

Abstract

Despite the record-breaking discovery, development and approval of vaccines and antiviral therapeutics such as Paxlovid, coronavirus disease 2019 (COVID-19) remained the fourth leading cause of death in the world and third highest in the United States in 2022. Here, we report the discovery and characterization of PF-07817883, a second-generation, orally bioavailable, SARS-CoV-2 main protease inhibitor with improved metabolic stability versus nirmatrelvir, the antiviral component of the ritonavir-boosted therapy Paxlovid. We demonstrate the in vitro pan-human coronavirus antiviral activity and off-target selectivity profile of PF-07817883. PF-07817883 also demonstrated oral efficacy in a mouse-adapted SARS-CoV-2 model at plasma concentrations equivalent to nirmatrelvir. The preclinical in vivo pharmacokinetics and metabolism studies in human matrices are suggestive of improved oral pharmacokinetics for PF-07817883 in humans, relative to nirmatrelvir. In vitro inhibition/induction studies against major human drug metabolizing enzymes/transporters suggest a low potential for perpetrator drug–drug interactions upon single-agent use of PF-07817883.
Introduction
Three significant novel human coronaviruses (SARS-CoV-1, MERS-CoV and SARS-CoV-2) have emerged in the last 20 years, in what appears to be an increasingly common occurrence.1 The impact of the SARS-CoV-2 outbreak of 2019 is devastating and ongoing, with COVID-19 now having caused almost 7 million recorded deaths globally as of December 2023.2 SARS-CoV-2 is a highly infectious, ribonucleic acid (RNA) beta coronavirus that can cause life-threatening respiratory disease in a small percentage of cases. The global, highly transmissible nature of the virus means that ongoing waves of infections continue to cause significant amounts of serious illness and death from COVID-19. The rapid discovery and development of COVID-19 vaccines and therapeutics significantly curtailed the potentially catastrophic global impact of COVID-19 disease.3 However, despite this there remains some unmet need in the treatment of COVID-19. Rapid development of viral resistance has rendered antibody therapies that target the SARS-CoV-2 spike protein ineffective.4,5 Oral antiviral therapies targeting the more conserved and less mutationally susceptible viral life-cycle proteins (i.e., the main protease (Mpro) or the RNA polymerase), such as Paxlovid (nirmatrelvir (1)/ritonavir, Figure 1)6 and molnupiravir,7 are available under various levels of authorization or approval globally. Intravenous agents such as remdesivir have also played a role as antiviral therapies in the pandemic response.8 A number of other oral antiviral agents remain under various stages of clinical investigation for COVID-19.9−11
Figure 1.
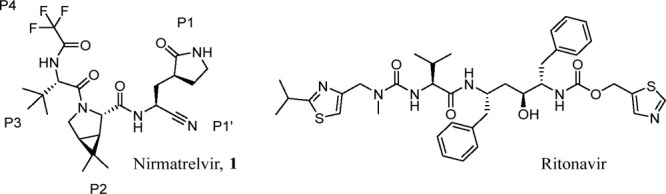
Structures of the SARS-CoV-2 Mpro inhibitor nirmatrelvir (1) and ritonavir, which is used as a booster of nirmatrelvir plasma concentrations in humans.
The SARS-CoV-2 Mpro (also known as 3C-like protease or 3CLpro) is a three-domain cysteine protease which features a Cys145-His41 catalytic dyad that cleaves the two large polyproteins first produced by the virus upon invasion of the human host cell. The clinical safety and efficacy data published for nirmatrelvir/ritonavir, the first in class Mpro inhibitor for COVID-19, shows the attractive nature of this viral target as the basis for an antiviral therapy.6 However, a small percentage of patients find themselves unable to take this drug at present, such as those who have extreme renal impairment and those whose medications are contraindicated with the ritonavir component of the therapy. Pharmacokinetic boosting of the systemic exposure of protease inhibitors via concomitant administration with inhibitors (e.g., ritonavir) of the major human constitutive CYP, i.e., CYP3A4/5 is well precedented in the clinic, since the vast majority of protease inhibitors are principally metabolized by CYP3A4/5.12 As the primary clearance pathway of 1 also involves oxidative metabolism by CYP3A4,12,13 low dose ritonavir (100 mg) was administered concomitantly with 1 (300 mg) in a twice daily dosing regimen over a 5-day dosing period, which resulted in unbound trough (Cmin) concentrations that were 5–10 times the cellular antiviral EC90,u.14 This well-tolerated level of target coverage was designed to maximize antiviral efficacy through Mpro inhibition, while minimizing the potential for drug-induced resistance in the longer term.
Our second-generation protease inhibitor work set out to discover a single agent that could achieve efficacious plasma concentrations similar to nirmatrelvir/ritonavir, but without the requirement for pharmacokinetic boosting and critically, possessed low potential for perpetrator drug–drug interactions of its own. We initially addressed a single-agent Mpro inhibitor by tackling the influential components of dose calculations: antiviral potency, metabolic clearance and oral absorption. Given the strong preclinical safety profile attained for nirmatrelvir (1), we decided to remain structurally close into this peptidomimetic chemotype.
Results
We have previously described the design and selection of P4 capping substituents (Figure 1) such as acetyl and methanesulfonyl in benzothiazole ketone and nitrile P1′ cysteine trap chemotypes and the profound effect of a P4 trifluoroacetamide on cellular antiviral activity, passive permeability and oral bioavailability (F) noted with 1.15 We also evaluated methylurea (2) and methylcarbamate (3) as alternative P4 groups (Table 1). These changes to P4 had clear effects on cellular antiviral activity and oxidative metabolic stability in human liver microsomes (HLMs), respectively, with minimal changes in the in vitro biochemical potency when compared to 1 (Table 1). Changes in in vitro passive permeability (Papp(AB)) as measured in Ralph Russ canine kidney (RRCK) cells16 appeared to be marginal for compounds 1–3 (Table 1). Consistent with the concomitant reduction in lipophilicity (governed by the shake flask LogD measurement),17 both 2 and 3 demonstrated improvements in the apparent intrinsic clearance (CLint,app) against CYP-catalyzed metabolism in human liver microsomes.13,16 However, the methyl urea 2, now containing four hydrogen bond donors, performed very poorly in the cellular antiviral assay in comparison to the methylcarbamate 3 or trifluoroacetamide 1. Given the similar biochemical activity but disparate antiviral activity for compound 2, it appeared that hydrogen bond count outweighed any marginal difference in RRCK Papp(AB) for these low permeability compounds with respect to the cell permeability component of the antiviral end point. From this point on, only methylcarbamates and trifluoroacetamides were pursued as P4 capping group options, as further changes were explored elsewhere in the peptidomimetic pharmacophore, seeking additional improvements in cellular antiviral potency and oxidative metabolic stability.
Table 1. Single-Point, P4 Cap Changes to Nirmatrelvir (1) to Investigate the Effect of a Methyl Urea or Carbamate on Biochemical Potency, Cellular Antiviral Activity, Passive Permeability, HLM Stability and LogD.

Compounds were tested for their ability to inhibit proteolytic activity of SARS-CoV-2 Mpro in a FRET assay. Compounds were tested up to 30 μM.
Compounds were tested for inhibition of SARS-CoV-2 induced cytopathic effect (CPE) as measured by cell viability using ATP as an end point in Vero E6 cells enriched for ACE2 receptor expression. A P-glycoprotein inhibitor, CP-100356 (EI), was added at 2 μM to inhibit the efflux of compounds from Vero E6 cells.
Absorptive passive permeability from apical to basolateral direction (Papp(AB)) was examined in the RRCK assay with a 30 min preincubation time and is reported as mean and standard deviation.16
CLint,app refers to total apparent intrinsic clearance obtained from scaling of in vitro half-lives in NADPH-supplemented human liver microsomes (HLM) (30 min incubations at 37 °C) and is reported as mean and standard deviation.18
LogD was measured at pH 7.4 using the previously described shake-flask method.17 All values reported are geometric mean values, with 95% confidence interval (CI) values and replicate numbers in parentheses.
As part of structure–activity relationship (SAR) explorations for a next-generation oral SARS-CoV-2 agent, we initially focused our attention on modifications to the P2 3.1.0 proline group from compound 1. This was primarily in an effort to improve metabolic stability by lowering the overall lipophilicity of compound 1 and mitigate the CYP3A4-mediated oxidation of the pendant geminal dimethyl group on the P2 3.1.0 proline motif in 1 that was noted in HLM.13Table 2 shows a matrix of three P2 prolines selected as 3.1.0 proline alternatives, while keeping P1′, P1 and P3 fixed and methylcarbamate and trifluoroacetamide as the only two variables in the P4 capping group (compounds 4–9). Regardless of the P4 capping group used, all three selections of alternate P2 fluoroalkoxy- or trifluoromethyl-substituted prolines furnished compounds of lower lipophilicity relative to 1 (LogD 1.9). All synthesized analogs contained three hydrogen bond donors and were biochemically potent against Mpro, which correlated with potency in the cellular antiviral assay. The only significant loss in biochemical and cellular potency was seen in compound 11 where a P3 isopropyl group, in combination with the P1 lactam, reduced activity. Of the proline substituents explored in P2, −OCHF2 (compounds 6 and 7) showed a trend for slightly weaker activity. Overall, this meant that with the relatively narrow spread in antiviral activity in the SAR explored, compounds were more likely going to be differentiated by their drug metabolism and solid form properties when it came to candidate selection. Relative to 1, all three P2 alternates (compounds 4–9) demonstrated reduced metabolic CLint,app values, which approached the lower limit of the high-throughput HLM stability assay. Substitution of the pyrrolidinone ring in 9 with a piperidinone ring (compound 10) led to measurable CLint,app in the HLM stability screen, whereas a P3 switch from tert-butyl to isopropyl (compound 11) resulted in loss of cellular antiviral potency. Compound 12, which featured the 6-membered piperidinone ring at P1 and an isopropyl substituent at P3, demonstrated a good balance of CLint,app and cellular antiviral activity.
Table 2. In Vitro Assessment of Biochemical Mpro Potency, Cellular Antiviral Activity, Passive Permeability, HLM Stability and LogD for P1, P2, P3 and P4 Changes to 1.

Compounds were tested for their ability to inhibit proteolytic activity of SARS-CoV-2 Mpro in a FRET assay. Compounds were tested up to 30 μM.
Compounds were tested for inhibition of SARS-CoV-2 induced cytopathic effect (CPE) as measured by cell viability using ATP as an end point in Vero E6 cells enriched for ACE2 receptor expression. A P-glycoprotein inhibitor, CP-100356 (EI), was added at 2 μM to inhibit the efflux of compounds from Vero E6 cells.
Absorptive passive permeability from apical to basolateral direction (Papp(AB)) was examined in the RRCK assay with a 30 min preincubation time and is reported as mean and standard deviation.16
CLint,app refers to total apparent intrinsic clearance obtained from scaling of in vitro half-lives in NADPH-supplemented human liver microsomes (30 min incubations at 37 °C) and is reported as mean and standard deviation.18
LogD was measured at pH 7.4 using the previously described shake-flask method.17
Based on the SAR analysis, compounds 5, 6, 9 and 12 were progressed into rat pharmacokinetic studies to gauge oral absorption characteristics of these relatively low permeability peptidomimetics. The pharmacokinetic parameters of compounds 5, 6 and 12 in rats after intravenous (i.v.) and oral (p.o.) administration are described in Table 3. Following i.v. administration, all three compounds demonstrated moderate plasma clearance (CLp) (25–35 mL/min/kg) and moderate steady state distribution volumes (Vd,ss) (0.55–1.05 L/kg), which resulted in terminal half-lives (t1/2) ranging from 1.9–6.0 h. Administration of amorphous 5 or 12 (10 mg/kg) or crystalline 6 (10 mg/kg) orally as a solution (compounds 5 and 12) or suspension (compound 6) in 0.5% (w/v) methyl cellulose containing 2% (v/v) Tween 80 to rats resulted in poor F (7.6–12%) and a low fraction of the oral dose absorbed (Fa × Fg) (12.2–20.2%). Oral administration (10 mg/kg) of 6 and 12 as a 50% spray dried dispersion (SDD) formulation did not improve absorption.
Table 3. Pharmacokinetics of Compounds 5, 6 and 12 in Ratsa.
| Compd | Route/Dose (mg/kg) | CLp (mL/min/kg) | Vd,ss (L/kg) | t1/2 (h) | CLrenal (mL/min/kg) | Cmax (ng/mL) | Tmax (h) | AUC0–24 (ng·h/mL) | Oral F (%) | Fa × Fgf (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 5 | i.v./1 | 25.4 (26.0, 24.9) | 0.87 (0.74, 1.0) | 6.0 (3.4, 8.5) | 8.1 (9.7, 6.5) | – | – | 650 (634, 666) | – | – |
| p.o./10b | – | – | – | – | 358 (317, 399) | 0.25 (0.25, 0.25) | 559 (519, 598) | 8.6 (8.0, 9.2) | 12.2 (11.4, 13.2) | |
| 6 | i.v./1 | 33.1 (33, 33.2) | 1.05 (0.486, 1.62) | 5.1 (0.98, 9.1) | 4.5 (6.2, 2.8) | – | – | 502 (505, 499) | – | – |
| p.o./10b,c | – | – | – | – | 272 (236, 308) | 0.50 (0.50, 0.50) | 599 (592, 605) | 11.9 (11.8, 12.1 | 20.2 (20.0, 20.4) | |
| p.o./10b,d | – | – | – | – | 92.8 (116, 69.6) | 0.75 (0.50, 1.0) | 212e (218, 206) | 4.2 (4.3, 4.1) | 7.2 (7.3, 6.9) | |
| 12 | i.v./1 | 35.6 (34.5, 36.7) | 0.55 (0.57, 0.54) | 1.9 (2.2, 1.7) | 7.6 (12.2, 2.9) | – | – | 467 (481, 453) | – | – |
| p.o./10b | – | – | – | – | 310 (203, 417) | 0.38 (0.5, 0.25) | 359 (296, 422) | 7.7 (6.3,9.0) | 14.7 (12.3, 17.6) | |
| p.o./10b,d | – | – | – | – | 177 (215, 138) | 0.38 (0.25, 0.50) | 380e (411, 349) | 8.1 (8.8, 7.5) | 15.8 (17.1, 14.5) |
Pharmacokinetic parameters were calculated from plasma concentration–time data and are reported as mean and individual values (n = 2). All pharmacokinetics studies were conducted in male Wistar–Han rats. Compounds 5 and 6 were dosed i.v. as a solution, in 10% DMSO/50% PEG400/40% water. Compound 12 was dosed i.v. as a solution in 10% (v/v) PEG400/90% (v/v) 23% (w/v) hydroxypropyl β-cyclodextrin (HPBCD) in water.
Oral pharmacokinetics studies were conducted in the fed state and using amorphous material unless otherwise indicated. Compounds were administered in 2% Tween 80 (v/v) in 0.5% (w/v) methyl cellulose in water either as a solution (compounds 5 and 12) or suspension (compound 6).
Crystalline.
50% ASD.
AUC0-∞.
The fraction of the oral dose absorbed (Fa × Fg) in rats was estimated using the following equation: Fa × Fg = F/(1 – CLblood/Q). CLp (after subtracting CLrenal) was converted into CLblood by dividing CLp by the rat blood to plasma ratio; compound 5 (0.821), compound 6 (0.998), compound 12 (0.777). Blood to plasma ratios were determined using previously described methods.13Q is hepatic blood flow (70 mL/min/kg) in the rat.
