

Abstract
ML-IAP (melanoma inhibitor of apoptosis) is a potent anti-apoptotic protein that is strongly up-regulated in melanoma and confers protection against a variety of pro-apoptotic stimuli. The mechanism by which ML-IAP regulates apoptosis is unclear, although weak inhibition of caspases 3 and 9 has been reported. Here, the binding to and inhibition of caspase 9 by the single BIR (baculovirus IAP repeat) domain of ML-IAP has been investigated and found to be significantly less potent than the ubiquitously expressed XIAP (X-linked IAP). Engineering of the ML-IAP-BIR domain, based on comparisons with the third BIR domain of XIAP, resulted in a chimeric BIR domain that binds to and inhibits caspase 9 significantly better than either ML-IAP-BIR or XIAP-BIR3. Mutational analysis of the ML-IAP-BIR domain demonstrated that similar enhancements in caspase 9 affinity can be achieved with only three amino acid substitutions. However, none of these modifications affected binding of the ML-IAP-BIR domain to the IAP antagonist Smac (second mitochondrial activator of caspases). ML-IAP-BIR was found to bind mature Smac with low nanomolar affinity, similar to that of XIAP-BIR2-BIR3. Correspondingly, increased expression of ML-IAP results in formation of a ML-IAP–Smac complex and disruption of the endogenous interaction between XIAP and mature Smac. These results suggest that ML-IAP might regulate apoptosis by sequestering Smac and preventing it from antagonizing XIAP-mediated inhibition of caspases, rather than by direct inhibition of caspases.
Keywords: apoptosis, inhibitor of apoptosis (IAP), melanoma inhibitor of apoptosis (ML-IAP), Smac/DIABLO (second mitochondrial activator of caspases/direct IAP binding protein with low pI), X-linked IAP (XIAP)
Abbreviations: Ac-LEHD-AFC, acetyl-Leu-Glu-His-Asp-7-amido-4-fluoromethylcourmarin; AVP-diPhe-FAM, 5-carboxyfluorescein-conjugated AVP-diphenylalanine-AKK; BIR, baculoviral IAP repeat; CARD, caspase recruitment domain; DIABLO, direct IAP binding protein with low pI; DTT, dithiothreitol; Hid-FAM, 5-carboxyfluorescein-conjugated AVPFAKK; IAP, inhibitor of apoptosis; IPTG, isopropyl β-D-thiogalactoside; ML-IAP, melanoma IAP; Ni-NTA, Ni2+-nitrilotriacetate; PDB, Protein Data Bank; RMSD, root mean square deviation; Smac, second mitochondrial activator of caspases; SPR, surface plasmon resonance; W310A etc., Trp310→Ala etc; XIAP, X-linked IAP
INTRODUCTION
IAP (inhibitor of apoptosis) proteins are generally thought to suppress apoptosis due, in part, to their ability to bind to and inhibit activated caspases, cytosolic cysteine/aspartate-specific-proteases, that are critical for the initiation and execution phases of apoptosis [1,2]. All IAP proteins contain one to three copies of the BIR (baculoviral IAP repeat) domain, a zinc-binding domain of approx. 80 amino acids, that are necessary for their interactions with a number of cytosolic target proteins, including activated caspases 3, 7, and 9, and natural IAP protein antagonists, such as Smac/DIABLO (second mitochondrial activator of caspases/direct IAP binding protein with low pI) and HtrA2/Omi. Different BIR domains, however, have differing affinities for these proteins, and thus distinct functions in the regulation of apoptosis. For instance, the second BIR domain of XIAP (X-linked IAP) together with the immediately preceding linker region (XIAP-BIR2) binds to and inhibits caspases 3 and 7 with inhibition constants in the ranges of 1–10 and 0.1–2 nM, respectively [3–5], whereas the third BIR domain of XIAP (XIAP-BIR3) specifically inhibits caspase 9 with an inhibition constant in the range of 10–20 nM [6–8]. By contrast, the single BIR domain of ML-IAP (melanoma IAP) has been shown to inhibit weakly caspases 3 and 9, but not caspase 7, although inhibition constants have not been reported [9].
