

ABSTRACT
The cortical epigenetic clock was developed in brain tissue as a biomarker of brain aging. As one way to identify mechanisms underlying aging, we conducted a GWAS of cortical age. We leveraged postmortem cortex tissue and genotyping array data from 694 participants of the Rush Memory and Aging Project and Religious Orders Study (ROSMAP; 11000,000 SNPs), and meta-analysed ROSMAP with 522 participants of Brains for Dementia Research (5,000,000 overlapping SNPs). We confirmed results using eQTL (cortical bulk and single nucleus gene expression), cortical protein levels (ROSMAP), and phenome-wide association studies (clinical/neuropathologic phenotypes, ROSMAP). In the meta-analysis, the strongest association was rs4244620 (p = 1.29 × 10−7), which also exhibited FDR-significant cis-eQTL effects for CD46 in bulk and single nucleus (microglia, astrocyte, oligodendrocyte, neuron) cortical gene expression. Additionally, rs4244620 was nominally associated with lower cognition, faster slopes of cognitive decline, and greater Parkinsonian signs (n ~ 1700 ROSMAP with SNP/phenotypic data; all p ≤ 0.04). In ROSMAP alone, the top SNP was rs4721030 (p = 8.64 × 10−8) annotated to TMEM106B and THSD7A. Further, in ROSMAP (n = 849), TMEM106B and THSD7A protein levels in cortex were related to many phenotypes, including greater AD pathology and lower cognition (all p ≤ 0.0007). Overall, we identified converging evidence of CD46 and possibly TMEM106B/THSD7A for potential roles in cortical epigenetic clock age.
KEYWORDS: Aging, epigenetics, brain, dementia
Introduction
Biomarkers of aging are critical tools for developing and testing interventions to delay aging-associated declines in health, as well as for assessing mechanisms underlying aging processes. Given the high prevalence and burden of dementia in older populations, biomarkers of brain aging have public health relevance [1].
Epigenetic clocks are among the most successful biomarkers of aging to date; these clocks use DNA methylation (DNAm) states across the genome to yield a ‘signature’ of aging, trained against chronological age or other proxies for health status [2]. Recently, a cortical epigenetic clock was developed, specifically using DNAm states in brain tissue [3]. This clock integrates 347 CpG sites into an ‘aging score’ and performs particularly well in distinguishing neurodegenerative phenotypes compared to clocks designed in blood or other tissues [3]. Identifying genes regulating cortical clock age is one pathway to discovering novel mechanisms underlying brain aging (although other factors which may be related to epigenetic age, such as environmental exposures, will be important to examine as well).
In our previous research, we calculated cortical clock age in postmortem prefrontal cortex specimens from deceased participants of the Religious Orders Study and Rush Memory and Aging Project, all of whom had been followed during life with annual clinical exams [4,5]. We observed strong associations of cortical clock age with multiple clinical and pathologic dementia-related phenotypes, as well as other aging-related phenotypes. Thus, to begin to understand the genetic architecture of cortical clock – which is one approach to potentially identify mechanisms at the intersection of aging and neurodegeneration – we conducted a genome-wide association study (GWAS) of cortical clock age in three large cohorts of aging, all with cortical brain specimens, available DNA methylation array data, and genome-wide array data.
Methods
Study Populations
Since epigenetic clock aging appears to differ in older than in younger persons [3,6], we focused our work here in three cohorts which primarily include older participants: Religious Orders Study (ROS), Rush Memory and Aging Project (MAP), and Brains for Dementia Research (BDR). The Religious Orders Study was initiated in 1994 [7] and includes older priests, nuns, and brothers from across the US, free of known dementia at the time of enrolment. Participants agreed to annual neurological exams, neuropsychological testing, and blood draw. Over 1,500 participants completed a baseline evaluation as of November 2023. The follow-up rate and autopsies exceed 90%. The Rush Memory and Aging Project was established in 1997 [7], with virtually identical design and data collection, and includes older men and women from across the Chicago metropolitan area, without known dementia at enrolment; over 2,300 participants completed a baseline evaluation as of November 2023. The follow-up rate exceeds 90% and the autopsy rate exceeds 80%. Both studies were approved by an Institutional Review Board of Rush University Medical Center. All participants signed an informed consent and Anatomical Gift Act for organ donation. More than 2000 autopsies have been obtained to date. The Brains for Dementia Research brain bank was established in 2008 in the United Kingdom, as a research tissue bank across six dementia research centres, with uniform procedures [8]. Participants were recruited from the community and provided informed consent for regular assessment and their consent to donate their brain for research upon death. At the time, these analyses were initiated, over 600 postmortem brains were available for research. The study was approved by the National Research Ethics Service.
Assessment of DNA methylation states and Epigenetic Clock Age
In ROSMAP brain specimens, DNA methylation was measured in tissue from the dorsolateral prefrontal cortex (DLPFC). Briefly, 100 mg frozen sections were thawed on ice, with the grey matter dissected from the white matter, as previously described in detail [9]. The Qiagen QIAamp DNA mini protocol was used for DNA isolation. DNA methylation profiles were generated using the Illumina Infinium HumanMethylation450 platform. In more recent work with these specimens, processing methods were updated compared to our previous publications [9]. The raw signal intensities were imported into the R statistical environment with functions from the methylumi package and further processed with the wateRmelon [10] package. Initial quality control assessment was performed using functions in the methylumi package to exclude samples with inefficient bisulfite conversion (<90%) as well as outliers. Further preprocessing was conducted using the wateRmelon package by applying a p-filter. Probes having more than 1% of samples with a detection p-value greater than 0.05 and a beadcount lower than 3 in more than 5% of samples were excluded. Finally, the filtered data were normalized with ‘dasen.’ Non-CpG SNP (single nucleotide polymorphism) probes, probes that had been reported to contain common (MAF > 5%) SNPs in the CG or single base extension position or probes that were non-specific or mismapped, were flagged and disregarded in the evaluation of our results. The resulting dataset for analysis here consisted of 730 samples with 423,841 probes each. At each probe, DNAm level was represented as a beta value, that is, the ratio of the methylated probe intensity to the sum of methylated and unmethylated probe intensities. The values ranged from 0 to 1, where a larger value indicates higher methylation.
In BDR, DNA was isolated from prefrontal cortex samples using the Qiagen AllPrep DNA/RNA 96 kit [3]. Genome-wide DNA methylation was profiled using the Illumina EPIC DNAm array, which interrogates >850,000 DNA methylation sites. All work was done in the R statistical environment, and processing used either wateRmelon or bigmelon packages. Briefly, the quality control pipeline included (i) checking signal intensities and excluding poorly performing samples, (ii) calculating a bisulphite conversion statistic for each sample, excluding those with a conversion rate <80%, (iii) using p-filter function to exclude samples with >1% of probes with a detection p-value >0.05 and probes with >1% of samples with detection p-value >0.05, and (iv) removing cross-hybridizing and SNP probes. The filtered data were normalized with ‘dasen.’ This resulted in 800,916 DNA methylation sites in 610 prefrontal cortex specimens.
The cortical clock was calculated using publicly available code (https://github.com/gemmashireby/CorticalCloc). Briefly, the original cortical clock was trained in cortex tissue, specifically including large samples of older participants, to focus on brain aging. The clock was trained only using 383,547 CpG sites that were common across the Illumina 450K and EPIC arrays. The cortical clock was trained against chronological age, and integrates 347 CpG sites into a brain age score.
Assessment of genome-wide array Data.
For the ROSMAP participants, genotyping was performed on either the Affymetrix GeneChip 6.0 platform (1,878 participants, 909,600 SNPs) or the Illumina OmniQuad Express platform (456 participants, 730,525 SNPs). DNA was extracted from whole blood, lymphocytes, or frozen brain tissue, as previously described [11]. To minimize population admixture, only self-declared non-Latino individuals of European ancestry were genotyped. Then, genotyping data from both platforms were processed using PLINK software, version 1.08p [12], with standard quality control (QC) metrics such as genotype success rate > 0.95, Hardy-Weinberg equilibrium p >0.001, and mishap test < 1 × 10 − 9, as previously described [11,13]. EIGENSTRAT was used with default settings to remove population outliers and to generate a genotype covariance matrix [14], and closely related participants were removed. After these QC steps, 1,709 individuals and 750,173 autosomal markers from the Affymetrix GeneChip 6.0 platform, and 382 individuals and 627,742 autosomal markers from the Illumina OmniQuad Express platform were used for imputation. Dosages for SNPs (>35 million) were imputed on the haplotype reference consortium (HRC) panel. Analyses filtered SNPs based on minor allele frequency (MAF) > 0.01 and imputation INFO score > 0.3. This yielded 11,507,242 SNPs for analysis.