The i.v. pharmacokinetics of 9 in rats (Table 4), which resulted in a moderate CLp and Vd,ss, were comparable to the parameters noted with compounds 5, 6 and 12. However, 9 distinguished itself from the other compounds in this species with respect to its oral pharmacokinetics. Administration of crystalline 9 (7.5 mg/kg) orally as a solution in 0.5% (w/v) methyl cellulose containing 2% (v/v) Tween 80 to rats resulted in rapid oral absorption (Tmax = 0.29 h) and relative improvements in oral F (22%) and Fa × Fg (42%). Encouraged by these findings, we studied the pharmacokinetics of 9 in male cynomolgus monkeys, which was the selected nonrodent toxicology species for this program.19 Following i.v. administration, 9 demonstrated a low CLp (6.9 mL/min/kg) and a moderate Vd,ss (0.66 L/kg), resulting in an elimination t1/2 of 2.8 h in monkeys. Oral administration of crystalline 9 (10 mg/kg) as a solution in 0.5% (w/v) methyl cellulose containing 2% (v/v) Tween 80 to monkeys also resulted in rapid oral absorption (Tmax = 0.38 h) and corresponding oral F and Fa × Fg of 30% and 37%, respectively (Table 4). Examination of the metabolic stability of compounds 5, 6, 9 and 12 in NADPH-supplemented rat liver microsomes (30 min incubations at 37 °C) revealed a general resistance toward metabolic turnover (CLint,app < 5.8 μL/min/mg, predicted hepatic clearance <11 mL/min/kg). In the case of 9, the predicted hepatic clearance (8.3 mL/min/kg), derived from scaling of the in vitro CLint,app (54.6 μL/min/mg) in NADPH-supplemented monkey liver microsomes (30 min incubations at 37 °C) aligned reasonably well with the observed CLp of 6.18 mL/min/kg (after subtracting the nonmetabolic renal clearance) in this species. A similar trend was also noted in corresponding metabolic stability studies with 1 in preclinical species (rats: predicted hepatic clearance = 5.8 mL/min/kg, observed CLp = 22.7 mL/min/kg; monkeys: predicted hepatic clearance = 25 mL/min/kg, observed CLp = 15.8 mL/min/kg).13
Table 4. Pharmacokinetics of 9 in Rats and Monkeysa.
| Species | Route/Dose (mg/kg) | CLp (mL/min/kg) | Vd,ss (L/kg) | t1/2 (h) | CLrenal (mL/min/kg) | Cmax (ng/mL) | Tmax (h) | AUC0–24 (ng·h/mL) | Oral F (%) | Fa × Fgc (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Rat | i.v./1 | 34.4 (34.4, 34.4) | 1.52 (1.67, 1.36) | 10 (9.0, 11.2) | 5.52 (3.25, 7.8) | 481 (481, 481) | ||||
| p.o./7.5b | – | 606 (665, 547) | 0.29 (0.083, 0.50) | 793 (704, 882) | 21.9 (19.5, 24.3) | 41.6 (37.19, 46.59) | ||||
| Monkey | i.v./1 | 6.88 (5.46, 8.29) | 0.657(0.591, 0.723) | 2.77 (2.87, 2.67) | 0.697 (0.0878, 1.31) | – | – | 2530 (3050, 2010) | – | – |
| p.o./10b | – | – | – | – | 1510 (1650, 1360) | 0.38 (0.25, 0.5) | 7380 (8160, 6590) | 29.2 (26.0, 32.3) | 37.3 (40.8, 34.1) |
Pharmacokinetic parameters were calculated from plasma concentration–time data and are reported as mean and individual values (n = 2). All pharmacokinetics studies were conducted in male Wistar–Han rats or cynomolgus monkeys. Compound 9 was dosed i.v. as a solution in 5% (v/v) PEG400/95% (v/v) 23% (w/v) HPBCD in water to monkeys and i.v. as a solution in 10% DMSO/30% PEG400/60% water to rats. Oral pharmacokinetics studies were conducted in the fed state.
Crystalline material administered as a solution in 2% Tween 80 (v/v) in 0.5% (w/v) methyl cellulose in water.
Fa × Fg was estimated using the following equation: Fa × Fg = F/(1 – CLblood/Q). CLp (after subtracting CLrenal) was converted into CLblood by dividing CLp by the BPR of compound 9 (monkey = 0.669, rat = 0.868). Q is hepatic blood flow (monkey = 44 mL/min/kg, rat = 70 mL/min/kg).
To examine the feasibility of attaining high systemic exposures in subsequent preclinical toxicology studies, the p.o. pharmacokinetics of 9 were further evaluated in p.o. dose-range finding studies in rats (Table 5). Oral administration (100 and 1000 mg/kg) of crystalline 9 formulated as a suspension in 0.5% (w/v) methyl cellulose in water containing 2% (v/v) Tween 80 or as a 75% SDD in 1% (v/v) Soluplus and 0.5% methyl cellulose in water resulted in rapid (Tmax 0.5–2.8 h) p.o. absorption and near or greater than dose-proportional increases in Cmax and greater than dose proportional increases in AUC when comparing the p.o. doses ranging from 7.5 → 100 → 1000 mg/kg (Tables 4 and 5). Systemic exposures (governed by Cmax and AUC) of 9 administered in crystalline form or as an SDD were generally comparable.
Table 5. Comparison of Single High-Dose p.o. Pharmacokinetics of 9 and Nirmatrelvir (1) in Ratsa.
| Compd | Dose (mg/kg) | Form | Cmax (ng/mL) | Tmax (h) | AUC0–24 (ng·h/mL) |
|---|---|---|---|---|---|
| 9 | 100 | Crystallineb | 7210 ± 2880 | 0.5 ± 0 | 15000 ± 1310 |
| 75% SDDc | 2830 ± 1150 | 1 ± 0 | 13400 ± 2950 | ||
| 1000 | Crystallineb | 52700 ± 7990 | 2.8 ± 2 | 500000 ± 211000 | |
| 75% SDDc | 59200 ± 16600 | 2.7 ± 1.2 | 565000 ± 124000 | ||
| Nirmatrelvir (1) | 10 | Crystalline Anhydrous Form 1d | 1450 ± 373 | 0.25 ± 0 | 2170 ± 1180f |
| 100 | Crystalline Anhydrous Form 1b | 5300 ± 1380 | 1.4 ± 1.0 | 18000 ± 8880 | |
| 10 | MTBEe | 1290 (851, 1730) | 1.5 (1.0, 2.0) | 3190 (1890, 4480) | |
| 100 | MTBEb | 29100 (32400, 25800) | 0.75 (1.0, 0.5) | 68300 (78200, 58400) | |
| 1000 | MTBEb | 88300 (75500, 101000) | 1.0 (1.0, 1.0) | 746500 (795000, 698000) | |
Pharmacokinetic parameters for 9 and 1 were generated from plasma-concentration time data and are reported as mean ± S.D. (n = 3) or mean and individual values (n = 2). Pharmacokinetic studies were performed in male Wistar–Han rats in the fed state.
Suspension in 2% (v/v) Tween 80 in of 0.5% (w/v) methylcellulose in water.
Suspension in 1% (v/v) Soluplus and 0.5% (w/v) methylcellulose in water.
Crystalline anhydrous form 1 of 1 administered as a solution in 2% (v/v) Tween 80/98% 0.5% (w/v) methyl cellulose in water.
Crystalline MTBE cosolvate of 1 administered as a solution in 10% (v/v) ethanol/10% (v/v) Capmul MCM/35% (v/v) PEG400/45% (v/v) Tween 80.
AUC0-∞.
Comparison of the low dose (≤10 mg/kg) p.o. rat pharmacokinetic parameters of crystalline 9 with the ones previously generated13 with the crystalline anhydrous Form 1 of nirmatrelvir (1) revealed considerable similarity (Tables 4 and 5). However, a corresponding crystalline methyl tert-butyl ether (MTBE) cosolvate of 1 had demonstrated greater p.o. absorption than crystalline nonsolvate of 1 in rats (see Table 5), particularly at doses (≥100 mg/kg) required to assess in vivo safety and was the preferred form for evaluation of the in vivo toxicological profile in both rats and monkeys.
The consistent performance of compound 9 in dose escalation studies up to 1000 mg/kg in both crystalline and 75% SDD forms contrasted with nirmatrelvir (1) which only showed vastly improved oral exposures when using the crystalline MTBE cosolvate at toxicological doses. This improved absorption profile for compound 9 is derived from a lower melting point, higher solubility and the lack of restricted rotation around the P2/3 amide bond, in contrast to 1 (Figure 2, Table 6). An evaluation of the crystal lattice and rotamer kinetics around the amide moiety was performed for 9 which does not contain the 3.1.0 proline framework present in 1. NMR solution conformations were determined in EXSY (EXchange SpectroscopY) experiments using a combination of residual dipolar couplings (RDCs), J-couplings and Nuclear Overhauser effects (NOEs). In contrast to compound 1, compound 9 displayed rapid conformational exchange even at room temperature, indicating a much lower barrier for syn/anti interconversion around the amide bond. From the NMR data, the t1/2 and energy barrier of rotation was determined for 9 (Figure 3). While both compounds (1 and 9) were rotamers, interconversion rates and energy barriers differed for 1 (including both the anhydrous and MTBE solvate forms) versus 9, while still being below formal atropisomerism designation levels (Table 6). From these data we expect 9 to have a dissolution profile more in line with its measured solubility, which is likely to manifest into an improved oral absorption pattern relative to crystalline anhydrous 1.
Figure 2.

Syn and anti rotamers of crystalline anhydrous 1 through restricted amide bond rotation versus compound 9 with a lower barrier to rotation.
Table 6. Key Molecular, Solid-State and NMR Characteristics for Compounds 1 and 9.
| Compound | Form | MP (°C) | Solubility (mg/mL) | Rotamer exchange in EXSYa | Torsion Rotation t1/2 (min)b | Energy of Rotation (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | Anhydrous | 192 | 1.2 | No | 10.54 | 20.4 |
| 1 | MTBE | 119 | 7.21 | No | 10.99 | 20.2 |
| 9 | Anhydrous | 167 | ∼2.5 | Yes | 0.21 | 18.9 |
With 300 ms mixing time at 298 K.
Extrapolated to 298 K.
Figure 3.

A series of EXSY spectra with an 80 ms mixing time were collected from 298–348 K for 9, to calculate the rotamer rate of exchange. The exchange peaks between the syn and anti rotamers were integrated at each temperature (A) and the exchange rate was plotted (B). From the exchange rate, an Eyring plot was calculated (C) and both the torsion rotation and energy of rotation were determined for the compound.
An X-ray cocrystal structure confirmed a highly similar binding mode of 9 to 1, including the formation of a covalent adduct between catalytic Cys145 and the P1′ nitrile. The primary difference observed in the cocrystal structure arose from the increased pucker enabled by the 4-trifluoromethylproline of 9 as it lacked the constrained cyclopropyl ring fusion found in 1. The trifluororomethyl group in 9 projects along a vector bisecting the gem-dimethylcyclopropane substitution in 1 to effectively fill the lipophilic P2 pocket (Figure 4). Critical H-bond networks with Glu166 and Phe140 were maintained. Overall, 9 retains an exceedingly high structural and conformational fidelity with this region of the endogenous SARS-CoV-2 Mpro polyprotein substrate, a design strategy we deliberately employed to further minimize the potential for drug-treatment induced antiviral resistance in the future.
Figure 4.

(A) X-ray crystal structure of SARS-CoV-2 Mpro complex with compound 9 (cyan) with H-bond network residues Glu166 and Phe140 highlighted (orange) (PDB: 8V4U). (B) Overlay of 9 (cyan) with nirmatrelvir (1, pale blue, PDB: 7RFS) where superposition is based on the protein binding pocket amino acids backbone heavy atoms. (C) Superposition of compound 9 (cyan) and substrate peptide nsp4 nsp5 (PDB: 7DVP, orange). Superposition is based on the protein binding pocket amino acids backbone heavy atoms.
The biochemical potency of 9 was tested across a human coronavirus panel (Figure 5A) and then in antiviral assays for SARS-CoV-1, MERS and 229E (Figure 5B). Compound 9 inhibited the SARS-CoV-1 Mpro (IC50 = 18 nM) and MERS Mpro (IC50 = 930 nM) activity, respectively, but had a much smaller disparity in the cell-based antiviral assays (SARS-CoV-1 EC50 = 157 nM and MERS EC50 = 158 nM). This antiviral activity, measured in the presence of the P-glycoprotein efflux inhibitor CP-100356 (EI) to suppress P-gp activity in the cell line used, was consistent across SARS-CoV-1, MERS and SARS-CoV-2 suggesting that like 1, 9 can be a considered a pan-human coronavirus Mpro inhibitor. Compound 9 had potent antiviral activity against a range of current and previous VOCs (variants of concern) including delta and omicron BA.1 across VeroE6/TMPRSS2 (Transmembrane protease, serine 2) cells (Figure 5C). Treatment of differentiated normal human bronchial epithelial (dNHBE) cells with compound 9 for 3 days led to inhibition of SARS-CoV-2 viral replication with EC50 and EC90 values of 34 nM and 70 nM respectively, as monitored by titration of virus harvested from the apical compartment using a 50% cell culture infective dose (CCID50) assay in Vero76 cells (Figure 5D). This critical dNHBE primary cell antiviral data served as the basis for calculating multiples of target coverage in our prediction of human dose for compound 9.
Figure 5.

(A) Compound 9 is a potent inhibitor of the proteolytic activity of SARS-CoV-2 Mpro as well as related human coronaviruses in FRET assays. Data shown are the mean ± SD from three independent experiments. (B) 9 demonstrates pan-coronavirus antiviral activity. Compound 9 inhibition in viral-induced CPE assays: SARS-CoV-1 in Vero E6 cells (in the presence of 2 μM EI CP-100356), h-CoV-229E in MRC-5 cells and MERS-CoV in Vero 81 cells (in the presence of 1 μM EI CP-100356). Data are shown as mean ± SD. CCID50 values were determined in all assays to be >100 μM. (C) Antiviral activity of compound 9 and remdesivir (positive control) against SARS-CoV-2 strains of VOC delta and omicron in VeroE6/TMPRSS2 cells. (D) Compound 9 demonstrates potent SARS-CoV-2 antiviral cellular activity in dNHBE cells. Compound 9 decreased SARS-CoV-2 viral replication (N = 3). Data shown are the geometric mean and 95% confidence intervals (CI).
The antiviral efficacy of 9 was also examined in a mouse-adapted SARS-CoV-2 animal model,20 following p.o. administration at doses of 100, 300 or 500 mg/kg BID as a 75% SDD throughout the duration of the four-day study, starting at 4 h post infection as well as at 500 mg/kg BID at 12 h post infection. Day 4 lung viral titers were evaluated by a CCID50 assay to assess whether compound 9 inhibited viral replication. At all doses, compound 9 prevented weight loss in comparison to the vehicle treated (0 mg/kg) group (Figure 6A). Oral treatment with compound 9 (n = 10/group) at 100, 300 or 500 mg/kg BID reduced viral lung titers by 1.3 log10 (p < 0.001), 2.6 log10 (p < 0.0001) or 3.1 log10 (p < 0.0001), respectively, compared to the vehicle treated group (Figure 6B). When dosed at 500 mg/kg BID, initiated at 12 h post infection, compound 9 reduced the lung virus titer by 2.7 log10 (p < 0.0001) (Figure 6B). The three dose groups were designed to target 0.3, 1 and 3× EC90,u at Cmin. In practice, unbound systemic concentrations for 9 at the top dose of 500 mg/kg BID achieved 2.9× EC90, Cmin (Figure 6C). Cumulative histopathological scoring of lungs from the vehicle-treated mice demonstrated evidence of increased perivascular inflammation, bronchial or bronchiolar epithelial degeneration or necrosis, bronchial or bronchiolar inflammation, cellular debris in alveolar lumen and alveolar inflammation and thickening of the alveolar septum compared to mice treated with 9 and mock-infected mice (Figure 6D). SARS-CoV-2 nucleocapsid protein immunohistochemical analysis to detect viral antigen levels in the lungs revealed that 9 inhibited virus replication in a dose-dependent manner compared to vehicle-treated and mock-infected mice (Figure 6E).
Figure 6.

Five mice per group were challenged intranasally with 1 × 105.0 50% CCID50 of SARS-CoV-2 MA10. Animals were orally administered 0, 100, 300 or 500 mg/kg BID of 9 throughout the duration of the study starting at 4 h post infection (hpi) or 500 mg/kg BID 9 starting at 12 hpi. Animals were euthanized at 4 days post infection (dpi) and lungs collected for virus titers. Data (for A–D) were compiled from two independent studies (n = 10 BALB/c mice). (A) Weight loss during infection. Mice were weighed daily. (B) Lung viral titer at 4 dpi. Lung titers are plotted as mean log10 CCID50/ml ± SEM. Dotted line represents the limit of detection for the CCID50 assay. (C) Twelve-hour compound 9 exposure levels of 100, 300 and 500 mg/kg doses in uninfected, orally treated mice. EC90 represented as determined in the day 3 dNHBE primary cell assay (D) Histopathology scores on a scale of 0 to 5, where 0 is a normal healthy lung and 5 is severe coalescing areas of necrosis and confluent areas of inflammation. (E) SARS-CoV-2 nucleocapsid protein immunohistochemistry. Shown are digital light microscopic scans of mouse lung tissue sections of mock-infected, 0, 100, 300, 500 and +12 h 500 mg/kg doses of 9-treated mice stained with SARS-CoV-2 nucleocapsid antibody. Data are scans from one study. Scale bars, 100 μm. Magnification is 20×.
Based on the observed low CLint,app of 9 (relative to 1) in HLM, the attractive oral pharmacokinetic characteristics in high dose rat toxicokinetic studies using crystalline material or a 75% ASD and the potent antiviral activity in a human physiological system (dNHBE cells) and in the mouse in vivo model, compound 9 (PF-07817883) was selected as a clinical candidate and profiled extensively for its safety profile and in vitro disposition characteristics including interactions with drug metabolizing enzymes and transporters.
