

Abstract
Alzheimer’s disease (AD) is an age-dependent neurodegenerative disease characterized by extracellular Amyloid Aβ peptide (Aβ) deposition and intracellular Tau protein aggregation. Glia, especially microglia and astrocytes are core participants during the progression of AD and these cells are the mediators of Aβ clearance and degradation. The microbiota-gut-brain axis (MGBA) is a complex interactive network between the gut and brain involved in neurodegeneration. MGBA affects the function of glia in the central nervous system (CNS), and microbial metabolites regulate the communication between astrocytes and microglia; however, whether such communication is part of AD pathophysiology remains unknown. One of the potential links in bilateral gut-brain communication is tryptophan (Trp) metabolism. The microbiota-originated Trp and its metabolites enter the CNS to control microglial activation, and the activated microglia subsequently affect astrocyte functions. The present review highlights the role of MGBA in AD pathology, especially the roles of Trp per se and its metabolism as a part of the gut microbiota and brain communications. We (i) discuss the roles of Trp derivatives in microglia-astrocyte crosstalk from a bioinformatics perspective, (ii) describe the role of glia polarization in the microglia-astrocyte crosstalk and AD pathology, and (iii) summarize the potential of Trp metabolism as a therapeutic target. Finally, we review the role of Trp in AD from the perspective of the gut-brain axis and microglia, as well as astrocyte crosstalk, to inspire the discovery of novel AD therapeutics.
Keywords: Age-dependent Alzheimer’s disease;, tryptophan metabolism, microglia and astrocyte crosstalk, microbiota-gut-brain axis, bioinformatics analysis
1. Introduction
Alzheimer’s disease (AD) is an age-dependent progressive neurodegenerative disease [1]. About 1 in 9 people age 65 and older have AD. The percentage of people with Alzheimer's dementia increases with age: 5.0% of people age 65 to 74, 13.1% of people age 75 to 84, and 33.2% of people age 85 and older have Alzheimer's dementia. AD is closely associated with the aging process. There is no cure for AD, which may partially reflect the lack of a clear AD pathogenesis. AD is characterized by extracellular Amyloid-β (Aβ) deposition and intracellular Tau-containing neurofibrillary tangles accumulation. Indeed, reports confirm that Aβ and Tau activities in human brain homogenates from Alzheimer’s patients [2, 3] and microglia and astrocytes are moderators of Aβ clearance and degradation that play critical roles in AD pathology. Interestingly, astrocytic degeneration is present in a subset of AD human brain samples [4]. Aging itself is the primary risk factor for the development of AD. Other critical factors affecting AD pathogenesis include genetics, environment, lifestyle habits, sleep, apolipoprotein E’s effect, viral and bacterial infection, and microbiota [5]. Microbial metabolites regulate the communication between astrocytes and microglia. Although such communication as part of AD pathophysiology is not well known. Aging affects the composition of the gut microbiota and microglia activity in neurodegenerative disorders like AD. Gut microbiota modulation and the communication between astrocytes and microglia represent a promising therapeutic strategy to counteract the progression of AD. Furthermore, recent advances in the field of aging biology, molecular biology, chemistry, and bioinformatics provide significant molecular insights into AD pathogenesis and promising therapeutics.
One of the potential links in bilateral gut-brain communication is tryptophan (Trp) metabolism. Trp enters the body via digestion of proteins in the gut. Subsequently, the small intestine absorbs this gut-derived Trp and its metabolites, such as nicotinamide adenosine dinucleotide (NAD), and then releases it into the peripheral circulation where it is transported through the large neutral amino acid transporter system (LAT1), crossing the blood brain barrier (BBB), and entering the central nervous system (CNS). After reaching the CNS, Trp acts on the aryl hydrocarbon receptor (AHR) to control the activation of astrocyte and microglia as well as to stimulate production of quinolinic acid (QUIN) and kynurenic acid (KYNA), respectively. QUIN is a direct activator of N-methyl-D-aspartate (NMDA) receptors that modulate the generation of ROS, lipid peroxidation, and release/uptake of glutamate. In AD pathology, indoleamine 2,3-dioxygenase (IDO) activity and production of QUIN increases in response to Aβ1-42 peptide in human macrophages and microglia. Also, in human AD brain specimen, the senile plaques show greater immunoreactivity to IDO1 and QUIN along with the other substances from reactive microglia and astrocytes. KYNA antagonizes the NMDA receptor as well as inhibits alpha 7 acetylcholine receptors, which regulate pre-synaptic glutamate release. The levels of KYNA decrease in the serum, red blood cells, and lumbar cerebrospinal fluid in AD patients [6]. QUIN and KYNA are responsible for the crosstalk between microglia and astrocytes, producing neurotoxic and neuroprotective effects, respectively. The present review highlights the role of microbiota-gut-brain axis (MGBA) in AD pathology, especially the roles of Trp per se and Trp metabolism as part of the gut microbiota and brain communications. We discuss the role of: (i) Trp and its derivatives in microglia-astrocyte crosstalk from a bioinformatics perspective; (ii) glia polarization in the microglia-astrocyte crosstalk and the involvement and interactions of inflammatory factors in AD pathology; and (iii) Trp in the glia crosstalk of gut-brain axis and summarizes Trp metabolism as a potential therapeutic target.
Tryptophan derivatives in AD
Role of indoleamine 2,3-dioxygenase (IDO)
Trp as well as its metabolism play vital roles in AD pathology. Trp metabolism is associated with aging and produces metabolites that control inflammation. The essential amino acid Trp is the precursor of serotonin (5-hydroxytryptamine (5-HT), which is widely distributed in both the peripheral nervous system (PNS) and the CNS [7]. There are four Trp metabolic pathways: (i) kynurenine (KYN), (ii) hydroxylation, (iii) decarboxylation, and (iv) transamination pathways. The major Trp metabolic pathway is the kynurenine pathway (KP), which produces various important Trp derivatives. The first step in the KP is the IDO-catalyzed Trp oxidation to formylkynurenine, which is then hydrolyzed to KYN [6, 8] (Fig. 1, 2). As the first and rate-limiting step in the KP of Trp metabolism, IDO is one of the potential players in AD pathogenesis. In 3xTg AD mice, IDO1 activity increases, which originates from the neurons and astrocytes [9]. Increased IDO activity leads to neuronal loss, and Tau protein, and Aβ accumulation via inflammatory pathways [7]. In a vicious cycle, IDO-mediated activation of inflammatory molecules give rise to pathological debris, which in turn increases the production of IDO [10]. Furthermore, the IDO inhibitor coptisine reduces serum IDO concentrations and leads to a decrease in microglia and astrocyte activation. As a result, coptisine treatment prevents neuron loss, reduces amyloid plaque formation, and ameliorates impaired cognition in the AβPP/PS1 transgenic AD mouse model [11]. Interestingly, both neurons and glia (astrocyte and microglia) produce IDO in the CNS [12]. Lipopolysaccharides (LPS) increase the production of IDO in microglia, leading to elevated levels of KYN along with an enhanced synthesis and secretion of the proinflammatory factors, tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) [13] (Fig. 1). IDO causes loss of neurons, activation of glia, accumulation of debris, and an increase - in the proinflammatory factors and Trp depletion. These anomalies together lead to the enhanced progression of AD. Besides IDO, TDO (Tryptophan-2, 3-dioxygenase) is another heme-containing enzyme that catalyzes the production of KYN. TDO expression level is significantly raised in AD and leads to further enhancement of neuroinflammation and neuro-pathological abnormalities [14].
Figure 1.

The metabolic routes of derivatives of tryptophan. The KYNA and QUIN pathways of major tryptophan (Trp) metabolism are depicted, which represent the neuroprotective branch and neurotoxic branch, respectively. The tryptophan-5-hydroxytryptophan (5-HTP), serotonin (5-HT), N-acetylserotonin (NAS), and melatonin pathways are shown as the minor pathways of Trp metabolism.
Roles of kynurenine (KYN) and kynurenic acid (KYNA)
Because KYN is one of the key intermediates after IDO catalyzed Trp oxidation in the KP pathway, the ratio of KYN/Trp is often used to evaluate IDO activation. AD patients possess elevated ratios of KYN/Trp [11, 15]. After KYN production, KYN metabolism diverges into two branches: (i) the neuroprotective branch and (ii) the neurotoxic branch (Fig. 1). By means of kynurenine aminotransferase II, KYN directly transforms into KYNA [16] (Fig. 1 and 2). Astrocyte primarily produce KYNA, which act as a neuroprotective agent in AD [17]. KYNA is an agonist of the AHR as well as acts as an endogenous antagonist of the NMDA receptor to prevent glutamate excitotoxicity in AD [6, 18, 19]. Astrocytic KYNA inhibits the α7 nicotinic acetylcholine receptors in neurons affording reduced levels of glutamate release (Fig. 1). KYNA-mediated interactions between astrocyte and neuron play a significant role in prefrontal mediated cognitive processes [20]. Glutamate serves as the energy source for neurons, and the shuttle of lactate between astrocytes and neurons can regulate the astrocyte glutamate uptake [21]. Due to these reasons, KYNA is a key Trp metabolic intermediate that mediates the neuron-astrocyte interaction, which regulates the extracellular glutamate concentration and related glutamatergic excitation process (Table 1). Besides KYNA-mediated astrocyte-neuron interactions, cytokines also serve as signals to connect the astrocytes and neurons. For example, interleukin-1β (IL-1β) is the core proinflammatory cytokine in AD, and astrocytic IL-1β causes an increase in Aβ accumulation in the neurons [22]. Blocking IL-1β production reduces the immune response [23, 24].
Figure 2.

Scheme of the four Trp metabolic pathways. The kynurenine pathway to produce KYNA and QUIN; the hydroxylation pathway to produce 5-HTP-5-HT-NAS-melatonin; the decarboxylation pathway to produce tryptamine; and the transamination pathways to produce Indole-3-propionic acid.
Table 1.
The pathology and function of derivatives of tryptophan in CNS.
| Derivatives of Tryptophan | Mechanism | Pathology/Function | References |
|---|---|---|---|
| Kynurenic acid | Neuroprotection. The antagonist for NMDA receptors and cholinergic α7 nicotine receptors. Agonist of AHR. Targeting GRP35. | Reducing glutamate and proinflammatory cytokines release. Inhibiting glial inflammatory response. Reducing excitatory neurotransmission. Inhibiting hippocampal plasticity. | [17, 20, 61-63] |
| Quinolinic acid | Neurotoxin. Activating NMDA receptors, Iron chelation, and ROS production. | Increasing oxidative stress and cytosolic Cu/Zn SOD disturbance. Increasing lipid peroxidation activity in synaptosome. Increasing peroxidized lipids in the striatum and hippocampus, lactate dehydrogenase (LDH) release, and neuronal nitric oxide synthase. Upregulating IL-6. Impairing Spatial learning and memory and depression. | [6, 15, 26, 28, 30, 32, 33, 64, 65] |
| Serotonin | Serotonin neuron activity, neurotransmitter, Triggering serotonin receptor in astrocytes and microglia. Activating the HPA axis. Neuroprotection. | Improving brain development. Regulating the basic physiological functions (mood, sleep, pain, aggression and sexual behavior, cognition, memory, and anxiety). | [34, 66] |
| N-acetyl Serotonin | Neuroprotectant. Antioxidant. Anti-apoptosis. | Improving cognition and depression. Protecting against Aβ induced neurotoxicity in vitro. Increasing neuroprotection. Inhibition of caspase-3 activation. Anti-autophagy dysfunction. Reducing oxidative stress. | [46-49, 52] |
| Melatonin | Antioxidant, Antiinflammation, Triggering neurons or glia MT1/MT2 receptors. Neuroprotection. | Supporting mitochondria. Regulating circadian amplitudes. Increasing neuroprotection. Decreasing excitotoxic agents. | [35, 38, 39, 67] |
| Tryptamine | Inhibitor of BACE1, neurotransmitter, and neuromodulator. Neurotoxicity. | Increasing axonal loss. Damaging mitochondria. Causing neurodegeneration. | [55, 56, 68] |
| Indole-3-propionic acid | Neuroprotection. Antioxidant. | Reducing Aβ accumulation Decreasing oxidative stress. Reducing the levels of proinflammatory cytokines and the activation of glial cells and astrocytes. Inhibiting NF-κB signaling. | [55, 57, 60] |
Role of quinolinic acid (QUIN)
On the neurotoxic branch, kynurenine 3-hydroxylase converts KYN to 3-hydroxykynurenine. Next, kynureninase transforms 3-hydroxykynurenine to 3-hydroxyanthranilic acid. Finally, 3-hydroxyanthranilate oxidase cleaves the aromatic ring and the resulting intermediate then re-cyclizes to produce QUIN. QUIN is the precursor of nicotinamide adenosine dinucleotide (NAD) [6, 8] (Fig. 1 and 2). The increase of the KYN/Trp ratio results in the elevation of QUIN [11, 15, 25]. QUIN and 3-hydroxykynurenine exhibit neurotoxic properties, and QUIN is involved in AD neuropathogenesis [26]. Although macrophages are the major producer of QUIN, microglia also produce QUIN but at a lesser quantity. QUIN produced by activated macrophages causes neurotoxicity through several pathways including activating NMDA receptors, inhibiting glutamate re-uptake, or increasing oxidative stress [6]. Increased QUIN is also present in peripheral mononuclear cells in Alzheimer's dementia [27].
