

SUMMARY
Crimean-Congo hemorrhagic fever virus can cause lethal disease in humans yet there are no approved medical countermeasures. Viral glycoprotein GP38, exclusive to Nairoviridae, is a target of protective antibodies and is a key antigen in preclinical vaccine candidates. Here, we isolate 188 GP38-specific antibodies from human survivors of infection. Competition experiments show that these antibodies bind across 5 distinct antigenic sites, encompassing 11 overlapping regions. Additionally, we show structures of GP38 bound with 9 of these antibodies targeting different antigenic sites. Although these GP38-specific antibodies are non-neutralizing, several display protective efficacy equal to or better than murine antibody 13G8 in two highly stringent rodent models of infection. Together, these data expand our understanding regarding this important viral protein and may inform the development of broadly effective CCHFV antibody therapeutics.
Graphical abstract
In brief
Crimean-Congo hemorrhagic fever virus is widespread across Africa, Asia, and Europe and causes severe disease in humans. Shin et al. report the isolation and characterization of GP38-specific antibodies from convalescent donors. Challenge experiments with authentic virus combined with structural studies provide insights into GP38 epitopes important for protection.
INTRODUCTION
Crimean-Congo hemorrhagic fever virus (CCHFV) is a member of the family Nairoviridae (Orthonairovirus genus) of the Bunyavirales order. Although infection by CCHFV is often asymptomatic in humans, severe hemorrhagic disease with fatality rates of 5%–40%—and sometimes as high as 80%—have been documented.1,2 Transmission of CCHFV to humans, as well as to domesticated and wild animals, occurs primarily through the bite of Hyalomma ticks.3–5 Direct contact with infected tissues, primarily due to contact with blood from infected livestock, can also result in transmission,6,7 and, although less common, nosocomial infections have been reported.8,9 The broad geographic range of Hyalomma ticks contributes to widespread outbreaks of CCHFV across at least three continents, including Europe, Asia, and Africa, where CCHFV is endemic.2,6,10,11
Proportionate to its extensive distribution, CCHFV exhibits considerable genetic diversity among geographically distinct isolates.6,12 Historically, CCHFV isolates were classified into six genotypes, or clades: I–III (endemic in Africa), IV (Asia), V (Europe I), and VI (Europe II).12–19 However, clade VI genotypes were recently reclassified into a separate and distinct species, Aigai virus, which infrequently causes severe disease.20 CCHFV has been recognized for its pandemic potential and, as of 2017, the World Health Organization has designated it a priority pathogen.21 Despite this designation, no specific approved medical countermeasures are currently available apart from the off-label use of the broad-spectrum antiviral ribavirin, but evidence for its efficacy against CCHFV is lacking.22
CCHFV has a tri-segmented negative-sense RNA genome. The genomic RNA segments are termed S (small), M (medium), and L (large), encoding for the nucleoprotein, the glycoprotein precursor complex (GPC), and the viral polymerase, respectively.23 The GPC undergoes a series of proteolytic cleavages and maturation to generate multiple structural glycoproteins (Gn and Gc) and non-structural glycoproteins (GP38, GP85, GP160, and mucin-like domain).24,25 GP38 is unique to members of the Nairoviridae family and is thought to play a crucial role in CCHFV pathogenesis and the maturation of viral particles.26 Crystal structures of CCHFV GP38 resolved in prior studies have shown the protein to have a novel fold consisting of an N-terminal three-helix bundle followed by a β sandwich.27,28 Some evidence points to GP38 localizing to the membrane of virus particles and the surface of infected cells.29 However, the specific function of GP38 and its role in pathogenesis remain unresolved.
Gc-specific neutralizing antibodies and GP38-specific non-neutralizing antibodies have been shown to be protective in animal models of infection.27–30 13G8, a non-neutralizing GP38-specific antibody of murine origin, has been characterized for its ability to protect mice against CCHFV-induced mortality and liver and spleen pathologies in both pre- and post-exposure studies.29 Furthermore, 13G8 has shown varied prophylactic potential against diverse isolates of CCHFV, including IbAr10200, Afg09, and Turkey2004.27–29 Two prior studies investigating the antibody responses to GP38 showed that antibodies target five discrete antigenic sites on CCHFV GP38.27,29 These include seven human GP38-specific antibodies, one of which was structurally characterized and determined to compete with 13G8, but it was shown to be poorly protective compared with 13G8.27 Given the unknown role of GP38 in viral pathogenesis and the limited understanding of which epitopes contribute to protection, we sought to isolate and characterize an extensive panel of human antibodies against GP38.
Here, the B cell repertoires of three human CCHF-convalescent donors from Uganda were mined for monoclonal antibodies (mAbs) specific for CCHFV GP38. A panel of 188 GP38-specific antibodies was isolated, binned into competition groups, and characterized for binding across several clinical isolates and for neutralization potency. Structural studies of select antibodies targeting each antigenic site were conducted to define epitopes across the surface of GP38. Subsequent animal challenge studies were performed to correlate protection with antigenic sites and gain insight into surfaces of GP38 that may be functionally important for pathogenesis.
RESULTS
Isolation of GP38-reactive antibodies from CCHF-convalescent donors
Peripheral blood mononuclear cells (PBMCs) were isolated from three human CCHF-convalescent donors from Uganda between 3 and 46 months post-infection (Table S1). All donors had detectable serum titers to GP38, relative to naive controls (Figure S1A). To sort memory B cells (MBCs) expressing GP38-reactive B cell receptors, PBMCs were stained with fluorescently conjugated recombinant IbAr10200 GP38 (rGP38)—expressed from a stably transfected Schneider 2 cell line—and a panel of fluorescently conjugated antibodies to cell-surface markers.
Of the total population of switched immunoglobulin (SwIg) B cells, 0.35%, 0.14%, and 0.11% were rGP38-reactive for donors 1, 5, and 6, respectively (Figure S1B). Flow analysis demonstrated that 63%–91% of GP38-reactive B cells from these donors were class switched (Figure 1A), indicative of an MBC response and of class-switch recombination dynamics consistent with the Gc-specific CCHFV response.30 Of this class-switched, GP38-reactive population, 50.0%, 15.8%, and 25.0% of the cells were CD27+ for donors 1, 5, and 6, respectively (Figures S1C and S1D), consistent with the varying levels of CD27 expression observed in the human MBC compartment.31–34 Because the majority of the GP38-reactive B cells were IgM− IgD−, only these SwIg B cells were isolated for further downstream analysis (Figures 1A and S1E). Isolated antibody genes from sorted B cells were amplified using VH and Vκ or Vλ single-cell PCR. In total, 254 paired VH/VL antibody genes were successfully cloned into an IgG1 isotype in a proprietary, engineered S. cerevisiae strain.
Figure 1. Isolation of monoclonal antibodies and genetic signatures of the B cell repertoire.
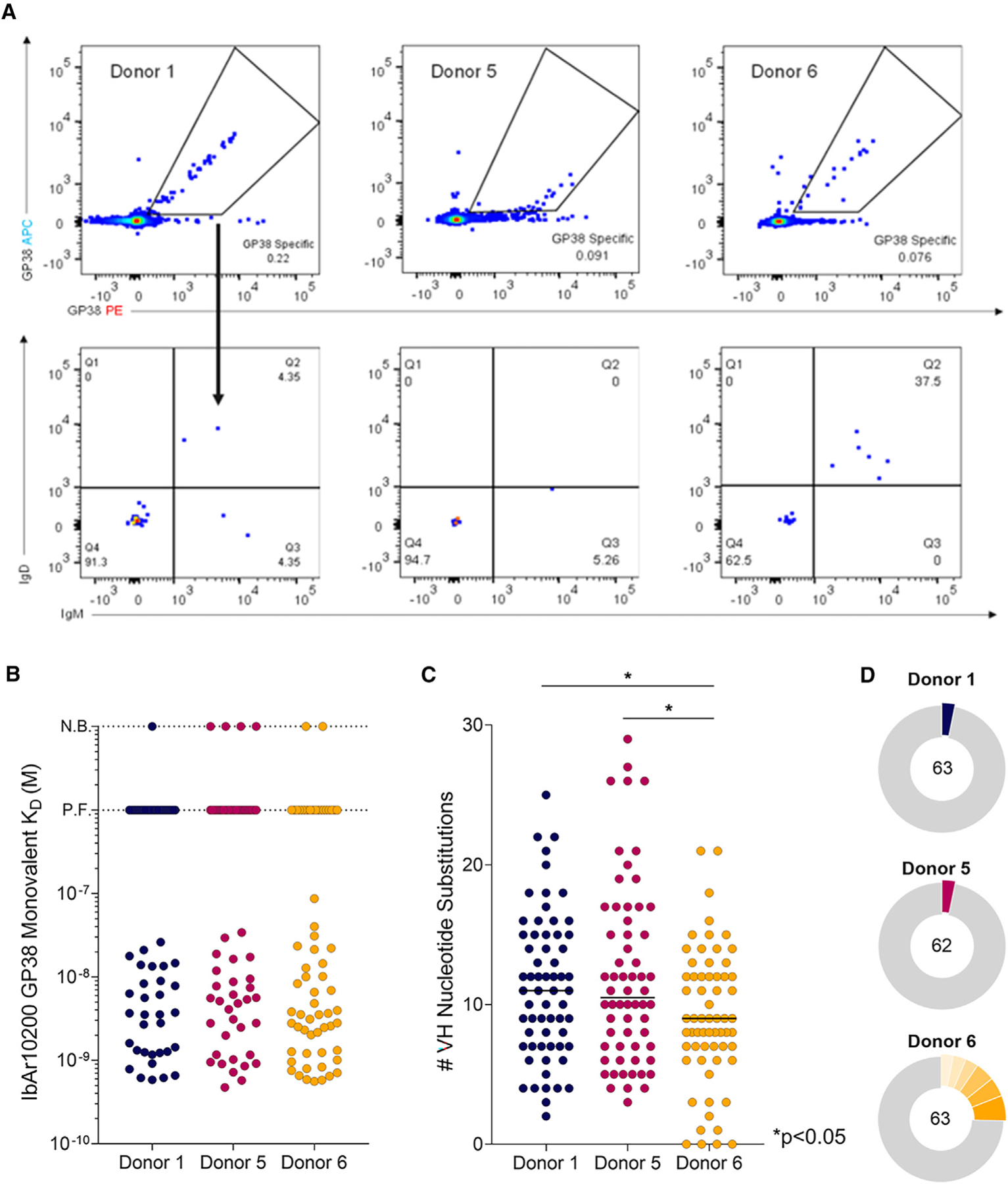
(A) Flow cytometric analysis of avid-rGP38 binding of B cells (top) and IgM and IgD expression on the surface of rGP38-reactive B cells (bottom). Donor 1 PBMCs were gated on CD3−CD8−CD14−CD16−PI−CD19+ lymphocytes; donors 5 and 6 PMBCs were gated on CD3−CD8−CD14−CD16−PI−CD19+CD20+ lymphocytes.
(B) Single concentration BLI binding analysis of 188 antibodies to IbAr10200 rGP38 protein. Dotted horizontal lines indicate antibodies for which no binding (N.B.) was detected or for which poor fits (P.F.) to the binding model were obtained.
(C) Analysis of VH nucleotide substitutions of each of the mAbs. Statistical comparison was performed using the Mann-Whitney test (*p < 0.05).
(D) Clonal lineage analysis of B cells from donors 1, 5, and 6. B cells with antibody sequences that had the same V heavy and V light germline gene usage and CDRH3s of the same length with >80% nucleotide sequence identity were considered to be clonally related. Colored slices represent the percentage of clones from each donor that are related. The total number of isolated mAbs from each donor is indicated in each corresponding circular diagram.
After expression and purification of this panel of mAbs, we assessed binding of the full-length IgGs to IbAr10200 rGP38 using biolayer interferometry (BLI). We found that 188 of the 254 purified mAbs bound to rGP38 in this assay (Figures S2A and S2B). To better understand the human immune response against GP38, we determined the affinities of these antigen-specific IgGs via BLI. A total of 181 mAbs had detectable monovalent binding to IbAr10200 rGP38 (Figure 1B). Of the 107 monovalent binders for which a 1:1 binding model could be fit, 78.5% (n = 84) had affinities better than 10 nM (Figures 1B and S2C). Antibodies isolated from donors 1, 5, and 6, displayed single-digit nanomolar median binding affinities against IbAr10200 rGP38 with median affinities of 3.5, 4.4, and 2.8 nM, respectively (Figure S2C). Taken together, these data indicate that convalescent CCHFV-infected donors can generate high-affinity, long-lived, GP38-specific antibody responses.
Genetic signatures of GP38-specific antibodies
We next assessed the specific genetic signatures associated with CCHF-convalescent donor antibody responses to GP38. Previous work has described CCHFV Gc-specific antibody responses, as well as genetic signatures typically observed in antibodies elicited by other primary viral infections or vaccinations.30,35–37 Somatic hypermutation (SHM)—a hallmark of affinity maturation—and clonal diversity are important metrics in the assessment of the quality of an antigen-specific antibody response following infection or immunization.38,39 Antibodies from the 3 donors had median values of SHM between 9 and 11 heavy-chain nucleotide substitutions (Figure 1C) and, in general, samples collected from donors with longer times between infection and blood donation contained B cells with higher levels of SHM (Figure 1C; Table S1). Paired heavy- and light-chain analyses demonstrated high levels of clonal diversity (3%–25% clonal relatedness) among antibodies cloned from all three donors (Figure 1D), similar to levels of diversity seen among B cells isolated from survivors of Ebola virus and SARS-CoV-2 infections.34,36 Interestingly, the higher clonal relatedness (25%) among GP38-reactive B cells cloned from donor 6 is in contrast with what was seen among Gc-specific MBCs (0% clonal relatedness) from the same donor.30 GP38-specific mAbs from all three donors had a similar distribution of heavy-chain complementarity-determining region three (CDRH3) lengths compared with the unselected human repertoire40 (Figure S3A). However, the donor 6 B cell response appears to be skewed toward clones with CDRH3 lengths of 13 and 21 amino acids, consistent with data showing that most of these clones arose from 2 distinct clonal expansions (Figure S3B).
We next sought to determine if specific V-genes were preferentially enriched in GP38 antibodies collected from these donors. Across all donors, sorted GP38-reactive B cells utilized VK1–39, VK3–20, and VL3–21 light-chain V-genes most often, at frequencies of 16%, 17%, and 26%, respectively (Figures S4A and S5). For each individual donor, greater than 50% of all sorted GP38-reactive B cells utilized these 3 light-chain V-genes (Figures S4B and S5). Heavy-chain V-gene usage was less skewed than light-chain V-gene usage; however, 13% of all cloned GP38-specific antibodies used VH3–48 and 15% used VH4–4 V-genes (Figures S4A and S5). VH3–48 predominantly paired with VL3–21 and VH4–4 paired with VK3–20 (Figure S4A). Although the VH3–48/VL3–21 pairing was seen across all donors, the VH4–4/VK3–20 pairing was a unique feature of the donor 6 response (Figure S4C). Collectively, our analysis shows that this isolated panel of GP38-specific antibodies is derived from a diverse population of B cells with a preference toward specific heavy- and light-chain V-genes.
GP38-specific antibodies recognize 11 overlapping antigenic regions
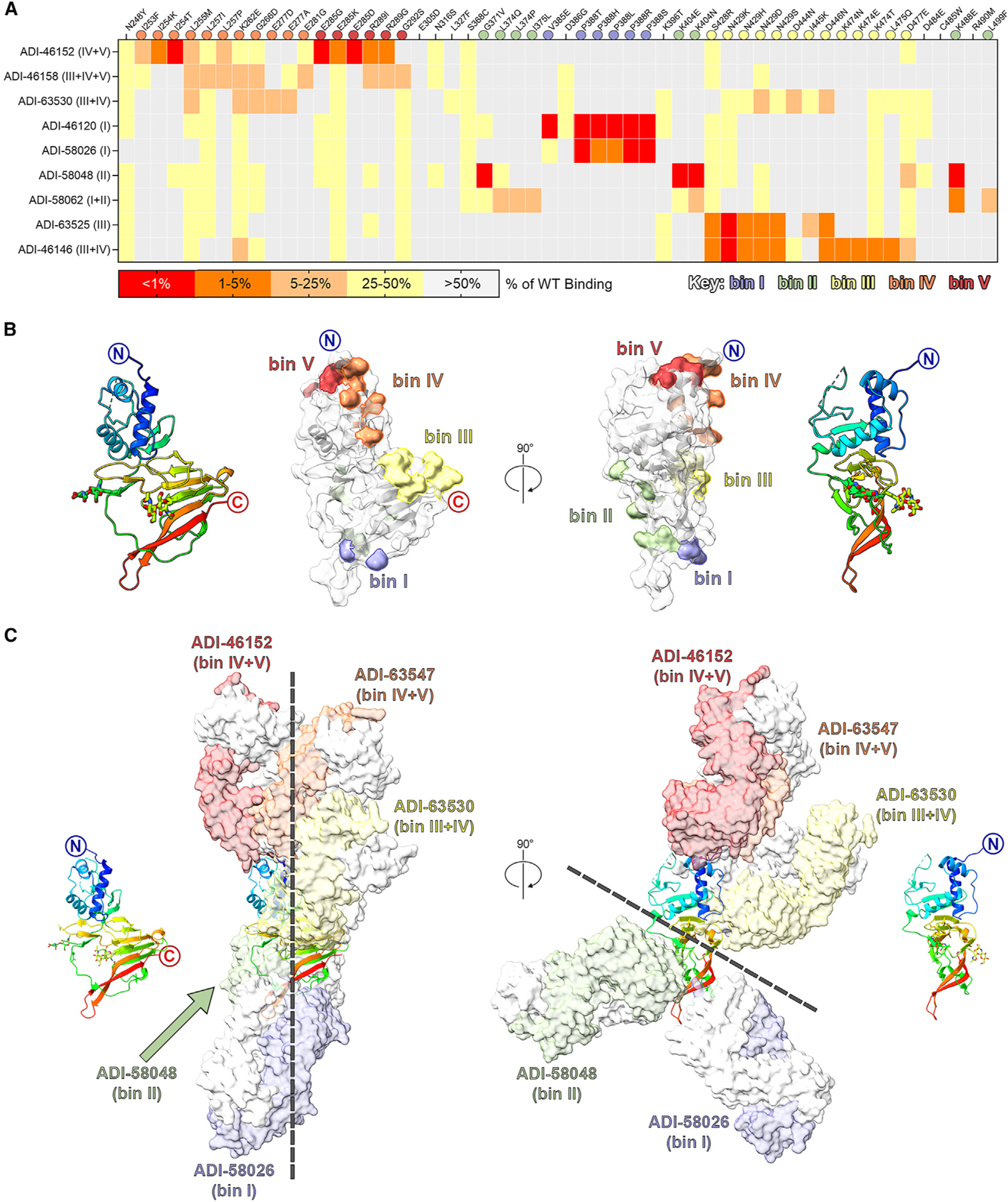
We conducted binding-competition assays to better understand where on GP38 the isolated antibodies bound. Because we lacked the capacity to cross-bin 188 mAbs (i.e., a 188 × 188 matrix), we down-selected our repertoire to 19 clones with disparate VH/VL germline pairings and CDRH3 sequences to perform multiple cross-competition experiments (Figure S6). From these experiments, we discovered 7 mAbs (ADI-46120, ADI-46146, ADI-46152, ADI-46158, ADI-46172, ADI-46174, and ADI-58048) that, when cross-binned in yeast-based competition assays, revealed the presence of 5 non-overlapping bins (Figure 2A), as has been described previously.27
Figure 2. Competition-binning profile of GP38 antibodies.
