

Abstract
The purpose of this protocol is to enable the user to produce a recombinant protein using the baculovirus-insect cell expression system.
1. THEORY
It is well known that there are many alternative systems that can be used for recombinant protein production, including bacteria, yeast, and mammalian cells, among others. Each system has its relative advantages and disadvantages. The oft-cited advantages of the baculovirus-insect cell expression system include its capacity for high-level protein production, its ability to provide eukaryotic protein modifications, its biosafety, and its relatively moderate cost (see Jarvis, 2009 for further details).
Recombinant proteins are often produced using the baculovirus-insect cell expression system (Pennock et al., 1984; Smith et al., 1983). Recombinant baculovirus vectors encoding a protein of interest can be produced using any one of several different approaches, which were recently reviewed (Jarvis, 2009). Subsequently, working stocks of the viral vector can be produced, quantified, and used to infect cultured lepidopteran insect cells. The recombinant baculovirus delivers the gene encoding the protein of interest and the infected cells ultimately produce the gene product. Detailed and comprehensive descriptions of the materials and methods used for insect cell culture and recombinant baculovirus isolation, purification, amplification, and infection can be found in classic ‘user manuals’ (O’Reilly et al., 1992; Summers and Smith, 1987) and a recent book (Murhammer, 2007) describing the baculovirus-insect cell expression system.
2. EQUIPMENT
Cell culture hood (e.g., class II biological safety cabinet)
Incubator and/or incubator-shaker (must be able to maintain 28 °C)
Compound microscope
Refrigerated tabletop (low-speed) centrifuge
Refrigerated high-speed centrifuge
Water bath (60 °C)
Water bath (30 °C)
Pipette aid
Micropipettor
6-well cell culture plates
Cell culture T-flasks
Cell culture shake flasks (Erlenmeyer)
Small (12 × 75 mm) glass test tubes
15-ml conical sterile polypropylene centrifuge tubes
50-ml conical sterile polypropylene centrifuge tubes
