

Abstract
Objective:
To characterize the clinical phenotype, with an emphasis on electrophysiologic findings, in a family with autosomal dominant retinitis pigmentosa caused by mutation in the recently identified KLHL7 gene.
Methods:
Eleven patients from a single family were selected from the Swedish retinitis pigmentosa register. Four patients had been examined 13 to 17 years earlier and underwent further ophthalmologic examination, including visual acuity, fundus inspection, Goldmann perimetry, full-field electroretinography (ERG), multifocal ERG, and optical coherence tomography. KLHL7 mutation was identified by sequence analysis.
Results:
In most examined family members, the fundus showed minor abnormalities. Full-field ERG demonstrated reduced cone and rod function, but rod responses were preserved in some patients late in life. Follow-up (≤17 years) demonstrated slowly progressive retinal degeneration. In an adolescent family member, cone and rod function was initially normal, but retinitis pigmentosa was confirmed by electrophysiology 17 years later. Optical coherence tomography and multifocal ERG demonstrated macular abnormalities of varying degree among family members. Genetic analysis revealed a heterozygous exon 6 change (c.458C>T) in 7 family members.
Conclusions:
Observed in 2 Scandinavian families to date, KLHL7 mutation has recently been associated with autosomal dominant retinitis pigmentosa. Clinical examination with long-term follow-up verified a phenotype with a varying degree of retinal photoreceptor dysfunction and, in some family members, with late onset and preserved rod function until late in life.
Clinical Relevance:
Patients with minor retinal abnormalities and normal ERG findings early in life can harbor an autosomal dominant form of retinitis pigmentosa with a varying degree of visual impediment. Some patients with late onset may retain night vision for many years.
Retinitis pigmentosa (RP) IS an inherited retinal dystrophy and a common cause of visual impediment among young patients world-wide. The first identified causative gene of this disorder was rhodopsin.1 Thereafter, several genes have been identified, possibly explaining the varying phenotypes regarding onset and type of visual effect.2 To date, 18 different genes are associated with autosomal dominant RP (adRP).3 A newly identified gene, KLHL7 (NCBI Entrez Gene 55975), was recently associated with adRP after linkage analysis in a large Scandinavian family.4 Other families, including a second Scandinavian family (family 101), carry KLHL7 mutation, but the ophthalmologic phenotype has not been described to date.
KLHL7 is widely expressed, including rod photoreceptors, and its biologic function is under investigation. Based on function of other Kelch-like family members, this protein is hypothesized to stabilize the formation of Cullin ubiquitin ligating enzyme (E3) ligase complexes in the retina, and failure to do so could lead to cellular accumulation of toxic proteins, resulting in retinal degeneration.4
Defects in this gene are responsible for an RP phenotype that is undescribed to date. We present herein the complete clinical characterization of a Swedish family with KLHL7 mutation.
METHODS
Eleven patients with adRP from a large family (family 101) in southern Sweden were selected from the Swedish RP register (Figure 1). Clinical examinations were performed in all 11 family members, 4 of whom had been examined earlier and were given reexamination with clinical follow-up ranging up to 17 years. The 4 previously examined patients underwent thorough ophthalmologic examinations, including best-corrected visual acuity, Schirmer test, slitlamp biomicroscopy, fundus photography, and Goldmann perimetry using the standardized targets I4e and V4e. Further electrophysiology and optical coherence tomography examinations were performed. Research procedures were performed in accord with institutional guidelines and the Declaration of Helsinki.
Figure 1.
