing the PFAS-exposed values by the vehicle values for each sex.
Atherosclerosis Analysis
Aortas were isolated from the mice and cleaned by removing adventitial tissue. The aortas were then sent to the University of Massachusetts for analysis by the Mouse Metabolic Phenotyping Center. The aortas were stained with Oil Red O and prepared for en face measurement. Bright-field images were obtained with a Nikon Stereo microscope (Nikon Instruments) equipped with a SPOT Insight camera (SPOT Imaging). Images were analyzed for atherosclerotic percentage using ImageJ software.59
Mesoplex Cytokine Analysis
Plasma samples collected following 7 wk of PFAS exposure were analyzed for levels of inflammatory biomarkers. Plasma biomarker levels were quantified using a subset of the U-PLEX Biomarker Group 1 (Mouse) multiplex assay (MSD) according to the manufacturer’s instructions. The assay was run using a MESO QuickPlex SQ 120 plate reader, and data analysis was conducted using the Discovery Workbench Desktop Analysis Software (version 4.0; Meso Scale Diagnostics).
RNA Sequencing
Total RNA was extracted from the ileum samples using TRI Reagent Solution (Thermo Fisher Scientific). Ilea were collected for male mice only, with female samples lacking owing to personnel oversight. The RNA purity and quantity were measured using a spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific). The Wayne State University Genome Sciences Core performed the RNA sequencing (RNA-seq). The quality of the RNA was assessed on a TapeStation (Agilent). The RNA integrity numbers (RINs) ranged from 3.6 to 7.2 (). The mRNA-seq library was prepared using the QuantSeq 3′ mRNA-Seq Library Prep Kit FWD (Lexogen). Libraries were assessed by the High Sensitivity D1000 (HS D1000) ScreenTape Assay (Agilent). Samples were sequenced on the NovaSeq system (Illumina). Reads were aligned to the mouse genome (Build mm10) and tabulated for each gene region.60,61 Differential gene expression analysis was used to compare transcriptome effects between treatment groups. Differential expression was analyzed using the analysis of variance (ANOVA)–like test in edgeR.62 The results are reported as the PFAS-exposure fold change from control, -value, the Benjamini–Hochberg–adjusted -value, false discovery rate (FDR)–adjusted -value, and the individual read counts by gene for each sample replicate. Significantly differentially expressed genes (|log fold change| ; -value ) were used to perform gene ontology enrichment analyses via the Database for Annotation, Visualization, and Integrated Discovery (DAVID) software.63 Differential expression was analyzed using the ANOVA-like test in edgeR.62 The RNA-seq data has been uploaded to the Gene Expression Omnibus (GEO) database with the reference number GSE263158.
Real-Time Quantitative Polymerase Chain Reaction Gene Expression Analysis
Total cellular RNA was extracted from liver tissue using TRI reagent. RNA quantity and purity were measured and validated via spectrophotometer (NanoDrop 2000, Thermo Fisher Scientific). Total RNA was used to synthesize complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (#4368814, Thermo Fisher Scientific). Reverse transcription was performed on a Bio-Rad T100 Thermal Cycler under the following conditions: a) 25°C for 10 min, b) 37°C for 2 h, c) 85°C for 5 min, and d) hold at 10°C. All analyzed genes and primer sequences were obtained from Integrated DNA Technologies and are presented in Table S2. For analysis of Cyp7a1 and Cyp27a1, Taqman Fast Advanced Master Mix (#4444557, Thermo Fisher Scientific) was used to perform real-time quantitative polymerase chain reaction (RT-qPCR) under the following conditions: a) 50°C for 2 min, b) 95°C for 20 s, c) 95°C for 3 s, and d) 60°C for 30 s (40 cycles). For all other genes, PowerUp SYBR Green Master Mix (#A25742, Thermo Fisher Scientific) was used to perform RT-qPCR under the following conditions: a) 50°C for 2 min, b) 95°C for 2 min, c) 95°C for 1 s, and d) 60°C for 30 s (40 cycles). All RT-qPCR was performed using the QuantStudio 6 Pro (Thermo Fisher Scientific). For all genes measured by RT-qPCR, the fold change was calculated as and normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The relative gene expression was set to 1 for the female vehicle samples as the reference samples.
Plasma Lipid and Hormone Measurements
Total plasma cholesterol levels were measured by colorimetric assay (Total Cholesterol E Assay Kit, #999-02601, Fujifilm Wako Diagnostics). Colorimetric assays were run on a CLARIOstar microplate reader (BMG Labtech) and results were analyzed using CLARIOstar MARS data analysis software (BMG Labtech). Samples were run in duplicate and the average percentage coefficient of variation between duplicate wells was . The lowest cholesterol standard was , and no samples fell outside the limit of detection (LOD). HDL and LDL/VLDL cholesterol fractions were separated by precipitation, and free and esterified cholesterol levels were measured, using the Cholesterol Assay Kit - HDL and LDL/VLDL, according to manufacturer protocol (Abcam, ab65390). Progesterone was measured by colorimetric assay (Enzo Life Sciences, ADI-900-011), according to manufacturer protocol. Follicle-stimulating hormone (FSH) was measured by colorimetric assay (Elabscience, E-EL-M0511), according to manufacturer protocol. Luteinizing hormone (LH) was measured by colorimetric assay (Elabscience, E-EL-M3053), according to manufacturer protocol. Fold change was calculated by dividing the measured PFAS-exposed values by the vehicle values for each sex.
Hepatic Bile Acid and Cholesterol Measurements
Liver tissues were homogenized in phosphate-buffered saline, incubated on ice for 30 min, and centrifuged at for 15 min at 4°C. Protein concentrations were determined by the Bradford assay (Quick Start Bradford Dye Reagent #5000205, Bio-Rad). Levels of hepatic cholesterol and bile acids were measured by colorimetric assays using the Total Cholesterol E Assay Kit (#999-02601, Fujifilm) and Total Bile Acid Assay Kit (#STA-631, Cell Biolabs), respectively, according to each manufacturer’s instructions. The levels of cholesterol and bile acids were normalized by protein concentrations.
Absolute Quantitation of Fecal and Circulating Bile Acids
Quantitation of bile acids was performed by the Michigan State University Mass Spectrometry Core. For the fecal samples, of fecal pellets were weighed and extracted with of acetonitrile (ACN):water (1:1 vol:vol) after spiking each sample with of d4-glycochenodeoxycholic acid (GCDCA-d4; internal standard). Samples were vortexed for 1 min, then mixed by shaking for 15 min, followed by centrifugation for 10 min, and the supernatants were collected to a new tube. The remaining pellet was reextracted with an additional of ACN:water (1:1 vol:vol). Samples were further diluted 50-fold prior to liquid chromatography–mass spectrometry (LC-MS) analysis. Each diluted sample was centrifuged for 10 min immediately prior to being transferred to glass LC-MS vials.
For the plasma samples, of plasma were mixed with of water and extracted with of ACN:water (1:1 vol:vol) after spiking each sample with of GCDCA-d4 (internal standard). Samples were vortexed for 1 min, then mixed by shaking for 15 min, followed by centrifugation for 10 min, and the supernatants were collected to a new tube. The remaining pellet was reextracted with an additional of ACN:water (1:1 vol:vol). The pooled supernatants were dried under vacuum (without heat) in a speedvac centrifuge and reconstituted in of ACN:water (1:1 vol:vol) by vortexing for 15 min, then transferring the entire supernatant to an LC-MS vial. Samples were centrifuged for 10 min immediately prior to being transferred to the autosampler.
LC-MS analysis was adapted from Swann et al.64 The LC-MS system consisted of a Shimadzu Prominence high-performance liquid chromatograph (HPLC) and an LTQ-Orbitrap Velos MS. Standards and samples were analyzed by LC-MS in the following condition: a) injection of ; b) flow: ; c) ; d) sample ; e) column: Phenomenex C18AQ (, , 80 Angstrom); and f) gradient. Chromatographic peak alignment, isotope correction, compound identification, and peak area quantification was performed using MAVEN metabolomics software.65 Peak areas were quantitated against the peak areas of the internal standard. Fold change was calculated by dividing measurements from PFAS-exposed mice by the measurements for vehicle control mice. Comparisons were done either as PFAS treatment with a sex or by overall PFAS treatment (regardless of sex), as indicated.
Absolute Quantitation of Circulating PFAS
Quantitation of absolute circulating PFAS levels were measured in plasma after 7 wk of PFAS exposure by the Wayne State University Lumigan Instrument Center, as previously reported.47 The circulating PFAS levels are presented in Table 1. Briefly, plasma sample extraction was completed using an Agilent Enhanced Matrix Removal (EMR)–Captiva lipid extraction 96-well plate. Samples were analyzed using a Thermo Scientific TSQ Altis with UltiMate 3000 ultra-HPLC. Mobile phases used were (A) ammonium acetate in LC-MS–grade water, and (B) LC-MS–grade ACN. PFAS were detected in negative ion mode using electrospray ionization. The LOD were as follows: PFOA , PFOS , PFHxS 0.46 ppb, PFNA , and GenX .
Table 1.
Circulating levels of individual PFAS after exposure of mice to the PFAS mixture.
| Absolute plasma PFAS levelsa | PFAS biomonitoring study (year of publication) | Measured PFAS levels (ng/mL) | ||||
|---|---|---|---|---|---|---|
| Biochemical Name | PFAS female average concentration (ng/mL) | PFAS male average concentration (ng/mL) | PFAS female vs. PFAS male (fold change) | Sex difference (-value)b | ||
| PFOA | 1.7 | 0.037* | Sakr et al. (2007)66 | Median (range): 494 (5–9,550) | ||
| Olsen and Zobel (2007)67 | Median (range): 950 (10–92,030) | |||||
| Girardi and Merler (2019)68 | Geometric mean (range): 4,048 (19–91,900) | |||||
| L-PFOS | 2.1 | 0.009* | Olsen et al. (2007)11 | Median (range): 626 (145–3,490) | ||
| Olsen and Zobel (2007)67 | Median (range): 450 (30–4,790) | |||||
| Lu et al. (2019)69 | Median (range): 909.3 (9.6–43,299) | |||||
| L-PFHxS | 1.4 | 0.19 | Olsen et al. (2007)11 | Median (range): 193 (16–1,295) | ||
| Lu et al. (2019)69 | Median (range): 785.2 ( to 1,226) | |||||
| PFNA | 1.6 | 0.10 | Wang et al. (2012)70 | Median (range): 5.7 (0.8–24.5) | ||
| Nilsson et al. (2010)71 | Median (range): 14.7 (0.8–163) | |||||
| HFPO-DA (GenX) | 0.9 | 0.88 | Petriello et al. (2022)72 | Median (range): 0.321 (0.06–1.00) | ||
Note: Ldlr, low density lipoprotein receptor; LOD, limit of detection; L-PFHxS, perfluorohexane sulfonic acid; L-PFOS, perfluorooctanesulfonic acid; PFAS, per- and polyfluoroalkyl substances; PFNA, perfluorononanoic acid; PFOA, perfluorooctanoic acid; HFPO-DA (GenX), hexafluoropropylene oxide dimer acid. *Represents statistical significance ().
total; data from 5 PFAS-exposed females and 5 PFAS-exposed males.
Significance between PFAS-exposed males and PFAS-exposed females were determined using -test; .
Western Blotting
Liver and ileal tissue were homogenized in radioimmunoprecipitation assay buffer. Samples were centrifuged at for 15 min at 4°C, and the supernatant was collected. Total protein levels were measured by the Bradford assay (Quick Start Bradford Dye Reagent #5000205, Bio-Rad). Twenty micrograms of total protein for each sample were separated by 10% sodium dodecyl sulfate–polyacrylamide gel and then transferred to a nitrocellulose membrane. For Western blot analysis, the membranes were incubated with antibodies to measures proteins of interest, as follows. ASBT was analyzed using anti-SLC10A2/ASBT rabbit polyclonal antibody (Abcam, #ab203205; diluted 1:1,000). The bile acid transporter sodium taurocholate cotransporting polypeptide (NTCP) was analyzed using anti-NTCP/SLC10A1 rabbit polyclonal antibody (Bioss, #bs-1958R; diluted 1:1,000). -actin was analyzed using anti; -actin (13E5) rabbit monoclonal antibody (Cell Signaling Technology, #4970; diluted 1:1,000). The secondary antibody used was anti-rabbit immunoglobulin G, horseradish peroxidase–linked antibody (Cell Signaling Technology, #7074; diluted 1:1,0,000). The membranes were blocked in Tris-buffered saline with Tween (TBST; 0.1% Tween) milk for 1 h at room temperature. The membranes were then incubated with primary antibody overnight at 4°C with rocking. The blots were then washed with TBST three times for 10 min at room temperature. Then the blots were incubated with secondary antibody for 1 h at room temperature. The blots were then again washed with TBST three times for 10 min at room temperature. Protein bands were detected using Pierce Enhanced Chemiluminescence (ECL) Western Blotting Substrate (Thermo Fisher Scientific, #32209). Protein bands were imaged using a ChemiDoc MP Imaging System (Bio-Rad). Quantification of protein levels was performed using ImageJ software.59
Statistical Analysis
RNA-seq of ileal tissue was performed by the Wayne State University Genome Sciences Core, and statistical analysis of the results was done with R statistical software (version 4.2.0; R Development Core Team). Spearman correlation analysis of circulating PFAS and cholesterol fractions was performed using JMP Statistical Software (version 15.2; SAS Institute Inc.). For data other than RNA-seq, comparisons of multiple groups were analyzed by two-way ANOVA and the Holm–Sidak post hoc test for multiple comparisons using SigmaPlot (version 14; Systat Software Inc.). Two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS. With past consultation from biostatisticians, a significant interaction term supersedes the main effects and can make their meaning unclear. We therefore have not included the main effects -values for any figure with a significant interaction. Graphed values are denoted as . A probability value of was considered statistically significant. Data that was nonnormally distributed was transformed. Statistics provided were based on transformed data and indicated in figure legends.
Results
PFAS Effects on Hepatic Toxicity
The present study investigated possible mechanisms linking exposure to a PFAS mixture with increased circulating cholesterol. To do this, we used male and female mice, which have high levels of LDL/VLDL cholesterol and aortic lesion development similar to humans when fed diets enriched with cholesterol.58 The mice were exposed to water containing a mixture of five environmentally relevant PFAS (PFOA, PFOS, PFNA, PFHxS, and GenX) for 7 wk.
To characterize hepatic toxicity in mice exposed to the PFAS mixture, liver sections of male and female mice were stained with H&E (Figure 1A). PFAS-exposed mice exhibited hepatocyte hypertrophy as indicated by the appearance of larger hepatocytes and lipid infiltration compared with vehicle control mice, especially in the female PFAS-exposed mice. By the end of the study, a significant PFAS:sex interaction in body weight was observed (Figure 1B,C). This interaction was driven by a significant difference in body weight in PFAS-exposed females (namely, lower body weight) compared with vehicle control females (), whereas body weight was not significantly different between male PFAS-exposed and vehicle control groups (; interaction ). Fat weight (reported as a percentage of body weight) was also found to be significantly lower in PFAS-exposed mice, especially females, after 4 and 7 wk of PFAS exposure (Figure S1). Circulating plasma levels of ALT were significantly higher in PFAS-exposed females compared with vehicle controls, whereas there was no significant difference in males (Figure 1D). Liver weight (reported as a percentage of body weight) was significantly greater after 7 wk of PFAS exposure in both females and males, with liver weight in females higher by -fold () and in males higher by -fold () (Figure 1E).
Figure 1.