The IAP-mediated inhibition of caspases can be countered by the mammalian mitochondrial protein Smac/DIABLO, which is released into the cytoplasm in response to pro-apoptotic stimuli [10,11]. The pro-apoptotic function of this IAP protein antagonist is dependent on a conserved 4-residue IAP protein-interaction motif (A-V/I-P/A-I/F/Y) found at the N-terminus of the mature post-translationally processed protein [12,13]. Structural studies have shown that the N-terminal peptide binds to a surface groove on the BIR domains, with the binding being stabilized by electrostatic interactions involving the conserved N-terminal alanine residue of the peptide, together with several intermolecular hydrogen bonds and hydrophobic interactions [8,14–17]. Smac/DIABLO-derived peptides have been shown to sensitize a number of different tumour cell lines to apoptosis induced by a variety of pro-apoptotic drugs [18–22].
Recent structures of XIAP-BIR2 in complex with caspase 3 or 7, and XIAP-BIR3 in complex with caspase 9, have revealed different mechanisms of caspase inhibition by these different BIR domains. The linker region preceding the BIR2 domain of XIAP binds tightly to the active site of caspases 3 and 7, thereby inhibiting the caspases by preventing substrate entry and catalysis [5,23,24]. Interactions are also seen between the processed N-terminus of the small subunit of caspase 3 and the peptide-binding groove of XIAP-BIR2 [5], although the question of whether these interactions are critical for caspase 3 binding or inhibition has yet to be settled [4,25]. By contrast, the peptide-binding groove of XIAP-BIR3 makes critical contacts with the N-terminus of the small subunit of caspase 9 [26]. Additional interactions between XIAP-BIR3 helices 3 and 5 and the caspase 9 homodimerization interface inhibit the caspase by trapping it in a catalytically inactive conformation [26]; activation of caspase 9 requires conformational changes that are induced by homodimerization [27,28].
In the present work ML-IAP-BIR binding to and inhibition of caspase 9 is investigated. In order to understand at a molecular level the observation that ML-IAP is a less potent inhibitor of caspase 9 than XIAP, a chimeric BIR domain was designed in which 11 residues correspond to those found in XIAP-BIR3, whereas the remainder correspond to ML-IAP-BIR. The chimeric protein binds to and inhibits caspase 9 significantly better than either of its parent BIR domains, but binds Smac-based peptides and mature Smac with affinities similar to those of native ML-IAP-BIR. Subsequent mutagenesis of ML-IAP-BIR shows that similar enhancement of affinity for caspase 9 can be achieved with just three amino acid substitutions. Finally, ML-IAP-BIR was shown to bind mature Smac with low nanomolar affinity similar to that of XIAP-BIR2-BIR3, suggesting that the potent anti-apoptotic effect of ML-IAP might be due to it antagonizing the XIAP–Smac interaction, rather than direct inhibition of caspase 9.
EXPERIMENTAL
Caspase 9, XIAP-BIR3, XIAP-BIR2-BIR3, and ML-IAP-BIR production
ΔCARD (caspase recruitment domain) human caspase 9 (lacking the first 138 residues) with alanine substitutions at residues 304–306 was produced as described previously [27]. The third BIR of XIAP (residues 241–348) was cloned into a pET15b vector (Novagen) to generate pet15bXIAPBIR3 and prepared as described previously [7]. Amino acids 124–356 of XIAP (designated XIAP-BIR2-BIR3) were cloned into a pET15b vector (Novagen) to generate pet15bXIAPBIR2BIR3. QuikChange® (Stratagene) mutagenesis was used to generate pet15bXIAPBIR2(C202AC213G)BIR3. ML-IAP-BIR (amino acids 63–179; designated MLBIR) was also subcloned into a pET15b vector (Novagen) for bacterial expression as described previously [15]. The above ML-IAP and XIAP mutants and deletions were also subcloned into pcDNA3.1 vector with a C-terminal FLAG tag for expression in mammalian cells. Plasmids expressing β-galactosidase, Myc–ML-IAP and Smac–FLAG have been described previously [9,18].