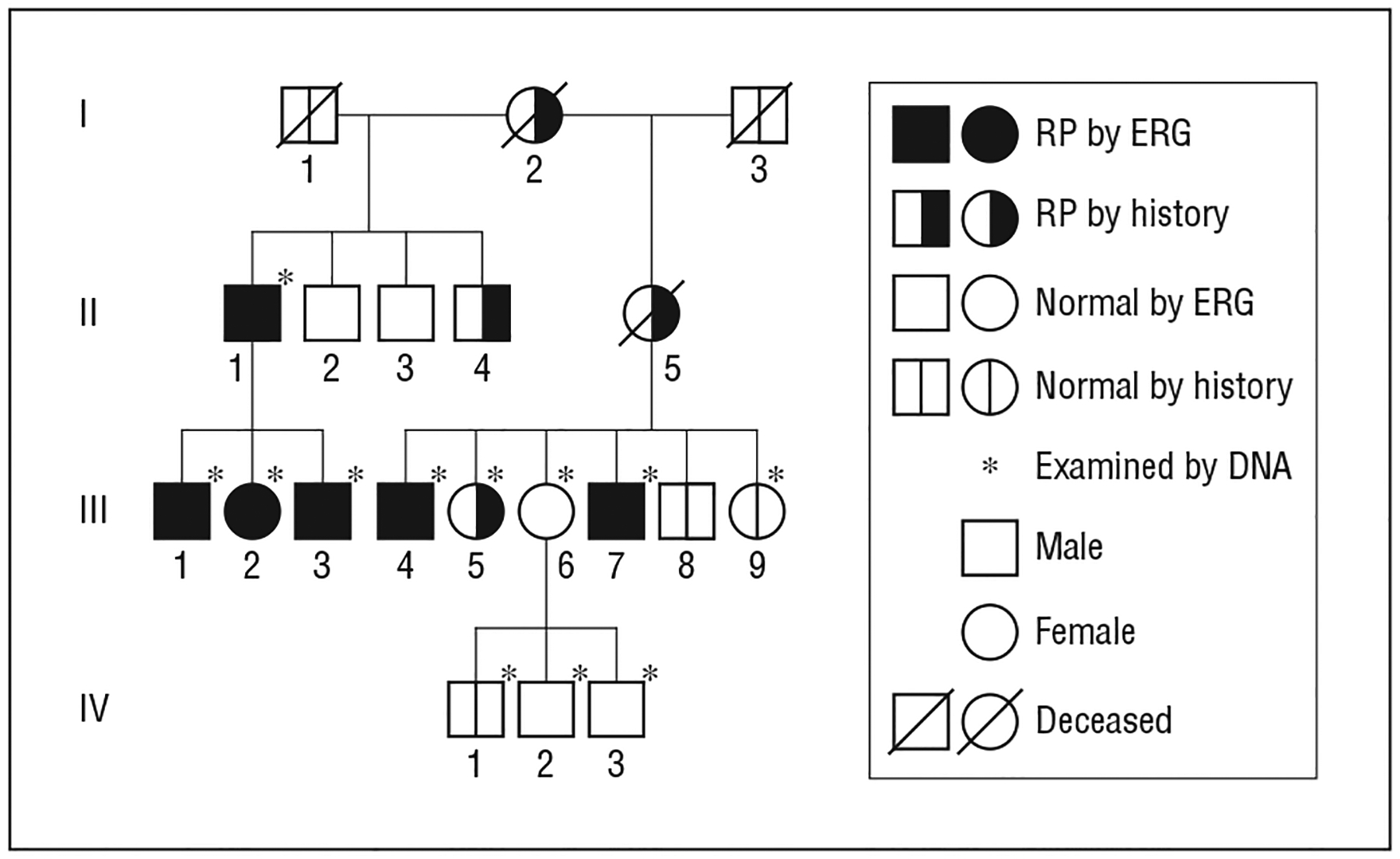
Autosomal dominant retinitis pigmentosa (RP) caused by KLHL7 mutation in family 101 from the Swedish retinitis pigmentosa register. Patient III-5 has RP by history and by fundus appearance. ERG indicates electroretinography.
Informed consent was obtained from patients, and policies of the appropriate institutional review board were followed. Blood samples for molecular genetic studies were obtained, and DNA was analyzed. KLHL7 exons 1 through 12 were sequenced as previously described.4 Two family members were newly screened for KLHL7 mutations in this study.