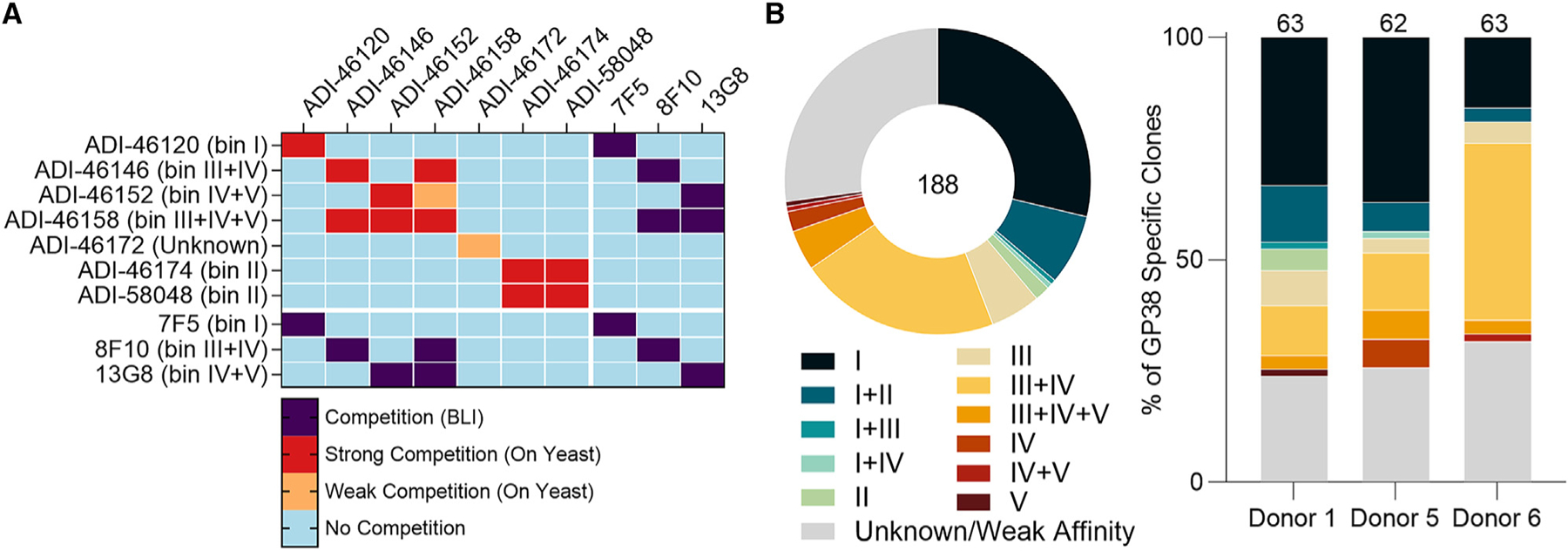
(A) Matrix of competition-binning experiments. For on-yeast competition experiments (top left quadrant), results are displayed with surface-presented IgGs on the y axis and competitive pre-complexed Fabs on the x axis. For BLI competition assays (the other three quadrants), binning was performed in an IgG vs. IgG format.
(B) Binning analysis of on-yeast competition assays of all 188 antibodies; each color represents 1 of 11 overlapping bins and the Unknown/Weak Affinity mAbs are shown in gray (Table S2). Distribution of overlapping bins of the antibody panel (left) and by each donor (right). Total number of mAbs is indicated in the circular diagram and total mAbs from each donor are indicated above each bar graph.
To gain a more granular understanding of the immunogenic surface of GP38, we performed a binning assay with our entire panel of 188 GP38-specific antibodies. We chose one antibody from each of the 5 non-overlapping antigenic sites to be run in competition against all 188 antibodies (i.e., a 188 × 5 matrix): ADI-46120, ADI-46146, ADI-46152, ADI-46158, and ADI-58048. The highest affinity antibody from each of the 5 non-overlapping bins was selected to provide the assay with the greatest discriminatory power. The results revealed that our panel of 188 GP38-specific mAbs fell into 11 overlapping bins (Figure 2B). Antibodies that only competed with one of the 5 representative antibodies were labeled as bin I (ADI-46120 competitor), II (ADI-58048 competitor), III (ADI-46146 competitor), IV (ADI-46158 competitor), or V (ADI-46152 competitor) and antibodies that competed with 1 or more of the 5 representative antibodies were labeled with 2 or more roman numerals (i.e., bin III+IV antibodies compete with both ADI-46146 and ADI-46158) (Table S2). Across all donors, the immune response consisted primarily of antibodies from bin I (n = 54) and bin III+IV (n = 40) (Figure 2B). Fifty of the 188 antibodies (26.6%) did not appear to compete with any of the 5 selected competitor antibodies (Figure 2B). Many of these antibodies likely appear non-competitive in yeast-based competition assays because of their weak affinity for IbAr10200 GP38; however, a subset did bind to IbAr10200 GP38 and may recognize unique antigenic sites (Data S1). We also conducted cross-competition assays with three previously characterized murine mAbs (7F5, 8F10, and 13G8). These experiments revealed that 7F5 is a bin I mAb as it competes with ADI-46120, 8F10 is a bin III+IV mAb as it competes with both ADI-46146 and ADI-46158, and 13G8 is a bin IV+V mAb as it competes with both ADI-46158 and ADI-46152 (Figure 2A). Collectively, these studies identify 11 overlapping regions on the GP38 surface targeted by human and murine antibodies.
Isolated GP38-specific antibodies are broadly reactive
Our initial binding studies used GP38 derived from CCHFV IbAr10200 (Figures 1 and S2), a clade III virus. However, this is a highly laboratory-passaged virus with little clinical relevance. Most confirmed reported cases of human infection are attributed to isolates from clades III, IV (Afg09, Oman, and M18-China), and V (Turkey2004 and Kosova-Hoti),1,6,18,41 and over the past few years new strains have emerged from areas where these clades are endemic.42,43 Therefore, we chose 5 clinically relevant isolates (Afg09, Turkey2004, Oman, Kosova-Hoti, and M18-China) in addition to IbAr10200 to determine the extent to which the 188 isolated GP38-specific antibodies bind to multiple clinically relevant and diverse isolates to assess their potential as therapeutic candidates. The GP38s of the aforementioned CCHFV isolates exhibit between 70% and 92% amino acid sequence similarity with IbAr10200 (Figure 3A). Sequence alignment of the 6 isolates reveals that much of the variation occurs in the variable loop (residues 322–341) with some additional variation observed within the extended loop (residues 377–394) (Figure S7). First, we used BLI to assess the monovalent affinity of each of the 188 mAbs at a single concentration to each of the 6 GP38 variants. mAbs for which the recorded response was greater than 0.05 response units were considered to bind to the respective rGP38 protein. These experiments revealed that 87% of the 188 GP38-specific mAbs bound GP38 derived from all 6 tested isolates and 8% across 5 of 6; the remaining 5% of mAbs bound GP38 derived from 4 or fewer isolates (Figure 3B). These high levels of cross-reactivity are comparable with those seen in the Gc-specific responses from the same donors.30 The single concentration BLI data was used to select high-affinity, cross-reactive clones with varying germline usage from discrete bins (Table S3). Antibody-drug developability metrics (i.e., polyreactivity, hydrophobic interaction chromatography, thermostability) (Table S4)44 were then run on these clones of interest and lead candidates were established for further study: ADI-58026 (bin I), ADI-58062 (bin I+II), ADI-58048 (bin II), ADI-63530 (bin III+IV), ADI-46138 (bin III+IV+V), and ADI-63547 (bin IV+V).
Figure 3. Analysis of antibody binding to GP38 proteins derived from six CCHFV isolates.
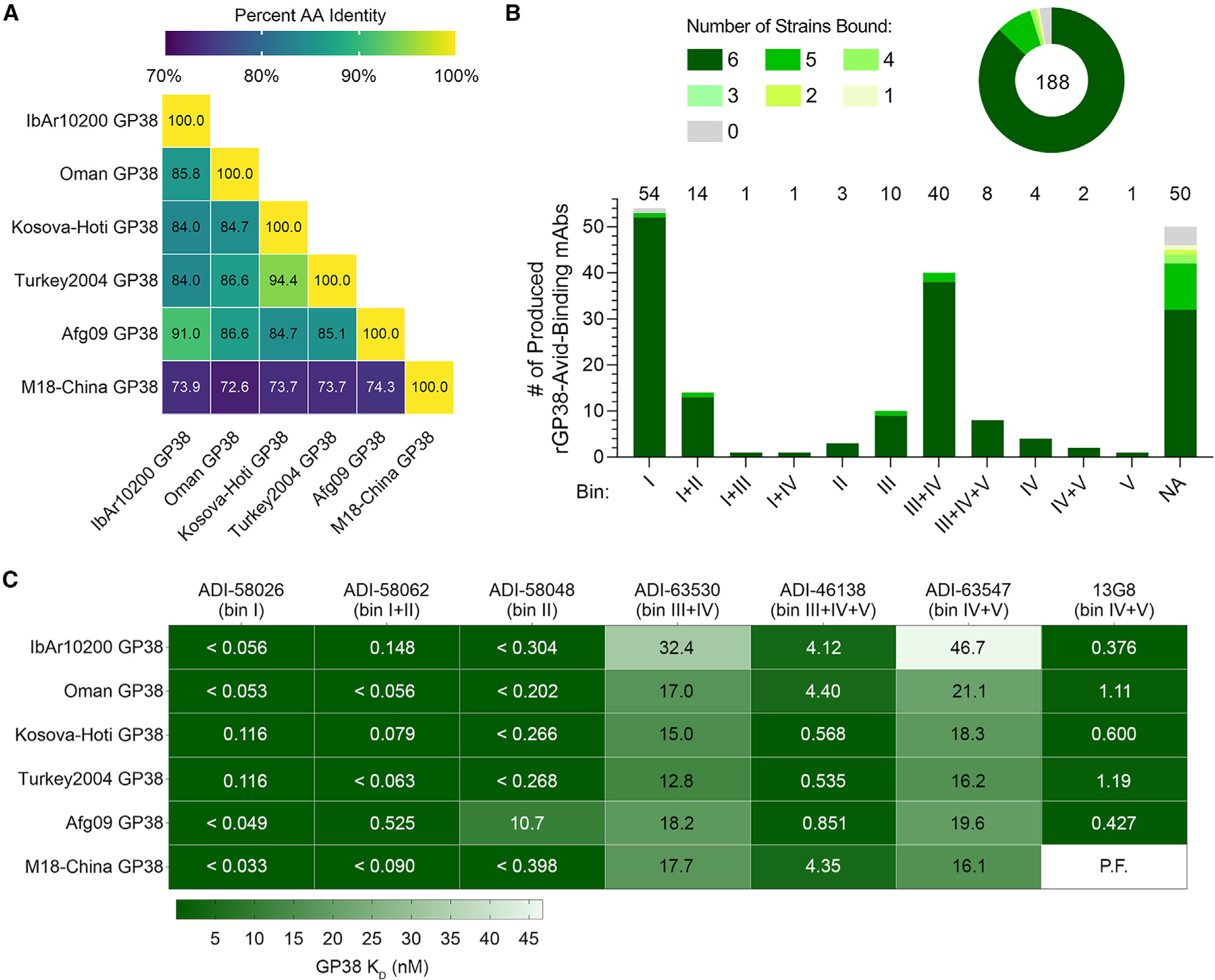
(A) Matrix of percent sequence identity of GP38 amino acid residues (AA) across six CCHFV isolates.
(B) Single concentration BLI binding analysis of 188 antibodies to the six rGP38 proteins as a whole panel (top) and broken down by bin (bottom). Shades of green represent the number of rGP38 proteins bound by a single antibody (from 0 in gray to 6 in darkest green). Total number of mAbs is indicated in the circular diagram and total mAbs from each bin are indicated above the bar graph.
(C) Carterra system HT-SPR binding analysis of six lead antibody candidates binding to six rGP38 proteins. The highest binding affinities are in dark green and the lowest binding affinities are in white. Calculated KD values appear in each rectangle of the heatmap; for samples that were off-rate limited, KD values are denoted as < the calculated KD. The one interaction for which a curve could not be fit is denoted as P.F.
To gain a more nuanced understanding of the cross-clade binding dynamics, we used the Carterra system to carry out multipoint KD measurements for the 6 lead antibodies as well as the previously described murine mAb 13G8. ADI-58026 (bin I) and ADI-58062 (bin I+II) bound to all 6 GP38 variants derived from clinical isolates with affinities better than 530 pM, and ADI-58048 (bin II) bound with an affinity less than 398 pM to 5 of 6 GP38 variants, but had an approximately 27-fold reduction in binding to Afg09-derived GP38 (Figure 3C; Table S5). Each mAb from bins III–V (ADI-63530, ADI-46138, and ADI-63547) bound to the 6 tested GP38 variants with affinities of 12.8–32.4, 0.54–4.4, and 16.2–46.7 nM, respectively (Figure 3C; Table S5). These 3 bins III–V mAbs all bound the 6 GP38 variants with affinities that were within 10-fold of their affinity to IbAr10200 GP38. Of these three antibodies, ADI-46138 exhibited the highest binding affinities, which were 3- to 30-fold higher than those determined for ADI-63530 and ADI-63547 (Figure 3C; Table S5). In addition, ADI-46138 (bin III+IV+V) and 13G8 (bin IV+V) bound to 5 GP38 variants with affinities within 11-fold of one another (Figure 3C; Table S5). Taken together, 95% of the 188 isolated GP38-specific antibodies bound to 5 or 6 GP38 variants derived from clinically relevant CCHFV isolates spanning diverse clades, and antibodies ADI-58026 (bin I) and ADI-58062 (bin I+II) bound these GP38 variants with picomolar affinities.
Antibodies targeting GP38 are non-neutralizing
The six lead GP38-specific mAbs were tested in a microneutralization assay utilizing transcription- and entry-competent virus-like particles (tecVLPs) bearing IbAr10200 GPC-derived proteins.30,45 None of the GP38-specific antibodies neutralized the tecVLPs in this assay (Figure 4A). Neutralization assays were also performed with authentic CCHFV, including the prototype IbAr10200 (clade III; Figure 4B) and clinically relevant isolates Afg09 (clade IV; Figure 4C), Turkey2004 (clade V; Figure 4D), and Oman (clade IV; Figure 4E) in SW-13 cells, a cell line relevant for CCHFV-infection that exhibits epithelial morphology.46 Again, none of the GP38-specific mAbs exhibited significant neutralization potency against the tested authentic viruses (Figures 4B–4E), consistent with previous reports.27,29,30,47 To determine whether neutralization potency was cell-type specific, a microneutralization assay was also conducted in VeroE6 cells with authentic viruses. Comparable with the results obtained in SW-13 cells, none of the GP38-specific mAbs afforded significant neutralization potency against any of the CCHFV isolates tested in VeroE6 cells (Figure S8). ADI-36121, a Gc-specific mAb previously shown to afford significant cross-clade neutralization efficacy against CCHFV,30 was utilized as a positive control and, as anticipated, potently neutralized tecVLPs (Figure 4A) and all isolates of authentic CCHFV tested in both SW-13 (Figures 4B–4E) and VeroE6 cells (Figure S8). Consistent with previously reported studies, our panel of GP38-specific antibodies was non-neutralizing under the conditions tested.27,29,30
Figure 4. CCHFV tecVLPs and authentic virus neutralization assays of GP38 antibodies.