For BDR participants, DNA was extracted from brain tissue, and the NeuroChip was used, a custom Illumina genotyping array with an extensive genome-wide backbone (n = 306,670 variants) and a custom content covering 179,467 variants specific to neurological diseases [15]. The quality control of the NeuroChip was completed in GenomeStudio (version 2.0, Illumina) and PLINK v1.9 [12]. Manual curation of SNP clustering performed by Genome Studio algorithms was conducted on all SNPs. SNPs with ambiguous clustering of the three genotypes were removed. Likewise, individual sample signals which lie ambiguously between genotype clusters were also removed. Average GenTrain score, cluster separation and SNP call frequency were 0.83, 0.85 and 0.996, respectively, in the exported PLINK compatible files. Genotype data was aligned to the GRCh37/hg19 reference genome. SNPs with a minor allele frequency of less than 1%, had genotype calls of less than 95% and had control samples that significantly deviated from Hardy-Weinberg Equilibrium (p < 0.0001) were removed. Imputation was done using the 1000 Genomes Project reference panel and yielded 6,607,832 SNPs for analysis here.
Across the ROSMAP and BDR datasets, there were 5,091,857 SNPs in common.
Genome-wide association analyses
We performed GWAS on cortical clock age in DLPFC in ROSMAP, using linear regression models with cortical clock age as the dependent variable and genotype as the independent variable; covariates included age at death, sex, GWAS chip, study, and the top three principal components derived from the genetic covariance matrix. Association testing was done using an additive model for dose. Due to our modest sample size, for statistical significance, we used a suggestive threshold of p < 10−5, as has been done previously [16]. We also removed SNPs if the beta estimate was greater than 30, as has been done in previous GWAS of epigenetic clocks [17], since such extreme values are not highly plausible.
We conducted a similar GWAS in the BDR data and meta-analysed results from ROSMAP and BDR, limited to the common SNPs across these cohorts; then, we used METAL to conduct fixed-effect meta-analysis combining results across the cohorts [18]. The larger sample size in this meta-analysis has important advantages in terms of the stability of findings. For all GWAS, we calculated lambda values and created Q–Q plots of the -log10 p-values.
In sensitivity analyses, since cell proportions in bulk tissue can be related to aging and DNAm, we also considered potential confounding by neuron proportion. We estimated the neuron proportion using the CETS algorithm [19], which utilizes DNAm states from the Illumina array. We added neuron proportion as a covariate to our models of SNPs and cortical clock age. In additional sensitivity analyses, we controlled for seven cell-type proportions (i.e., inhibitory neurons, excitatory neurons, astrocytes, endothelial cells, oligodendrocytes, oligodendrocyte precursor cells, microglia), using single-cell methylation sequencing as the reference with the Houseman deconvolution algorithm [20]. These were sensitivity analyses because we have consistently found in our previous DNAm research in brain tissue that control for cell-type proportion does not meaningfully change findings [4,5,9].
Clumping analysis
To assess the independent loci associated with clock age, we used PLINK to clump SNPs within the suggestive loci (250 kb), setting the linkage disequilibrium (LD) r2 >0.5 as a threshold [12]. The SNP with the most significant relation to clock age within each independent locus was selected as the leading SNP for that locus.
Functional annotation
We used FUMA (Functional Mapping and Annotation of Genome-wide Association Studies) [21] to examine the genetic foundations of cortical clock age. Specifically, we utilized the FUMA SNP2GENE function to annotate SNPs and map them to their respective genes, focusing on their biological functions. Our approach included positional mapping with a maximum distance of 10 kb from each SNP. We set the P-value threshold for lead SNPs at 10−5. Apart from these specific settings, we adhered to the standard configurations in SNP2GENE, such as an r2 threshold of 0.6 for identifying independent significant SNPs.
Expression quantitative trait loci (eQTL) and colocalization analyses
We queried eQTL data from BrainMeta [22]. This included eQTL mapping using RNA-seq data comprising 2,865 brain cortex samples obtained from 2,443 unrelated individuals of European ancestry with genome-wide SNP data. These RNA-seq data were sourced from seven cohorts, including ROSMAP.
In addition to considering eQTL in BrainMeta, we also conducted colocalization analyses to test if lead SNPs from either the ROSMAP GWAS or the ROSMAP/BDR meta-analysis could be linked to epigenetic age by dysregulation of gene expression. Specifically, we considered our GWAS hits in the context of the bulk RNA-Seq from cortex along with publicly available eQTL and splicing QTL (sQTL) from the Myeloid Cells in Neurodegenerative Diseases (MyND) [23] and Microglia Genomic Atlas (MiGA) [24] datasets of gene expression. For colocalization analyses, we used the COLOC package from Giambartolomei et al. [25], with default parameters. Our criteria for considering a signal as colocalized was posterior probability of 0.8 or higher.
In further analyses, we also used single nucleus RNA-Seq profiling available in 424 of the ROSMAP DLPFC specimens as a way to better understand possible biologic pathways [24]; this included eQTLs from seven cell types (astrocytes, endothelial cells, excitatory neurons, inhibitory neurons, microglia, oligodendrocytes, and oligodendrocyte precursor cells [OPC]). Briefly, grey matter was processed in batches, and 5000 nuclei from each batch were pooled, prepared, and sequenced using either Illumina HiSeqX at the Broad Institute’s Genomics Platform or Illumina NovaSeq 6000 at the New York Genome Center, with a target coverage of 1 million reads per channel [26,27]. The data were first processed using the CellRanger software (v6.0.0; 10× Genomics) and quality controlled. Each nucleus was assigned back to its participants based on its genotypes. Cell types of nuclei were determined, and pseudo-bulk matrices were created by summing counts per individual. Within each cell type, genes with CPM > 1 in 80% of samples were retained and normalized using tmm.voom.
Finally, we also considered colocalization across the GWAS results for lead SNPs with seven cell types from the ROSMAP snRNA-Seq to identify common causal variants for cortical clock age and cell-type gene expression.
Functional enrichment analyses
We used the platform snpXplorer to conduct functional analyses [28]. This included GO enrichment of biological processes and KEGG pathways, as well as an examination of any overlap of the SNPs and genes identified in our GWAS with previous associations reported in the GWAS catalog. We conducted two separate sets of functional analyses, one for the results of the ROSMAP GWAS and one for the meta-analysis of ROSMAP and BDR.
Analyses of aging phenotypes
As an additional approach to understand how SNPs of interest was related to function, we also examined the lead SNPs in relation to aging phenotypes available in ROSMAP. We first considered common postmortem neuropathologies, available in 1723 participants who also had SNP array data. Due to the broad range of SNP frequencies, including some with fairly low prevalence, we focused on the most common neuropathologies. This included AD pathologies (global Alzheimer’s disease pathology burden, amyloid-β load, PHFtau tangle density) and cerebrovascular pathologies (atherosclerosis and arteriolosclerosis, the two most common cerebrovascular pathologies in the ROSMAP cohorts); these neuropathologies are described in detail elsewhere [5,29]. In addition to neuropathologies, we also examined lead SNPs in relation to key clinical phenotypes, available in 1885 participants from annual clinical assessments prior to death; this included two cognitive phenotypes and two motor phenotypes, described in detail elsewhere [5]. Specifically, we examined the following: (i) global cognitive function at cohort baseline (the average of 17 cognitive tasks, such that higher scores represent better function), and slopes of cognitive decline from baseline to death; (ii) diagnosis of dementia as of death; (iii) motor function at baseline and over time (a combined score across 10 tests, such as grip strength and timed walking, such that higher scores represent better function); and (iv) Parkinsonian signs (the average of four domains of the 26-item modified United Parkinson’s Disease Rating Scale: bradykinesia, gait, rigidity, tremor, such that higher scores represent worse function).
Finally, we also had data available on proteins in DLPFC among 849 participants. Thus, as a further way to confirm functionality, we examined relations of relevant proteins to these same aging phenotypes (using similar methods as described above). Specifically, based on the gene annotation described above, we identified corresponding proteins for the leading SNPs, when available. The proteins were measured using a multiplex mass spectrometry-based proteomics approach with tandem mass tag (TMTs) to analyse frozen tissue samples in the DLPFC. Briefly, 100 mg frozen sections were thawed on ice, with the grey matter dissected from the white matter, as previously described in detail [30]. Details of the mass spectrometry – based proteomics, database searches, and quality control have been previously described [30]. To summarize, the samples were homogenized, and the protein concentration was determined. After protein digestion, isobaric TMT peptide labelling and high pH fractionation were performed. Fractions were then analysed by liquid chromatography-mass spectrometry. The resulting mass spectrometry spectra were searched against the UniProt human protein database, with individual protein abundance checked against the global internal standard. An additional data process included regressing out technical confounders. A total of 8425 proteins passed the final quality control.