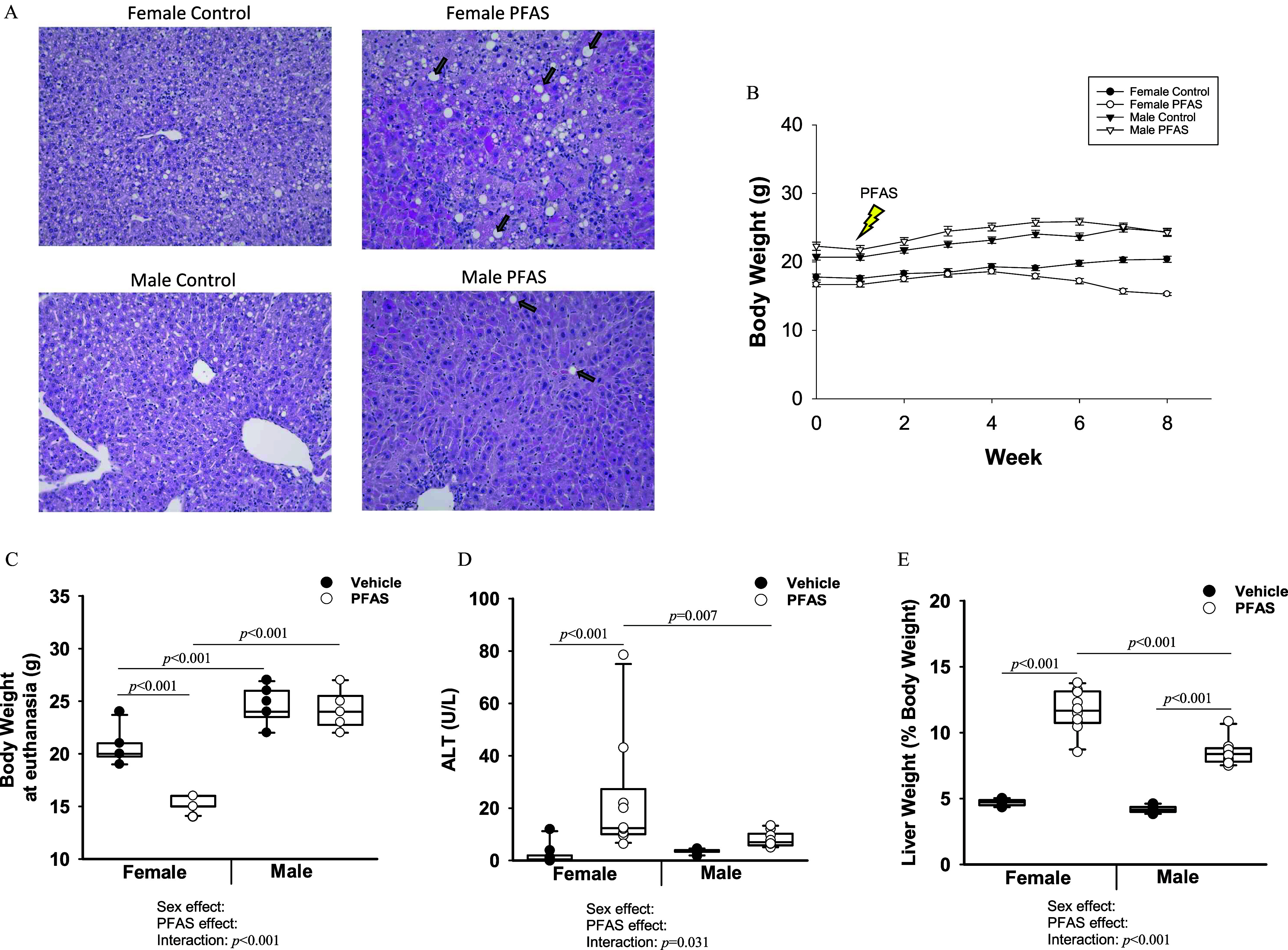
Measures of toxicity (liver weight, ALT, and histology; body weight) in mice exposed to the PFAS mixture. Male and female mice were exposed to vehicle water or the PFAS mixture for 7 wk. mice per treatment group. (A) Male and female liver sections were stained for H&E. Arrows indicate lipid droplets. Images were taken at magnification. (B) Weights for each mouse were recorded weekly. Graph represents the average weight over time. Error bars represent the standard error of the mean. (C) Body weights of mice measured at euthanasia. (D) ALT levels were measured in plasma collected after 7 wk of PFAS exposure. (E) Liver weight at euthanasia given as a percentage of total body weight. In (C–E), two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS (interaction ). The Holm–Sidak post hoc test was used for multiple comparisons. With past consultation from biostatisticians, a significant interaction term supersedes the main effects and can make their meaning unclear. We therefore have not included the main effects -values for any result with a significant interaction. Box plots represent the median values with upper and lower quartiles; whiskers extend to the 1st and 99th percentiles. Data are reported in Excel Tables S1–S3. Note: ALT, alanine aminotransferase; ANOVA, analysis of variance; H&E, hematoxylin and eosin; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances.
Circulating PFAS Levels and Dietary Intake and Excretion
After 7 wk of exposure to the PFAS mixture, the female PFAS-exposed mice had plasma PFAS levels at concentrations of 21.6, 20.1, 31.2, 23.5, and of PFOA, PFOS, PFHxS, PFNA, and GenX, respectively (Table 1). The male PFAS-exposed mice had plasma PFAS levels at concentrations of 12.9, 9.7, 23, 14.3, and of PFOA, PFOS, PFHxS, PFNA, and GenX, respectively (Table 1). Significant sex differences were observed for circulating levels of PFOA () and PFOS (), with higher levels of both measured in the female PFAS-exposed mice compared with PFAS-exposed males. Food intake was measured weekly throughout the study (Figure 2A). From week 2 onward, the male PFAS-exposed mice ate more than the female PFAS-exposed mice. Female food intake was not significantly affected due to PFAS exposure until after 6 wk, when the PFAS-exposed females ate less food compared with vehicle control females () (Figure 2A). Water intake was also measured weekly throughout the study (Figure 2B). At weeks 1, 4, and 6, the male PFAS-exposed mice drank significant more water than the female PFAS-exposed mice (Figure 2B). At none of the weekly time points were the female PFAS-exposed mice observed to drink more than their male counterparts.
Figure 2.
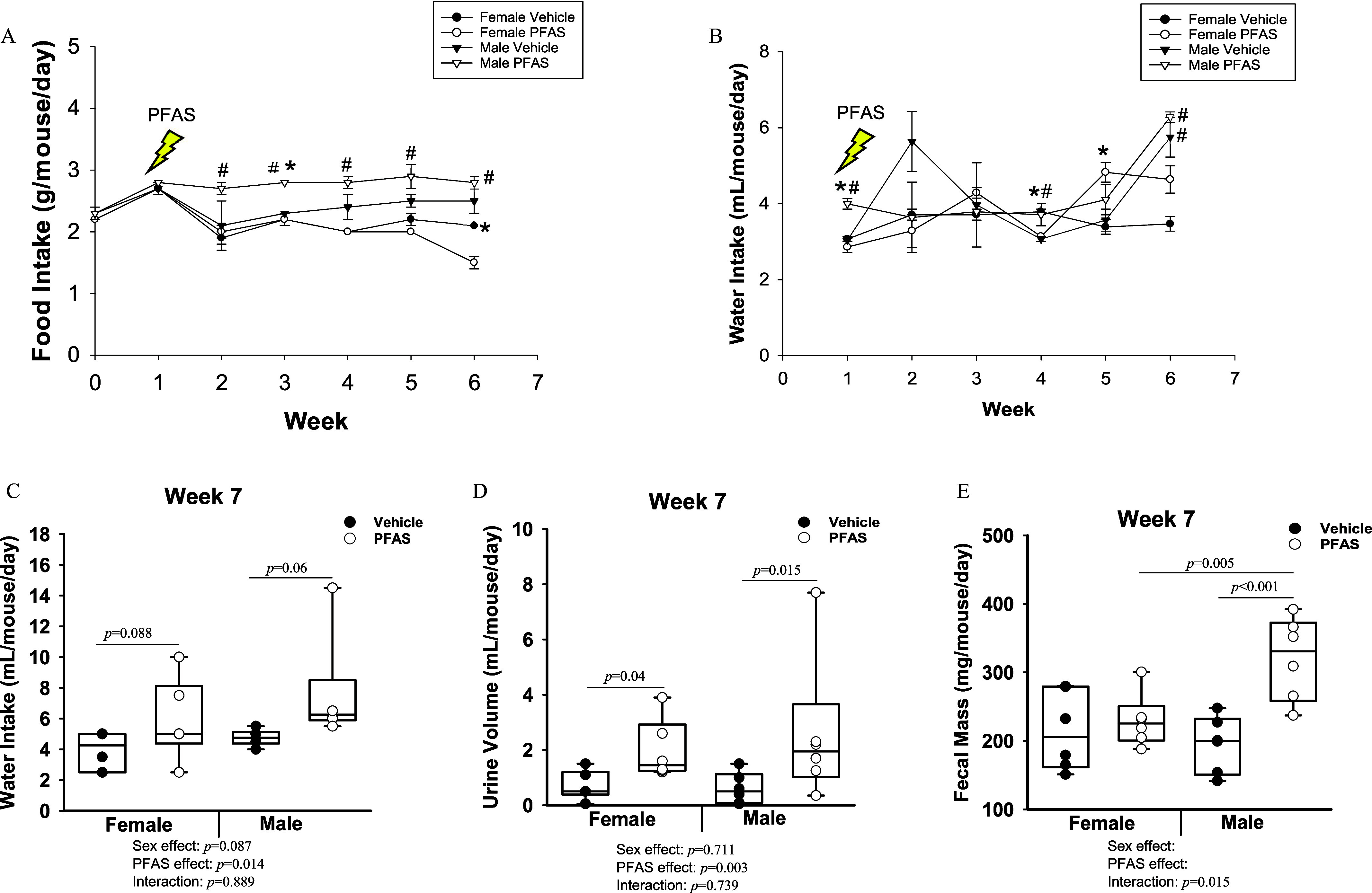
Exposure of mice to the PFAS mixture and food and water intake, urine volume, and fecal mass. (A) The average food intake per mouse per day was calculated weekly for all mice of each group (). (B) Changes in water intake from the beginning of PFAS exposure to 6 wk of PFAS exposure. Mice were placed into urine and feces collection cages after week 6: (), (), (), and (). The indicated statistics for week 4 refer to PFAS-exposed females compared with vehicle control females. (C) The average water intake per mouse over a 24-h period in week 7. The total amount of (D) urine and (E) feces excreted over a 24-h period in week 7 are presented. Statistical significance for (A,B) () was determined by -test and the error bars represent the standard error of the mean. # compared with opposite sex of the same treatment. * comparing treatment groups within same sex. In (C–E), two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS (interaction ). The Holm–Sidak post hoc test was used for multiple comparisons. With past consultation from biostatisticians, a significant interaction term supersedes the main effects and can make their meaning unclear. We therefore have not included the main effects -values for any result with a significant interaction. Box plots represent the median values with upper and lower quartiles; whiskers extend to the 1st and 99th percentiles. Data are reported in Excel Tables S4–S6. Note: ANOVA, analysis of variance; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances.
In addition, on the final week of PFAS exposure, six mice from each treatment group were moved into urine and feces collection cages. In these cages, PFAS-exposed mice had significantly greater water intake during this time (overall exposure effect ) (Figure 2C). Urine output volume was also measured over a 24-h collection period, and greater urine output was found in PFAS-exposed females () and in PFAS-exposed males (; overall exposure effect ) (Figure 2D). In addition, the amount of fecal mass excreted was significantly greater by in the male PFAS-exposed mice compared with male vehicle (), whereas female fecal excretion was not significantly different between PFAS-exposed () and vehicle control mice (interaction ) (Figure 2E).
PFAS Exposure and Circulating Cholesterol Levels
Plasma collected longitudinally throughout the study was analyzed for cholesterol levels. After 3 wk of PFAS exposure, total cholesterol levels in the female PFAS-exposed mice were significantly lower than those in vehicle control mice, with levels of in vehicle females and in PFAS-exposed females (). However, this effect was not observed in males (Figure 3A). After 5 wk of PFAS exposure, no significant differences were observed between PFAS-exposed and vehicle control mice of either sex (Figure 3B). However, after 7 wk of PFAS exposure, total cholesterol levels were significantly higher in female ( vs. ; ) and male ( vs. ; ) PFAS-exposed mice compared with vehicle control mice, respectively (Figure 3C).
Figure 3.
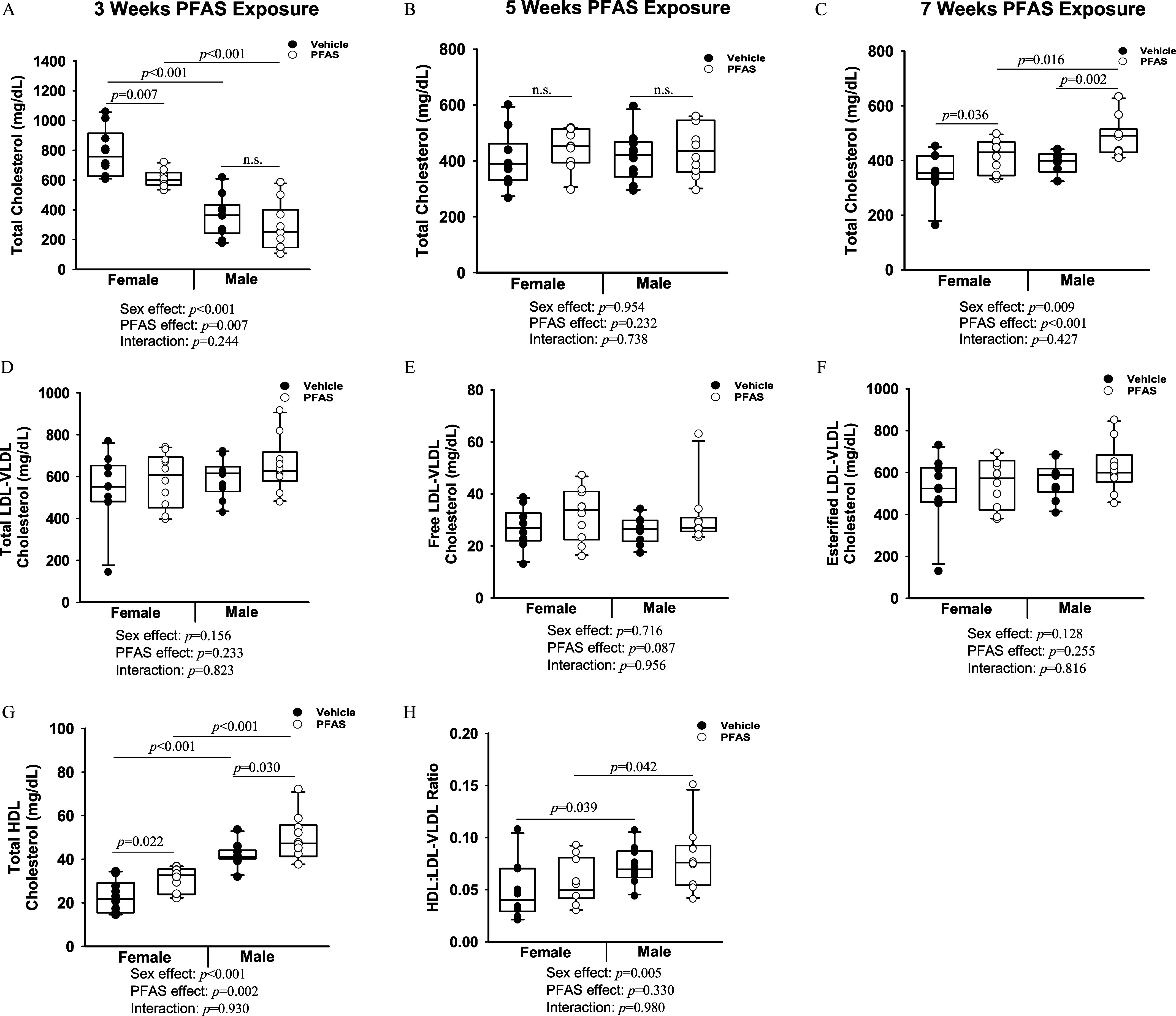
Exposure of mice to the PFAS mixture and cholesterol outcomes. Male and female mice were exposed to water containing the PFAS mixture for 7 wk. Blood was collected from mice in each treatment group and total cholesterol was measured after (A) 3 wk (unfasted), (B) 5 wk (unfasted), and (C) 7 wk (fasted) of PFAS exposure. The plasma collected at euthanasia was separated into different lipoprotein subfractions: (D) total LDL/VLDL cholesterol, (E) free LDL/VLDL, (F) esterified LDL/VLDL, and (G) total HDL cholesterol. (H) The ratio of HDL to LDL/VLDL is presented. Two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS (interaction ). The Holm–Sidak post hoc test was used for multiple comparisons. Box plots represent the median values with upper and lower quartiles; whiskers extend to the 1st and 99th percentiles. Data are reported in Excel Tables S7 and S8. Note: ANOVA, analysis of variance; HDL, high-density lipoprotein; LDL, low-density lipoprotein; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances; VLDL, very low-density lipoprotein.