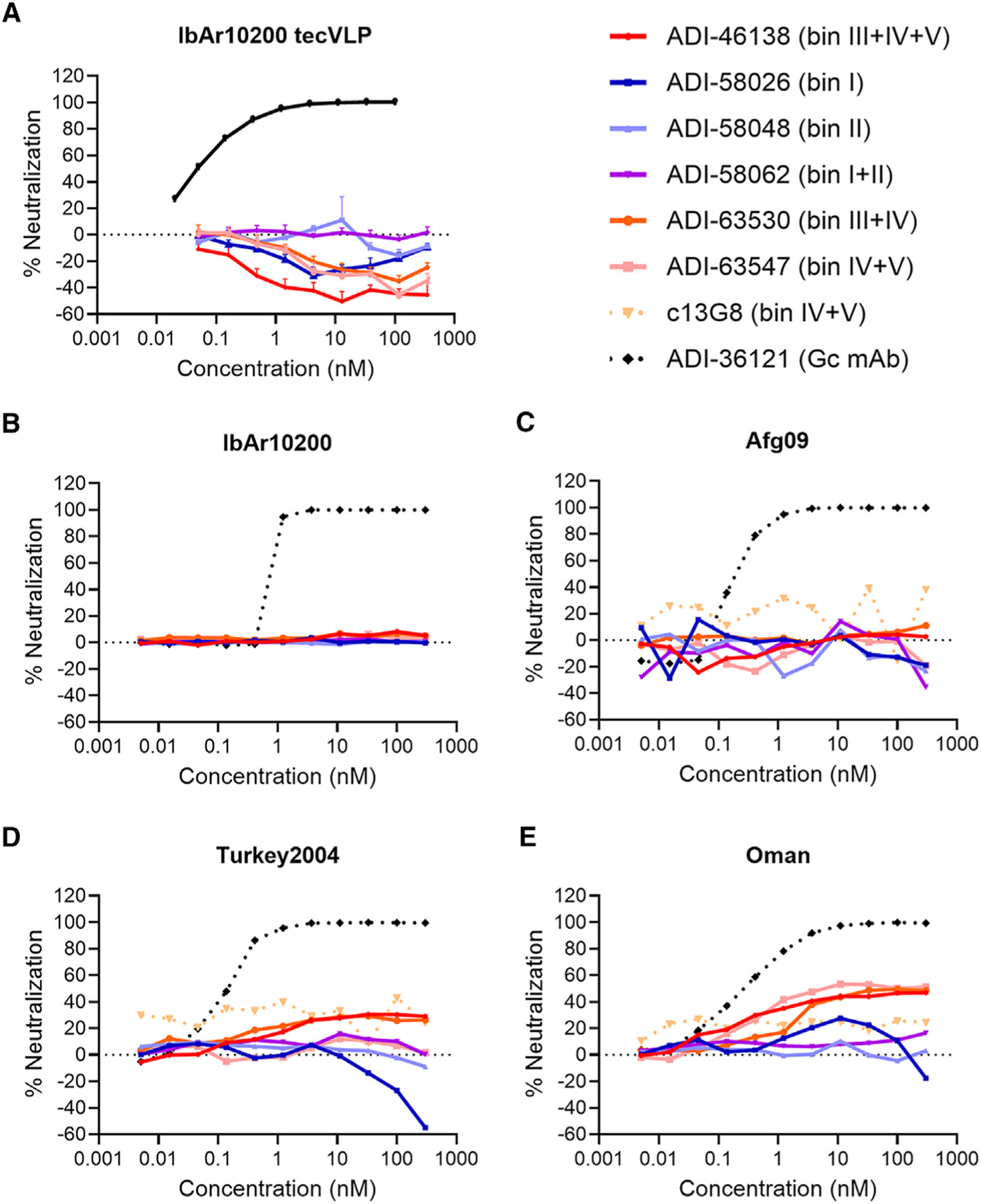
(A) Neutralization curves for CCHFV IbAr10200 tecVLPs, as measured by the reduction in luciferase activity compared with no-antibody treatment on Vero cells.
(B–E) Neutralization curves of the indicated mAbs against authentic (B) CCHFV IbAr10200, (C) CCHFV Afg09, (D) CCHFV Turkey2004, and (E) CCHFV Oman as measured by the reduction in infection compared with no-antibody treatment on SW-13 cells. The average of n = 3 replicates each from two independent experiments (n = 6 total) is shown for all neutralization curves.
Epitope mapping reveals two predominantly targeted regions on GP38
We set out to map the location of the antigenic sites on GP38 to correlate certain epitopes with protection and function. We employed a yeast surface display (YSD)-based mapping and structural characterization strategy utilizing select GP38-specific antibodies. A YSD library of GP38 single-amino-acid variants was generated to compare antibody binding between mutant and wild-type GP38. Nine antibodies representing 7 of the 11 overlapping bins successfully underwent YSD mapping to reveal critical residues on GP38 necessary for retaining antibody binding (Figure 5A). Critical residues that disrupted antibody binding by 75% or more were mapped onto the surface of IbAr10200 GP38 to represent the 5 discrete antigenic sites (Figures 5B and S9). These studies were complemented with structural studies of select antibodies to further characterize the antigenic sites.
Figure 5. Structural characterization of GP38-specific antibodies.
(A) Yeast-based mapping strategy of select antibodies to identify critical binding residues on GP38. The percentage of antibody binding retained by each GP38 variant is colored according to the key. Critical residues are defined as mutations that led to a binding disruption of 75% or more and are colored by the assigned antigenic site.
(B) Yeast-based critical residues mapped on the surface of GP38: bin I (blue, residues Val385, Pro388), bin II (green, residues Gly371, Leu374, Ile375, Lys404, Lys488, Leu499), bin III (yellow, residues Ser428-Ala429, Asp444-Asp446, Lys474-Leu475, Asp477), bin IV (orange, residues Ile253-Leu255, Leu257, Lys262, Gly266, Glu277, Glu281), bin V (red, residues Glu285, Arg289, Gly292).
(C) Composite structure of GP38 bound with representative antibodies. GP38 is shown as a rainbow ribbon and Fabs as molecular surfaces. Heavy chains are colored to represent the five non-overlapping bins, and light chains are white. Black dashed lines highlight the vertical alignment of Fabs along one plane (left) and the opposing binding directions to another plane (right).
To map the epitope of bin I antibodies, we determined a 5.0 Å resolution cryo-EM structure of ADI-58026 Fab (bin I) and ADI-63547 Fab (bin IV+V) bound to GP38 (Figure S10; Table S6). Due to the resolution of the cryo-EM reconstruction, we docked AlphaFold2 models of the Fabs into the maps to assess the epitopes. The docked ADI-58026 Fab binds near the extended loop and C-terminal β hairpin, in excellent agreement with bin I YSD critical residues Val385 and Pro388 (Figure 5C). To further characterize the bin I epitope, we complexed ADI-46143 Fab (bin I) to GP38 and determined a 2.6 Å resolution crystal structure (Rwork/Rfree = 0.177/0.217), which revealed that ADI-46143 Fab binds primarily to the extended loop, with additional contacts to the C-terminal β12-β13 hairpin, similar to ADI-58026 (Figures 6A and S10; Table S7). Pro388—a YSD-identified critical residue of bin I antibodies—is at the center of the ADI-46143 epitope (Figure 6A).
Figure 6. High-resolution structures of GP38-antibody complexes.
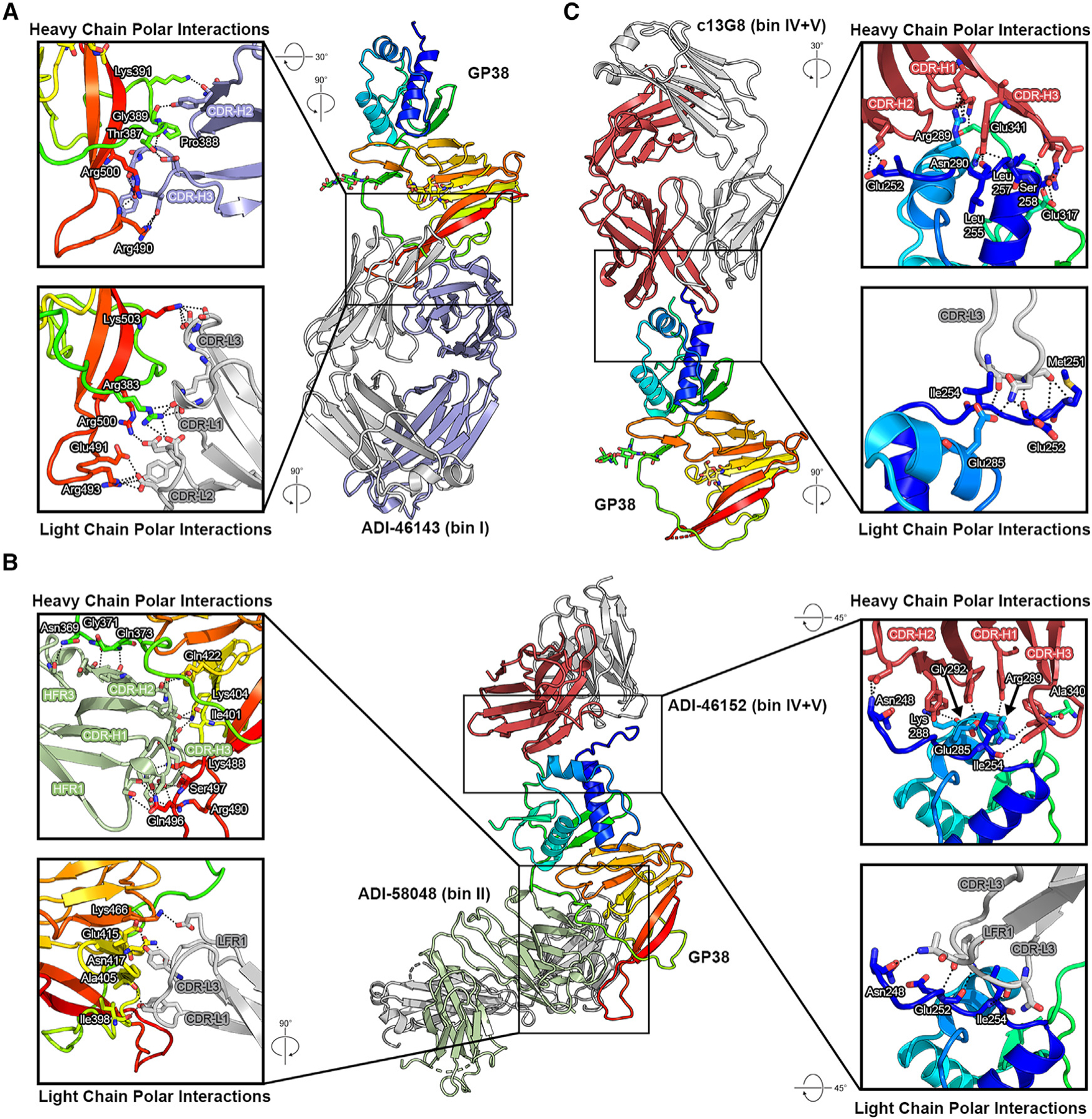
(A) Crystal structure of GP38 bound with ADI-46143 (bin I, blue) with heavy-chain interactions (top) and light-chain interactions (bottom).
(B) Cryo-EM structure of GP38 bound with ADI-58048 (bin II, green, left) and ADI-46152 (bin IV+V, red, right). Heavy-chain interactions (top left, top right) and light-chain interactions (bottom left, bottom right) are shown in the insets.
(C) Crystal structure of GP38 bound with c13G8 (bin IV+V, red) with heavy-chain interactions (top) and light-chain interactions (bottom). For all panels, heavy chains are colored, light chains are gray, polar interactions are indicated by black dashed lines, and GP38 residues are labeled in white text with a black outline.
To map the epitope of bin II antibodies, a complex of GP38 bound with ADI-58048 Fab (bin II) and ADI-46152 Fab (bin IV+V) was generated and a 3.8 Å resolution cryo-EM structure of the complex was determined (Figures 6B and S11; Table S6). ADI-58048 binds the β sandwich, including residues in the long loop connecting the C-terminal β12-β13 hairpin (Figures 5C, 6B, and S11). Three bin II YSD critical residues on GP38 (Gly371, Lys404, Lys488) are at the interface with the ADI-58048 heavy chain (Figure 6B). Lys404 and Lys488 are at the interface with the ADI-58048 CDRs, whereas Gly371 is located in the variable loop of GP38 and rests against the side of the VH domain. We also determined a 5.1 Å resolution cryo-EM structure of GP38 in complex with ADI-58062 Fab (bin I+II) and ADI-63530 Fab (bin III+IV) (Figure S12), which revealed that ADI-58062 binds to a similar epitope as ADI-58048 and that the antibodies would sterically clash, as expected for two bin II competitors.
To map the epitopes of antibodies that competed across bins III–V, we analyzed the aforementioned cryo-EM structures as well as determined a 5.8 Å resolution cryo-EM of GP38 in complex with ADI-46158 (bin III+IV+V) and ADI-46143 (bin I) (Figures 5C and S10–S13). Consistent with bin IV critical residues, ADI-46158 and ADI-63547 (bin IV+V) Fabs bind the three-helix bundle, primarily the first several N-terminal residues and the beginning of α helix 1 (Figures 5C, S10, and S13). Their epitopes predominantly target bin IV YSD critical residues while also contacting bin V YSD critical residues Glu285 and Arg289 on α helix 2. The ADI-63530 (bin III+IV) epitope spans both the three-helix bundle and β sandwich, consistent with bin III YSD critical residues Ser428-Ala429, Asp444-Asp446, Lys474-Leu475, and Asp477, which are in loops connecting strands β6-β7, β8-β9, and β11-β12 (Figures 5C and S12).
To further map the epitope of bin V antibodies, we selected two bin IV+V antibodies for structural studies: ADI-46152 and a humanized chimeric variant of 13G8 (c13G8).28 From the 3.8 Å resolution cryo-EM structure of GP38 bound with ADI-58048 Fab (bin II) and ADI-46152 Fab, the ADI-46152 heavy chain makes several contacts on α helix 2, N-terminal residues preceding α helix 1, and the variable loop, while the ADI-46152 light chain contacts N-terminal residues Asn248, Glu252, and Ile254, consistent with bin IV and V YSD residues (Figures 5C, 6B, and S11; Table S6). To resolve the epitope of 13G8, c13G8 Fab was complexed to GP38, and we determined a 1.8 Å resolution crystal structure (Rwork/Rfree = 0.200/0.215) (Figure 6C; Table S7). The structure revealed that c13G8 binds to the N-terminal three-helix bundle of GP38, consistent with the 3.6 Å structure determined by Durie et al.27 YSD critical residues identified on GP38 (Ser258, Arg289, and Asn290) interact with the c13G8 heavy chain and YSD critical residue Ile254 is also at the antibody interface (Figure 6C). Epitopes of c13G8 and ADI-46152 are highly overlapping and share two YSD critical residues (Ile254 and Arg289) (Figure S14). Compared with ADI-63547 (bin IV+V), ADI-46152 and c13G8 have shifted angles of approach that extend contacts to residues Glu317 and Ala340 within the bin V epitope.
To visualize the overall antigenic landscape, we generated a composite view of GP38 bound with Fabs ADI-58026 (bin I), ADI-58048 (bin II), ADI-63530 (bin III+IV), ADI-63547 (bin IV+V), and ADI-46152 (bin IV+V) (Figure 5C). These antibodies are representative of the five antigenic sites based on both YSD-based mapping and structural studies. The composite structure reveals that the antibodies approach GP38 along a similar plane. Furthermore, the antibodies bind predominately to two general regions: an N-terminal region containing bins III–V comprising the three-helix bundle and loops connecting adjacent β strands, and a region containing bins I and II comprising the extended loop and C-terminal β hairpin. These restricted binding modes may result in part from how GP38 is oriented on the virion or in complex with other proteins from the GPC.
Antibodies targeting epitope bins III, IV, and V afford partial therapeutic protection against a lethal CCHFV-IbAr10200 challenge
We next evaluated the therapeutic potential of our six lead GP38-specific antibodies in an immunocompromised rodent model of lethal CCHFV challenge: ADI-58026 (bin I), ADI-58048 (bin II), ADI-58062 (bin I+II), ADI-63530 (bin III+IV), ADI-63547 (bin IV+V), and ADI-46138 (bin III+IV+V). c13G8 (bin IV+V) was included as a benchmark for comparison with previously published studies. Type I interferon ɑ/β R−/− (IFNAR1−/−) mice48,49 were challenged with 100 PFU of CCHFV-IbAr10200 and subsequently treated with 1 mg of mAb per animal 1 and 4 days post-challenge (2 mg/mouse total), to replicate previous conditions testing 13G8 efficacy.27,29 As described previously, c13G8 afforded partial protection (40%) (Figures 7A–7C). Antibodies targeting GP38 epitope bins I (ADI-58026), II (ADI-58048), or I+II (ADI-58062) were minimally protective (20%–30% survival), and less so than that of c13G8. In contrast, antibodies targeting epitope bins III+IV+V (ADI-46138) and IV+V (ADI-63547) were similarly protective as c13G8 (40% survival). Moreover, antibody ADI-63530, targeting GP38 epitope bins III+IV, exhibited substantial protection (70%), which was greater than that observed for c13G8. Collectively, these data indicate that antibodies targeting GP38 epitope bins I and II are minimally protective, whereas antibodies targeting GP38 epitope bins III, IV, and V are most protective against a CCHFV-IbAr10200 lethal challenge. Interestingly, although antibodies targeting GP38 epitope bins III, IV, and V were more protective than the bin I and II targeting antibodies, the bin III, IV, and V specific antibodies displayed lower affinities compared with the bin I and II specific antibodies (Figures 3 and S15). These findings indicate that human mAbs targeting bins III, IV, and V on GP38 are equally, if not more, efficacious than the previously described murine mAb 13G8 against a lethal CCHFV-IbAr10200 challenge.
Figure 7. Protective efficacy of lead mAbs in two murine models of lethal CCHFV challenge.
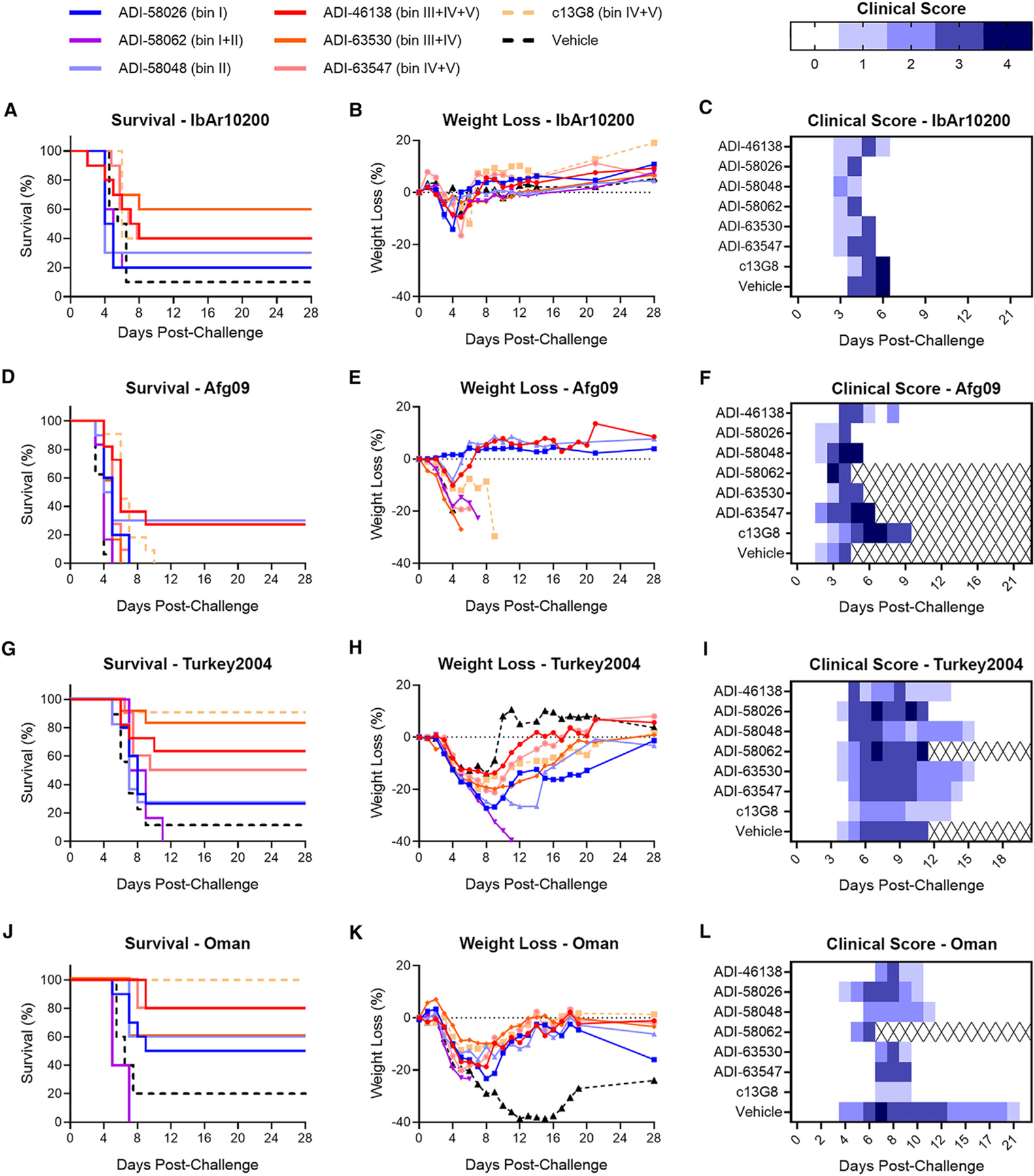
(A–C) IFNAR1−/− mice were treated with the indicated mAbs at 1 mg/mouse 1 and 4 days post-challenge (2 mg total; n = 10 mice per group) with IbAr10200. (A) Survival curves (vehicle vs. test mAb), (B) associated mean weight loss, and (C) clinical score data are shown.