Additionally, IDO and QUIN are detected in cortical microglia, astrocytes, and neurons. In the perimeter of senile plaques of the AD human hippocampus, high levels of IDO and QUIN are present in microglia and astrocytes [28]. Microglia produce QUIN. Glia and immune cells express IDO, which leads to QUIN production and increased inflammatory response in CNS [15]. Increased QUIN production positively correlates with the formation of Aβ and p-Tau protein [26, 28]. Aβ induces IDO expression and leads to a significantly increased level of QUIN in human macrophages and microglia, while in human astrocytes and macrophages, Aβ induces IL-1β expression [29, 30]. QUIN impairs spatial learning and memory [26] and downregulating QUIN improves the IDO-dependent depressive-like behaviors in the hippocampus of LPS-treated mice [31]. Astrocytes and neurons could uptake external QUIN to produce NAD [25]. In explaining QUIN’s neurotoxicity, Schiefer et al. report a substantial IL-6 upregulation in the rat striatum and the loss of striatal neurons following QUIN injection [32] (Table 1).
QUIN plays an important role in AD pathology. Firstly, it induces the production and accumulation of neuropathological substances [26, 28] due to the increase in oxidative stress and neuroinflammation that enhance the neuronal damage [15, 26]. It also disorders the metabolism of lactate in neurons that results in derailment of a variety of neurotransmitters [33]. These neuro-pathological alterations by QUIN induce loss of memory and further the progression of AD.
Role of melatonin
The KP accounts for 95% of Trp metabolism, and the other 3 minor pathways are responsible for 5% of Trp degradation [8, 34]. However, many important Trp metabolites are from the 3 minor pathways. For example, tryptophan hydroxylase hydroxylates Trp to produce 5-hydroxytryptophan, followed by conversion to serotonin by 5-hydroxytryptophan decarboxylase (Fig. 1 and 2). Next, serotonin N-acetyltransferase acetylates serotonin to afford N-acetylserotonin (NAS), which is methylated by hydroxyindole O-methyltransferase to produce melatonin as the final product [8, 35] (Fig. 1 and 2). Many of these molecules contribute to AD pathology and other neurological diseases [36].
Abnormal protein aggregates are the common pathological hallmarks for neurodegenerative diseases, including AD (Aβ peptides), Parkinson's disease (PD) (α-synuclein), Huntington's disease (HD) (Huntingtin), and Amyotrophic Lateral Sclerosis (ALS) (superoxide dismutase 1 or TAR-DNA binding protein-43). Melatonin, an anti-aging agent, as reported by us and others, supports the normal CNS function in HD through decreasing mutant huntingtin-mediated toxicity [37], and in AD through decreasing Aβ plaques, Tau protein hyperphosphorylation, supporting mitochondrial function and reducing oxidative stress and the inflammatory responses [38-41]. Furthermore, the neuroprotective effects of melatonin, another melatonin receptor agonist, arise through the upregulating of melatonin receptor type 1A (MT1) in HD [37] and ALS [42]. Interestingly, Sulkava et al. report higher levels of Aβ and Tau protein accumulations in AD patients with the rs12506228A variant, and this variant gene is located close to the MT1 gene. MT1 expression levels affect rs12506228A expression, while MT1 silencing leads to an elevated Aβ pathological process in culture neurons. Thus, MT1 is a gene responsible for regulating the amyloidogenic processing of amyloid precursor protein (APP) [39]. Mouse macrophage gene expression data, as analyzed by Kadena et al., show that melatonin attenuates immune responses and preferentially downregulates interferon regulatory factors, signal transducers, and activators of transcription-related signaling [43]. Recently, single-cell analysis and genetic fate mapping data reveal that a spectrum of macrophage/microglia exists in variety of phenotypic states. While the differentially expressed genes (DEGs) in the different macrophage phenotypes have been compared with the ImmGen database. The results demonstrate that both microglia and astrocyte display a multifunctional role in neuronal activity modulation in homeostatic and pathophysiological conditions [44].
Role of N-acetylserotonin (NAS)
NAS is a Trp methoxyindole derivative and may function as a melatonin receptor agonist. NAS is also an agonist of the tyrosine protein kinase B and exhibits antioxidant and neurotrophic effects [45, 46]. In cerebellar granule cell culture, NAS protects against Aβ induced neurotoxicity (Table 1) [47]. Additionally, NAS offers neuroprotection in experimental models of ischemic stroke [48, 49], retinal ischemia-reperfusion [50], hepatic ischemia-reperfusion injury, and hepatic cell apoptosis [45, 51]. NAS is being explored in developing treatments for age-associated cognitive decline and depression [52, 47] (Table 1).
DEG analysis and gene set enrichment assay (GSEA) of brain tissues with different levels of serotonin show that serotonin downregulates lipid metabolism-related genes [53]. Because lipids are the key components of neuronal structure and brain development [54], results from these DEG and GSEA analyses support the beneficial role of serotonin in neurons. Contrary to the neuroprotective effects of serotonin, NAS, and melatonin, some Trp metabolites, e.g., 5-hydroxyindoleacetic acid, 5-methoxytryptophol, and 5-hydroxytryptophol, cause neurotoxicity [35].
Roles of tryptamine and indole-3-propionic acid (IPA)
Decarboxylation and transamination reactions of Trp afford tryptamine and indole-3-propionic acid (IPA), respectively. IPA also can be transformed into KYNA [55, 56]. Tryptamine is an inhibitor of β-secretase, known as the β-site APP cleaving enzyme 1 (BACE1). The BACE1 enzyme is essential for the generation of β-amyloid. BACE1 catalyzes the initial cleavage of the APP to generate Aβ, which is the starting point of Aβ plaque formation [56]. Gut bacteria produce IPA from Trp. More specifically, IPA production completely depends on the presence of gut microflora and could be established by colonization with the bacterium Clostridium sporogenes [57]. The introduction of IPA producing enteric bacteria into the gastrointestinal tract is sufficient to introduce IPA into the host bloodstream [57]. Following absorption from the intestine and distribution to the brain, IPA offers a neuroprotective effect [58]. As a potent neuroprotective antioxidant and an inhibitor for Aβ fibril formation, IPA is prime for evaluation as a potential therapeutic for AD [59]. In humans, IPA treatment reduces Aβ accumulation levels and lessens oxidative stress [55]. IPA treatment also inhibits NF-κB signaling, reduces the levels of proinflammatory cytokines TNFα, IL-1β, and IL-6 in response to endotoxin in macrophages [60], and decreases the activation of glia and astrocytes. Taken together, Trp metabolic pathways are promising AD therapeutic targets.
Microglia and astrocyte crosstalk in AD
Sketch of microglia and astrocyte
Microglia and astrocyte are the major immune response cells in the CNS. Normally, they are responsible for improving tissue repair, maintaining homeostasis, and mitigating proinflammatory activity and tissue destruction [69]. Microglia, which serve as the primary immune cells of the CNS, regulate the innate immune functions of astrocytes, and determine the roles of reactive astrocytes ranging from neuroprotective to neurotoxic [70]. The Aβ-induced microglia increase astrocyte reactivity to express associated activated genes, such as C3, and Ggta1. Interestingly, NLY01, a glucagon-like peptide-1 receptors (GLP-1R) agonist, inhibits the microglia-induced activation of astrocyte. NLY01 can block Aβ-induced activation of microglia through GLP-1R and blocks the reactivity of astrocytes as well as reduces the accumulation of Aβ and preserves neurons in AD mouse models [71]. Thus, providing an example of functional interactions between microglia and astrocytes in relation to targeting these glial cells for possible AD therapy.
Not only microglia regulate the functions of astrocytes, but also astrocytes in turn regulate microglia functions and phenotypes. The IL-3 originating from the astrocyte triggers the IL-3R of microglia that results in increased pathological debris clearance by the microglia [72]. The reactive astrocytes also produce C3 via the NF-kB pathway induced by Aβ. The receptor, C3R, abundantly exists in the microglia and binding to C3R promotes microglia phagocytosis. Interestingly, both astrocytes and microglia secret C3, which affords a more anfractuous process [73].
Given the significant role of glia in the process of AD, astrocytes and microglia are potential therapeutic targets. For example, non-steroidal anti-inflammatory drugs (NSAIDs), such as indomethacin and flurbiprofen, reduce the pathological debris products in AD, in which the NSAIDs inhibit the enhancement of proinflammatory response via altering the activation of glia [74]. Additionally, NLY01 acts as a GLP-1R agonist to relieve the neurodegenerative disorders, including AD and PD. NLY01 blocks the debris-induced microglia activation and reduces the accumulation of reactive astrocyte [71].
Neuroinflammatory crosstalk of glia in AD
Activation of glia
Both microglia and astrocytes are major contributors of homeostasis in the CNS. The activation of microglia and astrocytes is the fundamental pathology in AD. However, dysregulation of normal function leads to cognitive deficits and neurodegeneration in pathogenesis of AD. Microglia can switch from the quiescent state to the disease-associated phenotype based on the analyses by both single-cell RNA sequencing (scRNA-Seq), as well as single-nucleus RNA sequencing, revealing subpopulations with distinct molecular and functional characteristics. Just like macrophage, the phenotypes of microglia also appear in different forms and are more complicated. For example, the disease-associated microglia (DAM) deliver a signal to recruit macrophages in the brain in neurological diseases, such as AD. In this disease, the DAM alters the gene expression of both homeostatic markers as well crucial pathological genes of cytokines and phagocytosis factors [75]. For instance, the gene expression of proliferation marker Ki67 changes in the 5xFAD model. In another example, TREM2 is a very important factor for microglial proliferation [76]. Furthermore, the microglia in AD can be separated into 4 types, DAM, IFN-I, MHC and proliferating types [77, 78]. Besides AD, microglia are also altered in other diseases or injuries of the CNS, such as Multiple sclerosis and traumatic brain injury. Additionally, a variety of microglial phenotypes exists in different brain regions as determined by a differential expression of genes. For example, CD68 and HLA-DR are highly expressed in white matter, while MHC-II is abundantly expressed in grey matter [79, 80]. Further, cytokines (e.g., IL-1, -12, -17, -18 and -23), chemokines (e.g., CCL-2, -12 and CXCL10) and other inflammatory molecules such as nitric oxide, reactive oxygen species, prostaglandin E2 and matrix metallopeptidase-9 and -10 are also significantly elevate in microglia during the neurotoxic phase. In contrast, alternatively activated subsets release anti-inflammatory cytokines (e.g., IL-4, -10 and -13), chemokines (e.g., CCL-17, -22, -24), and other neuroprotective molecules such as brain-derived neurotrophic factor, vascular endothelial growth factor (VEGF), insulin-like growth factor-1, nerve growth factor, and suppressor of cytokine signaling 3 when the microglia are in repairing and neuroprotective phase. There are two classes of astrocytes (i) protoplasmic astrocytes (which possess short projections, envelops synapses and present in the gray matter) and (ii) fibrous astrocytes (consisting of long projections having contact with Nodes of Ranvier located in the white matter). Like microglia, naïve/resting astrocytes also undergo reactive astrogliosis during inflammation and change their phenotype into three different forms depending on the pathological and recovering stages. The first type of astrocytes can switch to reactive astrocytes and become neurotoxic in neuroinflammatory conditions, causing impairment of synapses with increased level of C3. The second type of astrocytes (A2 reactive) adopt a neuroprotective status by repairing synapses with increased expression of S100 after ischemia. The third type belongs to the scar-forming astrocytes, which exhibit the characteristics of hypertrophy, cell proliferation with an overexpression of glial fibrillary acidic protein (GFAP) [81, 82]. However, the polarization and function of microglia are not categorical. The different phenotypes of microglia express a spectrum of activation patterns, not only absolute subtypes. Furthermore, the phenotypes of microglia are not stable, one phenotype can transform into another phenotype under the alteration of the surrounding microenvironment [81]. The activation and phenotypic transformation of astrocyte is microglia dependent. Released TNFα, IL-1β, and Complement component C1q (C1q) activate astrocytes through the downregulation of CXCR7 receptor and the PI3K-AKT pathway. In contrast, upregulating CXCR7 receptor by its agonist AMD3100 switches microglia and astrocytes into neuroprotective subtypes [82]. The gene transcriptome analyses of astrocytes show that 57 genes (e.g., C, guanine nucleotide binding protein 2; Guanylate Binding Protein 2, histocompatibility 2, D region locus 1; H2-D1 and Serping-1) are associated with neurotoxic function and 110 genes (e.g., S100 calcium-binding protein 10; pentraxin 3; sphingosine-1-phosphate receptor-3; and Tweak) are related to neuroprotective function of astrocytes [83]. In inflammatory conditions, the reactive astrocytes promote the activation of resting or quiescent microglia through the release of proinflammatory cytokines such as TNF-α changing their phenotype to proinflammatory microglia (Fig. 3A). In succession, these proinflammatory microglia activate resting astrocytes into reactive astrocytes that is modulated by the release of TNF-α and VEGF, leading to neuroinflammation. However, the production of anti-inflammatory cytokine TGF-α in the tissue microenvironment attenuates reactive astrocytes-mediated neuroinflammation (Fig. 3B). Normally, microglia maintain homeostatic environment in the CNS. The change in homeostatic status of the microglia solely depends on the stimuli, which can dictate microglia to either activate themselves to become proinflammatory type or switch to anti-inflammatory microglia and release TGF-β. The resting astrocytes, in response to the release of TGF-β change their state to anti-inflammatory astrocytes. In turn, these astrocytes produce IL-10 to reverse the proinflammatory state of the microglia. This crosstalk between microglia and astrocytes on the one hand initiates neuroinflammation and on the other hand resolves the neuroinflammatory response in the brain (Fig. 3C). Furthermore, the subtypes of glia depend on the glia situation in different physiology and pathology. For example, microglia express different features under different physiological or stimulated station, such as “satellite” microglia, Keratan sulfate proteoglycan (KSPG) microglia [84]. The subtypes of astrocytes express similar complexity because they play various functions in CNS). Astrocytes also vary in different regions of CNS. Results from a scRNA-Seq of large-scale integration analysis reveal that the different astroglia clusters distribute in different regions of the brain, such as cortex, hippocampus and midbrain. And the phenotypes of astrocytes also alter in different diseases, such as AD and PD [85]. The activation of AHR inhibits TNF-α expression in astrocytes, which indicates that AHR might be involved in astrocyte-mediated microglia polarization [86]. Acute hypoxia induces resting microglia polarization towards the proinflammatory phenotype through the activation of the NF-κB pathway, in which proinflammatory factors, e.g., TNF-α, IL-6, and CCL2, are increased, and antiinflammatory substances, e.g., IL-4 and IL-10, are decreased [87] (Fig. 2). The interaction between Trp and AHR increases TGF-α levels and decreases VEGF-β levels, via regulating the microglia NF-κB pathway, which is a primary inflammatory pathway [88].
