

Abstract
Interleukin-33 (IL-33), an epithelial cell-derived cytokine that responds rapidly to environmental insult, has a critical role in initiating airway inflammatory diseases. However, the molecular mechanism underlying IL-33 secretion following allergen exposure is not clear. Here, we found that two cell events were fundamental for IL-33 secretion after exposure to allergens. First, stress granule assembly activated by allergens licensed the nuclear-cytoplasmic transport of IL-33, but not the secretion of IL-33. Second, a neo-form murine amino-terminal p40 fragment gasdermin D (Gsdmd), whose generation was independent of inflammatory caspase-1 and caspase-11, dominated cytosolic secretion of IL-33 by forming pores in the cell membrane. Either the blockade of stress granule assembly or the abolishment of p40 production through amino acid mutation of residues 309–313 (ELRQQ) could efficiently prevent the release of IL-33 in murine epithelial cells. Our findings indicated that targeting stress granule disassembly and Gsdmd fragmentation could reduce IL-33-dependent allergic airway inflammation.
Allergic diseases such as asthma and hay fever (allergic rhinitis) are common diseases observed with increasing incidence, especially in developed countries. These diseases are usually exacerbated when patients are exposed to allergens in their environment, for example, asthma exacerbation induced by house dust mites (HDMs), molds, bacteria, plant pollen and animal dander. Certain enzymatically active components derived from allergens, including Der p and Der f from HDMs1,2, Asp f from Aspergillus oryzae3 or the subtilisin protease from Bacillus species4, induce the release of cytokines and promote allergic type 2 airway inflammation in vivo.
IL-33 is constitutively produced by cells in quiescent stromal tissue. Multiple nonimmune and immune cell types, including epithelial cells and macrophages, and natural killer T (NKT) cells and regulatory T (Treg) cells, can produce IL-33 (refs. 5,6). IL-33 belongs to the IL-1 cytokine family and functions as an alarmin that is released when barriers are disrupted. The most well-defined and important functions of IL-33 rely on its ST2-binding ability, which activates Myd88-mediated signaling pathways in ST2-expressing cells such as mast cells, group 2 innate lymphoid cells (ILC2s) and helper T type 2 (TH2) cells, contributing to systematic immune defenses and tissue repair7. Thus, the secretion of IL-33 is critical for regulating allergic inflammation.
Similar to other members of the IL-1 family, IL-33 lacks a secretion signal and cannot be released through the endoplasmic reticulum–Golgi secretory pathway or cross the cell membrane under physiological conditions8. IL-33 was reported to be released through cell death-associated membrane damage, including that induced by freeze–thaw cycles, cell lysis buffer, pore-forming toxin streptolysin O and oxidative stress9–12. These stimuli contribute to IL-33 release with the occurrence of cell death features such as lactate dehydrogenase (LDH) release or propidium iodide (PI) uptake. The secretion of IL-33 from living cells was also observed in cells exposed to mechanical stress, cockroaches, uric acid, HDMs and Alternaria allergens13,14. However, the precise mechanisms of the IL-33 secretion from living cells under allergen stimulation remain unknown, and the in-cell transportation of IL-33 has not been deciphered.
Stress granules (SGs) are highly dynamic cytosolic condensates that modulate cell survival in response to chronic and acute stressors, such as viral infection, heat shock and chemical treatments. Defects in the assembly and disassembly of SGs are linked to neurodegenerative diseases, aberrant antiviral responses and cancer15–17. SG assembly disrupts nucleocytoplasmic transport, suggesting that SGs might function in the transportation of nuclear damage-associated molecular patterns (DAMPs)18.
Here, we found that two events sequentially regulated the secretion of IL-33 after exposure to allergen proteases. The activation of phosphorylated eukaryotic initiation factor 2α (p-eIF2α)-independent SG assembly in epithelial cells controlled the transport of IL-33 from the nucleus into the cytoplasm. Subsequently, Gsdmd, a gasdermin family member that controls the release of inflammatory IL-1β produced through the caspase-1/8/11-dependent pathway19, was cleaved into a p40 N-terminal form (p40 NT-Gsdmd) through a caspase-independent mechanism. The generation of the p40 NT-Gsdmd fragment promoted the secretion of IL-33 from the cytosol into the extracellular space without the apparent occurrence of cell death. These observations provide two potential targets for interfering with IL-33-dependent inflammatory immune response following allergen protease exposure.
Results
Allergen stimulates IL-33 secretion and Gsdmd fragmentation.
Immunofluorescence staining in unstimulated murine lungs indicated the typical expression of IL-33 in alveolar type II (AT2) lung epithelial cells with surfactant protein C (SPC+) (Extended Data Fig. 1a). In humans, IL-33 was preferentially expressed in airway epithelium both in inflamed lungs from people with asthma and in noninflamed lung tissues (Extended Data Fig. 1b). Murine AT2 MLE-12 cells expressing IL-33 endogenously and human epithelial A549 cells expressing human influenza hemagglutinin (HA, the amino acid residues 98–106)-tagged human IL-33 with a carboxy-terminal fusion with green fluorescent protein (GFP) (A549-IL-33-GFP) were used to trace IL-33 secretion in vitro (Extended Data Fig. 2a,b). The smallest nuclear localization sequence (NLS) linked with GFP (A549-NLS-GFP) was also constructed for control analysis20. The IL-33-GFP signal in the nucleus was recorded for 30 min after exposure to 5 μg of papain with a live-cell imaging recording system (Fig. 1a and Supplementary Videos 1 and 2). IL-33-GFP was detected in A549-IL-33-GFP cells at 10 min and 30 min after papain stimulation, with a significant loss of GFP signal at 30 min. At the same time, NLS-GFP did not present a noticeable GFP fluorescence signal change in the nucleus (Fig. 1a,b), and this occurred concurrently with the appearance of two N-terminal (NT) fragments of Gsdmd near 40 kDa (Fig. 1c,d). This observation could be replicated in MLE-12 cells that ectopically expressed NT Flag-tagged mouse Gsdmd (MLE-12-Flag-Gsdmd) (Extended Data Fig. 2c,d). In A549-IL-33-GFP cells, papain exposure led to the generation of a p35 NT human gasdermin D (GSDMD) fragment (Extended Data Fig. 2e), also observed in A549 cells expressing NT Flag-tagged GSDMD (Extended Data Fig. 2e,f). Papain stimulated the release of IL-33 and the appearance of Gsdmd fragments in a time-dependent manner (Fig. 1e,f). Although papain induced the generation of two NT-Gsdmd fragments of around 40 kDa in molecular weight, the appearance of the bottom 40-kDa band did associate with IL-33 secretion and the upper 43-kDa band did not (Fig. 1c,e). The bottom 40-kDa fragment was thus defined as p40 NT-Gsdmd. Because Gsdmd cleavage usually leads to cell death, we investigated whether cell death happened after protease exposure. MLE-12 cells stimulated with papain for 30 min showed no significant PI uptake or annexin V staining (Fig. 1g) and no apparent death morphology at this stage (Supplementary Video 1) compared with tumor necrosis factor-α (TNF-α) and SM-164 (hereafter, TS) stimulated MLE-12 cells. Three hours after the removal of papain, papain-induced p40 NT-Gsdmd disappeared, and IL-33 secretion fell back to the baseline (Fig. 1h), indicating that the papain-induced generation of p40 NT-Gsdmd was reversible. Even with a higher papain stimulation dose (100 μg), LDH was nearly undetectable in the supernatant (Fig. 1i). The induction of phosphorylated mixed lineage kinase domain-like protein (p-Mlkl)-mediated necroptosis by the mixture of TNF-α, SM-164 and Z-VAD-FMK (hereafter, TSZ) or caspase-mediated apoptosis by TS did not contribute to IL-33 release in MLE-12 cells (Fig. 1i). These observations indicated that papain stimulation did not activate classical cell death pathways, such as apoptosis and necroptosis, but induced the activation of a p40 NT-Gsdmd fragment and IL-33 release.
Fig. 1 |. Allergen stimulates IL-33 secretion and Gsdmd fragmentation.
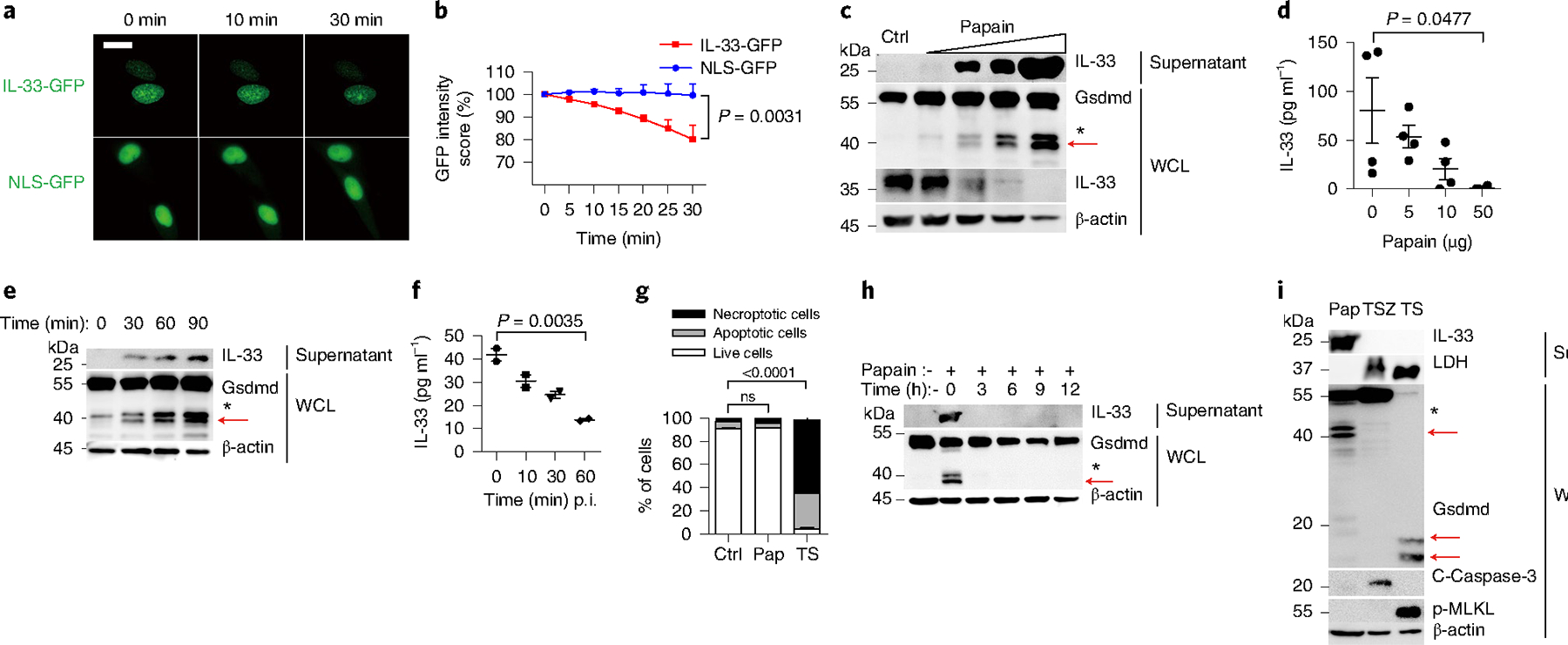
a, Live-cell imaging of IL-33-GFP and NLS-GFP in A549 cells exposed to 5 μg of papain for the indicated time. Scale bars, 15 μm. b, GFP intensity statistics (ImageJ) of IL-33-GFP expression and NLS-GFP expression as in a. c, Immunoblot analysis of IL-33 and Gsdmd in MLE-12 cells after papain (1 μg, 5 μg, 10 μg and 50 μg) exposure for 30 min. Ctrl, no stimulation. d, ELISA analysis of IL-33 in WCL of MLE-12 cells stimulated with papain for 30 min. e, Immunoblot analysis of IL-33 and Gsdmd in MLE-12 cells after 5 μg well−1 papain exposure for the indicated time. f, ELISA analysis of IL-33 in WCL from MLE-12 cells with 5 μg well−1 papain exposure for the indicated time. Representative of four independent experiments. p.i., post infection. g, Flow cytometry analysis of MLE-12 cell survival with indicated stimulations. Ctrl, no stimulation; Pap, exposure to 10 μg of papain for 30 min; TS, TNF-α+SM-164 stimulation for 12 h; ns, not significant. Live cell, PI−annexinV−; apoptotic cell, PI−annexinV+; necroptotic cell, PI+annexinV+. h, Immunoblot analysis of MLE-12 cells stimulated with papain (5 μg) for 30 min followed by removal of papain for the indicated time. i, Immunoblot analysis of MLE-12 cells stimulated with papain (Pap, 100 μg well−1) for 30 min, TSZ (TNF-α+SM-164+Z-VAD-FMK) for 12 h and TS (TNF-α+SM-164) for 12 h. Red arrows indicate the functional p40 NT-Gsdmd. Nonspecific fragments marked with an asterisk were not discussed in our work. Error bar, mean ± s.e.m. b, n = 2, using a two-tailed, unpaired Student’s t-test; d, n = 4, f, n = 2, g, n = 3, with Dunnett’s multiple comparisons test. All data are representative of at least three independent experiments.
Papain activates caspase-1/11-independent Gsdmd fragmentation.
Gsdmd can be cleaved by caspase-1 and caspase-11 through both the canonical and noncanonical activation of the inflammasome, which induces IL-1β release in multiple cell types, including macrophages21. Because macrophages can be an efficient source of IL-33 upon inflammatory stimulation5,22, we compared Gsdmd cleavage during caspase-1/11 and papain stimulation in murine bone marrow-derived macrophages (BMMs). After priming with bacterial lipopolysaccharides (LPS), which leads to the production of pro-IL-1β, we added papain, ATP or nigericin (Nig) as a second stimulatory signal. Stimulation with ATP or Nig generated the cleaved p20 caspase-1 and a 35-kDa fragment of NT-Gsdmd (Fig. 2a). The p35 NT-Gsdmd displays a high binding affinity to cell membrane-associated lipids with pore-forming capability23, promoted the release of IL-1β and LDH (Fig. 2a,b), and the release of LDH, suggesting that cell membrane rupture and cell death happened in this stage. In contrast, papain stimulation did not promote the activation of p20 caspase-1 (Fig. 2a) and did not permit the secretion of IL-1β into the supernatant (Fig. 2b), but induced the generation of p40 NT-Gsdmd and release of IL-33 in BMMs (Fig. 2a,b). This process did not result in apparent LDH release (Fig. 2a). Gsdmd-deficient BMMs showed impaired caspase-1-dependent Gsdmd cleavage and IL-1β secretion upon pyroptotic stimulation with LPS and ATP, and impaired release of IL-33 after papain stimulation (Fig. 2a–c), suggesting that Gsdmd processing was needed for IL-33 secretion. As reported21, caspase-1 and caspase-11 double-deficient BMMs did not generate the pyroptotic p35 NT-Gsdmd and secrete IL-1β after stimulation with LPS and ATP (Fig. 2d,e), but they generated p40 NT-Gsdmd and secreted IL-33 after stimulation with papain (Fig. 2d,f). MLE-12 cells were precultured with the pan-caspase inhibitor Z-VAD-FMK before papain stimulation. Neither Gsdmd cleavage nor IL-33 release was limited by Z-VAD-FMK treatment (Fig. 2g). These observations indicated that papain stimulated the concurrent generation of p40 NT-Gsdmd and secretion of IL-33 in BMMs and alveolar epithelial MLE-12 cells. The generation of p40 NT-Gsdmd was associated with IL-33 release through a caspase-1/11-independent pathway.