In all these analyses of aging phenotypes, for continuous phenotypes measured at death (e.g., global AD pathology), we used linear regression models; for continuous phenotypes measured repeatedly over time (e.g., global cognition), we used generalized linear mixed models. For categorical phenotypes (e.g., atherosclerosis severity, dementia diagnosis as of death), we used logistic regression models. All models controlled for age at death, sex, education, and cohort.
A flow chart summarizing our overall analytic approach is presented in Figure 1.
Figure 1.
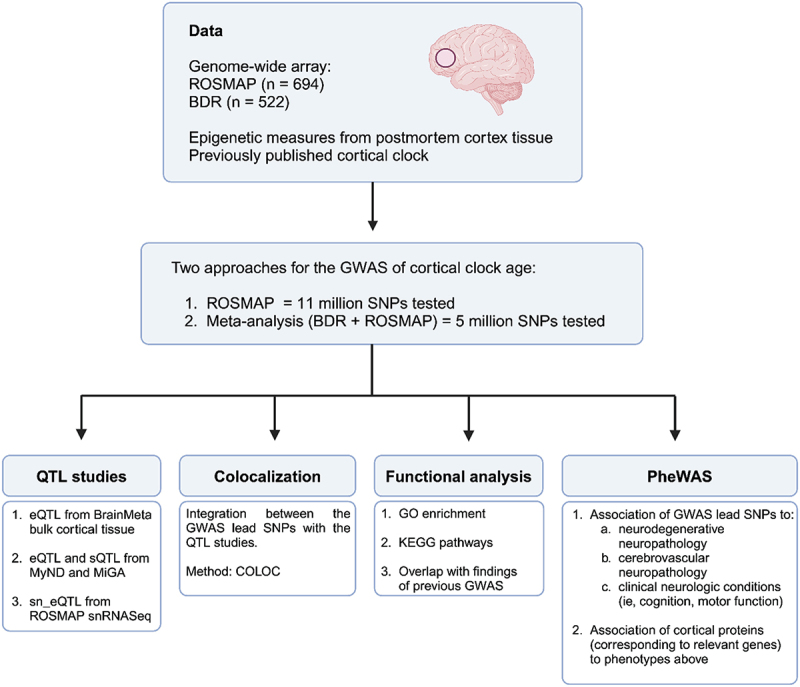
Flowchart of study design.
Data availability
ROSMAP resources can be requested at https://www.radc.rush.edu and www.synapse.org.
The BDR DNA methylation data have been deposited in the Dementias Platform UK (DPUK) data portal (https://portal.dementiasplatform.uk/CohortDirectory/Item?fingerPrintID=BDR) and the Gene Expression Omnibus (GEO) at accession number GSE197305.
Results
The 694 ROSMAP participants included in our GWAS had a mean age at death of 88 y; the mean cortical clock age was 86.5 (SD 6.7) y, slightly younger than the chronological age (Table 1). Approximately one-third of the ROSMAP sample was male, and participants were split fairly evenly across the ROS and MAP cohorts. Mean education was 16 y. Approximately 50% had pathologic AD, while 60% had clinical diagnosis of dementia as of death. The BDR cohort (n = 522) had a mean age at death of 83 (SD 9.0) y, and the mean cortical clock age was 84 y. Approximately half of the cohort was male, and mean education was approximately 13 y. Nearly half of the participants had pathologic AD, and about 60% had clinical dementia.
Table 1.
Characteristics of study cohorts.
| Characteristic | Religious Orders Study/Rush Memory and Aging Project (n = 694) Mean (SD) or % |
Brains for Dementia Research (n = 522) Mean (SD) or % |
|---|---|---|
| Mean age at death | 88.1 (6.7) | 83.4 (9.0) |
| Mean cortical clock age | 86.5 (6.0) | 84.0 (7.2) |
| Male | 37% | 53% |
| Mean years education | 16.4 (3.6) | 12.5 (3.5)* |
| Memory and Aging Project | 46% | n/a |
| Religious Orders Study | 54% | n/a |
| Pathologic Alzheimer’s disease | 62% | 43% |
| Clinical dementia | 58% | 59%* |
*Educational attainment was reported by 369 participants in the Brains for Dementia Research cohort, and clinical dementia status was known in 443 participants.
GWAS of cortical clock age
In our primary analyses, we used fixed-effect meta-analysis to combine the ROSMAP and BDR GWAS results, limited to the 5 million SNPs which were common after imputation to the Affymetrix GeneChip 6.0 and Illumina OmniQuad Express chip (from ROSMAP) as well as the NeuroChip custom array from BDR. We found 110 SNPs which met our suggestive criteria for statistical significance of p < 10−5 (Figure 2 and eTable 1). We found no evidence of genomic inflation for this meta-analysis (eFigure 1). After excluding SNPs based on the frequency of effect allele, there were 13 independent loci; Table 2 lists the 13 leading SNPs. The strongest association was for rs4244620 (p = 1.29 × 10−7) on chromosome 1, which was annotated with the gene CD46. CD46 gene encodes a type 1 membrane protein, which protects host cells from injury by complement and modulates T-cell activation [31].
Figure 2.
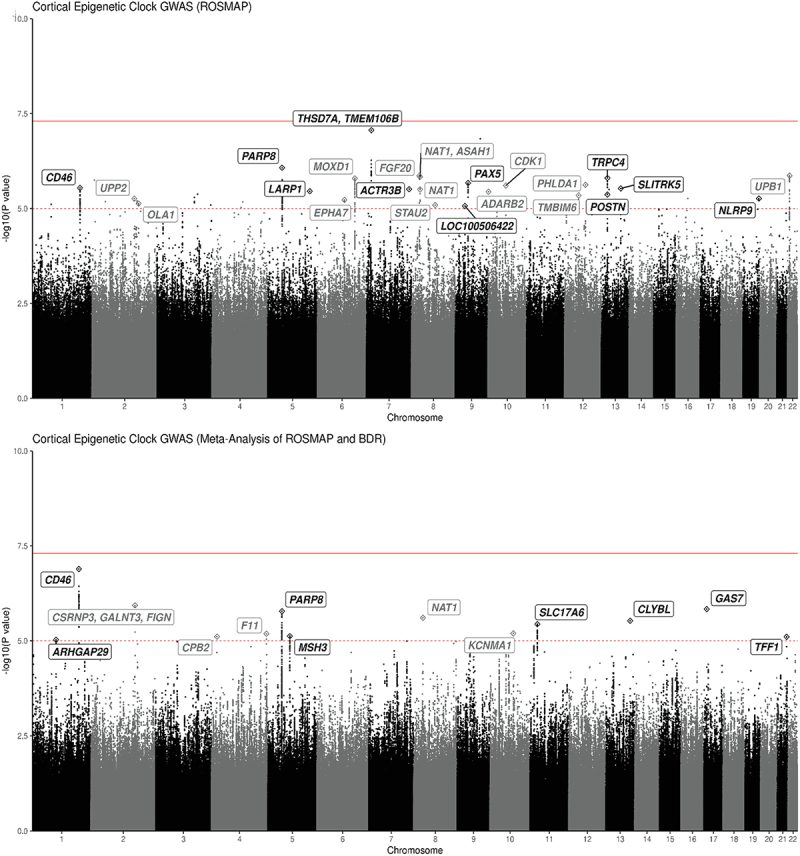
Manhattan plot: GWAS of cortical clock age for ROSMAP and Meta-analysis of ROSMAP/BDR.
* In the top panel, Manhattan plot shows the Cortical Epigenetic Clock GWAS results for ROSMAP. The bottom panel shows the GWAS results for the meta-analysis combining ROSMAP and BDR. Dashed red line represents a suggestive threshold of p = 10−5. The solid-line represents the genome-wide significance level of p = 5 × 10−8. Labels show the closest genes to GWAS lead SNPs with MAF >5%.
Table 2.