Figure 3.
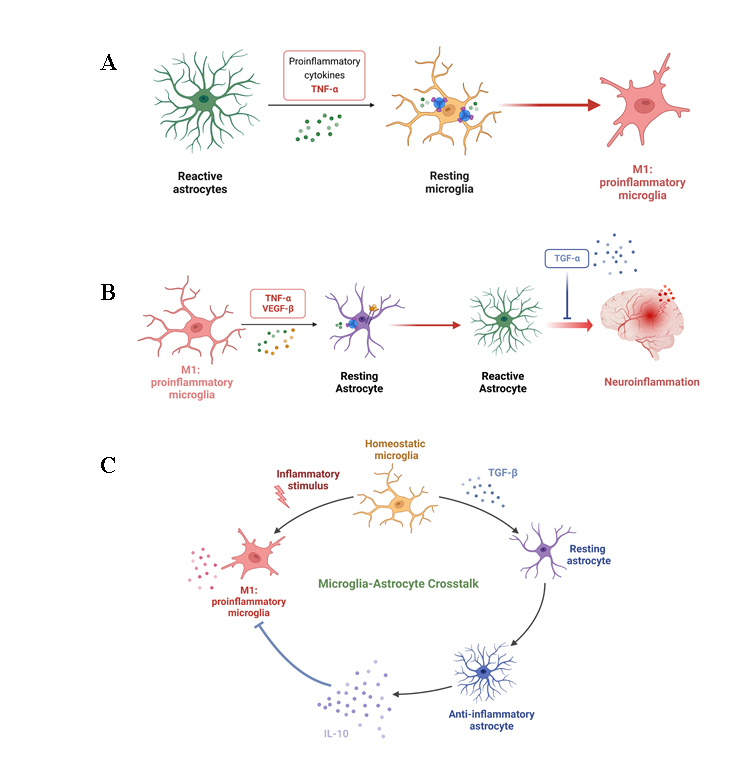
The interaction between microglia and astrocyte. (A) The reactive astrocyte promotes the activation of resting microglia via TNF-α, which switches them to proinflammatory microglia. (B) M1-type microglia induce the activation of resting astrocytes to convert them into reactive astrocyte, leading to neuroinflammation. In contrast, secretion of TGF-β inhibits neuroinflammatory response in the brain. The proinflammatory microglia induce neuroinflammation of astrocyte. (C) The circularity of interaction and crosstalk between microglia and astrocyte. This figure is created with BioRender.com.
The NF-κB pathway activation also upregulates TGF-α, which in turn promotes the glutamate transporter-1 production in astrocytes [89]. The reactivity of astrocyte is recognized as the one of basic pathological alterations of AD [90]. Glia cells, especially microglia and astrocyte, play a central role in orchestrating the neuroinflammation processes and neurodegeneration. Neuroinflammation serves as a bridge connecting microglia and astrocytes. Along these lines, dietary Trp metabolites, which are produced by microbiota in the gut, cross the blood brain barrier to activate microglia, which produces TGFα and VEGF-β by cross talking with astrocytes to change its transcriptional activity through AHR, subsequently modulating neuroinflammation [88]. The effects of Trp and its metabolites on phenotypic changes of microglia and astrocytes, and their crosstalk will clarify the roles of both glial cells in neurotoxicity and neuroprotection.
Role of VEGF
In astrocytes, VEGF-β stimulation upregulates proinflammatory factors (Fig. 3), e.g., TNF-α, IL-6, and nitric oxide synthase 2 (NOS2), while TGF-α upregulates anti-inflammatory factors such as IL-10 [88]. Astrocytes express VEGF receptor Flk-1, which responds to the VEGF stimulation to alter the number and shape of astrocytic branching and the distance between astrocytes [91]. VEGF stimulation of astrocytes also promotes BBB infiltration, which allows the inflammatory factors to cross the BBB and enter the CNS [92]. VEGF-β stimulation also increases astrocytic NOS2 expression by blocking AHR [86, 88]. As one of the iNOS, NOS2 produces nitric oxide (NO) to regulate inflammation and development. Blocking astrocytic NOS2 inhibits cell development and differentiation [93]. LPS/IFN-γ treatment upregulates NOS2, like other proinflammatory cytokine genes (CCL2, IL-6), and LPS and IFN-γ are the typical stimuli leading to the microglia/macrophage proinflammatory phenotype [94]. iNOS is usually used as the marker for the proinflammatory phenotype, which is highly expressed in AD [94, 95]. Upon VEGF stimulation, the activated astrocytes produce proinflammatory cytokines IL-1β, TNF-α, and IL-6, which induce neurotoxicity in neurodegeneration [86, 88, 96]. The presence of Aβ elevates astrocytic inflammatory mediators (e.g., TNF-α, IL-1β, iNOS, and cyclooxygenase 2 (COX-2)) [97]. Then, these mediators enhance Aβ accumulation or Tau protein deposits which leads to increased neurotoxicity due to the reduction of glia phagocytosis [98-100]. For example, TNF-α enhances Aβ accumulation and the activation of the NF-κB pathways, which result in iNOS up-regulation and oxidative stress [101].
Role of transforming growth factor-β (TGF-β)
Unlike VEGF, the TGF-β signal usually expresses anti-inflammatory action in astrocytes. Upregulation of the astrocytic TGF-β pathway inhibits immune cell accumulation, cytokine, and chemokine production [102]. As an anti-inflammatary factor to prevent AD, TGF-β expression level increases with age [103]. Astrocytic TGF-β inhibits the binding of Aβ to the synapses to maintain the synapse density. In AD, an increase in Aβ and loss of synapses are the primary hall markers [104]. Microglia originated-TGF-β stimulates astrocytes to produce IL-10, which is widely recognized as a protective factor to relieve inflammation [88, 105] (Fig. 3). Anti-inflammatory phenotype cells, such as microglia/macrophage, produce key anti-inflammatory cytokines (e.g., IL-10, IL-4, and IL-13) to relieve neuroinflammation [96, 105]. IL-4 and IL-13 protect against LPS-induced neuron injury by suppressing the production of proinflammatory cytokines (e.g., IL-8, IL-6, and TNF-α) and by reducing NO release [96].
Although TGF-β has been traditionally recognized as an anti-inflammatory cytokine, it also plays a proinflammatory role. For instance, TGF-β promotes the differentiation of Th17 cells, which in turn releases cytokines including IL-22 and IL-17 that are proinflammatory in nature. Hence, the TGF-β also indirectly acts as a proinflammatory cytokine, depending on the microenvironment [106].
Role of interleulin-10 (IL-10)
IL-10 is a significant mediator for the interactions between microglia and astrocyte in the CNS. Astrocytes express IL-10R1, and upon binding of IL-10 to IL-10R1 produce TGF-β. Then, the astrocytic TGF-β reduces the microglia activation [107]. The endothelial cells, as one important components of the BBB, stimulate microglia to produce IL-10 and iNOS under the deprivation of oxygen and glucose. In contrast, astrocyte, also a part of the BBB, does not alter the level of inflammatory cytokines [108]. Although IL-10 is an anti-inflammatory cytokine, some reports show that IL-10 might enhance pathological alteration in AD [109, 110]. As reported by Chakrabarty et al., IL-10 inhibits the microglia phagocytosis ability, which results in Aβ accumulation and exacerbation of memory loss [109]. Blocking the microglia IL-10/Stat3 signaling pathway increases the extent of Aβ phagocytosis, which suggests that inhibiting IL-10 might be a potential direction in developing AD therapies [110]. In contrast, an increase in IL-10 expression, inhibits the glia immune response and improves neurogenesis to alleviate memory disturbance without Aβ alteration [111] (Fig. 3).
Role of complement
The classically activated microglia produce cytokines (e.g., TNF-α, IL-1α, and C1q) and induce the A1 astrocyte form, which is a traditionally reactive phenotype. A decrease in A1 astrocytes activity causes a reduction in synaptogenesis and induction of neuronal and oligodendrocytes cell death [112-114]. A1 astrocytes are abundant in various human neurodegenerative diseases, including AD, HD, PD, and ALS [112]. Classically activated microglia produce early complement components (C1q, C1r, C1s, and C3); among them, C1q causes C3 cleavage [115]. LPS induces astrocyte polarization into the A1 phenotype, which typically expresses C3[112, 116]. The A1 astrocytes exhibit high C3 expression levels, and high C3 levels are present in some neurodegenerative diseases (e.g., PD and AD). The C3 positive astrocytes are near the Cluster of Differentiation 68 (CD68) positive microglia/macrophages. Because CD68 is a typical marker of activated microglia/macrophages [112, 117], it is suggested that activated microglia produce proinflammatory cytokines and complement components to promote the formation of the A1 type of astrocytes. Further, the interactions between C3 from A1 astrocytes and C3a receptor (C3aR) in microglia result in the loss of microglia phagocytosis activity [73, 112, 118]. Clarke et al. report that A1 astrocytic gene expression also alters with age. Complement C3, complement C4B, and cytokine CXCL10 increase with age, especially in the hippocampus and striatum [118]. A1 astrocytes lose their typical astrocytic protective effects and, at the same time, promote the immune response. Combining these factors leads to neurotoxicity [112-114, 118]. Aβ induces microglia to produce IL-1α, TNF-α, and C1q. The Aβ induced microglia conditioned medium induces astrocyte polarization towards the A1 phenotype. Therefore, inhibiting microglial inflammatory activity is one of the routes to reduce the damaging effects due to A1 type astrocytes. For instance, NLY01, a glucagon-like peptide-1 receptor, reduces the production of IL-1α, TNF-α, and C1q in microglia, which results in the reduction of A1 astrocytes. NLY01 protects dopaminergic neurons and improves the behavior in a PD animal model through the microglia-astrocyte interactions to modulate astrocyte polarization [113]. Additionally, milk fat globule epidermal growth factor 8 inhibits the A1/A2 alteration through NF-κB inhibition and PI3K-Akt activation [114].
Interactions between microglia and astrocyte also play a significant role in physiological and pathological processes in the CNS. Astrocyte and microglia crosstalk, as demonstrated by Lian et al., depends on the complement pathway in an AD mouse model [73]. C3, a protein of the immune system, is closely connected to cellular signal mediators between astrocytes and other cells in the CNS. Astrocytes and microglia are the primary sources of C3 and C3aR [73]. High levels of C3 expression are present in the brain and cerebrospinal fluid (CSF) of AD patients [119], and high levels of C3 affect the microglial inflammatory and phagocytotic functions [73]. IFN-γ treatment increases microglial C3 expression [115]. In Aβ-treated astrocytes and aged AD APP/PS1 mouse model, the astrocytic C3 level elevates, suggesting that the astrocytic complement system is involved in amyloid pathology in AD [73, 120]. Indeed, associated with Aβ formation, the activated astrocytes express high levels of C3 [120, 121], and exposure to Aβ triggers astroglial C3 release [119, 120]. As stimuli, Aβ increases the microglial immune response and reduces the ability of the microglia to clear the Aβ in AD [122].
C3 and its receptor C3aR are present within and around Aβ cerebral plaques and contribute to the phagocytosis and clearance of fibrillar Aβ by microglia in AD. C3 activates the microglia C3aR to mediate β-amyloid pathology and neuroinflammation [73, 115]. Upon triggering by C3, neurons also express C3aR [73, 120]. Interactions between neuronal C3aR and astrocytic C3 cause an inflammatory response leading to synaptic density loss and dendritic morphological alternation [120]. The C3-C3aR pathway activation reduces Aβ clearance, while the treatment with a C3aR antagonist SB290157 increases the Aβ clearance in C3 treated microglia [73]. In AD mice, the astrocytic C3-C3aR pathway results in Aβ accumulation via the NF-κB pathway [73], while astrocytes elevate C3 via the NF-κB pathway in AD mice. The NF-κB pathway produces various functional downstream factors, such as inflammatory factors and oxidative molecules [123, 124]. IκBα elimination causes C3 up-regulation [73, 120].