Fig. 2 |. Papain activates caspase-1/11-independent Gsdmd fragmentation.
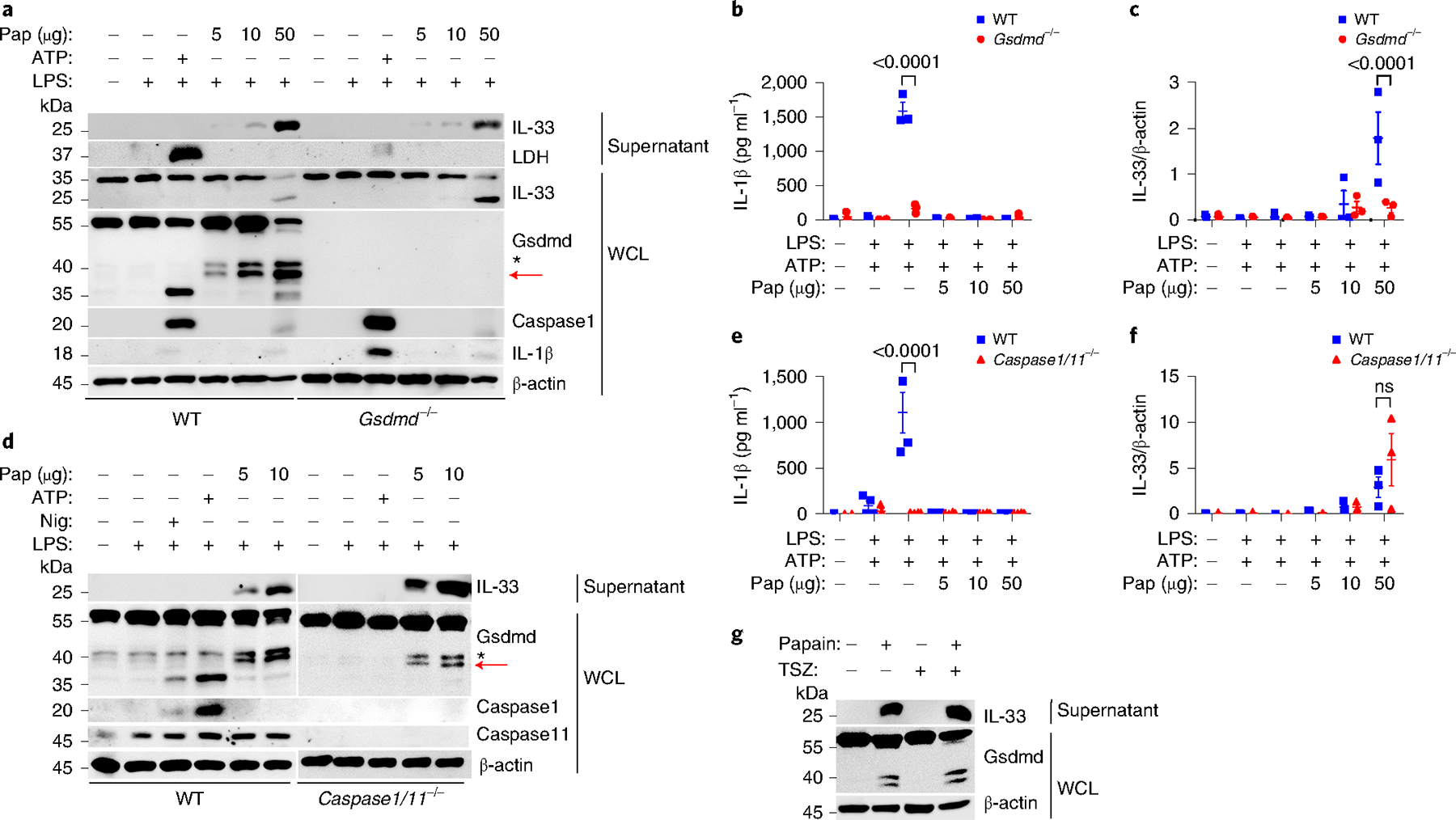
a, Immunoblot analysis of WCL and supernatants from wild-type (WT) and Gsdmd−/− BMMs treated with 200 ng ml−1 LPS for 4 h, 5 mM ATP for 30 min, and papain (5 μg well−1, 10 μg well−1 or 50 μg well−1) for 30 min. b, ELISA analysis of IL-1β in cell culture supernatants of wild-type and Gsdmd−/− BMMs as in a. c, ImageJ quantification of IL-33 (relative to the β-actin content in WCL) in the supernatants of wild-type and Gsdmd−/− BMMs as in a. d, Immunoblot analysis of WCL and culture supernatants derived from wild-type and Caspase1/11−/− BMMs treated with 200 ng ml−1 LPS for 4 h, 5 mM Nig or ATP for 30 min, and papain (5 μg well−1, 10 μg well−1 or 50 μg well−1) for 30 min. Wild-type and Caspase1/11−/− BMM samples were derived from the same experiment, and the gels and blots were processed in parallel. e, ELISA analysis of IL-1β in cell culture supernatants of wild-type and Caspase1/11−/− BMMs as in d. f, ImageJ quantification of IL-33 in cell culture supernatants of wild-type and Caspase1/11−/− BMMs as in d. g, Immunoblot analysis of MLE-12 cells and cell culture supernatants stimulated with or without papain (5 μg) for 30 min with the pretreatment of Z-VAD-FMK (20 μM) for 60 min and removal before papain stimulation. Red arrows indicate the cleaved functional NT-Gsdmd fragment. Nonspecific fragments marked with an asterisk were not discussed in our work. Error bar, mean ± s.e.m. b–f, n = 3, using Sidak’s multiple comparisons test. a–g, Data are representative or a summary of three independent experiments.
Gsdmd cleavage requires the protease activity of papain.
Papain is a proteolytic enzyme with cysteine protease activity. The p40 NT-Gsdmd and IL-33 release from MLE-12 cells were suppressed when papain was incubated with the irreversible cysteine protease inhibitor E-64 (Fig. 3a), or when papain was heat-inactivated at 100 °C for 10 min (Fig. 3a). When MLE-12 cells were incubated with other allergen-derived active enzymatic components, including those from the Aspergillus oryzae, Bacillus licheniformis, HDM extracts and the purified HDM proteases Der p and Der f at different concentrations, all tested allergen proteases (except for Alternaria alternata (Extended Data Fig. 3a)) activated the generation of p40 NT-Gsdmd and induced IL-33 secretion in a dose-dependent manner (Fig. 3b), suggesting that most allergen proteases share a common protease stress-sensing pathway leading to p40 NT-Gsdmd activation and IL-33 release. We did not detect any Gsdmd fragmentation and IL-33 release after stimulation with nonenzymatic allergens, including ovalbumin, alum, the toll-like receptor (TLR) 2/6 agonist fibroblast-stimulating lipopeptide-1 (FSL-1), the endoplasmic reticulum stress-inducer palmitic acid and the membrane-permeable poly-l-arginine (Fig. 3c). Other stressors—including oxidative stress induced by hydrochloric acid or hydrogen peroxide; chemical stress induced by inorganic salts of sodium, calcium or magnesium; peptide agonists of protease-activated receptors, including PAR2/1; and temperature-associated cold shock (4 °C) or heat shock (42–44 °C)—did not induce p40 NT-Gsdmd activation (Extended Data Fig. 3b–d). These results showed that lung epithelial cells possessed unique stress-sensing pathways for exogenous allergen proteases that lead to IL-33 secretion.
Fig. 3 |. Gsdmd cleavage requires the protease activity of papain.
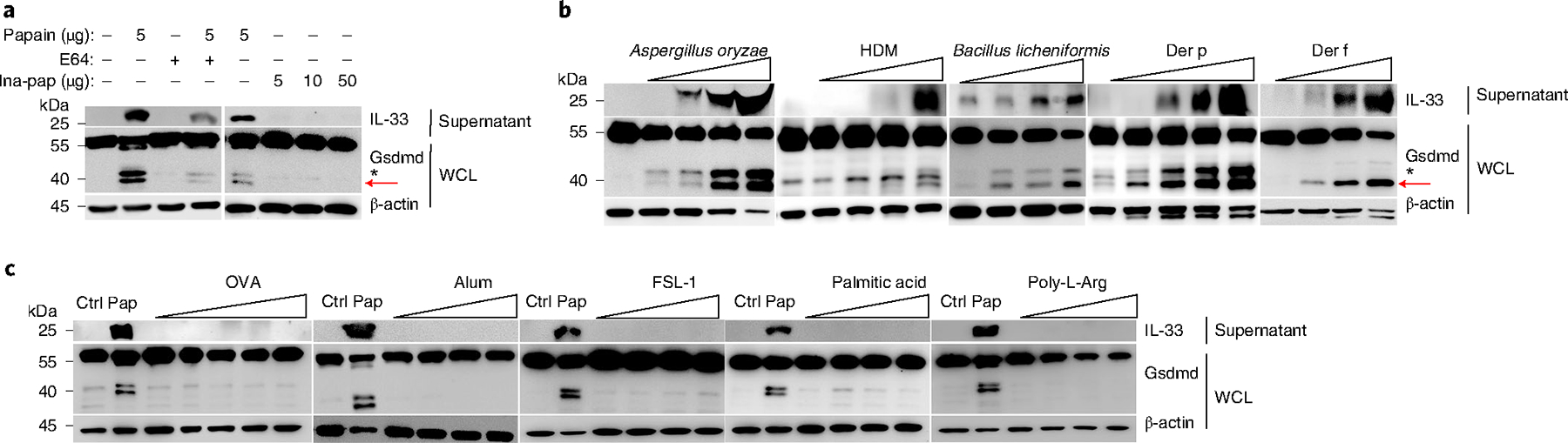
a, Immunoblot analysis of WCL and culture supernatants from MLE-12 cells treated with papain (5 μg) for 30 min, an E-64 pretreatment for 30 min before papain stimulation, and heat-inactivated papain (Ina-pap, 10 min for 100 °C) for 30 min. b, Immunoblot analysis of WCL and culture supernatants from MLE-12 cells treated as follows. Aspergillus oryzae: 0, 0.5, 5, 10, 50 (μl well−1); HDM: 1, 10, 20, 50, 100 (μg well−1); Bacillus licheniformis: 0.05, 0.1, 1, 5 (μg well−1); Der p: 0.01, 0.025, 0.25, 1, 2.5 (μg well−1); Der f: 0.01, 0.025, 0.25, 2.5 (μg well−1). Aspergillus oryzae and HDM were stimulated for 30 min; Bacillus licheniformis, Der p and Der f were stimulated for 60 min. c, Immunoblot analysis of MLE-12 cells treated with ovalbumin (OVA, 0.1–100 μg well−1) for 30 min, aluminum (Alum, 0.1–100 μg well−1) for 30 min, FSL-1 (1–500 ng ml−1 per well) for 6 h, palmitic acid (0.1–50 μM well−1) for 6 h, and poly-L-arginine (cationic polypeptides, 2.5–125 μg well−1) for 30 min. Red arrows indicate the neo-form of p40 NT-Gsdmd. Nonspecific fragments marked with an asterisk were not discussed in our work. a–c, Data are representative of three independent experiments.
Allergen proteases activate SG assembly.
To define whether SG assembly participated in allergen protease-induced IL-33 release in the airway epithelium, we analyzed the localization of the Ras-GTPase-activating protein G3BP1, which functions as a molecular switch that triggers the assembly of SGs by forming a multimer24 in A549-IL-33-GFP cells. Arsenite, an eIF2α phosphorylation-dependent stimulator25, induced typical G3BP1 puncta in the A549 cells (Fig. 4a), which indicated effective SG assembly. Following translocation to the cytoplasm, IL-33 colocalized with the G3BP1 puncta (Fig. 4a). Papain stimulation also induced the formation of G3BP1 puncta and the colocalization between G3BP1 puncta and IL-33 (Fig. 4a,b). Arsenite promoted the nucleocytoplasmic transfer of IL-33, but did not induce IL-33 secretion (Fig. 4a,c), whereas papain induced the direct secretion of IL-33 from the nucleus to the extracellular space (Fig. 4c). Next, we collected cell culture supernatants and whole-cell lysis (WCL) from murine airway epithelial MLE-12 cells stimulated with arsenite or papain, and used ultracentrifugation to isolate SGs26. In the arsenite-treated cells, IL-33 was associated with the SG condensates, similar to the SG core components such as TIA1, eIF4A1 and Hsp90 (Fig. 4d). At the same time, IL-33 was enriched in SG condensates and was detected in the supernatant in the papain-treated cells (Fig. 4d). Although papain stimulation induced TIA degradation in WCL when compared with unstimulated MLE-12 cells, TIA1 and other typical SG proteins (including eIF4A1 and Hsp90) were enriched in the SG fractions of papain-stimulated cells (Fig. 4d). Immunoprecipitation of G3BP1 from cell extracts also indicated an increased interaction between G3BP1 and IL-33 in papain-treated and arsenite-treated MLE-12 cells compared with unstimulated cells (Extended Data Fig. 3e). SG assembly is triggered by the bulk inhibition of translation initiation and can be dependent or independent of p-eIF2α27. Ectopically expressed IL-33 migrated from the nucleus to the cytosol in HEK293T cells after stimulation with arsenite, which is eIF2α dependent, or d-sorbitol, which is eIF2α independent (Extended Data Fig. 3f). Mass spectrometry analysis28 of the IL-33 interaction network with immunoprecipitation in HEK293T cells indicated that arsenite and papain stimulations promoted the enrichment of several common non-membrane-bound, organelle assembly-associated components, including G3BP1, KPNB1 and ATXN2L (Supplementary Table 1, Extended Data Fig. 4). Stimulation of MLE-12 cells with arsenite or d-sorbitol, or other stimuli reported to induce SG assembly, did not induce p40 NT-Gsdmd or the secretion of IL-33, even after extended (2 h) stimulation (Extended Data Fig. 5a–c). These observations indicated that IL-33 was transported into the cytoplasms and associated with the SG in papain-stimulated airway epithelial cells, but the assembly of SG did not contribute to IL-33 secretion.
Fig. 4 |. Allergen proteases activate SG assembly.