1.5-ml microcentrifuge tubes
Sterile, plastic serological pipettes
Micropipettor tips
Hemacytometer with cover slip
Plastic ziploc or seal-a-meal bags
Parafilm
3. MATERIALS
Sf9 insect cell line
TNM-FH insect cell medium
Baculovirus vectors [e.g., Autographa california nucleopolyhedrovirus (Ac NPV)]
Fetal bovine serum
Pluronic F-68
2× Grace’s insect cell medium
Trypan Blue, 0.4% solution
SeaPlaque agarose
Tris base
Sodium chloride (NaCl)
Nonidet P-40 (NP-40)
Sodium dodecyl sulfate (SDS)
SDS sample buffer
3.1. Solutions & buffers
Step 1 ‘Complete’ TNM-FH insect cell medium (for Sf9 cell culture)
| Component | Final concentrations | Stock | Amount/500 ml |
|---|---|---|---|
| TNM-FH medium⋆ | 90% | 450 ml | |
| Fetal bovine serum | 10% | 50 ml | |
| Pluronic-F68 | 0.1% (v/v) | 10% | 5 ml |
Also known as Grace’s insect cell medium-supplemented
Pluronic-F68, 10% (w/v)
| Dissolve 10 g Pluronic-F68 in 100 ml final volume of deionized water. |
| Sterilize by filtration |
‘Complete’ 2× Grace’s insect cell medium (for plaque overlays)
| Component | Final concentrations | Amount/500 ml |
|---|---|---|
| 2× Grace’s medium | 80% | 400 ml |
| Fetal bovine serum | 20% | 100 ml |
2% (w/v) SeaPlaque agarose
| Dissolve 2 g SeaPlaque agarose in 100 ml deionized water. |
| Sterilze by autoclaving |
Tris-buffered saline (TBS)
| Component | Final concentrations | Amount/500 ml |
|---|---|---|
| Tris base | 50 mM | 3.03 g |
| NaCl | 100 mM | 2.92 g |
Add water to dissolve salts, adjust to pH 7.4 with HCl, adjust final volume to 500 ml
Extraction Buffer 1 (mild protein extraction conditions)
| Component | Final concentrations | Amount/500 ml |
|---|---|---|
| Tris base | 50 mM | 3.03 g |
| NaCl | 100 mM | 2.92 g |
| Nonidet P-40 | 1% (v/v) | 5.00 ml |
Add water to dissolve salts, adjust to pH 8.0 with HCl, adjust final volume to 500 ml
Extraction Buffer 2 (harsh protein extraction conditions)
| Component | Final concentrations | Amount/500 ml |
|---|---|---|
| Tris base | 50 mM | 3.03 g |
| NaCl | 100 mM | 2.92 g |
| Sodium dodecyl sulfate | 1% (w/v) | 5.00 g |