Role of the microbiota-gut-brain axis in AD
Disorders of gut bacterial in AD
Alteration of the bacterial composition is one of the key features of the gut-brain axis (GBA) dysbiosis. The subsequent gut barrier disruption and systemic inflammation trigger neuroinflammation, BBB impairment, neural damage, and neurodegeneration [83]. Analyses of samples of early- and late-onset AD, using next-generation sequencing, show that self-antigen load may cause pathogenesis of AD because of the neuroinflammation [125]. For example, activation of HMGB1 signaling pathway is a causative factor of neuroinflammation in AD progression [126]. Alcohol-mediated neuroinflammation is also associated with AD and gut dysbiosis [127]. Substantial accumulating evidence supports the relationship between the MGBA and AD pathology, which includes the typical pathological alteration in the formation of extracellular Aβ plaques and intracellular neurofibrillary tangles. Further, in AD mice, the presence of the beneficial bacterial family Bifidobacteriaceae decreases and the pathogenic bacterial family Proteobacteria increases, at the phylum level, within the microbiota [83]. While in AD patients, negative correlations exist between bacterial diversities and concentrations of CSF Aβ biomarkers [128].
Table 2.
Different Spectrum of Microbiota and Microglia-Astrocyte Cross Talk in Alzheimer’s Disease.
| Microbiota | Pathology/Disorder | Mediators | Function/Animal Model | Ref. |
|---|---|---|---|---|
| Decreased Firmicutes, Verrucomicrobia, Proteobacteria and Actinobacteria Increased Bacteriodetes and Tenericutes | AD-like pathology | Indirect microglia-dependent actions | APPSPS1 (8 months old) mice | [129] |
| Lactobacillus johnsonii Muribaculum | Aggravation of neuroinflammatory response | Activated microglia and reduced motor activity | 3xTg-AD (FMT-AD) mice | [130] |
| Lactobacillus reuteri | Neuroinflammation | Ahr signaling in astrocytes and microglia | GFAP-Cre, and Ahrfl/fl mice | [86] |
| Toxoplasma gondii | Amelioration of β-amyloidosis | Ly6Chi Monocytes | AD mice | [131] |
| Sutterella (Betaproteobacteria) | AD pathology | Inflammation | Tg-APP/PS1 Transgenic mice (6 months old) | [132] |
| Erysipelotrichaceae | Tg-APP/PS1 Transgenic Mice (24 months old) | |||
| Bacteroides fragilis | Plaque deposition | Bacteroides colonization | Tg 2576 transgenic mice | [133] |
| Helicobacter pylori | Aβ accumulation | Outer membrane vesicles | AppNL-G-F AD mice | [134] |
| Porphyromonas gingivalis and Paenalcaligenes hominis | AD-like pathology | Outer membrane vesicles | C57BL/6J mice (14 months old) | [135] |
| Paenalcaligenes hominis | Cognitive function-impaired disorders, such as AD | Extracellular vesicles through blood and vagus nerve | Germ -free C57BL/6J mice | [136] |
| Porphyromonas gingivalis, Fusobacterium nucleatum, Borrelia burgdorferi and Chlamydia pneumoniae | AD pathology | Multiple pathogens Aβ fibrils and Tau tangles | AD mice | [137] |
| Aerococcus, Jeotgalicoccus, Blautia, Pseudomonas, Clostridiale and Ruminococcaceae | Accumulation of Aβ plaque | Extracellular vesicles | Tg-APP/PS1 transgenic Mice | [138] |
| Lachnospiraceae and S24-7 | Reduction in Aβ deposition | Microglia and Astrocytes | Aged APPSWE/PS1ΔE9 mice | [139] |
| Akkermanisa ans S24-7 | AD pathology | Engraftment | APP/PS1 Transgenic Mice | [140] |
| Bifidobacterium bifidum Lactobacillus plantarum | Reduced Aβ accumulation and enhanced cognitive function | FMT | AD Rat model | [141] |
| Proteobacteria, Verrucomicrobia, Akkenrmansia, Desulfovibrio, Bacteroidetes and Alloprevotella | Alzheimer’s disease like-pathogenesis | FMT | APP/PS1 Transgenic Mice | [142] |
In humans, eating habits affect the composition of gut microbiota and the microbiome plays an important role in aging and age-dependent disease. A double-blind clinical study, with mild cognitive impairment (MCI, the stage between the expected decline in memory and thinking that happens with age and the more serious decline of dementia) patients, found that the composition of the gut microbiota changes with 2 types of diets: 1) the American Heart Association Diet (AHAD), which consists of a low-fat and higher-carbohydrate meal, and 2) the Modified Mediterranean-Ketogenic Diet (MMKD), which has slightly higher carbohydrate consumption compared to the original ketogenic diet to permit increased intake of vegetables and fruits while emphasizing fats and proteins derived from healthy sources such as olive oil and fish [143]. The AHAD increases Mollicutes while the MMKD increases the abundance of Enterobacteriaceae and Akkermansia but decreases Bifidobacterium and Lachnobacterium [143]. MMKD, associated with increased CSF Aβ 42 and decreased tau, appears to suppress cognitive decline and improve CSF biomarkers in adults who were at risk for AD [143].
Proteobacteria and LPS
Proteobacteria is the main proinflammatory bacteria, causing various inflammatory diseases, e.g., diarrhea [144]. Escherichia coli is one of the members of the Gamma Proteobacteria Enterobacteriaceae family, and the inflammatory agent LPS is one of the constituents of its outer wall [145]. In AD patients, the blood LPS levels are 3-fold higher than that of healthy individuals. In addition, in the brain specimens of AD patients, results from 16s rRNA sequencing reveal that over 70% of bacteria are LPS+ α-proteobacteria [146]. In Wistar rats, LPS impairs spatial memory and causes hippocampal neuronal apoptosis [147]. CD14, an LPS receptor, enhances the phagocytosis of Aβ in microglia. Furthermore, in AD patients but not in control human subjects, a pronounced CD14 immunoreactivity on parenchymal microglia spatially correlates to characteristic AD lesion sites [148]. In AD pathology, LPS plays a bilateral role in having inflammatory effects while at the same time initiating the pathological process via clearing amyloid deposits. Injection of LPS into the brains of APP transgenic mice leads to the clearance of some pre-existing amyloid deposits. In a transgenic AD model, which contains the P301L mutation along with elevated levels of tau accumulation and production of Ser199/202.
TLRs are expressed on cell surface and in the intracellular space (e.g., endosomes) by many immune cells, including monocytes and macrophages, inside and outside the CNS. Within the CNS, in a similar way like macrophages, microglia also express TLRs, however, the expression of TLRs in astrocytes and their participation in the CNS inflammatory responses is still in debate. It is generally considered that astrocytes do not express TLRs. However, the expression of TLRs in astrocytes occurs following the depletion of microglia [149]. Apparently, in some cases, astrocytes do express TLRs but their expression after binding to ligands (e.g., LPS) does not trigger downstream signaling cascade, which usually involves key genes such as MyD88, NFB, TRIF, IRF3. In contrast, microglia express TLRs in response to LPS, DAMPs, and PAMPs as well as by IL-1α, TNFα, IL-6 and other proinflammatory cytokines, which indirectly induce astrocytes to transform to reactive or neurotoxic astrocytes. Most importantly, the crosstalk between the microglia and astrocytes is essential for inflammatory responses, which transpires through TLRs-mediated mechanisms.
LPS injection into the frontal cortex and hippocampus increases the expression levels of phosphorylation at Ser199/202 and Ser396 of tau protein, supporting the roles of proinflammatory LPS stimuli in promoting tau phosphorylation [150]. In contrast to the association between some proinflammatory bacteria and AD pathology, other gut bacteria might be protective. For example, probiotics are beneficial in protecting neurons against neurodegeneration. In AD mice, consumption of Bifidobacterium, a probiotic microorganism, improves the Y maze test outcomes, suggesting their potential roles in improving behavior and physiological processes of AD. The decrease in hippocampal inflammation and Aβ accumulation associate with the inhibition of cognitive AD impairment [151].
Short-chain fatty acids (SCFAs)
Small molecule SCFs in ones' diet modulate the composition of the gut flora in AD patients. A negative correlation exists between the concentrations of fecal SCFs (propionate and butyrate) and the CSF Aβ-42 concentration in MCI patients [143]. In individuals partaking in the MMKD diet, propionate and butyrate levels elevate, while lactate and acetate reduce. However, in patients with the AHAD diet, acetate and propionate levels increase, along with butyrate concentration decreasing [143]. SCFAs inhibit the Aβ1-40 interactions, which is key to the formation of Aβ1-40 dimers and trimers [152]. In fact, Aβ1-40 or Aβ1-42 peptide interactions are directly observed in vitro [153]. SCFAs also regulate inflammation, e.g., by regulating leukocytes and endothelial cells through the G protein coupled-receptor and histone deacetylase signaling pathways, SCFAs regulate the secretion of cytokines such as IL-10, IL-2, and TNF-α, as well as the leukocyte migrations [154]. In AD patients, leukocyte activation and the increase of proinflammatory factors are two key AD inflammatory events. Additionally, infrared spectroscopic analysis of peripheral mononuclear leukocytes in mild and moderate AD patients are distinguished from healthy controls [155].
Probiotics
Probiotics act on various benefits for many host organs, which is one of the important compositions of the gut-brain axis. Probiotics relieve some pathological processes in AD. Prebiotics as well as probiotics are used as preventive medicines to improve cognitive decline in AD, which can be caused by the alteration of commensal microbiota in the gut and dysfunction in gut-brain axis. Consumption of prebiotics and probiotics (synbiotics) together promote beneficial bacterial community in the gut. Recent clinical trials demonstrate the positive effects of probiotics for AD with a slowing down of disease progression [141]. For example, in a randomized controlled clinical trial, co-supplementation of a probiotic with selenium, in 79 patients, improves cognitive as well as metabolic functions. In other studies, preclinical and clinical, the results demonstrate that Bacillus or Bifidobacterium breve strain A1 metabolizes acetate, butyric, isovaleric, propionic, and valeric acids and promotes cognitive functions and inhibits deposition of Aβ via microglia and astrocytes mediated decreased inflammation in AD mice and AD patients [156]. Probiotics reduce oxidative stress and proinflammatory processes in AD rats [157]. The probiotics also restore the disorder of metabolism in AD mice. Oral probiotics enhance glucose uptake by affecting some associated metabolic molecules, such as glucose transporters, in 3xTg-AD [158]. The probiotics not only reduce inflammation and debris accumulation, but also decrease gut inflammation and gut permeability [159].
Xenobiotic receptors
Various studies report the multifaceted roles of the xenobiotic receptors, which include the AHR, pregnane X receptor (PXR), and constitutive androstane nuclear receptor (CAR), in the MGBA. Firstly, AHR acts as a mediator in the bi-directional communication between the brain and gut microbiota [160]. In the AD mice model, AHR activates occludin and claudin 1 and 4, thereby enhancing and restoring BBB integrity [161]. Secondly, in activated astrocytes, AHR suppresses neuroinflammation via dietary supplementation with the microbial metabolites of Trp (AHR ligands) [86]. PXR is a nuclear transcription factor referred to as the master maintainer of the barrier integrity in both the intestine and brain. While PXR-deficient mice exhibit “leaky” intestine symptoms. Ligand-activated PXR prevents leaky gut and maintains the intestinal epithelial barrier by stimulating the synthesis of junctional proteins [162]. Moreover, PXR knockout mice show decreased expression of tight junction protein ZO-1 in the cortex and cortical microvessels [163]. Activation of PXR in vivo tightens the BBB, which is mediated by microbiota derived-ligands. Thirdly, increasing evidence provides a basis to support the potential of CAR-mediated signaling between the gut and brain. Activation of CAR promotes mucosal healing by altering the natural milieu of the microbiota. Notably, CAR affects structural factors in the brain and the behavior of mice as CAR-null mice show impairment in recognition memory accompanied by neuronal and structural defects in the hippocampus and cortical area. Moreover, tight junction protein ZO-1 expression reduces in the cortical and hippocampal microvessels of the mice [164].
Trp controls the microglia-astrocyte crosstalk in AD - involvement of the microbiota-gut-brain axis
People with C3 deficiency are susceptible to bacterial infection [165]. Like other immune substances, Trp metabolism affects the complement system, in which dietary Trp levels modulate the expression of C3 [73].
For Trp metabolites, signaling through AHR constitutes the interface of MGBA. Ligands derived from microbial Trp metabolism play critical roles in intestinal homeostasis [166]. The gut flora strongly influences Trp metabolism. For instance, tryptophanase from Escherichia coli generates indole from Trp [167]. Several bacteria species convert Trp into indole and indole derivatives, including IPA, tryptamine, 3-methylindole (also known as skatol), indole-3-aldehyde (I3A), indoleacetic acid, indoleacrylic acid, indoleethanol, indolelactic acid [168]. Some of these derivatives are AHR ligands. For instance, Lactobacillus species metabolize Trp into I3A, which activates AHR in intestinal immune cells and induces cytochromes P450 1A1 and P450 1B1 expression [169]. Indole itself acts as a ligand for AHR and serotonin is an AHR ligand. Indole decreases mucosal inflammation and injury in animals [170].