Leading SNPs from GWAS of cortical clock: meta-analysis of ROSMAP and BDR.
| Chr:pos | Effect allele | Non-effect | EAF | beta | p-value | Gene annotation | ||
|---|---|---|---|---|---|---|---|---|
| Rs4844620 | 1:207980901 | A | G | 0.12 | 0.886 | 1.29E-07 | CD46 | Membrane cofactor 9, protects injury by complement, modulates T cell activation. inflammatory disorders, cancers |
| rs17187636 | 2:166309615 | A | G | 0.08 | 1.266 | 1.17E-06 | CSRNP3, GALNT3, FIGN | CSRNP3: DNA-binding transcription factor, apoptotic processes and transcriptional activity, related to education, bone density GALNT3: oligosaccharide biosynthesis, phosphate homeostasis FIGN: microtubule organization, locomotion, neuropathy |
| rs17743504 | 17:10080837 | A | T | 0.12 | 1.008 | 1.47E-06 | GAS7 | In purkinje neurons, intraocular pressure, glaucoma |
| Rs34403329 | 8:18029698 | A | G | 0.35 | −0.826 | 2.48E-06 | NAT1 | Folate catabolism, bladder cancer, urate, depression, blood pressure |
| Rs13180000 | 5:50132506 | A | T | 0.22 | −0.825 | 1.66E-06 | PARP8 | Protein ADP-ribosylase activity, diabetes, complement component C6 |
| rs9557340 | 13:100580750 | A | G | 0.32 | 0.713 | 2.97E-06 | CLYBL | Mitochondrial enzyme, regulation of cobalamin metabolic process, vitamin B12 levels |
| rs2593685 | 11:22391419 | T | C | 0.41 | 0.671 | 3.65E-06 | SLC17A6 | Neurotransmitter transporter, synaptic activity |
| rs4979892 | 10:79341734 | T | C | 0.46 | −0.626 | 6.39E-06 | KCNMA1 DLG5 |
KCNMA1: potassium channel, smooth muscle contraction, neurotransmitter release, neuron excitability, epilepsy DLG5: cell scaffolding, cell proliferation, dendritic spine formation, synaptogenesis |
| rs4253425 | 4:187205929 | A | C | 0.10 | 0.959 | 6.56E-06 | F11 CYP4V2 KLKB1 |
F11: coagulation factor XI, clotting, VTE, TPA levels |
| rs836815 | 5:79995116 | A | G | 0.30 | −0.679 | 7.52E-06 | MSH3 | DNA repair, cortical thickness, huntington disease, endometrial cancer |
| rs13105396 | 4:17047358 | T | C | 0.06 | 1.445 | 7.79E-06 | CPB2 | Hydrolyze c-terminal peptide bonds, downregulates fibrinolysis, complement cascade, thrombosis |
| rs79932664 | 21:43779709 | T | C | 0.09 | 1.038 | 7.87E-06 | TFF1 | Secretory protein |
| rs17111206 | 1:94646439 | A | G | 0.06 | 1.321 | 9.39E-06 | ARHGAP29 | Rho GTPase signaling, cleft palate |
In secondary analyses of our GWAS of cortical clock age in ROSMAP specimens, with a larger number of SNPs than available for the meta-analysis, we identified 304 SNPs meeting our predefined criteria for suggestive statistical significance (p < 10−5) (Figure 2 and eTable 2). We found no evidence of genomic inflation (eFigure 1). After excluding SNPs with effect allele frequency less than 5%, we identified ‘leading’ SNPs from 24 independent loci. Table 3 shows the 24 lead SNPs with the highest statistical significance for each independent loci. The top SNP was rs4721030, which was close to genome-wide significant (p = 8.64 × 10−8). This SNP was annotated with two nearby genes, TMEM106B (transmembrane protein 106B) and THSD7A (thrombospondin type 1 domain containing 7A) on chromosome 7. TMEM106B gene appears to be a key regulator of aging, including influences on microglial proliferation and survival and involvement in myelination and lysosomal pathways [32]. TMEM106B has been previously associated with Alzheimer’s dementia and other neurodegenerative phenotypes in multiple studies (i.e., TDP-43 proteinopathy, brain inflammation, and amyloid deposition in the brain) [30,32–37]; further, our own previous research in ROSMAP has found links of TMEM106B with LATE-NC and worse cognitive resilience to neuropathology [16,38].
Table 3.
Leading SNPs from GWAS of cortical clock: ROSMAP.
| Chr:pos | Effect allele | Non-effect | EAF | Beta | p-value | Gene annotation | ||
|---|---|---|---|---|---|---|---|---|
| rs4721030 | 7:12137067 | G | T | 0.09 | 8.728 | 8.64E-08 | THSD7A, TMEM106B | THSD7A: membrane-associated N-glycoprotein, neurovasculature, education TMEM106B: microglial survival/proliferation, myelination, lysosomal pathwyas, neurodegeneration |
| rs13180000 | 5:50132506 | A | T | 0.22 | −0.993 | 8.44E-07 | PARP8 | See above |
| Rs3788369 | 22:24892973 | G | A | 0.05 | 2.863 | 1.36E-06 | UPB1 | Pyrimidine degradation pathway |
| Rs11998660 | 8:16546686 | G | A | 0.14 | 8.594 | 1.37E-06 | FGF20 | Fibroblast growth factor, mitogenesis, cell survival, neurotrophic factor, CNS development and function |
| Rs12386903 | 8:18000966 | C | G | 0.35 | −1.770 | 1.47E-06 | NAT1, ASAH1 | ASAH1: acid ceramidase family, innate immune system, sphingolipid metabolism, cell signaling, seizures |
| Rs4943525 | 13:38206239 | T | C | 0.57 | 0.791 | 1.57E-06 | TRPC4 | Cation channels, endothelial permeability, vasodilation, neurotransmitter release, epilepsy |
| rs6925799 | 6:132467591 | G | A | 0.27 | 4.489 | 1.60E-06 | MOXD1 | Dopamine catabolic process, endoplasmic reticulum membrane protein, |
| rs7032313 | 9:36927081 | T | C | 0.52 | −0.763 | 2.13E-06 | PAX5 | Transcription factor, B lymphocytes |
| rs73133134 | 12:76420006 | T | C | 0.12 | −1.112 | 2.38E-06 | PHLDA1 | Apoptosis, neural development, IGF-1 |
| rs10761557 | 10:62523410 | G | A | 0.29 | 0.800 | 2.49E-06 | CDK1 | Protein kinase complex, M phase promoting factor |
| rs4844620 | 1:207980901 | A | G | 0.12 | 0.916 | 2.86E-06 | CD46 | See above |
| rs1343559 | 13:87264407 | G | C | 0.88 | −0.925 | 2.96E-06 | SLITRK5 | Membrane protein, homologous with neurotrophin receptors |
| rs77221864 | 7:152650529 | G | A | 0.06 | 1.508 | 3.09E-06 | ACTR3B | Actin-related proteins, cytoskeleton, cell motility, neuropathy |
| rs34403329 | 8:18029698 | A | G | 0.15 | −0.955 | 3.13E-06 | NAT1 | See above |
| rs7731137 | 5:154005654 | G | T | 0.57 | −0.885 | 3.50E-06 | LARP1 | RNA binding protein, mTORC1 complex, growth signals, nutrient availability |
| rs11250535 | 10:1504987 | G | C | 0.21 | −0.858 | 3.61E-06 | ADARB2 | RNA editing enzyme, diabetes, NFT, ALS |
| rs9315498 | 13:38099921 | A | C | 0.39 | −0.736 | 4.27E-06 | POSTN | Extracellular matrix protein, tissue development and regeneration, asthma |
| rs114109920 | 12:50159398 | A | G | 0.07 | 7.0979 | 4.51E-06 | TMBIM6 | Negative regulation of RNA metabolic processes and of intrinsic apoptotic signaling pathway |
| rs77378270 | 19:56229834 | C | T | 0.07 | −1.296 | 5.46E-06 | NLRP9 | Innate immune system, inflammation |
| rs116430246 | 2:158855781 | T | C | 0.08 | 1.881 | 5.47E-06 | UPP2 | Cleavage of uridine and deoxyuridine to uracil and ribose |
| rs1596489 | 6:92915192 | T | G | 0.16 | −0.951 | 5.95E-06 | EPHA7 | Protein tyrosine kinase family, axon guidance, brain development, cancers |
| rs6752755 | 2:175093901 | G | C | 0.15 | 0.882 | 7.42E-06 | OLA1 | GTPase protein family, Cancer (breast and ovarian) |
| rs55639005 | 8:74628306 | C | T | 0.12 | −0.920 | 8.12E-06 | STAU2 | dsDNA binding protein, transport of neuronal RNA from cell body to dendrite |
| rs10812360 | 9:26190219 | C | T | 0.55 | −0.737 | 8.56E-06 | LOC100506422 |
Most importantly, several SNPs were identified both in ROSMAP and in the meta-analysis of ROSMAP/BDR, suggesting consistent findings across the cohorts. In particular, rs4244620, was a leading SNP in both, including the strongest association in the meta-analysis. In addition, both sets of analyses pointed to rs34403329 on chromosome 8 (p = 2.48 × 10−6), which was annotated with two nearby genes, NAT1 (N-acetyltransferase 1) and ASAH1 (N-acylsphingosine amidohydrolase 1), and rs1318000 on chromosome 5 (p = 1.66 × 10−6), which was annotated to the gene PARP8 (poly[ADP ribose] polymerase family member 8). (Note: The SNP with the strongest p-value from the GWAS of ROSMAP, rs4721030, was not in the BDR data and thus was not be tested in the meta-analysis.)