FULL-FIELD ELECTRORETINOGRAPHY
Full-field electroretinography (ERG) was recorded (Nicolet Bio-medical Instruments, Madison, Wisconsin). Patients underwent dark adaptation for 40 minutes, and a bipolar contact lens (Hansen Labs, Coralville, Iowa) was placed on the cornea and a ground electrode on the forehead. Responses were obtained with a wide-band filter (−3 dB at 1 Hz and 500 Hz), stimulating with brief (30 microseconds) single full-field flashes of dim blue light (Wratten filters 47, 47A, and 47B [Kodak, Rochester, New York]) and white light (0.81 candela [cd]–seconds [cd-s]/m2 and 3.93 cd-s/m2). Cone responses were obtained with 30-Hz flickering white light (0.81 cd-s/m2) averaged from 20 sweeps. If responses measuring less than 10 μV were recorded with single white flashes, recordings were also obtained with computer averaging (30 flashes), a bipolar artifact rejecter, and a line frequency notch filter (50 Hz). To obtain small cone responses, stimulation then included 200 sweeps of 30-Hz flickering white light, and a digital or analog narrow band-pass filter was added to the system.5
MULTIFOCAL ERG
Multifocal ERG (mfERG) was recorded using a visual-evoked response imaging system (VERIS 4; EDI, San Mateo, California) developed by Sutter and Tran6 and by Bearse and Sutter.7 The stimulus matrix consisted of 103 hexagonal elements displayed on a screen in the infrared camera. The recording procedures were performed according to International Society for Clinical Electrophysiology of Vision guidelines for clinical mfERG.8 Recordings were monocular, and fixation was controlled using the infrared camera and illumination with infrared light from the recording electrode, with visualization of hexagonal elements over the retina. The first-order kernel P1 amplitudes and implicit times in ring areas 1 through 6 were calculated according to basic mfERG guidelines.8 Responses from ring areas 1 and 2 were averaged, noted as ring (1+2).
OPTICAL COHERENCE TOMOGRAPHY
Optical coherence tomography was performed (OCT-3; Zeiss Humphrey Instruments, Dublin, California). Single-line 5.0-mm images and macular thickness maps of 6.0-mm image length centered over the fovea were obtained.
RESULTS
Earlier sequence analysis of KLHL7 exon 6 in family 101 revealed a heterozygous c.458C>T mutation, encoding a predicted p.A153V change, which segregated with the adRP phenotype.4 Subsequently screened were 2 additional family members, one of whom carried the disease allele.
In 4 reexamined family members with KLHL7 mutation, visual acuity ranged from 20/200 to 20/20 (Table 1). The oldest patient had undergone bilateral cataract surgery. Goldmann perimetry in patients II-1, III-1, and III-2 demonstrated bilateral concentric constrictions of isopters for I4e and varying extent of pericentral scotomas for V4e, whereas paracentral scotomas for I4e were found in patient III-3 (Figure 2). Schirmer test results were within normal limits for all examined family members.
Table 1.
Visual Acuity and Multifocal ERG Findings in Right Eyes of 4 Reexamined Family Members With Autosomal Dominant Retinitis Pigmentosa Caused by KLHL7 Mutation
| Patient/Age, ya | Best-Corrected VA | Multifocal ERG Result | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 Amplitude, nV/degree2 | P1 Implicit Time, ms | ||||||||||
| Ring (1 + 2) | Ring 3 | Ring 4 | Ring 5 | Ring 6 | Ring (1 + 2) | Ring 3 | Ring 4 | Ring 5 | Ring 6 | ||
| II-1/69 | 20/200 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| III-1/38 | 20/20 | 19.3 | 6.4 | 5.1 | 4.7 | 3.5 | 33.3 | 34.2 | 36.7 | 37.5 | 39.2 |
| III-2/36 | 20/30 | 10.3 | 5.3 | 4.5 | 3.5 | 1.7 | 33.3 | 40.0 | 43.3 | 43.3 | 41.7 |
| III-3/34 | 20/20 | 51.1 | 25.5 | 13.4 | 8.3 | 6.4 | 30.0 | 30.8 | 31.7 | 35.0 | 35.8 |
| Normal, mean (2 SDs)b | … | 32.0 (23.8) |
22.4 (14.5) |
18.2 (10.8) |
16.1 (9.1) |
15.7 (9.2) |
27.0 (3.0) |
26.2 (2.9) |
25.7 (2.9) |
25.9 (2.9) |
26.3 (3.0) |
Abbreviations: ellipses, not applicable; ERG, electroretinography; ND, nondetectable; VA, visual acuity.