As a dioxin-binding protein, AHR is initially recognized as a regulator of dioxin and AHR causes immunosuppression after binding with dioxin. As a nuclear protein, AHR is a highly conserved transcription factor that affects aging and age-associated diseases (e.g., AD) by regulating many target genes [171]. KYN is one of the endogenous ligands of the AHR. Some natural ligands like KYN and KYNA (an AHR agonist with higher titer) also trigger AHR to affect differentiation and immunity [172]. For example, Benzo(a)pyrene, one of the AHR ligands, causes the alteration of the levels of neurotransmitters and leads to apoptosis as part of neurodegeneration [173]. AHR is involved in innate and adaptive immune responses [174].
AHR’s involvement in the immune response is also seen in its Connectivity Map, or CMap, using L1000 [175]. CMap is a resource that assists in identifying relationships between diseases, genes, and therapeutics. The top three CMap gene connections of AHR are shown in Table 3, and are PI4KIIB, APOBEC3, and CHMP2A. Any connection score over 95 means the connection is strong. PI4KIIB is implicated in the activation of early T cells via the CD3-T Cell Receptor signaling pathway [176]. The APOBEC3 gene is an interferon-stimulated gene whose expression increases in response to various stimuli, and thus the activation of innate immune responses that occurs upon infection, with many different viruses, could lead to increased APOBEC3 activity [177] (Fig. 4). Silencing of ESCRT-III-protein CHMP2A results in spontaneous necroptosis. As necroptosis actively shapes the immune response, ESCRT-III indirectly controls the immunogenicity of necroptotic dying cells [178].
Table 3.
Genes connected with AHR found using L1000.
| Gene Name | Connection Score | Description |
|---|---|---|
| PI4k2B | 98.45 | Phosphatidylinositol kinases |
| APOBEC3H | 96.85 | Apolipoprotein B mRNA editing enzymes |
| CHMP2A | 95.78 | Charged multivesicular body proteins |
Figure 4.
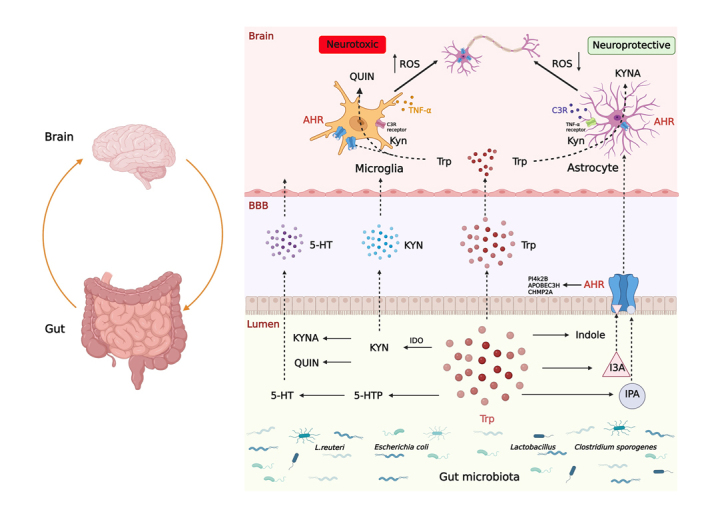
Tryptophan participates in microglia and astrocyte crosstalk in AD with the involvement of MGBA. The tryptophan (Trp) metabolism regulates the interaction between microglia and astrocyte. Molecules, such as C3, TNF-α, control the interaction. The C3 originates from microglia-primed astrocytes and the TNF-α is released from astrocytes-primed microglia. The interaction of microglia and astrocyte affects their functions (e.g., phagocytosis and generation of ROS) that are essential pathological processes in AD. The metabolism of gut flora produces small molecules, such as Trp. The metabolites of Trp and Trp affect the interaction between microglia and astrocyte via ROS pathway. Trp regulates three major metabolic pathways (e.g., 5-HT, KYN, and AHR ligand pathways). Alteration in gut microbiota composition, Trp synthesis and its related metabolites lead to change in the pattern of microbiome within the gut. In the brain, Trp acts on the AHR to stimulate the production of QUIN and KYNA. The activation of microglia and astrocyte through AHR increases and decreases ROS through the release of QUIN and KYNA, resulting in neurotoxic and neuroprotective effects, respectively. The top three CMap gene connections of AHR are PI4KIIB, APOBEC3, and CHMP2A. Trp controls the microglia-astrocyte crosstalk in AD with the involvement of MGBA mechanisms. This figure was created with BioRender.com.
AHR also plays a role in the glia immune response in both proinflammatory and anti-inflammatory processes. A Trp deficient diet induces gut microbiota dysbiosis and increases systemic inflammation [179]. Trp deficiency or IDO elevation activates the NF-κB pathway, which results in oxidative damage and proinflammatory responses [180, 181]. The basic function of microglia in AD is to clear pathological debris and control the inflammatory response. The increase of proinflammatory signaling inhibits the microglia’s ability to clear Aβ, while AHR might elevate the microglia’s phagocytosis capacity via inhibition of the microglia's proinflammatory responses [182, 183]. Microglial AHR activation, as reported by Lee et al., exerts bi-directional effects: the regulation of LPS-induced microglia inflammation depends on the availability of external AHR ligands [184]. In the absence of AHR ligands, LPS activates AHR via the MER1/2 pathway, and AHR knockout inhibits the microglia response, leading to proinflammation and neurotoxicity. As a result, in the absence of AHR ligands, the activation of microglial AHR exerts proinflammatory effects. On the contrary, in the presence of AHR ligands, the activation of AHR attenuates the LPS-induced microglia’s proinflammatory function by reducing LPS-induced AHR binding to the iNOS and TNF-α [184].
LPS and IFN-γ increase the AHR recruitment, while AHR also inhibits the NF-κB to reduce proinflammatory factors (eCCL2, Csf2, and NOS2) [86]. Astrocytic AHR limits CNS inflammation, as demonstrated in AHRfl/fl mice. The AHR deficient astrocytes produce more proinflammatory substances, such as CCL2 and IL-6, which promote the inflammatory response and immune cell recruitment [86]. These chemokines and cytokines are essential signaling factors in AD. Astrocytes also express these molecules and their receptors, which trigger the inflammatory chain reaction [100, 182].
Trp also affects the microglia-astrocyte crosstalk. Trp metabolites, such as IPA and I3A, activate microglia, which then initiate signaling through the AHR in astrocytes (Fig. 4). The dietary Trp also induces microglia to produce TGF-α and VEGF-β, and finally modulates the astrocyte transcriptional program through the AHR-mediated signaling [88]. The gut microbacterium L. reuteri degrades Trp to indole, which is then converted into many other derivatives, including IPA, I3A, and indole-3-sulfate. These indole derivatives cross through the BBB and interact with AHR in the CNS [86, 185].
Inhibiting either Trp synthesis or its uptake causes a decrease in Trp concentrations in the brain, which in turn results in emotional and behavioral changes [34]. Both neurons and glia express AHR, and the AHR ligands regulate AHR levels [86, 186]. AHR participates in various physiological and pathological processes that are highly relevant to intestinal homeostasis and CNS diseases, including AD. AHR serves as a critical node in MGBA through Trp metabolism in microbiota and related brain signaling by Trp metabolites [166].
Activation of AHR in neurons alters the caspase-3 activation, ROS expression, and LDH release [186]. As part of the growing interest in MGBA, AHR activation is also a potential therapeutic target to regulate the CNS inflammation [166]. Trp metabolites activate microglia, which then modulate astrocytes through AHR signaling to regulate the CNS inflammation [187]. The deficiency of astrocytic AHR leads to the expression of proinflammatory substances (e.g., IL-6, CXCL10, and CCL2 [86]. Microglial AHR signaling also regulates the immune response in the CNS [184]. Deletion of AHR increases microglial activation and affords increased levels of neuroinflammation and neurodegeneration [188, 189]. In microglial AHR deletion mice, astrocytes increase their expression of proinflammation and neurodegeneration genes (e.g., IL-1β, CCL2, and NOS2) [88] highlighting microglia and astrocyte crosstalk.
In summary, some microbiota species (e.g., Lactobacillus ssp., Bifidobacterium ssp., and Peptrostreptococcus Russellii) produce some AHR ligands (e.g., Trp derivative IPA and indole) to activate AHR [190]. The microglial AHR activation exerts proinflammatory effects and causes increased neuroinflammation and neurodegeneration. In contrast, astrocytic AHR activation limits CNS inflammation. In AD animal models, the activation of AHR in neurons alters the levels of apoptosis and oxidative stress, which alleviate cognitive deficits. Thus, AHR activation may offer solutions to developing AD therapies.
MGBA is a bidirectional link between the CNS and the enteric nervous system (ENS). ENS is a complex neuronal network. The gut saprophytic microflora can communicate with both the ENS and the CNS, e.g., the neurotransmitter signaling by Trp metabolites through MGBA. Serotonin is a neurotransmitter in both the CNS and the ENS. Both the brain and the gut produce serotonin. Some bacteria found in the gut microbiota synthesize serotonin [191]. Mucosal serotonin incites intestinal inflammation, and the reduction of serotonin and KYNA levels in the intestine is related to intestinal homeostasis disturbances [192].
Summary
Trp plays a vital role in regulating the GBA and is involved in neurodegenerative diseases such as AD, PD [193, 194], HD [195] as well as dementia [196]. Glia, especially the interaction between different glia, influence the pathological process of neurodegeneration. In this review, focusing on AD, we discuss the Trp metabolism and its roles in the glia pathology of AD, where Trp and its metabolites enter the CNS and modulate astrocytes-microglia crosstalk (Fig. 4). The importance of communication between glia and neuron is recognized. However, the connections between Trp and neurodegeneration remain elusive. Potential future research directions include a more comprehensive understanding of the roles of Trp and Trp metabolites in AD and aging biology; the AHR activation; targeting Trp metabolism and AHR in developing new therapies for AD; and the development of probiotics by modulating Trp metabolism. Continued preclinical and clinical studies will shed light on the crosstalk between microglia and astrocyte and MGBA. Such studies will elucidate the important role(s) of Trp as well as rationales for new therapeutic targets and strategies to treat AD, which afflicts millions of individuals worldwide.
Acknowledgments
This work was supported by the grants from the Brigham and Women’s Hospital (BRI Fund to Sustain Research Excellence to X.W.), the National Institute on Alcohol Abuse and Alcoholism (R21AA030087 to X.W.) and (Multi-PI R21AA029925 to M.A.S.K/S.L.C), and the National Natural Science Foundation of China (82174512 to Q.W. and 82174511 to L.X.). The authors thank Ning Xie, Ning Zhang, and Chenyu Li for their valuable assistance.
Funding Statement
This work was supported by the grants from the Brigham and Women’s Hospital (BRI Fund to Sustain Research Excellence to X.W.), the National Institute on Alcohol Abuse and Alcoholism (R21AA030087 to X.W.) and (Multi-PI R21AA029925 to M.A.S.K/S.L.C), and the National Natural Science Foundation of China (82174512 to Q.W. and 82174511 to L.X.). The authors thank Ning Xie, Ning Zhang, and Chenyu Li for their valuable assistance.
Author Contributions
This review was written by L.X., Q.W., P.L., G.A., M.W.G., M.A.S.K, S. Z., A. Z., and X.W., and designed by X.W.; L.X., Q.W., and K.L. All authors edited the manuscript or contributed to the figures. All authors have read and agreed to the published version of the manuscript. During the preparation of this work the author(s) used no tools, services, or AI-assisted technologies in the writing process.
Conflicts of Interest
The authors declare no conflict of interest.
Disclaimer
The views, information or content, and conclusions presented do not necessarily represent the official position or policy of, nor should any official endorsement be inferred on the part of, the Uniformed Services University, the Department of Defense, or the U.S. Government.