Compound 9 demonstrated no human off-target pharmacological activity in multiple in vitro broad profiling assay panels (representing and including G-protein coupled receptors, ion channels, transporters and enzymes) with the exception of two human cathepsins, cathepsin K (IC50 = 21 nM) and cathepsin S (IC50 = 33 nM) (Supplementary Table S4). Moreover, like 1, 9 did not cause mutagenicity in the Salmonella Ames (strains TA98, TA100, TA1535 and TA1537) and in vitro micronucleus assays (in Chinese hamster ovary or thymidine kinase heterozygote TK6 cells) in the absence or presence of metabolic activation (Aroclor-induced rat liver S9 fraction/NADPH). In addition, compound 9 demonstrated a favorable safety profile from a comprehensive battery of vitro safety tests, safety pharmacology studies and in 2-week GLP toxicity studies in two species (rats and monkeys).
In vitro metabolic profiling of 9 in NADPH-supplemented HLM and/or human hepatocytes principally revealed the formation of two monohydroxylated metabolites (M1 and M2) with a protonated exact mass (m/z+) of 506.2219 and 506.2221, respectively. Metabolite M1 (the major oxidative metabolite of 9) and M2 were observed in liver microsomes and hepatocytes across all preclinical species and human. In HLM, the formation of M1 and M2 were most significantly inhibited (>96%) upon coincubation with the selective CYP3A4/5 inhibitor ketoconazole, suggesting that the oxidative metabolism of 9 is principally mediated by human CYP3A4/5. There were no human unique metabolites of 9; all metabolites observed in human reagents were also detected in corresponding matrices from preclinical species, which were used for toxicity studies. Comparison of diagnostic fragment ions in the mass spectra for 9, M1 and M2 suggested that the two metabolites were derived from oxidations on the pyrrolidinone ring and the tert-butyl substituent, respectively (Supplementary Figures S3–S8). Purified biosynthetic standards of M1 and M2 were obtained from scaled-up incubations of 9 with NADPH-supplemented rabbit liver microsomes. One- and two-dimensional NMR spectroscopy studies on the purified metabolite samples revealed the site of modifications in M1 and M2 to be on the C5 methylene on the P1 5-membered lactam in 9 and one of the methyl groups on the P3 tert-butyl substituent, respectively (Supplementary Figures S6 and S8). Metabolite M1 was pharmacologically active and demonstrated potent inhibition (Ki = 13 nM) of SARS-CoV-2 Mpro activity in the recombinant biochemical assay. However, relative to 9, weaker cellular antiviral activity (compound 9: EC50 = 0.18 μM; M1: EC50 = 1.65 μM) was observed with M1 in SARS-CoV-2-infected VeroE6 cells. The phenomenon pertaining to the dramatically weaker cellular antiviral activity, relative to the biochemical inhibitory potency, which was also noted with the structurally related pyrrolidinone oxidative metabolite of nirmatrelvir, potentially occurs due to the decreased passive permeability of the CYP-generated hydroxylated metabolites.13
The availability of these biosynthesized metabolite standards also enabled the conduct of enzyme kinetics studies, including measurement of the formation rates of M1 and M2 to estimate CLint,app for 9 in NADPH-supplemented HLM using experimental protocols outlined for corresponding studies with 1.13 The total CLint,app for 9, based on the collective formation rates for M1 and M2 was measured to be 4.72 μL/min/mg (predicted hepatic clearance = 1.3 mL/min/kg), a value that was considerably lower than the one estimated for 1 (CLint,app = 28.8 μL/min/mg, predicted hepatic clearance = 6.3 mL/min/kg) using the substrate depletion assay protocol in HLM. It is noteworthy to point out that the major sites of metabolism and the metabolizing enzyme involved is virtually identical for 9 and 1.13 Consequently, the lower CLint,app for 9 (4.7 μL/min/mg), relative to 1 (28.8 μL/min/mg) most likely results from a reduction in lipophilicity of 9 (1 LogD = 1.9, 9 LogD = 0.9) and/or unfavorable interactions of 9 in the active site of CYP3A4.
Plasma protein binding determination using equilibrium dialysis21 revealed low binding of 9 (2 μM) to rat plasma with a corresponding unbound fraction (fu,p) value of 0.857. In contrast, 9 demonstrated concentration-dependent protein binding to monkey (fu,p = 0.121–0.965 at 0.3–300 μM) and trended toward concentration-dependent protein binding to human plasma at concentrations exceeding 10 μM (fu,p = 0.423–0.789 at 0.3–30 μM). In comparison, 1 exhibited moderate and concentration-independent plasma protein binding in rat (fu,p = 0.47–0.49), monkey (fu,p = 0.39–0.50) and human (fu,p = 0.30–0.33) across the concentration range (0.3–10 μM) evaluated.22 A potential cause(s) for the concentration-dependent plasma protein binding of 9 to monkey (and human) plasma is the saturation of binding to both serum albumin and α-1-acid glycoprotein as demonstrated previously with 1, which revealed concentration-dependent (2–200 μM) protein binding (fu,p = 0.024–0.69) in dog plasma. In the case of 1, mass spectrometric analysis of proteins following incubations with dog serum albumin did not reveal a mass shift change of 499.5 (molecular weight of 1), which ruled out the possibility of concentration-dependent plasma protein binding arising via covalent interactions between serum albumin and the nitrile group in 1.22
Compound 9 revealed no reversible inhibition (IC50 > 100 μM) of the major human CYP enzymes including CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 in HLM.18 Moreover, 9 also did not cause time-dependent inhibition of the catalytic activity of the major human CYPs in HLM, with no IC50 shift observed after a 30 min preincubation of 9 in HLM in the absence or presence of CYP cofactor NADPH.23
Examination of the induction potential of CYP3A4 messenger RNA (mRNA) and enzyme (midazolam-1′-hydroxylase) activity by 9 (0.3–300 μM) in human hepatocytes revealed weak induction of CYP3A4 mRNA and enzyme activity in a concentration-dependent fashion. Induction parameters (Indmax, Emax, EC50, Hill and linear slopes) for 9 are depicted in Supplemental Table 5. A cytotoxicity assay using Promega’s CellTiter 96 aqueous nonradioactive cell proliferation assay kit (Promega, WI) showed no reduction in hepatocyte cell viability after treatment with 9 at the tested concentrations. Projection of pharmacokinetic drug–drug interactions arising from the CYP3A induction phenomenon through mechanistic modeling of the predicted human pharmacokinetic parameters and the in vitro CYP3A4 induction data suggested little-to-no change in systemic exposure of midazolam, which is primarily metabolized by CYP3A4/5 (unpublished data).
In addition to the lack of inhibitory effects toward major human CYP enzymes, 9 was also devoid of potential reversible inhibition (IC50 > 100 μM) of the catalytic activities of major human uridine 5′-diphospho-glucuronsyl transferase (UGT) enzymes including UGT1A1, UGT1A4, UGT1A6, UGT1A9, UGT2B7 and UGT2B17 in HLM.24 The potential for inhibition of major human intestinal, hepatobiliary and renal drug transport proteins such as multidrug resistant 1 (MDR1) transporter, breast cancer resistant protein (BCRP), organic anion transporting polypeptides (OATPs), organic cation transporters (OCTs), organic anion transporters (OATs) and multidrug and toxic extrusion (MATE) transporters by 9 was also assessed in stably transfected cell-based transporter systems using established protocols and revealed no inhibitory effects (IC50’s > 250 μM) against all transporter proteins examined.
Synthetic strategies capitalized on the modular nature of the peptidomimetic chemotype. Compounds 4 and 5 were made via a route that allowed for late-stage variation of the P4 substituent (Scheme 1). The proline ethers at P2 were rapidly accessible from the corresponding, and readily available, suitably protected trans-4-hydroxy-l-proline 13. As such, trifluoromethyl methyl ether 14 was accessible in a single step from 13 using a silver-mediated oxidative trifluoromethylation25 in modest yields. Deprotection with HCl followed by a HATU coupling with Boc-l-tert-leucine afforded 16. Ester hydrolysis with lithium hydroxide gave 17 which was then coupled with glutamine mimetic 18(15) to afford tripeptide 19. Acidic deprotection provided the primary amine 20, which was subsequently trifluoroacetylated with TFAA to afford 21 or reacted with methyl chloroformate to give carbamate 22. Finally, dehydration of the primary amide to the corresponding nitrile was accomplished using Burgess reagent to give 4 and 5 respectively. Similarly, copper-catalyzed difluoromethylation of 13 provided concise access to 23.26 A similar deprotection–coupling sequence as before, led to 25. Basic hydrolysis to acid 26 was again followed by an amidation and deprotection to afford template 28 which could be diversified at the P4 cap to final targets. In this instance, a tandem trifluoroacetamide formation and dehydration of the primary amide was achieved using TFFA to provide 6 directly in good yield. Stepwise carbamate formation (29) followed by Burgess dehydration led to 7.
Scheme 1. Synthesis of Fluorinated Ether Analogs 4–7.

Reagents and conditions: (a) AgOTf, Selectfluor, KF, TMSCF3, 2-F-Py, EtOAc 29 °C, 21%; (b) 2-(fluorosulfonyl)difluoroacetic acid, CuI, CH3CN, 50 °C, 63%; (c) representative conditions: 4 N HCl in dioxane, 93%; (d) N-Boc-l-tert-leucine, HATU, i-Pr2NEt, DMF, 73–84%; (e) LiOH·H2O, CH3OH or THF, H2O, 0–25 °C, 87–92%; (f) 18, HATU, i-Pr2NEt, DMF, 0–25 °C 59–82%; (g) HCl in ethyl acetate, 94–100%; (h) TFAA, Et3N, DCM, 0–20 °C, 40%; (i) methylchloroformate, Et3N, DCM; (j) Burgess reagent, DCM, 21–26%; (k) TFAA, NMM, i-PrOAc, 0 °C, 73%.
In the case of 8 (Scheme 2), a variation on the previous synthetic sequence was used. Commercially available N-Boc-4-trans-trifluoromethyl proline 30 was coupled with the P1 fragment 18 first to afford 31. Acid-mediated Boc removal and standard coupling to Boc-l-tert-leucine provided 32 in 75% over two steps. Acidic deprotection followed by trifluoroacetaylation, this time using ethyl trifluoroacetate, and finally Burgess dehydration of the P1′ amide afforded 8 in 29% over the three steps.
Scheme 2. Synthesis of 8.

Reagents and conditions: (a) HATU, i-Pr2NEt, DMF, 93%; (b) 4 M HCl in dioxane; (c) N-Boc-l-tert-leucine, HATU, i-Pr2NEt, DMF, 75% over two steps; (d) CF3CO2CH2CH3, i-Pr2NEt, CH3OH; (e) Burgess reagent, DCM 29% over three steps.
While the previous sequence from Scheme 2 allowed effective access to CF3 proline P2 derivatives, we adopted a variation in our synthetic strategy in our preferred approach, particularly for larger scale preparation. As such the remaining compounds were made via Scheme 3. Compound 30 was deprotected and esterified in a single step using thionyl chloride in methanol to afford 33. Amide coupling with N-(methoxycarbonyl)-l-tert-leucine afforded 34, after which the ester was removed via saponification with lithium hydroxide to give 35 with high yields across all three steps. Coupling with 18 as before provided 36 which upon dehydration with TFAA afforded 9. Access to the corresponding oxo-piperidine congener fragment at P1 was accomplished in two steps. Specifically, direct aminolysis of methyl ester 37 provided the primary amide 38, which was deprotected using HCl to afford 39. Subsequent coupling to 35 followed by dehydration gave the ring expanded variant 10. Compounds 11 and 12 were made in analogous fashion using N-(methoxycarbonyl)-l-valine in the first amidation of the sequence.
Scheme 3. Synthesis of 9–12.

Reagents and conditions: (a) SOCl2, CH3OH 96%; (b) (methoxycarbonyl)-l-valine or (methoxycarbonyl)amino)-l-tert-leucine, HATU, i-Pr2NEt, DMF, CH3CN, 77–93%; (c) LiOH, CH3OH, H2O, 90%; (d) representative conditions: 18 or 39, HATU, NMM, DMF, acetonitrile 76–94%; (e) Representative conditions: TFAA, NMM, i-PrOAc, 40–53% over two steps; (f) NH3, MeOH, 68%; (g) HCl, EtOAc 90%.
Conclusions
Starting from nirmatrelvir (1), and with an initial focus on optimizing antiviral potency and LogD-driven CYP3A-mediated metabolic clearance, primarily through changes to P2 and P4, we identified molecules with improved metabolic stability without compromising passive permeability or cellular antiviral activity. Compound 9 (PF-07817883) went on to show high solubility, oral absorption and dose-dependent antiviral in vivo efficacy. Compared to Paxlovid, 9 was predicted to have a little-to-no potential for perpetrator drug–drug interactions of its own, based on studies utilizing human reagents. The distinctive, polar nature of 9 (LogD 0.9) compared to ritonavir-boosted protease inhibitors such as 1 (LogD 1.9), lopinavir (LogD 3.8) and paritaprevir (LogD 3.1) is of note. PF-07817883 (9) has been nominated as a clinical candidate for the treatment of COVID-19 and has entered clinical trials (NCT05580003, NCT05799495). Results from the first-in-human pharmacokinetics, safety and tolerability studies as well as investigations into the drug–drug interaction potential of 9 will be published in due course.
Experimental Section
General Methods for Compound Synthesis/Analysis
All reactants, reagents and solvents were obtained from commercial sources and used without further purification. Except where otherwise noted, reactions were run under an inert atmosphere of nitrogen gas using anhydrous solvents at room temperature (∼23 °C). The terms “concentrated” and “evaporated” refer to the removal of solvent at reduced pressure on a rotary evaporator with a water bath temperature not exceeding 60 °C. Reactions were monitored by thin layer chromatography (TLC) performed on Analtech, Inc. silica gel GF 250 μm plates or Merck silica gel plates (60 F254) and were visualized with UV light (254 nm) and/or KMnO4 staining or by UPLC-MS (Waters Acquity, ESCI (ESI +/–, APCI +/−)). Flash chromatography was carried out using a CombiFlash system from Teledyne Isco; Biotage SNAP or Redisep Rf silica columns were used.
All biologically tested compounds were determined to be >95% purity by LC-MS and NMR methods described below. In vivo material was determined to be >99% purity by analytical HPLC. Nuclear magnetic resonance (NMR) spectra were collected using a 600 MHz Bruker Avance III spectrometer, a 400 MHz Bruker Avance III spectrometer or a 400 MHz JEOL ECZ spectrometer. Chemical shifts (δ in ppm) for 1H spectra are reported relative to the residual solvent signals: 7.26 ppm for chloroform-d, 2.50 ppm for dimethyl sulfoxide-d6, and 3.31 ppm for methanol-d4 using the δ, multiplicity, coupling constant(s) in Hz and integration. The multiplicities are denoted as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and br s, broad singlet. Chemical shifts (δ in ppm) for 13C spectra are reported relative to the residual solvent signals: 77.5 ppm for chloroform-d, 39.5 ppm for dimethyl sulfoxide-d6. All tested compounds and most intermediates present as conformational isomers (rotamers) in 1H NMR spectra. For compounds 1 and 9, all 1H and 13C assignments were determined using a combination of the following NMR spectra: 1H, 13C, COSY, NOESY, HSQC and HMBC. The NMR experiments were collected on either a Bruker Avance III NMR instrument operating at 600.13 MHz for 1H and 150.90 MHz for 13C using a 5 mm TCI helium cryoprobe probe equipped with a z-axis gradient using Topspin 3.5pl7 or a Bruker Avance Neo NMR instrument operating at 500.05 MHz for 1H, 125.74 MHz for 13C and 470.47 MHz for 19F using a 5 mm Prodigy nitrogen cryoprobe probe with a z-axis gradient using Topspin 4.2. Chemical shifts were referenced to residual solvent (dimethyl sulfoxide-d6) and all spectra were assigned using MestreNova software version 14.0. Unless otherwise specified, only the major conformational isomer is described. High-resolution mass spectrometry (HRMS) data were gathered on a Sciex TripleTOF 5600+ (Sciex, Ontario, Canada) with DuoSpray ionization source. The LC instrument includes an Agilent (Agilent Technologies, Wilmington, DE) 1200 binary pump, Agilent 1200 autosampler, Agilent 1200 column compartment, and Agilent 1200 DAD. The instrument acquisition and data handling were done with Sciex Analyst TF version 1.7.1. Prior to acquisition the instrument was calibrated with less than 5 ppm accuracy. During acquisition, a calibration run was performed initially and after every 5 injections using the Sciex positive polarity tuning mix. Elution conditions: column, Waters Xselect HSS T3, 2.1 × 30 mm, 2.5 μm particle size; column temperature, 60 °C; solvent A, water (0.1% formic acid); solvent B, acetonitrile (0.1% formic acid); gradient: initial 5% B, hold for 0.10 min, 5–95% B in 2.8 min, 95–5% B in 0.20 min, 3.5 min total run time; flow rate, 0.8 mL/min. TOF conditions, ESI in positive mode: spray chamber: gas 1 and 2 at 60, curtain gas at 40; temperature, 600 °C; ion spray voltage, 5500 V; declustering potential, 100; collision energy, 10. The acquisition is done in TOF MS mode with a range of 100–2000 amu with accumulation time of 0.20 s. ESI in negative mode: spray chamber: gas 1 and 2 at 60, curtain gas at 40; temperature, 600 °C; ion spray voltage, −4500 V; declustering potential, −100; collision energy, −10. The acquisition is done in TOF MS mode with a range of 100–2000 amu with accumulation time of 0.20 s.