At the time of the presented data.
Values at the Department of Clinical Sciences, Ophthalmology, Lund University, Lund, Sweden, among 30 eyes of 21 individuals (age range, 8–55 years [mean age, 38 years]).
Figure 2.

Full-field electroretinography (right eye) demonstrating the rate of progression during a follow-up period of 13 to 17 years. Shown for each patient are Goldmann perimetry results (right eye) from the most recent full-field electroretinography examinations.
FUNDUS APPEARANCE
In early stages of adRP, as in patient III-3 (Figure 3), normal fundus appearance was seen on ophthalmoscopy. With disease progression, scattered bone corpuscular pigment, waxy pale disc, thinning of the retina, and attenuated retinal blood vessels were typically observed in all 4 quadrants bilaterally and were symmetric, as in patient II-1, in whom perifoveal atrophy was also seen.
Figure 3.
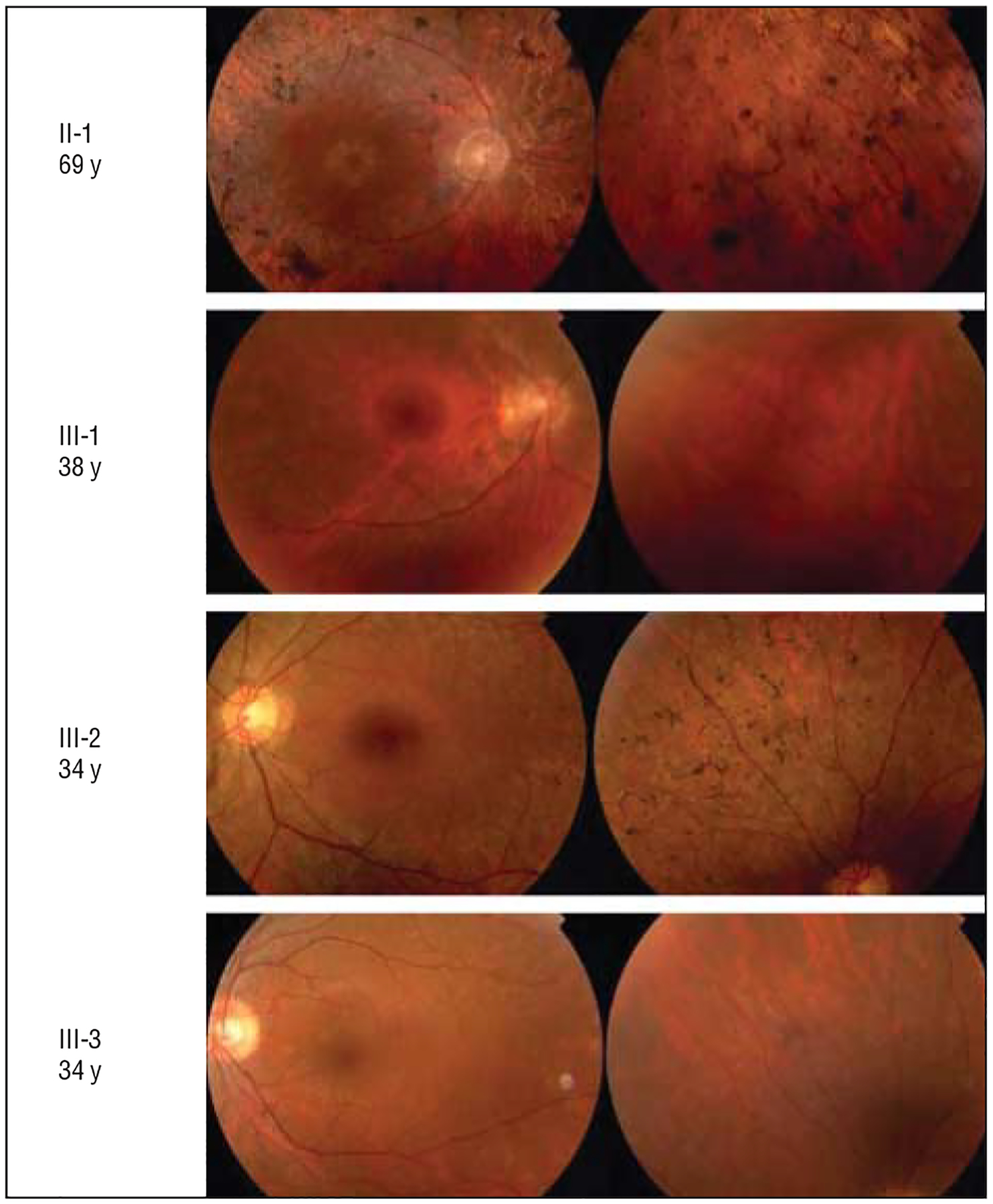
Varying fundus appearance in 4 reexamined members of family 101 with KLHL7 mutation. The right eye is shown for 2 patients at the top, and the left eye is shown for 2 patients at the bottom.