The plasma HDL and LDL/VLDL lipoprotein fractions were also analyzed. Total LDL/VLDL cholesterol did not significantly differ in mice exposed to the PFAS mixture compared with those exposed to vehicle (Figure 3D). The free LDL/VLDL cholesterol fraction demonstrated a trend toward being higher in PFAS-exposed mice, but was not statistically significant () (Figure 3E). The esterified LDL/VLDL cholesterol fraction did not differ significantly (Figure 3G). The HDL cholesterol fraction was significantly higher in PFAS-exposed females ( in vehicle vs. in the PFAS-exposed group; ), and in PFAS-exposed males ( in vehicle vs. in PFAS-exposed group; ) (Figure 3G). The HDL:LDL/VLDL ratio did not differ significantly with PFAS exposure (Figure 3H).
Circulating PFAS levels were correlated with LDL/VLDL and HDL cholesterol fractions (Table S3). There were significant positive correlations between PFOS and free VLDL/LDL cholesterol () and significant inverse correlations with HDL cholesterol (). Similar trends were observed for circulating PFOA. No significant correlations between LDL/VLDL or HDL cholesterol with other PFAS were observed (Table S3).
Inflammatory Markers and Atherosclerotic Lesion Development
Finally, circulating levels of inflammatory cytokines were measured after 7 wk of PFAS exposure (Figure 4A–C; Figure S2A–E). Circulating levels of tumor necrosis factor- (TNF-) demonstrated a significant PFAS:sex interaction (interaction ), driven by lower levels in female PFAS-exposed mice () and higher levels in male PFAS-exposed mice (), compared with relative controls (Figure 4A). Circulating levels of monocyte chemoattractant protein-1 (MCP-1) also demonstrated a significant PFAS:sex interaction (interaction ), driven by lower levels in female PFAS-exposed mice () and higher levels in male PFAS-exposed mice approaching significance (), compared with relative controls (Figure 4B). Circulating levels of interleukin-6 (IL-6) demonstrated a PFAS:sex interaction approaching significance (interaction ), driven by higher levels in female PFAS-exposed mice (), whereas IL-6 levels in males were not significantly different (Figure 4C). The other measured cytokines did not demonstrate a significant effect due to PFAS exposure (Figure S2). Finally, en face analysis was completed on a subset of aortic arches. No significant differences were recorded at this early time point of atherosclerotic development (Figure 4D,E).
Figure 4.
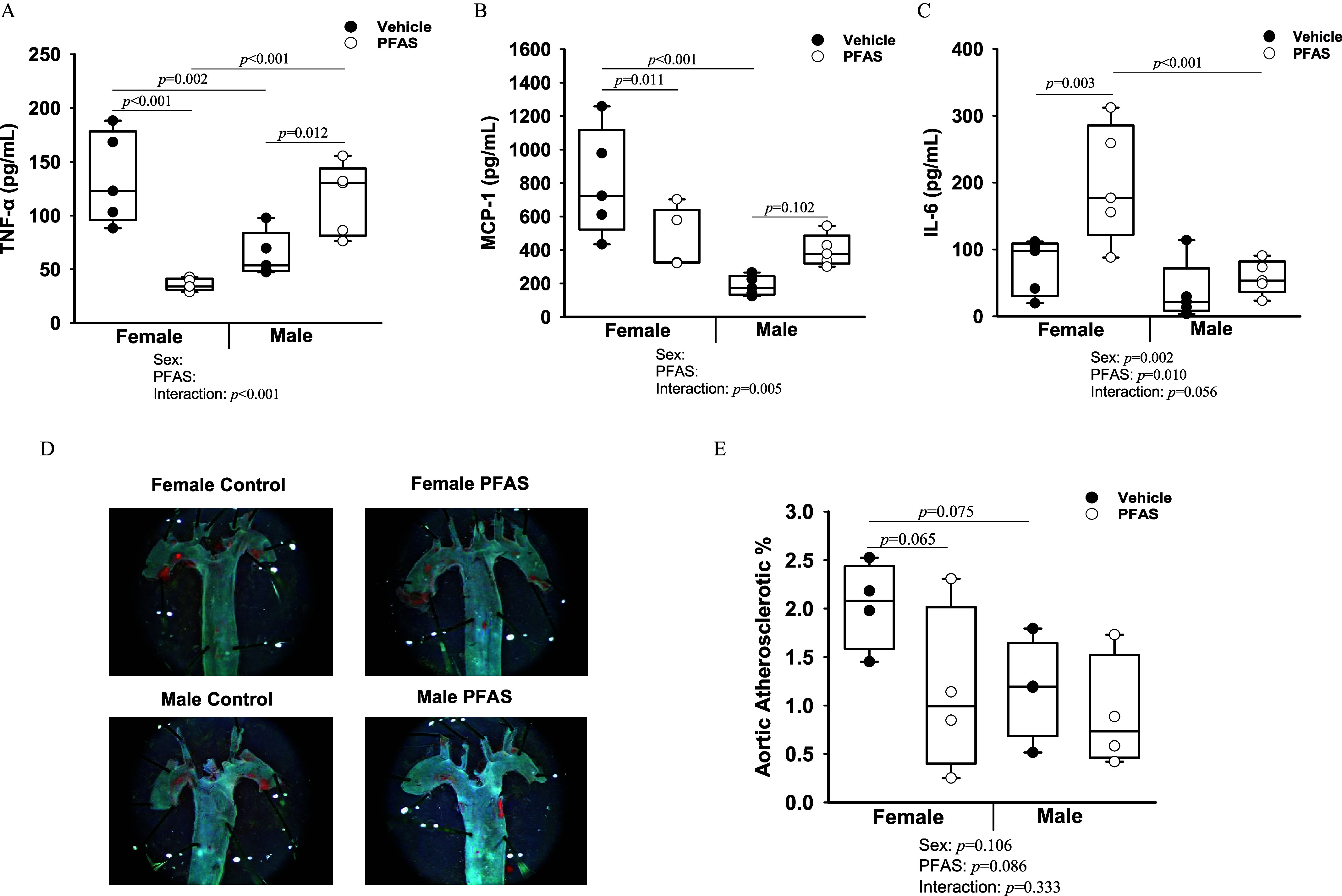
Exposure of mice to the PFAS mixture for 7 wk and inflammatory and atherosclerotic outcomes. Circulating cytokine protein levels for (A) TNF-, (B) MCP-1, and (C) IL-6 were measured from plasma collected at euthanasia from mice of each treatment group (). (D) En face aortas were stained with Oil Red O for mice per treatment group and representative images of stained aortas are shown. (E) The percentage of staining was quantified to determine aortic atherosclerotic percentage. Two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS (interaction ). The Holm–Sidak post hoc test was used for multiple comparisons. With past consultation from biostatisticians, a significant interaction term supersedes the main effects and can make their meaning unclear. We therefore have not included the main effects -values for any result with a significant interaction. Box plots represent the median values with upper and lower quartiles; whiskers extend to the 1st and 99th percentiles. Data are reported in Excel Tables S9 and S10. Note: ANOVA, analysis of variance; IL, interleukin; Ldlr, low density lipoprotein receptor; MCP, monocyte chemoattractant protein; PFAS, per- and polyfluoroalkyl substances; TNF, tumor necrosis factor.
Circulating Bile Acid Levels
Circulating levels of total bile acids were significantly higher in the PFAS-exposed mice compared with vehicle control mice (Figure 5). Female total bile acid levels were in vehicle control females and in PFAS-exposed females (), whereas male total bile acid levels were in vehicle control males and in PFAS-exposed males (; overall exposure effect ; interaction ). Specific circulating primary and secondary bile acids, along with many of their conjugated forms, were measured after 7 wk of PFAS exposure (Table 2). Plasma levels of the primary bile acids—cholic acid (CA), taurocholic acid (TCA), glycocholic acid (GCA), taurochenodeoxycholic acid (TCDCA), -muricholic acid (MCA), tauro--muricholic acid (TMCA), and taurocholic acid 3-sulfate (TCA-3-)—were all significantly higher in PFAS-exposed mice, ranging from 1.77- to 6.63-fold higher than those of vehicle control mice (overall exposure effect for all) (Table 2). The only primary bile acid included in our measurements that was lower in PFAS-exposed animals was chenodeoxycholic acid (CDCA), which was nearly absent in PFAS-exposed mice compared with vehicle control mice (overall exposure effect ). In addition, the bile acids glycochenodeoxycholic acid (GCDCA), cholic acid 7-sulfate (CA-7-), and ursodeoxycholic acid 3-sulfate (UDCA-3-) demonstrated a significant interaction between PFAS exposure and sex, driven by higher levels in the PFAS-exposed females (Table 2). The bile acids MCA, UDCA-3-, and deoxycholic acid disulfate (DCA-3-) also demonstrated a significant interaction between PFAS exposure and sex, which was driven by higher levels in PFAS-exposed males (Table 2).
Figure 5.

Total circulating bile acids in mice exposed to the PFAS mixture. Blood was collected from mice in each treatment group and total bile acid levels were measured in the plasma after 7 wk of PFAS exposure. Nonnormally distributed data was transformed prior to statistical analysis. Two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS (interaction ). The Holm–Sidak post hoc test was used for multiple comparisons. Box plots represent the median values with upper and lower quartiles; whiskers extend to the 1st and 99th percentiles. Data are reported in Excel Table S11. Note: ANOVA, analysis of variance; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances.
Table 2.
Circulating levels of individual plasma bile acids after exposure of mice to the PFAS mixture.
| Plasma bile acidsa | Female vehicle () pg/µL | Female PFAS () pg/µL | Male vehicle () pg/µL | Male PFAS () pg/µL |
|---|---|---|---|---|
| Primary bile acids | ||||
| Cholic acid (CA) | * | |||
| Chenodeoxycholic acid (CDCA) | * | # | * | |
| Taurocholic acid (TCA) | * | # | *,# | |
| Glycocholic acid (GCA) | * | # | *,# | |
| Glycochenodeoxycholic acid (GCDCA)b | * | # | ||
| Taurochenodeoxycholic acid (TCDCA) | * | # | *,# | |
| -Muricholic acid (MCA) | * | * | ||
| -Muricholic acid (MCA)b | # | * | ||
| Tauro--Muricholic acid (TMCA) | * | # | *,# | |
| Cholic acid 7-sulfate (CA-7-)b | * | # | # | |
| Taurocholic acid 3-sulfate (TCA-3-) | *,# | |||
| Secondary bile acids | ||||
| Lithocholic acid (LCA) | ||||
| Taurolithocholic acid (TLCA) | * | # | ||
| Taurodeoxycholic acid (TDCA) | # | |||
| Ursodeoxycholic acid (UDCA)b | # | * | ||
| Ursodeoxycholic acid 3-sulfate (UDCA-3-)b | * | # | ||
| Deoxycholic acid disulfate (DCA-di-)b | # | * | ||
Note: Average absolute plasma bile acid levels () for each treatment group are listed as . Statistical significance for the interaction and main effects of PFAS exposure and sex between all treatment groups (male vehicle, male PFAS, female vehicle, female PFAS) was determined using two-way ANOVA and the Holm–Sidak post hoc test for multiple comparisons. ANOVA, analysis of variance; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances; SEM, standard error of the mean . * compared with respective vehicle treatment. # compared with opposite sex of the same treatment.
total; male vehicle, male PFAS, female vehicle, and female PFAS.
PFAS:sex interaction determined by two-way ANOVA using SigmaPlot.
Hepatic Levels of Total Cholesterol and Bile Acids
Hepatic levels of total cholesterol were significantly lower in both male and female PFAS-exposed mice compared with vehicle control mice after 7 wk of PFAS exposure (Figure 6A). Total cholesterol levels were significantly lower by 0.40-fold in female PFAS-exposed mice () and 0.36-fold in male PFAS-exposed mice (; overall exposure effect ; interaction ). Expression of the rate-limiting enzyme in cholesterol synthesis, 3-hydroxy-3-methylglutaryl coenzyme A reductase (Hmgcr), demonstrated a significant PFAS:sex interaction, which was driven by a significant 0.42-fold lower level in the PFAS-exposed females () and a 0.67-fold lower level in the PFAS-exposed males () (interaction ) (Figure 6B). General pathways for both endogenous cholesterol synthesis and bile acid synthesis are depicted in a schematic in Figure 6C.
Figure 6.