(D–L) STAT1−/− mice were challenged with (D–F) CCHFV-Afg09, (G–I) CCHFV-Turkey2004, or (J–L) CCHFV-Oman and then treated with 0.2 mg/mouse of mAb or vehicle 30 min post-exposure (n = 5–6 mice per study; represented by 2 replicate studies). (D, G, and J) Survival curves. (E, H, and K) Associated mean weight loss. (F, I, and L) Clinical scores are defined as: 1 = decreased grooming and/or ruffled fur; 2 = subdued behavior when un-stimulated; 3 = lethargy, hunched posture, and/or subdued behavior even when stimulated; 4 = bleeding, unresponsiveness, severe weakness, or inability to walk. Mice scoring a 4 were considered moribund and were humanely euthanized based on IACUC-approved criteria (denoted as X over white).
ADI-46138 and ADI-58048 provide cross-clade protection in a stringent lethal mouse model of infection
Having demonstrated protective efficacy for ADI-63530 against CCHFV-IbAr10200 challenge (Figures 7A–7C), we tested its cross-clade protective efficacy against Afg09, Turkey2004, and Oman in STAT1−/− (signal transducer and activator of transcription 1 knockout) mice. STAT1−/− mice are more susceptible to a broad range of CCHFV isolates compared with IFNAR1−/− mice,50 and were therefore used to assess broad-spectrum efficacy. STAT1−/− mice were challenged with either 100 PFU of Afg09 or with 1,000 PFU of Turkey2004 or Oman and subsequently treated with 1 mg/mouse of ADI-63530, ADI-58062, c13G8, or vehicle 1 and 4 days post-challenge (2 mg/mouse total). ADI-58062 was included in these studies to investigate the extent to which protection correlates with binding affinity, as it exhibited the highest binding affinities against Afg09, Oman, and Turkey2004 of all lead mAbs (Figure 3D). Overall, survival was relatively poor regardless of the mAb used for treatment (Figure S16), suggesting that these mAbs cannot provide significant protection under more stringent challenge conditions.
Considering the poor survival observed in the previous study, a third challenge study was conducted utilizing less-stringent infection conditions to gain a better understanding of the relationship between cross-clade protective efficacy breadth and GP38-specific antibody bin. Each of our six lead candidates, in addition to c13G8, was tested in this study. Previous results have shown that 13G8 is 80%–100% protective against a CCHFV-Turkey2004 challenge in STAT1−/− mice when given 30 min post-exposure at a dose of 0.25 mg.28 To enhance the stringency, mice were treated with a slightly lower dose of 0.2 mg/mouse. STAT1−/− mice were challenged with either 100 PFU of Afg09 or 1,000 PFU of Turkey2004 or Oman and subsequently treated with 0.2 mg/mouse of our 6 lead mAbs 30 min post-challenge.
Although none of the c13G8-treated mice survived challenge with CCHFV-Afg09 (Figure 7D), 90% and 100% of the c13G8-treated mice survived challenge against CCHFV-Turkey2004 (Figure 7G) and CCHFV-Oman (Figure 7J), respectively. Only two antibodies, one targeting bin III+IV+V (ADI-46138) and the other targeting bin II (ADI-58048), were partially protective against all tested viruses; CCHFV-Afg09 (27% and 30%, Figures 7D and 7E), CCHFV-Turkey2004 (~64% and 27%, respectively; Figure 7G and 7H), and CCHFV-Oman (80% and 60%, respectively; Figure 7J and 7K). While other antibodies from bins III–V, including ADI-63530 (bin III+IV) and ADI-63547 (bin IV+V), were not protective against CCHFV-Afg09 (Figures 7D and 7E), they were broadly protective against CCHFV-Turkey2004 (~83% and 45%, respectively; Figures 7G and 7H) and CCHFV-Oman (60% and 80%, respectively; Figures 7J and 7K), similar to what was observed for c13G8. Apart from ADI-58048 (bin II), other mAbs from bins I–II (ADI-58062 and ADI-58026) demonstrated minimal-to-no cross-clade protection. Relative to CCHFV-Agf09 (Figure 7F), overall survival across all mAbs was greater against CCHFV-Turkey2004 and CCHFV-Oman, although a prolonged course of disease for CCHFV-Turkey2004 and CCHFV-Oman was observed whereby animals exhibited clinical signs of disease ranging from days 4 to 15 (Figure 7I) and 4 to 11 (Figure 7L). Taken together, these data show that antibodies targeting epitope bins III–V (ADI-46138, ADI-63530, ADI-63547, and c13G8) exhibit the best protection across isolates, including CCHFV-IbAr10200, Turkey2004, and Oman. However, antibodies targeting bins I–II (namely ADI-58048 and ADI-58026) elicit some cross-protection, albeit less than that of bins III–V antibodies. Furthermore, although there appears to be an inverse correlation across isolates between protective efficacy and binding affinity (i.e., lower-affinity antibodies were more protective), this relationship is not statistically significant (Figure S15). Overall, ADI-46138 (bin III+IV+V) and ADI-58048 (bin II) emerged as lead GP38-specific mAbs by providing partial protection against all four CCHFV isolates tested (IbAr10200, Afg09, Turkey2004, and Oman).
DISCUSSION
GP38 is a validated target for the development of mAb-based therapeutics and vaccines.28,29,51 Moreover, isolation of protective mAbs from human survivors of infection has been shown to be a promising approach for the development of therapeutics against a number of different viruses.30,52–60 Herein, we isolated and characterized a large panel of GP38-specific mAbs from human survivors of CCHFV infection in Uganda. Several of these mAbs, particularly ADI-46138 and ADI-58048, were found to be as protective as, or more so than, the previously described murine mAb 13G8 against multiple CCHFV isolates in our animal model systems. Further study of these lead candidates could give insight into regions on the GP38 surface that are important for pathogenesis.
Previous reports have determined the presence of 5 distinct antigenic sites on GP38.27,29,61 Utilizing our sizable antibody panel, we confirmed, structurally mapped, and characterized each of the 5 distinct antigenic sites and described the existence of 11 novel overlapping antibody competition ‘‘bins’’ that span the GP38 protein (Figures 2, 3, 5, and 6; Table S2). Although antibodies bind across GP38, we observed two distinct binding regions: one comprising the N-terminal three-helix bundle and adjacent loops from the β sheet (bins III–V), and the other comprising the extended loop and C-terminal β hairpin of β12–13 (bins I–II) (Figure 5C). Interestingly, we observed variation in protection between epitope bins such that the antibodies targeting bins III–V were overall more protective than the antibodies targeting bins I–II (Figure 7). Paired with affinity data (Figure 3), these results suggest that higher affinity mAbs are not necessarily the most protective (Figure S15). Similarly, although previously described human mAb CC5-17 has a higher affinity to GP38 than does 13G8, it was poorly protective.27 Moreover, previous studies have demonstrated that non-neutralizing protective antibodies often function through Fc-mediated mechanisms62–66; in fact, reports have characterized a partial contribution of Fc-mediated functions in the protection provided by 13G8.28,29 One possibility is that mAbs from varied epitope bins differentially engage Fc receptors and complement factors, an observation seen in the studies of filoviruses and influenza viruses.67–69 Taken together, these data suggest that binding affinity, and even epitope bin, do not exclusively determine protective efficacy provided by GP38-specific mAbs. However, in this work, only a single antibody from each bin was selected for further in vitro and in vivo characterization, limiting our ability to draw definitive conclusions regarding the relationship between epitope bin and protection, warranting additional follow-up studies utilizing multiple antibodies from each epitope bin.
CCHFV is the most genetically divergent of the arboviruses.2,6,10 GP38, in particular, exhibits high diversity among lineages. Sequence diversity of GP38 has been cited as the reason for the poor cross-clade efficacy of 13G8.29 Along with variable protection between antigenic sites, we observed variable protection within overlapping epitope bins across the divergent isolates (Figure 7). Our knowledge regarding GP38 function and its contribution(s) to pathogenesis is limited. Therefore, a plausible explanation for the observed differences in mAb efficacy across isolates in vivo is rooted in the unidentified pathogenic functions of GP38 and the ability of these mAbs to limit these functions. Epitope-bin-specific protection could be explained by a potential structural role for GP38. GP38 has been speculated to form a complex with Gn on the virion surface, acting as the head region of the attachment protein, as suggested by an AlphaFold2-predicted model.70 In this model, the epitopes of bins I and II are near the GP38-Gn interface while those of bins III–V are predicted to be orientated away from Gn, potentially making the bin III–V epitopes more accessible for antibodies to bind and mediate protection. A more thorough investigation into the association of GP38 and Gn is needed to resolve the structural relevance of GP38 on the viral surface and further scrutinize the implications on epitope accessibility for GP38-specific mAbs. Further uncovering the pathogenic functions of GP38 will strengthen our understanding of the mechanisms of protection utilized by our panel of GP38-specific mAbs.
Cocktails of mAbs have shown promise for the broad-spectrum treatment of diverse viral isolates.52,53,71–73 Earlier work in the context of Ebola virus infection demonstrated that ‘‘enabling pairs’’ of neutralizing and non-neutralizing mAbs can result in potent neutralization and complete protection, even though neither antibody alone was able to provide complete protection.74 Neutralizing Gc-specific antibodies have been isolated from human survivors of infection and developed into a bi-specific mAb, DVD-121-801, resulting in robust post-exposure protection against a lethal CCHFV-IbAr10200 challenge.30 Although DVD-121-801 exhibits potent neutralization across multiple clades of CCHFV, weaker neutralization was observed for clade V isolates Kosova-Hoti and Turkey2004, suggesting that in vivo potency against clade V isolates may be impacted, although it has not been experimentally tested. In the context of this study, future work should consider combining potent Gc-specific mAbs, such as DVD-121-801, with GP38-specific mAbs (e.g., ADI-46138) to improve potency and maximize cross-clade protective efficacy. Combining multiple GP38-specific mAbs targeting different epitope regions could also be a useful approach for broadening efficacy and increasing potency. The wealth of structural data and characterization pertaining to antigenic sites across the GP38 protein described in this study should facilitate efforts to identify optimal mAb pairings as well as inform vaccine development.
Limitations of the study
Our study is limited in scope by the number of convalescent donors and the fact that all donors were male. Collection from more donors of diverse populations and geographical locations would provide further understanding of the human immune response following CCHFV infection. As the strains that infected our donors are unknown, we used rGP38 from the lab-derived IbAr10200 strain as sorting bait. Potential incompatibility between donor responses and sorting bait may have limited the discovery of clade- or isolate-specific mAbs. Although our panel is larger than any described previously for GP38-specific mAb panels, our study is limited in that we were only able to characterize a subset of the antibody panel for structural and protection studies. Furthermore, while immunocompromised mouse models of CCHFV infection are widely utilized and accepted for the initial down-selection and characterization of mAbs in vivo, studies in other animal models that more faithfully recapitulate human CCHFV infection (e.g., non-human primates) are needed to progress lead antibody candidates to combat human infection. Lastly, additional functional studies of GP38 along with characterization of effector functions provided by GP38-specific antibodies will garner insight into the potential mechanistic role of GP38 in CCHFV pathogenesis and support the use of GP38-specific antibodies as therapeutics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jason S. McLellan (jmclellan@austin.utexas.edu).
Materials availability
Materials will be made available upon request under an MTA.
Data and code availability
Models of CCHFV GP38 bound to Fabs of GP38-specific mAbs have been deposited at Worldwide Protein DataBank (wwPDB) under accession codes 8VVK, 8VVL, and 8VVW. Cryo-EM maps have been deposited at Electron Microscopy DataBank under accession codes EMD-43604, EMD-43551, EMD-43552, and EMD-43553. Depositions are available as of the date of publication. The sequence data for the GP38-specific mAbs cannot be deposited in a public repository because they are the subject of pending provisional patent applications. To request access, contact the lead contact, Jason S. McLellan, and the sequences will be made available upon request under an MTA. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Patient recruitment and ethics statement
CCHFV convalescent donors were recruited as described previously.28,30 Briefly, donors with documented clinical history of CCHFV infection between 2013 and 2017 in Agago and Nakaseke districts, Uganda were recruited through the Uganda virus Research Institute, Entebbe, Uganda. The study was approved by the Helsinki committees of Uganda Virus Research Institute (UVRI), Entebbe, Uganda (reference number GC/127/13/01/15); Soroka Hospital, Beer Sheva, Israel (protocol number 0263-13-SOR); and the Ugandan National Council for Science and Technology (UNCST) (registration number HS1332). Written informed consent was obtained and a personal health questionnaire was completed for each donor who participated in this study. Study participants were adult Black, East African, males ages 68, 35, and 30, respectively (Table S1), and were not related. Data related to patient ancestry and socioeconomic status was not included under local IRB and thus not collected. All experiments were performed in accordance with the relevant guidelines and regulations.
Cell lines
VeroE6 and Vero cells, immortalized epithelial cell lines isolated from the kidney of an adult female African grivet monkey (RRID:CVCL-0574 and CVCL-0059, respectively), were obtained from the American Type Culture Collection (ATCC). SW-13 cells, a cell line isolated from the adrenal gland and cortex of a 55-year-old female patient with carcinoma (RRIDD:CCL-105), were obtained from ATCC. BSR-T7 cells (RRID:CVCL_RW96), generated by stable T7 RNA polymerase expression in BHK-21 cells, were a kind gift from K.-K. Conzelmann. The parent cell line (RRID: CVCL_1915) was isolated from the kidney of a 1-day-old male golden hamster. All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM; ThermoFisher Scientific) enriched with 10% fetal bovine serum (Bio-Techne), 1% GlutaMAX (ThermoFisher Scientific), and 1% penicillin-streptomycin (ThermoFisher Scientific). All cell lines were maintained in a 37°C incubator supplied with 5% CO2. Cell lines were not authenticated following purchase.
Viruses
The authentic CCHFV isolates CCHFV-IbAr10200, CCHFV-Afg09-2990 (labeled as ‘Afg09’), CCHFV-Turkey2004, and Oman-199809166 (labeled as ‘Oman’) were used in this study.
Animal models
3–8-week-old male and female B6.129S(Cg)-Stat1tm1Dlv/J mice (STAT1−/−; strain #012606; The Jackson Laboratory)50,91 and 5–8-week-old male and female B6(Cg)-Ifnar1tm1.2Ees/J mice (IFNAR−/−; strain #028288; Charles River),48,49 ranging in weight from 17 to 27 g, were used in animal challenge experiments. A wealth of evidence shows that there is no influence of sex on CCHFV infection in the rodent models utilized.27,28,30,48–50,92 These animals had previously never undergone experimentation and were confirmed to be free of contaminating bacterial or viral pathogens by the vendor. Animals were randomly allocated to experimental groups. Animals were provided with food and water ad libitum and housed in individually ventilated cages.
Murine challenge studies were conducted under Institutional Animal Care and Use Committee (IACUC)-approved protocols in compliance with the Animal Welfare Act, PHS Policy, and other applicable federal statutes and regulations. The facilities where these studies were conducted (USAMRIID) are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALAC) and adhere to the principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2013. IACUC-approved euthanasia criteria were defined as follows: mouse displays severely hunched posture, inability or reluctance to move, appears weak (staggering when moving around cage), or has labored breathing.
METHOD DETAILS
rGP38 serum ELISA
High-binding half-area plates (Greiner Bio-One) were coated with 50 μL of IbAr10200 rGP38 at 5 μg/mL. Plates were incubated overnight at 4°C. Plates were then blocked with 100 μL of 5% BSA/PBS and flicked to remove liquid. Serum was serially diluted 5-fold in PBS. 50 μL of each dilution was added to plates and incubated for 1 h at room temperature. Plates were washed 3X with PBS plus 0.05% Tween 20 (PBST). Anti-Human-HRP (Invitrogen) was diluted 1:5000 in 1% BSA/PBS. 50 μL of the diluted solution was added to plates and incubated for 1 h at room temperature. Plates were again washed 3X with PBST. 50 μL of KPL Blue Sure Substrate (Seracare) was added to plates. Plates were incubated for 5 min at room temperature, and the reaction was stopped with 50 μL of 2 N H2SO4. OD450 was measured with a PerkinElmer EnVision multimode plate reader. Data were plotted and analyzed using GraphPad Prism Software V9.5.1; a Sigmoidal, 4PL curve was fit to interpolate data.