Add water to dissolve salts, adjust to pH 8.0 with HCl, adjust final volume to 500 ml
4. PROTOCOL
4.1. Duration
| Preparation | Variable, about 1 week to 1 month |
|---|---|
| Protocol | About 4–5 weeks |
4.2. Preparation
| Produce or acquire a recombinant baculovirus |
| Acquire Sf9 cells and establish a suspension culture |
| Routinely subculture the Sf9 cells to keep them healthy |
Sterilize 1.5-ml microcentrifuge tubes and pipette tips by autoclaving.
See Fig. 13.1 for the flowchart of the complete protocol.
Figure 13.1.
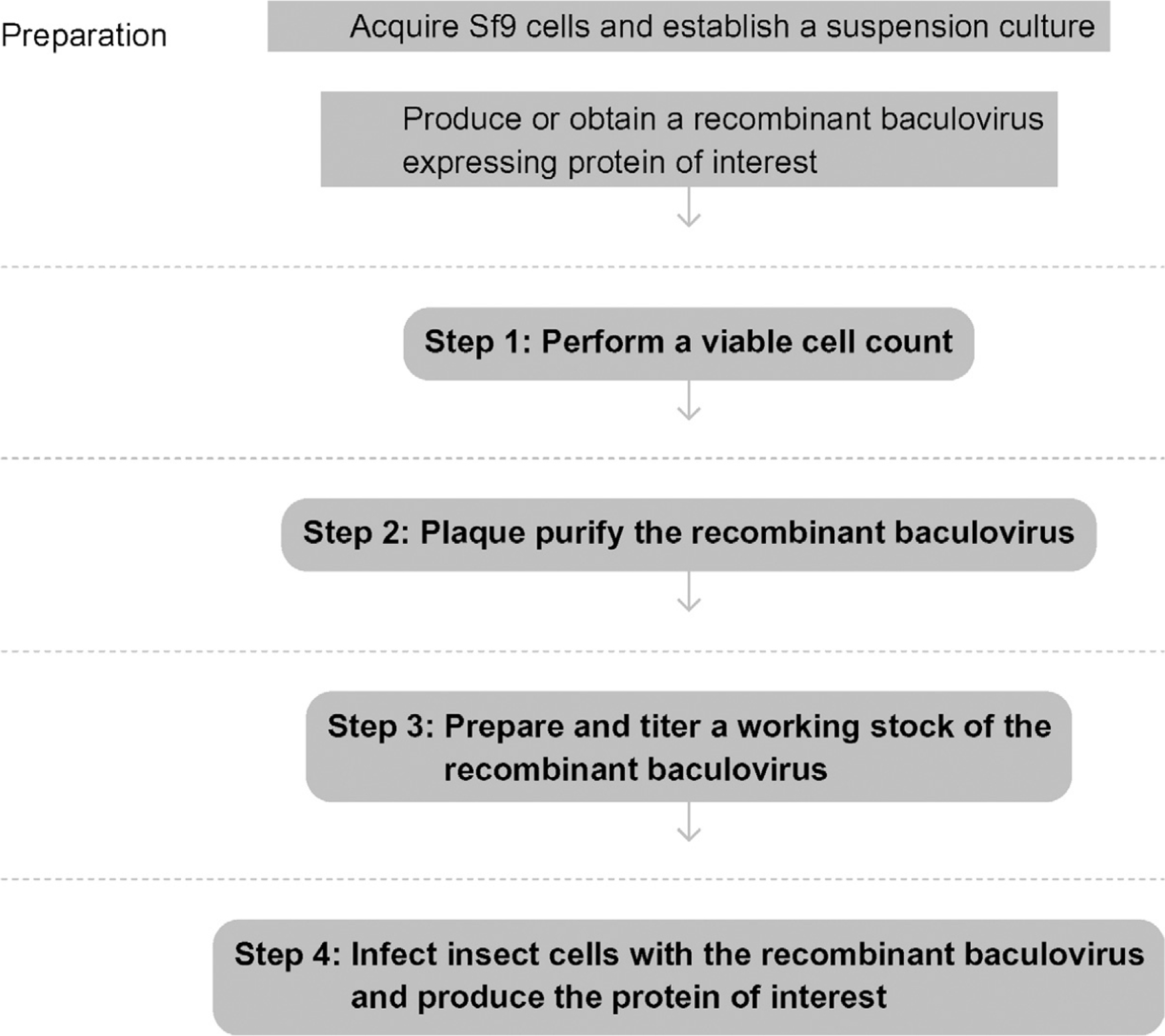
Flowchart of the complete protocol, including preparation.
5. STEP 1 PERFORM A VIABLE CELL COUNT
5.1. Overview
Viable cell counts must be performed prior to plaque purification of the recombinant baculovirus and subsequent protein production runs.
5.2. Duration
30 min
-
1.1
Remove the master cell culture from the incubator and take to a biological safety cabinet.
-
1.2
Aseptically remove 1.0 ml of the cell suspension using a 5-ml pipette.
-
1.3
Transfer exactly 0.5 ml of the cell suspension to a 12× 75 mm test tube.
-
1.4
Aseptically add exactly 1.4 ml of culture medium using a 5-ml pipette.
-
1.5
Use a micropipettor to add 0.1 ml of 0.4% Trypan Blue solution.
-
1.6
Cover the tube with parafilm and mix by gentle inversion.
-
1.7
Place the glass cover slip on top of the microscopic grid in the center of the hemacytometer.
-
1.8
Use a micropipette to transfer a drop (~12–15 μl) of the cell suspension into the slots on each side of the hemacytometer.
-
1.9
Place the hemacytometer on the stage of the compound microscope.
-
1.10
Count the unstained cells in all four corners (16 squares) of the grid on each side of the hemacytometer (the cells stained dark blue are dead).
-
1.11
Divide the total number of cells by two to calculate the average cell density (= #× 104 cells ml−1).
5.3. Tip
By diluting the cells from the master culture 1:4, as described earlier, you may simply divide the total number of cells counted by 2 to obtain the cell density (expressed as the average cell count × 104cells ml−1).
See Fig. 13.2 for the flowchart of Step 1.
Figure 13.2.