FULL-FIELD ERG
Patient III-3 demonstrated reduced rod response amplitudes, well-preserved 30-Hz flicker amplitudes, but some-what prolonged implicit times (Table 2 and Figure 2). When his sister (patient III-2) was examined at the same age (34 years), similar rod responses and markedly prolonged 30-Hz flicker implicit times were observed. Their older brother (patient III-1), examined at age 38 years, had almost no detectable isolated rod responses when stimulated with dim blue light, and 30-Hz flickering cone responses showed reduced amplitudes and clearly prolonged implicit times. Full-field ERG after 24-hour dark adaptation of the right eye was also performed in patient III-1, but no improvement was seen in rod response. In their father (patient II-1), 30-Hz flicker with narrow band-pass filter resulted in scarcely measurable amplitudes.
Table 2.
Full-Field Electroretinography Findings Among Family Members
| Patient, Age, y | Blue Light | White Lighta | 30-Hz Flickerb | |||||
|---|---|---|---|---|---|---|---|---|
| Amplitude, μV | Amplitude, μV | Amplitude, μV | Implicit Time, ms | |||||
| OD | OS | OD | OS | OD | OS | OD | OS | |
| II-1 | ||||||||
| 56 | ND | ND | ND | ND | 2 | 1 | 44.2 | 42.8 |
| 69 | ND | ND | ND | ND | 0.4c | 0.2c | 58.5c | 53.3c |
| III-1 | ||||||||
| 21 | 20 | 51 | 78 | 123 | 24 | … | 36.0 | … |
| 38 | ND | 10 | 33 | 57 | 13 | 19 | 41.0 | 39.1 |
| 38d | ND | … | 39 | … | 2 | … | 40.7 | … |
| III-2 | ||||||||
| 20 | 66 | 56 | 102 | 129 | 50 | 52 | 34.0 | 33.6 |
| 34 | 36 | 38 | 87 | 87 | 34 | 37 | 38.8 | 37.1 |
| III-3 | ||||||||
| 17 | 70 | 105 | 160 | 238 | 111 | 133 | 31.2 | 32.0 |
| 34 | 44 | 23 | 117 | 76 | 44 | 31 | 34.5 | 36.0 |
| III-4 | ||||||||
| 43 | ND | ND | 7 | 7 | 5c | 7c | 43.2c | 45.2c |
| III-7 | ||||||||
| 51 | ND | ND | 33 | 27 | 19 | 17 | 41.9 | 41.3 |
| Normal, mean (2 SDs)e | 167 (105) | 308 (144) | 63 (42) | 29.2 (3.6) | ||||
Abbreviations: ellipses, not examined; ND, nondetectable.
For 0.81 candela [cd]–seconds [cd-s]/m2.
Without background illumination.
With narrow band-pass filter.
Right eye dark-adapted for 24 hours.
Values at the Department of Clinical Sciences, Ophthalmology, Lund University, Lund, Sweden, among 56 individuals (age range, 6–72 years [mean age, 31 years]).