In sensitivity analyses, in models controlling for neuron proportion, we did not find any meaningful differences in the relations of leading SNPs to cortical clock age (eTable 3). Results were also largely consistent with analyses controlled for seven different cell-type proportions (eTable 4), which were estimated using single-cell methylation sequencing as the reference.
Expression quantitative trait loci (eQTL)
Next, we leveraged expression QTLs derived from bulk RNA-Seq data in 2,865 cortex specimens from BrainMeta, to examine the leading SNPs in meta-analysis and in ROSMAP GWAS (Figure 3a). In the 13 lead SNPs from the ROSMAP/BDR meta-analysis, we found that five SNPs (rs4844620, rs17187637, rs4979892, rs4253425, rs836815) exhibited an FDR-significant cis-eQTL effect (Figure 3a) in 8 transcripts in the ROSMAP bulk transcriptomic data. From the leading SNPs in ROSMAP, we found that five SNPs (rs4844620, rs6752755, rs7032313, rs12386903, rs3788369) exhibited an FDR-significant cis-eQTL effect in five transcripts (CD46, OLA1, PAX5, ASAH, ADORA2A) (Figure 3a). Overall, transcripts included genes involved in a range of pathways, including inflammation and immune response (CD46, [31] PAX5, [39] ADORA2A [40], energetics (OLA1 [41]), signal transduction (DLG5 [42]), clotting (F11 [43]) and DNA repair (MSH3 [44]).
Figure 3.
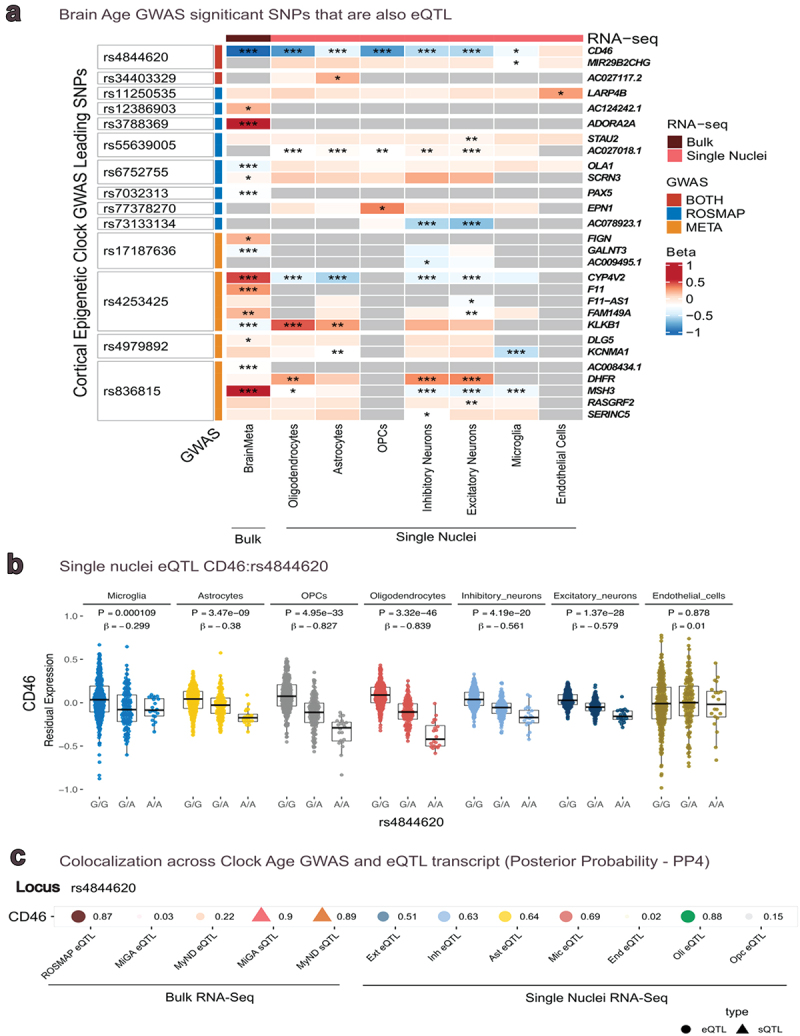
Expression quantitative trait Loci** in panel a, Heatmap show the GWAS lead SNPs that are FDR significant eQTL in bulk brain RNAseq (BrainMeta), and sn-RNAseq cell specific eQTL. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. The strongest relation of SNP to expression level was identified for rs4844620 with CD46. In panel b, we show CD46 expression levels across genotypes of rs4844620 in 7 cell types from ROSMAP single nucleus RNAseq. This was the strongest finding for the snRnaseq results. In panel c, colocalization analyses demonstrate the likelihood that a SNP is causal for both cortical clock age and for expression levels. The value to the right of each point is the posterior probability (PP) that the SNP is related to clock age and to the QTL; PP > 0.8 is standardly considered as high likelihood the SNP is causal for both traits. ROSMAP eQTL are from frontal cortex; MiGA are from primary monocytes; MyND are primary microglia; single cell eQTL are from ROSMAP frontal cortex.
We also examined eQTLs in seven cell types from the single nucleus RNA-Seq in ROSMAP cortex (Figure 3b and eTable 5). Most interestingly, we found that rs4844620 exhibited significant eQTL effects for CD46 gene expression in six cell types, such that each additional effect allele was associated with lower CD46 expression (Figure 3b). The strongest associations were observed in oligodendrocyte precursor cells (p = 4.95 × 10−33) and in oligodendrocytes (p = 3.32 × 10−46). For the SNPs from the ROSMAP/BDR meta-analysis, there were no further significant single nucleus eQTLs beyond those for rs4844620.
Colocalization across clock age and quantitative trait loci
To better evaluate whether lead SNPs may be associated with both clock age as well as gene expression, we examined colocalization for the lead SNPs and five different QTL datasets (Figure 3c), with a focus on immune systems: eQTL from our ROSMAP DLPFC and eQTL and sQTL from MyND (sorted monocytes) and MiGA (microglia). We found that rs4844620 (posterior probability > 0.8) colocalized across clock age, CD46 eQTL in ROSMAP brain tissue as well as CD46 sQTL in both monocytes and microglia. This suggests that rs4844620 is a common causal variant of clock age as well as the three different QTLs, providing support for a role of the CD46 gene and immune pathways in DNA methylation aging in brain.
We also conducted colocalization analyses across our lead SNPs and the seven cell types in the ROSMAP single-nucleus eQTL (Figure 3c). We found that observed associations with cortical clock age and with CD46 expression in oligodendrocytes appeared to be due to the single colocalizing variant rs4844620 (PP > 0.8). Thus, the CD46 gene with respect to oligodendrocyte biology may have particular relevance.
Functional analysis
When we used snpXplorer to examine biologic processes that may underlie cortical clock age, in the lead SNPs, there were no significant findings after correction for multiple comparisons. Nonetheless, two pathways were nominally significant (eTable 6): drug metabolism (p = 0.009) and pyrimidine nucleoside catabolic processes (p = 0.04).
With snpXplorer, we also identified SNPs and genes that overlapped with previous GWAS association studies. We found that rs4844620 was reported in previous GWAS of age-related macular degeneration. In addition, several genes from our GWAS were previously related to type 2 diabetes and to waist circumference, as well as to depression and cognitive function. These results may suggest that metabolism or metabolic dysregulation underlie DNA methylation age in brain.
Aging phenotypes
In our last steps, to help identify clinical relevance, we examined if each leading SNP was associated with aging phenotypes measured in the ROSMAP cohorts, after controlling for age at death, sex, education, and cohort (eTable 7). It is difficult to determine appropriate correction for multiple comparisons, since many of these aging outcomes are correlated with each other; we considered nominally significant findings for any phenotype (Figure 4a) and focus here on the strongest results. Perhaps most interesting, rs4844620 (which was identified in the ROSMAP GWAS, the ROSMAP/BDR meta-analysis, and exhibited cis-eQTL effects for CD46 in the bulk RNA-Seq and snRNA-Seq, as described above), was associated with lower level of baseline cognition (β=-0.10, p = 0.030), faster slopes of cognitive decline (β=-0.01, p = 0.007), and with greater level of Parkinsonian signs at baseline (β = 0.11, p = 0.04), indicating pleiotropic relations with cognitive and motor phenotypes. Among additional leading SNPs, rs9557340 was related to multiple neuropathologies and to cognition. Specifically, this SNP was associated with higher global AD pathologic burden (β = 0.04, p = 0.0047), greater β-amyloid load (β = 0.007, p = 0.014), greater tau tangle density (β = 0.11, p = 0.0022), higher odds of dementia (odds ratio = 1.17, p = 0.016), and to faster slopes of cognitive decline (β=-0.10, p = 0.014).
Figure 4.