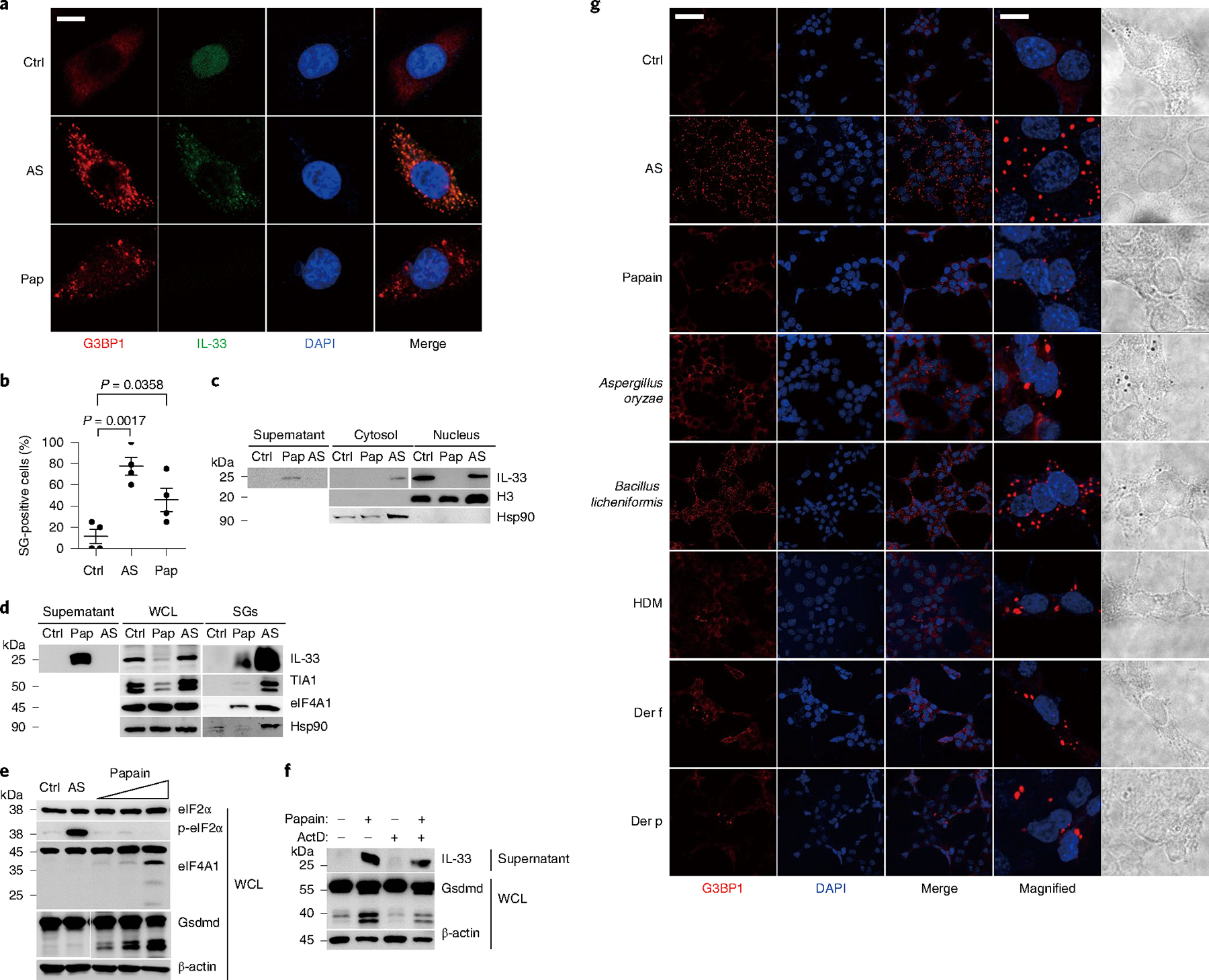
a, Immunofluorescent analysis of A549-IL-33-GFP cells treated with Ctrl (without stimulation), 0.5 mM arsenite (AS) for 1 h, and 5 μg of papain (Pap) for 30 min. b, Quantification analysis of SG-positive cells (G3bp1 foci) as in a with the SG counter plugin in ImageJ. Error bar, mean ± s.e.m. n = 4, using Sidak’s multiple comparisons test. c, Immunoblot analysis of extracted nuclear and cytoplasmic proteins as in a. d, Immunoblot analysis of WCL, culture supernatants and SG components from MLE-12 cells treated with Ctrl, 0.5 mM arsenite for 1 h, and 10 μg of papain for 30 min. e, Western blot analysis of cells stimulated with different amounts of papain (5 μg well−1, 10 μg well−1 and 50 μg well−1) for 30 min, 0.5 mM arsenite for 1 h, and Ctrl. f, Immunoblot analysis of WCL and culture supernatants of MLE-12 cells with pretreatment of actinomycin D (ActD, 2 μM) for 3 h following the stimulation by 10 μg of papain for 30 min. g, Immunofluorescent analysis of G3BP1 puncta in MLE-12 cells treated with Ctrl, 0.5 mM arsenite, 5 μg well−1 papain, 1 μl well−1 Aspergillus oryzae, 0.1 μg well−1 Bacillus licheniformis, 10 μg well−1 HDM, 0.025 μg well−1 Der f and 0.025 μg well−1 Der p. Scale bars: a, 10 μm; g, 50 μm; magnified g, 10 μm. a–g, Data are representative of three independent experiments.
Unlike arsenite, which promoted eIF2α phosphorylation when compared with unstimulated cells, stimulation with papain did not alter the amount of eIF2α or p-eIF2α, but increased the degradation of eIF4A1, which inhibits SG assembly29, when compared with arsenite-treated or unstimulated MLE-12 cells (Fig. 4e). Treatment with integrated stress response inhibitor (ISRIB), which inhibits SG assembly downstream of eIF2α30, did not affect p40 NT-Gsdmd activation and IL-33 release (Extended Data Fig. 5d). Actinomycin D, a potent inhibitor of both cell-intrinsic and cell-extrinsic EIF4A inhibitors that helps to relieve the function of EIF4A31–34, impaired IL-33 secretion and blunted p40 NT-Gsdmd fragmentation in MLE-12 cells (Fig. 4f), indicating that papain stimulation triggered a p-eIF2α-independent SG assembly pathway. All tested allergen proteases activated SG assembly into G3BP1 puncta in MLE-12 cells (Fig. 4g). These data indicated that protease-activated SG assembly was necessary for controlling the nucleocytoplasmic transport of IL-33.
p40 NT-Gsdmd contributes to IL-33 secretion.
Next, we investigated whether p40 NT-Gsdmd facilitated the extracellular secretion of cytosolic IL-33. We used the online pro-protein conversion prediction tool ProP 1.0 to predict potential cleavage sites in Gsdmd (ref. 35). Aside from the amino acid (aa) sites within the aa 1–276, which is generated by caspase-1/8/11 cleavage, we obtained six putative cleavage sites in arginine (R) and lysine (K), among which R311, K394 and K409 were chosen for further analysis (Extended Data Fig. 6). We constructed three murine Gsdmd fragments (Gsdmd1–311, Gsdmd1–394 and Gsdmd1–409) with a Flag-tagged N terminus, the caspase-1/11-processed Gsdmd1–276 as a positive control and the full-length form of Gsdmd (GsdmdFL) as a nonfunctional control (Fig. 5a). After 12 h of expression in HEK293T cells, Gsdmd1–311 induced less LDH release than pyroptotic Gsdmd1–276 (Fig. 5b). To further define their function in controlling IL-33 release, we cotransfected these fragments and C-terminal HA-tagged mature IL-33 (aa 109–266, without the N-terminal nuclear localization signal peptide) into HEK293T cells. Compared with GsdmdFL, the expression of pyroptotic Gsdmd1–276 and Gsdmd1–311 induced IL-33 release with high efficiency, whereas the other expressed fragments did not (Fig. 5c,d). Next, we introduced point mutations and deletions close to R311 into full-length mouse Gsdmd and lentivirally expressed them in MLE-12 cells. We found that amino acid mutation or deletion of residues 309–313 (ELRQQ) blocked the generation of p40 NT-Gsdmd and prevented IL-33 release in papain-stimulated MLE-12 cells (Fig. 5e), which suggested that this sequence was necessary for the generation of p40 NT-Gsdmd.
Fig. 5 |. truncated Gsdmd 311 fragment promotes IL-33 release.
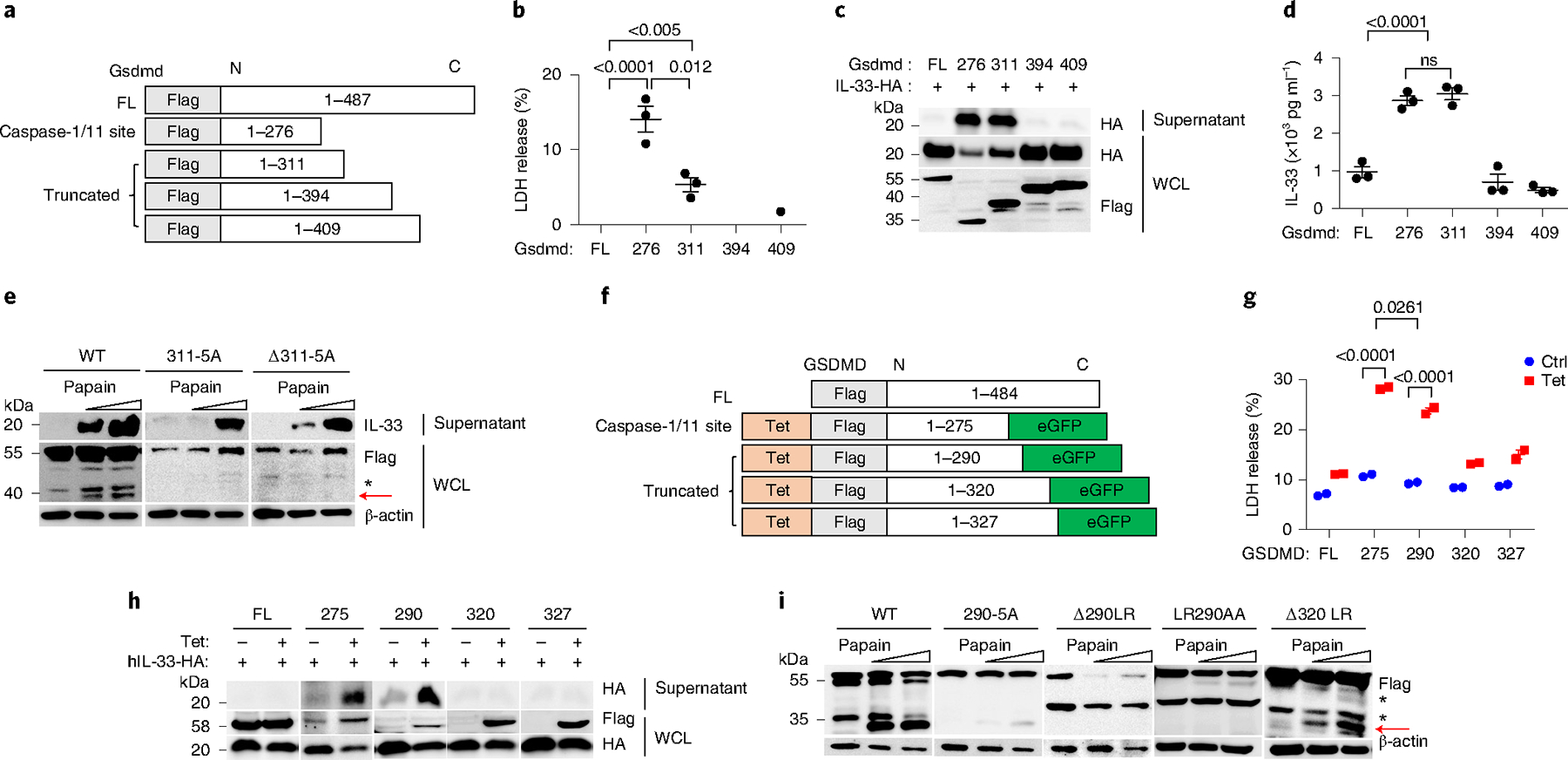
a, Schematic showing truncated Gsdmd mutants with an NT Flag tag. b, LDH release assay of culture supernatants from HEK293T cells expressed with truncated Gsdmd fragments for 12 h. c, Immunoblot analysis of WCL and culture supernatants from HEK293T cells expressed with murine C-terminal HA-tagged mature IL-33 and GsdmdFL, Gsdmd1–276, Gsdmd1–311, Gsdmd1–394 and Gsdmd1–409 in HEK293T. d, ELISA analysis of IL-33 in supernatants as in c. e, Immunoblot analysis of WCL and culture supernatants from MLE-12 cells expressed with Gsdmd mutations following papain stimulation for 30 min. 311–5A, substitution of 309–313 (ELRQQ) to AAAAA; Δ311–5A, deletion of 309–313 (ELRQQ). f, A truncation scheme of human GSDMD with NT Flag-tagged. A tetracycline-responsive sequence (Tet) was added to control the expression of the GSDMD fragments. eGFP, enhanced GFP. g, LDH release assay of culture supernatants from HEK293T cells with 2 μg ml−1 tetracycline-induced expression of truncated GSDMD fragments for 12 h. Error bar, mean ± s.e.m. n = 2, Sidak’s multiple comparisons test, and two-way analysis of variance (ANOVA). Data are representative of four independent experiments. h, Immunoblot analysis of WCL and culture supernatants from HEK293T cells coexpressed with truncated GSDMD fragments and human C-terminal HA-tagged mature IL-33 for 12 h with tetracycline (2 μg ml−1). i, Immunoblot analysis of WCL from GSDMD−/− HeLa cells expressed with different GSDMD mutants, following 1 h with different doses of papain stimulation. 290–5A, substitution of 288–292 (GLRAE) to AAAAA; Δ290LR, deletion of 289–290 (LR); LR290AA, substitution of 289–290 (LR) to AA; Δ320LR, deletion of 319–320 (LR). Error bar, mean ± s.e.m. b,d, n = 3, Dunnett’s multiple comparisons test. a–i, Data are representative of at least three independent experiments.
To identify the specific cleavage site of the p35 NT-GSDMD fragment in human cells, we cloned the GSDMD fragments GSDMD1–290, GSDMD1–320 and GSDMD1–327 and the pyroptosis-mediating fragment GSDMD1–275 into tetracycline-responsive expression vectors expressing GFP (Fig. 5f) and expressed them in HEK293T cells. The 35-kDa GSDMD1–290 induced LDH to release at lower levels than the pyroptotic GSDMD1–275 fragment (Fig. 5g). Following tetracycline-induced expression, GSDMD1–290 presented high efficiency in promoting human mature IL-33 release into the supernatant, similar to GSDMD1–275, and other constructs did not possess this function (Fig. 5h). To confirm the specific GSDMD cleavage site, we constructed Flag-tagged GSDMD expression vectors with several mutations close to aa 290 and transfected them in GSDMD-deficient HeLa cells. Mutation of aa 288–292 (GLRAE) into AAAAA blocked the generation of the p35 NT-GSDMD following papain exposure (Fig. 5i), suggesting that the cleavage site for p35 NT-GSDMD was located in this region. Deleting both R290 and L289 also effectively prevented the generation of p35 NT-GSDMD (Fig. 5i). Together, these results indicated that human GSDMD1–290 promoted the secretion of cytosolic IL-33 into the supernatant.
Gsdmd contributes to type 2 inflammatory immune responses.
To determine the expression of GSDMD in asthmatic human lung tissue, we collected bronchial samples from 17 people with asthma and 6 people without asthma (Extended Data Fig. 7). Asthmatic lung tissue presented increased inflammatory cell infiltration, as well as hyperplasia of the mucous glands, compared with the nonasthmatic controls with hematoxylin and eosin (H&E) staining (Fig. 6a). GSDMD was mainly expressed in the pulmonary airway epithelium of both noninflamed and inflamed asthmatic lung tissues (Fig. 6a) and presented typical expression in areas with mucous gland hyperplasia (Fig. 6a). Expression of GSDMD in the lung tissues also showed significant positive correlation with the amount of IL-33 secreted into the bronchoalveolar lavage (BAL) and the amount of IgE in the serum from people with asthma (Fig. 6b,c), suggesting that higher GSDMD expression in the lung tissue in people with asthma might contribute to increased IL-33 secretion and potentially promote the allergic immune response. In mice, Gsdmd expression was weak in the unstimulated wide-type murine airway epithelium, including peribronchial and AT2 cells (Fig. 6d). Intranasal HDM stimulation increased bronchial immune cell infiltration and significantly upregulated the expression of Gsdmd in the lung tissue in mice (Fig. 6d–f). These observations indicated the association between Gsdmd expression and type 2 inflammation in humans and mice.