MULTIFOCAL ERG
In patient III-3, the youngest of the 4 reexamined family members and with the least advanced disease, mfERG showed well-preserved amplitudes corresponding to the foveal area but affected amplitudes and prolonged implicit times in the outer ring areas (Table 1 and Figure 4). Patients III-1 and III-2 had diminished remaining responses within the central areas and prolonged implicit times. In patient II-1, with the most advanced disease, no definitive responses were detected on mfERG.
Figure 4.
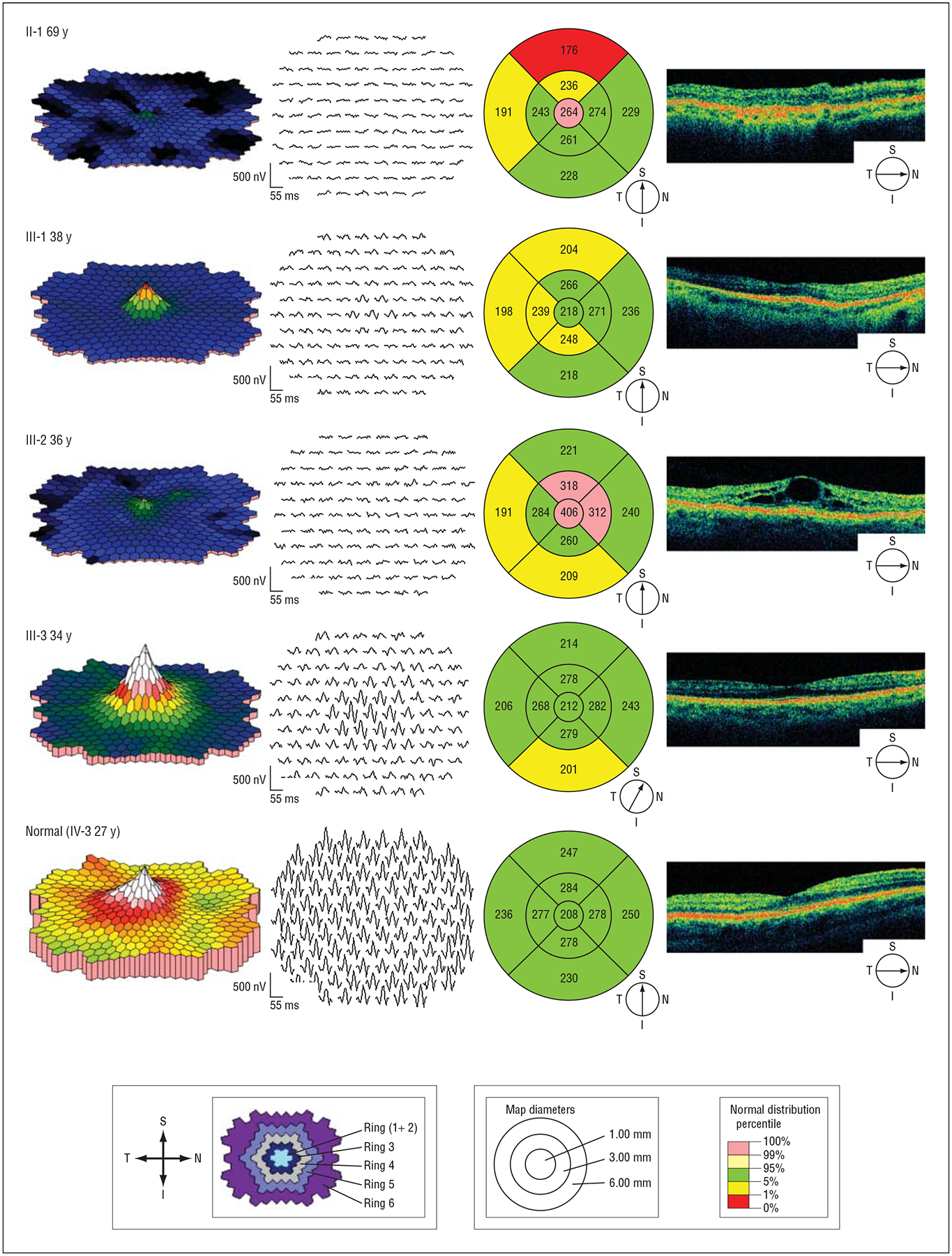
Multifocal electroretinography and optical coherence tomography results of retinal function and morphologic structure in the macular region. Shown are 4 reexamined members from family 101 with KLHL7 mutation and 1 family member without the mutation. Multifocal electroretinography, trace arrays, and plots are retinal views from right eyes. Shown at the bottom left are multifocal electroretinography definitions of ring areas 1 through 6. Optical coherence tomography, line images, and macular thickness maps are from right eyes. Shown at the bottom right are optical coherence tomography definitions of macular thickness maps (in microns). I indicates inferior; N, nasal, S, superior; and T, temporal.