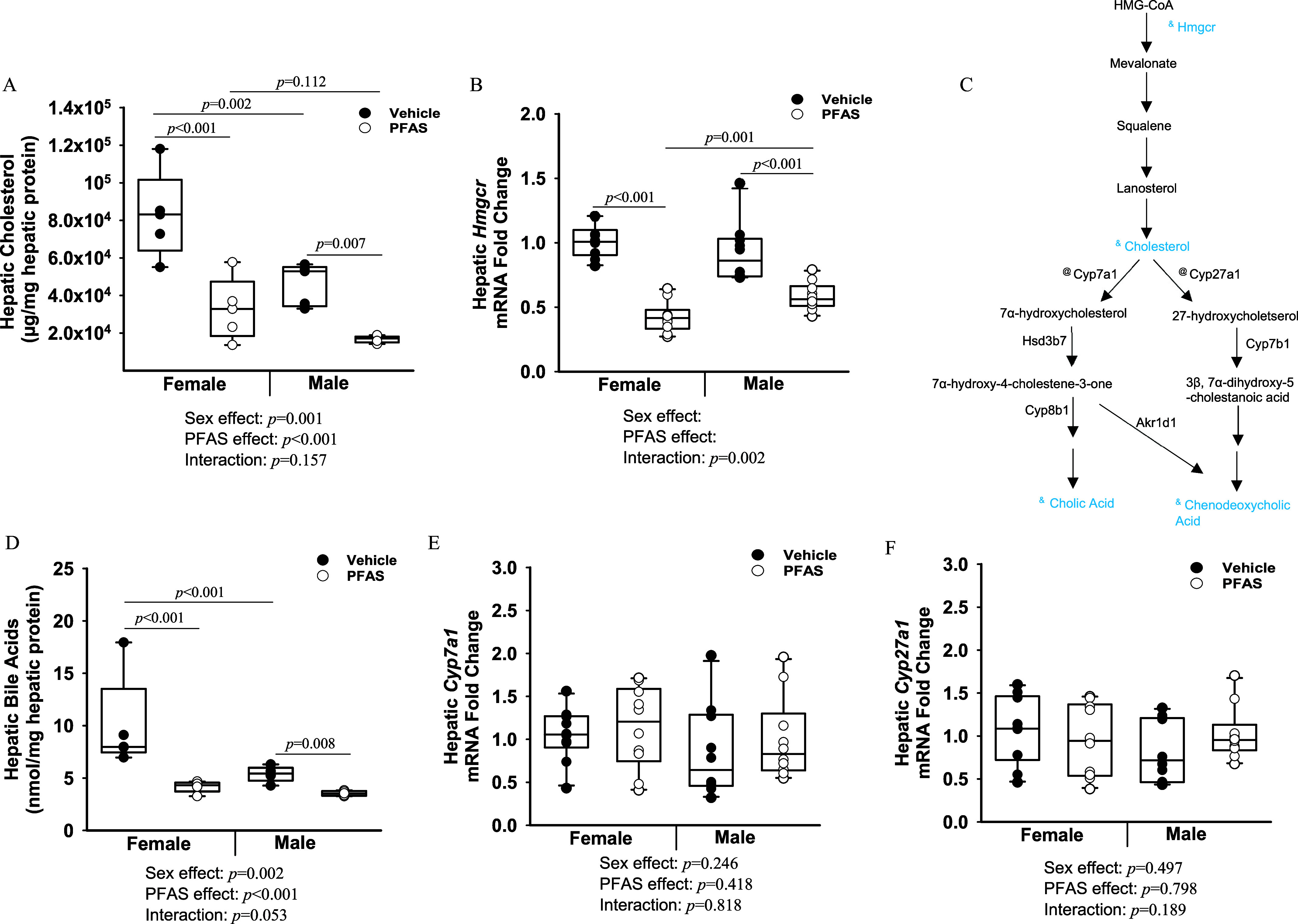
Exposure to the PFAS mixture, hepatic cholesterol and bile acid levels, and synthesis pathways. (A) Hepatic protein was isolated from mice from each treatment group and hepatic total cholesterol levels were measured. (B) Total hepatic RNA ( mice from each treatment group) was isolated, and gene expression levels of Hmgcr were determined by RT-qPCR. (C) Diagram of hepatic cholesterol and bile acid synthesis pathways. Steps preceded by “&” represent a significant reduction, whereas those measured and not significantly affected are preceded by “@.” (D) Hepatic total bile acid levels. Gene expression levels of (E) Cyp7a1, and (F) Cyp27a1 were determined by RT-PCR. GAPDH was used as a housekeeping gene. Two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS (interaction ). The Holm–Sidak post hoc test was used for multiple comparisons. With past consultation from biostatisticians, a significant interaction term supersedes the main effects and can make their meaning unclear. We therefore have not included the main effects -values for any figure with a significant interaction. Box plots represent the median values with upper and lower quartiles; whiskers extend to the 1st and 99th percentiles. Data are reported in Excel Tables S12 and S13. Note: ANOVA, analysis of variance; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; PFAS, per- and polyfluoroalkyl substances; RT-qPCR, real-time quantitative polymerase chain reaction.
Total hepatic bile acid levels demonstrated a PFAS:sex interaction approaching significance (interaction ) (Figure 6D). This was driven by 0.42-fold lower level in the female PFAS-exposed mice (), whereas male PFAS-exposed mice were lower by 0.65-fold (). Transcriptional regulation of Cyp7a1 (Figure 6E) and Cyp27a1 (Figure 6F), two rate-limiting enzymes in bile acid synthesis, were measured by RT-qPCR. Neither Cyp7a1 (overall exposure effect ) nor Cyp27a1 (overall exposure effect ) were differentially expressed in PFAS-exposed mice compared with vehicle control mice.
Hepatic Bile Acid Transporters
After 7 wk of PFAS exposure, the effects on transcriptional regulation of several hepatic bile acid transporters were measured by RT-qPCR. Many of the following hepatic bile acid transporters are summarized in Figure 7A, as well as their roles in hepatic influx and efflux and the observed PFAS-mediated effects. Specifically, expression of hepatic Abcc3, Abcc4, Slc10a1, and Ephx1 (Figure 7B–D; Figure S3) all demonstrated a significant interaction between PFAS exposure and sex. The PFAS:sex interaction for Abcc3 was driven by significantly higher expression in PFAS-exposed male mice (), whereas the difference in females was not quite significant () (Figure 7B). The PFAS:sex interaction for Abcc4 was driven by a greater fold difference in expression in PFAS-exposed males as opposed to PFAS-exposed females when compared with their relative vehicle controls (Figure 7C). The PFAS:sex interaction for Slc10a1 was driven by a larger fold difference in PFAS-exposed females as opposed to PFAS-exposed males when compared with their relative vehicle controls (Figure 7D). Conversely, transcriptional expression of hepatic Slc10a2, Abcb11, and Ost were all significantly lower by 0.14- to 0.45-fold compared with vehicle controls due to overall PFAS exposure (Figure 7E; Figure S3). Hepatic protein levels of NTCP, coded by Slc10a1, were measured via Western blot and quantified relative to -actin (Figure S3). Although no significant differences were seen for female mice, hepatic NTCP levels in the male mice were significantly up-regulated 2.4-fold in the PFAS-exposed males compared with male vehicle control mice ().
Figure 7.
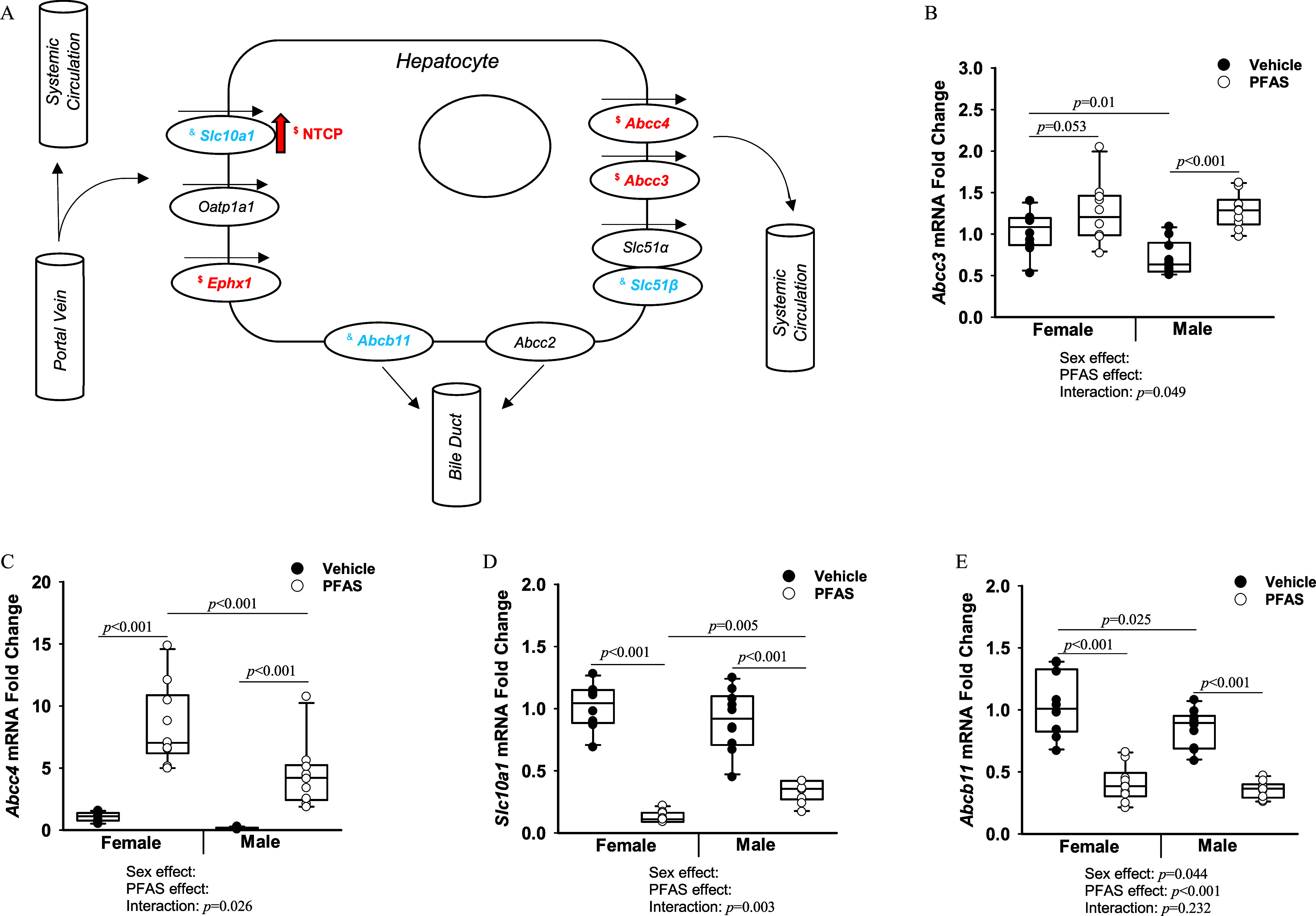
Hepatic bile acid transporters in mice exposed to the PFAS mixture. Total hepatic RNA from mice from each treatment group was isolated. (A) Diagram of PFAS-induced effects on hepatic bile acid transporters. Transcriptional changes are italicized. NTCP protein changes are indicated by arrow. “$” indicates a significant relative fold change increase (). “&” indicates a significant relative fold change decrease (). Expression levels of the transporters (B) Abcc3, (C) Abcc4, (D) Slc10a1, and (E) Abcb11 were determined by RT-qPCR. GAPDH was used as a housekeeping gene. Two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS (interaction ). The Holm–Sidak post hoc test was used for multiple comparisons. With past consultation from biostatisticians, a significant interaction term supersedes the main effects and can make their meaning unclear. We therefore have not included the main effects -values for any result with a significant interaction. Box plots represent the median values with upper and lower quartiles; whiskers extend to the 1st and 99th percentiles. Data are reported in Excel Table S14. Note: ANOVA, analysis of variance; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Ldlr, low density lipoprotein receptor; NTCP, sodium taurocholate cotransporting polypeptide; PFAS, per- and polyfluoroalkyl substances; RT-qPCR, real-time quantitative polymerase chain reaction.
Ileal Transcriptome and Bile Acid Transporters after PFAS Exposure in Male Mice
Transcriptomic analyses of the ileum were performed in the male mice. A total of 36 genes were found to be significantly up-regulated and 34 genes were found significantly down-regulated in PFAS-exposed males (|log fold change| ; FDR ) (Table S4). Gene ontology enrichment analyses were completed using the DAVID software. The top 15 significantly enriched pathways due to PFAS exposure, determined by the lowest Benjamini–Hochberg–adjusted -values, are depicted in Figure 8A and include the acute inflammatory response, fatty acid metabolic process, and lipid metabolic process. The alterations in gene expression due to PFAS exposure for the specific genes within the acute inflammatory response and the lipid metabolic process are listed in Table S5. Transcriptomic sequencing data was next analyzed specifically for gene expression of ileal bile acid transporters. Many of the ileal bile acid influx and efflux transporters are depicted in Figure 8B. Effects on gene expression levels of several ileal bile acid transporters are presented in Table 3. Transcriptional expression of ileal Slc10a2, Slc51, Slc51 were all significantly up-regulated between 1.28- to 1.55-fold due to PFAS exposure (Table 3). Protein levels of the ASBT, encoded by Slc10a2, were also significantly higher. Ileal protein levels of ASBT were measured via Western blot (Figure 8C) and quantified relative to -actin (Figure 8D). Ileal levels of ASBT protein were significantly higher by 4-fold in males due to PFAS exposure ().
Figure 8.
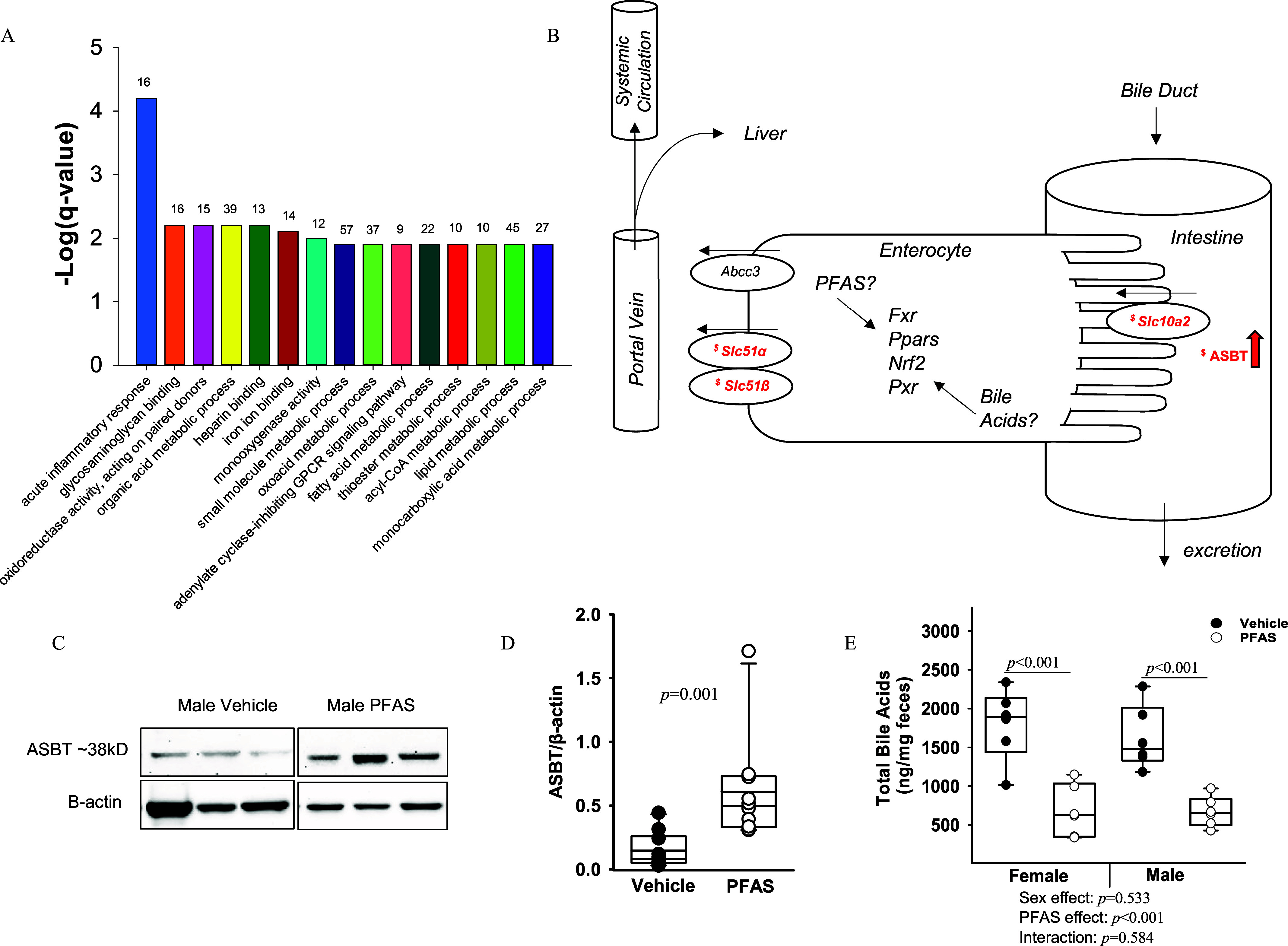
Ileal transcriptome and bile acid transporters in male mice exposed to the PFAS mixture. Total ileal RNA was isolated from males from the vehicle control and PFAS-exposed groups. (A) Gene ontology analysis results are graphically depicted, with the top 15 significant pathways listed as determined by the lowest Benjamini–Hochberg–adjusted -values. The number of genes within each pathway is listed above. (B) Diagram of PFAS-induced effects on ileal bile acid transporters. Transcriptional changes are italicized. ASBT protein changes are indicated by arrow. “$” indicates a significant relative fold change increase (). (C) Western blot analysis of ASBT protein in the ileum. (D) Quantification of band intensity for ASBT protein relative to -actin. Statistical significance for Western blot quantitation was determined by Mann–Whitney rank sum test (). (E) Fecal excretion of total bile acids was also measured via LC-MS in mice from each treatment group. Nonnormally distributed data was transformed prior to statistical analysis. Two-way ANOVA was used to analyze both main effects (i.e., sex, PFAS), as well as the interaction between sex and PFAS (interaction ). The Holm–Sidak post hoc test was used for multiple comparisons. Box plots represent the median values with upper and lower quartiles; whiskers extend to the 1st and 99th percentiles. Data are reported in Excel Tables S15–S17. Note: ANOVA, analysis of variance; ASBT, apical sodium-dependent bile acid transporter; GPCR, G protein-coupled receptor; LC-MS, liquid chromatography–mass spectrometry; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances.