1-(tert-Butyl) 2-methyl (2S,4R)-4-(trifluoromethoxy)pyrrolidine-1,2-dicarboxylate (14)
Two parallel reactions were set up as described and then combined for workup and isolation.25 Silver triflate (4.19 g, 16.3 mmol), Selectfluor (2.89 g, 8.15 mmol) potassium fluoride (1.00 g, 17.3 mmol) and 13 (N-Boc-trans-4-hydroxy-l-proline methyl ester, 1.0 g, 4.08 mmol) were combined in a sealed tube. The flask was purged with argon. Ethyl acetate (20 mL) was added, followed by the addition of (Trifluoromethyl)trimethylsilane (2.32 g, 16.3 mmol) and 2-fluoropyridine (1.58 g, 16.3 mmol). The reaction mixture was stirred under argon at 29 °C for 16h. The two parallel reactions were combined, filtered through a Celite pad, and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 15% ethyl acetate in petroleum ether) provided 14 (539 mg, 21%) as an oil. Material does not ionize for LCMS. 1H NMR(400 MHz, DMSO-d6) d 5.22–4.97 (m, 1H), 4.35–4.26 (m, 1H), [3.69 (s) and 3.66 (s), total 3H], 3.64–3.60 (m, 2H), 2.58–2.46 (m, 1H), 2.34–2.19 (m, 1H), [1.41 (s) and 1.35 (s), total 9H]. Prominent (∼5:3) conformational isomers. 19F NMR (376 MHz, DMSO-d6) d −57.05.
Methyl (2S,4R)-4-(trifluoromethoxy)pyrrolidine-2-carboxylate, hydrochloride salt (15)
To a solution of 14 (778 mg, 2.48 mmol) in DCM (8 mL) at 0 °C was added hydrogen chloride in ethyl acetate (1 N, 8 mL, 8 mmol). The mixture was then stirred for 2.5h then concentrated in vacuo to obtain 15 (622 mg, > 99.9%) as a pale-yellow solid. The material was used in the next step without any further purification. LCMS m/z 214.2 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.94 (s, 2H), 4.57 (dd, J = 8.7, 7.1 Hz, 1H), 3.77 (s, 3H), 3.65 (dd, J = 12.2, 8.9 Hz, 1H), 3.55–3.42 (m, 1H), 3.27 (dd, J = 12.2, 7.2 Hz, 1H), 2.49–2.31 (m, 2H).
Methyl (2S,4R)-1-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-4-(trifluoromethoxy)pyrrolidine-2-carboxylate (16)
A solution of N-Boc-l-tert-leucine (530 mg, 2.29 mmol) in DMF (8 mL) was cooled to 0 °C then treated with HATU (958 mg, 2.52 mmol). After the reaction mixture had been stirred for 10 min, 15 (572 mg, 2.29 mmol) was added, followed by dropwise addition of N,N-diisopropylethylamine (1.40 mL, 8.02 mmol). Stirring was continued and the reaction was allowed to warm to room temperature where it was kept for 3 days. The mixture was poured into water (20 mL) and extracted with ethyl acetate (3 × 12 mL). The combined organic layers were washed with 1 M hydrochloric acid (15 mL), 1 M aqueous sodium carbonate (15 mL), saturated sodium chloride (15 mL) then dried over sodium sulfate and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 20% ethyl acetate in petroleum ether) provided 16 (712 mg, 73%) as a gum. LCMS m/z 449.1 [M+Na]+. 1H NMR (400 MHz, DMSO-d6) ä = 6.83 (br d, J = 8.8 Hz, 1H), 5.17 (br s, 1H), 4.39 (br t, J = 8.7 Hz, 1H), 4.26–4.10 (m, 2H), 3.85 (br d, J = 10.8 Hz, 1H), 3.64(s, 3H), 2.54–2.53 (m, 1H), 2.25–2.14 (m, 1H), 1.37 (s, 9H), 0.95 (s, 9H).
(2S,4R)-1-((S)-2-((tert-Butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-4-(trifluoromethoxy)pyrrolidine-2-carboxylic acid (17)
A solution of 16 (760 mg 1.78 mmol) in methanol (7 mL) was cooled was cooled to 0 °C. Lithium hydroxide monohydrate (165 mg, 3.92 mmol) in water (1.96 mL) was added slowly, and the resulting mixture was stirred for 20 min. The mixture was removed from the cooling bath, allowed to warm to room temperature and stirred for 2.5h then acidified with 1 M hydrochloric acid to pH∼2. The resulting solids were collected by filtration and dried to afford 17 (674.8 mg, 92%) as a white solid. LCMS m/z 413.2 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 12.71 (s, 1H), 6.79 (d, J = 9.4 Hz, 1H), 5.15 (s, 1H), 4.29 (t, J = 8.8 Hz, 1H), 4.22–4.06 (m, 2H), 3.81 (d, J = 12.5 Hz, 1H), 2.57–2.43 (m, 1H, assumed, obscured by solvent), 2.26–2.10 (m, 1H), 1.36 (s, 9H), 0.94 (s, 9H).
tert-Butyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-4-(trifluoromethoxy)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (19)
A stirred solution of 17 (400 mg 0.970 mmol) in DMF (4 mL) was cooled to 0 °C. HATU (406 mg, 1.07 mmol) was added and the resulting mixture was stirred for 10 min. 18 (201 mg, 0.970 mmol) was added, followed by dropwise addition of N,N-diisopropylethylamine (439 mg, 3.39 mmol). After addition, the mixture was stirred at room temperature for 16h. The mixture was poured into water (10 mL) and extracted with chloroform/isopropanol (3 × 8 mL). The combined organic layers were washed with 1 M hydrochloric acid (10 mL), 1 M aqueous sodium carbonate (10 mL), saturated sodium chloride (10 mL) then dried over sodium sulfate and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 8% methanol in DCM) provided 19 (450 mg, 82%) as a white solid. LCMS m/z 566.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.32 (d, J = 8.8 Hz, 1H), 7.54 (s, 1H), 7.31 (s, 1H), 7.06–7.01 (m, 1H), 6.79 (d, J = 9.3 Hz, 1H), 5.15 (s, 1H), 4.45 (t, J = 8.5 Hz, 1H), 4.27 (ddd, J = 12.3, 8.9, 3.5 Hz, 1H), 4.10 (t, J = 12.8 Hz, 2H), 3.83 (d, J = 12.7 Hz, 1H), 3.18–2.98 (m, 2H), 2.56–2.46 (m, 1H, assumed, obscured by solvent), 2.39 (dd, J = 13.9, 7.3 Hz, 1H), 2.22–2.04 (m, 2H), 2.02–1.90 (m, 1H), 1.69–1.55 (m, 1H), 1.54–1.43 (m, 1H), 1.36 (s, 9H), 0.92 (s, 9H).
(2S,4R)-N-((S)-1-Amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-(trifluoromethoxy)pyrrolidine-2-carboxamide hydrochloride salt (20)
A stirred solution of 19 (450 mg, 0.796 mmol) in DCM (5 mL) was cooled to 0 °C. Hydrogen chloride in ethyl acetate (1 N, 10 mL, 10 mmol) was added. The mixture was allowed to warm to room temperature and then stirred for 2.5h. Removal of solvents in vacuo afforded 20 (375 mg, ≤ 94%) as pale-yellow solid. The crude material was used directly in the next step.
(2S,4R)-N-((S)-1-Amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-1-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)-4-(trifluoromethoxy)pyrrolidine-2-carboxamide (21)
A stirred solution of 20 in DCM (5 mL) was cooled to 0 °C. Triethylamine (161 mg, 1.59 mmol) was added dropwise followed by TFAA (126 mg 0.598 mmol) dropwise, to the reaction solution at 0 °C. The reaction was allowed to warm to room temperature, stirred for 3h, then quenched with water (15 mL) and extracted with DCM (3× 10 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (10 mL) and saturated sodium chloride (2× 5 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo to afford a crude yellow solid. Silica gel chromatography (Gradient: 0% to 12% MeOH in DCM) afforded 21 (89.7 mg, 40%) as a white solid. LCMS m/z 584.2 [M+Na]+. 1H NMR (400 MHz, DMSO-d6) δ 9.52 (d, J = 8.6 Hz, 1H), 8.35 (d, J = 9.0 Hz, 1H), 7.56 (s, 1H), 7.36 (s, 1H), 7.05 (s, 1H), 5.17 (s, 1H), 4.57–4.46 (m, 2H), 4.35–4.27 (m, 1H), 4.02 (d, J = 12.2 Hz, 1H), 3.87 (dd, J = 12.2, 3.1 Hz, 2H), 3.20–2.99 (m, 2H), 2.58–2.36 (m, 1H), 2.23–2.05 (m, 2H), 2.01–1.90 (m, 1H), 1.72–1.59 (m, 1H), 1.54–1.43 (m, 1H), 0.99 (s, 9H).
(2S,4R)-N-((S)-1-Cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-1-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)-4-(trifluoromethoxy)pyrrolidine-2-carboxamide (4)
To a stirred solution of 21 in DCM (3 mL) was added Burgess reagent (189 mg 0.793 mmol) at room temperature. After 16h, the reaction was diluted with water (10 mL) and extracted with chloroform/isopropanol (4:1, 3× 15 mL). The combined organic layers were washed with 1 M aqueous sodium carbonate (15 mL), saturated sodium chloride (15 mL) then dried over sodium sulfate, filtered and concentrated. Silica gel chromatography (Gradient: 0% to 5% methanol in DCM) afforded 4 (18.5 mg, 21%) as a white solid. HRMS calc for C21H27F6N5O5 [M+H]+: 544.1989; found: 544.1994. 1H NMR (600 MHz, DMSO-d6) δ 9.51 (d, J = 8.5 Hz, 1H), 9.09 (d, J = 8.7 Hz, 1H), 7.69 (s, 1H), 5.20 (t, J = 4.0 Hz, 1H), 4.98 (ddd, J = 11.2, 8.7, 4.9 Hz, 1H), 4.51 (d, J = 8.6 Hz, 1H), 4.35 (dd, J = 9.7, 7.3 Hz, 1H), 4.06 (d, J = 12.3 Hz, 1H), 3.89 (dd, J = 12.3, 3.5 Hz, 1H), 3.14 (t, J = 9.1 Hz, 1H), 3.05 (td, J = 9.3, 7.3 Hz, 1H), 2.49–2.45 (m, 1H), 2.42 (dd, J = 14.2, 7.3 Hz, 1H), 2.19–2.07 (m, 3H), 1.75–1.68 (m, 2H), 0.98 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 177.58, 170.51, 168.07, 156.90 (q, J = 37.0 Hz), 121.15 (q, J = 255.0 Hz), 119.58, 114.89 (q, J = 287.9 Hz), 78.33 (d, J = 2.5 Hz), 58.12, 57.93, 53.87, 37.78, 36.67, 35.05, 35.01, 34.37, 26.89, 26.06. 19F NMR (470 MHz, DMSO-d6) δ −56.95, −72.92.
Methyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-4-(trifluoromethoxy)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (22)
A stirred solution of 20 (175 mg, 0.349 mmol) in DCM (5 mL) was cooled to 0 °C. Triethylamine (176 mg, 1.74 mmol) was added, followed by dropwise addition methyl chloroformate (49.4 mg, 0.523 mmol) in DCM (1 mL). The resulting mixture was allowed to warm to room temperature and stir for 3h. The reaction solution was diluted with water (10 mL). The reaction mixture was extracted with a mixture of chloroform:2-propanol (4:1, 3 × 10 mL) and washed with 1 M aqueous sodium carbonate (2 × 20 mL), dried over sodium sulfate and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 15% methanol in DCM) provided 22 (43 mg, 24%) as a white solid. LCMS m/z 524.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (d, J = 8.7 Hz, 1H), 7.53 (s, 1H), 7.31 (d, J = 17.5 Hz, 2H), 7.03 (s, 1H), 5.16 (s, 1H), 4.50–4.41 (m, 1H), 4.33–4.22 (m, 1H), 4.14–4.01 (m, 2H), 3.86 (d, J = 11.7 Hz, 1H), 3.52 (d, J = 3.6 Hz, 3H), 3.22–2.96 (m, 2H), 2.73–2.29 (m, 1H), 2.14 (s, 2H), 1.96 (t, J = 13.1 Hz, 1H), 1.70–1.56 (m, 1H), 1.48 (t, J = 12.3 Hz, 1H), 1.29–1.09 (m, 1H), 0.94 (s, 9H).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)carbamoyl)-4-(trifluoromethoxy)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (5)
To a stirred solution of 22 (43 mg (0.082 mmol) in DCM (3 mL), Burgess reagent (97.9 mg, 0.411 mmol) was added and the mixture was stirred for 16h. Dilution with water (10 mL) followed by extraction with a mixture of chloroform:2-propanol (4:1, 3 × 15 mL). Combined organics were washed with 1 M aqueous sodium carbonate (15 mL), saturated sodium chloride (15 mL) then dried over sodium sulfate and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 5% methanol in DCM) provided 5 (9.1 mg, 22%) as a white solid. HRMS calc for C21H30F3N5O6 [M+H]+: 506.2221; found: 506.2232. 1H NMR (600 MHz, DMSO-d6) δ 9.05 (d, J = 8.7 Hz, 1H), 7.66 (s, 1H), 7.32 (d, J = 8.7 Hz, 1H), 5.19 (s, 1H), 4.96 (ddd, J = 11.2, 8.7, 4.9 Hz, 1H), 4.35–4.28 (m, 1H), 4.14–4.09 (m, 1H), 4.08 (s, 1H), 3.88 (dd, J = 12.0, 3.1 Hz, 1H), 3.52 (s, 3H), 3.13 (t, J = 9.2 Hz, 1H), 3.04 (td, J = 9.3, 7.2 Hz, 1H), 2.58–2.51 (m, 1H), 2.39 (dd, J = 13.8, 6.8 Hz, 1H), 2.22–2.13 (m, 1H), 2.13–2.06 (m, 1H), 1.76–1.65 (m, 2H), 0.93 (s, 9H). Residual solvent observed. 13C NMR (151 MHz, DMSO-d6) δ 177.58, 172.17, 170.66, 170.01, 156.98, 121.17 (q, J = 256.0, 254.9 Hz), 119.63, 78.39 (d, J = 1.5 Hz), 59.26, 57.84, 53.65, 51.50, 37.71, 36.61, 35.03, 34.63, 34.28, 26.90, 26.67, 26.15. Residual solvent observed. 19F NMR (470 MHz, DMSO-d6) δ −56.88.
1-(tert-Butyl) 2-methyl (2S,4R)-4-(difluoromethoxy)pyrrolidine-1,2-dicarboxylate (23)
To a solution of 13 (20.7 g, 84.4 mmol) in acetonitrile (450 mL), copper(I) iodide (3.21 g, 16.9 mmol) was added and the reaction was heated to 50 °C. A solution of 2,2-difluoro-2-(fluorosulfonyl)acetic acid (18.0 g 101 mmol) in acetonitrile (50 mL) was added dropwise over a period of 1h. The reaction was then heated at 50 °C for an additional 30 min, after which the reaction was judged complete by TLC and evaporated. The reaction mixture was suspended in ethyl acetate and filtered, then concentrated in vacuo to afford a light-yellow oil. After combining with similar crude material from a 12.2 mmol scale reaction, the crude was purified via chromatography on silica gel (0–35% ethyl acetate in petroleum ether) to afford 23 (17.9 g, 63%) as a colorless oil. MS m/z 196.2 [M+H–CO2tBu]+. 1H NMR (400 MHz, DMSO-d6) δ 6.77 (t, J = 75.4 Hz, 1H), 4.87–4.78 (m, 1H), 4.31–4.19 (m, 1H), 3.70 (s, 3H), 3.63–3.48 (m, 2H), 2.54–2.34 (m, 1H), 2.23–2.10 (m, 1H), 1.35 (s, 9H). Conformational isomers present.
Methyl (2S,4R)-4-(difluoromethoxy)pyrrolidine-2-carboxylate hydrochloride (24)
To a solution of 23 (10 g, 33.866 mmol) in dioxane (25 mL) was added 4 M hydrogen chloride in dioxane (42 mL. 169 mmol, 5 equiv). The colorless solution was stirred for 3h, then concentrated in vacuo. The residue was suspended in diethyl ether (200 mL) and stirred for 45 min at which point excess diethyl ether was decanted off. Remaining solids were rinsed with diethyl ether and then dried under vacuum to afford 24 (7.33 g, 93%) as a white solid LCMS m/z 196.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 2H), 6.80 (t, J = 74.9 Hz, 1H), 5.03–4.95 (m, 1H), 4.51 (dd, J = 10.5, 7.7 Hz, 1H), 3.76 (s, 3H), 3.58 (dd, J = 13.0, 5.0 Hz, 1H), 3.40–3.31 (m, 1H), 2.46–2.28 (m, 2H).