OPTICAL COHERENCE TOMOGRAPHY
In patient III-2, optical coherence tomography demonstrated bilateral cystoid macular edema (Figure 4). In all patients, areas of reduced thickness consistent with degeneration of the retinal layers were observed.
FOLLOW-UP PERIOD
The 4 reexamined family members had undergone previous assessment (visual acuity examination, slitlamp biomicroscopy, Goldmann perimetry, and full-field ERG) 13 to 17 years earlier, and some had been examined more than once (Table 2 and Figure 2). Goldmann perimetry results in patient III-3 using the same standardized targets were bilaterally normal at age 17 years, and full-field ERG responses at that examination were within normal limits, with remarkably high 30-Hz flicker amplitudes. Patient III-2 demonstrated mild RP when she was examined at age 20 years, and successive examinations showed slow progression. At a follow-up examination, she was diagnosed as having acute angle-closure glaucoma and was treated with laser and medical therapy. In patient III-1, examination at age 21 years demonstrated markedly reduced rod responses and prolonged 30-Hz flicker implicit times. Patient II-1 underwent electrophysiologic examination for the first time at age 56 years; his visual acuity was 20/30, and RP was apparent.
COMMENT
Rhodopsin was the first identified genetic defect in RP, and several adRP genes have been described since then. Some have been associated with diverse phenotypes and with varying rates of retinal degeneration. Mutations in the peripherin/RDS gene are often associated with less progressive disorders and sometimes with atypical findings, including early macular involvement.9 In contrast, RP10 correlates to IMDPH1 gene mutations, which have been associated with RP and with long-standing residual function in the central part of the macula as verified by mfERG.10
Other RP variants have been described, such as late-onset RP in families with rhodopsin mutations (ie, RHO90), but these patients often demonstrate early rod dysfunction comparable to nyctalopia.11 Similarly, KLHL7 gene mutations seem to be associated with a specific RP phenotype, despite some diversity. In the family with KLHL7 mutation described herein, follow-up ranging up to 17 years demonstrated normal visual function in adolescents, verified by full-field ERG, and long-standing rod function among some older family members.
Although the specific function of KLHL7 protein in the photoreceptors or other retinal cell layers is unknown, the clinical phenotype described in this article is similar to other forms of RP with a varying degree of night vision deterioration. In contrast to families demonstrating Bothnia dystrophy or fundus flavimaculatus who manifest normal rod function after prolonged darkness, the phenotype associated with the KLHL7 mutation shows no recovery.12 Early and often rapid alteration of rod function has been a cornerstone in the classic form of adRP, but the phenotype we describe differs because of its late onset, preserved rod function late in life among some family members, and similarity to a slowly progressive neurodegenerative disorder.
The physiologic function of KLHL7 protein is not well understood, but it is widely expressed, including rod photoreceptors, and encodes a protein of the BTB-Kelch family that is implicated in ubiquitinylation through Cullin E3 ligases. We hypothesize that KLHL7 acts in the ubiquitin proteosomal pathway and that mutations affect its proper function. Failure to ubiquitinate a target protein could result in accumulation of the substrate, leading to long-term cellular toxicity within the photoreceptors.4 A second BTB-Kelch protein, gigaxonin, is also mutated in the neurodegenerative disorder giant axonal neuropathy.13
No other clinical implications of altered function of KLHL7 have been reported, but antibodies have been detected in patients with Sjögren syndrome.14 In the present family, Schirmer test revealed no signs of dry eye, which is associated with Sjögren syndrome.