Table 3.
Expression of ileal bile acid transporters after exposure of male mice to the PFAS mixture.
| Ileal bile acid Transportersa | Male vehicle RNA-seq counts () | Male PFAS RNA-seq counts () | Overall PFAS exposure (fold change) | Overall PFAS exposure (-value) | Overall PFAS exposure (FDR-adjusted -value) |
|---|---|---|---|---|---|
| Slc10a1 (NTCP) | 0.44 | 0.15 | 0.72 | ||
| Slc10a2 (ASBT) | 1.55* | 0.014* | |||
| Abcc2 (MRP2) | 1.05 | 0.57 | 1.00 | ||
| Abcc3 (MRP3) | 1.28 | 0.039 | 0.48 | ||
| Abcc4 (MRP4) | 1.36 | 0.074 | 0.60 | ||
| Abcb11 (BSEP) | 0.85 | 0.89 | 1.00 | ||
| Ephx1 (mEH) | 0.94 | 0.57 | 1.00 | ||
| Slc51 (OST) | 1.30* | * | |||
| Slc51 (OST) | 1.47* | * |
Note: Statistical significance was determined by -test using R statistical software (version 4.2.0; R Developmental Core Team). FDR, false discovery rate; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances; RNA-seq, RNA sequencing; SEM, standard error of the mean. *Represents significant change relative to vehicle control ().
total; male vehicle and male PFAS.
Bile Acid Excretion after PFAS Exposure
Bile acid levels were measured in the feces of our mice during the final week of the study. Total fecal bile acid levels were lower by 0.38-fold in PFAS-exposed females () and were lower by 0.41-fold in PFAS-exposed males () compared with those in the respective vehicle control mice (Figure 8E). The fecal levels of many individual primary and secondary bile acids were also measured and are presented in Table 4. Plasma levels of the primary and secondary bile acids—CA, TCA, TCDCA, MCA, -muricholic acid (MCA), TMCA, chenodeoxycholic acid-sulfate (CDCA-), lithocholic acid (LCA), deoxycholic acid (DCA), taurodeoxycholic acid (TDCA), UDCA, tauroursodeoxycholic acid (TUDCA), and DCA-3-—were all significantly lower in PFAS-exposed mice, ranging from 0.05 to 0.59 (overall exposure effect for all) (Table 4). In addition, six bile acids—CDCA, GCDCA, CA-7-, TCA-3-, UDCA-3-, and DCA-di-—demonstrated a significant interaction between PFAS exposure and sex. For CDCA, and GCDCA, the PFAS:sex interaction was driven by a greater difference in bile acid levels between PFAS-exposed and vehicle control males. For TCA-3- and DCA-di-, the PFAS:sex interaction was driven by a greater difference in bile acid levels between PFAS-exposed females and vehicle controls. For CA-7- and UDCA-3-, the PFAS:sex interaction was driven by lower levels in PFAS-exposed females and higher levels in males overall.
Table 4.
Fecal bile acid levels after exposure of mice to the PFAS mixture.
| Fecal bile acidsa | Female vehicle () ng/mg | Female PFAS () ng/mg | Male vehicle () ng/mg | Male PFAS () ng/mg |
|---|---|---|---|---|
| Primary bile acids | ||||
| Cholic acid (CA) | * | * | ||
| Chenodeoxycholic acid (CDCA)b | * | *,# | ||
| Taurocholic acid (TCA) | * | # | ||
| Taurochenodeoxycholic acid (TCDCA) | * | # | * | |
| Chenodeoxycholic acid-sulfate (CDCA-) | * | |||
| -Muricholic acid (MCA) | * | # | * | |
| -Muricholic acid (MCA) | * | # | *,# | |
| Tauro--Muricholic acid (TMCA) | * | # | ||
| Cholic acid 7-sulfate (CA-7-)b | * | # | # | |
| Taurocholic acid 3-sulfate (TCA-3-)b | * | # | ||
| Secondary bile acids | ||||
| Lithocholic acid (LCA) | * | # | # | |
| Deoxycholic acid (DCA) | * | # | *,# | |
| Taurodeoxycholic acid (TDCA) | * | # | * | |
| Ursodeoxycholic acid (UDCA) | * | # | *,# | |
| Tauroursodeoxycholic acid (TUDCA) | * | |||
| Ursodeoxycholic acid 3-sulfate (UDCA-3-)b | * | # | *,# | |
Note: Average absolute fecal bile acid levels (ng/mg) for each treatment group are listed as . Statistical significance for the interaction and main effects of PFAS exposure and sex between all treatment groups (male vehicle, male PFAS, female vehicle, female PFAS) was determined using two-way ANOVA and the Holm–Sidak post hoc test for multiple comparisons; . ANOVA, analysis of variance; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances; SEM, standard error of the mean. * compared with respective vehicle treatment. # compared with opposite sex of the same treatment.
total; male vehicle, male PFAS, female vehicle, and female PFAS.
PFAS:sex interaction determined by two-way ANOVA using SigmaPlot.
Gene expression analyses of hepatic and ileal tissues were analyzed via RT-qPCR or RNA-seq, respectively. Differences in ileal gene expression of several nuclear receptors and downstream mediators are presented in Table 5. Interestingly, in our mice, ileal farnesoid X receptor (FXR) expression was higher in PFAS-exposed male mice, whereas small heterodimer partner (SHP) expression was not different. Downstream of FXR, ileal Fgf15 expression was also not significantly different in PFAS-exposed mice () (Table 5). Ileal peroxisome proliferator-activated receptor delta/beta (PPAR) was significantly higher in PFAS-exposed animals, as was its downstream target aspartoacylase (Aspa). On the other hand, the nuclear receptor PPAR was significantly lower, along with downstream mediator Hsd17b11. Other ileal nuclear receptors, including Nr1i2 (PXR), Nr1i3 (CAR), Nr1i1 (VDR), Nfe2l2 (NRF2), and Pparγ, were not significantly different in PFAS-exposed mice, nor were the selected downstream targets (Table 5). In the liver, FXR expression was significantly down-regulated in PFAS-exposed mice (Table S6). Hepatic SHP expression demonstrated a significant PFAS:sex interaction, which was driven by significantly lower levels of SHP in females (), whereas levels in males were not significantly different (). Hepatic CAR expression also demonstrated a significant PFAS:sex interaction, driven by significantly lower levels in PFAS-exposed females (), whereas levels in males were not significantly different () (Table S6). Finally, hepatic Gstm1 expression also exhibited a significant PFAS:sex interaction, which was driven by significantly higher levels in PFAS-exposed females (), whereas levels in males were not significantly different ().
Table 5.
Ileal nuclear receptor signaling after exposure of male mice to the PFAS mixture.
| Gene name | Male vehicle RNA-seq counts () | Male PFAS RNA-seq counts () | Overall PFAS exposure (fold change) | Overall PFAS exposure (-value) | Overall PFAS exposure (FDR-adjusted -value) |
|---|---|---|---|---|---|
| Nuclear receptor signalinga | |||||
| Nr1h4 (FXR) | 1.48* | * | |||
| mFgf15 | 0.74 | 0.12 | 0.68 | ||
| Nr0b2 (SHP) | 0.54 | 0.34 | 0.95 | ||
| Nr1i2 (PXR) | 1.14 | 0.04 | 0.48 | ||
| Cyp3a11 | 0.82 | 0.25 | 0.88 | ||
| Nr1i3 (CAR) | 0.26 | 0.08 | 0.62 | ||
| Cyp2b10 | 0.69 | 0.095 | 0.64 | ||
| Nr1i1 (VDR) | 0.97 | 0.57 | 1.0 | ||
| Nfe2l2 (NRF2) | 0.86 | 0.02 | 0.31 | ||
| Keap1 | 0.88 | 0.03 | 0.43 | ||
| Gstm1 | 1.05 | 0.50 | 1.0 | ||
| Hmox1 | 1.60 | 0.16 | 0.73 | ||
| Ppar | 0.72* | 0.01* | |||
| Fbp2 | 1.00 | 0.99 | 1.0 | ||
| Hsd17b11 | 0.73* | * | |||
| Ppar | 1.32 | 0.21 | 0.84 | ||
| Adipoq | 1.28 | 0.14 | 0.71 | ||
| Ppar/ | 1.52* | * | |||
| Aspa | 1.33* | 0.003* | |||
Note: Statistical significance was determined by -test using R statistical software (version 4.2.0; R Development Core Team). FDR, false discovery rate; Ldlr, low density lipoprotein receptor; PFAS, per- and polyfluoroalkyl substances; RNA-seq, RNA sequencing; SEM, standard error of the mean. *Represents significant change relative to vehicle control ().
total, male vehicle and male PFAS.
Estrous Cycle Hormones after PFAS Exposure
The estrous cycle was not controlled for in our female mice. Because of this, circulating levels of FSH, LH, and progesterone were measured in the plasma of our female mice after 7 wk of PFAS exposure (Figure S4A–C). PFAS exposure did not significantly impact the circulating concentrations of these three hormones. The variability of FSH was not significant (Figure S4A). Most LH measurements were below the LOD. The progesterone levels measured in the PFAS-exposed females did not have significant variance (Figure S4C). However, variance of progesterone in the vehicle control females was observed.
Discussion
Epidemiological studies have repeatedly determined a significant association between PFAS exposure and hypercholesterolemia, but the specific mechanisms involved are not well understood.39–42 Previous studies have additionally shown that PFAS exposure is significantly associated with alterations of bile acid profiles, suggesting alteration of bile acid synthesis, transport, and reuptake as logical mechanisms of action.46–49 In the present study, we investigated mechanisms linking exposure to a PFAS mixture with effects on cholesterol and bile acid metabolism in a mouse model with high levels of LDL/VLDL cholesterol and aortic lesion development similar to humans.58 Compared with vehicle control mice, those exposed to the PFAS mixture had higher circulating cholesterol and bile acid levels, as well as differential expression of critical transporters of the enterohepatic circulation, including ileal ASBT levels.
Previous studies have shown that PFAS exposure is associated with liver injury, such as hypertrophy, steatosis, and MASLD (formerly nonalcoholic fatty liver disease).27,73–75 Histological examination of the mouse livers showed greater injury and lipid accumulation in PFAS-exposed mice, especially in the PFAS-exposed females (Figure 1A). Body weight at study end was significantly lower in the PFAS-exposed females, whereas weights of PFAS-exposed males were not significantly different from vehicle control mice (Figure 1C), most likely owing to a similar loss in fat weight (Figure S1). In addition, serum ALT levels and liver weights were significantly higher in both male and female PFAS-exposed mice (Figure 1D), further supporting PFAS-induced liver injury, especially in females.
The individual circulating PFAS concentrations averaged between , suggesting the present exposure scenario results in PFAS levels above those typically seen in highly exposed populations (Table 1).11,66–72 However, circulating PFOA concentrations in our mice were within range of some occupational exposure studies, which measured PFOA levels in humans as high as (Table 1).67 In addition, circulating PFOS levels in our mice were also within range of an occupational PFAS exposure cohort in China analyzed by Lu et al.,69 which measured PFOS levels as high as . PFHxS had the highest circulating levels in our study and were times higher than those measured in the Lu et al. cohort.69 PFNA and GenX concentrations in our study were an order of magnitude higher than maximum levels of those measured in PFAS biomonitoring studies (Table 1), although occupational studies focusing on manufacturers of PFNA and GenX are lacking.
Similar to liver toxicity, there were significant sex differences in circulating levels of PFOA and PFOS, with higher levels measured in female PFAS-exposed mice compared with males (Table 1). Although differences in water intake, which would impact PFAS exposure levels, was observed in our study, at no time point did the PFAS-exposed females drink more water than the PFAS-exposed males. In fact, PFAS-exposed males drank significantly more of the PFAS water than the PFAS-exposed females on weeks 1, 4, and 6 (Figure 2B). Because PFOA and PFOS concentrations were higher in females, it is unlikely that differences in water intake between sexes is responsible for these differences in the PFAS-exposed mice. However, male PFAS-exposed mice excreted significantly more feces than the females (Figure 2E), which could also affect PFAS exposure levels, leading to the lower PFOA and PFOS concentrations observed in the male mice (Table 1). Significant sex differences were not measured in plasma levels of PFHxS, PFNA, or GenX, though, indicating that although differences in excretion may be a contributing factor for PFOA and PFOS, excretion differences alone do not fully explain the sex differences in PFAS concentrations.
After 3 wk of PFAS exposure, total circulating cholesterol levels were lower in the PFAS-exposed mice compared with vehicle controls (Figure 3A), which could be due to initial PPAR activation.76–79 However, this effect was transient, and by 7 wk of PFAS exposure, total circulating cholesterol levels were higher in both male and female PFAS-exposed mice (Figure 3C). Cholesterol is primarily synthesized in the liver and is transported through plasma via various lipoproteins, including LDL, HDL, VLDL, and intermediate-density lipoprotein (IDL) cholesterol and chylomicrons.80 Of these lipoproteins, LDL is the major carrier of cholesterol in human plasma,81 whereas HDL is a key mediator of reverse cholesterol transport.82 PFAS exposure has repeatedly been associated with elevated cholesterol levels, especially in the total and LDL cholesterol fractions.39–43 We found that the free LDL/VLDL cholesterol fraction demonstrated an upward trend in PFAS-exposed mice () (Figure 3D), which could be an indication of atherogenic risk.83 In addition, HDL levels were slightly higher in both male and female PFAS-exposed mice (Figure 3G). Although PFAS usually have been associated with increases in non-HDL cholesterol fractions,40,41,84 a few studies have shown PFHxS associated with increased HDL in humans.43,85 However, the overall HDL values were low and do not make up for the differences in total cholesterol (Figure 3). Therefore, it is likely that HDL was not driving the increase in total cholesterol. Of the five PFAS in our mixture, PFOA and PFOS were the only two with significant correlations with cholesterol—positively associated with free or esterified LDL/VLDL and inversely correlated with HDL levels (Table S3). It is possible that PFOA and PFOS could be the specific PFAS driving the observed PFAS-induced effects in cholesterol levels, but a more in-depth analysis of lipoprotein fractions is called for, especially given the low sample size used in the correlations.
Elevated levels of circulating cholesterol play an important role in the progression of inflammatory atherosclerotic plaques.36–38 When plasma samples were also analyzed for inflammatory cytokines, circulating levels of TNF- and MCP-1 were found to be lower in PFAS-exposed females, whereas they were higher in PFAS-exposed males compared with vehicle controls (Figure 4). Of note, female vehicle control mice had much higher baseline levels of both TNF- and MCP-1 compared with male vehicle control mice, suggesting that females may already be in a higher inflammatory state. However, the aortas contained only 1%–2% aortic lesions at this time point, with no significant PFAS-associated differences (Figure 4D,E). A recent study by Wang et al. observed that exposure of hyperlipidemic mice to PFOS for 12 wk accelerated atherosclerotic progression,86 suggesting the need for longer exposure studies to examine atherosclerosis.