Fig. 6 |. Gsdmd contributes to type 2 inflammatory immune responses.
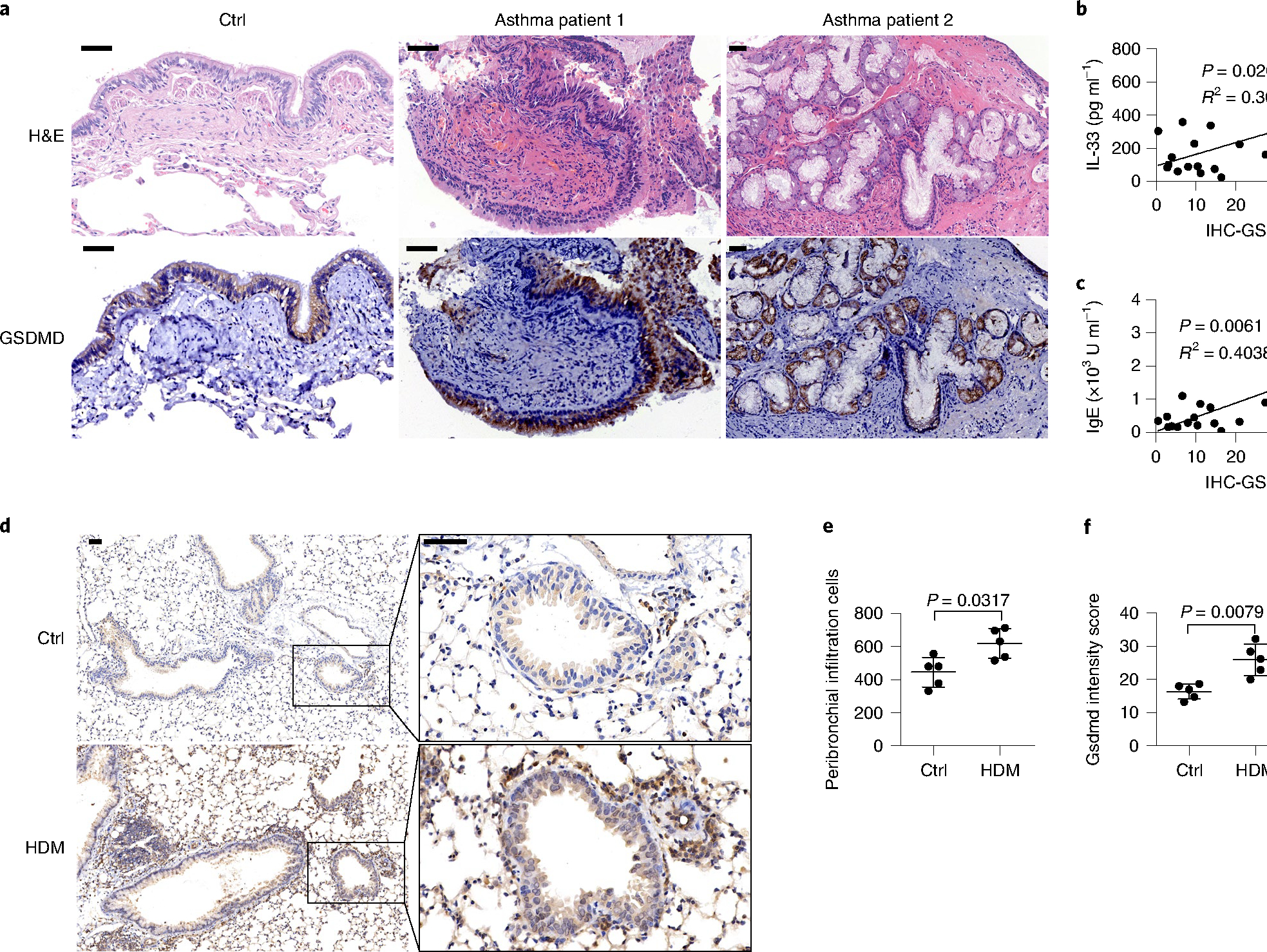
a, Microscopy images of H&E staining and immunohistochemical (IHC) staining of human GSDMD (brown) in bronchial tissue from people with and without asthma. b, IL-33 production in the BALF versus GSDMD expression in lung tissues of people with asthma. c, Serum IgE versus GSDMD expression in lung tissues of people with asthma. b,c, n = 17, two-tailed Pearson correlation. Linear regression curves fitted to these data are provided, alongside P values and R2 values. d, Microscopy imaging of immunohistochemically stained mouse Gsdmd protein in lung tissues. BALB/c mice were administered with HDM or PBS intranasally for up to 3 weeks to induce asthma symptoms. e, Histological evaluation of peribronchial inflammatory cell infiltration as in d. f, Statistical analysis of the Gsdmd protein intensity scores as in d. Error bar, mean ± s.e.m. e,f, n = 5, using the two-tailed Mann–Whitney U-test. Scale bars: a,d, 50 μm. a–f, Data are representative of at least three independent experiments.
Gsdmd modulates IL-33 release in vivo.
Multiple inflammatory cytokines contribute to the initiation and development of airway inflammation, among which epithelial cell-derived IL-33, IL-25 and thymic stromal lymphopoietin (TSLP) were shown to be important in initiating type 2 inflammation36. To investigate the contribution of IL-33 in vivo during allergen protease exposure, we evaluated the secretion of inflammatory cytokines in the BAL fluid (BALF) after papain exposure in mice. Using a multiple-cytokine detection system, we analyzed the amount of interferon-γ (IFN-γ), IL-10, IL-13, IL-17A, IL-25, IL-1β, IL-33, IL-4, IL-5 and IL-9 in the BALF 3 h after papain inhalation. IL-33 had a markedly high production (Fig. 7a), whereas IL-25 and TSLP displayed no significant change after papain exposure (Fig. 7b). Kinetic analysis indicated that the secretion of IL-33 into the BALF peaked at 3 h after papain exposure (Fig. 7b), indicating that IL-33 was released early during lung inflammation.
Fig. 7 |. Gsdmd modulates IL-33 release in vivo.
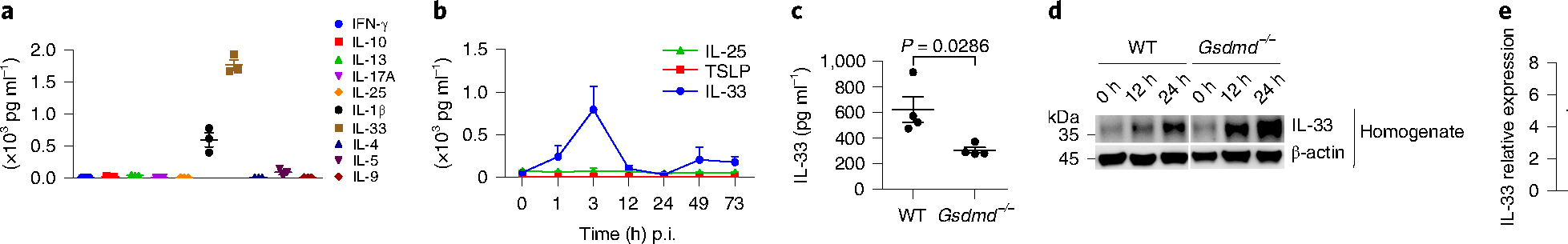
a, Cytokine analysis of BALF from wild-type BALB/c mice with intranasal papain (5 μg in 40 μl PBS) stimulation for 3 h. b, ELISA analysis of IL-33, IL-25 and TSLP in the BALF after a single dose of papain (5 μg) exposed for different lengths of time. c, ELISA analysis of IL-33 in the BALF from wild-type and Gsdmd−/− mice 3 h after papain exposure. d, Immunoblot analysis of IL-33 in the lung homogenates with single-dose papain (5 μg) exposed for 12 and 24 h. e, IL-33 mRNA expression in lung tissue from wild-type and Gsdmd−/− mice. Error bar, mean ± s.e.m. a,b, n = 3, c, n = 4, e, n = 6, using two-tailed Mann–Whitney U-test. Data are representative of three independent experiments with similar results.
Analysis of BALF from wild-type and Gsdmd-deficient (Gsdmd−/−) mice 3 h after papain exposure indicated that Gsdmd deficiency inhibited the early release of IL-33 (Fig. 7c,d) and resulted in elevated IL-33 retained in the lung tissue homogenates, with no effect on the expression of IL-33 messenger RNA after papain stimulation (Fig. 7e). To determine whether Gsdmd interfered with IL-33-ST2 signaling, we intraperitoneally challenged Gsdmd-deficient mice with recombinant murine mature IL-33 (rIL-33, 25 μg) for 4 continuous days. rIL-33 induced an inflammatory immune response in both wild-type and Gsdmd−/− mice, which showed no significant difference in inflammatory cell infiltration (Extended Data Fig. 8a–e) and IL-5+ or IL-13+ ILC2s (Extended Data Fig. 8f–h), indicating that Gsdmd did not interfere with ST2 signaling. These results indicated that Gsdmd controlled cellular events before IL-33 secretion.
Gsdmd modulated HDM-induced chronic airway inflammation.
To further investigate the function of Gsdmd in allergic airway inflammation, we administered HDM extracts intranasally for 16 noncontinuous days in Gsdmd−/− and wild-type mice to induce chronic asthmatic airway inflammation. We found less inflammatory infiltration into the bronchus, less mucus production in the airways (Fig. 8a) and lower numbers of infiltrated eosinophils and ILC2s in the lungs (Fig. 8b and Extended Data Fig. 9a,b) in the Gsdmd−/− mice than in the wild-type mice. The secretion of inflammatory cytokines, including IL-5 and IL-13, in BALF was also significantly reduced in the Gsdmd−/− mice compared with the wild-type mice (Fig. 8c). In the papain-induced acute airway inflammation model (5 μg of papain for 5 continuous days), Gsdmd−/− mice had blunted airway inflammation infiltration (Fig. 8d–f). The infiltration of alveolar eosinophil and ILC2s was decreased (Fig. 8g), and the numbers of activated IL-5-producing or IL-13-producing ILC2s were significantly lower (Fig. 8h) in the Gsdmd−/− mice than in the wild-type mice. The secretion of the inflammatory cytokines IL-5 and IL-13 into the BALF was also impaired (Fig. 8i). Together, these data indicated that Gsdmd deficiency in mice alleviated both HDM-induced chronic and papain-induced acute airway inflammatory responses by limiting the secretion of IL-33, suggesting that Gsdmd played a fundamental role in the pathogenesis of type 2 immunity in vivo.
Fig. 8 |. Gsdmd modulates HDM-induced chronic airway inflammation.
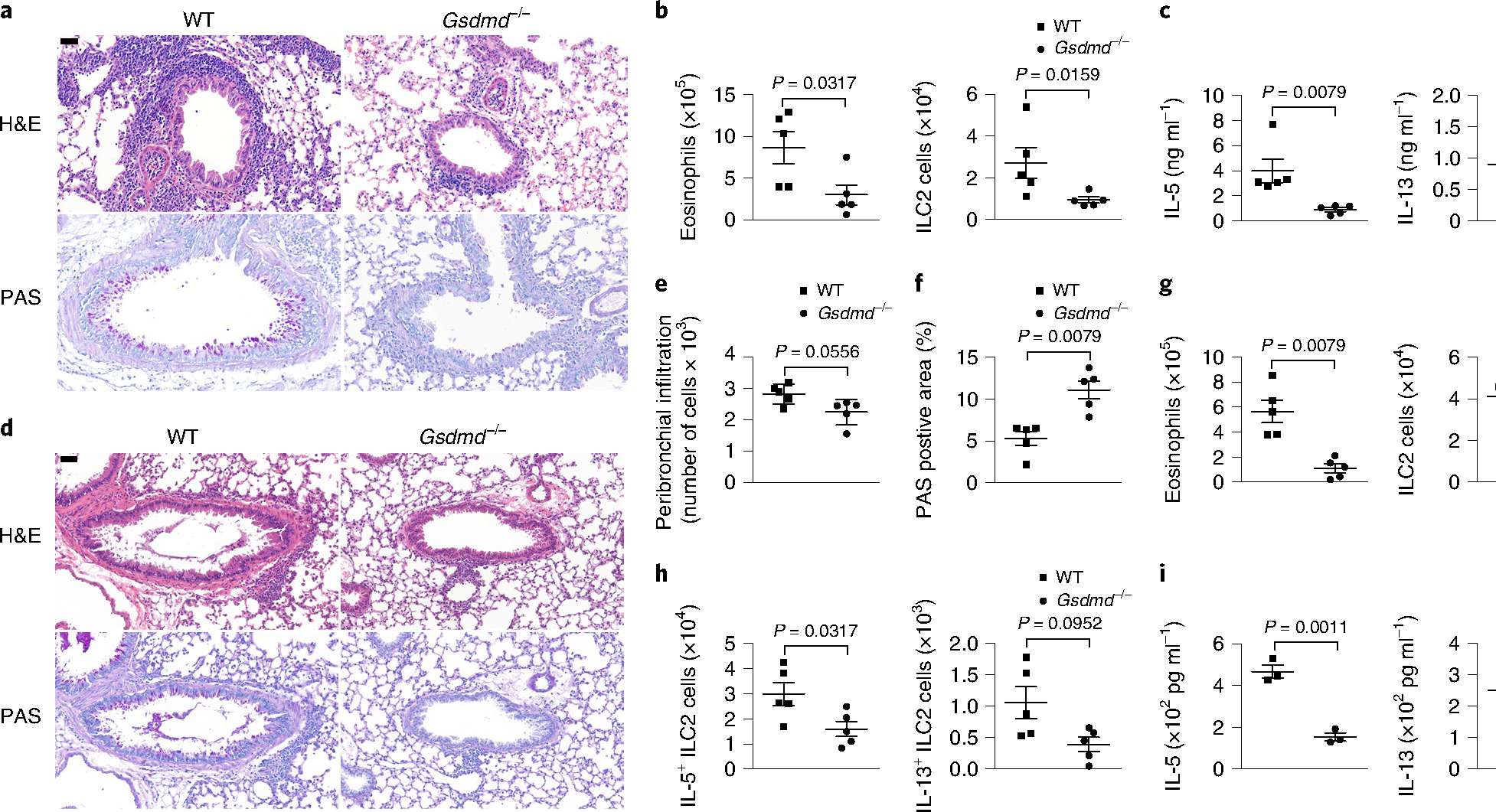
a, Representative H&E staining and periodic acid–Schiff (PAS) staining of lung tissue sections from wild-type and Gsdmd−/− mice treated with intranasal HDM for 16 noncontinuous days. b, Flow cytometry analysis of cell numbers of SiglecF+CD11c+ eosinophils in the BALF and Lin−CD90.2+ST2+ ILC2s in the lungs as in a. c, ELISA analysis of IL-5 and IL-13 in the BALF from HDM-treated wild-type and Gsdmd−/− mice as in a. d, Representative H&E staining and PAS staining of lung sections wild-type and Gsdmd−/− mice treated with intranasal papain for 5 continuous days. e, Histological evaluation of peribronchial inflammatory cell infiltration as in d. f, Quantification of PAS-positive area in d with ImageJ. g, Flow cytometry analysis of cell numbers of SiglecF+CD11c+ eosinophils in the BALF and Lin−CD90.2+ST2+ ILC2s in the lungs as in d. h, Flow cytometry analysis of cell numbers of IL-5+-expressing and IL-13+-expressing ILC2s as in d. i, ELISA analyses of IL-5 and IL-13 in the BALF as in d. Scale bars: b,d, 50 μm. Error bar, mean ± s.e.m. b,c,e–h, n = 5, using the two-tailed Mann–Whitney U-test. i, n = 3, using the two-tailed Holm–Sidak test. Data are representative of two independent experiments with similar results.