Single B cell sorting
B cells were eluted from PBMCs using a MACS Human B Cell isolation kit (Miltenyi Biotec). B cells were stained with rGP38 (IbAr10200) that had been tetramerized at 25 nM using Streptactin-PE (IBA Lifesciences) and Streptactin-APC (IBA Lifesciences). B cells were simultaneously stained with rGP38-Streptactin-PE and rGP38-Streptactin-APC tetramers for 1 h on ice. Cells were washed twice in buffer (PBS, FBS, EDTA). Next, B cells were stained with a panel of antibodies. Donor 1 PBMCs were stained with a cocktail of anti-human CD3 PerCP-Cy5.5 (Biolegend), CD8 PerCP-Cy5.5 (Biolegend), CD14 PerCP-Cy5.5 (Invitrogen), CD16 PerCP-Cy5.5 (Biolegend), propidium iodide (PI) (Invitrogen), CD19 PE-Cy7 (Biolegend), CD27 BV510 (BD Biosciences), IgM BV711 (BD Biosciences), IgD BV421 (Biolegend), IgG BV605 (BD Biosciences), and IgA AF488 (Abcam). Donor 5 and 6 PBMCs were stained with a cocktail of anti-human CD3 PerCP-Cy5.5 (Biolegend), CD8 PerCP-Cy5.5 (Biolegend), CD14 PerCP-Cy5.5 (Invitrogen), CD16 PerCP-Cy5.5 (Biolegend), PI (Invitrogen), CD19 PE-Cy7 (Biolegend), CD20 PE-Cy7 (Biolegend), CD27 BV510 (BD Biosciences), IgM AF488 (Biolegend), and IgD BV421 (Biolegend). B cells were washed twice in buffer and run on a FACS Aria Fusion Cytometer (BD Biosciences). B cells were sorted into Super Script III reaction buffer (ThermoFisher Scientific) in 96-well Costar plates and frozen at −80°C.
Amplification of antibody variable genes
cDNA was synthesized using SuperScript III Reverse Transcriptase (ThermoFisher Scientific). Antibody VH and VL genes were amplified following previously designed methods.77 Gene amplification with HotStartTaq Plus Polymerase (Qiagen) was carried out in two steps. IgG-, IgA-, IgM-specific primers were used in the first reaction. Primers with 5' and 3' homology domains, specific to plasmids used for cloning into an engineered strain of S. cerevisiae, were used in the second reaction.
Cloning into engineered S. Cerevisiae
Amplified variable genes were transformed into S. cerevisiae through the lithium acetate method.75 One colony of engineered S. cerevisiae was inoculated in yeast extract-peptone-dextrose medium for 14–16 h. Yeast were washed twice in dH2O and resuspended in dH2O (67 μL). Resuspended yeast were mixed with variable gene product (10 μL of unpurified VH and 10 μL of unpurified Vκ or Vλ product), digested plasmid (200 ng), 50% w/v polyethylene glycol 3350 (240 μL), 1 M lithium acetate, and boiled salmon sperm DNA (10 μL). Contents of the transformation were incubated at 42°C. After a 45-min incubation, yeast were washed twice with dH2O, resuspended in selective growth medium, and grown for 48 h at 30°C.
Expression and purification of IgG and Fab
Production in yeast
Full length IgG1 and Fabs were produced and purified as previously described.93 Briefly, cultures were grown in 24-well plates for 6 days at 30°C and 80% relative humidity with shaking at 650 rpm on a Multitron Shaking Incubator (Infors HT). Cultures were centrifuged to obtain supernatants, which were purified by Protein A chromatography. Bound IgGs were eluted with 200 mM acetic acid (pH 3.5), 50 mM NaCl and neutralized with 1/8 v/v 2 M HEPES (pH 8.0). IgGs were buffer exchanged into PBS (pH 7.0) and stored for later use.
To produce Fabs, IgGs were papain-digested for 2 h at 30°C. The reaction was quenched with iodoacetamide. The material was passed over a Protein A column to remove undigested IgGs and Fc domains. The flow-through was collected and Fabs were purified using CaptureSelect IgG-CH1 affinity resin (ThermoFisher Scientific). 200 mM acetic acid (pH 3.5), 50 mM NaCl was used to elute Fabs, which were neutralized with 1/8 v/v 2 M HEPES (pH 8.0). Fabs were buffer exchanged into PBS (pH 7.0) and stored for later use.
Production in mammalian cells
For IgGs used for in vitro and in vivo studies, and later used to produce Fabs for structural studies (ADI-58048, ADI-46143, ADI-46138, ADI-46158, and 13G8), genes encoding the variable regions were ordered as gBlocks (Integrated DNA Technologies) with a 15-base-pair 5’ overlap to a murine IgKVIII secretion signal and a 15-base-pair 3’ overlap to the appropriate constant region (human kappa, human lambda or human IgG1). The variable regions were cloned into pCDNA 3.4 (ThermoFisher Scientific) vectors previously constructed with a mouse IgKVIII signal sequence and each constant region. In-Fusion enzyme (Takara Bio) was used to insert the gBlocks between the secretion signal and the constant region.
Antibodies were transiently expressed in ExpiCHO cells (ThermoFisher Scientific) following the high-titer protocol for CHO Expifectamine (ThermoFisher Scientific). Cultures were centrifuged 9–10 days after transfection, and the supernatants were filtered and loaded onto a HiTrap MabSelect SuRe affinity column (Cytiva) using an AKTA Pure FPLC system. The column was washed with 10 column volumes of PBS pH 7.2 and antibodies were eluted with Pierce IgG elution buffer (ThermoFisher Scientific). Fractions containing the antibody were combined and neutralized to ~ pH 7 with 1 M Tris pH 7.8.
To produce Fabs of ADI-58048, ADI-46143, ADI-46158, and c13G8 used in structural studies, purified IgG was digested with LysC at a 1:2000 M ratio of LysC:IgG overnight at 37°C. A cOmplete Protease Inhibitor Cocktail tablet (Sigma-Aldrich) was dissolved into the reaction before loading the digested IgG mixture over a CaptureSelect IgG-CH1 affinity resin (ThermoFisher Scientific) to bind the Fabs. The column was washed with 1X PBS followed by elution of the Fabs with 100 mM glycine pH 3.0 into a neutralization buffer of 100 mM Tris pH 8.0.
For IgGs used to produce Fabs for structural studies (ADI-46152, ADI-58026, ADI-58062, ADI-63530, and ADI-63547), the heavy and light chain variable regions were cloned into Igγ1 and either human Igκ or Igλ vectors, respectively. To later generate Fabs from the IgG, a human rhinovirus (HRV) 3C protease site was present at the hinge region of the heavy chain in the Igγ1 vector. Plasmids encoding both the heavy chain and light chain for each antibody were co-transfected into FreeStyle 293-F cells (Invitrogen) using polyethylenimine. Secreted IgG was purified from the culture supernatants via Pierce Protein A Plus Agarose resin (ThermoFisher Scientific). The IgG eluent was further purified via SEC with a HiLoad 16/600 Superdex 200 column (GE Healthcare Biosciences) in 2 mM Tris pH 8.0, 200 mM NaCl, and 0.02% NaN3.
To produce Fabs for structural studies, purified IgG was bound to Pierce Protein A Plus Agarose resin (ThermoFisher Scientific) and washed with 1X PBS. The IgG-bound Protein A resin was removed from the column holder and added to a conical tube with 1X PBS buffer and 10% w/w HRV 3C protease and nutated on a rotating shaker for 2 h at 23°C. Following the cleavage reaction, the Fc domains remained bound to the Protein A resin and the Fabs were collected in the nutated flow-through. Purified Fabs were stored for later use.
Biolayer interferometry binding analysis
For all experiments, a Fortébio Octet HTX (Sartorius) was used. All steps of the experiments were performed at 25°C with an orbital shaking speed of 1,000 rpm and all reagents were formulated in PBSF (PBS with 0.1% w/v BSA). For avid binding experiments, biotinylated rGP38 at 100 nM was loaded onto streptavidin biosensors for 10–40 s, providing load levels of 0.30–0.40 nm. The sensors were then soaked for 30 min in PBSF, dipped in 100 nM IgG for 180 s, and dipped into PBSF for 180 s to measure dissociation. For monovalent binding, IgGs were loaded onto AHC biosensors (0.6–1.2 nm) at 100 nM for 30 min. Considering that antigens contained a twin-strep-tag, the sensors were blocked with 100 μM biocytin for 10 min to saturate any remaining streptavidin binding sites. Sensors were incubated for 60 s in PBSF to establish a baseline. Next, sensors were dipped in 100 nM antigen for 180 s followed by PBSF for 180 s to measure dissociation. Data for which binding responses were greater than 0.05 nm were aligned, interstep-corrected to the association step, and subsequently fit to a 1:1 binding model using Fortébio Octet Data Analysis, v 11.1.
Surface plasmon resonance binding analysis
For all experiments, the Carterra LSA (Carterra USA) was used. Kinetic analysis was conducted in HBS-ET running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.01% Tween 20) (Carterra USA) at 25°C. The standard amine coupling step was conducted in 25 mM MES buffer (Carterra USA) with 0.05% Tween 20. The sample compartment was maintained at a temperature of 20°C for the duration of the experiment.
Standard amine coupling (1:1 EDC:NHS) was used to covalently couple a goat anti-human Fc antibody (Jackson ImmunoResearch) to the HC30M chip; the chip was then blocked with 1.0 M ethanolamine pH 8.5. Next, antibodies (100 nM in running buffer) were flowed for 5 min over discrete regions of interest on the chip surface. Once the antibody samples were captured to the sensor surface, kinetic measurements were collected in cycles. For a given antigen, the loaded biosensor array was first exposed to running buffer (60 s), then three blank buffer injections (300 s association and 300 s dissociation). This was followed by a series of four antigen injections (300 s association and 3000 s dissociation) of increasing concentration (1.56–100 nM). At the end of each cycle, all surfaces were regenerated via two 30 s injections of 10 mM glycine, pH 1.7.
All kinetic data were reference subtracted using interspot reference surfaces evenly distributed throughout the biosensor surface array. The data were then y axis aligned, x axis aligned, corrected for baseline drift using a minimum baseline drift parameter of 4 RU, and blank subtracted from the leading (third of three) blank injection. Sensorgrams were filtered using a minimum spike height of 5 RU and width of 3 points before being cropped, beginning just after the start of the association and ending just before the end of the dissociation. The processed sensorgrams were then fit to a 1:1 binding model with floating T0 using the Carterra LSA Kinetics Software version 1.7.1.3055 (Carterra, USA).
Antibody competition assays
Biolayer interferometry
For all experiments, a Fortébio Octet HTX (Sartorius) was used. All steps of the experiments were performed at 25°C with an orbital shaking speed of 1,000 rpm and all reagents were formulated in PBSF (PBS with 0.1% w/v BSA). IgGs were loaded onto AHC biosensors (0.7–1.5 nm) at 100 nM for 30–600 s, providing load levels of 1.0–1.3 nm. The sensors were blocked for 10 min with an inert human antibody at 0.5 mg/mL to fill unoccupied binding sites and then were equilibrated for 30 min in PBSF. To check for cross-interactions on the protein surface, prior to binding analysis, the sensors were dipped in 300 nM control antibody for 90 s. After a baseline step in PBSF for 60 s, the sensors were exposed first to antigen (100 nM) for 180 s, then to control antibody (300 nM) for an additional 180 s. Data were then y axis normalized and interstep-corrected using Fortébio Octet Data Analysis, v 11.1. Binding of the secondary antibody indicates a non-competitor (unoccupied epitope), whereas no binding indicates a competitor antibody (epitope blocking).
Yeast presentation
Biotinylated CCHFV GP38 (IbAr10200; 50 nM) was incubated with a 20-fold excess of anti-CCHFV-GP38 Fab (1 μM) for 30 min at room temperature. Pre-complexed biotinylated CCHFV GP38 and Fab mixtures were incubated with yeast expressing full-length anti-CCHFV-GP38 IgG for 5 min at room temperature. Yeast were washed two times with PBSF (PBS with 0.1% w/v BSA) to remove any unbound GP38-Fab complexes. Samples were incubated for 30 min on ice with a cocktail of streptavidin Alexa Fluor 633 (Invitrogen; to detect bound GP38), goat F(ab')2 anti-human kappa FITC and goat F(ab')2 anti-human lambda FITC (SouthernBiotech; to detect antibody expression), and PI (Invitrogen; to detect cell viability). After staining, samples were run on a FACSCanto II flow cytometer (BD Biosciences). Competition levels were assessed by calculating the fold reduction between a known non-competitive isotype control IgG and an IgG of interest; bound GP38 levels were normalized to light chain expression. The following equation was used to calculate the fold reduction with mean fluorescence intensity (MFI); Fold Reduction = (AF633 MFI/FITC MFI)No-competition/(AF633 MFI/FITC MFI)Competition. Antibodies with a calculated fold reduction greater than 10 were considered competitive with the pre-complexed Fab.
CCHFV GP38 yeast display and epitope mapping
Display of CCHFV GP38 on the surface of yeast
The sequence encoding GP38 from the CCHFV-IbAr10200 GP gene (GenBank Accession: NC_005300.2) was inserted into a plasmid containing an N-terminal HA tag-G4S linker and a G4S- HA tag C-terminal linker. The plasmid was transformed and expressed as previously described.30
CCHFV GP38 library construction
PCR was carried out with an error-prone polymerase (Agilent, GeneMorph II Random Mutagenesis Kit) to create a randomly mutagenized GP38 library as previously described.94
Titration of anti-GP38 mAbs
Antibodies used in epitope mapping studies were titrated against yeast displaying GP38 to adequately calculate EC50s and EC80s for each antibody. Yeast were induced to express non-mutagenized GP38 as noted above. Antibodies were titrated from 100 nM in 2-fold, 12-point serial dilutions. Once an OD600 of 0.1 was achieved, the non-mutagenized GP38-expressing yeast were mixed with each antibody dilution and incubated on ice for 1 h. Yeast cells were washed two times with PBSF and subsequently stained for 30 min on ice with a cocktail of anti-HA APC antibody (Biolegend, Clone: 16B12, dilution 1:100), goat F(ab')2 anti-human IgG PE (SouthernBiotech, dilution 1:100), and PI (Invitrogen, 1:100 dilution). After staining, samples were run on a FACSCanto II flow cytometer (BD Biosciences). PE MFIs were plotted against antibody concentrations; EC50 and EC80 concentrations were calculated using GraphPad Prism 9.
Sorting of mutant GP38 libraries
The mutant GP38 library and non-mutagenized GP38-expressing yeast were induced as noted above. Both the mutant GP38 library and non-mutagenized GP38-expressing yeast were incubated with a solution of each mAb at its respective EC80 for 1 h on ice. Cells were washed two times in PBSF and further stained with anti-HA APC, anti-human IgG PE, and PI (as described above). Cells were washed and run on a FACSAria (BD Biosciences). Mutagenized GP38 clones that showed reduced binding to each antibody of interest were sorted and cultured in synthetic complete (SC) media minus tryptophan (4% dextrose, 0.1 M sodium phosphate, pH 6.3) for further rounds of selection. The same selection strategy was applied to cultured cells from the first round of selection to carry out a second round of selection. A third and final round of selection occurred; the final selection was a positive selection used to remove any mutagenized clones that were global knock-outs. Cultured cells from the second round of selection were stained with a panel of anti-GP38 antibodies of non-overlapping epitopes to the antibody used in the first round of selection. Cells that bound the non-competing anti-GP38 antibodies were sorted and plated on complete minimal media glucose agar plates minus tryptophan (Teknova). For each library, 100 clones were picked and sequenced.
Analysis of single GP38 mutants
Unique clones that came out of selections were induced as described above. GP38 wild-type control clones were induced along-side the clones from selections. Clones were stained with each antibody of interest as well as with an isotype control antibody. Next, clones were stained with each antibody at its respective EC50 for 1 h on ice. Yeast were washed twice with PBSF. Cells were washed two times in PBSF and further stained with anti-HA (hemagglutinin) APC, anti-human IgG PE, and PI (as described above). Samples were run on a FACSCanto II flow cytometer (BD Biosciences). Percent loss of binding was calculated utilizing the following equation; % of WT Binding = [((IgG MFI/HA MFI)MUT – (IgG MFI/HA MFI)BACK)/((IgG MFI/HA MFI)WT – (IgG MFI/HA MFI)BACK)] × 100. Clones with less than 25% of wild-type binding for a specific antibody were considered to have a mutation critical for binding.
Cloning, expression, and purification of GP38
Recombinant CCHFV GP38 proteins were produced from the following isolates: Oman-199809166 (UniProt: A0A0U3C6Q7), Kosova-Hoti (UniProt: B2BSL7), 200406546-Turkey (UniProt: A0A0U2SQZ0), Afg09-2990 (UniProt: E5FEZ4), and 79121M18 (UniProt: D4NYK3). Gene fragments (Integrated DNA Technologies) of each isolate’s MLD-GP38 sequence encoding for residues 1–515, as denoted by CCHFV IbAr10200 strain GPC numbering, were codon-optimized for human cell expression (GenScript Codon Optimization Tool). Gene fragments were each cloned into a paH eukaryotic expression vector with a C-terminal HRV 3C protease cleavage site, an 8x HisTag, and a Twin-Strep-tag. The plasmid for CCHFV strain IbAr10200 GP38 was previously reported.28 To ensure cleavage of the MLD from GP38, a pCDNA3.1 plasmid encoding for furin was co-transfected with each clinical GP38 plasmid at a mass ratio of 1:9 furin:GP38. The two plasmids were transiently transfected into FreeStyle 293 cells (Invitrogen) using polyethylenimine followed by treatment with 5 μM kifunensine to ensure uniform high-mannose glycosylation. Soluble GP38 was secreted from the harvested medium and purified via Ni-NTA resin (Thermo Scientific HisPur Ni-NTA Resin). GP38 proteins were further purified via SEC using a Superdex 200 Increase 10/300 GL (GE Healthcare Biosciences) in 2 mM Tris pH 8.0, 200 mM NaCl, and 0.02% NaN3.
Crystallization and data collection
GP38 + ADI-46143 Fab
GP38 (from CCHFV-IbAr10200) was incubated at room temperature for 20 min with a 1.2-fold molar excess of ADI-46143 Fab and the complex was purified by SEC on a Superdex 200 Increase 10/300 GL (GE Healthcare Biosciences) in 2 mM Tris pH 8.0, 50 mM NaCl, and 0.02% NaN3. The GP38-ADI-46143 Fab complex (4.1 mg/mL) underwent crystallization trials via the sitting-drop vapor diffusion method. The crystal from which the diffraction data were obtained was grown in 9.3% w/v PEG 3350, 12.2% v/v isopropanol, 0.2 M ammonium citrate pH 7.5 at a protein:buffer ratio of 1:1. The crystal was looped with 20% ethylene glycol as a cryoprotectant and flash frozen in liquid nitrogen. The 19-ID beamline (Advanced Photon Source; Argonne National Laboratories) was used to collect the X-ray diffraction data to 2.6 Å resolution.