Methyl (2S,4R)-1-((S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-4-(difluoromethoxy)pyrrolidine-2-carboxylate (25)
To a 0 °C solution of N-Boc-l-tert-leucine (7.55 g, 31.6 mmol) and 24 (7330.0 mg, 31.65 mmol) in DMF (50.0 mL) was added HATU (14.9 g, 38.0 mmol) followed by dropwise addition of N,N-diisopropylethylamine (12.7 g, 98.1 mmol, 17 mL) over 10 min. Five minutes after addition was complete, the cooling bath was removed and the reaction was stirred for 1h, then poured into water (300 mL). The reaction mixture was extracted with ethyl acetate, then washed with water, then saturated sodium chloride, dried over magnesium sulfate and concentrated in vacuo. Silica gel chromatography (10–30% ethyl acetate in heptanes) afforded 25 (10.85 g, 84%) as a colorless oil. LCMS m/z 309.3 [M+H–CO2tBu]+. 1H NMR (400 MHz, DMSO-d6) δ 6.76 (t, J = 75.5 Hz, 1H), 6.72 (d, J = 10.3 Hz, 1H), 4.94–4.80 (m, 1H), 4.35 (t, J = 8.5 Hz, 1H), 4.13 (d, J = 9.3 Hz, 1H), 3.98 (dd, J = 11.8, 2.7 Hz, 1H), 3.88–3.77 (m, 1H), 3.63 (s, 3H), 2.38 (dd, J = 14.0, 8.0 Hz, 1H), 2.12 (ddd, J = 13.8, 9.2, 4.6 Hz, 1H), 1.37 (s, 9H), 0.94 (s, 9H).
(2S,4R)-1-((S)-2-((tert-Butoxycarbonyl)amino)-3,3-dimethylbutanoyl)-4-(difluoromethoxy)pyrrolidine-2-carboxylic acid (26)
To a 0 °C solution of 25 (1200 mg, 2.9 mmol in methanol (2.5 mL) and THF (2.5 mL) was added lithium hydroxide monohydrate (247 mg, 5.88 mmol) in water (2.5 mL). The cooling bath was removed and the reaction was stirred 2h. Dilution with water (20 mL) and acidification with 1 M hydrochloric acid (8 mL) followed. The reaction mixtures were extracted with ethyl acetate (3× 20 mL). The combined organics were dried over sodium sulfate, then concentrated in vacuo to afford 26 (1010 mg, 87%) as a white solid. LCMS m/z 395.5 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 12.63 (s, 1H), 6.75 (t, J = 75.5 Hz, 1H), 6.67 (d, J = 9.3 Hz, 1H), 4.87 (s, 1H), 4.26 (t, J = 8.4 Hz, 1H), 4.13 (d, J = 9.3 Hz, 1H), 3.96 (d, J = 11.7 Hz, 1H), 3.79 (dd, J = 11.8, 4.0 Hz, 1H), 2.37 (dd, J = 14.1, 8.1 Hz, 1H), 2.10 (ddd, J = 13.8, 9.1, 4.7 Hz, 1H), 1.37 (s, 9H), 0.94 (s, 9H).
tert-Butyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-4-(difluoromethoxy)yrrolidine-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (27)
To a stirred solution of 26 (500 mg, 1.27 mmol) in DMF (10 mL) at 0 °C was added HATU (530 mgg, 1.39 mmol). After 15 min, 18 (217 mg, 1.27 mmol) was added to the reaction mixture followed by dropwise addition of N,N-diisopropylethylamine (492 mg, 3.80 mmol). The cooling bath was removed and the reaction was stirred for 2h. The reaction mixture was poured into water (20 mL) and extracted with ethyl acetate (2 × 15 mL). Combined organics were washed with brine, dried over sodium sulfate and concentrated in vacuo. Silica gel chromatography (0–10% methanol in DCM) afforded 27 (410 mg, 59% yield) as a white solid. LCMS m/z 548.6 [M+H+]. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (d, J = 8.9 Hz, 1H), 7.55 (s, 1H), 7.35–7.26 (m, 1H), 7.10–7.01 (m, 1H), 6.76 (t, J = 75.6 Hz, 1H), 6.72 (d, J = 9.2 Hz, 1H), 4.87 (s, 1H), 4.42 (t, J = 8.0 Hz, 1H), 4.27 (ddd, J = 12.4, 8.9, 3.5 Hz, 1H), 4.10 (d, J = 9.3 Hz, 1H), 3.93 (d, J = 11.7 Hz, 1H), 3.82–3.73 (m, 1H), 3.40–3.27 (m, 1H), 3.19–2.97 (m, 2H), 2.37–2.22 (m, 1H), 2.22–2.10 (m, 1H), 2.08–1.88 (m, 2H), 1.70–1.55 (m, 1H), 1.54–1.40 (m, 1H), 1.36 (s, 9H), 0.92 (s, 9H).
(2S,4R)-N-((S)-1-Amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-(difluoromethoxy)pyrrolidine-2-carboxamide (28)
A solution of 27 (400 mg, 0.73 mmol) in ethyl acetate (5 mL) was cooled to 0 °C. Hydrogen chloride (4 M in ethyl acetate, 10 mL) was added. The cooling bath was removed and the reaction was stirred for 2h, then concentrated in vacuo to afford 28 (360 mg, > 99%) as a white solid. The crude material was used without purification. LCMS m/z 448.2 [M+H]+.
(2S,4R)-N-((S)-1-Cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-4-(difluoromethoxy)-1-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)pyrrolidine-2-carboxamide (6)
A slurry of 28 (1040 mg, ≤ 2.1 mmol) in isopropyl acetate was cooled to 0 °C. NMM (2.43 g, 24.0 mmol, 2.6 mL) was added dropwise at a rate such that the internal temp remained below 3 °C, followed by dropwise addition of TFAA (2.3 g, 11 mmol, 1.5 mL) maintaining an internal temperature <10 °C. Once the addition was completed, the resulting light slurry was stirred at 0 °C for 1h. The reaction was quenched with dropwise addition of methanol (1 mL). The mixture was removed from the cooling bath and stirred at room temperature for 30 min. Following addition of ammonia in methanol (7 N, 35.5 mg, 2.08 mmol, 0.30 mL) the reaction was stirred for an additional 30 min. The reaction mixture was diluted by MTBE/ethyl acetate (1:1, 60 mL), washed by water (25 mL), then 1 N hydrochloric acid/saturated sodium chloride (2:1, 30 mL), saturated sodium chloride (25 mL), saturated sodium bicarbonate/saturated sodium chloride (2:1, 30 mL). The separated organic phase was dried over magnesium sulfate, concentrated to obtain 943 mg crude which was combined with 74 mg crude from a second small scale reaction. The combined material was slurried in 12 mL of 10% ethyl acetate:MTBE at 50 °C overnight. After filtration and drying, 6 (903 mg, 73% combined yield) was obtained as a white solid. HRMS calc for C21H28F5N5O5 [M+H]+: 526.2083; found: 526.2089. 1H NMR (600 MHz, DMSO-d6) δ 9.50 (d, J = 8.7 Hz, 1H), 9.07 (d, J = 8.7 Hz, 1H), 7.68 (s, 1H), 6.78 (t, J = 75.2 Hz, 1H), 4.98 (ddd, J = 11.2, 8.7, 4.8 Hz, 1H), 4.93 (s, 1H), 4.53 (d, J = 8.8 Hz, 1H), 4.32 (dd, J = 9.2, 7.5 Hz, 1H), 3.90 (d, J = 11.8 Hz, 1H), 3.85 (dd, J = 11.7, 3.8 Hz, 1H), 3.14 (t, J = 9.1 Hz, 1H), 3.05 (td, J = 9.3, 7.4 Hz, 1H), 2.49–2.42 (m, 1H), 2.32 (dd, J = 13.7, 7.4 Hz, 1H), 2.21–2.12 (m, 1H), 2.12–2.01 (m, 2H), 1.77–1.64 (m, 2H), 0.98 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 177.60, 170.75, 167.94, 156.79 (q, J = 37.1 Hz), 119.62, 116.94 (t, J = 256.1 Hz), 115.85 (q, J = 287.5 Hz), 74.53 (t, J = 3.6 Hz), 58.14, 57.93, 54.09, 37.70, 36.66, 35.26, 35.22, 34.40, 26.88, 26.08. 19F NMR (470 MHz, DMSO-d6) δ −72.89, −81.15 (dd, J = 75.3, 5.7 Hz).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-4-(difluoromethoxy)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (29)
A solution of 28 (164 mg, 0.34 mmol) in DCM (5 mL) was cooled to 0 °C. Triethylamine (103 mg, 1.0 mmol) was added, followed by methyl chloroformate (72 mg, 0.75 mmol). The cooling bath was removed and reaction was stirred for 2h. The reaction was quenched with dropwise addition of saturated sodium carbonate (30 mL), then extracted with DCM (3 × 20 mL). The combined organics washed with brine, dried over sodium sulfate, then concentrated in vacuo to afford crude 29 (84 mg) as a white solid. LCMS m/z 528.3 [M+Na]+. The material was used directly in subsequent step.
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)carbamoyl)-4-(difluoromethoxy)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (7)
29 (84 mg, 0.17 mmol) was dissolved in DCM (5 mL). Burgess reagent was added and the reaction was stirred for 3h. Additional Burgess reagent (59.4 mg, 0.25 mmol) was charged and the reaction was stirred for 1h then diluted with water (5 mL). The reaction mixture was extracted with DCM (3 × 10 mL). The combined organics were washed with saturated sodium bicarbonate (10 mL) then saturated sodium chloride (2 × 10 mL), dried over sodium sulfate and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 5% methanol in DCM) afforded 7 (21 mg, 26% over two steps) as a white solid. HRMS calc. for C21H31F2N5O6 [M+H]+: 488.2315; found: 488.2318. 1H NMR (600 MHz, DMSO-d6) δ 9.03 (d, J = 8.7 Hz, 1H), 7.66 (s, 1H), 7.29 (d, J = 9.0 Hz, 1H), 6.78 (t, J = 75.3 Hz, 1H), 4.96 (ddd, J = 11.3, 8.7, 4.8 Hz, 1H), 4.92 (s, 1H), 4.28 (t, J = 8.2 Hz, 1H), 4.12 (d, J = 9.0 Hz, 1H), 3.93 (d, J = 11.7 Hz, 1H), 3.83 (dd, J = 11.5, 3.7 Hz, 1H), 3.52 (s, 3H), 3.12 (t, J = 9.1 Hz, 1H), 3.04 (q, J = 9.3 Hz, 1H), 2.56–2.50 (m, 1H), 2.29 (dd, J = 13.4, 7.3 Hz, 1H), 2.17 (ddd, J = 13.4, 11.4, 4.2 Hz, 1H), 2.10 (dd, J = 12.9, 6.9 Hz, 1H), 2.04 (ddd, J = 13.6, 9.6, 4.6 Hz, 1H), 1.76–1.61 (m, 2H), 0.93 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 177.61, 170.93, 169.89, 156.93, 119.69, 116.95 (t, J = 256.0 Hz), 74.57, 59.16, 58.03, 53.80, 51.51, 37.64, 36.60, 35.29, 34.77, 34.31, 26.91, 26.17. 19F NMR (470 MHz, DMSO-d6) δ −81.09 (d, J = 50.0 Hz), −81.25 (d, J = 49.8 Hz).
tert-Butyl (2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-4-(trifluoromethyl)pyrrolidine-1-carboxylate (31)
To a −30 °C mixture of 30 ((4R)-1-(tert-butoxycarbonyl)-4-(trifluoromethyl)-l-proline, 429 mg, 1.51 mmol) and 18 (346 mg, 1.67 mmol) in DMF (7.8 mL) was added N,N-diisopropylethylamine (0.791 mL, 4.54 mmol), followed by HATU (633 mg, 1.66 mmol). The reaction mixture was allowed to warm to 0 °C over 1h, whereupon it was diluted with aqueous sodium bicarbonate solution (30 mL) and extracted with a mixture of 2-butanol and DCM (9:1, 3 × 7 mL). The combined organic layers were concentrated in vacuo and purified via silica gel chromatography (Gradient: 0% to 100% methanol in DCM), affording 31 (613 mg, 93%) as an off-white foam. LCMS m/z 459.3 [M+Na]+. 1H NMR (400 MHz, DMSO-d6),: d 8.33–8.18 (m, 1H), [7.65 (br s) and 7.59 (br s), total 1H], [7.39 (br s) and 7.27 br (s), total 1H], 7.05 (br s, 1H), 4.38–4.28 (m, 1H), 4.28–4.17 (m, 1H), 3.46–3.36 (m, 1H), 2.02–1.89 (m, 1H), 1.80–1.45 (m, 2H), [1.39 (s) and 1.32 (s), total 9H]. Prominent conformational isomers: roughly 55:45.
tert-Butyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (32)
A mixture of 31 (242 mg, 0.554 mmol) and hydrogen chloride in dioxane (4 M; 2 mL, 8 mmol) was stirred at room temperature for 5 min, whereupon the reaction mixture was concentrated in vacuo. The resulting deprotected material was combined with N-Boc-l-tert-leucine (128 mg, 0.553 mmol) and HATU (232 mg, 0.610 mmol) in DMF (2 mL), and then cooled to −30 °C. N,N-Diisopropylethylamine (0.290 mL, 1.66 mmol) was added, and the reaction mixture was warmed to 0 °C over 1h. After addition of aqueous sodium bicarbonate solution, the resulting mixture was extracted three times with ethyl acetate. The combined organic layers were concentrated in vacuo and purified via silica gel chromatography (Gradient: 0% to 30% methanol in DCM), affording 32 (230 mg, 75%) as a solid. LCMS m/z 550.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.28 (d, J = 8.8 Hz, 1H), 7.55 (s, 1H), 7.29 (s, 1H), 7.03 (s, 1H), 6.77 (d, J = 9.1 Hz, 1H), 4.48 (t, J = 7.1 Hz, 1H), 4.24 (ddd, J = 12.2, 8.7, 3.5 Hz, 1H), 4.12 (d, J = 9.3 Hz, 1H), 4.02–3.84 (m, 1H), 3.20–2.93 (m, 2H), 2.50–2.35 (m, 1H), 2.31–2.20 (m, 1H), 2.19–2.05 (m, 2H), 1.94 (ddd, J = 13.6, 12.0, 3.6 Hz, 1H), 1.69–1.55 (m, 1H), 1.50 (ddd, J = 13.7, 11.6, 3.7 Hz, 1H), 1.36 (s, 9H), 1.29–1.18 (m, 2H), 0.93 (s, 9H).
(2S,4R)-N-((S)-1-Cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)-1-((S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl)-4-(trifluoromethyl)pyrrolidine-2-carboxamide (8)
A mixture of 32 (230 mg, 0.418 mmol) and a hydrogen chloride in dioxane (4 M; 2 mL, 8 mmol) was stirred at room temperature for 5 min, whereupon the reaction mixture was concentrated in vacuo. The resulting deprotected material was combined with ethyl trifluoroacetate (595 mg, 4.19 mmol) and N,N-diisopropylethylamine (0.219 mL, 1.26 mmol) in methanol (1.0 mL). After the reaction mixture had been stirred at room temperature for 30 min, additional ethyl trifluoroacetate (60 mg, 0.422 mmol) was added, and stirring was continued for 30 min. Aqueous sodium bicarbonate solution was then added, and the resulting mixture was extracted three times with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, concentrated in vacuo, and dissolved in DCM (3 mL). To this was added Burgess reagent (299 mg, 1.25 mmol), and the reaction mixture was stirred for 2h, whereupon it was treated with additional Burgess reagent (100 mg, 0.420 mmol) and allowed to stir for a further 30 min. Dilute aqueous sodium carbonate solution was then added, and the mixture was extracted twice with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification via supercritical fluid chromatography (Column: Princeton Dinitrophenyl, 10 × 250 mm, 5 μm; Mobile phase: 9:1 carbon dioxide/methanol; Back pressure: 120 bar; Flow rate: 80 mL/min) afforded material that was then slurried in heptane (2.0 mL) at 50 °C for 2h, cooled to room temperature, and collected via filtration, providing 8 (64 mg, 29%) as a solid. HRMS calc for C21H27F6N5O4 [M+H]+: 528.2030; found: 528.2040. 1H NMR (600 MHz, DMSO-d6) δ 9.48 (d, J = 8.5 Hz, 1H), 9.06 (d, J = 8.6 Hz, 1H), 7.68 (s, 1H), 4.96 (ddd, J = 11.1, 8.6, 4.9 Hz, 1H), 4.56 (d, J = 8.5 Hz, 1H), 4.37 (t, J = 7.4 Hz, 1H), 3.98 (dd, J = 11.2, 7.6 Hz, 1H), 3.93 (dd, J = 11.2, 4.7 Hz, 1H), 3.33 (s, 2H), 3.14 (t, J = 9.1 Hz, 1H), 3.04 (td, J = 9.3, 7.2 Hz, 1H), 2.33 (ddd, J = 13.7, 8.4, 5.5 Hz, 1H), 2.20–2.02 (m, 3H), 1.76–1.62 (m, 2H), 0.99 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 177.52, 170.77, 167.82, 156.91 (q, J = 36.9 Hz), 127.06 (q, J = 278.3 Hz), 119.56, 115.85 (q, J = 288.3, 287.8, 287.2 Hz), 58.72, 57.85, 47.03, 41.27 (q, J = 28.0 Hz), 39.30, 37.78, 36.68, 34.86, 34.27, 28.24, 26.86, 26.06. 19F NMR (470 MHz, DMSO-d6) δ −70.29 (d, J = 9.7 Hz), −72.92.