Although there was no lesion development at this 7-wk time point, total circulating cholesterol levels were still higher in the PFAS-exposed mice (Figure 3C). Cholesterol levels can be affected in a variety of ways. One mechanism involves modulation of endogenous cholesterol synthesis. In our study, hepatic cholesterol levels were significantly lower in both the male and female PFAS-exposed mice (Figure 6A). Endogenous cholesterol synthesis can be regulated transcriptionally, with Hmgcr being the rate-limiting enzyme.87 We observed that transcriptional regulation of Hmgcr was significantly down-regulated in both male and female PFAS-exposed mice (Figure 6B), indicating that PFAS-induced transcriptional induction of endogenous cholesterol synthesis was likely not responsible for the elevated circulating cholesterol.
The main route of cholesterol catabolism and removal from the body is through its conversion into bile acids.44,88,89 PFAS exposure has been shown to alter bile acid profiles46,47,49 and metabolism.56,57,90 In our study, circulating bile acid levels were significantly higher in both male and female PFAS-exposed mice compared with vehicle control mice, with PFAS-exposed females often having significantly higher total bile acid levels compared with males (Figure 5 and Table 2). Conversely, hepatic bile acid levels were significantly lower in PFAS-exposed mice () (Figure 6D). However, this effect did not appear to be driven by PFAS-induced transcriptional effects in bile acid synthesis pathways. Two pathways are responsible for the majority of bile acid synthesis. The first, known as the classical pathway, is catalyzed in the rate-limiting step by cholesterol-7-hydroxylase (Cyp7a1).91 The alternative pathway is catalyzed by sterol-27-hydroxylase (Cyp27a1).91 Elevated levels of bile acids usually trigger negative feedback mechanisms, leading to down-regulation of Cyp7a1 and Cyp27a1 in the liver.92,93 Decreased levels of these enzymes due to PFAS exposure have been observed before both in vitro and in vivo.84,94,95 No significant differences were seen in either Cyp7a1 and Cyp27a1 between PFAS-exposed and vehicle control animals, indicating that transcriptional regulation of the conversion of cholesterol to bile acids does not explain the elevated cholesterol or bile acid levels in the plasma (Figure 6E,F).
In addition to synthesis, bile acid levels can also be modulated via changes to their transport through the enterohepatic circulation.45 Gene expression levels of the hepatic transporters multidrug resistance protein 3 (MRP3), coded for by Abcc3, were higher in PFAS-exposed males (Figure 7B), whereas MRP4, coded for by Abcc4, was higher in both male and female PFAS-exposed mice (Figure 7C). Given that both MRP3 and MRP4 participate in bile acid efflux into the circulation, our results suggests that up-regulation of hepatic efflux transporters could also be a mechanism for increased circulating bile acid levels.96 The bile acid transporter sodium taurocholate cotransporting polypeptide (NTCP), coded by Slc10a1, is highly expressed on the basolateral membrane of hepatocytes.97 NTCP is responsible for of bile acid uptake into the liver, including the major taurine- and glycine-conjugated bile acids.52,98 In our mice, hepatic expression of Slc10a1, the gene encoding NTCP, was significantly lower in PFAS-exposed mice of both sexes (Figure 7D). However, NTCP protein levels were significantly higher in male mice exposed to PFAS, but not in females (Figure S3).
Several previous studies have explored the relationship between PFAS exposure and impacts on bile acid metabolism.56,57,90 However, our study is distinct in its analysis of circulating, hepatic, and fecal bile acids to get an overall idea of the effect of PFAS on bile acids, as well as their impacts on the ileum. Gene ontology enrichment analysis performed on the ilea of our male mice, revealed highly significant differences in several pathways due to PFAS exposure, including the lipid metabolic process and the acute inflammatory pathway, which can be linked to atherosclerosis (Figure 8A).99 Within the lipid metabolic process pathway, we found lower expression of many genes related to fatty acid metabolism, including Acot1/2 and Cyp4a (Table S5). In addition, many differentially expressed genes were observed in the ilea of PFAS-exposed male mice (Table S4), including the significant up-regulation of serum amyloid A1 (Saa1), which has been linked to cholesterol metabolism and atherosclerosis100,101 and is a novel biomarker for cardiovascular disease.102,103
Bile acid transporters are highly expressed in the ileum as part of the reabsorption process. The enterohepatic circulation is very efficient, with of bile acids being reabsorbed in the ileum, occurring primarily via ASBT-mediated uptake from the intestinal lumen into the ileal enterocytes.104,105 Inhibition of ASBT has been shown to increase the fecal excretion of bile acids and to decrease the amount of bile acids returning to the liver.106,107 Furthermore, a study by Iwaki et al. demonstrated that concomitant treatment of mice with the bile acid sequestrant, colestyramine, and the ASBT inhibitor, elobixibat, reduced LDL cholesterol levels as well as plaque formation rates.108 In our study, transcriptional regulation of ASBT, as well as protein levels, were significantly higher in PFAS-exposed male mice (Figure 8D and Table 3). Interestingly, ASBT has been shown able to transport both PFAS and bile salts from the intestines into enterocytes in vitro.56 Within the ileal enterocyte, efflux transporters then move bile acids into the portal vein. Gene expression of the ileal efflux transporters MRP3 and OST/ were up-regulated in our PFAS-exposed mice (Table 3). This up-regulation, together with the observed lower amounts of bile acids excreted in the feces (Figure 8E), suggests that PFAS exposure led to increased reabsorption and lower excretion of bile acids. Interestingly, the male PFAS-exposed mice produced significantly more fecal mass compared with vehicle control mice, whereas female fecal mass was not affected by PFAS (Figure 2E). These alterations in fecal mass are not mirrored by concurrent increases in food intake, nor by the amounts of bile acids excreted through the feces (Figures 2A and 8E).
Bile acid transporters are governed by mechanisms of transcriptional control that act through nuclear receptor pathways.92,109,110 The effects of PFAS exposure on these pathways may be due directly to PFAS, or indirectly through PFAS-induced alterations in signaling mediators, including bile acids. The main pathway responsible for bile acid regulation is via FXR, which senses elevated bile acid levels and activates negative feedback mechanisms.111 In our study, transcriptional expression of ileal FXR was significantly up-regulated in PFAS-exposed mice, whereas SHP and downstream target Fgf15 expression were unaffected (Table 5). However, previous studies have shown that FXR negatively regulated ileal ASBT levels in mice via SHP.112,113 In addition, ileal Ppar and downstream target Hsd17b11 were also down-regulated (Table 5), although previous studies have found that PPAR ligands can activate ASBT.114 Overall, our results suggest that normal bile acid signaling pathways are modulated when exposed to PFAS, and the observed effects do not by themselves explain ASBT levels.
There are certain limitations present throughout the present study. First, the human relevance of the mouse model bears consideration. Although VLDL levels measured in mice fed the Clinton/Cybulsky diet58 are lower than those observed in mice on other atherogenic diets,115 VLDL levels are still higher than those observed in humans.115 In addition, human epidemiological studies reproducibly support a positive association of specific PFAS with increased LDL cholesterol, but not with an increase in HDL cholesterol. In our study, we observed significantly higher levels of HDL cholesterol, but not to the extent that would explain our observed increases in total cholesterol. Given that our measures of cholesterol fractions were done using separation by precipitation, here we have not considered the chylomicron or chylomicron remnant lipoprotein fractions, which are produced in the intestines, impacted by fasting, and may also contribute to atherosclerosis as a significant transporter of cholesterol to the atherosclerotic plaque.116 Thus, future studies using fast protein liquid chromatography and other methods, such as LipoPrint, to measure all cholesterol subfractions will be valuable as a more accurate analysis of what lipoprotein fractions are driving the total cholesterol increase. Furthermore, in this study we did not observe differences in the expression of Cyp7a1 in PFAS-exposed animals, which has reproducibly been shown in mouse studies of similar duration,47,84 as well as with human-relevant in vitro models.90,94,95 In addition, the mouse model has been shown to be predisposed to hepatic lipid accumulation and steatosis when fed an atherogenic diet.117 Our vehicle control mice did exhibit early hepatic lipid accumulation, thus it is possible that the exacerbation due to PFAS, especially in females, could have been impacted by the genotype. It has also been shown that some PFAS are excreted via the urine,57,118 so future studies should address urinary excretion as well as fecal. In addition, PFAS have been shown to accumulate in different tissues not analyzed here, such as bile and kidneys.57
Another limitation is the lack of control for the estrous cycle in the female mice. Possible effects from the estrous cycles are especially interesting given that female sex hormones are proposed to be protective against atherosclerosis.119 Previous studies have found that estrogens can significantly increase hepatic VLDL production,120 whereas progestins promote VLDL clearance.121,122 Estrogen has also been found to promote reverse cholesterol transport and the uptake of cholesterol by HDL.123–126 It is possible that fluctuations in these hormones could obscure PFAS-induced effects on VLDL and HDL cholesterol levels. Furthermore, rodent studies have shown that the estrous cycle can modulate expression of Cyp7a1 and Cyp27a1,124 which could also add variability to PFAS-induced modulation of bile acids. Our PFAS measurements also provide a limitation to our study, given that PFAS levels in vehicle control mice were below the LODs of our plasma LC/MS methodology, so correlative analyses only included plasma PFAS concentration data from PFAS-treated mice. In addition, the PFAS-exposed males generally ate more food than the PFAS-exposed females throughout the study. This could lead to some effects being attributable to the reduced plane of nutrition and therefore secondary to the PFAS exposure, such as with observed modulation of hepatic Hmgcr expression and ABCC4 levels, which have both been shown to be impacted by overconsumption.127–129 Finally, ilea were only collected for the males and should be studied in females as well in future studies, especially given that sexual dimorphisms of ASBT and other ileal transporters have been observed.130,131
Our study showed that PFAS exposure in mice led to higher levels of circulating cholesterol and bile acids, as well as reduced fecal excretion of bile acids. These effects may result from a modulation of critical transporters of the enterohepatic circulation, such as ileal ASBT levels. These findings expand our understanding of the effects of PFAS exposure on cholesterol and bile acid metabolism and may have important implications for human health and cardiometabolic disease.
Supplementary Material
Acknowledgments
This research was supported in part by the National Institute of Environmental Health Sciences [P30ES020957, P42ES030991, R01ES034407-01A1 (all to M.P.)] and the Office of the Vice President for Research at Wayne State University (to M.P.). The salary of Dr. Roth was supported by the National Institute of Environmental Health Sciences [ES034638-01A1 (to K.R.)]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conclusions and opinions are those of the individual authors and do not necessarily reflect the policies or views of EHP Publishing or the National Institute of Environmental Health Sciences.
References
- 1.Clara M, Scheffknecht C, Scharf S, Weiss S, Gans O. 2008. Emissions of perfluorinated alkylated substances (PFAS) from point sources—identification of relevant branches. Water Sci Technol 58(1):59–66, PMID: 18653937, 10.2166/wst.2008.641. [DOI] [PubMed] [Google Scholar]
- 2.Nickerson A, Maizel AC, Kulkarni PR, Adamson DT, Kornuc JJ, Higgins CP. 2020. Enhanced extraction of AFFF-associated PFASs from source zone soils. Environ Sci Technol 54(8):4952–4962, PMID: 32200626, 10.1021/acs.est.0c00792. [DOI] [PubMed] [Google Scholar]
- 3.Trier X, Granby K, Christensen JH. 2011. Polyfluorinated surfactants (PFS) in paper and board coatings for food packaging. Environ Sci Pollut Res Int 18(7):1108–1120, PMID: 21327544, 10.1007/s11356-010-0439-3. [DOI] [PubMed] [Google Scholar]
- 4.Drage DS, Sharkey M, Berresheim H, Coggins M, Harrad S. 2023. Rapid determination of selected PFAS in textiles entering the waste stream. Toxics 11(1):55, PMID: 36668781, 10.3390/toxics11010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, de Voogt P, et al. . 2011. Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr Environ Assess Manag 7(4):513–541, PMID: 21793199, 10.1002/ieam.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadia M, Kunz M, Ter Laak T, De Jonge M, Schriks M, van Wezel AP. 2023. Forever legacies? Profiling historical PFAS contamination and current influence on groundwater used for drinking water. Sci Total Environ 890:164420, PMID: 37236451, 10.1016/j.scitotenv.2023.164420. [DOI] [PubMed] [Google Scholar]
- 7.Domingo JL, Nadal M. 2017. Per- and polyfluoroalkyl substances (PFASs) in food and human dietary intake: a review of the recent scientific literature. J Agric Food Chem 65(3):533–543, PMID: 28052194, 10.1021/acs.jafc.6b04683. [DOI] [PubMed] [Google Scholar]
- 8.Liddie JM, Schaider LA, Sunderland EM. 2023. Sociodemographic factors are associated with the abundance of PFAS sources and detection in U.S. community water systems. Environ Sci Technol 57(21):7902–7912, PMID: 37184106, 10.1021/acs.est.2c07255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL. 2007. Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ Health Perspect 115(11):1596–1602, PMID: 18007991, 10.1289/ehp.10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conder JM, Hoke RA, De Wolf W, Russell MH, Buck RC. 2008. Are PFCAs bioaccumulative? A critical review and comparison with regulatory criteria and persistent lipophilic compounds. Environ Sci Technol 42(4):995–1003, PMID: 18351063, 10.1021/es070895g. [DOI] [PubMed] [Google Scholar]
- 11.Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, et al. . 2007. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect 115(9):1298–1305, PMID: 17805419, 10.1289/ehp.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindstrom AB, Strynar MJ, Libelo EL. 2011. Polyfluorinated compounds: past, present, and future. Environ Sci Technol 45(19):7954–7961, PMID: 21866930, 10.1021/es2011622. [DOI] [PubMed] [Google Scholar]
- 13.Brase RA, Mullin EJ, Spink DC. 2021. Legacy and emerging per- and polyfluoroalkyl substances: analytical techniques, environmental fate, and health effects. Int J Mol Sci 22(3):995, PMID: 33498193, 10.3390/ijms22030995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, Cousins IT, Scheringer M, Hungerbuehler K. 2015. Hazard assessment of fluorinated alternatives to long-chain perfluoroalkyl acids (PFAAs) and their precursors: status quo, ongoing challenges and possible solutions. Environ Int 75:172–179, PMID: 25461427, 10.1016/j.envint.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Guo H, Sheng N, Guo Y, Wu C, Xie W, Dai J. 2021. Exposure to GenX and its novel analogs disrupts fatty acid metabolism in male mice. Environ Pollut 291:118202, PMID: 34562693, 10.1016/j.envpol.2021.118202. [DOI] [PubMed] [Google Scholar]