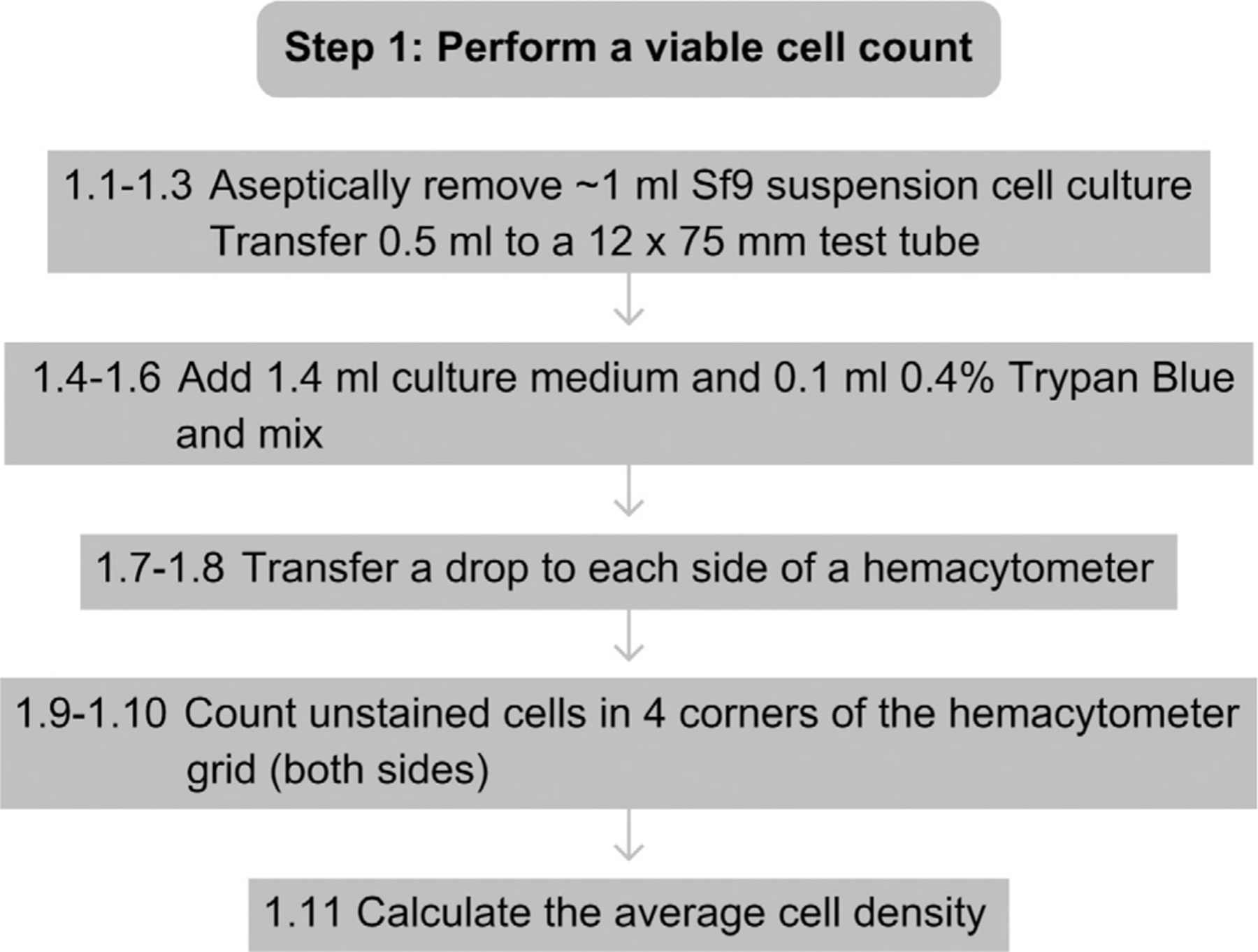
Flowchart for Step 1.
6. STEP 2 PLAQUE PURIFY THE RECOMBINANT BACULOVIRUS
6.1. Overview
Plaque purify the baculovirus stock you have produced or obtained from another laboratory to ensure that you are starting with a clonal recombinant baculovirus isolate.
6.2. Duration
7–10 days
-
2.1
Based on the cell count from Step 1, calculate the volume needed to obtain a total of 4.5 × 106 cells. This is enough to seed one 6-well plate at 0.75 × 106 cells/well.
-
2.2
Aseptically transfer that volume from the master cell culture to a sterile, 15-ml conical centrifuge tube.
-
2.3
Pellet the cells by low-speed centrifugation (~200 ×g) for 2 min in a tabletop centrifuge.
-
2.4
Aseptically decant the supernatant medium and dispose.
-
2.5
Aseptically resuspend the cell pellet by gentle trituration with a sterile pipette in 12 ml of serum-free TNM-FH medium.
-
2.6
Aseptically dispense 2 ml of the cell suspension into each well of the 6-well plate (seeding density of 0.75 × 106 Sf9 cells per well).
-
2.7
Incubate the cells for about an hour at 28 °C to allow the cells to attach to the plastic.