Methyl (2S,4R)-4-(trifluoromethyl)pyrrolidine-2-carboxylate hydrochloride (33)
The reaction was carried out in six parallel batches: To a solution of 30 (400 g, 1.41 mol) in methanol (3.2 L) was added dropwise thionyl chloride (386.42 g, 3.25 mol, 235.62 mL) over a period 1 h, while keeping the reaction temperature between 0 and 5 °C. After reagent addition, the reaction was warmed to room temperature and left to stir for 16h. [Note: Off-gassing was observed and was passed through 8N aqueous sodium hydroxide trap.] Each reaction was concentrated and triturated with MTBE (2 L) for 1 h. The batches were combined and filtered. The filter cake was washed with MTBE (2 L × 2), dried under vacuum to give 33 (1.9 kg, 96% yield) as a white solid. LCMS m/z 195.8 [M+H]+. 1H NMR (400 MHz, DMSO) δ 9.94 (s, 1H), 4.58 (dd, J = 8.7, 7.1 Hz, 1H), 3.78 (s, 3H), 3.66 (dd, J = 12.2, 8.9 Hz, 1H), 3.49 (qt, J = 9.2, 6.9 Hz, 1H), 3.28 (dd, J = 12.2, 7.2 Hz, 1H), 2.48–2.30 (m, 2H).
Methyl N-(methoxycarbonyl)-3-methyl-l-valyl-(4R)-4-(trifluoromethyl)-l-prolinate (34)
The reaction was carried out in six parallel batches: To a solution of 33 (315.5 g, 1.35 mol) in DMF (2860 mL) and acetonitrile (640 mL) was N-(methoxycarbonyl)-l-tert-leucine (281.08 g, 1.49 mol), followed by N,N-diisopropylethylamine (558.54 g, 4.32 mol, 752.75 mL) then portionwise addition of HATU (564.85 g, 1.49 mol) keeping the reaction at 0–5 °C. After reagent addition, the mixture was warmed to room temperature and left to stir for 16h. The reaction mixture was quenched with water (1.5 L) and extracted with ethyl acetate (3× 1 L). The combined organics were washed with brine (2 L × 4), dried over magnesium sulfate, filtered and concentrated under vacuum to give the crude product. Silica gel chromatography (0–25% ethyl acetate in petroleum ether) afforded 34 (2.78 kg, 93% yield) as white solid. MS m/z 369.4 [M+H]+. 1H NMR (400 MHz, DMSO–D6) δ 7.23 (d, J = 8.6 Hz, 1H), 4.47 (dd, J = 9.1, 4.9 Hz, 1H), 4.14 (d, J = 8.7 Hz, 1H), 3.91 (q, J = 10.6, 9.3 Hz, 2H), 3.60 (s, 3H), 3.48 (d, J = 9.2 Hz, 3H), 2.33 (dt, J = 15.6, 8.5 Hz, 1H), 2.16 (ddd, J = 13.3, 7.9, 5.2 Hz, 1H), 0.93 (s, 9H), 0.85 (s, 1H).
N-(Methoxycarbonyl)-3-methyl-l-valyl-(4R)-4-(trifluoromethyl)-l-proline (35)
The reaction was carried out in eight parallel batches: To a solution of 34 (347.5 g, 943.40 mmol) in THF (975 mL), methanol (97.5 mL) and water (975 mL) was added lithium hydroxide monohydrate (98.97 g, 2.36 mol) in portions keeping the reaction at 0–5 °C. The reaction was then allowed to warm to room temperature for 1h. The reaction mixture was cooled to 0 °C then acidified by gradual addition of hydrochloric acid (3 M, 786.17 mL, 2.5 equiv) to pH = ∼2. The reaction mixture was extracted with ethyl acetate/2-methylTHF (v/v= 2:1, 1 L × 3). The combined organics were washed with saturated sodium chloride (500 mL), dried over magnesium sulfate, and filtered. The filtrates from each reaction were combined and concentrated to give crude 35 as white solid. The solid was suspended in isopropyl acetate (970 mL), n-heptane (6.05 L) was added, and the mixture was heated to 40 °C for 30 min to give a white slurry. Additional n-heptane (12.1 L) was added at 40 °C, then the mixture was heated to 70 °C for 1 h. Heat was discontinued and the mixture was allowed to cool to 20 °C, then cooled to 0–5 °C for 30 min with an ice water bath. The mixture was filtered. The filter cake was washed with isopropyl acetate/n-heptane (20:1, 1 L × 2), dried under vacuum to give 35 (2.5 kg, 90% yield) as white solid. MS m/z 377.3 [M+Na]+. 1H NMR (400 MHz, DMSO) δ 12.74 (s, 1H), 7.25 (d, J = 8.8 Hz, 1H), 4.42 (dd, J = 9.0, 5.0 Hz, 1H), 4.18 (d, J = 8.9 Hz, 1H), 3.95 (d, J = 7.0 Hz, 2H), 3.54 (s, 3H), 2.41–2.29 (m, 1H), 2.18 (ddd, J = 13.3, 7.8, 5.1 Hz, 1H), 0.98 (s, 9H), 0.90 (s, 1H).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (36)
The reaction was carried out in six parallel batches: To a suspension of 35 (410 g, 1.16 mol) in methyl ethyl ketone (2.5 L) was added (96.42 g, 867.86 mmol) in one portion, followed by dropwise triethylamine (175.64 g, 1.74 mol, 241.59 mL) at 20 °C. During the addition of triethylamine, the temperature rose from 20 to 25 °C and the reaction turned into a clear solution. To the reaction mixture was added 18 (276.33 g, 1.33 mol) in one portion, followed by EDC (332 g, 1.74 mol) in one portion. The reaction was stirred at 20 °C for 16 h. The reaction was diluted with ethyl acetate (50 mL) followed by addition of 14% aq. sodium chloride (1.6 L) then stirred for 5 min to give a clear solution, then separated. The organic layer was washed with 14% aqueous sodium chloride (1.6 L), dried over sodium sulfate, filtered and concentrated in vacuo to give 36 (4180 g, crude) as yellow solid. MS m/z 508.2
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (9)
The reaction was carried out in ten batches: To a solution of 36 (418 g, 824 mmol) in isopropyl acetate (2.5 L) was added NMM (333 g, 329.5 mmol, 362 mL), followed by dropwise TFAA (346 g, 164.7 mmol, 229 mL) at 15–20 °C under N2. During addition of TFAA, the reaction was exothermic. After addition, the reaction was stirred at 15–20 °C for 1 h. A solution of ammonium hydroxide (93.94 g, 693.59 mmol, 102.7 mL, 26%) in water (1200 mL) was added gradually. During the addition, the temperature rose from 20 to 25 °C. After quennching, the reaction was stirred at room temperature for 1 h. The organic layer washed twice with water (1200 mL, then 800 mL). The organic layers from the ten batches were combined, dried over magnesium sulfate, filtered, and concentrated to 5 L volume. To the residue was added cyclopentyl methyl ether (12.5 L) followed by concentration under vacuum at 50 °C to ∼3 L volume. To the residue was added further cyclopentyl methyl ether (12.5 L) and concentrated under vacuum at 50 °C to ∼2.5 L volume. The mixture was further diluted with cyclopentyl methyl ether (10 L), heated to 60 °C, where n-heptane (2.5 L) was then added in portions, maintaining the temperature at 60 °C. The mixture was stirred at 60 °C for 1 h, then cooled to 20 °C over 6 h. The mixture was filtered. The filter cake was washed with cyclopentyl methyl ether/n-heptane (v/v = 5:1, 2 L × 3) then dried in oven at 50 °C for 48 h to give 9 (1.81 kg, 53% yield over two steps) as off-white solid. HRMS calc. for C21H30F3N5O5 [M+H]+: 490.2272; found: 490.2259. 1H NMR (600 MHz, DMSO-d6) δ 9.03 (d, J = 8.6 Hz, 1H), 7.66 (s, 1H), 7.28 (d, J = 8.7 Hz, 1H), 4.95 (ddd, J = 11.1, 8.6, 5.0 Hz, 1H), 4.36 (t, J = 7.3 Hz, 1H), 4.14 (d, J = 8.7 Hz, 1H), 3.96 (d, J = 6.7 Hz, 2H), 3.52 (s, 3H), 3.40 (td, J = 13.9, 12.4, 7.1 Hz, 1H), 3.13 (t, J = 9.1 Hz, 1H), 3.03 (td, J = 9.3, 7.3 Hz, 1H), 2.45 (ddt, J = 14.3, 10.5, 4.2 Hz, 1H), 2.29 (dt, J = 13.9, 8.1 Hz, 1H), 2.16 (ddd, J = 13.5, 11.2, 4.3 Hz, 1H), 2.08 (tt, J = 13.3, 7.0 Hz, 2H), 1.73–1.64 (m, 2H), 0.94 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 177.51, 170.95, 169.86, 157.01, 127.31 (q, J = 280.8 Hz), 119.60, 58.98, 58.60, 51.53, 46.78, 41.28 (q, J = 28.0 Hz), 37.74, 36.64, 34.44, 34.18, 28.24, 26.88, 26.15. 19F NMR (470 MHz, DMSO-d6) δ −70.15 (d, J = 9.7 Hz).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (9) Prepared as a 75% Loaded Spray-Dried Dispersion
Compound 9 was dissolved with hydroxypropyl methylcellulose acetate succinate (HPMCAS-M) polymer in a volatile organic solvent and the resulting solution was atomized concurrently with an inert drying gas to afford a 75% Compound 9:HPMCAS-M spray-dried dispersion (SDD). The 75% loaded SDD of compound 9 was used for in vivo work at 99.6% purity (see Supporting Information for HPLC trace).
tert-Butyl ((S)-1-amino-1-oxo-3-((S)-2-oxopiperidin-3-yl)propan-2-yl)carbamate (38)
The reaction was set up in three parallel batches. To a solution of 37 (methyl (S)-2-((tert-butoxycarbonyl)amino)-3-((S)-2-oxopiperidin-3-yl)propanoate, 144.3 g, 480.44 mmol, 1 equiv) in methanol (300 mL) was added ammonia in methanol (7 M, 3.00 L, 43.71 equiv). The reaction mixture was stirred 16h at 35 °C. The crude residues were combined with a previous (44.8 g) batch and purified by silica gel chromatography (0–7% methanol in DCM) to afford 38 (281 g, 68% yield) as a yellow solid. MS m/z 286.1 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.46 (s, 1H), 7.24 (s, 1H), 6.97 (s, 1H), 6.92 (d, J = 8.5 Hz, 1H), 3.97–3.84 (m, 1H), 3.14–3.05 (m, 2H), 2.20–1.98 (m, 2H), 1.95–1.83 (m, 1H), 1.81–1.68 (m, 2H), 1.53 (ddt, J = 13.5, 8.9, 4.9 Hz, 2H), 1.37 (s, 9H).
(S)-2-Amino-3-((S)-2-oxopiperidin-3-yl)propanamide hydrochloride (39)
To a solution of 38 (66 g, 231.30 mmol) in ethyl acetate (300 mL) was added hydrogen chloride in ethyl acetate (4 M, 660.00 mL, 11.41 equiv) at 0–5 °C. Then the reaction mixture was then stirred for 16h at room temperature. The reaction mixture was concentrated in vacuo. The crude product combined with additional material (317 g) from an earlier reaction. The combined crude product was triturated by MTBE (1000 mL) at 25 °C for 16 h, then filtered. The filter cake was dried in vacuum to give crude product (336 g, crude, 90%) as a white solid. A 100 g portion of this material was further purified via slurry in 5% isopropanol/water (1040 mL, 10 V) for 90 min. Filtration followed by drying under vacuum afforded 39 (57.1 g, overall yield 52%) MS m/z 186.1 [M+H]+. 1H NMR (600 MHz, DMSO-d6) δ 8.37 (s, 3H), 8.03 (s, 1H), 7.83 (s, 1H), 7.55 (s, 1H), 3.83 (dd, J = 9.5, 4.4 Hz, 1H), 3.25–2.94 (m, 2H), 2.46–2.37 (m, 1H), 2.09 (ddd, J = 14.5, 9.5, 7.4 Hz, 1H), 1.99–1.91 (m, 1H), 1.80–1.66 (m, 2H), 1.66–1.55 (m, 1H), 1.49–1.38 (m, 1H).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopiperidin-3-yl)propan-2-yl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (40)
N,N-diisopropylethylamine (383 mg, 2.96 mmol, 0.516 mL) was added to a slurry of 35 (300 mg, 0.847 mmol), 39 (0.198 g, 0.808 mmol) and 2-hydroxypyridine 1-oxide (70.6 mg, 0.635 mmol) in methyl ethyl ketone (3.0 mL, c = 0.3 m) at 25 °C. EDC (211 mg, 1.10 mmol) was added, then the reaction slurry was stirred overnight. The reaction was quenched with 1 N aqueous hydrochloric acid/saturated aqueous sodium chloride (1:1, 20 mL) then extracted with 50 mL of ethyl acetate. The resulting organic layer was washed with 20 mL of saturated aqueous sodium chloride and with saturated aqueous sodium bicarbonate/saturated aqueous sodium chloride (1:1, 20 mL) then dried over magnesium sulfate. The combined organics were filtered and concentrated in vacuo. Following an azeotrope with heptane, the procedure yielded crude 40 (340 mg, 77%) as a glass which was used in the next step without further purification. MS m/z 522.5 [M+H]+.
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopiperidin-3-yl)ethyl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (10)
NMM (406 mg, 4.01 mmol, 0.44 mL) was added to a slurry of 40 (332.0 mg, 0.57 mmol) in isopropyl acetate (2.5 mL, c = 0.23 m) at −5 °C followed by dropwise addition of TFAA (361 mg, 1.72 mmol, 0.24 mL) via syringe over 5 min. The resulting solution was stirred at 0 °C for 1 h then quenched with methanol (147 mg, 4.58 mmol, 0.2 mL). The resulting light-yellow solution was allowed to warm to room temperature and stirred for 15 min. Ammonia in methanol (7 N, 39.0 mg, 2.29 mmol, 0.3 mL) was then added to the reaction mixture and stirred for 30 min. The resulting reaction mixture was washed with 10 mL of 1 N hydrochloric acid/saturated aqueous sodium chloride (1:1, 10 mL) and extracted with MTBE (60 mL). The combined organics were washed with saturated aqueous sodium chloride (20 mL), then saturated aqueous sodium bicarbonate/saturated aqueous sodium chloride (1:1, 20 mL), and dried over magnesium sulfate before filtering and concentration in vacuo. The residue was dissolved in 1 mL of methyl acetate at 50 °C followed by dropwise addition of 1.5 mL of heptane. Stirring at 50 °C for 72 h led to solvent evaporation and left a solid remaining. The solid was suspended in 2.5 mL of 1:1 ethyl acetate/heptane then heated to 50 °C and stirred for 1 h followed by 1 h of stirring at 25 °C, whereupon the solid was collected by filtration, rinsed with 25% ethyl acetate/heptane (2× 2 mL) then dried under vacuum for 1 h to yield 10 (118 mg, 40%) as a white solid. HRMS calc. for C22H32F3N5O5 [M+H]+: 504.2428; found: 504.2412. 1H NMR (600 MHz, DMSO-d6) δ 8.98 (d, J = 8.3 Hz, 1H), 7.50 (s, 1H), 7.24 (d, J = 8.7 Hz, 1H), 4.99 (ddd, J = 10.8, 8.4, 5.7 Hz, 1H), 4.44–4.31 (m, 1H), 4.14 (d, J = 8.7 Hz, 1H), 3.95 (d, J = 6.4 Hz, 2H), 3.52 (s, 3H), 3.44–3.35 (m, 1H), 3.07 (tdd, J = 9.7, 8.6, 7.3, 3.7 Hz, 2H), 2.35 (ddd, J = 13.5, 10.8, 5.4 Hz, 1H), 2.28 (qd, J = 11.7, 10.0, 4.3 Hz, 2H), 2.07 (dt, J = 13.6, 7.2 Hz, 1H), 1.93–1.79 (m, 1H), 1.78–1.70 (m, 1H), 1.68 (dq, J = 12.9, 4.5 Hz, 1H), 1.62–1.52 (m, 1H), 1.34 (qd, J = 11.5, 3.4 Hz, 1H), 0.95 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 171.94, 170.88, 169.80, 157.01, 127.09 (q, J = 278.2 Hz), 119.74, 59.02, 58.53, 51.54, 46.78, 41.15, 37.72, 36.49, 34.49, 34.45, 28.29, 26.20, 25.67, 21.25. 19F NMR (470 MHz, DMSO-d6) δ −70.21 (d, J = 9.6 Hz).