Discussion
Here, we show that two cellular events regulated IL-33 secretion sequentially in the context of allergen protease exposure. First, the activation of SG assembly in airway epithelial cells licensed the nucleocytoplasmic transport of IL-33 but did not control its secretion. Second, the cleavage of Gsdmd at aa 309–313 (ELRQQ) generated p40 NT-Gsdmd, which promoted the cytosolic IL-33 to cross the membrane into the extracellular space. Our investigation also noted that GSDMD had a relatively high expression in the human airway epithelium and was associated with increased IL-33 secretion into BALF in people with asthma, suggesting that GSDMD may be involved in the pathogenesis of asthma.
We show that various allergen-derived proteases, including plant-derived papain, fungal allergen Aspergillus oryzae, HDMs, HDM-derived Der p and Der f, and subtilisin protease from Bacillus species, activated SG assembly in airway epithelium, which was fundamental for the translocation of IL-33 from the nucleus into the cytosol. SGs undergo fusion, fission and flow in mammalian cell cytosol cycles, and their assembly is highly dynamic37. The activation of various cell death signaling pathways in lung airway epithelial cells, including caspase-mediated apoptosis (stimulated with TS) and p-Mlkl-dependent necroptosis (stimulated with TSZ), was insufficient to induce IL-33 release in airway epithelial cells, suggesting that IL-33 secretion was independent of these pathways. In our model, we observed the cessation of IL-33 secretion and the loss of Gsdmd fragmentation within 3 h after removing allergen protease, which indicated the dynamic regulation of IL-33 secretion through SG assembly in living cells.
Gsdmd-mediated pore formation has been reported in several cell types, including immune cells such as macrophages and neutrophils, and stromal cells such as nasal epithelial cells38,39. The cleavage of Gsdmd at D276 by caspase-1, caspase-8 or caspase-11 has been reported during various viral and bacterial infections. However, caspase-1 and caspase-11 double deficiency and the pan-caspase inhibitor Z-VAD-FMK did not inhibit the release of IL-33 and the generation of p40 NT-Gsdmd, indicating that this process was independent of the caspase family. Mutations in the aa 309–313 (ELRQQ) in Gsdmd efficiently prevented the cleavage of Gsdmd into the functional p40 fragment and blocked IL-33 release in the airway epithelial cells. Similarly, the human residues L289–R290 were needed to generate the functional NT-GSDMD fragment. Thus, the cleavage of leucine and arginine might provide clues for enzymes associated with this process. The enzymes involved in the allergen protease stress-sensing pathway that contributed to the generation of p40 NT-Gsdmd have yet to be determined.
It was previously reported that arsenite-induced activation of SG assembly did not contribute to Gsdmd fragmentation in BMMs, and stimulation with arsenite significantly inhibited NLRP3 inflammasome activation and cell death in BMMs16. These observations supported that SG assembly is independent of Gsdmd fragmentation, and this is consistent with our observation in airway epithelial cells.
Allergens from Alternaria were shown to trigger the RIPK1–caspase-8 ripoptosome, which engaged caspase-3 and caspase-7 and contributed to intracellular maturation and the release of IL-33 in epithelial cells40. Although we did not observe caspase-3 activation and cell death during early exposure (30 min) to papain, sustained allergen protease stimulation can contribute to the release of DAMPs, including ATP, from damaged cells, further promoting cell death in bystander cells1,41,42. We observed IL-33 secretion without Gsdmd fragmentation in MLE-12 cells exposed to Alternaria for 30 min, indicating that Alternaria uniquely activated a Gsdmd-independent IL-33 secretion pathway. Therefore, additional mechanisms might regulate IL-33 secretion under various stimulations.
Our data highlight a unique role of Gsdmd in controlling cytokine release by switching the two different cleaved forms. The pyroptotic p35 NT-Gsdmd fragment reacts to classic caspase-1/8/11 cleavage that contributes to IL-1β release under typical type 1 inflammatory contexts. The p40 NT-Gsdmd fragment responds to environmental allergen protease stimulation permitting IL-33 secretion without apparent cell death, thus contributing to type 2 disease pathogenesis. Each fragment reacts to different inflammatory cues and amplifies different downstream immune cell activation events.
online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41590-022-01255-6.
Methods
Mice.
C57BL/6 wild-type mice were purchased from the Shanghai Laboratory Animal Center. Gsdmd−/− mice on the C57BL/6J background were from F. Shao, and caspase-1 and caspase-11 double-deficient mice were kindly provided by G. Meng (Institut Pasteur of Shanghai). Mice of age 6–10 weeks were used for all experiments; all animals were age-matched and sex-matched, and then randomized into different groups. All mice were maintained in specific pathogen-free animal facilities at the Animal Care Facility of the Chinese Academy of Sciences and used according to protocols approved by the Institutional Animal Care and Use Committee.
Induction of allergic airway inflammation.
For the induction of acute type 2 airway inflammation, C57BL/6 wild-type and Gsdmd−/− mice were anaesthetized by isoflurane inhalation, treated intranasally with papain (5 μg) in 40 μl PBS every day for 5 days, and euthanized for analysis on day 6. For the induction of chronic type 2 airway inflammation, HDMs (50 μg) were applied intranasally for 3 continuous days, and the mice were allowed to rest for 11 days and then treated with HDMs (10 μg) for 4 continuous days. The mice were sacrificed for analysis 24 h after the final intranasal treatment. rIL-33 (25 μg) was administered intranasally for 4 continuous days to induce acute airway inflammation.
Primary cells isolation.
BMMs were prepared by flushing bone marrow from the femurs and tibias of C57BL/6 mice following the removal of red blood cells using ammonium chloride. Cells were seeded at a density of 1 × 106 cells per well in 24-well plates in DMEM supplemented with 20 ng ml−1 murine macrophage colony-stimulating factor (M-CSF). The cell culture medium was refreshed every 2 days, and cells were collected on day 6.
Allergen extracts and proteases.
HDM extracts were obtained from Greer Laboratories. The purified natural HDM proteases Natural Der p 1 (NA-DP1–1) and Natural Der f 1 (NA-DF1–1) were obtained from Indoor Biotechnologies. A protease from Aspergillus oryzae (Sigma-Aldrich, P6110), papain from papaya latex (Sigma-Aldrich, P3125), subtilisin A protease from Bacillus licheniformis (Sigma-Aldrich, P5380) and Alternaria alternata (Greer Laboratories) were used as sources of purified allergen proteases. All allergen extracts and proteases were reconstituted in PBS (1 mg ml−1) and stored at −80 °C. The Aspergillus oryzae protease (500 U g−1; 4 °C) was used with ten-fold dilutions in PBS.
Generation of constitutive IL-33-expressing or GSDMD-expressing cells.
To generate cells with stable constitutive overexpression of IL-33 or GSDMD, a lentivirus was generated by transiently transfecting HEK293T cells with the pLVX-IL-33-HA-GFP plasmid, the Flag-GSDMD-Puro plasmid or the empty vector with an improved packaging plasmid and the VSdVG plasmid. Then, A549/mle-12 cells were infected with lentiviral supernatants in the presence of polybrene (5 μg ml−1) for 12 h. The following day, puromycin selection was applied to generate pools of cells with stable overexpression. Following at least 1 week of puromycin treatment, single-cell clones were generated using limiting dilution.
Cleavage of endogenous GSDMD with allergen extracts and proteases.
GSDMD-deficient HeLa cells were kindly provided by F. Shao. Cells seeded in 24-well plates were grown to form monolayers and then exposed to increasing amounts of allergen extracts and proteases (up to 50 μg for papain and Bacillus licheniformis, up to 10 μg for Der p and Der f, up to 200 μg for HDMs, and up to 5 μl for Aspergillus oryzae; ~1 × 106 cells) in 300 μl well−1 DMEM supplemented with 1% FBS and cultured for 30 min at 37 °C. The cells were collected for analysis by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblotting, and the supernatants were collected for enzyme-linked immunosorbent assay (ELISA) detection or analyzed with SDS–PAGE after precipitation with FreeZone Freeze Dryers.
SG isolation.
MLE-12 cells were plated in 10 cm tissue culture dishes and grown to form monolayers of 80–90% confluence on the day of collection. Cells were stimulated with the following stressors under 1% FBS DMEM culture medium: 200 μg of papain per dish for 30 min at 37 °C, and 0.5 mM arsenite for 1 h at 37 °C. Media were aspirated, and cells were washed with 5 ml of fresh media. Cells were scraped and collected into a 15 ml Falcon tube. Then, cells were pelleted at 1,500g for 3 min at room temperature (22–26 °C). Media were aspirated, and the pellet was flash-frozen using liquid N2. The pellet was then thawed on ice for 5 min. We resuspended the pellet in 1 ml of SG lysis buffer (50 mM Tris HCl pH 7.4, 100 mM potassium acetate, 2 mM magnesium acetate, 0.5 mM dithiothreitol (DTT), 50 μg ml−1 heparin, 0.5% NP-40, 1:5,000 antifoam B, one cOmplete Mini EDTA-free protease inhibitor tablet 50 per ml of lysis buffer) containing 1 U μl−1 RnaseIN right before lysis, and we syringed the lysis buffer with a 25G × 5/8” needle on ice. Five passages through a needle are normally sufficient to lyse cells. After lysis, the cells were spun at 1,000g for 5 min at 4 °C to pellet cell debris. The pellet was discarded, and the supernatant was transferred to a 1.5-ml microcentrifuge tube, which was spun at 18,000g for 20 min at 4 °C. Supernatants were removed, and the pellet was resuspended in 1 ml of SG lysis buffer and spun at 18,000g for 20 min at 4 °C. The supernatant was discarded, and the pellet was resuspended in 300 μl of SG lysis buffer and spun at 850g for 2 min at 4 °C. The supernatant was transferred to a new 1.5-ml microcentrifuge tube. The supernatant represents the SG core enriched fraction. Quantification analyses of SGs were performed using the SG counter plugin in ImageJ (https://imagej.nih.gov/ij/plugins/stress_granule_counter). The parameters for A549 cells were adjusted as (default): number of smoothes, 70; number of smoothes after subtraction, 4; threshold, 2,000; numbers of counted particles >2 were taken as positive.
Antibodies, reagents and plasmids.
Anti-HA 1:1,000 (16B12, HA.11, Covance, CO-MMS-101R), anti-GSDMDC1 0.04–0.4 μg ml−1 (Novus Biologicals, NBP2–33422) and mouse anti-GSDMD 1:1,000 (EPR20859, Abcam, ab219800) antibodies were used. We used a mouse anti-IL-33 propeptide antibody 1 μg ml−1 (R&D Systems, AF5010-SP); an anti-IL-33 antibody 1:1,000 (mouse clone Nessy-1, Alexis Biochemicals, ALX-804–840); an anti-IL-33-Cter antibody 1:1,000 (Alexis Biochemicals, AT-110); an anti-IL-33 (human) monoclonal antibody 1:2,000–1:5,000 (IL33305B, Cayman Chemical, 10809); a donkey anti-sheep IgG NorthernLights NL557-conjugated antibody 1:200 (R&D Systems, NL010); a sheep IgG HRP-conjugated antibody 1:1,000 (R&D Systems, HAF016); an anti-HA tag antibody 1:1,000 (16B12, Abcam, ab130275); an anti-Caspase-1 mouse antibody 1:1,000 (Casper-1, AdipoGen Life Sciences, AG-20B-0042-C100); an anti-mouse IL-1β/IL-1F2 antibody 0.25 μg ml−1 (R&D Systems, AF-401-NA); an anti-Cleaved Caspase-3 (Asp175) antibody 1:1,000 (Cell Signaling Technology, 9661S); an anti-MLKL (phospho S345) antibody 1:1,000 (EPR9515(2), Abcam, ab196436); an anti-LDH rabbit monoclonal antibody 1:1,000–1:5,000 (Beyotime Biotechnology, AF1660); Caspase-11 (17D9) Rat mAb 1:1,000 (Cell Signaling Technology, 14340); a goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 555 4 μg ml−1 (Invitrogen, A-21428); a goat anti-mouse IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor Plus 488 1–10 μg ml−1 (Invitrogen, A32723); an anti-Flag M2 antibody 1:200 (Monoclonal (M2), Sigma-Aldrich, F4049); eIF2α Rabbit pAb 1:500–1:1,000 (ABclonal, A0764); TIA1 Rabbit pAb 1:500–1:2,000 (ABclonal, A12517); eIF4A1 Rabbit pAb 1:500–1:2,000 (ABclonal, A5294); G3BP1 Rabbit mAb 1:500–1:2,000 (ABclonal, A3968); G3BP2 Rabbit pAb 1:500–1:2,000 (ABclonal, A6026); p-eIF2α (Ser51) (D9G8) XP Rabbit mAb 1:1,000 (Cell Signaling Technology, 3398); an anti-actin antibody 1:5,000 (Sigma-Aldrich, A2066); Anti-Prosurfactant protein C 1 μg ml−1 (Abcam, ab90716) and Hsp90α/β 1:100–1:1,000 (H-114, Santa Cruz Biotechnology, sc-7947).
4,6-diamidino-2-phenylindole (DAPI) for nucleic acid staining (Sigma-Aldrich, D9542–5MG), Z-VAD (Selleck Chemicals, S7023), puromycin (Invitrogen, A1113803), LPS (Sigma-Aldrich, L2630), the Pierce BCA Protein Assay Kit for protein quantification (Thermo Scientific, 23227), GSK2606414 (MedChemExpress, HY-18072), sodium arsenite (Sigma-Aldrich, S7400) and the cysteine protease inhibitor E-64 (Sigma-Aldrich, E3132) were obtained. The following ELISA kits were used for cytokine detection: Human IL-33 Quantikine (R&D Systems, D3300B), Mouse IL-33 DuoSet (R&D Systems, DY3626), Mouse IL-1β/IL-1F2 DuoSet (R&D Systems, Y401–05) and Mouse IL-6 DuoSet (R&D Systems, DY406). The KOD -Plus- Mutagenesis Kit (TOYOBO, SMK-101) was used to create GSDMD constructs with different point mutations. The Necroptosis Inducer Kit with TSZ and the Apoptosis Inducer Kit (TNF-α + SM-164) (Beyotime Biotechnology, C1058S and C0006S) were used, as well as nuclear and cytoplasmic protein extraction kits (Beyotime Biotechnology, P0027; Merck Millipore, 539790).
Complementary DNA for human GSDMD (HG25207-UT), human IL-33 (HG10368-CY) and mouse IL-33 (MG50118-CY) were purchased from Sino Biological. Mouse Gsdmd was amplified from reverse-transcribed cDNA derived from BMMs. A lentiviral vector encoding a doxycycline-inducible GFP-fusion Flag-tagged GSDMD construct was modified in a Lenti-iCas9-neo vector (Addgene, 85400). The improved lentiviral packaging plasmid was a gift from J. Zhong (Institut Pasteur of Shanghai).
Quantitative real-time PCR.