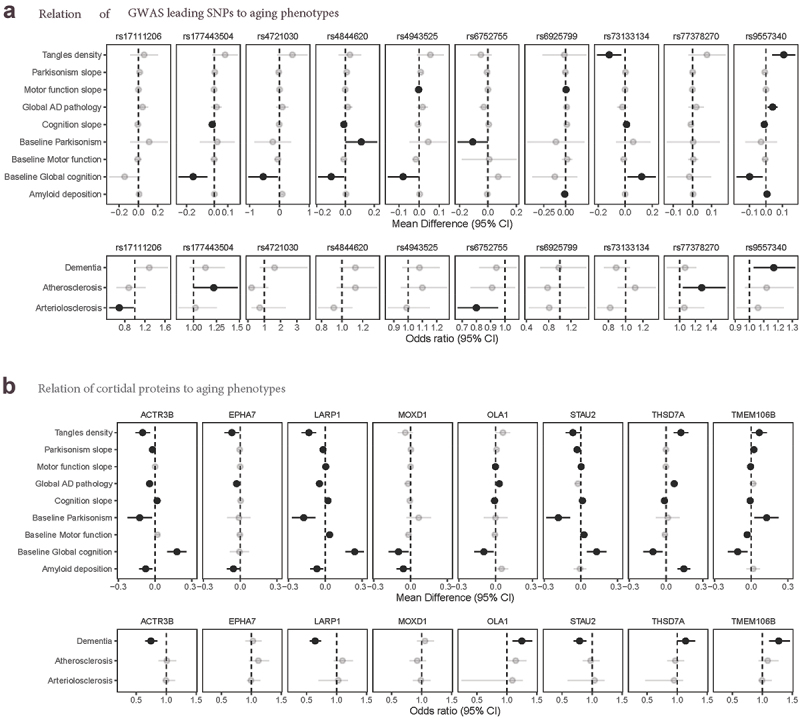
Relations of SNPs and cortical proteins to aging Phenotypes**, panel a: we show relations of leading SNPs to aging phenotypes, for SNPs with nominally significant relations with phenotypes. For continuous outcomes, we show mean differences from linear or linear mixed effects models; for categorical outcomes, we show odds ratios from logistic regression models. All models controlled for age at death, sex, and education. Of particular interest here, rs4844620 was related to lower baseline cognition (p = 0.031), steeper slopes of cognitive decline over time (p = 0.007), higher baseline levels of Parkinsonian signs (p = 0.046). Additionally, rs9557340 was related to more global AD neuropathology (p = 0.0047), more amyloid-β load (p = 0.014), greater PHFtau tangle density (p = 0.0022), lower baseline cognition (p = 0.014) and higher odds of dementia (p = 0.0038). panel b: we were able to examine protein levels in frontal cortex, based on annotated genes in our GWAS (14 genes of interest had corresponding protein levels in our proteomics data). We show here results for proteins which reached nominal significance for phenotypes. Of particular interest here, higher levels of the protein THSD7A in prefrontal cortex were related to more global AD pathology (p < 10−5), more amyloid-β load (p < 10−5), greater PHFtau tangle density (p < 10−5), lower baseline cognition (p = 0.007), steeper slopes of cognitive decline (p = 0.001) and higher odds of dementia (p = 0.046). Higher levels of TMEM106B protein were related to greater PHFtau tangle density (p = 0.03), more parkinsonism at baseline (p = 0.01) and steeper slopes over time (p = 0.0001), lower motor function at baseline (p = 0.001) and steeper slopes (p < 10−5), lower baseline cognition (p = 0.007), steeper slopes of cognitive decline (p = 0.04) and higher odds of dementia (p = 0.0004). Solid colors indicate significant association.
Finally, using available proteomics data in over 800 ROSMAP participants, we were able to select 14 proteins measured in DLPFC (ACTR3B, ASAH1, CD46, EPHA7, LARP1, MOXD1, OLA1, NAT1, SLITRK5, POSTN, STAU2, TMEM106B, THSD7A, TMBIM6), which corresponded to annotated genes or eQTLs for lead SNPs in our GWAS. We examined the relation of these 14 proteins with neuropathologies, cognitive, and motor phenotypes, after controlling for age at death, sex, education, and cohort (eTable 8). Again, we considered any nominally significant relation of protein level to phenotypes, and focus here on the strongest findings (Figure 4B). Notably, our GWAS SNP rs4721030 on chromosome 7 was annotated with two genes, TMEM106B and THSD7A; we found broad relations of both proteins to most of the aging phenotypes we examined. In particular, TMEM106B protein levels were strongly related to odds of dementia (p = 0.0004) and lower level of cognitive function at baseline (p = 0.007), as well as to worse motor function (motor function at baseline, p = 0.001; slopes of decline, p < 10−5) and more Parkinsonian signs (baseline, p = 0.01; slopes, p < 10−5). THSD7A protein levels were strongly to greater global AD pathology, to greater amyloid-β load and PHFtau tangle density (all p < 10−5), as well as to lower level of cognitive function at baseline (p = 0.007) and faster slopes of cognitive decline (p = 0.001).
Discussion
We conducted a GWAS of cortical epigenetic clock age calculated in brain tissue, among 1,340 older, deceased participants from three cohorts; we further leveraged transcriptomic, proteomic, and phenotypic data from participants, as well as publicly available data, to identify converging evidence of genes underlying cortical epigenetic clock age. In particular, our results supported several genes, especially CD46, as candidates of interest with potentially broad roles in cortical clock age.
Several previous studies have also conducted GWAS of epigenetic clocks in both peripheral blood and brain tissue [45–48]. Consistent with our findings, the largest GWAS of five clocks, calculated in blood samples (n = 40,000),45 prioritized CD46 as a possible determinant of epigenetic age from the Horvath clock. Additionally, CLYBL (Citramalyl-CoA Lyase) was prioritized as a predictor of the PhenoAge clock; we found that a loci (rs9557340) in the CLYBL gene was related to cortical clock age and was strongly related to AD neuropathologies and to cognition – suggesting clinical relevance of CLYBL gene to brain aging. However, in a GWAS (n = 1100) of the Horvath clock in brain tissue (which included some of our ROSMAP participants) [47], there was no overlap of their primary findings in prefrontal cortex with ours for the cortical clock; these samples spanned a much wider age range than ours, and there may be differences in genetic correlates of aging at different ages. Nonetheless, overall, there is some evidence that specific genes may have systemic effects on biologic aging.
In terms of our own findings, CD46 gene encodes the membrane cofactor protein, an ubiquitously expressed complement regulatory protein. CD46 protects host cells from injury by complement and appears to link innate and adaptive immune responses. The complement system regulates central nervous system function, and complement-mediated neuroinflammation may be involved in a range of aging-related neurodegenerative disorders, from cognitive impairment to age-related macular degeneration [49]. Other genes in the complement system are strongly related to dementia, including CR1, C1S, CD33, and TREM2; interestingly, CR1, a well-established genetic risk factor for dementia, is a receptor for complement C3b and C4b proteins, and CD46 mediates inactivation of C3b and C4b, pointing to potential mechanistic pathways [50]. Further, in our results, rs4844620 appeared to be a common causal SNP across epigenetic age and eQTLs for CD46 in cortex tissue, as well as in primary monocytes and microglial samples, supporting the role of CD46 in immune response in the brain. In addition, our single nucleus data from DLPFC further identified eQTL effects for CD46 in oligodendrocytes, and colocalization of this SNP with epigenetic age and CD46 expression in oligodendrocytes. This may support recent data that oligodendrocytes could have immunomodulatory properties [51]. Overall, there is compelling evidence, from our study and others, to support CD46 as a target for further research.
In additional results from the GWAS of ROSMAP alone, the strongest loci in relation to clock age in our GWAS was rs4721030 on chromosome 7, which was associated with older epigenetic age, and nearly reached genome-wide significance. We note that the beta value we found for this SNP in relation to clock age was 8.7, which is much larger than many of our other findings, indicating more caution is warranted in interpreting this result. For context, the mean beta value in our 304 SNPs of interest in ROSMAP was 2.6 (SD 2.5); that is, the beta for rs4721030 was 2.4 standard deviations greater than the mean beta in our research here, which is large although not an extreme outlier. Further, we could not examine this SNP in the meta-analysis, since it was not present in the BDR data, even after imputation, likely due to the highly specialized array used by BDR. For these reasons, this finding should be carefully considered and interpreted cautiously. Nonetheless, the SNP was annotated with two genes, TMEM106B and THSD7A. Previous work has implicated the TMEM106B gene in neurodegenerative phenotypes, especially TDP-43 pathology [38]. THSD7A is a soluble form of membrane-associated N-glycoprotein, produced by cells of endothelial and neuronal origin. In zebrafish, THSD7A may have a role in the neurovasculature [52], and THSD7A has been related to educational attainment [53]; in recent research using GWAS summary statistics, TMEM106B/THSD7A were reported as pleiotropically related to dementia and major depression disorder [54]. Interestingly, we found associations of higher THSD7A/TMEM106B protein levels in ROSMAP DLPFC with a wide array of aging phenotypes, from AD pathologies to lower cognitive function to worse motor function. Thus, our findings in GWAS as well as in cortical protein levels, together with the existing literature, may suggest that TMEM106B and THSD7A have relevance to brain aging.