Full-length ML-IAP production
The cDNA encoding full-length ML-IAP plus an N-terminal His6 tag was cloned into the pET15b(+) expression vector (Novagen). ML-IAP was expressed in Escherichia coli strain BL21(DE3)pLysS. Expression of ML-IAP was induced with 0.5 mM IPTG (isopropyl β-D-thiogalactoside) for 5 h when cells had reached a D600=0.5. The protein was purified by affinity chromatography using chelating Sepharose (Pharmacia) charged with NiSO4 according to the manufacturer's instructions. Eluted ML-IAP protein was >95% pure, as judged by SDS/PAGE. The protein concentration of purified proteins was determined from the A280 based on the molar absorption coefficients calculated from the Edelhoch relationship [29].
Chimeric and mutant protein production
The pet15bMLBIR vector used for production of wild-type MLBIR was modified in a two-step PCR based approach to generate MLXBIR3SG. Firstly, amino acids 160 –179 of MLBIR were replaced with amino acids 336–348 of XIAP-BIR3 to give the plasmid pet15bMLXBIR3. Secondly, Ser150 of MLBIR was mutated to glycine to give pet15bMLXBIR3SG. The pet15bMLBIR vector was also modified to produce a C-terminally truncated version of MLBIR that has the same number of amino acids as MLXBIR3 and MLXBIR3SG (designated MLBIR-Q). An amino acid sequence alignment of the original ML-IAP-BIR construct (MLBIR) and XIAP-BIR3, is shown in Figure 1(A); the C-terminally truncated variant of MLBIR used in the present work (MLBIR-Q) ends at residue Gln172. The chimeric variants are illustrated schematically in Figure 1(B).
Figure 1. Protein amino acid sequences and constructs.

(A) Amino acid sequence alignment of MLBIR and XIAP-BIR3. The secondary structure of MLBIR is indicated above the sequence. Residues indicated with an asterisk are within 4 Å of the Smac-based peptide in the MLBIR–AVPIAQKSE complex structure [15]. Sequence identity between the wild-type ML-IAP construct and XIAP-BIR3 is indicated. MLBIR-Q corresponds to a seven residue C-terminal truncation of MLBIR (i.e. the C-terminal residue of MLBIR-Q is Gln172). The residues shown in bold italics in the sequence of XIAP-BIR3 are those that were exchanged into MLBIR-Q to produce the chimeric constructs MLXBIR3 and MLXBIR3SG. (B) Schematic representation of the chimeric variants. Residues present in wild-type MLBIR-Q are grey, whereas those present in wild-type XIAP-BIR3 are black. (C) Schematic representation of double and triple point mutants.
pet15bMLBIRQ and pet15bXIAPBIR3 were further mutated in a two-step PCR approach which incorporated the desired changes into internal PCR primers. The following double and triple mutants were generated using this strategy: pet15bMLBIRQ-S150G-Q167H, pet15bMLBIRQ-S150G-E168L, pet15bMLBIRQ-Q167H-E168L, pet15bMLBIRQ-S150G-Q167H-E168L and pet15bXIAPBIR3-G326S-H343Q-L344E. These mutants are illustrated schematically in Figure 1(C).
One litre cultures of E. coli strain BL21(DE3) transformed with pet15bMLXBIR3SG were induced with 1 mM IPTG for 4 h at 30 °C in the presence of 50 μM zinc acetate. Cells were pelleted and resuspended in 50 ml/l Buffer A [50 mM Tris (pH 8.0), 300 mM NaCl, 5 mM 2-mercaptoethanol, 0.5 mM PMSF, 2 mM benzamidine] with 5 mM imidazole. Cells were homogenized, microfluidized and centrifuged. Lysate was passed over Ni-NTA (Ni2+-nitrilotriacetate)–agarose (Qiagen) and eluted in Buffer A containing 300 mM imidazole. Finally, protein was passed over a Superdex 75 gel filtration (Pharmacia) column in buffer containing 50 mM Tris/HCl (pH 7.6), 200 mM NaCl, 5 mM DTT (dithiothreitol), 0.5 mM PMSF, 2 mM benzamidine, 50 μM zinc acetate. Protein was concentrated and stored at −80 °C. Samples of MLBIR-Q, MLXBIR3 and MLBIR-Q double and triple mutants were prepared similarly.