Total RNA was extracted from tissues and cells using TRIzol (Invitrogen). The purified RNA was quantified and reverse-transcribed using the ReverTraAce qPCR RT Kit (TOYOBO). The expression levels of mRNA transcripts were calculated relative to the expression level of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the 2−ΔΔCT method.
Flow cytometry.
Lung cell suspensions were obtained by digesting the lungs with 0.5 mg ml−1 collagenase I for 60 min at 37 °C and mechanical dissociation through a 70-μm cell strainer. Red blood cells were lysed after digestion by applying ammonium–chloride–potassium (ACK) lysis buffer for 2 min. Leukocytes were obtained by density gradient centrifugation with a 40:80% Percoll (GE Healthcare) gradient. Cells in the lungs and BALF were counted with a hemocytometer. Single-cell suspensions were stained with combinations of the following antibodies: anti-CD4 (clone GK1.5, eBioscience), anti-SiglecF (clone E50–2440, BD Biosciences), anti-CD45R (clone RA3–6B2, BD Biosciences), anti-NK1.1 (clone PK136, eBioscience), anti-CD3 (clone 2C11, eBioscience), anti-CD11b (clone M1/70, eBioscience), anti-Ter119 (clone Ter119, eBioscience), anti-Ly-6G (clone RB6–8C5, eBioscience), anti-FcεR1 (clone MAR-1, eBioscience), anti-CD11c (clone N418, eBioscience), anti-CD90.2 (clone 53–2.1, BD Biosciences), anti-CD127 (clone A7R34, eBioscience) and anti-IL-33R (T1/ST2, clone DJ8, MD Biosciences). Eosinophils were characterized with the markers SiglecF and CD11c using the antibodies identified above. ILC2s were identified as lineage markers (Lin−: TCRγ/δ−CD45R−NK1.1−CD3−CD11b−CD11c−Ter119−Ly6G−FcεR1−Gr1−) CD90.2+ST2+ cells. Single-cell suspensions were incubated with unlabeled, purified anti-Fc receptor blocking antibodies (anti-CD16/CD32) before staining with fluorochrome-conjugated antibodies. Dead cells were excluded by using the fixable viability dye eFluor 780 (eBioscience). Samples were acquired on a CytoFLEX flow cytometer (Beckman Coulter), and the data were analyzed with FlowJo v10 (BD Biosciences).
Live-cell imaging.
A549-IL-33-HA epithelial cells were seeded in four-chamber dishes (Cellvis, D35C4–20-1.5-N), and live-cell imaging experiments were performed 12 h later. For Hoechst colocalization experiments, cells were pretreated with Hoechst 33342 DNA dye (1 μg ml−1) for 10 min prior to imaging in DMEM (Gibco, 31053028) supplemented with 10% FBS using a Nikon A1R LUN-V inverted confocal microscope with a ×60/1.27NA water objective.
Human samples.
Clinical information is summarized in Extended Data Fig. 5 (accession no. PJ2019–015).
Tissue collection.
All tissues were collected after subjects provided informed consent, with the approval of tissue-specific protocols by the Medical Ethical Committee of the Academic Medical Centre, Affiliated Hospital, Institute of Respiratory Diseases, Guangdong Medical College, Zhanjiang, China. Inflamed lung tissue samples were obtained from people with asthma. Uninflamed control lung tissue samples were obtained from adult patients undergoing lung tumor surgery or lung transplantation surgery; tissue samples were obtained at an appropriate distance from the tumor. BALF samples were obtained from patients undergoing bronchoscopy for diagnostic purposes.
Statistical analysis.
Statistical significance was analyzed as described in the figure legends, and statistical analyses were performed using GraphPad Prism 6.
Extended Data
Extended Data Fig. 1 |. IL-33 is mainly expressed in airway epithelium cells both in humans and mice.
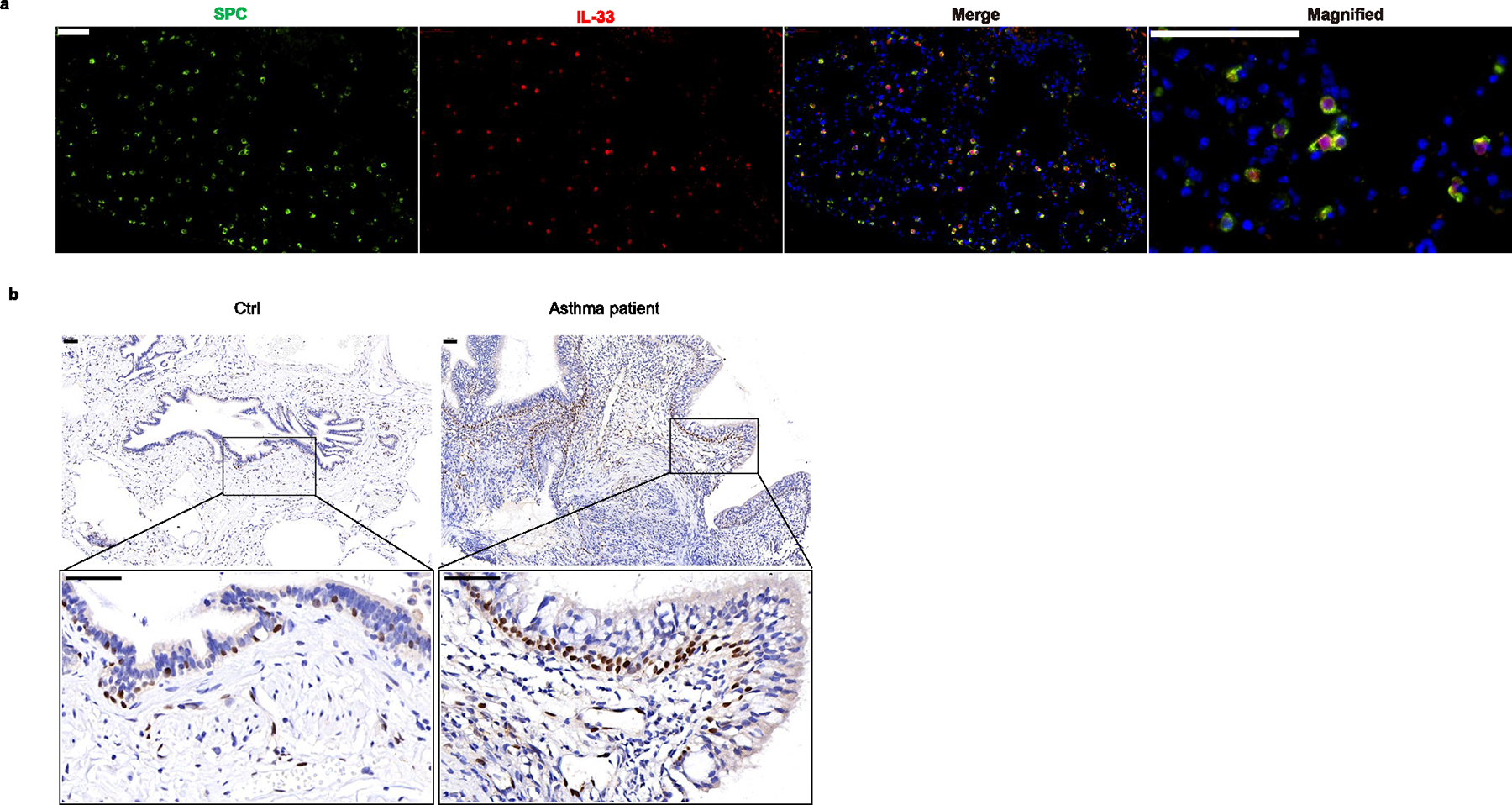
a, Microscopy of the immunofluorescent (IF) stained SPC (green) expressing AT2 cells and IL-33(red) expressing cells in lung tissue from WT Balb/c mice without any stimulations. b, Representative microscopy of the immunohistochemically (IHC) stained IL-33 (brown) in airway epithelium respective in inflamed asthma patients and non–inflamed lung tissues. Scale bars 50 μm. Data are representative of two independent experiments.
Extended Data Fig. 2 |. Construction of IL-33 and Gsdmd constitutively expressive airway epithelial cells.
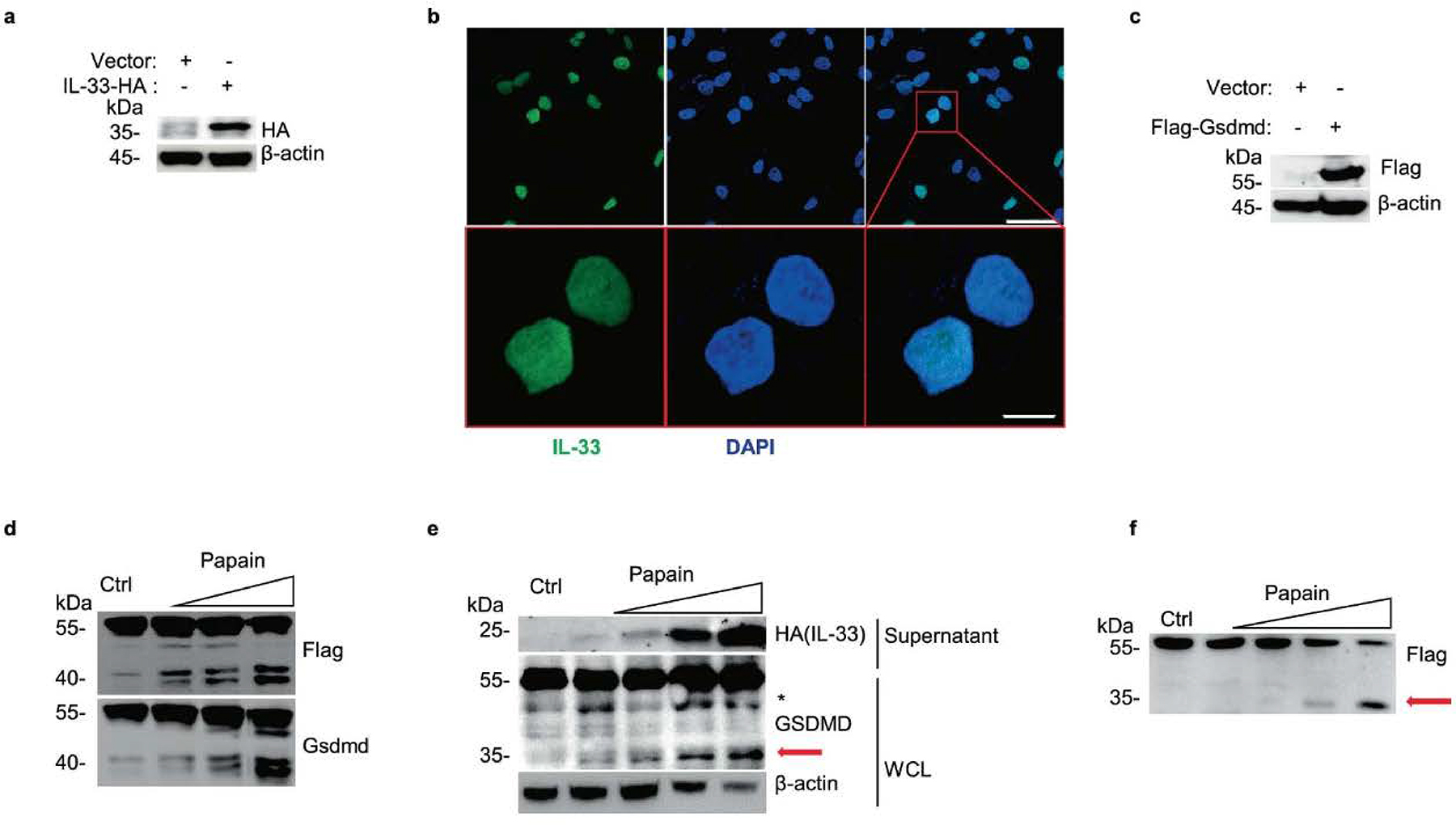
a, Immunoblot analysis of whole-cell lysis (WCL) of constructed C-terminal HA-tagged IL-33 expressing A549 with Lenti-virus overexpression. b, Microscopy of the immunofluorescence stained IL-33 (green) and DAPI (blue) in A549-IL-33 cells without stimulations. Scale bars (50 μm upper, 10 μm below). c, Immunoblot analysis of constructed N-terminal Flag-tagged Gsdmd expressing MLE-12. d, Immunoblot analysis of MLE-12-flag-Gsdmd cells treated with papain (0 μg, 5 μg, 10 μg and 50 μg per well) for 30 min. e, Immunoblot analysis of WCL and culture supernatants of constructed IL-33 expressing A549 after treatment with papain (0 μg, 1 μg, 5 μg and 10 μg) for 30 min. f, Immunoblot analysis of WCL of N-terminal Flag-tagged GSDMD expressing A549 cells exposed to papain (0 μg, 5 μg, 10 μg and 50 μg) for 30 min. The red arrow marked fragment represents the activated form of p35 NT-GSDMD. Asterisk marked nonspecific fragment were not discussed in our work. Data are representative of two independent experiments with similar results.
Extended Data Fig. 3 |. Stimulators that do not contribute to Gsdmd fragmentation and IL-33 release.