GP38 + c13G8 Fab
GP38 (from CCHFV-IbAr10200) was incubated at room temperature for 20 min with a slight molar excess of c13G8 Fab and the complex was purified by SEC on a HiLoad 16/600 Superdex 200 column (GE Healthcare Biosciences) in 2 mM Tris pH 8.0, 200 mM NaCl, and 0.02% NaN3. The GP38-c13G8 Fab complex (9.8 mg/mL) underwent crystallization trials through the sitting-drop vapor diffusion method. The crystal used to obtain the diffraction data was grown in 2 M ammonium sulfate, 0.1 M Bis-Tris pH 5.5, 0.01 M cobalt chloride hexahydrate at a protein:buffer ratio of 2:1. The crystal was looped with 20% ethylene glycol as a cryoprotectant and flash frozen in liquid nitrogen. The 19-ID beamline (Advanced Photon Source; Argonne National Laboratories) was used to collect the X-ray diffraction data to 1.8 Å resolution.
Structure determination, building, and refinement
Diffraction data from the 19-ID beamline were processed using the CCP4 software,78 indexed and integrated in iMOSFLM,79 and scaled and merged in Aimless.80 Both crystal structures were phased using PhaserMR81 using PDB ID: 6VKF for GP38 and AlphaFold2 models for Fabs.87 Structures were then refined and built using COOT82 and Phenix.83 The GP38 + c13G8 crystal structure was refined to a final Rwork/Rfree of 20.0%/21.5% (Table S7). The GP38+ADI-46143 crystal structure was refined to a final Rwork/Rfree of 17.7%/21.7% (Table S7). The crystal structures were displayed in PyMOL84 and protein-protein interactions were determined by PDBePISA.89
Cryo-EM sample preparation and data collection
GP38-Fabs complex preparation
For the GP38+ADI-58026 Fab+ADI-63547 Fab complex, a 0.4 mg/mL complex was prepared by combining purified IbAr10200 GP3828 with a 1.8-fold molar excess of each Fab followed by incubation for 30 min at room temperature in 2 mM Tris pH 8.0, 200 mM NaCl, 0.02% NaN3, and 0.03% amphipol A8-35. For the GP38+ADI-58062 Fab+ADI-63530 Fab complex, a 0.4 mg/mL complex was prepared by combining purified IbAr10200 GP3828 with a 1.8-fold molar excess of each Fab followed by incubation for 30 min at room temperature in 2 mM Tris pH 8.0, 200 mM NaCl, 0.02% NaN3, and 0.03% amphipol A8-35.
Two complexes were prepared by using purified IbAr10200 GP3828 complexed with a 1.2- to 1.5-fold molar excess of ADI-46152 Fab and ADI-58048 Fab or ADI-46143 Fab and ADI-46158 Fab. Complexes were incubated for 20 min at 23°C before further purification via SEC on a Superdex 200 Increase 10/300 GL (GE Healthcare Biosciences) in 2 mM Tris pH 8.0, 200 mM NaCl, and 0.02% NaN3. The GP38+ADI-46152+ADI-58048 complex was used at a concentration of 0.5 mg/mL and the GP38+ADI-46143+ADI-46158 was at a concentration of 0.4 mg/mL.
Cryo-EM data collection
A3 μL aliquot of each complex was applied to a Quantifoil 1.2/1.3 Cu300 grid that was glow discharged for 25 s at 15 mAmps (PELCO easiGlow Glow Discharge Cleaning System). A Vitrobot Mark IV (ThermoFisher Scientific) was used to plunge freeze the grids at 10°C and 100% humidity with a blot time of 3.5 s, blot force of −4, blot total of 1, and wait time of 2 s. 2,504 micrographs for the GP38+ADI-58026 Fab+ADI-63547 Fab complex, 3,647 micrographs for the GP38+ADI-46152 Fab+ADI-58048 Fab complex, 2,962 micrographs for the GP38+ADI-58062 Fab+ADI-63530 Fab complex, and 1,485 micrographs for the GP38+ADI-46143 Fab+ADI-46158 Fab complex, were collected using a FEI Titan Krios equipped with a K3 detector (Gatan). Data were collected with a 30° tilt at a magnification of 105,000x, corresponding to a calibrated pixel size of 0.81 Å /pixel and a total electron dose of 80 e−/Å .2 Statistics for each data collection are in Table S6.
Cryo-EM data processing, building, and refinement
On-the-fly data processing was performed in cryoSPARC Live,85 and included motion correction, defocus estimation, micrograph curation, particle picking, particle extraction, and particle curation through iterative streaming 2D classification. Data processing and refinement of all datasets were performed using cryoSPARC v3.2 and subsequent versions. The initial building or docked model for GP38 was PDB ID: 6VKF and the models for Fabs were generated by AlphaFold2.87 Protein-protein interactions were determined by PDBePISA89 and visualized using UCSF ChimeraX.90 Statistics for each dataset are in Table S6.
For the GP38+ADI-58026 Fab+ADI-63547 Fab complex, several rounds of 2D classification and ab initio reconstruction were performed to refine the particle stack for the complex with two Fabs bound to GP38, as the lower binding affinity for ADI-63547 led to heterogeneity in the Fab occupancy. After volumes were refined for the complex bound with two Fabs, the volume underwent homogeneous and non-uniform refinement before another round of non-uniform refinement using particles from the extracted particle stack. The dataset underwent two rounds of heterogeneous, homogeneous, and non-uniform refinements. Duplicate particles were then removed followed by a non-uniform refinement. The final map was sharpened using DeepEMhancer.88 The EM processing pipeline is summarized in Figure S10.
For the GP38+ADI-46152 Fab+ADI-58048 Fab complex, selected particles underwent ab initio 3D reconstruction followed by heterogeneous refinement. For the best class, homogeneous and non-uniform refinements were performed, then curated particles were further refined using another round of heterogeneous refinement. The best class underwent homogeneous and non-uniform refinement, followed by extracting the curated particles without Fourier cropping and removing duplicate particles with non-uniform refinements between each step. The final volume was sharpened using DeepEMhancer.88 The model was built iteratively using PHENIX,83 COOT,82 and ISOLDE.86 The EM processing pipeline is summarized in Figure S11.
For the GP38+ADI-58062 Fab+ADI-63530 Fab complex, extracted particles underwent two rounds of 2D classification to generate a curated particle stack. Particles were further processed using ab initio 3D reconstruction and heterogeneous refinement. From the best class, a non-uniform refinement was conducted before extracting the particles without Fourier crop followed by another round of non-uniform and heterogeneous refinements. Next, the best class underwent homogeneous refinement and non-uniform refinement before duplicate particles were removed. Lastly, a non-uniform refinement was performed on the resulting map before the map was sharpened using DeepEMhancer.88 The EM processing pipeline is summarized in Figure S12.
For the GP38+ADI-46143 Fab+ADI-46158 Fab complex, extracted particles were curated via 2D classification followed by iterative rounds of ab initio reconstruction, heterogeneous refinement, homogeneous refinement, and non-uniform refinement. In some steps, volumes obtained from the processing of a smaller initial particle stack were used. After a final non-uniform refinement, the maps were processed with DeepEMhancer.88 The EM processing pipeline is summarized in Figure S13.
Polyreactivity assay
A polyreactivity assay was carried out as previously described.76 Briefly, soluble cytosolic protein (SCP) and soluble membrane protein (SMP) preps were extracted from Chinese hamster ovary (CHO) cells and were biotinylated using an NHS-LC-Biotin kit (ThermoFisher Scientific). Yeast displaying IgGs on their surface were incubated with biotinylated SCP and SMP preps at a 1:10 dilution in PBSF (PBS with 0.1% w/v BSA) and incubated on ice for 20 min. Yeast cells were then washed two times in PBSF and further stained with a cocktail of ExtraAvidin-R-PE (Sigma Aldrich, dilution 1:50), anti-human kappa FITC (Southern Biotech, dilution 1:100), anti-human lambda FITC (Southern Biotech, dilution 1:100), and PI (Invitrogen, dilution 1:100) for 20 min on ice. Yeast were again washed two times and samples were analyzed on a BD FACSCanto II flow cytometer (BD Biosciences).
Hydrophobic interaction chromatography (HIC)
HIC assays were carried out as previously described.95 Briefly, antibodies were diluted in a solution of 1.8 M ammonium sulfate and 0.1 M sodium phosphate pH 6.5 (phase A solution) to achieve a final concentration of 1.0 M ammonium sulfate. A linear salt gradient from phase A solution to the same solution without ammonium sulfate (phase B solution) was set up on a Sepax Proteomix HIC butyl-NP5 column; the gradient was run for 20 min at a flow rate of 1.0 mL/min. The UV absorbance at 280 nm was monitored to obtain peak retention times.
Thermostability assay
Thermal melting (Tm) measurements of the Fabs were carried out as previously described using differential scanning fluorescence (DSF).96 Briefly, 20 μL of 1 mg/mL antibody sample was mixed with 10 μL of 20X SYPRO orange. The CFX Real-Time System (BioRad) was used to scan the plate from 40°C to 95°C at a rate of 0.25°C/min. Subsequently, BioRad analysis software was used to calculate TmApp from the primary derivative of the raw data.
Generation of tecVLPs
The amino acid sequence for the IbAr10200 GPC was derived from GenBank M-segment sequences with an accession number NC_005300. Transcription- and entry-competent virus-like particles (tecVLPs) were generated as described previously.30,45 Briefly, BSR-T7 cells were transfected with plasmids encoding the T7 polymerase, a minigenome expressing Nano-Glo Luciferase, and the CCHFV nucleoprotein (NP), glycoprotein complex (GPC), and polymerase (L). 15 h post-transfection, transfection medium was removed and replaced with fresh DMEM growth media. 48 h post-transfection, tecVLP-containing supernatants were collected, clarified by low-speed centrifugation, and pelleted by ultracentrifugation at 25,000 x g for 2.5 h. Pelleted tecVLPs were resuspended in DMEM overnight and stored at −80°C overnight prior to use.
Neutralization assays against CCHFV tecVLPs
Neutralization by candidate mAbs against CCHFV IbAr10200 tecVLPs were assessed in Vero cells, maintained as described above and previously.30 In brief, antibodies were diluted to starting concentrations of 350 nM (anti-GP38 mAbs) or 100 nM (anti-Gc mAbs) and subsequently serially diluted 3-fold in complete DMEM. TecVLPs, at an amount empirically determined such that the luciferase signal in target cells was approximately 500-fold over background, were then incubated with antibodies for 1 h at 4°C. After 1 h, antibody/tecVLP mixtures were added to Vero cells in triplicate and incubated for 16 h. Following infection, luciferase signal was assayed using Nano-Glo Luciferase assay system (Promega) and the signal for each mAb tested was normalized to a no-antibody control.
Neutralization assays against authentic CCHFV
Neutralization assays were conducted similarly to what was described previously, with modifications.28,30 Briefly, CCHFV-IbAr10200, CCHFV-Afg09, CCHFV-Turkey2004, or CCHFV-Oman were incubated with serial 3-fold dilutions of mAbs (at a starting concentration of 500 nM) for 1 h at 37°C. The antibody-virus mixture was added to monolayers of VeroE6 or SW-13 cells in a 96-well plate at a final multiplicity of infection of 1 (IbAr10200 and Afg09) or 0.3 (Turkey2004 and Oman) and incubated for 1 h at 37°C. Infection medium was then removed, and fresh cell culture medium without mAb was added. 24 (IbAr10200 and Afg09) or 48 h (Turkey2004 and Oman) post infection, culture medium was removed, and plates were submerged in 10% formalin and plates were fixed for at least 24 h at 4°C. Plates were removed from formalin and permeabilized with 0.2% Triton X- for 10 min at room temperature and treated with blocking buffer (5% milk). Infected cells were detected by consecutive incubation with CCHFV-specific antibody 9D5 (3 μg/mL; BEI NR-40270) and secondary detection antibody (goat anti-mouse) conjugated to Alexa Fluor 488 (1:2000 dilution; Invitrogen). Percent infection was determined using the Cytation5 high-content imaging instrument and data analysis was performed using the or Gen5.11 software (BioTek).
Murine challenge studies
Therapeutic IbAr10200 study
5–8-week-old male and female IFNAR−/− mice (Charles River) were exposed intraperitoneally (IP) to 100 PFU of CCHFV-IbAr10200. Mice were treated IP with 1 mg of indicated mAb, or an equivalent volume (200 μL) of phosphate-buffered saline (PBS) vehicle 24 h (+1 day) and 96 h (+4 day) post-exposure, for a total of 2 mg of mAb per mouse. Animals were observed daily for clinical signs of disease and morbidity for 28 days. Mice were scored on a 4-point grading scale, where a 1 was defined by decreased grooming and/or ruffled fur, a 2 defined by subdued behavior when un-stimulated, a 3 defined by lethargy, hunched posture, and/or subdued behavior even when stimulated, and a 4 defined by bleeding, unresponsiveness, severe weakness, or inability to walk. Mice scoring a 4 were considered moribund and were humanely euthanized based on IACUC-approved criteria. Daily observations were increased to a minimum of twice daily while mice were exhibiting clinical signs of disease (clinical score = 3).