There are substantial strengths to our work here. We assembled a sample of older individuals for this GWAS of cortical clock age; the uniform older ages of participants may be particularly important for evaluating the genetic architecture of cortical clock age, as there is substantial evidence that epigenetic clocks function differently at older than at younger ages [3,6]. We also had available a wide array of genomic and phenotypic data; this enabled us to validate and prioritize genes of interest despite the absence of a replication dataset. Limitations of our study should also be considered. Most importantly, our sample size was small for detecting genome-wide significant findings. However, many SNPs achieved a predetermined, ‘suggestive’ threshold of 10−5, which we [16] and others [55] have previously used. Further, to prioritize candidate loci, we were able to validate results with a range of additional information, from gene expression (both bulk and single-cell expression) to protein levels in brain tissue and evaluation of neuropathologic and clinical aging phenotypes. Thus, we were able to confirm findings via multiple lines of converging evidence. Nonetheless, we likely missed important SNPs which did not meet even our suggestive threshold. Second, the DNAm states as well as the transcriptomic and proteomic data were all derived from cortex. Thus, we may have missed genes that are primarily relevant in other brain regions. However, we focus on DLPFC given its critical role in a wide range of higher order human behaviour; additionally, evidence suggests a good correlation of epigenetic age across brain regions [4,47]. Finally, another limitation is our primarily non-Latino white participants, across all three cohorts. Evidence indicates that neurodegenerative diseases of aging, such as dementia, differ across diverse groups; for example, older African Americans appear to have higher risk of dementia, and Latinx individuals appear to develop dementia at younger ages, than non-Hispanic whites. [56,57]. Thus, future research is clearly needed in minoritized older participants to better understand brain aging in groups who often have worse health.
Supplementary Material
Acknowledgments
This work was supported by the National Institute on Aging under Grants R01AG69904, P30AG10161, P30AG72975, R01AG15819, R01AG17917, R01AG36042, U01AG61356, U01AG46152, and U01AG46161/U01AG61357 (Accelerating Medicine Partnership). DNA methylation data generated in the Brains for Dementia Research (BDR) cohort were supported by the Alzheimer’s Society and Alzheimer’s Research UK. The BDR is jointly funded by Alzheimer’s Research UK and the Alzheimer’s Society in association with the Medical Research Council. Dr Mill was supported by the Medical Research Council under Grant M008924.
ROSMAP resources can be requested at https://www.radc.rush.edu and www.synapse.org
BDR data are available at Dementias Platform UK (DPUK, https://portal.dementiasplatform.uk) and through the Medical Research Council (https://mrc.ukri.org/research/facilities-and-resources-for-researchers/brain-banks/).
Funding Statement
The work was supported by the Alzheimer’s Society Alzheimer’s Research UK Medical Research Council [M008924]; National Institute on Aging [U01AG46161, U01AG61357]; National Institute on Aging [P30AG10161, R01AG15819, R01AG17917, U0146152]; National Institute on Aging [R01AG36042]; National Institute on Aging [R01AG69904, P30AG72975].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2024.2392050
References
- [1].Alzheimer’s Association . 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023;19(4). doi: 10.1002/alz [DOI] [PubMed] [Google Scholar]
- [2].Jylhava J, Pedersen NL, Hagg S.. Biological age predictors. EBioMedicine. 2017;21:29–18. doi: 10.1016/j.ebiom.2017.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shireby GL, Davies JP, Francis PT, et al. Recalibrating the epigenetic clock: implications for assessing biological age in the human cortex. Brain. 2020;143(12):3763–3775. doi: 10.1093/brain/awaa334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Grodstein F, Lemos B, Yu L, et al. Characteristics of epigenetic clocks across blood and brain tissue in older women and men. Front Neurosci. [2021 Jan 7];14:555307. doi: 10.3389/fnins.2020.555307 PMID: 33488342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Grodstein F, Lemos B, Yu L, et al. The association of epigenetic clocks in brain tissue with brain pathologies and common aging phenotypes. Neurobiol Dis. 2021. Sep;157:105428. doi: 10.1016/j.nbd.2021.105428 Epub 2021 Jun 19. PMID: 34153464; PMCID: PMC8373772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].El Khoury LY, Gorrie-Stone T, Smart M, et al. Systematic underestimation of the epigenetic clock and age acceleration in older subjects. Genome Biol. 2019;20(1):283. doi: 10.1186/s13059-019-1810-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bennett DA, Buchman AS, Boyle PA, et al. Religious orders study and rush memory and aging project. J Alzheimer’s Disease. 2018;64(s1):S161–S189. doi: 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Francis PT, Costello H, Hayes GM, et al. Brains for dementia research: evolution in a longitudinal brain donation cohort to maximize current and future value. J Alzheimers Dis. 2018;66(4):1635–1644. doi: 10.3233/JAD-180699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17(9):1156–1163. doi: 10.1038/nn.3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pidsley R, Wong Cc Y, Volta M, et al. A data-driven approach to preprocessing illumina 450K methylation array data. BMC Genomics. [2013 May 1];14(1):293. doi: 10.1186/1471-2164-14-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bennett DA, Yu L, De Jager PL. Building a pipeline to discover and validate novel therapeutic targets and lead compounds for Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):617–630. doi: 10.1016/j.bcp.2014.01.037 PMID: 24508835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795 PMID: 17701901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].De Jager PL, Shulman JM, Chibnik LB, et al. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol Aging. 2012;33(5):1017e1–101715. doi: 10.1016/j.neurobiolaging.2011.09.033 PMID: 22054870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Patterson NJ, Plenge RM, Weinblatt ME, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904±9. doi: 10.1038/ng1847 PMID: 16862161. [DOI] [PubMed] [Google Scholar]
- [15].Young J, Gallagher E, Koska K, et al. Genome-wide association findings from the brains for dementia research cohort. Neurobiol Aging. 2021. Nov;107:159–167. doi: 10.1016/j.neurobiolaging.2021.05.014 Epub 2021 Jun 26. PMID: 34183186. [DOI] [PubMed] [Google Scholar]
- [16].White CC, Yang HS, Yu L, et al. Identification of genes associated with dissociation of cognitive performance and neuropathological burden: multistep analysis of genetic, epigenetic, and transcriptional data. PLOS Med. [2017 Apr 25];14(4):e1002287. doi: 10.1371/journal.pmed.1002287 PMID: 28441426; PMCID: PMC5404753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lu AT, Xue L, Salfati EL, et al. GWAS of epigenetic aging rates in blood reveals a critical role for TERT. Nat Commun. [2018 Jan 26];9(1):387. doi: 10.1038/s41467-017-02697-5 PMID: 29374233; PMCID: PMC5786029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. [2010 Sep 1];26(17):2190–2191. doi: 10.1093/bioinformatics/btq340 Epub 2010 Jul 8. PMID: 20616382; PMCID: PMC2922887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8(3):290–302. doi: 10.4161/epi.23924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yap CX, Vo DD, Heffel MG, et al. Brain cell-type shifts in Alzheimer’s disease, autism, and schizophrenia interrogated using methylomics and genetics. Sci Adv. [2024 May 24];10(21):eadn7655. doi: 10.1126/sciadv.adn7655 Epub 2024 May 23. PMID: 38781333; PMCID: PMC11114225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Watanabe K, Taskesen E, van Bochoven A, et al. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi: 10.1038/s41467-017-01261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qi T, Wu Y, Fang H, et al. Genetic control of RNA splicing and its distinct role in complex trait variation. Nat Genet. 2022;54(9):1355–1363. doi: 10.1038/s41588-022-01154-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Navarro E, Udine E, de Paiva Lopes K, et al. Dysregulation of mitochondrial and proteolysosomal genes in Parkinson’s disease myeloid cells. Nat Aging. 2021. Sep;1(9):850–863. doi: 10.1038/s43587-021-00110-x Epub 2021 Sep 14. PMID: 35005630; PMCID: PMC8728893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lopes KP, Snijders GJL, Humphrey J, et al. Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies. Nat Genet. 2022. Jan;54(1):4–17. doi: 10.1038/s41588-021-00976-y Epub 2022 Jan 6. PMID: 34992268; PMCID: PMC9245609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genet. [2014 May 15];10(5):e1004383. doi: 10.1371/journal.pgen.1004383 PMID: 24830394; PMCID: PMC4022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fujita M, Gao Z, Zeng L, et al. Cell-subtype specific effects of genetic variation in the aging and Alzheimer cortex. bioRxiv. 2022. doi: 10.1101/2022.11.07.515446. [DOI] [PubMed] [Google Scholar]