- 16.Wu S, Xie J, Zhao H, Sanchez O, Zhao X, Freeman JL, et al. . 2023. Pre-differentiation GenX exposure induced neurotoxicity in human dopaminergic-like neurons. Chemosphere 332:138900, PMID: 37172627, 10.1016/j.chemosphere.2023.138900. [DOI] [PubMed] [Google Scholar]
- 17.Gannon SA, Fasano WJ, Mawn MP, Nabb DL, Buck RC, Buxton LW, et al. . 2016. Absorption, distribution, metabolism, excretion, and kinetics of 2,3,3,3-tetrafluoro-2-(heptafluoropropoxy)propanoic acid ammonium salt following a single dose in rat, mouse, and cynomolgus monkey. Toxicology 340:1–9, PMID: 26743852, 10.1016/j.tox.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Fenton SE, Ducatman A, Boobis A, DeWitt JC, Lau C, Ng C, et al. . 2021. Per- and polyfluoroalkyl substance toxicity and human health review: current state of knowledge and strategies for informing future research. Environ Toxicol Chem 40(3):606–630, PMID: 33017053, 10.1002/etc.4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steenland K, Winquist A. 2021. PFAS and cancer, a scoping review of the epidemiologic evidence. Environ Res 194:110690, PMID: 33385391, 10.1016/j.envres.2020.110690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barry V, Winquist A, Steenland K. 2013. Perfluorooctanoic acid (PFOA) exposures and incident cancers among adults living near a chemical plant. Environ Health Perspect 121(11–12):1313–1318, PMID: 24007715, 10.1289/ehp.1306615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vieira VM, Hoffman K, Shin H-M, Weinberg JM, Webster TF, Fletcher T. 2013. Perfluorooctanoic acid exposure and cancer outcomes in a contaminated community: a geographic analysis. Environ Health Perspect 121(3):318–323, PMID: 23308854, 10.1289/ehp.1205829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waterfield G, Rogers M, Grandjean P, Auffhammer M, Sunding D. 2020. Reducing exposure to high levels of perfluorinated compounds in drinking water improves reproductive outcomes: evidence from an intervention in Minnesota. Environ Health 19(1):42, PMID: 32321520, 10.1186/s12940-020-00591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song X, Tang S, Zhu H, Chen Z, Zang Z, Zhang Y, et al. . 2018. Biomonitoring PFAAs in blood and semen samples: investigation of a potential link between PFAAs exposure and semen mobility in China. Environ Int 113:50–54, PMID: 29421407, 10.1016/j.envint.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 24.Lam J, Koustas E, Sutton P, Johnson PI, Atchley DS, Sen S, et al. . 2014. The Navigation Guide—evidence-based medicine meets environmental health: integration of animal and human evidence for PFOA effects on fetal growth. Environ Health Perspect 122(10):1040–1051, PMID: 24968389, 10.1289/ehp.1307923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gleason JA, Post GB, Fagliano JA. 2015. Associations of perfluorinated chemical serum concentrations and biomarkers of liver function and uric acid in the US population (NHANES), 2007–2010. Environ Res 136:8–14, PMID: 25460614, 10.1016/j.envres.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Jin R, McConnell R, Catherine C, Xu S, Walker DI, Stratakis N, et al. . 2020. Perfluoroalkyl substances and severity of nonalcoholic fatty liver in children: an untargeted metabolomics approach. Environ Int 134:105220, PMID: 31744629, 10.1016/j.envint.2019.105220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costello E, Rock S, Stratakis N, Eckel SP, Walker DI, Valvi D, et al. . 2022. Exposure to per- and polyfluoroalkyl substances and markers of liver injury: a systematic review and meta-analysis. Environ Health Perspect 130(4):046001, PMID: 35475652, 10.1289/EHP10092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batzella E, Girardi P, Russo F, Pitter G, Da Re F, Fletcher T, et al. . 2022. Perfluoroalkyl substance mixtures and cardio-metabolic outcomes in highly exposed male workers in the Veneto Region: a mixture-based approach. Environ Res 212(pt A):113225, PMID: 35390304, 10.1016/j.envres.2022.113225. [DOI] [PubMed] [Google Scholar]
- 29.Wen Z-J, Wei Y-J, Zhang Y-F, Zhang Y-F. 2023. A review of cardiovascular effects and underlying mechanisms of legacy and emerging per- and polyfluoroalkyl substances (PFAS). Arch Toxicol 97(5):1195–1245, PMID: 36947184, 10.1007/s00204-023-03477-5. [DOI] [PubMed] [Google Scholar]
- 30.Dunder L, Salihovic S, Varotsis G, Lind PM, Elmståhl S, Lind L. 2023. Plasma levels of per- and polyfluoroalkyl substances (PFAS) and cardiovascular disease—results from two independent population-based cohorts and a meta-analysis. Environ Int 181:108250, PMID: 37832261, 10.1016/j.envint.2023.108250. [DOI] [PubMed] [Google Scholar]
- 31.ATSDR (Agency for Toxic Substances and Disease Registry). 2021. Toxicological Profile for Perfluoroalkyls. Atlanta, GA: ATSDR, US Department of Health and Human Services. https://www.atsdr.cdc.gov/ToxProfiles/tp200.pdf [accessed 8 January 2023]. [Google Scholar]
- 32.Roth GA, Huffman MD, Moran AE, Feigin V, Mensah GA, Naghavi M, et al. . 2015. Global and regional patterns in cardiovascular mortality from 1990 to 2013. Circulation 132(17):1667–1678, PMID: 26503749, 10.1161/CIRCULATIONAHA.114.008720. [DOI] [PubMed] [Google Scholar]
- 33.Mensah GA, Roth GA, Fuster V. 2019. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and beyond. J Am Coll Cardiol 74(20):2529–2532, PMID: 31727292, 10.1016/j.jacc.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 34.Bhatnagar A. 2017. Environmental determinants of cardiovascular disease. Circ Res 121(2):162–180, PMID: 28684622, 10.1161/CIRCRESAHA.117.306458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Münzel T, Sørensen M, Gori T, Schmidt FP, Rao X, Brook FR, et al. . 2017. Environmental stressors and cardio-metabolic disease: part II–mechanistic insights. Eur Heart J 38(8):557–564, PMID: 27460891, 10.1093/eurheartj/ehw294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kronmal RA, Cain KC, Ye Z, Omenn GS. 1993. Total serum cholesterol levels and mortality risk as a function of age. A report based on the Framingham data. Arch Intern Med 153(9):1065–1073, PMID: 8481074, 10.1001/archinte.1993.00410090025004. [DOI] [PubMed] [Google Scholar]
- 37.Bui AL, Horwich TB, Fonarow GC. 2011. Epidemiology and risk profile of heart failure. Nat Rev Cardiol 8(1):30–41, PMID: 21060326, 10.1038/nrcardio.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumric M, Urlic H, Bozic J, Vilovic M, Ticinovic Kurir T, Glavas D, et al. . 2023. Emerging therapies for the treatment of atherosclerotic cardiovascular disease: from bench to bedside. Int J Mol Sci 24(9):8062, PMID: 37175766, 10.3390/ijms24098062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mora AM, Fleisch AF, Rifas-Shiman SL, Woo Baidal JA, Pardo L, Webster TF, et al. . 2018. Early life exposure to per- and polyfluoroalkyl substances and mid-childhood lipid and alanine aminotransferase levels. Environ Int 111:1–13, PMID: 29156323, 10.1016/j.envint.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson JW, Hatch EE, Webster TF. 2010. Exposure to polyfluoroalkyl chemicals and cholesterol, body weight, and insulin resistance in the general U.S. population. Environ Health Perspect 118(2):197–202, PMID: 20123614, 10.1289/ehp.0901165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakr CJ, Leonard RC, Kreckmann KH, Slade MD, Cullen MR. 2007. Longitudinal study of serum lipids and liver enzymes in workers with occupational exposure to ammonium perfluorooctanoate. J Occup Environ Med 49(8):872–879, PMID: 17693785, 10.1097/JOM.0b013e318124a93f. [DOI] [PubMed] [Google Scholar]
- 42.Steenland K, Tinker S, Frisbee S, Ducatman A, Vaccarino V. 2009. Association of perfluorooctanoic acid and perfluorooctane sulfonate with serum lipids among adults living near a chemical plant. Am J Epidemiol 170(10):1268–1278, PMID: 19846564, 10.1093/aje/kwp279. [DOI] [PubMed] [Google Scholar]
- 43.Liu B, Zhu L, Wang M, Sun Q. 2023. Associations between per- and polyfluoroalkyl substances exposures and blood lipid levels among adults—a meta-analysis. Environ Health Perspect 131(5):056001, PMID: 37141244, 10.1289/EHP11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Insull W Jr. 2006. Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: a scientific review. South Med J 99(3):257–273, PMID: 16553100. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez FJ. 2012. Nuclear receptor control of enterohepatic circulation. Compr Physiol 2(4):2811–2828, PMID: 23720266, 10.1002/cphy.c120007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salihović S, Dickens AM, Schoultz I, Fart F, Sinisalu L, Lindeman T, et al. . 2020. Simultaneous determination of perfluoroalkyl substances and bile acids in human serum using ultra-high-performance liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 412(10):2251–2259, PMID: 31760452, 10.1007/s00216-019-02263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roth K, Yang Z, Agarwal M, Liu W, Peng Z, Long Z, et al. . 2021. Exposure to a mixture of legacy, alternative, and replacement per- and polyfluoroalkyl substances (PFAS) results in sex-dependent modulation of cholesterol metabolism and liver injury. Environ Int 157:106843, PMID: 34479135, 10.1016/j.envint.2021.106843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sinisalu L, Yeung LWY, Wang J, Pan Y, Dai J, Hyötyläinen T. 2021. Prenatal exposure to poly-/per-fluoroalkyl substances is associated with alteration of lipid profiles in cord–blood. Metabolomics 17(12):103, PMID: 34816353, 10.1007/s11306-021-01853-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sen P, Qadri S, Luukkonen PK, Ragnarsdottir O, McGlinchey A, Jäntti S, et al. . 2022. Exposure to environmental contaminants is associated with altered hepatic lipid metabolism in non-alcoholic fatty liver disease. J Hepatol 76(2):283–293, PMID: 34627976, 10.1016/j.jhep.2021.09.039. [DOI] [PubMed] [Google Scholar]
- 50.Charach G, Grosskopf I, Rabinovich A, Shochat M, Weintraub M, Rabinovich P. 2011. The association of bile acid excretion and atherosclerotic coronary artery disease. Therap Adv Gastroenterol 4(2):95–101, PMID: 21694811, 10.1177/1756283X10388682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagasaka H, Yorifuji T, Egawa H, Yanai H, Fujisawa T, Kosugiyama K, et al. . 2005. Evaluation of risk for atherosclerosis in Alagille syndrome and progressive familial intrahepatic cholestasis: two congenital cholestatic diseases with different lipoprotein metabolisms. J Pediatr 146(3):329–335, PMID: 15756213, 10.1016/j.jpeds.2004.10.047. [DOI] [PubMed] [Google Scholar]
- 52.Dawson PA, Lan T, Rao A. 2009. Bile acid transporters. J Lipid Res 50(12):2340–2357, PMID: 19498215, 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shneider BL. 2001. Intestinal bile acid transport: biology, physiology, and pathophysiology. J Pediatr Gastroenterol Nutr 32(4):407–417, PMID: 11396803, 10.1002/j.1536-4801.2001.tb07287.x. [DOI] [PubMed] [Google Scholar]
- 54.Dawson PA. 2011. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol (201):169–203, PMID: 21103970, 10.1007/978-3-642-14541-4_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dietschy JM. 1968. Mechanisms for the intestinal absorption of bile acids. J Lipid Res 9(3):297–309, PMID: 5646181, 10.1016/S0022-2275(20)43096-2. [DOI] [PubMed] [Google Scholar]
- 56.Zhao W, Zitzow JD, Ehresman DJ, Chang S-C, Butenhoff JL, Forster J, et al. . 2015. Na+/taurocholate cotransporting polypeptide and apical sodium-dependent bile acid transporter are involved in the disposition of perfluoroalkyl sulfonates in humans and rats. Toxicol Sci 146(2):363–373, PMID: 26001962, 10.1093/toxsci/kfv102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao H, Zhou Z, Hu Z, Wei C, Li J, Wang L, et al. . 2022. Effect of enterohepatic circulation on the accumulation of per- and polyfluoroalkyl substances: evidence from experimental and computational studies. Environ Sci Technol 56(5):3214–3224, PMID: 35138827, 10.1021/acs.est.1c07176. [DOI] [PubMed] [Google Scholar]
- 58.Teupser D, Persky AD, Breslow JL. 2003. Induction of atherosclerosis by low-fat, semisynthetic diets in LDL receptor-deficient C57BL/6J and FVB/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement). Arterioscler Thromb Vasc Biol 23(10):1907–1913, PMID: 12907460, 10.1161/01.ATV.0000090126.34881.B1. [DOI] [PubMed] [Google Scholar]
- 59.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675, PMID: 22930834, 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anders S, Pyl PT, Huber W. 2015. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics 31(2):166–169, PMID: 25260700, 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. . 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21, PMID: 23104886, 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140, PMID: 19910308, 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57, PMID: 19131956, 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 64.Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, et al. . 2011. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci USA 108(suppl 1):4523–4530, PMID: 20837534, 10.1073/pnas.1006734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clasquin MF, Melamud E, Rabinowitz JD. 2012. LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr Protoc Bioinformatics 37(1):14.11.1–14.11.23, PMID: 22389014, 10.1002/0471250953.bi1411s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakr CJ, Kreckmann KH, Green JW, Gillies PJ, Reynolds JL, Leonard RC. 2007. Cross-sectional study of lipids and liver enzymes related to a serum biomarker of exposure (ammonium perfluorooctanoate or APFO) as part of a general health survey in a cohort of occupationally exposed workers. J Occup Environ Med 49(10):1086–1096, PMID: 18000414, 10.1097/JOM.0b013e318156eca3. [DOI] [PubMed] [Google Scholar]
- 67.Olsen GW, Zobel LR. 2007. Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (PFOA) concentrations in fluorochemical production workers. Int Arch Occup Environ Health 81(2):231–246, PMID: 17605032, 10.1007/s00420-007-0213-0. [DOI] [PubMed] [Google Scholar]
- 68.Girardi P, Merler E. 2019. A mortality study on male subjects exposed to polyfluoroalkyl acids with high internal dose of perfluorooctanoic acid. Environ Res 179(pt A):108743, PMID: 31542491, 10.1016/j.envres.2019.108743. [DOI] [PubMed] [Google Scholar]
- 69.Lu Y, Gao K, Li X, Tang Z, Xiang L, Zhao H, et al. . 2019. Mass spectrometry-based metabolomics reveals occupational exposure to per- and polyfluoroalkyl substances relates to oxidative stress, fatty acid β-oxidation disorder, and kidney injury in a manufactory in China. Environ Sci Technol 53(16):9800–9809, PMID: 31246438, 10.1021/acs.est.9b01608. [DOI] [PubMed] [Google Scholar]
- 70.Wang J, Zhang Y, Zhang W, Jin Y, Dai J. 2012. Association of perfluorooctanoic acid with HDL cholesterol and circulating miR-26b and miR-199-3p in workers of a fluorochemical plant and nearby residents. Environ Sci Technol 46(17):9274–9281, PMID: 22862179, 10.1021/es300906q. [DOI] [PubMed] [Google Scholar]
- 71.Nilsson H, Kärrman A, Westberg H, Rotander A, van Bavel B, Lindström G. 2010. A time trend study of significantly elevated perfluorocarboxylate levels in humans after using fluorinated ski wax. Environ Sci Technol 44(6):2150–2155, PMID: 20158198, 10.1021/es9034733. [DOI] [PubMed] [Google Scholar]