Therapeutic Afg09, Oman, and Turkey2004 study
3–8-week-old male and female STAT1−/− mice (The Jackson Laboratory) were exposed IP to 100 PFU of CCHFV-Afg09 or 1000 PFU of CCHFV-Turkey2004 or CCHFV-Oman. For the second challenge study (Figure S16), mice were either treated IP with 1 mg of indicated mAb, or an equivalent volume (200 μL) of PBS vehicle 24 h (+1 day) and 96 h (+4 day) post-exposure, for a total of 2 mg of mAb per mouse. For the third challenge study (Figure 7), mice were treated IP with 0.2 mg of indicated mAb or an equivalent volume (200 μL) of PBS vehicle 30 min post-exposure. Animals were observed daily for clinical signs of disease and morbidity for 28 days. Mice were scored on a 4-point grading scale as described above. Daily observations were increased to a minimum of twice daily while mice were exhibiting signs of disease (clinical score = 3). Mice scoring a 4 were considered moribund and were humanely euthanized based on IACUC-approved criteria.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details, including the number of replicates (n), measures of precision, and the statistical test used for each experiment can be found in the corresponding figure legends and in the results section. For analysis of VH nucleotide substitutions (Figure 1C), the data were not normally distributed and therefore the statistical comparison was performed using the Mann-Whitney Test with *p < 0.05. For analysis of the correlation between percent survival and KD (Figure S15), the R-squared value was calculated by Spearman’s correlation coefficient. All statistical analyses were conducted in GraphPad Prism Software V9.5.1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| c13G8 | Mishra et al.28 | N/A |
| Monoclonal anti-GP38 patient derived antibodies | This paper | N/A |
| Anti-CCHFV nucleocapsid mAb clone 9D5 | BEI Resources | Cat. #NR-40270 |
| Goat anti-human Fc HRP | Invitrogen | Cat. #A18817 |
| Anti-Human CD3 PerCp-Cy5.5 (clone UCHT1) | BioLegend | Cat. #300430; RRID: AB_893299 |
| Anti-Human CD8 PerCp-Cy5.5 (clone SK1) | BioLegend | Cat. #344710; RRID: AB_2044010 |
| Anti-Human CD14 PerCp-Cy5.5 (clone 61D3) | Invitrogen | Cat. #45-0149-42; RRID: AB_1518736 |
| Anti-Human CD16 PerCp-Cy5.5 (clone B73.1) | BioLegend | Cat. #360712; RRID: AB_2562955 |
| Anti-Human CD19 PE-Cy7 (clone HIB19) | BioLegend | Cat. #302216; RRID: AB_314246 |
| Anti-Human CD27 BV510 (clone M-T271) | BD Biosciences | Cat. #740167; RRID: AB_2739920 |
| Anti-Human IgM BV711 (clone UCH-B1) | BD Biosciences | Cat. #747877; RRID: AB_2872339 |
| Anti-Human IgD BV421 (clone IA6-2) | BioLegend | Cat. #348226; RRID: AB_2561619 |
| Anti-Human IgG BV605 (clone G18-145) | BD Biosciences | Cat. #563246; RRID: AB_2738092 |
| Anti-human IgA DyLight® 488 (clone EPR5367-76) | Abcam | Cat. #ab98553; RRID: AB_10672542 |
| Anti-human CD20 PE-Cy7 (clone 2H7) | BioLegend | Cat. #302311; RRID: AB_314259 |
| Anti-human IgM AF488 (MHM-88) | BioLegend | Cat. # 314533; RRID: AB_2566486 |
| Goat Anti-human Fc Antibody | Jackson ImmunoResearch | Cat. #109-001-008; RRID: AB_2337530 |
| Anti-CCHFV pre-Gn mAb clone 13G8 | BEI Resources | Cat. #NR-40294 |
| Anti-CCHFV pre-Gn mAb clone 7F5 | BEI Resources | Cat. #NR-40281 |
| Anti-CCHFV pre-Gn mAb clone 8F10 | BEI Resources | Cat. #NR-40282 |
| Goat F(ab')2 Anti-Human Kappa FITC | SouthernBiotech | Cat. #2062-02; RRID: AB_2795737 |
| Goat F(ab')2 Anti-Human Lambda FITC | SouthernBiotech | Cat. #2072-02; RRID: AB_2795767 |
| Anti-HA.11 Epitope Tag Antiobdy APC (clone 16B12) | BioLegend | Cat. #901523; RRID: AB_2734657 |
| Goat F(ab')2 anti-human IgG PE | SouthernBiotech | Cat. #2042-09; RRID: AB_2795662 |
| Bacterial and virus strains | ||
| CCHFV-IbAr10200 | USAMRIID | Institute stock |
| CCHFV-Afg09-2990 | USAMRIID | Institute stock |
| CCHFV-Turkey2004 | USAMRIID | Institute stock |
| CCHFV-Oman-199809166 | USAMRIID | Institute stock |
| Biological samples | ||
| PBMCs from CCHFV convalescent donors | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| In-Fusion enzyme | Takara Bio | Cat. #638949 |
| Pierce IgG elution buffer | ThermoFisher Scientific | Cat. #21009 |
| Lys-C Endoproteinase | ThermoFisher Scientific | Cat. #90051 |
| cOmplete™ Protease Inhibitor Cocktail tablet | Roche | Cat. #4693132001 |
| Polyethylenimine (PEI 25K™) | Polysciences | Cat. #23966-1 |
| Kifunensine | GlycoSyn | Cat. #FC-034 |
| rGP38 (CCHFV IbAr10200) | Mishra et al.28 | N/A |
| rGP38 (CCHFV Oman) | This Paper | N/A |
| rGP38 (CCHFV Kosova-Hoti) | This Paper | N/A |
| rGP38 (CCHFV Turkey2004) | This Paper | N/A |
| rGP38 (CCHFV Afg09) | This Paper | N/A |
| rGP38 (CCHFV M18-China) | This Paper | N/A |
| KPL Blue Sure Substrate | Seracare | Cat. #52-00-01 |
| Streptactin-PE | IBA Lifesciences | Cat. #6-5000-001 |
| Streptactin-APC | IBA Lifesciences | Cat. #6-5010-001 |
| Propidium Iodide | Invitrogen | Cat. #P3566 |
| 10% NP-40 | Thermo Scientific | Cat. #85124 |
| RNaseOUT | Invitrogen | Cat. #10777019 |
| SuperScript III 5X First Strand Reaction Buffer | ThermoFisher Scientific | Cat. #18057018 |
| SuperScript III Reverse Transcriptase | ThermoFisher Scientific | Cat. #18080044 |
| HotStartTaq Plus Polymerase | QIAGEN | Cat. #203646 |
| Yeast Extract-Peptone-Dextrose Medium | Gietz and Woods75 | N/A |
| Salmon Sperm DNA | Invitrogen | Cat. #15632011 |
| Polyethylene Glycol 3350 | Sigma Aldrich | Cat. #202444 |
| Lithium Acetate | Sigma Aldrich | Cat. #517992 |
| Streptavidin Alexa Fluor 633 | Invitrogen | Cat. #S11223 |
| Soluble Cytosolic Protein | Xu et al.76 | N/A |
| Soluble Membrane Protein | Xu et al.76 | N/A |
| ExtraAvidin-R-PE | Sigma | Cat. #E4011 |
| Critical commercial assays | ||
| MACS B Cell Isolation Kit II, Human | Miltenyi Biotec | Cat. #130-091-151 |
| Streptavidin Biosensors | Sartorius | Cat. #18-5021 |
| Anti-Human IgG Fc Capture Biosensors | Sartorius | Cat. #18-5064 |
| Anti-Mouse IgG Fc Capture Biosensors | Sartorius | Cat. #18-5090 |
| Octet Kinetics Buffer 10x | Sartorius | Cat. #18-1105 |
| GeneMorph II Random Mutagenesis Kit | Agilent | Cat. #200550 |
| EZ-Link Sulfo-NHS-LC Biotin | ThermoFisher Scientific | Cat. #A39257 |
| Deposited data | ||
| CCHFV IbAr10200 GP38 | Mishra et al.28 | PDB: 6VKF |
| Model of CCHFV GP38 bound to ADI-46143 Fab | This Paper | PDB: 8VVK |
| Model of CCHFV GP38 bound to c13G8 Fab | This Paper | PDB: 8VVL |
| Model of CCHFV GP38 bound to ADI-46152 and ADI-58048 Fabs | This Paper | PDB: 8VWW |
| CryoEM map of CCHFV GP38 bound to ADI-46152 and ADI-58048 Fabs | This Paper | EMDB: EMD-43604 |
| CryoEM map of CCHFV GP38 bound with ADI-46143 and ADI-46158 Fabs | This Paper | EMDB: EMD-43551 |
| CryoEM map of CCHFV GP38 bound with ADI-58062 and ADI-63530 Fabs | This Paper | EMDB: EMD-43552 |
| CryoEM map of CCHFV GP38 bound with ADI-58026 and ADI-63547 Fabs | This Paper | EMDB: EMD-43553 |
| Experimental models: Cell lines | ||
| ExpiCHO Expression System Kit | Gibco | Cat. #A29133 |
| FreeStyle 293-F cells | Gibco | Cat. #R79007 |
| Vero E6 | ATCC | Cat. #CRL-1586; RRID:CVCL_XD71 |
| Vero | ATCC | Cat. #CCL-81; RRID: CVCL_0059 |
| S. cerevisiae: Strain background: EBY100 | ATCC | Cat. #MYA-4941 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129S(Cg)-Stat1tm1Dlv/J | Jackson Labs | Cat. #012606 |
| Mouse: Type 1 interferon a/β receptor KO | Jackson Labs | RRID:MMRRC Strain 032405-JAX |
| Oligonucleotides | ||
| Single B Cell Primers for PCR | Tiller et al.77 | N/A |
| Recombinant DNA | ||
| Plasmid: pCAGGS-NP | Zivcec et al.45 | N/A |
| Plasmid: pCAGGS-L | Zivcec et al.45 | N/A |
| Plasmid: pCAGGS-GPC | Zivcec et al.45 | N/A |
| Plasmid: pSMART-LCK-Luc (minigenome) | Zivcec et al.45 | N/A |
| Plasmid: pCAGGS-T7 | Zivcec et al.45 | N/A |
| Software and algorithms | ||
| CCP4 | Winn et al.78 | RRID:SCR_007255 |
| GraphPad Prism Software V9.5.1 | GraphPad Software | https://graphpad.com/ |
| iMosflm | Battye et al.79 | RRID:SCR_014217 |
| AIMLESS | Evans and Murshudov80 | RRID:SCR_015747 |
| Phaser | McCoy et al.81 | RRID:SCR_014219 |
| Coot | Emsley and Cowtan82 | RRID:SCR_01422 |
| Phenix | Adams et al.83 | RRID:SCR_014224 |
| PyMOL | DeLano et al.84 | RRID: SCR_000305 |
| cryoSPARC | Punjani et al.85 | RRID:SCR_016501 |
| ISOLDE | Tristan Croll86 | https://tristanic.github.io/isolde/ |
| AlphaFold Protein Structure Database | Varadi et al.87 | RRID:SCR_023662 |
| DeepEMhancer | Sanchez-Garcia et al.88 | https://github.com/rsanchezgarc/deepEMhancer |
| PDBePISA | Krissinel and Henrick89 | RRID:SCR_015749 |
| UCSF ChimeraX | Goddard et al.90 | RRID:SCR_015872 |
| FlowJo | FlowJo/BD Biosciences | RRID:SCR_008520 |
| FCS Express | De Novo Software | RRID:SCR_016431 |
| Fortébio Octet Data Analysis Software V11.1 | Sartorius | RRID:SCR_023267 |
| Carterra LSA Kinetics Software V1.7.3055 | Carterra | https://carterra-bio.com/applications/kinetics-software/ |
| Bio-Rad CFX Manager 3.1 | Bio-Rad | Cat. # 1845000; RRID:SCR_017251 |
| Other | ||
| HiTrap MabSelect SuRe affinity column | Cytiva | Cat. #11-0034-94 |
| CaptureSelect™ IgG-CH1 affinity resin | ThermoFisher Scientific | Cat. #194320010 |
| Pierce™ Protein A Plus Agarose resin | ThermoFisher Scientific | Cat. #22812 |
| HisPur™ Ni-NTA Resin | Thermo Scientific | Cat. # 88223 |
| HiLoad 16/600 Superdex 200 column | Cytivia | Cat. #28-9893-35 |
| Superdex 200 Increase 10/300 GL | Cytiva | Cat. #28-9909-44 |
| CCHFV Oman-199809166 sequence | UniProt | Accession # A0A0U3C6Q7 |
| CCHFV Kosova-Hoti sequence | UniProt | Accession #B2BSL7 |
| CCHFV 200406546-Turkey sequence | UniProt | Accession # A0A0U2SQZ0 |
| CCHFV Afg09-2990 sequence | UniProt | Accession #E5FEZ4 |
| CCHFV 79121M18 sequence | UniProt | Accession #D4NYK3 |
| PELCO easiGlow™ Glow Discharge Cleaning System | Ted Pella | Cat. #91000 |
| Vitrobot Mark IV | ThermoFisher Scientific | N/A |
| Expifectamine CHO Transfection Kit | ThermoFisher Scientific | Cat. #A29129 |
| Quantifoil R 1.2/1.3 300 Mesh, Cu | Electron Microscopy Sciences | Cat. #Q350CR1.3 |
Highlights.
188 monoclonal antibodies against CCHFV GP38 isolated from human survivors
Isolated antibodies are non-neutralizing and target 11 overlapping sites on GP38
Structural characterization of 9 antibodies targeting diverse epitopes
Antibodies targeting specific regions afford therapeutic efficacy
ACKNOWLEDGMENTS
All IgGs were sequenced by Adimab’s Molecular Core, and yeast-expressed mAbs and Fabs were produced by Adimab’s High Throughput Expression group. Biolayer interferometry binding experiments with IgGs were performed by Adimab’s Protein Analytics group. We thank Christina Spiropoulu, Eric Bergeron, and Marko Zivcec at the Centers for Disease Control and Prevention for kindly providing the plasmids and protocols necessary to generate tecVLPs. The following reagents were obtained from the Joel M. Dalrymple-Clearance J. Peters USAMRIID Antibody Collection through BEI Resources, NIAID, NIH: Monoclonal Anti-Crimean-Congo Hemorrhagic Fever Virus Pre-Gn Glycoprotein, Clone 13G8 (produced in vitro), NR-40294; Monoclonal Anti-Crimean-Congo Hemorrhagic Fever Virus Pre-Gn Glycoprotein, Clone 7F5 (produced in vitro), NR-40281; Monoclonal Anti-Crimean-Congo Hemorrhagic Fever Virus Pre-Gn Glycoprotein, Clone 8F10 (produced in vitro), NR-40282; and Monoclonal Anti-Crimean-Congo Hemorrhagic Fever Virus Nucleocapsid Protein, Clone 9D5 (produced in vitro), NR-40270. We thank members of all of our groups and the Prometheus consortium for their feedback on preliminary versions of the manuscript. We would like to thank Drs. Axel Brilot and Evan Schwartz at the Sauer Structural Biology Laboratory at UT Austin for assistance with cryo-EM data collection. We would also like to thank Kandis Cogliano for her project management support. Research was supported by NIAID of the National Institutes of Health (NIH) under award no. U19AI142777 (Centers of Excellence in Translational Research) to K.C., L.M.W., J.M.D., J.S.M., L.Z., and A.S.H., award no. R01AI152246 to K.C. and J.S.M, and The Welch Foundation under award no. F-0003-1962064 awarded to J.S.M. We acknowledge The University of Texas at Austin College of Natural Sciences and award RR160023 of the Cancer Prevention and Research Institute of Texas for support of the EM facility at the University of Texas at Austin. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center (SBC) at the Advanced Photon Source. SBC-CAT is operated by UChicago Argonne, LLC, for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The content is solely the responsibility of the authors and does not necessarily represent the official views of our institutions or funders. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the US Army.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114502.
DECLARATION OF INTERESTS
E.C., J.L., and N.T.P. are employees and shareholders of Adimab, LLC. O.S.S., V.H., M.D., D.P.M., and L.M.W. are shareholders of Adimab.
REFERENCES
- 1.Spengler JR, Bente DA, Bray M, Burt F, Hewson R, Korukluoglu G, Mirazimi A, Weber F, and Papa A (2018). Second International Conference on Crimean-Congo Hemorrhagic Fever. Antiviral Res 150, 137–147. 10.1016/j.antiviral.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spengler JR, Bergeron É, and Spiropoulou CF (2019). Crimean-Congo hemorrhagic fever and expansion from endemic regions. Curr. Opin. Virol 34, 70–78. 10.1016/j.coviro.2018.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spengler JR, Estrada-Peña A, Garrison AR, Schmaljohn C, Spiropoulou CF, Bergeron É, and Bente DA (2016). A chronological review of experimental infection studies of the role of wild animals and livestock in the maintenance and transmission of Crimean-Congo hemorrhagic fever virus. Antiviral Res 135, 31–47. 10.1016/j.antiviral.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spengler JR, Bergeron É, and Rollin PE (2016). Seroepidemiological Studies of Crimean-Congo Hemorrhagic Fever Virus in Domestic and Wild Animals. PLoS Negl. Trop. Dis 10, e0004210. 10.1371/journal.pntd.0004210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nurettin C, Engin B, Sukru T, Munir A, Zati V, and Aykut O (2022). The Seroprevalence of Crimean-Congo Hemorrhagic Fever in Wild and Domestic Animals: An Epidemiological Update for Domestic Animals and First Seroevidence in Wild Animals from Turkiye. Vet. Sci 9, 462. 10.3390/vetsci9090462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bente DA, Forrester NL, Watts DM, McAuley AJ, Whitehouse CA, and Bray M (2013). Crimean-Congo hemorrhagic fever: history, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antiviral Res 100, 159–189. 10.1016/j.antiviral.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Atim SA, Niebel M, Ashraf S, Vudriko P, Odongo S, Balinandi S, Aber P, Bameka R, Ademun AR, Masembe C, et al. (2023). Prevalence of Crimean-Congo haemorrhagic fever in livestock following a confirmed human case in Lyantonde district, Uganda. Parasit. Vectors 16, 7. 10.1186/s13071-022-05588-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smego RA Jr., Sarwari AR, and Siddiqui AR (2004). Crimean-Congo Hemorrhagic Fever: Prevention and Control Limitations in a Resource-Poor Country. Clin. Infect. Dis 38, 1731–1735. 10.1086/421093. [DOI] [PubMed] [Google Scholar]
- 9.Conger NG, Paolino KM, Osborn EC, Rusnak JM, Günther S, Pool J, Rollin PE, Allan PF, Schmidt-Chanasit J, Rieger T, and Kortepeter MG (2015). Health care response to CCHF in US soldier and nosocomial transmission to health care providers, Germany, 2009. Emerg. Infect. Dis 21, 23–31. 10.3201/eid2101.141413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belobo JTE, Kenmoe S, Kengne-Nde C, Emoh CPD, Bowo-Ngandji A, Tchatchouang S, Sowe Wobessi JN, Mbongue Mikangue CA, Tazokong HR, Kingue Bebey SR, et al. (2021). Worldwide epidemiology of Crimean-Congo Hemorrhagic Fever Virus in humans, ticks and other animal species, a systematic review and meta-analysis. PLoS Negl. Trop. Dis 15, e0009299. 10.1371/journal.pntd.0009299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Messina JP, Pigott DM, Golding N, Duda KA, Brownstein JS, Weiss DJ, Gibson H, Robinson TP, Gilbert M, William Wint GR, et al. (2015). The global distribution of Crimean-Congo hemorrhagic fever. Trans. R. Soc. Trop. Med. Hyg 109, 503–513. 10.1093/trstmh/trv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deyde VM, Khristova ML, Rollin PE, Ksiazek TG, and Nichol ST (2006). Crimean-Congo hemorrhagic fever virus genomics and global diversity. J. Virol 80, 8834–8842. 10.1128/jvi.00752-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anagnostou V, and Papa A (2009). Evolution of Crimean-Congo Hemorrhagic Fever virus. Infect. Genet. Evol 9, 948–954. 10.1016/j.meegid.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 14.Carroll SA, Bird BH, Rollin PE, and Nichol ST (2010). Ancient common ancestry of Crimean-Congo hemorrhagic fever virus. Mol. Phylogenet. Evol 55, 1103–1110. 10.1016/j.ympev.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Chamberlain J, Cook N, Lloyd G, Mioulet V, Tolley H, and Hewson R (2005). Co-evolutionary patterns of variation in small and large RNA segments of Crimean-Congo hemorrhagic fever virus. J. Gen. Virol 86, 3337–3341. 10.1099/vir.0.81213-0. [DOI] [PubMed] [Google Scholar]