-
2.8
Meanwhile, aseptically prepare a tenfold dilution series (10−1 to 10−6) with your virus stock, using complete TNM-FH as the diluent. Add 2.25 ml complete TNM-FH to each tube, add 0.25 ml virus stock to the first tube, mix, and then serially transfer 0.25 ml diluted virus to the next tube.
-
2.9
After the cells have attached, aseptically remove the medium from each well and add 2.0 ml of each virus dilution to separate wells of the 6-well plate.
-
2.10
Incubate the 6-well plate for 1 h at 28 °C.
-
2.11
Meanwhile, melt 10 ml of 2% agarose per 6-well plate in a microwave oven and equilibrate in a 60 °C water bath (you need 1.5 ml of agarose per well).
-
2.12
Warm 10 ml of complete 2× Grace’s medium in a 30 °C water bath (you need 1.5 ml of 2× Grace’s medium per well).
-
2.13
Aseptically mix equal volumes of the melted, cooled agarose and complete 2× Grace’s medium (a total of 3.0 ml per well) and swirl to mix thoroughly.
-
2.14
Use a pipette to aseptically remove the viral inoculum from each well.
-
2.15
Check to make sure that the overlay mixture is cool to the touch (~40 °C) and then add 3 ml per well and allow the overlay to harden for ~15 min.
-
2.16
Seal the 6-well plate in a plastic ziploc or seal-a-meal bag and incubate upside down for 7–10 days at 28 °C.
-
2.17
Remove the 6-well plate from the bag, place upside down under a dissecting microscope, and look for viral plaques.
-
2.18
Circle a well-isolated plaque (or several if you have any doubts about the original virus stock) and aseptically core it using a sterile cotton-plugged Pasteur pipette or a micropipettor and plastic tip.
-
2.19
Transfer the agarose plug into 1 ml of complete TNM-FH in a sterile 1.5-ml microcentrifuge tube.
-
2.20
Vortex vigorously to elute the plaque-purified virus, designate as a PP1 (plaque-purified once) stock.
6.3. Tip
It is best to seed the Sf9 cells into 6-well plates in serum-free TNM-FH because they attach better than when seeded in complete TNM-FH.
6.4. Tip
Altering the cell seeding density can influence the size and quality of baculovirus plaque formation. It is typical to adjust these densities slightly on an ad hoc basis.
6.5. Tip
Work quickly when you remove the viral inocula and overlay the infected cell cultures with the agarose-Grace’s mixture. The cells dry out quickly and if this happens, they will die and you will see cleared areas (typically crescent-shaped) in your assays. If you see these clearings, remove the inocula and replace with the overlay from only 2–3 wells at a time.
6.6. Tip
When you add the overlay, tip the plate and dribble the viscous fluid down the side of the well, rather than adding it directly to the middle of the infected cell monolayer.
6.7. Tip
If you have any doubts about the original virus stock, it is prudent to pick several independent plaques and examine the genetic structure of the eluted virus (e.g., by PCR) to make sure that it still contains the intact gene of interest.
6.8. Tip
If you do not obtain well-isolated plaques, it is prudent to pick, elute, and plaque purify the eluted virus a second time (PP2).
See Fig. 13.3 for the flowchart of Step 2.
Figure 13.3.