- 72.Petriello MC, Mottaleb MA, Serio TC, Balyan B, Cave MC, Pavuk M, et al. . 2022. Serum concentrations of legacy and emerging per- and polyfluoroalkyl substances in the Anniston Community Health Surveys (ACHS I and ACHS II). Environ Int 158:106907, PMID: 34763231, 10.1016/j.envint.2021.106907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han R, Hu M, Zhong Q, Wan C, Liu L, Li F, et al. . 2018. Perfluorooctane sulphonate induces oxidative hepatic damage via mitochondria-dependent and NF-κB/TNF-α-mediated pathway. Chemosphere 191:1056–1064, PMID: 28939271, 10.1016/j.chemosphere.2017.08.070. [DOI] [PubMed] [Google Scholar]
- 74.Nakagawa T, Ramdhan DH, Tanaka N, Naito H, Tamada H, Ito Y, et al. . 2012. Modulation of ammonium perfluorooctanoate-induced hepatic damage by genetically different PPARα in mice. Arch Toxicol 86(1):63–74, PMID: 21499893, 10.1007/s00204-011-0704-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qazi MR, Abedi MR, Nelson BD, DePierre JW, Abedi-Valugerdi M. 2010. Dietary exposure to perfluorooctanoate or perfluorooctane sulfonate induces hypertrophy in centrilobular hepatocytes and alters the hepatic immune status in mice. Int Immunopharmacol 10(11):1420–1427, PMID: 20816993, 10.1016/j.intimp.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 76.Takacs ML, Abbott BD. 2007. Activation of mouse and human peroxisome proliferator-activated receptors (α, β/δ, γ) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci 95(1):108–117, PMID: 17047030, 10.1093/toxsci/kfl135. [DOI] [PubMed] [Google Scholar]
- 77.Rosen MB, Lee JS, Ren H, Vallanat B, Liu J, Waalkes MP, et al. . 2008. Toxicogenomic dissection of the perfluorooctanoic acid transcript profile in mouse liver: evidence for the involvement of nuclear receptors PPARα and CAR. Toxicol Sci 103(1):46–56, PMID: 18281256, 10.1093/toxsci/kfn025. [DOI] [PubMed] [Google Scholar]
- 78.Gervois P, Torra IP, Fruchart JC, Staels B. 2000. Regulation of lipid and lipoprotein metabolism by PPAR activators. Clin Chem Lab Med 38(1):3–11, PMID: 10774955, 10.1515/CCLM.2000.002. [DOI] [PubMed] [Google Scholar]
- 79.Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. 1998. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 98:2088–2093, PMID: 9808609, 10.1161/01.cir.98.19.2088. [DOI] [PubMed] [Google Scholar]
- 80.Duan Y, Gong K, Xu S, Zhang F, Meng X, Han J. 2022. Regulation of cholesterol homeostasis in health and diseases: from mechanisms to targeted therapeutics. Signal Transduct Target Ther 7(1):265, PMID: 35918332, 10.1038/s41392-022-01125-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Itabe H. 1998. Oxidized phospholipids as a new landmark in atherosclerosis. Prog Lipid Res 37:181–207, PMID: 9829125, 10.1016/s0163-7827(98)00009-5. [DOI] [PubMed] [Google Scholar]
- 82.Tosheska Trajkovska K, Topuzovska S. 2017. High-density lipoprotein metabolism and reverse cholesterol transport: strategies for raising HDL cholesterol. Anatol J Cardiol 18:149–154, PMID: 28766509, 10.14744/AnatolJCardiol.2017.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abe RJ, Abe J-I, Nguyen MTH, Olmsted-Davis EA, Mamun A, Banerjee P, et al. . 2022. Free cholesterol bioavailability and atherosclerosis. Curr Atheroscler Rep 24(5):323–336, PMID: 35332444, 10.1007/s11883-022-01011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schlezinger JJ, Puckett H, Oliver J, Nielsen G, Heiger-Bernays W, Webster TF. 2020. Perfluorooctanoic acid activates multiple nuclear receptor pathways and skews expression of genes regulating cholesterol homeostasis in liver of humanized PPARα mice fed an American diet. Toxicol Appl Pharmacol 405:115204, PMID: 32822737, 10.1016/j.taap.2020.115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Starling AP, Engel SM, Whitworth KW, Richardson DB, Stuebe AM, Daniels JL, et al. . 2014. Perfluoroalkyl substances and lipid concentrations in plasma during pregnancy among women in the Norwegian Mother and Child Cohort Study. Environ Int 62:104–112, PMID: 24189199, 10.1016/j.envint.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang D, Tan Z, Yang J, Li L, Li H, Zhang H, et al. . 2023. Perfluorooctane sulfonate promotes atherosclerosis by modulating M1 polarization of macrophages through the NF-κB pathway. Ecotoxicol Environ Saf 249:114384, PMID: 36512850, 10.1016/j.ecoenv.2022.114384. [DOI] [PubMed] [Google Scholar]
- 87.Burg JS, Espenshade PJ. 2011. Regulation of HMG-CoA reductase in mammals and yeast. Prog Lipid Res 50(4):403–410, PMID: 21801748, 10.1016/j.plipres.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sato R. 2020. Recent advances in regulating cholesterol and bile acid metabolism. Biosci Biotechnol Biochem 84(11):2185–2192, PMID: 32657638, 10.1080/09168451.2020.1793658. [DOI] [PubMed] [Google Scholar]
- 89.Luo J, Yang H, Song B-L. 2020. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol 21(4):225–245, PMID: 31848472, 10.1038/s41580-019-0190-7. [DOI] [PubMed] [Google Scholar]
- 90.Andersen ME, Hagenbuch B, Apte U, Corton JC, Fletcher T, Lau C, et al. . 2021. Why is elevation of serum cholesterol associated with exposure to perfluoroalkyl substances (PFAS) in humans? A workshop report on potential mechanisms. Toxicology 459:152845, PMID: 34246716, 10.1016/j.tox.2021.152845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lorbek G, Lewinska M, Rozman D. 2012. Cytochrome P450s in the synthesis of cholesterol and bile acids—from mouse models to human diseases. FEBS J 279(9):1516–1533, PMID: 22111624, 10.1111/j.1742-4658.2011.08432.x. [DOI] [PubMed] [Google Scholar]
- 92.Chiang JYL. 2009. Bile acids: regulation of synthesis. J Lipid Res 50(10):1955–1966, PMID: 19346330, 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Russell DW. 2003. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72:137–174, PMID: 12543708, 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 94.Behr A-C, Kwiatkowski A, Ståhlman M, Schmidt FF, Luckert C, Braeuning A, et al. . 2020. Impairment of bile acid metabolism by perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) in human HepaRG hepatoma cells. Arch Toxicol 94(5):1673–1686, PMID: 32253466, 10.1007/s00204-020-02732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beggs KM, McGreal SR, McCarthy A, Gunewardena S, Lampe JN, Lau C, et al. . 2016. The role of hepatocyte nuclear factor 4-alpha in perfluorooctanoic acid- and perfluorooctanesulfonic acid-induced hepatocellular dysfunction. Toxicol Appl Pharmacol 304:18–29, PMID: 27153767, 10.1016/j.taap.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kruh GD, Belinsky MG. 2003. The MRP family of drug efflux pumps. Oncogene 22(47):7537–7552, PMID: 14576857, 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- 97.Watashi K, Urban S, Li W, Wakita T. 2014. NTCP and beyond: opening the door to unveil hepatitis B virus entry. Int J Mol Sci 15(2):2892–2905, PMID: 24557582, 10.3390/ijms15022892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hagenbuch B, Meier PJ. 1994. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest 93(3):1326–1331, PMID: 8132774, 10.1172/JCI117091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Spagnoli LG, Bonanno E, Sangiorgi G, Mauriello A. 2007. Role of inflammation in atherosclerosis. J Nucl Med 48(11):1800–1815, PMID: 17942804, 10.2967/jnumed.107.038661. [DOI] [PubMed] [Google Scholar]
- 100.Jahangiri A, de Beer MC, Noffsinger V, Tannock LR, Ramaiah C, Webb NR, et al. . 2009. HDL remodeling during the acute phase response. Arterioscler Thromb Vasc Biol 29(2):261–267, PMID: 19008529, 10.1161/ATVBAHA.108.178681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eriksen N, Benditt EP. 1980. Isolation and characterization of the amyloid-related apoprotein (SAA) from human high density lipoprotein. Proc Natl Acad Sci USA 77(11):6860–6864, PMID: 6161374, 10.1073/pnas.77.11.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shridas P, Tannock LR. 2019. Role of serum amyloid A in atherosclerosis. Curr Opin Lipidol 30(4):320–325, PMID: 31135596, 10.1097/MOL.0000000000000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ji A, Trumbauer AC, Noffsinger VP, de Beer FC, Webb NR, Tannock LR, et al. . 2023. Serum amyloid A augments the atherogenic effects of cholesteryl ester transfer protein. J Lipid Res 64(5):100365, PMID: 37004910, 10.1016/j.jlr.2023.100365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xiao L, Pan G. 2017. An important intestinal transporter that regulates the enterohepatic circulation of bile acids and cholesterol homeostasis: the apical sodium-dependent bile acid transporter (SLC10A2/ASBT). Clin Res Hepatol Gastroenterol 41(5):509–515, PMID: 28336180, 10.1016/j.clinre.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 105.Balakrishnan A, Polli JE. 2006. Apical sodium dependent bile acid transporter (ASBT, SLC10A2): a potential prodrug target. Mol Pharm 3(3):223–230, PMID: 16749855, 10.1021/mp060022d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wong BS, Camilleri M, McKinzie S, Burton D, Graffner H, Zinsmeister AR. 2011. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am J Gastroenterol 106(12):2154–2164, PMID: 21876564, 10.1038/ajg.2011.285. [DOI] [PubMed] [Google Scholar]
- 107.Tiessen RG, Kennedy CA, Keller BT, Levin N, Acevedo L, Gedulin B, et al. . 2018. Safety, tolerability and pharmacodynamics of apical sodium-dependent bile acid transporter inhibition with volixibat in healthy adults and patients with type 2 diabetes mellitus: a randomised placebo-controlled trial. BMC Gastroenterol 18(1):3, PMID: 29304731, 10.1186/s12876-017-0736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iwaki M, Kessoku T, Tanaka K, Ozaki A, Kasai Y, Kobayashi T, et al. . 2023. Combined, elobixibat, and colestyramine reduced cholesterol toxicity in a mouse model of metabolic dysfunction-associated steatotic liver disease. Hepatol Commun 7(11):e0285, PMID: 37902528, 10.1097/HC9.0000000000000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang S, Zhou J, Wu W, Zhu Y, Liu X. 2023. The role of bile acids in cardiovascular diseases: from mechanisms to clinical implications. Aging Dis 14(2):261–282, PMID: 37008052, 10.14336/AD.2022.0817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fiorucci S, Distrutti E, Carino A, Zampella A, Biagioli M. 2021. Bile acids and their receptors in metabolic disorders. Prog Lipid Res 82:101094, PMID: 33636214, 10.1016/j.plipres.2021.101094. [DOI] [PubMed] [Google Scholar]
- 111.Eloranta JJ, Kullak-Ublick GA. 2008. The role of FXR in disorders of bile acid homeostasis. Physiology (Bethesda) 23:286–295, PMID: 18927204, 10.1152/physiol.00020.2008. [DOI] [PubMed] [Google Scholar]
- 112.Matsubara T, Li F, Gonzalez FJ. 2013. FXR signaling in the enterohepatic system. Mol Cell Endocrinol 368(1–2):17–29, PMID: 22609541, 10.1016/j.mce.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen F, Ma L, Dawson PA, Sinal CJ, Sehayek E, Gonzalez FJ, et al. . 2003. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J Biol Chem 278(22):19909–19916, PMID: 12456679, 10.1074/jbc.M207903200. [DOI] [PubMed] [Google Scholar]
- 114.Jung D, Fried M, Kullak-Ublick GA. 2002. Human apical sodium-dependent bile salt transporter gene (SLC10A2) is regulated by the peroxisome proliferator-activated receptor α. J Biol Chem 277(34):30559–30566, PMID: 12055195, 10.1074/jbc.M203511200. [DOI] [PubMed] [Google Scholar]
- 115.Yin W, Carballo-Jane E, McLaren DG, Mendoza VH, Gagen K, Geoghagen NS, et al. . 2012. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J Lipid Res 53(1):51–65, PMID: 22021650, 10.1194/jlr.M019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tomkin GH, Owens D. 2012. The chylomicron: relationship to atherosclerosis. Int J Vasc Med 2012:784536, PMID: 22007304, 10.1155/2012/784536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Joven J, Rull A, Ferré N, Escolà-Gil JC, Marsillach J, Coll B, et al. . 2007. The results in rodent models of atherosclerosis are not interchangeable: the influence of diet and strain. Atherosclerosis 195(2):e85–e92, PMID: 17651742, 10.1016/j.atherosclerosis.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 118.Hartmann C, Jamnik T, Weiss S, Göß M, Fareed Y, Satrapa V, et al. . 2023. Results of the Austrian Children’s Biomonitoring Survey 2020 part A: per- and polyfluorinated alkylated substances, bisphenols, parabens and other xenobiotics. Int J Hyg Environ Health 249:114123, PMID: 36738493, 10.1016/j.ijheh.2023.114123. [DOI] [PubMed] [Google Scholar]
- 119.Palmisano BT, Zhu L, Stafford JM. 2017. Role of estrogens in the regulation of liver lipid metabolism. Adv Exp Med Biol 1043:227–256, PMID: 29224098, 10.1007/978-3-319-70178-3_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schaefer EJ, Foster DM, Zech LA, Lindgren FT, Brewer HB Jr, Levy RI. 1983. The effects of estrogen administration on plasma lipoprotein metabolism in premenopausal females. J Clin Endocrinol Metab 57(2):262–267, PMID: 6408108, 10.1210/jcem-57-2-262. [DOI] [PubMed] [Google Scholar]
- 121.Kissebah AH, Harrigan P, Wynn V. 1973. Mechanism of hypertriglyceridaemia associated with contraceptive steroids. Horm Metab Res 5(3):184–190, PMID: 4351691, 10.1055/s-0028-1093969. [DOI] [PubMed] [Google Scholar]
- 122.Magkos F, Mittendorfer B. 2009. Gender differences in lipid metabolism and the effect of obesity. Obstet Gynecol Clin North Am 36(2):245–265, PMID: 19501312, 10.1016/j.ogc.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 123.Lopez D, McLean MP. 2006. Estrogen regulation of the scavenger receptor class B gene: anti-atherogenic or steroidogenic, is there a priority? Mol Cell Endocrinol 247(1–2):22–33, PMID: 16297529, 10.1016/j.mce.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 124.Della Torre S, Mitro N, Fontana R, Gomaraschi M, Favari E, Recordati C, et al. . 2016. An essential role for liver ERα in coupling hepatic metabolism to the reproductive cycle. Cell Rep 15(2):360–371, PMID: 27050513, 10.1016/j.celrep.2016.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, et al. . 2012. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125(15):1905–1919, PMID: 22508840, 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fukata Y, Yu X, Imachi H, Nishiuchi T, Lyu J, Seo K, et al. . 2014. 17β-Estradiol regulates scavenger receptor class BI gene expression via protein kinase C in vascular endothelial cells. Endocrine 46(3):644–650, PMID: 24347243, 10.1007/s12020-013-0134-5. [DOI] [PubMed] [Google Scholar]
- 127.Angelin B, Backman L, Einarsson K, Eriksson L, Ewerth S. 1982. Hepatic cholesterol metabolism in obesity: activity of microsomal 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Lipid Res 23(5):770–773, PMID: 7119574, 10.1016/S0022-2275(20)38111-6. [DOI] [PubMed] [Google Scholar]
- 128.Ståhlberg D, Rudling M, Angelin B, Björkhem I, Forsell P, Nilsell K, et al. . 1997. Hepatic cholesterol metabolism in human obesity. Hepatology 25(6):1447–1450, PMID: 9185766, 10.1002/hep.510250623. [DOI] [PubMed] [Google Scholar]
- 129.Kulkarni SR, Xu J, Donepudi AC, Wei W, Slitt AL. 2013. Effect of caloric restriction and AMPK activation on hepatic nuclear receptor, biotransformation enzyme, and transporter expression in lean and obese mice. Pharm Res 30(9):2232–2247, PMID: 23949303, 10.1007/s11095-013-1140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baars A, Oosting A, Lohuis M, Koehorst M, El Aidy S, Hugenholtz F, et al. . 2018. Sex differences in lipid metabolism are affected by presence of the gut microbiota. Sci Rep 8(1):13426, PMID: 30194317, 10.1038/s41598-018-31695-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Steegenga WT, Mischke M, Lute C, Boekschoten MV, Pruis MG, Lendvai A, et al. . 2014. Sexually dimorphic characteristics of the small intestine and colon of prepubescent C57BL/6 mice. Biol Sex Differ 5:11, PMID: 25243059, 10.1186/s13293-014-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.