References
- [1].Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, et al. (2021). Alzheimer's disease. Lancet, 397:1577-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jiang H, Esparza TJ, Kummer TT, Brody DL (2021). Unbiased high-content screening reveals Abeta- and tau-independent synaptotoxic activities in human brain homogenates from Alzheimer's patients and high-pathology controls. PLoS One, 16:e0259335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, et al. (2013). Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol, 73:104-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sadick JS, O'Dea MR, Hasel P, Dykstra T, Faustin A, Liddelow SA (2022). Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer's disease. Neuron, 110:1788-1805 e1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y (2020). Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease. Mol Neurodegener, 15:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kincses ZT, Toldi J, Vecsei L (2010). Kynurenines, neurodegeneration and Alzheimer's disease. J Cell Mol Med, 14:2045-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Meier-Stephenson FS, Meier-Stephenson VC, Carter MD, Meek AR, Wang Y, Pan L, et al. (2022). Alzheimer's disease as an autoimmune disorder of innate immunity endogenously modulated by tryptophan metabolites. Alzheimers Dement (N Y), 8:e12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Badawy AA (2017). Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res, 10:1178646917691938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu W, Nicolazzo JA, Wen L, Chung R, Stankovic R, Bao SS, et al. (2013), Expression of tryptophan 2,3-dioxygenase and production of kynurenine pathway metabolites in triple transgenic mice and human Alzheimer's disease brain. PloS One 8:e59749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang YS, Ogbechi J, Clanchy FI, Williams RO, Stone TW (2020). IDO and kynurenine metabolites in peripheral and CNS disorders. Front Immunol, 11:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yu D, Tao BB, Yang YY, Du LS, Yang SS, He XJ, et al. (2015). The IDO inhibitor coptisine ameliorates cognitive impairment in a mouse model of Alzheimer's disease. J Alzheimers Dis, 43:291-302. [DOI] [PubMed] [Google Scholar]
- [12].Wang J, Wang B, Jiang L, Zhou K, Yang GY, Jin K (2020). The effect of IDO on neural progenitor cell survival under oxygen glucose deprivation. Front Cell Neurosci, 14:581861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang Y, Lawson MA, Dantzer R, Kelley KW (2010). LPS-induced indoleamine 2,3-dioxygenase is regulated in an interferon-gamma-independent manner by a JNK signaling pathway in primary murine microglia. Brain Behav Immun, 24:201-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu CP, Pan ZZ, Luo DY (2016). TDO as a therapeutic target in brain diseases. Metab Brain Dis, 31:737-47. [DOI] [PubMed] [Google Scholar]
- [15].Widner B, Leblhuber F, Walli J, Tilz GP, Demel U, Fuchs D (2000). Tryptophan degradation and immune activation in Alzheimer's disease. J Neural Transm (Vienna), 107:343-353. [DOI] [PubMed] [Google Scholar]
- [16].Mieszkowski J, Brzezinska P, Stankiewicz B, Kochanowicz A, Niespodzinski B, Reczkowicz J, et al. (2022). Direct effects of vitamin D supplementation on ultramarathon-induced changes in kynurenine metabolism. Nutrients, 14:4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gonzalez-Sanchez M, Jimenez J, Narvaez A, Antequera D, Llamas-Velasco S, Martin AH, et al. (2020). Kynurenic acid levels are increased in the CSF of Alzheimer's disease patients. Biomolecules, 10:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Makhaeva GF, Lushchekina SV, Boltneva NP, Sokolov VB, Grigoriev VV, Serebryakova OG, et al. (2015). Conjugates of gamma-carbolines and phenothiazine as new selective inhibitors of butyrylcholinesterase and blockers of NMDA receptors for Alzheimer disease. Sci Rep, 5:13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Modoux M, Rolhion N, Mani S, Sokol H (2021). Tryptophan metabolism as a pharmacological target. Trends Pharmacol Sci, 42:60-73. [DOI] [PubMed] [Google Scholar]
- [20].Wu HQ, Pereira EF, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R (2010). The astrocyte-derived alpha7 nicotinic receptor antagonist kynurenic acid controls extracellular glutamate levels in the prefrontal cortex. J Mol Neurosci, 40:204-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Perea G, Gomez R, Mederos S, Covelo A, Ballesteros JJ, Schlosser L, et al. (2016). Activity-dependent switch of GABAergic inhibition into glutamatergic excitation in astrocyte-neuron networks. elife, 5:e20363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Scarabino D, Peconi M, Broggio E, Gambina G, Maggi E, Armeli F, et al. (2020). Relationship between proinflammatory cytokines (Il-1beta, Il-18) and leukocyte telomere length in mild cognitive impairment and Alzheimer's disease. Exp Gerontol, 136:110945. [DOI] [PubMed] [Google Scholar]
- [23].Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, et al. (2011). Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer's disease model. J Immunol, 187:6539-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xie L, Lai Y, Lei F, Liu S, Liu R, Wang T (2015). Exploring the association between interleukin-1beta and its interacting proteins in Alzheimer's disease. Mol Med Rep, 11:3219-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liang Y, Xie S, He Y, Xu M, Qiao X, Zhu Y, et al. (2022). Kynurenine pathway metabolites as biomarkers in Alzheimer's disease. Dis Markers, 2022:9484217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rahman A, Rao MS, Khan KM (2018). Intraventricular infusion of quinolinic acid impairs spatial learning and memory in young rats: a novel mechanism of lead-induced neurotoxicity. J Neuroinflammation, 15:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Busse M, Hettler V, Fischer V, Mawrin C, Hartig R, Dobrowolny H, et al. (2018). Increased quinolinic acid in peripheral mononuclear cells in Alzheimer's dementia. Eur Arch Psychiatry Clin Neurosci, 268:493-500. [DOI] [PubMed] [Google Scholar]
- [28].Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM (2005). Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer's disease hippocampus. Neuropathol Appl Neurobiol, 31:395-404. [DOI] [PubMed] [Google Scholar]
- [29].Guillemin GJ, Williams KR, Smith DG, Smythe GA, Croitoru-Lamoury J, Brew BJ (2003). Quinolinic acid in the pathogenesis of Alzheimer's disease. Adv Exp Med Biol, 527:167-176. [DOI] [PubMed] [Google Scholar]
- [30].Guillemin GJ, Smythe GA, Veas LA, Takikawa O, Brew BJ (2003). A beta 1-42 induces production of quinolinic acid by human macrophages and microglia. Neuroreport, 14:2311-2315. [DOI] [PubMed] [Google Scholar]
- [31].Parrott JM, Redus L, O'Connor JC (2016). Kynurenine metabolic balance is disrupted in the hippocampus following peripheral lipopolysaccharide challenge. J Neuroinflammation, 13:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schiefer J, Topper R, Schmidt W, Block F, Heinrich PC, Noth J, et al. (1998). Expression of interleukin 6 in the rat striatum following stereotaxic injection of quinolinic acid. J Neuroimmunol, 89:168-176. [DOI] [PubMed] [Google Scholar]
- [33].Braidy N, Grant R, Adams S, Brew BJ, Guillemin GJ (2009). Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox Res, 16:77-86. [DOI] [PubMed] [Google Scholar]
- [34].Jenkins TA, Nguyen JC, Polglaze KE, Bertrand PP (2016). Influence of tryptophan and serotonin on mood and cognition with a possible role of the Gut-Brain Axis. Nutrients, 8:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Duleu S, Mangas A, Sevin F, Veyret B, Bessede A, Geffard M (2010). Circulating antibodies to IDO/THO pathway metabolites in Alzheimer's disease. Int J Alzheimers Dis, 2010:501541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang X (2019). Molecules in the tryptophan-5-hydroxytryptophan-serotonin-NAS-melatonin/6-hydroxymelatonin sulfate-melatonin receptor 1A pathway in neurological diseases. Application US15/974,184. [Google Scholar]
- [37].Wang X, Sirianni A, Pei Z, Cormier K, Smith K, Jiang J, et al. (2011). The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J Neurosci, 31:14496-14507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dragicevic N, Copes N, O'Neal-Moffitt G, Jin J, Buzzeo R, Mamcarz M, et al. (2011). Melatonin treatment restores mitochondrial function in Alzheimer's mice: a mitochondrial protective role of melatonin membrane receptor signaling. J Pineal Res, 51:75-86. [DOI] [PubMed] [Google Scholar]
- [39].Sulkava S, Muggalla P, Sulkava R, Ollila HM, Peuralinna T, Myllykangas L, et al. (2018). Melatonin receptor type 1A gene linked to Alzheimer's disease in old age. Sleep, 41:zsy103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luo F, Sandhu AF, Rungratanawanich W, Williams GE, Akbar M, Zhou S, et al. (2020). Melatonin and autophagy in aging-related neurodegenerative diseases. Int J Mol Sci, 21:7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang X (2009). The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci Ther, 15:345-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang Y, Cook A, Kim J, Baranov SV, Jiang J, Smith K, et al. (2013). Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis, 55:26-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kadena M, Kumagai Y, Vandenbon A, Matsushima H, Fukamachi H, Maruta N, et al. (2017). Microarray and gene co-expression analysis reveals that melatonin attenuates immune responses and modulates actin rearrangement in macrophages. Biochem Biophys Res Commun, 485:414-420. [DOI] [PubMed] [Google Scholar]
- [44].Lin JD, Nishi H, Poles J, Niu X, McCauley C, Rahman K, et al. (2019). Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight, 4:e124574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jiang J, Yu S, Jiang Z, Liang C, Yu W, Li J, et al. (2014). N-acetyl-serotonin protects HepG2 cells from oxidative stress injury induced by hydrogen peroxide. Oxid Med Cell Longev, 2014:310504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Oxenkrug G (2005). Antioxidant effects of N-acetylserotonin: possible mechanisms and clinical implications. Ann N Y Acad Sci, 1053:334-347. [DOI] [PubMed] [Google Scholar]
- [47].Bachurin S, Oxenkrug G, Lermontova N, Afanasiev A, Beznosko B, Vankin G, et al. (1999). N-acetylserotonin, melatonin and their derivatives improve cognition and protect against beta-amyloid-induced neurotoxicity. Ann N Y Acad Sci, 890:155-166. [DOI] [PubMed] [Google Scholar]
- [48].Zhou H, Wang J, Jiang J, Stavrovskaya IG, Li M, Li W, et al. (2014). N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. J Neurosci, 34:2967-2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Luo C, Yang Q, Liu Y, Zhou S, Jiang J, Reiter RJ, et al. (2019). The multiple protective roles and molecular mechanisms of melatonin and its precursor N-acetylserotonin in targeting brain injury and liver damage and in maintaining bone health. Free Radic Biol Med, 130:215-233. [DOI] [PubMed] [Google Scholar]
- [50].Liu J, Zhang N, Zhang M, Yin H, Zhang X, Wang X, et al. (2021). N-acetylserotonin alleviated the expression of interleukin-1beta in retinal ischemia-reperfusion rats via the TLR4/NF-kappaB/NLRP3 pathway. Exp Eye Res, 208:108595. [DOI] [PubMed] [Google Scholar]
- [51].Yu S, Zheng J, Jiang Z, Shi C, Li J, Du X, et al. (2013). Protective effect of N-acetylserotonin against acute hepatic ischemia-reperfusion injury in mice. Int J Mol Sci, 14:17680-17693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Oxenkrug G, Ratner R (2012). N-acetylserotonin and aging-associated cognitive impairment and depression. Aging Dis, 3:330-338. [PMC free article] [PubMed] [Google Scholar]
- [53].Park S, Kim Y, Lee J, Lee JY, Kim H, Lee S, et al. (2021). A systems biology approach to investigating the interaction between serotonin synthesis by tryptophan hydroxylase and the metabolic homeostasis. Int J Mol Sci, 22:2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bruce KD, Zsombok A, Eckel RH (2017). Lipid processing in the brain: A key regulator of systemic metabolism. Front Endocrinol (Lausanne), 8:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Goetzl EJ, Schwartz JB, Abner EL, Jicha GA, Kapogiannis D (2018). High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann Neurol, 83:544-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jiaranaikulwanitch J, Govitrapong P, Fokin VV, Vajragupta O (2012). From BACE1 inhibitor to multifunctionality of tryptoline and tryptamine triazole derivatives for Alzheimer's disease. Molecules, 17:8312-8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A, 106:3698-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang LS, Davies SS (2016). Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med, 8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bendheim PE, Poeggeler B, Neria E, Ziv V, Pappolla MA, Chain DG (2002). Development of indole-3-propionic acid (OXIGON) for Alzheimer's disease. J Mol Neurosci, 19:213-217. [DOI] [PubMed] [Google Scholar]
- [60].Zhao ZH, Xin FZ, Xue Y, Hu Z, Han Y, Ma F, et al. (2019). Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp Mol Med, 51:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Moroni F, Cozzi A, Sili M, Mannaioni G (2012). Kynurenic acid: a metabolite with multiple actions and multiple targets in brain and periphery. J Neural Transm (Vienna), 119:133-139. [DOI] [PubMed] [Google Scholar]
- [62].Kozak R, Campbell BM, Strick CA, Horner W, Hoffmann WE, Kiss T, et al. (2014). Reduction of brain kynurenic acid improves cognitive function. J Neurosci, 34:10592-10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Potter MC, Elmer GI, Bergeron R, Albuquerque EX, Guidetti P, Wu HQ, et al. (2010). Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology, 35:1734-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Santamaria A, Galvan-Arzate S, Lisy V, Ali SF, Duhart HM, Osorio-Rico L, et al. (2001). Quinolinic acid induces oxidative stress in rat brain synaptosomes. Neuroreport, 12:871-874. [DOI] [PubMed] [Google Scholar]
- [65].Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP (1996). Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J, 320 (Pt 2):595-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].O'Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF (2015). Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res, 277:32-48. [DOI] [PubMed] [Google Scholar]