- 16.Grard G, Drexler JF, Fair J, Muyembe JJ, Wolfe ND, Drosten C, and Leroy EM (2011). Re-emergence of Crimean-Congo hemorrhagic fever virus in Central Africa. PLoS Negl. Trop. Dis 5, e1350. 10.1371/journal.pntd.0001350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hewson R, Chamberlain J, Mioulet V, Lloyd G, Jamil B, Hasan R, Gmyl A, Gmyl L, Smirnova SE, Lukashev A, et al. (2004). Crimean-Congo haemorrhagic fever virus: sequence analysis of the small RNA segments from a collection of viruses world wide. Virus Res 102, 185–189. 10.1016/j.virusres.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 18.Lukashev AN, Klimentov AS, Smirnova SE, Dzagurova TK, Drexler JF, and Gmyl AP (2016). Phylogeography of Crimean Congo Hemorrhagic Fever Virus. PLoS One 11, e0166744. 10.1371/journal.pone.0166744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherifi K, Cadar D, Muji S, Robaj A, Ahmeti S, Jakupi X, Emmerich P, and Krüger A (2014). Crimean-Congo Hemorrhagic Fever Virus Clades V and VI (Europe 1 and 2) in Ticks in Kosovo, 2012. PLoS Negl. Trop. Dis 8, e3168. 10.1371/journal.pntd.0003168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papa A, Marklewitz M, Paraskevopoulou S, Garrison AR, Alkhovsky SV, Avšič-Županc T, Bente DA, Bergeron É, Burt F, Di Paola N, et al. (2022). History and classification of Aigai virus (formerly Crimean–Congo haemorrhagic fever virus genotype VI). J. Gen. Virol 103, 001734. 10.1099/jgv.0.001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehand MS, Al-Shorbaji F, Millett P, and Murgue B (2018). The WHO R&D Blueprint: 2018 review of emerging infectious diseases requiring urgent research and development efforts. Antiviral Res 159, 63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson S, Henschke N, Maayan N, Mills I, Buckley BS, Kakourou A, and Marshall R (2018). Ribavirin for treating Crimean Congo haemorrhagic fever. Cochrane Database Syst. Rev 6, CD012713. 10.1002/14651858.CD012713.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zivcec M, Scholte FE, Spiropoulou CF, Spengler JR, and Bergeron É (2016). Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus. Viruses 8, 106. 10.3390/v8040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez AJ, Vincent MJ, Erickson BR, and Nichol ST (2006). Crimean-congo hemorrhagic fever virus glycoprotein precursor is cleaved by Furin-like and SKI-1 proteases to generate a novel 38-kilodalton glycoprotein. J. Virol 80, 514–525. 10.1128/jvi.80.1.514-525.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez AJ, Vincent MJ, and Nichol ST (2002). Characterization of the glycoproteins of Crimean-Congo hemorrhagic fever virus. J. Virol 76, 7263–7275. 10.1128/jvi.76.14.7263-7275.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freitas N, Enguehard M, Denolly S, Levy C, Neveu G, Lerolle S, Devignot S, Weber F, Bergeron E, Legros V, and Cosset FL (2020). The interplays between Crimean-Congo hemorrhagic fever virus (CCHFV) M segment-encoded accessory proteins and structural proteins promote virus assembly and infectivity. PLoS Pathog 16, e1008850. 10.1371/journal.ppat.1008850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durie IA, Tehrani ZR, Karaaslan E, Sorvillo TE, McGuire J, Golden JW, Welch SR, Kainulainen MH, Harmon JR, Mousa JJ, et al. (2022). Structural characterization of protective non-neutralizing antibodies targeting Crimean-Congo hemorrhagic fever virus. Nat. Commun 13, 7298. 10.1038/s41467-022-34923-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishra AK, Moyer CL, Abelson DM, Deer DJ, El Omari K, Duman R, Lobel L, Lutwama JJ, Dye JM, Wagner A, et al. (2020). Structure and Characterization of Crimean-Congo Hemorrhagic Fever Virus GP38. J. Virol 94, e02005–19. 10.1128/jvi.02005-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golden JW, Shoemaker CJ, Lindquist ME, Zeng X, Daye SP, Williams JA, Liu J, Coffin KM, Olschner S, Flusin O, et al. (2019). GP38-targeting monoclonal antibodies protect adult mice against lethal Crimean-Congo hemorrhagic fever virus infection. Sci. Adv 5, eaaw9535. 10.1126/sciadv.aaw9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fels JM, Maurer DP, Herbert AS, Wirchnianski AS, Vergnolle O, Cross RW, Abelson DM, Moyer CL, Mishra AK, Aguilan JT, et al. (2021). Protective neutralizing antibodies from human survivors of Crimean-Congo hemorrhagic fever. Cell 184, 3486–3501.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrews SF, Chambers MJ, Schramm CA, Plyler J, Raab JE, Kanekiyo M, Gillespie RA, Ransier A, Darko S, Hu J, et al. (2019). Activation Dynamics and Immunoglobulin Evolution of Pre-existing and Newly Generated Human Memory B cell Responses to Influenza Hemagglutinin. Immunity 51, 398–410.e5. 10.1016/j.immuni.2019.06.024. [DOI] [PubMed] [Google Scholar]
- 32.Glass DR, Tsai AG, Oliveria JP, Hartmann FJ, Kimmey SC, Calderon AA, Borges L, Glass MC, Wagar LE, Davis MM, and Bendall SC (2020). An Integrated Multi-omic Single-Cell Atlas of Human B Cell Identity. Immunity 53, 217–232.e5. 10.1016/j.immuni.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wec AZ, Haslwanter D, Abdiche YN, Shehata L, Pedreño-Lopez N, Moyer CL, Bornholdt ZA, Lilov A, Nett JH, Jangra RK, et al. (2020). Longitudinal dynamics of the human B cell response to the yellow fever 17D vaccine. Proc. Natl. Acad. Sci. USA 117, 6675–6685. 10.1073/pnas.1921388117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakharkar M, Rappazzo CG, Wieland-Alter WF, Hsieh C-L, Wrapp D, Esterman ES, Kaku CI, Wec AZ, Geoghegan JC, McLellan JS, et al. (2021). Prolonged evolution of the human B cell response to SARS-CoV-2 infection. Sci. Immunol 6, eabg6916. 10.1126/sciimmunol.abg6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogers TF, Goodwin EC, Briney B, Sok D, Beutler N, Strubel A, Nedellec R, Le K, Brown ME, Burton DR, and Walker LM (2017). Zika virus activates de novo and cross-reactive memory B cell responses in dengue-experienced donors. Sci. Immunol 2, eaan6809. 10.1126/sciimmunol.aan6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, et al. (2016). Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 351, 1078–1083. 10.1126/science.aad5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, et al. (2008). Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671. 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schramm CA, and Douek DC (2018). Beyond Hot Spots: Biases in Antibody Somatic Hypermutation and Implications for Vaccine Design. Front. Immunol 9, 1876. 10.3389/fimmu.2018.01876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elsner RA, and Shlomchik MJ (2020). Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 53, 1136–1150. 10.1016/j.immuni.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Briney B, Inderbitzin A, Joyce C, and Burton DR (2019). Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566, 393–397. 10.1038/s41586-019-0879-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoogstraal H (1979). The epidemiology of tick-borne Crimean-Congo hemorrhagic fever in Asia, Europe, and Africa. J. Med. Entomol 15, 307. 10.1093/jmedent/15.4.307. [DOI] [PubMed] [Google Scholar]
- 42.Kong Y, Yan C, Liu D, Jiang L, Zhang G, He B, and Li Y (2022). Phylogenetic analysis of Crimean-Congo hemorrhagic fever virus in inner Mongolia, China. Ticks Tick. Borne. Dis 13, 101856. 10.1016/j.ttbdis.2021.101856. [DOI] [PubMed] [Google Scholar]
- 43.Guo R, Shen S, Zhang Y, Shi J, Su Z, Liu D, Liu J, Yang J, Wang Q, Hu Z, et al. (2017). A new strain of Crimean-Congo hemorrhagic fever virus isolated from Xinjiang, China. Virol. Sin 32, 80–88. 10.1007/s12250-016-3936-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. (2017). Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 114, 944–949. 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zivcec M, Metcalfe MG, Albariño CG, Guerrero LW, Pegan SD, Spiropoulou CF, and Bergeron É (2015). Assessment of Inhibitors of Pathogenic Crimean-Congo Hemorrhagic Fever Virus Strains Using Virus-Like Particles. PLoS Negl. Trop. Dis 9, e0004259. 10.1371/journal.pntd.0004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai S, Deng F, Wang H, and Ning Y (2021). Crimean-Congo Hemorrhagic Fever Virus: Current Advances and Future Prospects of Antiviral Strategies. Viruses 13, 1195. 10.3390/v13071195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zivcec M, Guerrero LIW, Albariño CG, Bergeron É, Nichol ST, and Spiropoulou CF (2017). Identification of broadly neutralizing monoclonal antibodies against Crimean-Congo hemorrhagic fever virus. Antiviral Res 146, 112–120. 10.1016/j.antiviral.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bereczky S, Lindegren G, Karlberg H, Akerström S, Klingström J, and Mirazimi A (2010). Crimean-Congo hemorrhagic fever virus infection is lethal for adult type I interferon receptor-knockout mice. J. Gen. Virol 91, 1473–1477. 10.1099/vir.0.019034-0. [DOI] [PubMed] [Google Scholar]
- 49.Zivcec M, Safronetz D, Scott D, Robertson S, Ebihara H, and Feldmann H (2013). Lethal Crimean-Congo hemorrhagic fever virus infection in interferon a/b receptor knockout mice is associated with high viral loads, proinflammatory responses, and coagulopathy. J. Infect. Dis 207, 1909–1921. 10.1093/infdis/jit061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bente DA, Alimonti JB, Shieh WJ, Camus G, Ströher U, Zaki S, and Jones SM (2010). Pathogenesis and immune response of Crimean-Congo hemorrhagic fever virus in a STAT-1 knockout mouse model. J. Virol 84, 11089–11100. 10.1128/jvi.01383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suschak JJ, Golden JW, Fitzpatrick CJ, Shoemaker CJ, Badger CV, Schmaljohn CS, and Garrison AR (2021). A CCHFV DNA vaccine protects against heterologous challenge and establishes GP38 as immunorelevant in mice. NPJ Vaccines 6, 31. 10.1038/s41541-021-00293-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bornholdt ZA, Herbert AS, Mire CE, He S, Cross RW, Wec AZ, Abelson DM, Geisbert JB, James RM, Rahim MN, et al. (2019). A Two-Antibody Pan-Ebolavirus Cocktail Confers Broad Therapeutic Protection in Ferrets and Nonhuman Primates. Cell Host Microbe 25, 49–58.e5. 10.1016/j.chom.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wec AZ, Bornholdt ZA, He S, Herbert AS, Goodwin E, Wirchnianski AS, Gunn BM, Zhang Z, Zhu W, Liu G, et al. (2019). Development of a Human Antibody Cocktail that Deploys Multiple Functions to Confer Pan-Ebolavirus Protection. Cell Host Microbe 25, 39–48.e5. 10.1016/j.chom.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bornholdt ZA, Ndungo E, Fusco ML, Bale S, Flyak AI, Crowe JE Jr., Chandran K, and Saphire EO (2016). Host-Primed Ebola Virus GP Exposes a Hydrophobic NPC1 Receptor-Binding Pocket, Revealing a Target for Broadly Neutralizing Antibodies. mBio 7, e02154. 10.1128/mBio.02154-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chapman NS, Zhao H, Kose N, Westover JB, Kalveram B, Bombardi R, Rodriguez J, Sutton R, Genualdi J, LaBeaud AD, et al. (2021). Potent neutralization of Rift Valley fever virus by human monoclonal antibodies through fusion inhibition. Proc. Natl. Acad. Sci. USA 118, e2025642118. 10.1073/pnas.2025642118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M, et al. (2016). Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 351, 1339–1342. 10.1126/science.aad5224. [DOI] [PubMed] [Google Scholar]
- 57.Engdahl TB, Kuzmina NA, Ronk AJ, Mire CE, Hyde MA, Kose N, Josleyn MD, Sutton RE, Mehta A, Wolters RM, et al. (2021). Broad and potently neutralizing monoclonal antibodies isolated from human survivors of New World hantavirus infection. Cell Rep 35, 109086. 10.1016/j.celrep.2021.109086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mittler E, Wec AZ, Tynell J, Guardado-Calvo P, Wigren-Byström J, Polanco LC, O’Brien CM, Slough MM, Abelson DM, Serris A, et al. (2022). Human antibody recognizing a quaternary epitope in the Puumala virus glycoprotein provides broad protection against orthohantaviruses. Sci. Transl. Med 14, eabl5399. 10.1126/scitranslmed.abl5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quiroz JA, Malonis RJ, Thackray LB, Cohen CA, Pallesen J, Jangra RK, Brown RS, Hofmann D, Holtsberg FW, Shulenin S, et al. (2019). Human monoclonal antibodies against chikungunya virus target multiple distinct epitopes in the E1 and E2 glycoproteins. PLoS Pathog 15, e1008061. 10.1371/journal.ppat.1008061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.West BR, Wec AZ, Moyer CL, Fusco ML, Ilinykh PA, Huang K, Wirchnianski AS, James RM, Herbert AS, Hui S, et al. (2019). Structural basis of broad ebolavirus neutralization by a human survivor antibody. Nat. Struct. Mol. Biol 26, 204–212. 10.1038/s41594-019-0191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bertolotti-Ciarlet A, Smith J, Strecker K, Paragas J, Altamura LA, McFalls JM, Frias-Stäheli N, García-Sastre, Schmaljohn CS, and Doms RW (2005). Cellular localization and antigenic characterization of crimean-congo hemorrhagic fever virus glycoproteins. J. Virol 79, 6152–6161. 10.1128/jvi.79.10.6152-6161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Earnest JT, Holmes AC, Basore K, Mack M, Fremont DH, and Diamond MS (2021). The mechanistic basis of protection by non-neutralizing anti-alphavirus antibodies. Cell Rep 35, 108962. 10.1016/j.celrep.2021.108962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gunn BM, Yu WH, Karim MM, Brannan JM, Herbert AS, Wec AZ, Halfmann PJ, Fusco ML, Schendel SL, Gangavarapu K, et al. (2018). A Role for Fc Function in Therapeutic Monoclonal Antibody-Mediated Protection against Ebola Virus. Cell Host Microbe 24, 221–233.e5. 10.1016/j.chom.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ilinykh PA, Huang K, Santos RI, Gilchuk P, Gunn BM, Karim MM, Liang J, Fouch ME, Davidson E, Parekh DV, et al. (2020). Non-neutralizing Antibodies from a Marburg Infection Survivor Mediate Protection by Fc-Effector Functions and by Enhancing Efficacy of Other Antibodies. Cell Host Microbe 27, 976–991.e11. 10.1016/j.chom.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lewis GK, Pazgier M, Evans DT, Ferrari G, Bournazos S, Parsons MS, Bernard NF, and Finzi A (2017). Beyond Viral Neutralization. AIDS Res. Hum. Retroviruses 33, 760–764. 10.1089/aid.2016.0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sicca F, Neppelenbroek S, and Huckriede A (2018). Effector mechanisms of influenza-specific antibodies: neutralization and beyond. Expert Rev. Vaccines 17, 785–795. 10.1080/14760584.2018.1516553. [DOI] [PubMed] [Google Scholar]
- 67.Krammer F, and Palese P (2015). Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov 14, 167–182. 10.1038/nrd4529. [DOI] [PubMed] [Google Scholar]
- 68.Saphire EO, Schendel SL, Fusco ML, Gangavarapu K, Gunn BM, Wec AZ, Halfmann PJ, Brannan JM, Herbert AS, Qiu X, et al. (2018). Systematic Analysis of Monoclonal Antibodies against Ebola Virus GP Defines Features that Contribute to Protection. Cell 174, 938–952.e13. 10.1016/j.cell.2018.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lux A, Yu X, Scanlan CN, and Nimmerjahn F (2013). Impact of immune complex size and glycosylation on IgG binding to human FcgRs. J. Immunol 190, 4315–4323. 10.4049/jimmunol.1200501. [DOI] [PubMed] [Google Scholar]
- 70.Guardado-Calvo P, and Rey FA (2021). The Viral Class II Membrane Fusion Machinery: Divergent Evolution from an Ancestral Heterodimer. Viruses 13, 2368. 10.3390/v13122368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dyer O (2019). Two Ebola treatments halve deaths in trial in DRC outbreak. BMJ 366, l5140. 10.1136/bmj.l5140. [DOI] [PubMed] [Google Scholar]
- 72.Herbert AS, Froude JW, Ortiz RA, Kuehne AI, Dorosky DE, Bakken RR, Zak SE, Josleyn NM, Musiychuk K, Jones RM, et al. (2020). Development of an antibody cocktail for treatment of Sudan virus infection. Proc. Natl. Acad. Sci. USA 117, 3768–3778. 10.1073/pnas.1914985117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pascal KE, Dudgeon D, Trefry JC, Anantpadma M, Sakurai Y, Murin CD, Turner HL, Fairhurst J, Torres M, Rafique A, et al. (2018). Development of Clinical-Stage Human Monoclonal Antibodies That Treat Advanced Ebola Virus Disease in Nonhuman Primates. J. Infect. Dis 218, S612–S626. 10.1093/infdis/jiy285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Howell KA, Brannan JM, Bryan C, McNeal A, Davidson E, Turner HL, Vu H, Shulenin S, He S, Kuehne A, et al. (2017). Cooperativity Enables Non-neutralizing Antibodies to Neutralize Ebolavirus. Cell Rep 19, 413–424. 10.1016/j.celrep.2017.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gietz RD, and Woods RA (2006). Yeast transformation by the LiAc/SS Carrier DNA/PEG method. Methods Mol. Biol 313, 107–120. 10.1385/1-59259-958-3:107. [DOI] [PubMed] [Google Scholar]
- 76.Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu T-Y, Torrey J, Thomas J, Bobrowicz P, Vásquez M, et al. (2013). Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel 26, 663–670. 10.1093/protein/gzt047. [DOI] [PubMed] [Google Scholar]
- 77.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, and Wardemann H (2008). Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 329, 112–124. 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67,235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Battye TG, Kontogiannis L, Johnson O, Powell HR, and Leslie AG (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr 67, 271–281. 10.1107/s0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr 69, 1204–1214. 10.1107/s0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCoy AJ (2007). Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr 63, 32–41. 10.1107/s0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- 83.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.DeLano WL (2002). Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 40, 82–92. [Google Scholar]
- 85.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryo-SPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 86.Croll TI (2018). ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol 74, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al. (2022). Alpha-Fold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50, D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanchez-Garcia R, Gomez-Blanco J, Cuervo A, Carazo JM, Sorzano COS, and Vargas J (2021). DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol 4, 874. 10.1038/s42003-021-02399-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krissinel E, and Henrick K (2007). Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol 372, 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 90.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bowick GC, Airo AM, and Bente DA (2012). Expression of interferon-induced antiviral genes is delayed in a STAT1 knockout mouse model of Crimean-Congo hemorrhagic fever. Virol. J 9, 122. 10.1186/1743-422x-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garrison AR, Moresco V, Zeng X, Cline CR, Ward MD, Ricks KM, Olschner SP, Cazares LH, Karaaslan E, Fitzpatrick CJ, et al. (2024). Nucleocapsid protein-specific monoclonal antibodies protect mice against Crimean-Congo hemorrhagic fever virus. Nat. Commun 15, 1722. 10.1038/s41467-024-46110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wec AZ, Wrapp D, Herbert AS, Maurer DP, Haslwanter D, Sakharkar M, Jangra RK, Dieterle ME, Lilov A, Huang D, et al. (2020). Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science 369, 731–736. 10.1126/science.abc7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mata-Fink J, Kriegsman B, Yu HX, Zhu H, Hanson MC, Irvine DJ, and Wittrup KD (2013). Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. J. Mol. Biol 425, 444–456. 10.1016/j.jmb.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Estep P, Caffry I, Yu Y, Sun T, Cao Y, Lynaugh H, Jain T, Vásquez M, Tessier PM, and Xu Y (2015). An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies. mAbs 7, 553–561. 10.1080/19420862.2015.1016694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.He F, Woods CE, Becker GW, Narhi LO, and Razinkov VI (2011). High-throughput assessment of thermal and colloidal stability parameters for monoclonal antibody formulations. J. Pharm. Sci 100, 5126–5141. 10.1002/jps.22712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Models of CCHFV GP38 bound to Fabs of GP38-specific mAbs have been deposited at Worldwide Protein DataBank (wwPDB) under accession codes 8VVK, 8VVL, and 8VVW. Cryo-EM maps have been deposited at Electron Microscopy DataBank under accession codes EMD-43604, EMD-43551, EMD-43552, and EMD-43553. Depositions are available as of the date of publication. The sequence data for the GP38-specific mAbs cannot be deposited in a public repository because they are the subject of pending provisional patent applications. To request access, contact the lead contact, Jason S. McLellan, and the sequences will be made available upon request under an MTA. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.