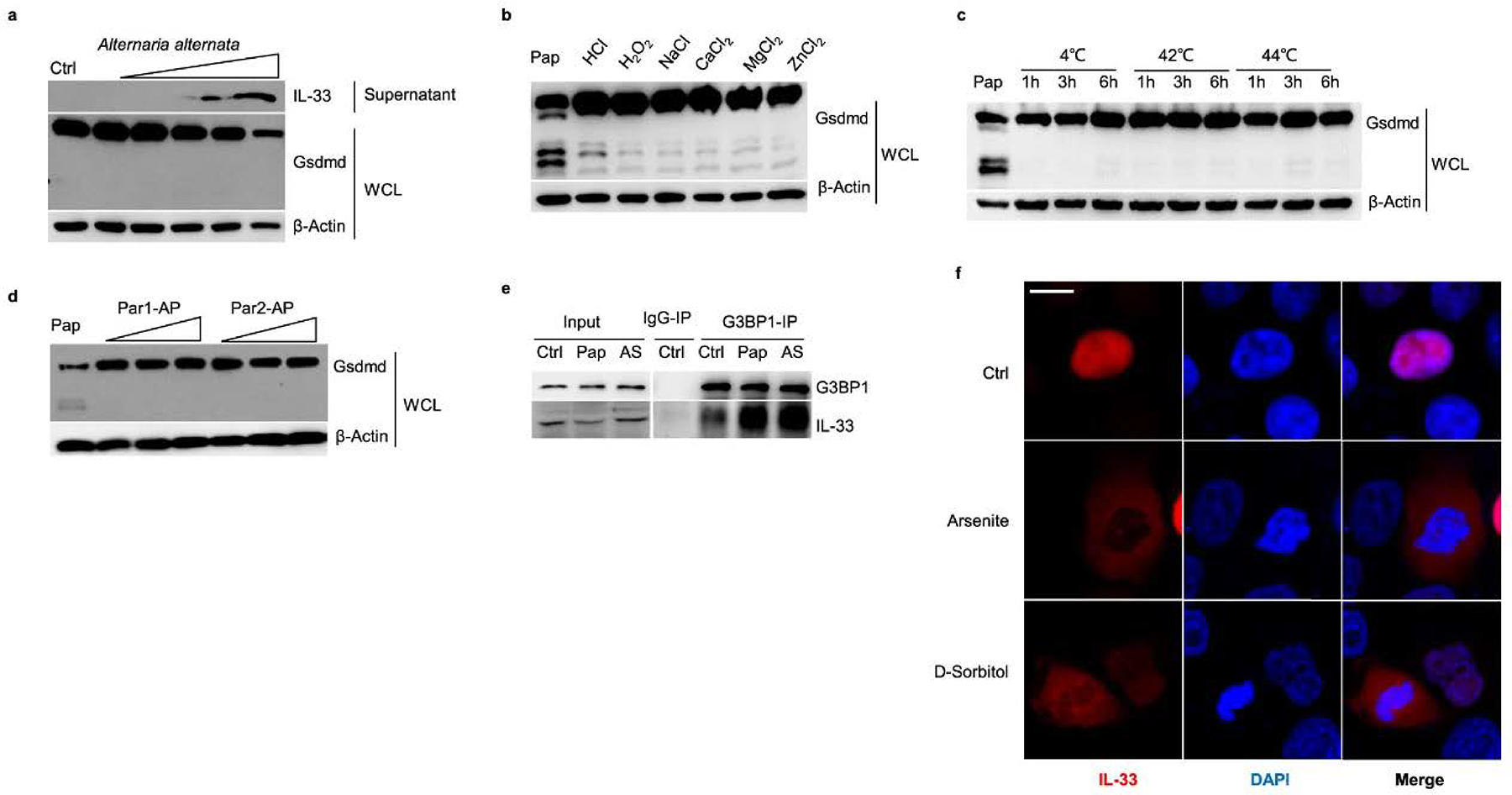
a, Immunoblot analysis of whole-cell lysis (WCL) and culture supernatants of MLE-12 cells treated with A. alternata (0.1, 1, 5, 10, 50 μg/well) for 30 min. b, Immunoblot analysis of WCL of MLE-12 cells treated with Ctrl (without stimulation); Pap (10 μg/well papain) for 30 min; 500 mM HCl, 500 mM H2O2, 5 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, and 5 mM ZnCl2 for 1 h. c, Immunoblot analysis of WCL from MLE-12 cells treated with Pap (10 μg/well papain for 30 min), and the temperatures of 4, 42 and 44 °C for 1–6 h. d, Immunoblot analysis of WCL of MLE-12 cells treated with Ctrl (without stimulation); Pap (10 μg/well papain for 30 min); agonist for PAR-1 (TFLLR-NH2 trifluoroacetate salt, 0.1–50 μg/mL for 1 h), and agonist for PAR-2 (2-furoyl-LIGRLO-amide, 0.1–50 μg/mL for 1 h). e, Immunoblot analysis of G3BP1-immunoprecipitation samples from MLE-12 cells treated with Ctrl (without stimulations), Pap (10 μg/mL papain for 30 min), and AS (0.5 mM arsenite for 1 h). f, Immunofluorescent analyses of IL-33 from HEK293T cells with C-terminal HA-tagged IL-33 expression following the treatment of arsenite (0.5 mM) and d-sorbitol (0.5 mM) for 1 h. Data are representative of two independent experiments with similar results.
Extended Data Fig. 4 |. Stress granule assembly is involved in IL-33 transportation under allergen protease stimulation.
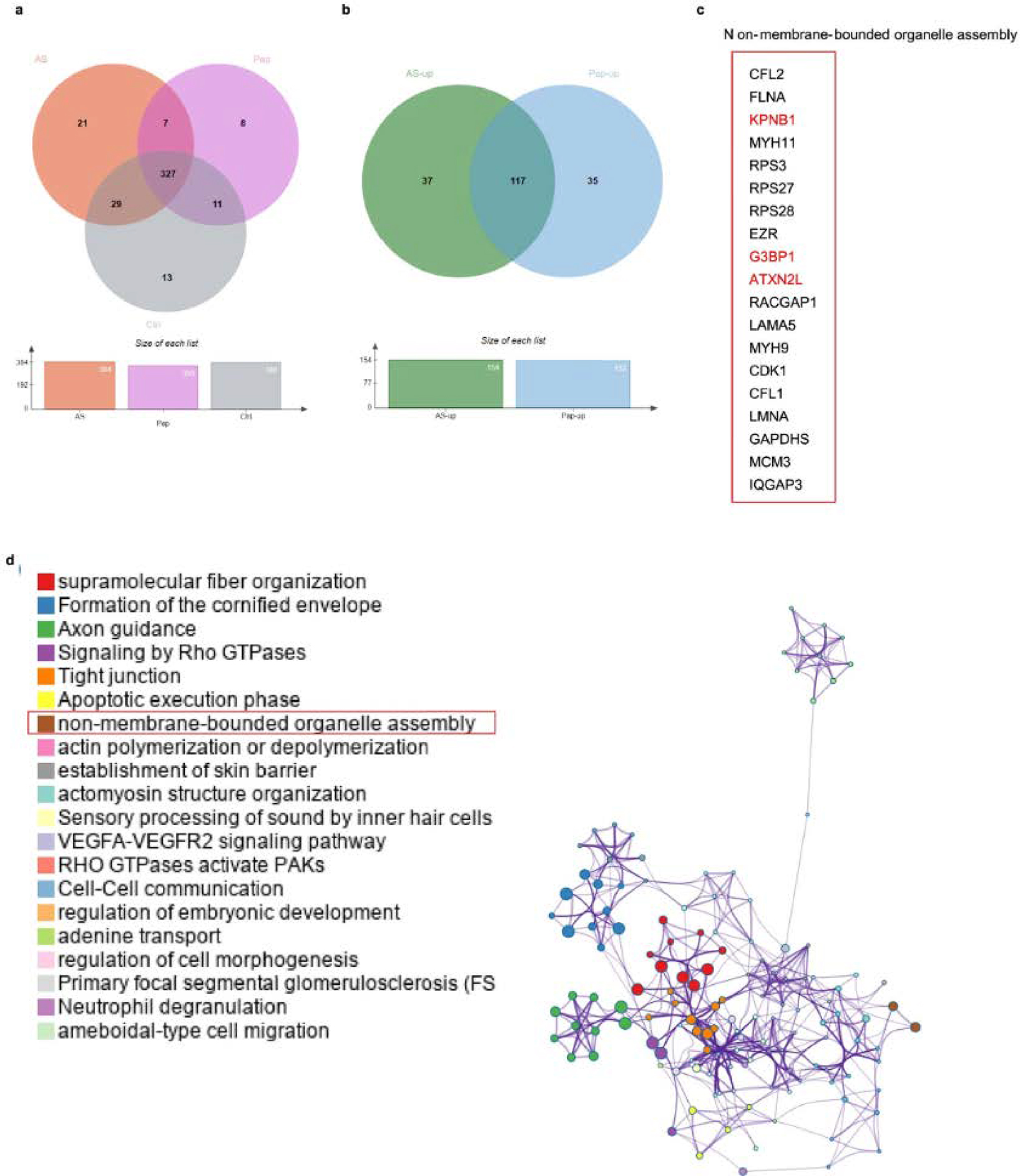
a, Venn diagrams of mass spectrometry analyzed IL-33 interactome from HEK293T cells expressed with C-terminal HA-tagged human IL-33 following the treatment of Ctrl (without stimulations), Pap (10 μg/mL papain for 30 min), and AS (0.5 mM arsenite exposure for 1 h). b, Venn diagrams of proteins with up-regulated interaction with IL-33 under papain and AS stimulation as in a. c, Common proteins associated with non-membrane-bounded organelle assembly were observed with up-regulated interaction with IL-33 as in a. d, Metascape analyzed protein clusters enriched with IL-33 under papain (10 μg/mL for 30 min) exposure context.
Extended Data Fig. 5 |. Stress granule assembly-associated stimulators do not contribute to Gsdmd fragmentation and IL-33 release.
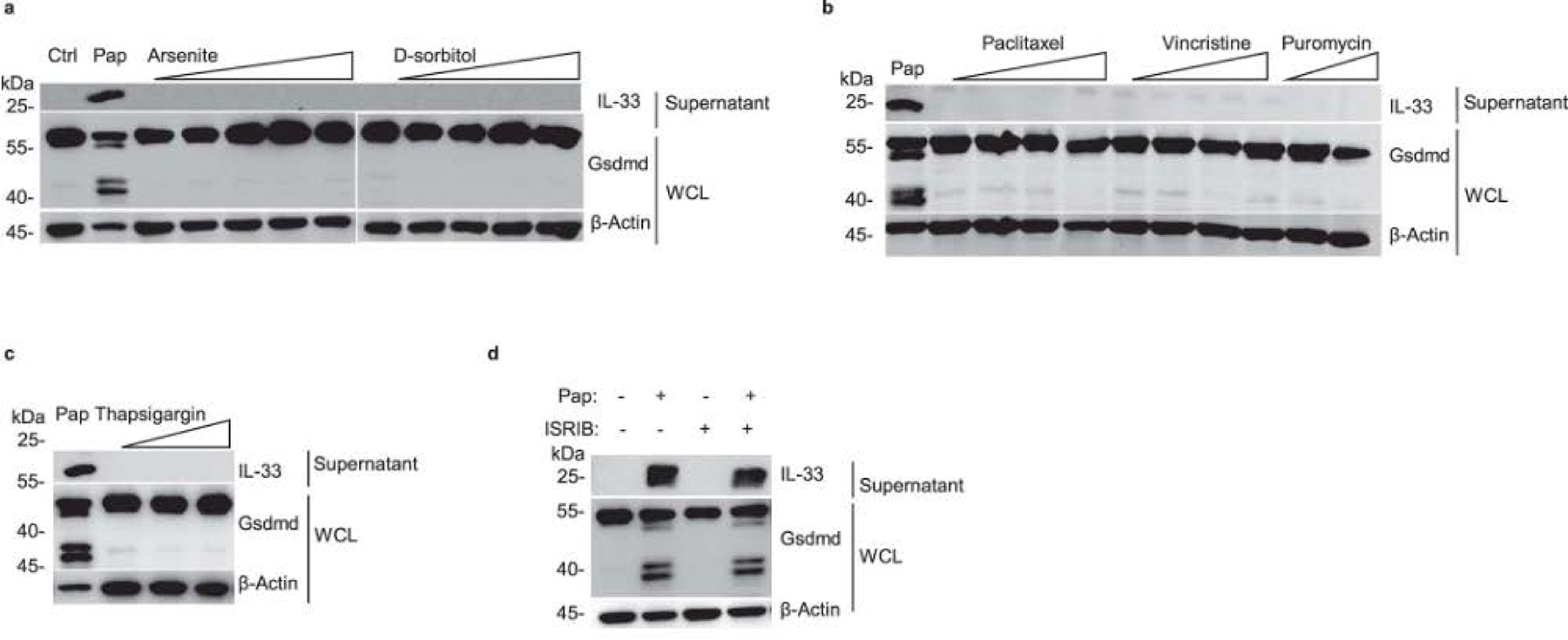
a–c, Immunoblot analysis of whole-cell lysis (WCL) and culture supernatants of MLE-12 cells treated with following stimulants for 2 h, a arsenite: 0.05, 0.5,1, 5, 50 mM; d-sorbitol: 0.01, 0.05, 0.1, 0.5 mM; b paclitaxel: 0.1, 1, 5, 50 M; vincristine: 0.1, 1, 5, 50 μM; puromycin: 1, 20 μg/mL; c thapsigargin: 0.1, 1, 10 μg/mL. d, Immunoblot analysis of whole-cell lysis (WCL) and culture supernatants of MLE-12 cells with a pre-treatment of ISRIB (10 μM) or 30 min following with the stimulation of 20 μg/mL papain. Data are representative of two independent experiments with similar results.
Extended Data Fig. 6 |. Predicted propeptide cleavage sites of mGsdmd with ProP-1.0.

Amino acid sequence of murine Gsdmd were analyzed with pro-protein converting prediction online tools ProP-1.0 for predicting of possible cleave sites (https://services.healthtech.dtu.dk/service.php?ProP-1.0). Predicted cleave site with different cleaving score were presented in the table. Sites in black dashed box were those possible for predicted p40 fragment, red colored sites were chosen for functional analyzing.
Extended Data Fig. 7 |. Human sample’s information.
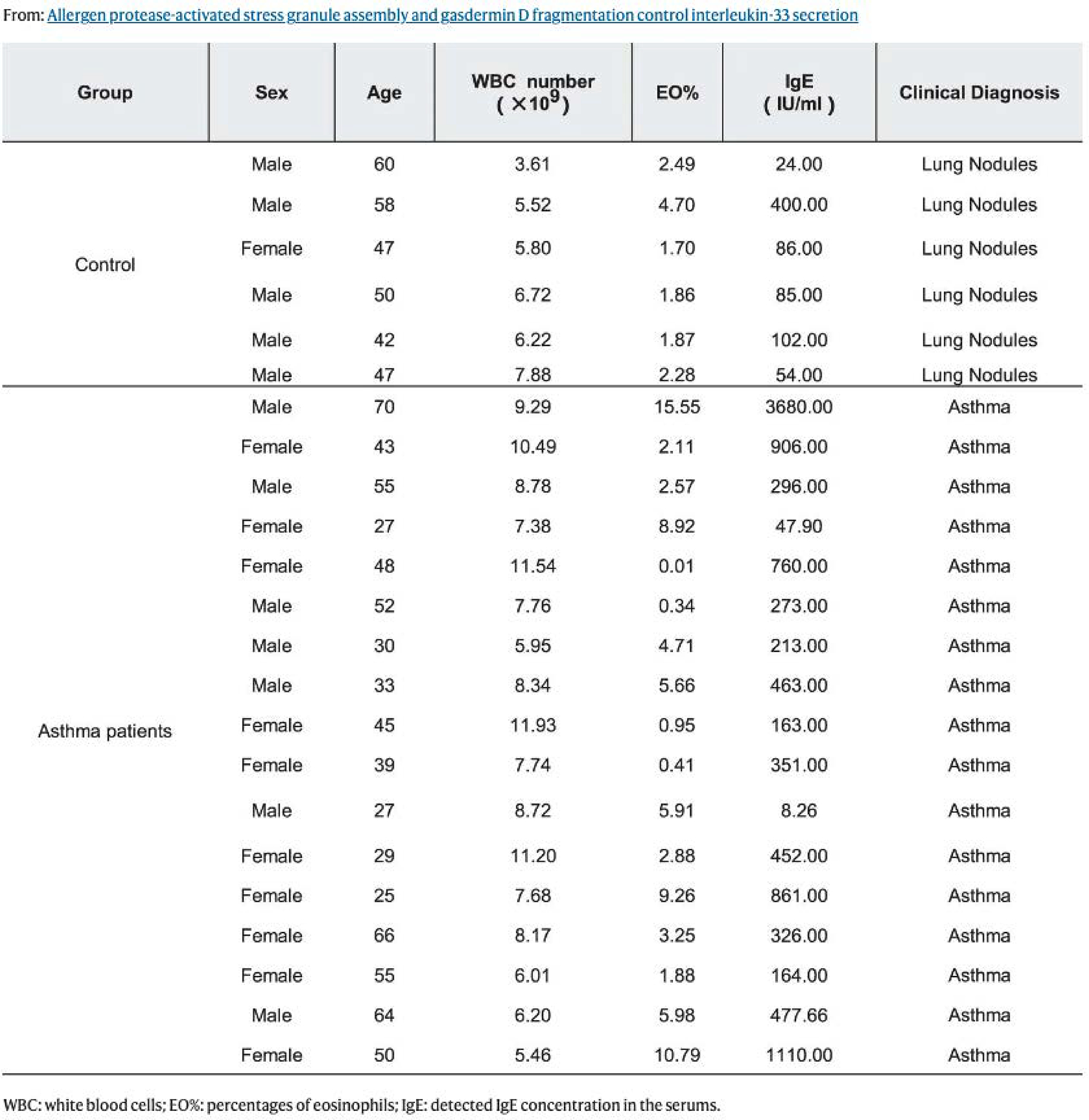
WBC: white blood cells; EO%: percentages of eosinophils; IgE: detected IgE concentration in the serums.