Smac production
A PCR product containing amino acids 56–239 (precursor numbering) of Smac was cloned into the XbaI/XhoI sites of pet21b+ (Novagen), generating a C-terminal His6-tag fusion protein. Pet21bSMAC was transformed into E. coli strain BL21(DE3) competent cells (Stratagene). Overnight cultures were diluted 1:100 and grown at 37 °C in Luria–Bertani media with 50 μg/ml carbenicillin to a D600 of 0.8 with vigorous shaking. IPTG was added to a final concentration of 1 mM and cultures were grown overnight at 16 °C. Cell pellets were resuspended in Buffer A with 5 mM imidazole and placed on ice for 30 min. Cells were homogenized, microfluidized and centrifuged at 26900 g for 45 min. Supernatant was loaded on to a Ni-NTA–agarose column (Qiagen), washed with 10 column volumes Buffer A with 10 mM imidazole, and eluted with 10 column volumes Buffer A with 300 mM imidazole. Fractions containing Smac protein were pooled, concentrated and loaded on to a Superdex 200 gel filtration column (Pharmacia) equilibrated with 50 mM Tris/HCl (pH 7.6), 300 mM NaCl, 0.5 mM PMSF, 2 mM benzamidine and 5 mM DTT. Fractions containing Smac protein were pooled and dialysed against 3 changes of buffer containing 50 mM Tris/HCl (pH 7.6), 0.5 mM PMSF, 2 mM benzamidine and 5 mM DTT. Dialysed sample was loaded on to a Q-Sepharose FF column (Pharmacia) and eluted over a 10 column volume gradient from zero to 1 M NaCl in buffer 50 mM Tris/HCl (pH 7.6), 0.5 mM PMSF, 2 mM benzamidine and 5 mM DTT. MS confirmed that the resulting Smac protein was equivalent to mature processed Smac, in agreement with previous reports [30].
Cell culture, immunoprecipitations and apoptosis assays
HEK-293T cells and MCF7 human breast carcinoma cells were cultured using standard procedures. Apoptosis assays and immunoprecipitations were performed as described previously [9,18]. The primary antibodies used were anti-FLAG M2 (Sigma–Aldrich), anti-Myc (Covance), anti-caspase 9 (Pharmingen), anti-XIAP (BD Transduction Laboratories) and anti-Smac (ProSci Incorporated).
Determination of caspase 9 inhibitory constants
Recombinant ΔCARD caspase 9 (300 nM final concentration in the assay) was pre-activated in salt-free caspase buffer [20 mM Pipes, 10 mM EDTA, 20 mM 2-mercaptoethanol, 0.1% (w/v) CHAPS and 10% (w/v) sucrose, pH 7.2] for 15–30 min at 37 °C. Following this, a range of inhibitor concentrations were pre-incubated with the enzyme for 20 min at 37 °C. The assay was started by the addition of Ac-LEHD-AFC (acetyl-Leu-Glu-His-Asp-7-amido-4-fluoromethylcourmarin; 100 μM final concentration) and measured kinetically for 30 min using an Fmax Fluorescence Plate Reader (Molecular Devices) at an excitation wavelength of 405 nm and an emission wavelength of 510 nm. Reaction mixtures were thermostatically controlled at 37 °C. The individual Ki values for the inhibitors [I] were determined from the uninhibited substrate hydrolysis rate (vo) and the inhibited rates (vi), so that a plot of (vo/vi)−1 against [I] gives Ki(app), the equilibrium inhibition constant in the presence of substrate [31]. Alternatively, Ki(app) was determined using non-linear regression analysis of vi/vo plotted against [I], the program GraphPad Prism 2.0, and equation 1:
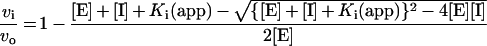 |
(1) |
Peptide-binding assays
Polarization experiments were performed on an Analyst HT 96-384 (Molecular Devices). Samples for fluorescence polarization affinity measurements were prepared by addition of 1:2 or 1:3 serial dilutions of MLBIR, MLBIR-Q, MLXBIR3, MLXBIR3SG, XIAP-BIR3 or XIAP-BIR2-BIR3 in polarization buffer [50 mM Tris/HCl (pH 7.2), 120 mM NaCl, 1% bovine globulins, 5 mM DTT and 0.05% octylglucoside] to 5-carboxyfluorescein-conjugated AVPFAKK (Hid-FAM), in the case of the ML constructs, or AVP-diphenylalanine-AKK (AVP-diPhe-FAM) in the case of the XIAP constructs, at 5 nM final concentration. The reactions were read after an incubation time of 10–30 min at room temperature with standard cut-off filters for the fluorescein fluorophore (λexcitation=485 nm; λemission=530 nm) in 96-well black HE96 plates (Molecular Devices). Fluorescence polarization values were plotted as a function of the protein concentration, and the EC50 values were obtained by fitting the data to a 4-parameter equation using Kaleidagraph software (Synergy software, Reading, PA, U.S.A.). The apparent Kd values were determined from the EC50 values.