- [67].Lacoste B, Angeloni D, Dominguez-Lopez S, Calderoni S, Mauro A, Fraschini F, et al. (2015). Anatomical and cellular localization of melatonin MT1 and MT2 receptors in the adult rat brain. J Pineal Res, 58:397-417. [DOI] [PubMed] [Google Scholar]
- [68].Paley EL, Perry G, Sokolova O (2013). Tryptamine induces axonopathy and mitochondriopathy mimicking neurodegenerative diseases via tryptophanyl-tRNA deficiency. Curr Alzheimer Res, 10:987-1004. [DOI] [PubMed] [Google Scholar]
- [69].Wu Y, Eisel ULM (2023). Microglia-astrocyte communication in Alzheimer's disease. J Alzheimers Dis, 95:785-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bettcher BM, Tansey MG, Dorothee G, Heneka MT (2021). Peripheral and central immune system crosstalk in Alzheimer disease - a research prospectus. Nat Rev Neurol, 17:689-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Park JS, Kam TI, Lee S, Park H, Oh Y, Kwon SH, et al. (2021). Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer's disease. Acta Neuropathol Commun, 9:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].McAlpine CS, Park J, Griciuc A, Kim E, Choi SH, Iwamoto Y, et al. (2021). Astrocytic interleukin-3 programs microglia and limits Alzheimer's disease. Nature, 595:701-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H (2016). Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer's disease. J Neurosci, 36:577-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Fakhoury M (2018). Microglia and astrocytes in alzheimer's disease: implications for therapy. Curr Neuropharmacol, 16:508-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chen Y, Colonna M (2021). Microglia in Alzheimer's disease at single-cell level. Are there common patterns in humans and mice? J Exp Med, 218:e20202717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, et al. (2018). Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer's disease not evident in mouse models. Cell Rep, 22:832-847. [DOI] [PubMed] [Google Scholar]
- [77].Ellwanger DC, Wang S, Brioschi S, Shao Z, Green L, Case R, et al. (2021). Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A, 118:e2017742118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Saura CA, Deprada A, Capilla-Lopez MD, Parra-Damas A (2023). Revealing cell vulnerability in Alzheimer's disease by single-cell transcriptomics. Semin Cell Dev Biol, 139:73-83. [DOI] [PubMed] [Google Scholar]
- [79].Pettas S, Karagianni K, Kanata E, Chatziefstathiou A, Christoudia N, Xanthopoulos K, et al. (2022). Profiling microglia through single-cell RNA sequencing over the course of development, Cells, 11:2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ochocka N, Kaminska B (2021). Microglia diversity in healthy and diseased brain: insights from single-cell omics. Int J Mol Sci, 2:3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hashemiaghdam A, Mroczek M (2020). Microglia heterogeneity and neurodegeneration: the emerging paradigm of the role of immunity in Alzheimer's disease. J Neuroimmunol, 341:577185. [DOI] [PubMed] [Google Scholar]
- [82].Li T, Liu T, Chen X, Li L, Feng M, Zhang Y, et al. (2020). Microglia induce the transformation of A1/A2 reactive astrocytes via the CXCR7/PI3K/Akt pathway in chronic post-surgical pain. J Neuroinflammation, 17:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kowalski K, Mulak A (2019). Brain-Gut-Microbiota Axis in Alzheimer's disease. J Neurogastroenterol Motil, 25:48-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang J, He W, Zhang J (2023). A richer and more diverse future for microglia phenotypes. Heliyon, 9:e14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Qian Z, Qin J, Lai Y, Zhang C, Zhang X (2023). Large-scale integration of single-cell RNA-Seq data reveals astrocyte diversity and transcriptomic modules across six central nervous system disorders. Biomolecules, 13:692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. (2016). Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med, 22:586-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zhang F, Zhong R, Li S, Fu Z, Cheng C, Cai H, et al. (2017). Acute hypoxia induced an imbalanced M1/M2 activation of microglia through NF-kappaB signaling in Alzheimer's disease mice and wild-type littermates. Front Aging Neurosci, 9:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao CC, Ardura-Fabregat A, et al. (2018). Microglial control of astrocytes in response to microbial metabolites. Nature, 557:724-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Qian K, Jiang X, Liu ZQ, Zhang J, Fu P, Su Y, et al. (2023). Revisiting the critical roles of reactive astrocytes in neurodegeneration. Mol Psychiatry, 28:2697-2706. [DOI] [PubMed] [Google Scholar]
- [90].Preman P, Alfonso-Triguero M, Alberdi E, Verkhratsky A, Arranz AM (2021). Astrocytes in Alzheimer's disease: pathological significance and molecular pathways. Cells, 10:540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lenzer-Fanara JR, Li T, Salerni EA, Payen F, Croll SD (2017). VEGF treatment during status epilepticus attenuates long-term seizure-associated alterations in astrocyte morphology. Epilepsy Behav, 70:33-44. [DOI] [PubMed] [Google Scholar]
- [92].Hu Y, Zheng Y, Wang T, Jiao L, Luo Y (2022). VEGF, a key factor for blood brain barrier injury after cerebral ischemic stroke. Aging Dis, 13:647-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Christie IN, Theparambil SM, Braga A, Doronin M, Hosford PS, Brazhe A, et al. (2023). Astrocytes produce nitric oxide via nitrite reduction in mitochondria to regulate cerebral blood flow during brain hypoxia. Cell Rep, 42:113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bai RX, Chen XZ, Ren JF, Hu L, Li H, Wang H, et al. (2022). Toxoplasma gondii rhoptry protein (TgROP18) enhances the expression of pro-inflammatory factor in LPS/IFN-gamma-induced murine BV2 microglia cells via NF-kappaB signal pathway. Acta Trop, 235:106650. [DOI] [PubMed] [Google Scholar]
- [95].Xie LH, Wu QF, Tang Y, Zhuang ZQ, Zhao N, Huang B, Yu SG (2018). Study on electroacupuncture promoting the polarization of M2 phenotype microglia cells in hippocampus of AD model rats. Chinese Arch of Trad Med, 33:1816-1820. [Google Scholar]
- [96].Tang Y, Le W (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol, 53:1181-1194. [DOI] [PubMed] [Google Scholar]
- [97].Valles SL, Dolz-Gaiton P, Gambini J, Borras C, Lloret A, Pallardo FV, et al. (2010). Estradiol or genistein prevent Alzheimer's disease-associated inflammation correlating with an increase PPAR gamma expression in cultured astrocytes. Brain Res, 1312:138-144. [DOI] [PubMed] [Google Scholar]
- [98].Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. (2012). TLR2 is a primary receptor for Alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. J Immunol, 188:1098-1107. [DOI] [PubMed] [Google Scholar]
- [99].Belkhelfa M, Rafa H, Medjeber O, Arroul-Lammali A, Behairi N, Abada-Bendib M, et al. (2014). IFN-gamma and TNF-alpha are involved during Alzheimer disease progression and correlate with nitric oxide production: a study in Algerian patients. J Interferon Cytokine Res, 34:839-847. [DOI] [PubMed] [Google Scholar]
- [100].Liu C, Cui G, Zhu M, Kang X, Guo H (2014). Neuroinflammation in Alzheimer's disease: chemokines produced by astrocytes and chemokine receptors. Int J Clin Exp Pathol, 7:8342-8355. [PMC free article] [PubMed] [Google Scholar]
- [101].Jazvinscak Jembrek M, Orsolic N, Mandic L, Sadzak A, Segota S (2021). Anti-oxidative, anti-inflammatory and anti-apoptotic effects of flavonols: targeting Nrf2, NF-kappaB and p53 pathways in neurodegeneration. Antioxidants (Basel), 10:1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cekanaviciute E, Dietrich HK, Axtell RC, Williams AM, Egusquiza R, Wai KM, et al. (2014). Astrocytic TGF-beta signaling limits inflammation and reduces neuronal damage during central nervous system Toxoplasma infection. J Immunol, 193:139-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Caraci F, Battaglia G, Bruno V, Bosco P, Carbonaro V, Giuffrida ML, et al. (2011). TGF-beta1 pathway as a new target for neuroprotection in Alzheimer's disease. CNS Neurosci Ther, 17:237-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Diniz LP, Tortelli V, Matias I, Morgado J, Bergamo Araujo AP, Melo HM, et al. (2017). Astrocyte transforming growth factor beta 1 protects synapses against abeta oligomers in Alzheimer's disease model. J Neurosci, 37:6797-6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].da Silva MD, Bobinski F, Sato KL, Kolker SJ, Sluka KA, Santos AR (2015). IL-10 cytokine released from M2 macrophages is crucial for analgesic and anti-inflammatory effects of acupuncture in a model of inflammatory muscle pain. Mol Neurobiol, 51:19-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].van der Veeken J, Campbell C, Pritykin Y, Schizas M, Verter J, Hu W, et al. (2022), Genetic tracing reveals transcription factor Foxp3-dependent and Foxp3-independent functionality of peripherally induced Treg cells. Immunity. 55:1173-1184 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Norden DM, Fenn AM, Dugan A, Godbout JP (2014). TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia, 62:881-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Xing C, Li W, Deng W, Ning M, Lo EH (2018). A potential gliovascular mechanism for microglial activation: differential phenotypic switching of microglia by endothelium versus astrocytes. J Neuroinflammation, 15:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, et al. (2015). IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron, 85:519-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Guillot-Sestier MV, Doty KR, Gate D, Rodriguez J Jr, Leung BP, Rezai-Zadeh K, et al. (2015). Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron, 85:534-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].O'Neil SM, Hans EE, Jiang S, Wangler LM, Godbout JP (2022). Astrocyte immunosenescence and deficits in interleukin 10 signaling in the aged brain disrupt the regulation of microglia following innate immune activation. Glia, 70:913-934. [DOI] [PubMed] [Google Scholar]
- [112].Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541:481-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, et al. (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med, 24:931-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Xu X, Zhang A, Zhu Y, He W, Di W, Fang Y, et al. (2018). MFG-E8 reverses microglial-induced neurotoxic astrocyte (A1) via NF-kappaB and PI3K-Akt pathways. J Cell Physiol, 234:904-914. [DOI] [PubMed] [Google Scholar]
- [115].Linnartz B, Kopatz J, Tenner AJ, Neumann H (2012). Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci, 32:946-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. (2012). Genomic analysis of reactive astrogliosis. J Neurosci, 32:6391-6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci, 16:1211-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA (2018). Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A, 115:E1896-E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, et al. (2019). Complement C3 Is activated in human AD brain and Is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep, 28:2111-2123 e2116. [DOI] [PubMed] [Google Scholar]
- [120].Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, et al. (2015). NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron, 85:101-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Sekar S, McDonald J, Cuyugan L, Aldrich J, Kurdoglu A, Adkins J, et al. (2015). Alzheimer's disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol Aging, 36:583-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Prokop S, Miller KR, Heppner FL (2013). Microglia actions in Alzheimer's disease. Acta Neuropathol, 126:461-477. [DOI] [PubMed] [Google Scholar]
- [123].Xie L, Jiang C, Wang Z, Yi X, Gong Y, Chen Y, et al. (2016). Effect of huperzine A on abeta-induced p65 of astrocyte in vitro. Biosci Biotechnol Biochem, 80:2334-2337. [DOI] [PubMed] [Google Scholar]
- [124].Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro M, Iradi A, Obrador E, Ortega A, et al. (2015). Astrocytes protect neurons from Abeta1-42 peptide-induced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-gamma and SIRT-1. Int J Med Sci, 12:48-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Huang P, Yang YH, Chang YH, Chang SL, Chou MC, Lai CL, et al. (2020). Association of early-onset Alzheimer's disease with germline-generated high affinity self-antigen load. Transl Psychiatry, 10:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Alabed S, Zhou H, Sariyer IK, Chang SL (2021). Meta-Analysis of Methamphetamine modulation on amyloid precursor protein through HMGB1 in Alzheimer's disease. Int J Mol Sci, 22:4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Liu X, Vigorito M, Huang W, Khan MAS, Chang SL (2022). The Impact of Alcohol-Induced dysbiosis on diseases and disorders of the central nervous system. J Neuroimmune Pharmacol, 17:131-151. [DOI] [PubMed] [Google Scholar]
- [128].Nagpal R, Neth BJ, Wang S, Mishra SP, Craft S, Yadav H (2020). Gut mycobiome and its interaction with diet, gut bacteria and alzheimer's disease markers in subjects with mild cognitive impairment: A pilot study. EBioMedicine, 59:102950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Bairamian D, Sha S, Rolhion N, Sokol H, Dorothee G, Lemere CA, et al. (2022). Microbiota in neuroinflammation and synaptic dysfunction: a focus on Alzheimer's disease. Mol Neurodegener, 17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Soriano S, Curry K, Wang Q, Chow E, Treangen TJ, Villapol S (2022). Fecal microbiota transplantation derived from Alzheimer's disease mice worsens brain trauma outcomes in wild-type controls. Int J Mol Sci, 23:4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mohle L, Israel N, Paarmann K, Krohn M, Pietkiewicz S, Muller A, et al. (2016). Chronic Toxoplasma gondii infection enhances beta-amyloid phagocytosis and clearance by recruited monocytes. Acta Neuropathol Commun, 4:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Bauerl C, Collado MC, Diaz Cuevas A, Vina J, Perez Martinez G (2018). Shifts in gut microbiota composition in an APP/PSS1 transgenic mouse model of Alzheimer's disease during lifespan. Lett Appl Microbiol, 66:464-471. [DOI] [PubMed] [Google Scholar]