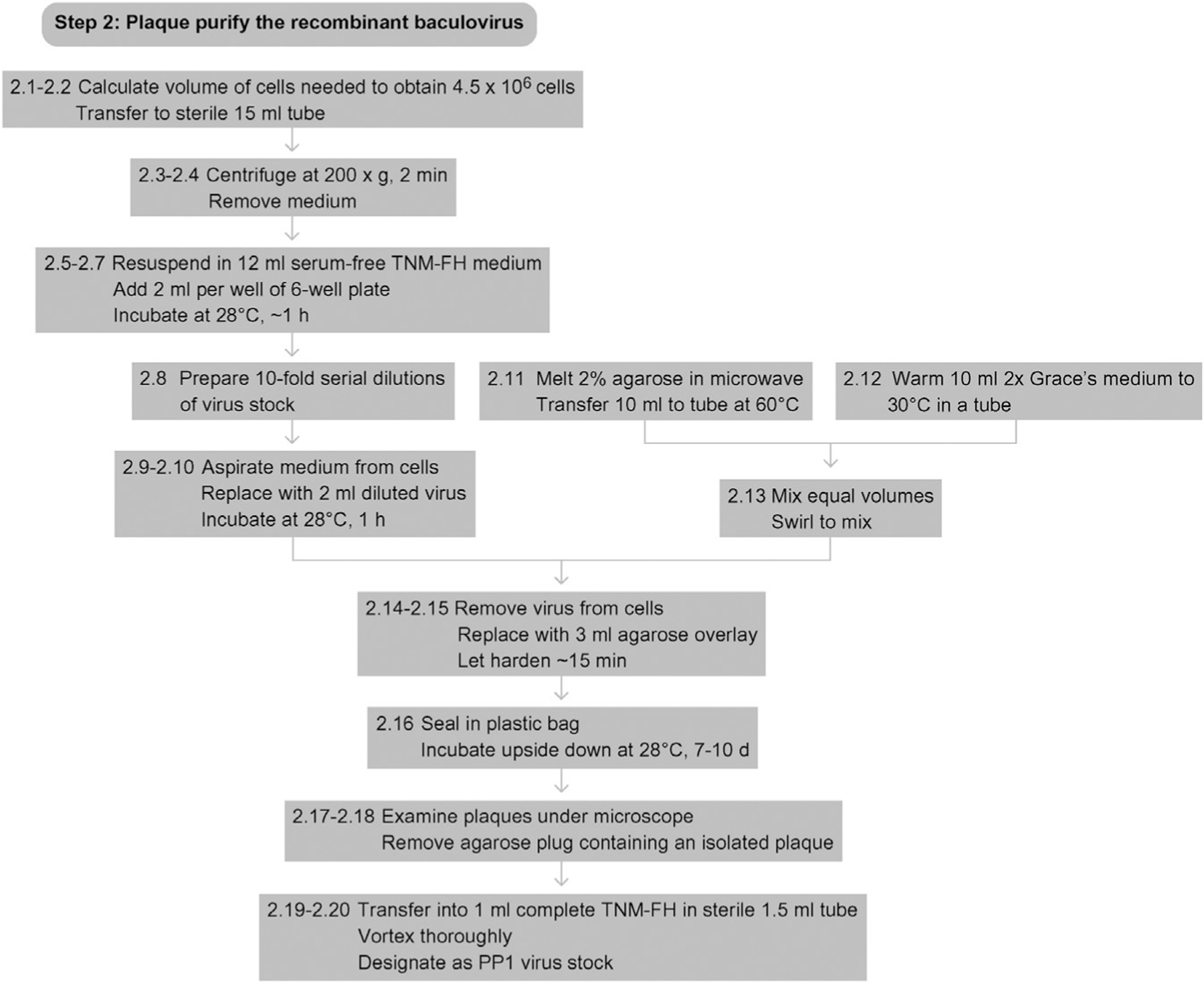
Flowchart for Step 2.
7. STEP 3 PREPARE AND TITER A WORKING STOCK OF THE RECOMBINANT BACULOVIRUS
7.1. Overview
The PP1 baculovirus stock must be amplified and the number of infectious particles quantified in preparation for subsequent infection(s) performed to produce the recombinant protein of interest.
7.2. Duration
About 12–14 days
-
3.1
Perform a viable cell count on the Sf9 master cell culture.
-
3.2
Calculate the volume needed for a total of 25 × 106 cells.
-
3.3
Aseptically transfer that volume of cells into a 100-ml shake flask and adjust the final volume to 50 ml with complete TNM-FH.
-
3.4
Incubate the culture in the shaking incubator at 125 rpm overnight at 28 °C.
-
3.5
Aseptically transfer 0.1 ml of the viral plaque eluant from Step 2.20 into the cell culture.