Competition experiments were performed by addition of the ML-IAP-BIR, BIR chimera constructs or XIAP-BIR2-BIR3 at 0.2 μM or XIAP-BIR3 protein at 0.5 μM to wells containing 5 nM of the Hid-FAM or AVP-diPhe-FAM probes, as well as 1:3 serial dilutions of the Smac-9mer peptide (AVPIAQKSE) or mature Smac protein antagonists in the polarization buffer. Samples were read after a 10–30 min incubation. Fluorescence polarization values were plotted as a function of the antagonist concentration, and the IC50 values were obtained by fitting the data to a 4-parameter equation using Kaleidagraph software (Synergy Software). Inhibition constants (Ki) for the antagonists were determined from the IC50 values [32].
SPR (surface plasmon resonance) measurements
All SPR measurements were performed at room temperature using an Applied Biosystems 8500 Analyzer. Smac, MLBIR, MLBIR3SG and XIAP-BIR2-BIR3 were each printed on to a bare gold surface in triplicate, at concentrations of 500, 250, 125, 62.5 and 31 μg/ml, using a Genomics Solutions GeneMachine Omnigrid Microspotter. BSA (100 μg/ml) and PBS were also spotted on to the chip surface as controls. The chip was exposed to 1 mM thiolated poly(ethylene glycol) methoxyl ether (2 kDa) in 8500 Running Buffer [10 mM sodium phosphate (pH 7.4), 138 mM NaCl, 2.7 mM KCl, 0.05% Tween-20] for 35 min. To collect kinetic data, the chip was equilibrated using 8500 Running Buffer for 2 h, exposed to 10 nM Smac in running buffer containing 0.5 mM DTT for 30 min to measure kon, and 8500 Running Buffer for 2 h to measure koff. All flow rates were 200 μl/min. All data were reference corrected and fit globally, with a mass transport correction, to a 1:1 interaction model using Applied Biosystems Data Analysis software package version 1.0.
MLXBIR3SG complex crystallization
Purified MLXBIR3SG protein (20 –25 mg/ml) was mixed with synthetic peptide (AVPW or AVPIAQKSE) in a 1:2 protein/peptide ratio. Peptides were reconstituted from freeze-dried powder in 10 mM Mes, pH 5.5; final peptide concentration (20 or 30 mg/ml) was verified by A280 in the case of AVPW and estimated from dry mass in the case of AVPIAQKSE. The MLXBIR3SG–peptide complexes were mixed with crystallization well solution [100 mM Bistris, pH 6, 200 mM lithium sulphate, 20–25% (w/v) poly(ethylene glycol) 3350] in a ratio of 1 μl of protein complex to 1 μl of well solution. Hanging or sitting drops of the mixed solutions were then allowed to equilibrate by vapour diffusion against a reservoir of the well solution. Tetragonal bipyramidal crystals typically appeared in a few days, and grew to full size (typically 0.1–0.2 mm long) in a week.