One family member demonstrated macular edema. A 2008 study15 verified that cystoid macular edema can be detected in about 50% of examined eyes among families with adRP.
In conclusion, KLHL7 mutation has recently been associated with an autosomal dominant form of RP. To our knowledge, our study is the first to report clinical examinations that include full-field ERG among patients with this mutation. Our study demonstrates a varying degree of photoreceptor degeneration during long-term follow-up, with some family members manifesting late onset and preserved rod function until late in life.
Funding/Support:
This study was supported by the Faculty of Medicine, Lund University; by grants from the Swedish Medical Research Council, Foundation Fighting Blindness, and Swedish Association of the Visually Impaired; and by intramural support from the National Eye Institute, National Institutes of Health.
Footnotes
Financial Disclosure: None reported.
REFERENCES
- 1.Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88(20):9370–9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125(2):151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.RetNet: Retinal Information Network. http://www.sph.uth.tmc.edu/Retnet/. Accessed March 13, 2010.
- 4.Friedman JS, Ray JW, Waseem N, et al. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa Am J Hum Genet. 2009;84 (6):792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andréasson SO, Sandberg MA, Berson EL. Narrow-band filtering for monitoring low-amplitude cone electroretinograms in retinitis pigmentosa. Am J Ophthalmol. 1988;105(5):500–503. [DOI] [PubMed] [Google Scholar]
- 6.Sutter EE, Tran D. The field topography of ERG components in man, I: the photopic luminance response. Vision Res. 1992;32(3):433–446. [DOI] [PubMed] [Google Scholar]
- 7.Bearse MA Jr, Sutter EE. Imaging localized retinal dysfunction with the multifocal electroretinogram. J Opt Soc Am A Opt Image Sci Vis. 1996;13(3):634–640. [DOI] [PubMed] [Google Scholar]
- 8.Hood DC, Bach M, Brigell M, et al. ISCEV guidelines for clinical multifocal electroretinography (2007 edition). Doc Ophthalmol. 2008;116(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ekström U, Andréasson S, Ponjavic V, et al. A Swedish family with a mutation in the peripherin/RDS gene (Arg-172-Trp) associated with a progressive retinal degeneration. Ophthalmic Genet. 1998;19(3):149–156. [DOI] [PubMed] [Google Scholar]
- 10.Schatz P, Ponjavic V, Andreasson S, McGee TL, Dryja TP, Abrahamson M. Clinical phenotype in a Swedish family with a mutation in the IMPDH1 gene. Ophthalmic Genet. 2005;26(3):119–124. [DOI] [PubMed] [Google Scholar]
- 11.Sieving PA, Richards JE, Naarendorp F, Bingham EL, Scott K, Alpern M. Dark-light: model for nightblindness from the human rhodopsin Gly-90→Asp mutation. Proc Natl Acad Sci U S A. 1995;92(3):880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gränse L, Abrahamson M, Ponjavic V, Andreasson S. Electrophysiological findings in two young patients with Bothnia dystrophy and a mutation in the RLBP1 gene. Ophthalmic Genet. 2001;22(2):97–105. [DOI] [PubMed] [Google Scholar]
- 13.Allen E, Ding J, Wang W, et al. Gigaxonin-controlled degradation of MAP1B light chain is critical to neuronal survival. Nature. 2005;438(7065):224–228. [DOI] [PubMed] [Google Scholar]
- 14.Uchida K, Akita Y, Matsuo K, et al. Identification of specific autoantigens in Sjögren’s syndrome by SEREX. Immunology. 2005;116(1):53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hajali M, Fishman GA, Anderson RJ. The prevalence of cystoid macular oedema in retinitis pigmentosa patients determined by optical coherence tomography. Br J Ophthalmol. 2008;92(8):1065–1068. [DOI] [PubMed] [Google Scholar]