Methyl (2S,4R)-1-((methoxycarbonyl)-l-valyl)-4-(trifluoromethyl)pyrrolidine-2-carboxylate (41)
To a solution of 33 (5000 mg, 25.36 mmol) and N-(methoxycarbonyl)-l-valine (4530 mg, 25.4 mmol) in dimethylformamide (45 mL, c = 0.56 m) at 0 °C was added HATU (11.6 g, 30.4 mmol) followed by a dropwise addition of N,N-diisopropylethylamine (9830 mg, 76.1 mmol, 13.3 mL) over 5 min. The mixture was stirred for 5 min at 0 °C before removing cooling bath. Stirring was continued at for 20h, whereupon the reaction was poured onto 0.5 M aqueous HCl, then extracted with ethyl acetate. The organic layer was washed with water and saturated aqueous sodium chloride solution, dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification via silica gel chromatography (gradient 30–60% ethyl acetate in heptane) afforded 41 (6.9 g, 77%) as a colorless oil. MS m/z 355.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.87–1.01 (m, 6 H) 1.85–1.97 (m, 1 H) 2.13–2.27 (m, 1 H) 2.29–2.45 (m, 1 H) 3.40 (br dd, J = 16.39, 8.20 Hz, 1 H) 3.52 (s, 3 H) 3.64 (s, 3 H) 3.92–4.02 (m, 3 H) 4.52 (dd, J = 8.98, 4.29 Hz, 1 H) 7.43–7.60 (m, 1 H).
(2S,4R)-1-((Methoxycarbonyl)-l-valyl)-4-(trifluoromethyl)pyrrolidine-2-carboxylic acid (42)
Lithium hydroxide (1.0 g, 40 mmol, 40 mL, 1 M aq) was added to a stirred solution 41 (6900 mg, 19.47 mmol) in THF (50 mL) and methanol (50 mL). The reaction mixture was stirred for 1 h, whereupon the mixture was concentrated in vacuo to remove organic solvents. The residue was diluted further with water and adjusted to pH 3 with 1N hydrochloric acid then extracted with ethyl acetate. The organic layer was washed with water, then saturated aqueous sodium chloride solution and dried over magnesium sulfate, filtered and concentrated in vacuo to yield 42 (6.1 g, 91%) as a colorless foam. MS m/z 341.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 12.75 (s, 1H), 7.48 (d, J = 8.4 Hz, 1H), 4.42 (dd, J = 9.1, 4.4 Hz, 1H), 4.14–3.88 (m, 3H), 3.52 (s, 3H), 3.44–3.30 (m, 1H), 2.41–2.25 (m, 1H), 2.17 (ddd, J = 13.5, 7.8, 4.5 Hz, 1H), 1.93 (ddd, J = 13.3, 8.7, 6.6 Hz, 1H), 0.95–0.80 (m, 6H).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl)carbamate (43)
To a stirred solution of 42 (380 mg, 1.12 mmol) and 18 (255 mg, 1.23 mmol) in DMF (6 mL) at 0 °C was added HATU (467, 1.23 mmol) followed by N,N-diisopropylethylamine (649 mg, 5.02 mmol). The cooling bath was removed and the reaction was stirred for 18h. Dilution with water (20 mL) and extraction with chloroform/isopropanol = 4:1 (4 × 30 mL) followed. The combined organics were dried over sodium sulfate and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 15% methanol in DCM) afforded 43 (420 mg, 76%) as a white solid. MS m/z 494.3 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (d, J = 8.7 Hz, 1H), 7.56 (br s, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.31 (br s, 1H), 7.04 (br s, 1H), 4.51 (dd, J = 8.4, 4.9 Hz, 1H), 4.23 (ddd, J = 12.2, 8.6, 3.7 Hz, 1H), 3.94 (d, J = 7.7 Hz, 2H), 3.68–3.55 (m, 0H), 3.51 (s, 3H), 3.18–2.97 (m, 3H), 2.34–2.03 (m, 2H), 1.98–1.83 (m, 1H), 1.68–1.41 (m, 2H), 1.33–1.20 (m, 4H), 0.88 (dd, J = 6.7, 3.8 Hz, 6H).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopyrrolidin-3-yl)ethyl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl)carbamate (11)
43 (420 mg, 0.851 mmol) was dissolved in DCM (10 mL). Burgess reagent (608 mg, 2.55 mmol) was added and the reaction was stirred for 30h. Dilution with water and extraction with DCM (3 × 20 mL) followed. The combined organics were washed with brine (20 mL), dried over sodium sulfate and then concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 10% MeOH in DCM) gave 11 (220 mg, 54%) as a white solid. HRMS calc. for C20H28F3N5O5 [M+H]+: 476.2115; found: 476.2098. 1H NMR (600 MHz, DMSO-d6) δ 9.02 (d, J = 8.4 Hz, 1H), 7.68 (s, 1H), 7.51 (d, J = 8.3 Hz, 1H), 4.95 (ddd, J = 10.6, 8.5, 5.6 Hz, 1H), 4.37 (dd, J = 8.3, 5.5 Hz, 1H), 4.01–3.92 (m, 3H), 3.51 (s, 3H), 3.47–3.36 (m, 1H), 3.14 (t, J = 9.1 Hz, 1H), 3.04 (td, J = 9.3, 7.1 Hz, 1H), 2.50–2.42 (m, 1H), 2.29 (dt, J = 14.9, 8.2 Hz, 1H), 2.24–2.02 (m, 3H), 1.89 (ddd, J = 13.6, 6.8, 2.8 Hz, 1H), 1.82–1.57 (m, 2H), 0.87 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 177.63, 170.97, 170.70, 156.82, 127.06 (q, J = 278.3, 278.1, 276.9 Hz), 119.56, 58.59, 57.83, 51.50, 46.17, 41.17 (presumed q, J = 27.6 Hz), 37.92, 36.80, 33.86, 29.82, 28.25, 27.08, 18.63, 18.56. 19F NMR (470 MHz, DMSO-d6) δ −70.08 (d, J = 9.5 Hz).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-amino-1-oxo-3-((S)-2-oxopiperidin-3-yl)propan-2-yl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl)carbamate (44)
To a 0 °C solution of 42 (3.0 g, 8.8 mmol) and 39 (4.40 g, 11.7 mmol) in acetonitrile (47 mL) and DMF (4 mL) was added HATU (4.26 g, 11.2 mmol) followed by NMM (3.21 g, 31.7 mmol, 3.5 mL). The solution was allowed to warm to room temperature and stirred overnight. Solids were filtered off, then the filtrate was added to half saturated aqueous sodium chloride solution and extracted three times with a 1:1:1 mixture of MEK/2-propanol/ethyl acetate. The combined organic extracts were washed with saturated aqueous sodium chloride solution then dried over sodium sulfate and concentrated in vacuo. The residue was treated with minimal DCM and the resulting solids were filtered off. The subsequent filtrate was purified via silica gel chromatography (gradient 0–20% methanol in DCM). Collected fractions were concentrated in vacuo and the residue was triturated in heptane, then filtered and dried under high vacuum to yield 44 (4.2 g, 94%) as a white solid. MS m/z 508.5 [M+H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.37 (s, 1H), 7.27–7.22 (m, 1H), 7.01 (s, 1H), 4.50 (dd, J = 8.4, 4.8 Hz, 1H), 4.25 (ddd, J = 12.4, 8.7, 3.7 Hz, 1H), 3.99–3.90 (m, 3H), 3.51 (s, 3H), 3.15–3.02 (m, 3H), 2.72 (s, 1H), 2.37–2.06 (m, 3H), 1.98–1.85 (m, 2H), 1.72–1.62 (m, 1H), 1.61–1.46 (m, 2H), 1.32–1.20 (m, 1H), 0.88 (d, J = 6.7 Hz, 6H).
Methyl ((S)-1-((2S,4R)-2-(((S)-1-cyano-2-((S)-2-oxopiperidin-3-yl)ethyl)carbamoyl)-4-(trifluoromethyl)pyrrolidin-1-yl)-3-methyl-1-oxobutan-2-yl)carbamate (12)
NMM (8.37 g, 82.8 mmol, 9.10 mL) was added to a slurry of 44 (4.20 g, 8.28 mmol) in isopropyl acetate (55.2 mL, c = 0.15 m) at −5 °C followed by the dropwise addition of TFAA (6.95 g, 33.1 mmol, 4.60 mL) via syringe (fuming observed). The reaction mixture was stirred at 0 °C for 45 min, then 2 mL of methanol was added followed by the addition of ammonia in methanol (7 N, 0.169 g, 9.93 mmol, 1.42 mL). The reaction mixture was then allowed to warm to room temperature while stirring for 15 min, whereupon the reaction was quenched with half saturated aqueous sodium chloride solution and extracted with ethyl acetate. The organic layer was washed with 1:1 aqueous 1N hydrochloride acid/saturated sodium chloride, 1:1 saturated aqueous sodium bicarbonate/saturated aqueous sodium chloride, then saturated aqueous sodium chloride. The resulting organic layer was dried over sodium sulfate, filtered and concentrated in vacuo to yield 2.49 g of a residue that was triturated in diethyl ether for 30 min then filtered. The hygroscopic solid was immediately dissolved in DCM and dried over sodium sulfate, filtered, and concentrated in vacuo. Purified via silica gel chromatography (0–10% gradient of methanol in DCM). Fractions that contained an impurity were repurified via silica gel chromatography (0–10% gradient methanol in DCM) then clean fractions from both column chromatographies were combined and concentrated in vacuo to yield 12 (1.61 g, 40%) as a white solid. HRMS calc. for C21H30F3N5O5 [M+H]+: 490.2272; found: 490.2256. 1H NMR (600 MHz, DMSO-d6) δ 9.01 (d, J = 8.4 Hz, 1H), 7.53–7.48 (m, 2H), 5.00 (ddd, J = 10.8, 8.5, 5.9 Hz, 1H), 4.37 (dd, J = 8.3, 5.4 Hz, 1H), 4.04–3.91 (m, 3H), 3.51 (s, 3H), 3.45–3.35 (m, 1H), 3.14–3.02 (m, 2H), 2.41–2.23 (m, 3H), 2.08 (ddd, J = 13.2, 7.6, 5.4 Hz, 1H), 1.90 (tdt, J = 11.6, 6.7, 3.9 Hz, 1H), 1.86–1.80 (m, 1H), 1.78–1.63 (m, 2H), 1.63–1.49 (m, 1H), 1.41–1.25 (m, 1H), 0.92–0.84 (m, 6H). 13C NMR (151 MHz, DMSO-d6) δ 172.05, 170.90, 170.67, 156.82, 127.02 (q, J = 278.1 Hz), 119.75, 58.54, 57.89, 51.50, 46.17, 41.16, 41.15 (q, J = 27.6 Hz), 37.70, 36.44, 34.38, 29.81, 28.51–27.73 (m), 25.69, 21.16, 18.73, 18.57. 19F NMR (470 MHz, DMSO-d6) δ −70.09 (d, J = 9.5 Hz).
Biochemical Determination for Human Coronavirus Mpro Assays
The respective human coronavirus Mpro in assay buffer (20 mM Tris-HCl, pH 7.3, 100 mM NaCl, 1 mM EDTA, 5 mM TCEP) and 0.1% BSA was added to assay-ready plates containing compound. The enzymatic reaction was then immediately initiated with the addition of 5 μL substrate in assay buffer. Final concentrations of respective protease and substrate are shown in the table below. Initial rates were measured by following the fluorescence of the cleaved substrate using a Spectramax (Molecular Devices) fluorescence plate reader in the kinetic format.
Cellular Antiviral Activity
The ability of compounds to inhibit viral induced cytopathic effect (CPE) against human coronaviruses (SARS-CoV-1, SARS-CoV-2, hCoV-229E, MERS) was assessed by monitoring cell viability using two different assay end points in VeroE6, MRC-5 or Vero81 cells.
VeroE6 cells that are enriched for hACE2 expression were batch inoculated with SARS-CoV-2 (USA_WA1/2020) at a multiplicity of infection (MOI) of 0.002 in a BSL-3 lab (Southern Research Institute). Virus-inoculated cells are then added to assay ready compound plates at a density of 4,000 cells/well in DMEM containing 2% heat inactivated FBS. Cells were incubated for 3 days at 37 °C with 5% CO2, a time at which virus induced CPE is 95% in the untreated, infected control conditions.
MRC-5 cells at a density of 20,000 cells/well were incubated overnight in MEM containing 5% FBS at 37 °C and 5% CO2. (Wuxi AppTech). Following addition of test compounds, HCoV-229E virus (ATCC VR-740) (200 TCID50) was added at concentrations which correspond to a multiplicity of infection (MOI) of 0.0007 to MRC-5 cells. Cells were incubated for 3 days at 35 °C with 5% CO2.
Vero81 (ATCC CCL-81) cells were batched inoculated with MERS (EMC/2012) at M.O.I. ∼ 0.01 in a BSL-3 lab (Southern Research Institute). Virus inoculated cells were then added to assay ready compound plates at a density of 4,000 cells/well in DMEM containing 2% heat inactivated FBS. Following a 4-day incubation at 37 °C with 5% CO2, a time at which virus-induced CPE is 90 to 95% in the untreated, infected control conditions.
Cell viability was evaluated using Cell Titer-Glo (Promega), according to the manufacturer’s protocol, which quantitates ATP levels. Cytotoxicity of the compounds was assessed in parallel in assay ready compound plates with noninfected cells in a BSL-2 lab.
Test compound(s) were tested either alone or in the presence of the P-glycoprotein (P-gp) inhibitor, CP-100356 at indicated concentrations. The inclusion of CP-100356 was to assess if the test compound(s) are being effluxed out of cells due to endogenous expression of P-glycoprotein in the cell line. Percent effect at each concentration of test compound was calculated based on the values for the no virus control wells and virus containing control wells on each assay plate. The concentration required for a 50% response (EC50) value was determined from these data using a 4-parameter logistic model. EC50 curves were fit to a Hill slope of 3 when >3 and the top dose achieved ≥50% effect. If cytotoxicity was detected at greater than 30% effect, the corresponding concentration data was eliminated from the EC50 determination. For cytotoxicity plates, a percent effect at each concentration of test compound was calculated based on the values for the cell only control wells and hyamine or no cell containing control wells on each assay plate. The CC50 value was calculated using a 4-parameter logistic model. A therapeutic index was then calculated by dividing the CC50 value by the EC50 value.
Antiviral activity for compound 9 was evaluated in dNHBE cells in a BSL-3 facility. The dNHBE cells (EpiAirway) were procured from MatTek Corporation (Ashland, MA) and were grown on trans-well inserts consisting of approximately 1.2 × 106 cells in MatTek’s proprietary culture medium (AIR-100-MM) added to the basolateral side, with the apical side exposed to a humidified 5% CO2 environment at 37 °C. On day 1, dNHBE cells were infected with SARS-CoV-2 strain USA-WA1/2020 at a MOI of approximately 0.0015 CCID50 per cell, and PF-07817883 (9) treatment was carried out by inclusion of drug dilutions in basolateral culture media. At day 3 and day 5, virus released into the apical compartment was harvested by the addition of 0.4 mL culture media. The virus titer was then quantified by infecting Vero76 cells in a standard end point dilution assay and virus dose that was able to infect 50% of the cell cultures (CCID50 per ml) was calculated.27 To determine the EC50 and EC90, the CCID50/ml values were normalized to that of no drug control as a percentage of inhibition and plotted against compound concentration in GraphPad Prism software by using four-parameter logistic regression.