MLXBIR3SG X-ray data collection and structure determination
The dataset for the AVPIAQKSE complex was collected at the Cornell High Energy Synchrotron Source. Data statistics are listed in Table 1. The starting model for refinement of the structure was derived from a crystal in which the AVPW peptide had been exchanged by soaking with a peptidomimetic antagonist; this structure was solved by molecular replacement using the wild-type ML-IAP-BIR structure [PDB (Protein Data Bank) code 1OXN] as a search model and refined to 1.3 Å (1 Å=0.1 nm) using Refmac5 [33]. The R-factor and free R-factor of this high-resolution structure are 0.142 and 0.151 respectively; details of this structure determination will be published elsewhere.
Table 1. X-ray data collection and refinement statistics.
Numbers in parentheses are for the highest resolution shell.
| Parameter | Value |
|---|---|
| Antagonist | AVPIAQKSE |
| Resolution (Å) | 50–1.7 (1.77–1.71) |
| Unique reflections | 31742 (2793) |
| Rsym* | 0.115 (0.664) |
| I/σ(I) | 21.4 (2.2) |
| Redundancy | 12.0 (7.2) |
| Rcryst† | 0.213 |
| Rfree‡ | 0.250 |
| Bond RMSD (Å) | 0.011 |
| Angle RMSD (°) | 1.2 |
* Rsym=Σ|I−〈I〉|/ΣI.
† Rcryst=Σ||Fobs|−|Fcalc||/Σ|Fobs|.
‡ Rfree=Rcryst for a random 5% of reflections.
The high-resolution structure, stripped of the peptidomimetic and all water molecules within 10 Å of it, was refined against the AVPIAQKSE dataset (using the same cross-validation test set as the high-resolution structure), and the peptide placed into clear difference density visible in an Fo−Fc map. Additional cycles of positional, anisotropic B-factor and translation-libration-screw (TLS) refinement using Refmac5, and automated water addition and removal using the program Arp/wArp [34], yielded the final structure for which statistics are reported in Table 1. No manual rebuilding of the MLXBIR3SG protein was necessary as compared with the high-resolution peptidomimetic complex. The first four residues of the AVPIAQKSE peptide are clearly visible, and weak electron density suggests the positions of residues 5 and 6.
RESULTS AND DISCUSSION
ML-IAP inhibits caspase 9 activity less potently than XIAP
To investigate the interactions of ML-IAP and XIAP with caspase 9, apparent inhibition constants for full-length ML-IAP, MLBIR-Q (ML-IAP residues 63–172), and XIAP-BIR3 were determined. The caspase 9 inhibition constant [Ki(app)] for XIAP-BIR3 was found to be 13 nM, in agreement with previous reports [7,8]. By contrast, the Ki(app) values for the ML-IAP constructs were found to be 250–350-fold weaker (Table 2). In order to understand at a molecular level why ML-IAP is a less potent inhibitor of caspase 9 than XIAP a chimeric ML-IAP/XIAP-BIR domain was designed, on the basis of existing structural and functional data, that might have affinity for processed caspase 9 similar to that of XIAP-BIR3, while retaining the peptide- and Smac-binding characteristics of native ML-IAP.
Table 2. Caspase 9 inhibition and peptide binding affinity of BIR domains.
| Protein | Caspase 9 inhibition Ki(app) (μM) | Fluorescence polarization probe Kd (μM) | Smac 9-mer Ki (μM) | Smac Ki (μM) |
|---|---|---|---|---|
| MLBIR | 3.2* | 0.021† | 0.20 | 0.026 |
| MLBIR-Q | 4.5 | 0.020† | 0.35 | 0.054 |
| MLXBIR3 | 0.96 | 0.021† | ||
| MLXBIR3SG | ≪0.010 | 0.030† | 0.34 | 0.059 |
| MLBIR-Q(S150G/Q167H) | 2.1 | |||
| MLBIR-Q(Q167H/E168L) | 2.4 | |||
| MLBIR-Q(S150G/E168L) | 0.04 | |||
| MLBIR-Q(S150G/Q167H/E168L) | ≪0.010 | |||
| XIAP-BIR3 | 0.013 | 0.051‡ | 0.67 | 0.26 |
| XIAP-BIR2-BIR3 | 0.030‡ | 0.85 | 0.021 |
* Ki(app) for full-length ML-IAP.
† Hid-FAM fluorescence polarization probe.