Extended Data Fig. 8 |. Gsdmd deficiency does not interfere with IL-33/St2 downstream events in mice.
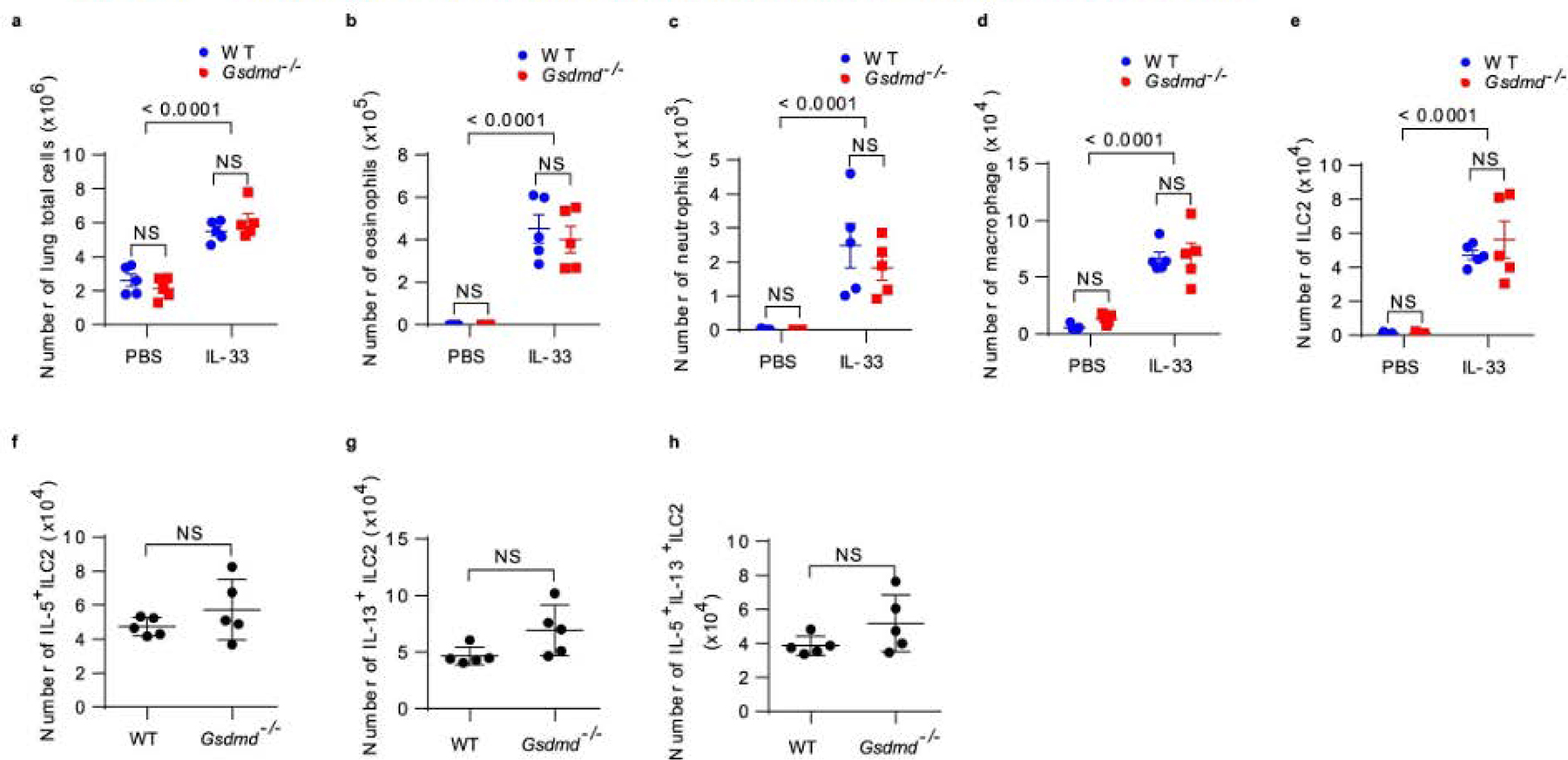
a, Flow cytometry analyzed cell numbers of lung total from wide-type and Gsdmd−/− mice treated with recombinant murine IL-33 (or PBS for the control group) for continuous 4 days. b, Flow cytometry analyzed SiglecF+CD11c− eosinophils from BAL fluids as in a. c, Flow cytometry analyzed SiglecF-CD11c−CD11b+ neutrophils from BAL fluids as in a. d, Flow cytometry analyzed SiglecF+CD11c+ macrophages from BAL fluids as in a. e, Flow cytometry analysis of Lin−CD90.2+ST2+ ILC2 from mice lungs as in a. Flow cytometry analyzed cell numbers of IL-5+ ILC2 (f), IL-13+ ILC2 (g) and IL-5+ IL-13+ ILC2 (h) in mice lungs as in a. Results are depicted as means ± SEM, n = 5 using the two-tailed, Mann–Whitney test, and Dunnett’s multiple comparisons test. Data are representative of two independent experiments with similar results.
Extended Data Fig. 9 |. Flow-gating strategies of mice eosinophils and ILC2 cells in mice.
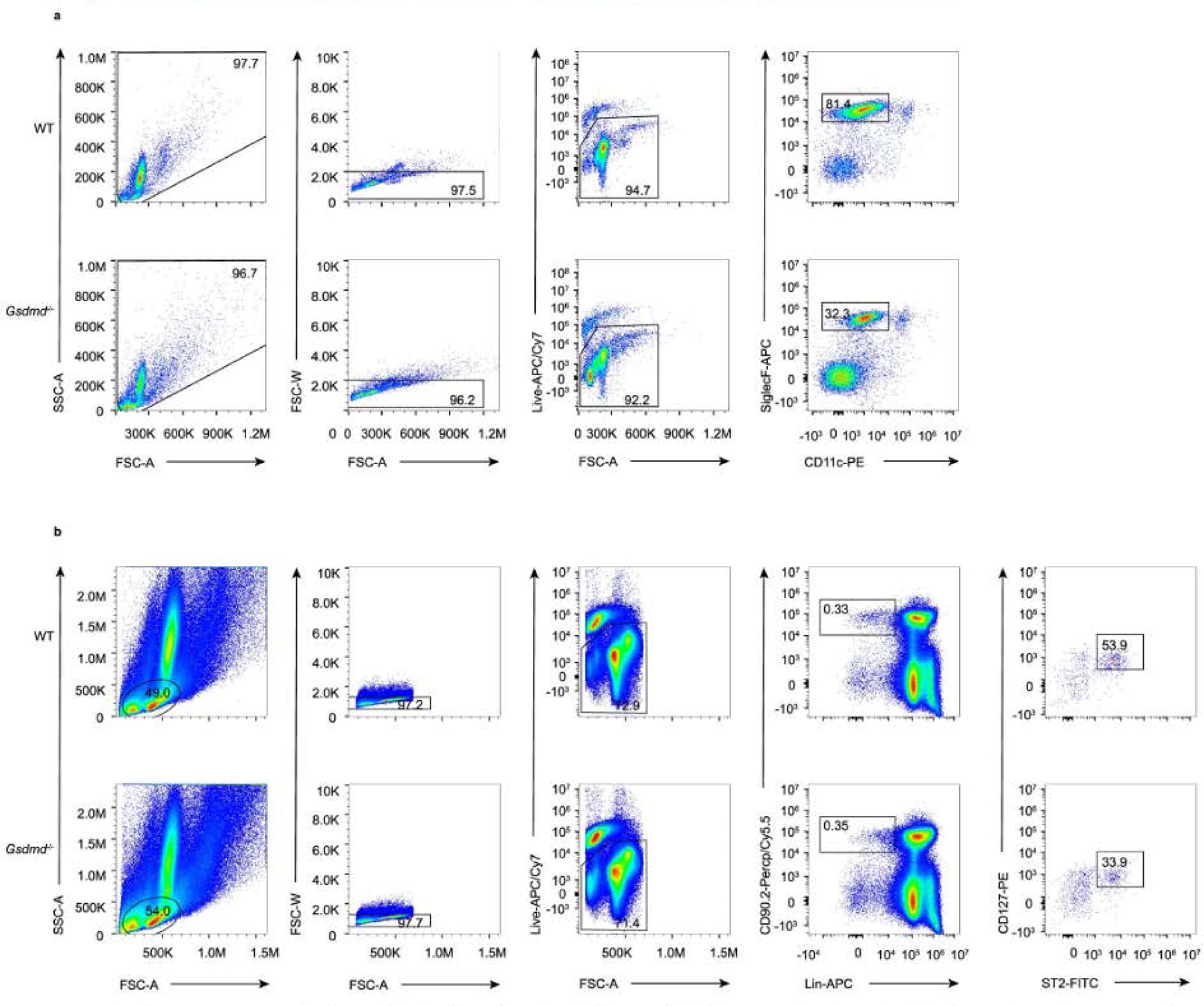
a, Representative flow cytometry gated scheme of Siglec F+CD11c+ eosinophils in BAL fluid of WT C57BL/6 mice and Gsdmd−/− mice. b, Gating strategy of Lin−CD90.2+ST2+ ILC2 in the lungs.
Supplementary Material
Acknowledgements
We thank F. Shao (National Institute of Biological Sciences, China) for helpful suggestions on the study and for providing Gsdmd-deficient mice and cell lines. This work was supported by the Ministry of Science and Technology of China (2018YFA0507402), the National Natural Science Foundation of China (81761128009 and 32000667), Shanghai Science and Technology Innovation Action (21ZR1470600), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2022264), the Affiliated Hospital of Guangdong Medical University ‘Clinical Medicine+’ CnTech Co-operation Project (CLP2021B001 and CLP2021B017), the Discipline Construction Project of Guangdong Medical University (4SG21231G), the Project of Zhanjiang City (2018A01025, 2020A01016 and 2021A05052) and the Natural Science Foundation of Guangdong Province, China (2021A1515011062 and 2022A1515011731).
Footnotes
Competing interests
The authors declare no competing interests.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended data is available for this paper at https://doi.org/10.1038/s41590-022-01255-6.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41590-022-01255-6.
Peer review information Nature Immunology thanks Hirohito Kita and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ioana Visan, in collaboration with the Nature Immunology team.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD033460. Source data are provided with this paper.
References
- 1.Dai X et al. House dust mite allergens induce interleukin 33 (IL-33) synthesis and release from keratinocytes via ATP-mediated extracellular signaling. Biochim. Biophys. Acta, Mol. Basis Dis. 1866, 165719 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Alvarez MJ et al. Effect of current exposure to Der p 1 on asthma symptoms, airway inflammation, and bronchial hyperresponsiveness in mite-allergic asthmatics. Allergy 55, 185–190 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Namvar S et al. Aspergillus fumigatus proteases, Asp f 5 and Asp f 13, are essential for airway inflammation and remodelling in a murine inhalation model. Clin. Exp. Allergy 45, 982–993 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Florsheim E et al. Integrated innate mechanisms involved in airway allergic inflammation to the serine protease subtilisin. J. Immunol. 194, 4621–4630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorski SA, Hahn YS & Braciale TJ Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. 9, e1003615 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatzioannou A et al. An intrinsic role of IL-33 in Treg cell–mediated tumor immunoevasion. Nat. Immunol. 21, 75–85 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamkanfi M & Dixit VM IL-33 raises alarm. Immunity 31, 5–7 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Garlanda C, Dinarello CA & Mantovani A The interleukin-1 family: back to the future. Immunity 39, 1003–1018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Travers J et al. Chromatin regulates IL-33 release and extracellular cytokine activity. Nat. Commun. 9, 3244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lüthi AU et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31, 84–98 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi S et al. Tumor-initiating cells establish an IL-33–TGF-β niche signaling loop to promote cancer progression. Science 369, eaay1813 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaczmarek A, Vandenabeele P & Krysko DV Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Hara K et al. Airway uric acid is a sensor of inhaled protease allergens and initiates type 2 immune responses in respiratory mucosa. J. Immunol. 192, 4032–4042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kakkar R, Hei H, Dobner S & Lee RT Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 287, 6941–6948 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Protter DSW & Parker R Principles and properties of stress granules. Trends Cell Biol. 26, 668–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samir P et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolozin B & Ivanov P Stress granules and neurodegeneration. Nat. Rev. Neurosci. 20, 649–666 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K et al. Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173, 958–971.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Raab M et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Nabe T et al. Production of interleukin (IL)-33 in the lungs during multiple antigen challenge-induced airway inflammation in mice, and its modulation by a glucocorticoid. Eur. J. Pharmacol. 757, 34–41 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Liu Z et al. Crystal structures of the full-length murine and human gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity 51, 43–49.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang P et al. G3BP1 Is a tunable switch that triggers phase separation to assemble stress granules. Cell 181, 325–345.e28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szaflarski W et al. Vinca alkaloid drugs promote stress-induced translational repression and stress granule formation. Oncotarget 7, 30307–30322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wheeler JR, Jain S, Khong A & Parker R Isolation of yeast and mammalian stress granule cores. Methods 126, 12–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Advani VM & Ivanov P Stress granule subtypes: an emerging link to neurodegeneration. Cell. Mol. Life Sci. 77, 4827–4845 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma J et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tauber D et al. Modulation of RNA condensation by the DEAD-box protein eIF4A. Cell 180, 411–426.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sidrauski C, McGeachy AM, Ingolia NT & Walter P The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife 4, e05033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel A et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Carayol N et al. Suppression of programmed cell death 4 (PDCD4) protein expression by BCR-ABL-regulated engagement of the mTOR/p70 S6 kinase pathway. J. Biol. Chem. 283, 8601–8610 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilmore S et al. Targeted inhibition of eIF4A suppresses B-cell receptor-induced translation and expression of MYC and MCL1 in chronic lymphocytic leukemia cells. Cell. Mol. Life Sci. 78, 6337–6349 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho JJD et al. Proteomics reveal cap-dependent translation inhibitors remodel the translation machinery and translatome. Cell Rep. 37, 109806 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duckert P, Brunak S & Blom N Prediction of proprotein convertase cleavage sites. Protein Eng., Des. Sel. 17, 107–112 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Barnes PJ Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 18, 454–466 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Wheeler JR, Matheny T, Jain S, Abrisch R & Parker R Distinct stages in stress granule assembly and disassembly. eLife 5, e18413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu T et al. NOD-like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J. Allergy Clin. Immunol. 144, 777–787.e9 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Sollberger G et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 3, eaar6689 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Brusilovsky M et al. Environmental allergens trigger type 2 inflammation through ripoptosome activation. Nat. Immunol. 22, 1316–1326 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Souza CO et al. Extracellular ATP induces cell death in human intestinal epithelial cells. Biochim. Biophys. Acta, Gen. Subj. 1820, 1867–1878 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Srisomboon Y, Squillace DL, Maniak PJ, Kita H & O’Grady SM Fungal allergen-induced IL-33 secretion involves cholesterol-dependent, VDAC-1-mediated ATP release from the airway epithelium. J. Physiol. 598, 1829–1845 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD033460. Source data are provided with this paper.