- [27].Green GS, Fujita M, Yang H-S, et al. Cellular dynamics across aged human brains uncover a multicellular cascade leading to Alzheimer’s disease. Cellular Dyn Across Aged Hum Brains Uncover a Multicell Cascade Leading To Alzheimer’s Disease. bioRxiv. 2023;19(S24). doi: 10.1101/2023.03.07.531493 [DOI] [Google Scholar]
- [28].Tesi N, van der Lee S, Hulsman M, et al. snpXplorer: a web application to explore human snp-associations and annotate snp-sets. Nucleic Acids Res. [2021 Jul 2];49(W1):W603–612. doi: 10.1093/nar/gkab410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wilson RS, Arnold SE, Schneider JA, et al. The relationship between cerebral Alzheimer’s disease pathology and odour identification in old age. J Neurol, Neurosurg Psychiatry. 2007;78:30–35. doi: 10.1136/jnnp.2006.099721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yu L, Tasaki S, Schneider JA, et al. Cortical proteins associated with cognitive resilience in community-dwelling older persons. JAMA Psychiatry. [2020 Jul 1];77(11):1–9. doi: 10.1001/jamapsychiatry.2020.1807 PMID: 32609320; PMCID: PMC7330835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Riley-Vargas RC, Gill DB, Kemper C, et al. CD46: expanding beyond complement regulation. Trends Immunol. 2004;25(9):496–503. doi: 10.1016/j.it.2004.07.004 [DOI] [PubMed] [Google Scholar]
- [32].Yang HS, White CC, Klein HU, et al. Genetics of gene expression in the aging human brain reveal TDP-43 proteinopathy pathophysiology. Neuron. [2020 Aug 5];107(3):496–508.e6. doi: 10.1016/j.neuron.2020.05.010 Epub 2020 Jun 10. PMID: 32526197; PMCID: PMC7416464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54(4):412–436. doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nicholson AM, Rademakers R. What we know about TMEM106B in neurodegeneration. Acta Neuropathol. 2016. Nov;132(5):639–651. doi: 10.1007/s00401-016-1610-9 Epub 2016 Aug 20. PMID: 27543298; PMCID: PMC5074873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Feng T, Lacrampe A, Hu F. Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol. 2021. Mar;141(3):327–339. doi: 10.1007/s00401-020-02246-3 Epub 2021 Jan 1. PMID: 33386471; PMCID: PMC8049516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang T, Pang W, Feng T. TMEM106B regulates microglial proliferation and survival in response to demyelination. Sci Adv. 2023;9(18):eadd2676. doi: 10.1126/sciadv.add2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schweighauser M, Arseni D, Bacioglu M, et al. Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature. 2022;605(7909):310–314. doi: 10.1038/s41586-022-04650-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yu L, De Jager PL, Yang J, et al. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology. [2015 Mar 3];84(9):927–934. doi: 10.1212/WNL.0000000000001313 Epub 2015 Feb 4. PMID: 25653292; PMCID: PMC4351662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Emelyanov AV, Kovac CR, Sepulveda MA, et al. The interaction of Pax5 (BSAP) with Daxx can result in transcriptional activation in B cells. J Biol Chem. [2002 Mar 29];277(13):11156–11164. doi: 10.1074/jbc.M111763200 Epub 2002 Jan 17. PMID: 11799127. [DOI] [PubMed] [Google Scholar]
- [40].Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414(6866):916–920. doi: 10.1038/414916a PMID 11780065. [DOI] [PubMed] [Google Scholar]
- [41].Koller-Eichhorn R, Marquardt T, Gail R, et al. Human OLA1 defines an ATPase subfamily in the obg family of gtp-binding proteins. J Biol Chem 2007. 2007;282(27):19928–19937. doi: 10.1074/jbc.M700541200 PMID 1743088. [DOI] [PubMed] [Google Scholar]
- [42].Song XQ, Li Q, Zhang J. A double-edged sword: DLG5 in diseases. Biomed Pharmacother. 2023. Jun;162:114611. doi: 10.1016/j.biopha.2023.114611 Epub 2023 Mar 29. PMID: 37001186. [DOI] [PubMed] [Google Scholar]
- [43].Emsley J, Pa M, Gailani D. Structure and function of factor XI. Blood. [2010 Apr 1];115(13):2569–2577. doi: 10.1182/blood-2009-09-199182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kleczkowska HE, Marra G, Lettieri T, et al. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001;15(6):724–736. doi: 10.1101/gad.191201 PMC 312660. PMID 11274057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McCartney DL, Min JL, Richmond RC, et al. Genome-wide association studies identify 137 genetic loci for DNA methylation biomarkers of aging. Genome Biol. [2021 Jun 29];22(1):194. doi: 10.1186/s13059-021-02398-9 PMID: 34187551; PMCID: PMC8243879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gibson J, Russ TC, Clarke TK, et al. A meta-analysis of genome-wide association studies of epigenetic age acceleration. PLOS Genet. [2019 Nov 18];15(11):e1008104. doi: 10.1371/journal.pgen.1008104 PMID: 31738745; PMCID: PMC6886870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lu AT, Hannon E, Levine ME, et al. Genetic architecture of epigenetic and neuronal ageing rates in human brain regions. Nat Commun. [2017 May 18];8(1):15353. doi: 10.1038/ncomms15353 PMID: 28516910; PMCID: PMC5454371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lu AT, Hannon E, Levine ME, et al. Genetic variants near MLST8 and DHX57 affect the epigenetic age of the cerebellum. Nat Commun. [2016 Feb 2];7(1):10561. doi: 10.1038/ncomms10561 PMID: 26830004; PMCID: PMC4740877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fatoba O, Itokazu T, Yamashita T. Complement cascade functions during brain development and neurodegeneration. FEBS J. 2022;289(8):2085–2109. doi: 10.1111/febs.15772 [DOI] [PubMed] [Google Scholar]
- [50].Daskoulidou N, Shaw B, Torvell M, et al. Complement receptor 1 is expressed on brain cells and in the human brain. Glia. 2023;71(6):1–14. doi: 10.1002/glia.24355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].l K, Castelo-Branco G, Castelo-Branco G. Crossing boundaries: interplay between the immune system and oligodendrocyte lineage cells. Sem Cell Dev Biol. 2021;116:45–52. doi: 10.1016/j.semcdb.2020.10.013 [DOI] [PubMed] [Google Scholar]
- [52].Liu LY, Lin MH, Lai ZY, et al. Motor neuron-derived Thsd7a is essential for zebrafish vascular development via the notch-dll4 signaling pathway. J Biomed Sci. [2016 Aug 2];23(1):59. doi: 10.1186/s12929-016-0277-9 PMID: 27484901; PMCID: PMC4971630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee JJ, Wedow R, Okbay A, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet. [2018 Jul 23];50(8):1112–1121. doi: 10.1038/s41588-018-0147-3 PMID: 30038396; PMCID: PMC6393768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Santos FCD, Mendes-Silva AP, Nikolova Y, et al. Genetic overlap between mood disorders and Alzheimer’s disease: could it explain the clinical and epidemiological overlap of these disorders? Alzheimer’s Dement. 2021;17:e053913. doi: 10.1002/alz.053913 [DOI] [Google Scholar]
- [55].Lin X, Chan KK, Huang YT, et al. Genetic determinants for leisure-time physical activity. Med Sci Sports Exerc. 2018. Aug;50(8):1620–1628. doi: 10.1249/MSS.0000000000001607 PMID: 29538177; PMCID: PMC6087666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Barnes LL, Bennett DA. Alzheimer’s disease in African Americans: risk factors and challenges for the future. Health Aff (Millwood). 2014. Apr;33(4):580–586. doi: 10.1377/hlthaff.2013.1353 PMID: 24711318; PMCID: PMC4084964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hale JM, Schneider DC, Mehta NK, et al. Cognitive impairment in the U.S.: lifetime risk, age at onset, and years impaired. SSM Popul Health. 2020. Mar 31;11:100577. doi: 10.1016/j.ssmph.2020.100577 Erratum in SSM Popul Health. 2020 Dec10;12: 100715. PMID: 32300635; PMCID: PMC7 153285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ROSMAP resources can be requested at https://www.radc.rush.edu and www.synapse.org.
The BDR DNA methylation data have been deposited in the Dementias Platform UK (DPUK) data portal (https://portal.dementiasplatform.uk/CohortDirectory/Item?fingerPrintID=BDR) and the Gene Expression Omnibus (GEO) at accession number GSE197305.