- [133].Cox LM, Schafer MJ, Sohn J, Vincentini J, Weiner HL, Ginsberg SD, et al. (2019). Calorie restriction slows age-related microbiota changes in an Alzheimer's disease model in female mice. Sci Rep, 9:17904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Xie J, Cools L, Van Imschoot G, Van Wonterghem E, Pauwels MJ, Vlaeminck I, et al. (2023). Helicobacter pylori-derived outer membrane vesicles contribute to Alzheimer's disease pathogenesis via C3-C3aR signalling. J Extracell Vesicles, 12:e12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Gong T, Chen Q, Mao H, Zhang Y, Ren H, Xu M, et al. (2022). Outer membrane vesicles of Porphyromonas gingivalis trigger NLRP3 inflammasome and induce neuroinflammation, tau phosphorylation, and memory dysfunction in mice. Front Cell Infect Microbiol, 12:925435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Lee KE, Kim JK, Han SK, Lee DY, Lee HJ, Yim SV, et al. (2020). The extracellular vesicle of gut microbial Paenalcaligenes hominis is a risk factor for vagus nerve-mediated cognitive impairment. Microbiome, 8:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Vigasova D, Nemergut M, Liskova B, Damborsky J (2021). Multi-pathogen infections and Alzheimer's disease. Microb Cell Fact, 20:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Park JY, Choi J, Lee Y, Lee JE, Lee EH, Kwon HJ, et al. (2017). Metagenome analysis of bodily microbiota in a mouse model of Alzheimer disease using bacteria-derived membrane vesicles in blood. Exp Neurobiol, 26:369-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Minter MR, Hinterleitner R, Meisel M, Zhang C, Leone V, Zhang X, et al. (2017). Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APP(SWE)/PS1(DeltaE9) murine model of Alzheimer's disease. Sci Rep, 7:10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, et al. (2017). Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep, 7:41802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Naomi R, Embong H, Othman F, Ghazi HF, Maruthey N, Bahari H (2021). Probiotics for Alzheimer's disease: a systematic review. Nutrients, 14:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Sun J, Xu J, Ling Y, Wang F, Gong T, Yang C, et al. (2019). Fecal microbiota transplantation alleviated Alzheimer's disease-like pathogenesis in APP/PS1 transgenic mice. Transl Psychiatry, 9:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Nagpal R, Neth BJ, Wang S, Craft S, Yadav H (2019). Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer's disease markers in subjects with mild cognitive impairment. eBioMedicine, 47:529-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Fischer Walker CL, Sack D, Black RE (2010). Etiology of diarrhea in older children, adolescents and adults: a systematic review. PLoS Negl Trop Dis, 4:e768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Zhan X, Stamova B, Jin LW, DeCarli C, Phinney B, Sharp FR (2016). Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology, 87:2324-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Branton WG, Ellestad KK, Maingat F, Wheatley BM, Rud E, Warren RL, et al. (2013). Brain microbial populations in HIV/AIDS: alpha-proteobacteria predominate independent of host immune status. PLoS One, 8:e54673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Zarifkar A, Choopani S, Ghasemi R, Naghdi N, Maghsoudi AH, Maghsoudi N, et al. (2010). Agmatine prevents LPS-induced spatial memory impairment and hippocampal apoptosis. Eur J Pharmacol, 634:84-88. [DOI] [PubMed] [Google Scholar]
- [148].Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, et al. (2005). LPS receptor (CD14): a receptor for phagocytosis of Alzheimer's amyloid peptide. Brain, 128:1778-1789. [DOI] [PubMed] [Google Scholar]
- [149].Barbierato M, Facci L, Argentini C, Marinelli C, Skaper SD, Giusti P (2013). Astrocyte-microglia cooperation in the expression of a pro-inflammatory phenotype. CNS Neurol Disord Drug Targets, 12:608-618. [DOI] [PubMed] [Google Scholar]
- [150].Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, et al. (2010). LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation, 7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kobayashi Y, Sugahara H, Shimada K, Mitsuyama E, Kuhara T, Yasuoka A, et al. (2017). Therapeutic potential of Bifidobacterium breve strain A1 for preventing cognitive impairment in Alzheimer's disease. Sci Rep, 7:13510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Ho L, Ono K, Tsuji M, Mazzola P, Singh R, Pasinetti GM (2018). Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer's disease-type beta-amyloid neuropathological mechanisms. Expert Rev Neurother, 18:83-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Rahimi F, Maiti P, Bitan G (2009). Photo-induced cross-linking of unmodified proteins (PICUP) applied to amyloidogenic peptides. [J] Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Vinolo MA, Rodrigues HG, Nachbar RT, Curi R (2011). Regulation of inflammation by short chain fatty acids. Nutrients, 3:858-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Carmona P, Molina M, Calero M, Bermejo-Pareja F, Martinez-Martin P, Alvarez I, et al. (2012). Infrared spectroscopic analysis of mononuclear leukocytes in peripheral blood from Alzheimer's disease patients. Anal Bioanal Chem, 402:2015-2021. [DOI] [PubMed] [Google Scholar]
- [156].V DA, Sarnataro D (2021). Probiotics, prebiotics and their role in Alzheimer's disease. Neural Regen Res, 16:1768-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Mehrabadi S, Sadr SS (2020). Assessment of probiotics mixture on memory function, inflammation markers, and oxidative stress in an Alzheimer's disease model of rats. Iran Biomed J, 24:220-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Bonfili L, Cecarini V, Gogoi O, Berardi S, Scarpona S, Angeletti M, et al. (2020). Gut microbiota manipulation through probiotics oral administration restores glucose homeostasis in a mouse model of Alzheimer's disease. Neurobiol Aging, 87:35-43. [DOI] [PubMed] [Google Scholar]
- [159].Kaur H, Nagamoto-Combs K, Golovko S, Golovko MY, Klug MG, Combs CK (2020). Probiotics ameliorate intestinal pathophysiology in a mouse model of Alzheimer's disease. Neurobiol Aging, 92:114-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Korecka A, Dona A, Lahiri S, Tett AJ, Al-Asmakh M, Braniste V, et al. (2016). Bidirectional communication between the Aryl hydrocarbon Receptor (AhR) and the microbiome tunes host metabolism. NPJ Biofilms Microbiomes, 2:16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Andrysik Z, Prochazkova J, Kabatkova M, Umannova L, Simeckova P, Kohoutek J, et al. (2013). Aryl hydrocarbon receptor-mediated disruption of contact inhibition is associated with connexin43 downregulation and inhibition of gap junctional intercellular communication. Arch Toxicol, 87:491-503. [DOI] [PubMed] [Google Scholar]
- [162].Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, et al. (2014). Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity, 41:296-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Boussadia B, Lakhal L, Payrastre L, Ghosh C, Pascussi JM, Gangarossa G, et al. (2018). Pregnane X receptor deletion modifies recognition memory and electroencephalographic activity. Neuroscience, 370:130-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Boussadia B, Gangarossa G, Mselli-Lakhal L, Rousset MC, de Bock F, Lassere F, et al. (2016). Lack of CAR impacts neuronal function and cerebrovascular integrity in vivo. Exp Neurol, 283:39-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Lachmann P (1975). Genetics of the complement system. J Med Genet, 12:372-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ma N, He T, Johnston LJ, Ma X (2020). Host-microbiome interactions: the aryl hydrocarbon receptor as a critical node in tryptophan metabolites to brain signaling. Gut Microbes, 11:1203-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Li G, Young KD (2013). Indole production by the tryptophanase TnaA in Escherichia coli is determined by the amount of exogenous tryptophan. Microbiology (Reading), 159:402-410. [DOI] [PubMed] [Google Scholar]
- [168].Roager HM, Licht TR (2018). Microbial tryptophan catabolites in health and disease. Nat Commun, 9:3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Jin UH, Lee SO, Sridharan G, Lee K, Davidson LA, Jayaraman A, et al. (2014). Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol Pharmacol, 85:777-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Whitfield-Cargile CM, Cohen ND, Chapkin RS, Weeks BR, Davidson LA, Goldsby JS, et al. (2016). The microbiota-derived metabolite indole decreases mucosal inflammation and injury in a murine model of NSAID enteropathy. Gut Microbes, 7:246-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Brinkmann V, Ale-Agha N, Haendeler J, Ventura N (2019). The aryl hydrocarbon receptor (AhR) in the aging process: another puzzling role for this highly conserved transcription factor. Front Physiol, 10:1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Lee HU, McPherson ZE, Tan B, Korecka A, Pettersson S (2017). Host-microbiome interactions: the aryl hydrocarbon receptor and the central nervous system. J Mol Med (Berl), 95:29-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Gao D, Wu M, Wang C, Wang Y, Zuo Z (2015). Chronic exposure to low benzo[a]pyrene level causes neurodegenerative disease-like syndromes in zebrafish (Danio rerio). Aquat Toxicol, 167:200-208. [DOI] [PubMed] [Google Scholar]
- [174].Shinde R, McGaha TL (2018). The aryl hydrocarbon receptor: connecting immunity to the microenvironment. Trends Immunol, 39:1005-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. (2017). A next generation connectivity map: L1000 platform and the first 1,000,000 Profiles. Cell, 171:1437-1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Sinha RK, Bojjireddy N, Kulkarni D, Ratheesh A, Chiplunkar SV, Gude R, et al. (2013). Type II phosphatidylinositol 4-kinase beta is an integral signaling component of early T cell activation mechanisms. Biochimie, 95:1560-1566. [DOI] [PubMed] [Google Scholar]
- [177].Stavrou S, Ross SR (2015). APOBEC3 proteins in viral immunity. J Immunol, 195:4565-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Tonnus W, Gembardt F, Hugo C, Linkermann A (2017). Die later with ESCRT! oncotarget, 8:41790-41791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Yusufu I, Ding K, Smith K, Wankhade UD, Sahay B, Patterson GT, et al. (2021). A tryptophan-deficient diet induces gut microbiota dysbiosis and increases systemic inflammation in aged mice. Int J Mol Sci, 22:5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Jiang WD, Wen HL, Liu Y, Jiang J, Kuang SY, Wu P, et al. (2015). The tight junction protein transcript abundance changes and oxidative damage by tryptophan deficiency or excess are related to the modulation of the signalling molecules, NF-kappaB p65, TOR, caspase-(3,8,9) and Nrf2 mRNA levels, in the gill of young grass carp (Ctenopharyngodon idellus). Fish Shellfish Immunol, 46:168-180. [DOI] [PubMed] [Google Scholar]
- [181].Robinson CM, Hale PT, Carlin JM (2006). NF-kappa B activation contributes to indoleamine dioxygenase transcriptional synergy induced by IFN-gamma and tumor necrosis factor-alpha. Cytokine, 35:53-61. [DOI] [PubMed] [Google Scholar]
- [182].Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, et al. (2011). Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer's disease. Curr Alzheimer Res, 8:67-80. [DOI] [PubMed] [Google Scholar]
- [183].Nordengen K, Kirsebom BE, Henjum K, Selnes P, Gisladottir B, Wettergreen M, et al. (2019). Glial activation and inflammation along the Alzheimer's disease continuum. J Neuroinflammation, 16:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Lee YH, Lin CH, Hsu PC, Sun YY, Huang YJ, Zhuo JH, et al. (2015). Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia, 63:1138-1154. [DOI] [PubMed] [Google Scholar]
- [185].Mu Q, Tavella VJ, Luo XM (2018). Role of Lactobacillus reuteri in human health and diseases. Front Microbiol, 9:757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Szychowski KA, Wnuk A, Kajta M, Wojtowicz AK (2016). Triclosan activates aryl hydrocarbon receptor (AhR)-dependent apoptosis and affects Cyp1a1 and Cyp1b1 expression in mouse neocortical neurons. Environ Res, 151:106-114. [DOI] [PubMed] [Google Scholar]
- [187].Nicolas GR, Chang PV (2019). Deciphering the chemical lexicon of Host-Gut Microbiota interactions. Trends Pharmacol Sci, 40:430-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Kim SY, Yang HJ, Chang YS, Kim JW, Brooks M, Chew EY, et al. (2014). Deletion of aryl hydrocarbon receptor AHR in mice leads to subretinal accumulation of microglia and RPE atrophy. Invest Ophthalmol Vis Sci, 55:6031-6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Chen WC, Chang LH, Huang SS, Huang YJ, Chih CL, Kuo HC, et al. (2019). Aryl hydrocarbon receptor modulates stroke-induced astrogliosis and neurogenesis in the adult mouse brain. J Neuroinflammation, 16:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Ghiboub M, Verburgt CM, Sovran B, Benninga MA, de Jonge WJ, Van Limbergen JE (2020). Nutritional therapy to modulate tryptophan metabolism and aryl hydrocarbon-receptor signaling activation in human diseases. Nutrients, 12:2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Gheorghe CE, Martin JA, Manriquez FV, Dinan TG, Cryan JF, Clarke G (2019). Focus on the essentials: tryptophan metabolism and the microbiome-gut-brain axis. Curr Opin Pharmacol, 48:137-145. [DOI] [PubMed] [Google Scholar]
- [192].Keszthelyi D, Troost FJ, Jonkers DM, Kruimel JW, Leue C, Masclee AA (2013). Decreased levels of kynurenic acid in the intestinal mucosa of IBS patients: relation to serotonin and psychological state. J Psychosom Res, 74:501-504. [DOI] [PubMed] [Google Scholar]
- [193].Hatano T, Saiki S, Okuzumi A, Mohney RP, Hattori N (2016). Identification of novel biomarkers for Parkinson's disease by metabolomic technologies. J Neurol Neurosurg Psychiatry, 87:295-301. [DOI] [PubMed] [Google Scholar]
- [194].Anderson G, Seo M, Berk M, Carvalho AF, Maes M (2016). Gut permeability and microbiota in Parkinson's disease: role of depression tryptophan Catabolites, Oxidative and nitrosative stress and melatonergic pathways. Curr Pharm Des, 22:6142-6151. [DOI] [PubMed] [Google Scholar]
- [195].Dehhaghi M, Kazemi Shariat Panahi H, Guillemin GJ (2019). Microorganisms, tryptophan metabolism, and kynurenine pathway: a complex interconnected loop influencing human health status. Int J Tryptophan Res, 12:1178646919852996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Cheah IK, Feng L, Tang RMY, Lim KHC, Halliwell B (2016). Ergothioneine levels in an elderly population decrease with age and incidence of cognitive decline; a risk factor for neurodegeneration? Biochem Biophys Res Commun, 478:162-167. [DOI] [PubMed] [Google Scholar]