-
3.6
Incubate the culture in the shaker-incubator, shaking at 125 rpm, at 28 °C until you see clear signs of infection (usually 3–5 days).
-
3.7
Aseptically transfer the medium from the shake flask to a 50-ml conical centrifuge tube.
-
3.8
Centrifuge for 10 min at ~1000 ×g in a tabletop centrifuge.
-
3.9
Aseptically remove the supernatant, and place it in a fresh tube or bottle.
-
3.10
Designate this as the PP1P1 (plaque purified once, passaged once) virus stock. Wrap the tube or bottle with aluminum foil to protect the virus from light.
-
3.11
Seed one or more 6-well plates with Sf9 cells for plaque assays, as described under Step 2.
-
3.12
Aseptically prepare a tenfold dilution series (10−1 to 10−6) of the PP1P1 virus stock as described in Step 2.8.
-
3.13
Inoculate individual wells in the 6-well plate(s) in duplicate with 2 ml of the diluted virus and complete the plaque assay, as described under Step 2.
-
3.14
Determine the titer of your PP1P1 virus stock in plaque-forming units (pfu)/ml by counting plaques and multiplying the average number of plaques by the dilution factor (reciprocal of the dilution), then dividing by 2.
7.3. Tip
Sf9 cells are well infected with a recombinant baculovirus when they are swollen, the nuclei are enlarged, the nuclei have a very grainy (‘broken glass’) appearance, and/or when they begin to lyse. The timing depends upon the virus and the amount of virus in the inoculum, but is typically 3–5 days.
7.4. Tip
Baculovirus stocks can also be produced using adherent Sf9 cell cultures in plastic T-flasks. Seeding densities are typically 2.5, 7.5, or 15 × 106cells/flask in 5, 10, or 15 ml of complete TNM-FH for 25, 75, or 150 cm2T-flasks, respectively.
7.5. Tip
Baculovirus stocks can be amplified through further passages of the PP1P1 stock (e.g., to produce PP1P2 and PP1P3 stocks), but genetic diversity, including the appearance of defective interfering particles, is more likely to arise with increasing passages.
See Fig. 13.4 for the flowchart of Step 3.
Figure 13.4.

Flowchart for Step 3.
8. STEP 4 INFECT INSECT CELLS WITH THE RECOMBINANT BACULOVIRUS AND PRODUCE THE PROTEIN OF INTEREST
8.1. Overview
Use the working (PP1P1) baculovirus stock produced in Step 3 to infect an insect cell culture and produce the recombinant protein of interest. In lab jargon, this is the ‘production run.’
8.2. Duration
About 1 week
-
4.1
Perform a viable cell count on the Sf9 master cell culture.
-
4.2
Calculate the volume needed for a total of 25 × 106 cells.
-
4.3
Aseptically transfer that volume of cells into a 100-ml shake flask and adjust the final volume to 50 ml with complete TNM-FH.
-
4.4
Incubate the culture in the shaking incubator, shaking at 125 rpm, overnight at 28 °C.
-
4.5
Perform a viable cell count and calculate the average cell density (it should double to ~50 × 106 cells ml−1 overnight).
-
4.6
Using the titer determined in Step 3, calculate the volume of PP1P1 virus stock needed for a multiplicity of infection of ~2–5 pfu/cell.
-
4.7
Aseptically transfer that volume of the virus stock into the cell culture.
-
4.8
Incubate the culture in the shaker-incubator, shaking at 125 rpm, at 28 °C for 2–3 days.
-
4.9
Transfer the shake flask culture to a 50-ml conical centrifuge tube.
-
4.10
Centrifuge for 5 min at ~1000 ×g in a tabletop centrifuge.
-
4.11
Remove the supernatant, and place it in a fresh tube or bottle.
-
4.12
If your protein of interest is a secreted protein, retain and further process the supernatant as the source of recombinant protein from your production run.
-
4.13
If your protein of interest is an intracellular or membrane-bound protein, dispose the supernatant and gently wash the infected cells by resuspending the pellet in ice-cold Tris-buffered saline.