Mouse-Adapted SARS-CoV-2 Infection and Treatment Studies
The in vivo infection studies were performed in an animal biosafety level 3 (ABSL3) facility in the AAALAC-accredited Laboratory Animal Research Center at Utah State University. The study procedures were conducted with approval by the Institutional Animal Care and Use Committee. A total of 40 BALB/c mice (Charles River, 8 week old female, n = 5 mice/group) were divided into 8 groups: group 1: untreated, uninfected (mock), group 2: untreated, infected 0 mg/kg compound 9); group 3: 100 mg/kg BID compound 9 ; group 4: 300 mg/kg BID compound 9, group 5: 500 mg/kg BID compound 9, group 6: 500 mg/kg BID compound 9, treatment starting at 12 h post infection (5). i.p.) injection of ketamine/xylazine (50 mg/kg/5 mg/kg) and inoculated intranasally (i.n.) with 1 × 105 50% cell culture infectious dose (CCID50) of SARS-CoV-2 MA10 (90 mL/nares). The mouse adapted MA10 virus20 was provided by Professor Ralph Baric (University of North Carolina). For oral (p.o.) administration, compound 9 (ASD form) was solubilized in in 1% (w/v) solplus and 0.5% (w/v) methylcellulose A4M in deionized water by Geometric dilution. Mice were dosed BID × 4 days beginning at 4 h post infection. Mice were weighed daily starting at day 0 until end of study to measure infection-associated weight loss. At 4 dpi, mice were euthanized by isoflurane inhalation. The lungs were collected and placed in 1 mL PBS and stored at −80 °C for evaluation of lung virus titers or collected for histopathology as described below. For virus titer assays, serial log10 dilutions of 1.0 mL lung tissue homogenates were performed in quadruplicate on confluent monolayers of Vero 76 cells seeded in 96-well microplates. The cells were incubated at 37 °C and 5% CO2 for 6 days and then scored for cytopathic effect (CPE) using a light microscope. Virus lung titer (CCID50/ml (Log10) was calculated by linear regression using the Reed-Muench method.27
Lung Immunohistochemistry Assessment
For immunohistochemistry staining of SARS-CoV-2 nucleocapsid protein, 4 mm sections were obtained from formalin-fixed, paraffin-embedded lung tissue and immunostained using the Leica Biosystems Bond automated stainer (performed at Histowiz, Inc., Brooklyn). Epitope retrieval was performed using citrate-based pH 6 solution for 20 min at 95 °C for heat-induced epitope retrieval (HIER). The tissue sections were then incubated with background eraser blocking reagent (Leica Biosystems) for 10 min to prevent nonspecific binding. Next, the tissue sections were incubated for 30 min with SARS-CoV-2 (COVID-19) nucleocapsid antibody [HL448] (GTX635686, GeneTex) at a 1:10,000 dilution, followed by incubation for 30 min with DAB rabbit secondary reagents (Bond Polymer Refine Detection, DS9800, Leica Biosystems). The slides were visualized using an Aperio AT2 slide scanner (Leica Biosystems).
Lung Histopathology Assessment
To assess virus-induced damage to the lungs of SARS-CoV-2 MA10-infected mice, mice were euthanized at 4 dpi and lung lobes were collected for virus titer evaluation or left lobes were fixed in 4% paraformaldehyde at 4 °C for histopathology. Fixed lung lobes were shipped to an external histology laboratory (Histowiz, Inc. (study 2) or M.D. Anderson (study 1)) for processing and blinded evaluation by an experienced veterinary pathologist and yielded similar results. Group samples (n = 5) from study 2 were processed as one H&E-stained slide from each lung specimen. Each lung sample was evaluated using a semiquantitative analysis using four parameters: perivascular inflammation, bronchial or bronchiolar epithelial degeneration or necrosis, bronchial or bronchiolar inflammation, and alveolar inflammation. A 5-point scoring system for assessment of epithelial degeneration/necrosis and inflammation was utilized: 0-within normal limits; 1-mild; scattered cell necrosis/vacuolation, few/scattered inflammatory cells, 2-moderate; multifocal vacuolation or sloughed/necrotic cells, thin layer of inflammatory cells, 3-marked; multifocal/segmental necrosis, epithelial loss/effacement, thick layer of inflammatory cells, 4-severe; coalescing areas of necrosis; parenchymal effacement, confluent areas of inflammation. A total pathology score was calculated for each mouse by adding the individual histopathological scores.
Statistics and Figures
All graphs were generated using GraphPad Prism. The statistical analysis was performed as follows. For the body weight, the % initial data from day 1 to 4, the slope of each dose group was compared to that of the placebo group (0 mg/kg compound 9) via mixed model analysis. Raw (unadjusted) p-values are reported. For the lung virus log10 titer, and histopathology score data each dose group was compared to that of the 0 mg/kg group via one-way ANOVA. Raw (unadjusted) p-values are reported.
Acknowledgments
Eugene Kadar (bioanalysis), Zhiwu Lin and Nathaniel Johnson (CYP induction studies), Jian Lin (drug metabolism), Gabrielle Gualtieri (CYP inhibition studies), Sarah Lazzaro, Mark West, Sangwoo Ryu and Sumathy Mathialagan (transporter IC50 determinations), Gregory Walker (NMR) and Jessica Gremminger (PCAG) are acknowledged for their contributions.
Glossary
Abbreviations Used
- ACE2
angiotensin converting enzyme 2 (receptor)
- ATP
adenosine triphosphate
- AUC
area under the plasma concentration–time curve
- BID
bis in die (twice a day)
- Boc
tert-butyloxycarbonyl
- CCID
cell culture infective dose
- COSY
correlation spectroscopy
- CYP
cytochrome P450
- DCM
dichloromethane, CH2Cl2
- DMEM
Dulbecco’s modified Eagle medium
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- FRET
fluorescence resonance energy transfer
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HMBC
heteronuclear multiple bond correlation
- HOPO
2-hydroxypyridine 1-oxide
- HPBCD
2-hydroxylpropyl-β-cyclodextrin
- HPLC
high-performance liquid chromatography
- HSQC
heteronuclear single quantum coherence
- K
kelvin
- LCMS
liquid chromatography–mass spectrometry
- MERS
Middle East respiratory syndrome
- NADPH
nicotinamide adenine dinucleotide phosphate
- NMM
N-methylmorpholine
- NMR
nuclear magnetic resonance
- NOESY
nuclear Overhauser effect spectroscopy
- PEG
polyethylene glycol
- SDD
spray-dried dispersion
- TFAA
trifluoroacetic acid anhydride
- TMS
trimethylsilyl
Data Availability Statement
The coordinates and structure factors have been deposited with the RCSB Protein Data Bank under accession code 8V4U.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c02469.
Use of the IMCA-CAT beamline 17-ID (or 17-BM) at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research used resources at the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA-CAT) beamline 17-ID, supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
The authors declare the following competing financial interest(s): Conflict of Interest Disclosure:C.M.N.A., J.T.A., L.A., M.A., B.M.B., M.A.B., B.B., L.M.B., R.D.C., N.R.C., A.C., K.J.C., A.D., L.D., H.E., K.A.F., R.F., S.S.G., S.E.G., S.R.G., A.S.K., E.K., L.F.L., G.H.L., Y.L., W.L., L.A.M.A., S.N., R.S.O., D.R.O., N.C.P., D.K.R., M.R.R., H.A.R., S.S., M.F.S., J.G.S., R.S., C.M.S., J.B.T., P.R.V., L.W., Q.Y., I.Y., Y.Z. are employees of Pfizer and some of the authors are shareholders in Pfizer Inc.. B.L.H. and S.A.G received funding from Pfizer to support in vivo studies and dHNBE assays.
The version of this paper that was published ASAP April 30, 2024, contained a labeling error in Figure 2 and a spelling error in the name of author Emi Kimoto. The revised version was reposted May 4, 2024.
Supplementary Material
References
- Zhu Z.; Lian X.; Su X.; Wu W.; Marraro G. A.; Zeng Y. From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir Res. 2020, 21 (1), 224. 10.1186/s12931-020-01479-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO COVID-10 Dashboard. https://covid19.who.int/ (accessed October 31, 2023).
- Toussi S. S.; Hammond J. L.; Gerstenberger B. S.; Anderson A. S. Therapeutics for COVID-19. Nat. Microbiol 2023, 8 (5), 771–786. 10.1038/s41564-023-01356-4. [DOI] [PubMed] [Google Scholar]
- Taylor P. C.; Adams A. C.; Hufford M. M.; de la Torre I.; Winthrop K.; Gottlieb R. L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol 2021, 21 (6), 382–393. 10.1038/s41577-021-00542-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox M.; Peacock T. P.; Harvey W. T.; Hughes J.; Wright D. W.; Willett B. J.; Thomson E.; Gupta R. K.; Peacock S. J.; Robertson D. L.; Carabelli A. M. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat. Rev. Microbiol 2023, 21 (2), 112–124. 10.1038/s41579-022-00809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond J.; Leister-Tebbe H.; Gardner A.; Abreu P.; Bao W.; Wisemandle W.; Baniecki M.; Hendrick V. M.; Damle B.; Simon-Campos A.; Pypstra R.; Rusnak J. M. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N. Eng. J. Med. 2022, 386 (15), 1397–1408. 10.1056/NEJMoa2118542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F.; Feng Q.; Chen Z. Efficacy and Safety of Molnupiravir Treatment for COVID-19: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Int. J. Antimicrob. Agents 2023, 62 (2), 106870. 10.1016/j.ijantimicag.2023.106870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; Kalil A. C.; Hohmann E.; Chu H. Y.; Luetkemeyer A.; Kline S.; Lopez de Castilla D.; Finberg R. W.; Dierberg K.; Tapson V.; Hsieh L.; Patterson T. F.; Paredes R.; Sweeney D. A.; Short W. R.; Touloumi G.; Lye D. C.; Ohmagari N.; Oh M.-d.; Ruiz-Palacios G. M.; Benfield T.; Fatkenheuer G.; Kortepeter M. G.; Atmar R. L.; Creech C. B.; Lundgren J.; Babiker A. G.; Pett S.; Neaton J. D.; Burgess T. H.; Bonnett T.; Green M.; Makowski M.; Osinusi A.; Nayak S.; Lane H. C. Remdesivir for the Treatment of Covid-19 - Final Report. N. Engl. J. Med. 2020, 383 (19), 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Su H.; Shang W.; Zhou F.; Zhang Y.; Zhao W.; Zhang Q.; Xie H.; Jiang L.; Nie T.; Yang F.; Xiong M.; Huang X.; Li M.; Chen P.; Peng S.; Xiao G.; Jiang H.; Tang R.; Zhang L.; Shen J.; Xu Y. Structure-based development and preclinical evaluation of the SARS-CoV-2 3C-like protease inhibitor simnotrelvir. Nat. Commun. 2023, 14 (1), 6463. 10.1038/s41467-023-42102-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman R. L.; Kalla R. V.; Babusis D.; Pitts J.; Barrett K. T.; Chun K.; Du Pont V.; Rodriguez L.; Moshiri J.; Xu Y.; Lee M.; Lee G.; Bleier B.; Nguyen A. Q.; O’Keefe B. M.; Ambrosi A.; Cook M.; Yu J.; Dempah K. E.; Bunyan E.; Riola N. C.; Lu X.; Liu R.; Davie A.; Hsiang T. Y.; Dearing J.; Vermillion M.; Gale M. Jr.; Niedziela-Majka A.; Feng J. Y.; Hedskog C.; Bilello J. P.; Subramanian R.; Cihlar T. Discovery of GS-5245 (Obeldesivir), an Oral Prodrug of Nucleoside GS-441524 That Exhibits Antiviral Efficacy in SARS-CoV-2-Infected African Green Monkeys. J. Med. Chem. 2023, 66 (17), 11701–11717. 10.1021/acs.jmedchem.3c00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukae H.; Yotsuyanagi H.; Ohmagari N.; Doi Y.; Sakaguchi H.; Sonoyama T.; Ichihashi G.; Sanaki T.; Baba K.; Tsuge Y.; Uehara T. Efficacy and Safety of Ensitrelvir in Patients With Mild-to-Moderate Coronavirus Disease 2019: The Phase 2b Part of a Randomized, Placebo-Controlled, Phase 2/3 Study. Clin Infect Dis 2023, 76 (8), 1403–1411. 10.1093/cid/ciac933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renjifo B.; van Wyk J.; Salem A. H.; Bow D.; Ng J.; Norton M. Pharmacokinetic enhancement in HIV antiretroviral therapy: a comparison of ritonavir and cobicistat. AIDS Rev. 2015, 17 (1), 37–46. [PubMed] [Google Scholar]
- Eng H.; Dantonio A. L.; Kadar E. P.; Obach R. S.; Di L.; Lin J.; Patel N. C.; Boras B.; Walker G. S.; Novak J. J.; Kimoto E.; Singh R. S. P.; Kalgutkar A. S. Disposition of Nirmatrelvir, an Orally Bioavailable Inhibitor of SARS-CoV-2 3C-Like Protease, across Animals and Humans. Drug Metab. Dispos. 2022, 50 (5), 576–590. 10.1124/dmd.121.000801. [DOI] [PubMed] [Google Scholar]
- Singh R. S. P.; Toussi S. S.; Hackman F.; Chan P. L.; Rao R.; Allen R.; Van Eyck L.; Pawlak S.; Kadar E. P.; Clark F.; Shi H.; Anderson A. S.; Binks M.; Menon S.; Nucci G.; Bergman A. Innovative Randomized Phase I Study and Dosing Regimen Selection to Accelerate and Inform Pivotal COVID-19 Trial of Nirmatrelvir. Clin Pharmacol Ther 2022, 112 (1), 101–111. 10.1002/cpt.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374 (6575), 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y.; Federico J. J. 3rd; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-Kattan A. F.; Liston T. E.; Troutman M. D. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci. 2011, 100 (11), 4974–85. 10.1002/jps.22674. [DOI] [PubMed] [Google Scholar]
- Stopher D.; McClean S. An improved method for the determination of distribution coefficients. J. Pharm. Pharmacol 2011, 42 (2), 144. 10.1111/j.2042-7158.1990.tb05373.x. [DOI] [PubMed] [Google Scholar]
- Walsky R. L.; Obach R. S. Validated assays for human cytochrome P450 activities. Drug Metab. Dispos. 2004, 32 (6), 647–60. 10.1124/dmd.32.6.647. [DOI] [PubMed] [Google Scholar]
- Sathish J. G.; Bhatt S.; DaSilva J. K.; Flynn D.; Jenkinson S.; Kalgutkar A. S.; Liu M.; Manickam B.; Pinkstaff J.; Reagan W. J.; Shirai N.; Shoieb A. M.; Sirivelu M.; Vispute S.; Vitsky A.; Walters K.; Wisialowski T. A.; Updyke L. W. Comprehensive Nonclinical Safety Assessment of Nirmatrelvir Supporting Timely Development of the SARS-COV-2 Antiviral Therapeutic, Paxlovid. Int. J. Toxicol 2022, 41 (4), 276–290. 10.1177/10915818221095489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist S. R.; Dinnon K. H. 3rd; Schafer A.; Tse L. V.; Okuda K.; Hou Y. J.; West A.; Edwards C. E.; Sanders W.; Fritch E. J.; Gully K. L.; Scobey T.; Brown A. J.; Sheahan T. P.; Moorman N. J.; Boucher R. C.; Gralinski L. E.; Montgomery S. A.; Baric R. S. A mouse-adapted SARS-CoV-2 induces acute lung injury and mortality in standard laboratory mice. Cell 2020, 183 (4), 1070–1085. 10.1016/j.cell.2020.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker M. J.; Clark T. H.; Williams J. A. Development and validation of a 96-well equilibrium dialysis apparatus for measuring plasma protein binding. J. Pharm. Sci. 2003, 92 (5), 967–74. 10.1002/jps.10332. [DOI] [PubMed] [Google Scholar]
- Greenfield S. R.; Eng H.; Yang Q.; Guo C.; Byrnes L.; Dantonio A.; West G.; Di L.; Kalgutkar A. S. Species differences in plasma protein binding of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease inhibitor nirmatrelvir. Xenobiotica 2023, 53 (1), 12–24. 10.1080/00498254.2023.2183158. [DOI] [PubMed] [Google Scholar]
- Orr S. T.; Ripp S. L.; Ballard T. E.; Henderson J. L.; Scott D. O.; Obach R. S.; Sun H.; Kalgutkar A. S. Mechanism-based inactivation (MBI) of cytochrome P450 enzymes: structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks. J. Med. Chem. 2012, 55 (11), 4896–933. 10.1021/jm300065h. [DOI] [PubMed] [Google Scholar]
- Walsky R. L.; Bauman J. N.; Bourcier K.; Giddens G.; Lapham K.; Negahban A.; Ryder T. F.; Obach R. S.; Hyland R.; Goosen T. C. Optimized assays for human UDP-glucuronosyltransferase (UGT) activities: altered alamethicin concentration and utility to screen for UGT inhibitors. Drug Metab. Dispos. 2012, 40 (5), 1051–65. 10.1124/dmd.111.043117. [DOI] [PubMed] [Google Scholar]
- Liu J. B.; Xu X. H.; Qing F. L. Silver-Mediated Oxidative Trifluoromethylation of Alcohols to Alkyl Trifluoromethyl Ethers. Org. Lett. 2015, 17 (20), 5048–51. 10.1021/acs.orglett.5b02522. [DOI] [PubMed] [Google Scholar]
- Levchenko K.; Datsenko O. P.; Serhiichuk O.; Tolmachev A.; Iaroshenko V. O.; Mykhailiuk P. K. Copper-Catalyzed O-Difluoromethylation of Functionalized Aliphatic Alcohols: Access to Complex Organic Molecules with an OCF2H Group. J. Org. Chem. 2016, 81 (14), 5803–13. 10.1021/acs.joc.6b00628. [DOI] [PubMed] [Google Scholar]
- Reed L. J.; Muench H. A simple method of estimating fifty percent endpoints. Am. J. Hygiene 1938, 27 (3), 493–497. 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors have been deposited with the RCSB Protein Data Bank under accession code 8V4U.