-
4.14
Gently centrifuge the cells (~200 ×g) to repellet and repeat the wash Steps (4.13–4.14).
-
4.15
Resuspend the washed infected cell pellet in a minimal volume of ice-cold extraction buffer 1 and incubate for 10 min on ice.
-
4.16
Centrifuge the extract for 10 min at ~10 000 ×g at 4 °C.
-
4.17
Harvest the supernatant and use as the source of solubilized recombinant protein from your production run.
-
4.18
Alternatively, extract the resulting pellet on ice with an ice-cold, harsher buffer, such as extraction buffer 2, with sonication or trituration through a fine gauge needle.
-
4.19
Centrifuge the extract for 10 min at ~10 000 ×g at 4 °C.
-
4.20
Harvest the supernatant and use it as the source of solubilized recombinant protein from your production run.
8.3. Tip
It is useful to perform preliminary experiments to determine the time of maximal recombinant protein production, with minimal degradation, for each protein of interest. While the best time to harvest is typically 48–72 h after infection, there are many different variables, including insect cell type, cell culture system (adherent vs. suspension), cell density, culture scale, culture medium, recombinant baculovirus stock (passage level, titer, age), and the nature of the protein produced that influence production parameters obtained for any given protein of interest.
8.4. Tip
A wide variety of methods can be used to assess recombinant protein production in the baculovirus-insect cell expression system. For example, one can compare total protein profiles extracted from infected cells or infected cell-free media by SDS-PAGE (see One-dimensional SDS-Polyacrylamide Gel Electrophoresis (1D SDS-PAGE)), with wild-type baculovirus-infected cells as controls. Detection methods include protein staining (e.g., Coomassie Brilliant Blue, see Coomassie Blue Staining), radiolabeling, and immunoblotting (see Western Blotting using Chemiluminescent Substrates), among others. One can also perform comparative enzyme or other biological activity assays if an assay is available for the protein of interest.
8.5. Tip
While Step 4 describes a production run for a 100-ml shake flask culture, the production format is highly variable and can be scaled up or down.
8.6. Tip
Production runs can be performed using adherent Sf9 cell cultures in plastic T-flasks, as described Tips under Step 3, or using stirred tank, airlift, or WAVE bioreactors, as described elsewhere (Murhammer, 2007). The variety of formats that can be used allows one to scale production runs up or down across a wide range of possibilities.
8.7. Tip
If your protein of interest is a secreted protein, it is best to use a serum- or protein-free insect cell culture for the production run, as described elsewhere (Murhammer, 2007).
8.8. Tip
If your protein of interest is an intracellular or membrane-bound protein, it might be necessary to determine extraction conditions empirically. Step 4 describes two commonly used sequential extraction methods, but many other extraction procedures can be utilized.
See Fig. 13.5 for the flowchart of Step 4.
Figure 13.5.

Flowchart for Step 4.
REFERENCES
Referenced Literature
- Jarvis DL (2009). Baculovirus-insect cell expression systems. Methods in Enzymology, 463, 191–222. [DOI] [PubMed] [Google Scholar]
- Murhammer DW (2007). Molecular Biology (vol. 388). Methods, In Baculovirus and Insect Cell Expression Protocols (2nd ed.). Clifton, UK: Humana Press. [Google Scholar]
- O’Reilly DR, Miller LK, & Luckow VA (1992). Baculovirus Expression Vectors. New York: W.H. Freeman and Company. [Google Scholar]
- Pennock GD, Shoemaker C, & Miller LK (1984). Strong and regulated expression of Escherichia coli beta-galactosidase in insect cells with a baculovirus vector. Molecular and Cellular Biology, 4(3), 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GE, Summers MD, & Fraser MJ (1983). Production of human beta interferon in insect cells infected with a baculovirus expression vector. Molecular and Cellular Biology, 3(12), 2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers MD, & Smith GE (1987). A manual of methods for baculovirus vectors and insect cell culture procedures. Tx. Ag. Expt. Stn. Bull. No. 1555. [Google Scholar]