‡ AVP-diPhe-FAM fluorescence polarization probe.
Chimera construction
Previous structure and mutagenesis analysis of XIAP-BIR3 revealed that the binding sites for Smac and caspase 9 are overlapping but not identical [7,8,14]. For instance, the three mutations W310A (Trp310→Ala), E314S and H343A were each shown to abolish inhibition of caspase 9 activity [7], suggesting that these residues may directly contact caspase 9, whereas only two of these three mutations (W310A and E314S) significantly reduced binding to a fluorescein-labelled Smac peptide [8]. Subsequent structures of XIAP-BIR3 in complex with Smac or a Smac-based peptide showed that XIAP residues Trp310 and Glu314 form part of the peptide-binding site, whereas His343 is located at the C-terminal end of the fifth α-helix and is remote from the peptide-binding site [8,14]. Consistent with this, the H343A mutant binds the Smac-based peptide with wild-type affinity [8].
Comparison of the amino acid sequences of MLBIR and XIAP-BIR3 shows that the region of least sequence identity (for the structured portions of these BIR domains; residues Gly78–Ser171 for ML-IAP) corresponds to the fifth α-helix that includes His343 of XIAP (Figure 1). By contrast, residues that define the Smac-peptide-binding site are highly conserved between the two BIR domains. Inspection of the respective three-dimensional structures of MLBIR and XIAP-BIR3 indicates that the structures, including the peptide-binding sites, are very similar [15]. In particular, the packing of helix-5 to the BIR domains is similar, despite the lack of sequence conservation.
This analysis suggested that helix-5 of XIAP-BIR3 might contain, in addition to His343, other determinants of caspase 9 binding specificity. To ascertain if the approx. 300-fold lower potency for inhibition of caspase 9 activity by ML-IAP relative to XIAP (Table 2) is due to residues in helix-5 that differ between the two proteins, a chimeric protein construct was made in which residues 160–172 of MLBIR-Q were replaced with residues 336 –348 of XIAP-BIR3 (Figure 1B). This construct, MLXBIR3, is approx. 5-fold better at inhibiting caspase 9 than the wild-type MLBIR-Q construct, but is still approx. 70-fold less potent than XIAP-BIR3 (Table 2), suggesting that there are other important caspase 9 binding determinants specific to XIAP-BIR3 outside of helix-5.
Indeed, the recent crystal structure of the complex between XIAP-BIR3 and caspase 9 [26] revealed that in addition to residues from helix-5 and the peptide-binding site, residues from the C-terminal end of helix-3 and the immediately following loop in XIAP also contact caspase 9. Of the important contacts identified in this region of XIAP-BIR3, Gly326 is not conserved in ML-IAP (the corresponding residue being Ser150). Mutation of this glycine residue to glutamate in XIAP-BIR3 resulted in a loss of caspase 9 inhibition, presumably due to the introduction of a steric clash at the XIAP–caspase 9 interface [26]. To determine if the side chain of Ser150 is interfering with the binding of MLXBIR3 to caspase 9, the S150G mutation was introduced into MLXBIR3 (Figure 1B). This construct, MLXBIR3SG, was found to inhibit caspase 9 with a Ki(app) too low to measure accurately (≪10 nM), and is thus a more potent caspase 9 inhibitor than either XIAP-BIR3 [Ki(app)=13 nM] or ML-IAP [Ki(app)=3–5 μM] (Table 2).
Caspase 9 binding
To better understand the mechanism by which the MLXBIR3 and MLXBIR3SG chimeric proteins inhibit caspase 9, binding to caspase 9 in cells was investigated. When over-expressed, caspase 9 undergoes autocatalytic processing and it is the processed form that interacts with the XIAP and ML-IAP-BIR domains [13]. Accordingly, MLBIR and XIAP-BIR3 co-immunoprecipitate processed caspase 9, but not its zymogen precursor. Western blotting revealed that MLXBIR3SG is significantly more efficient in binding caspase 9 than wild-type MLBIR or MLXBIR3 (Figure 2A). By contrast, the caspase 9 binding efficiency of MLXBIR3SG appears to be similar to that of XIAP-BIR3.
Figure 2. Caspase 9 binding by BIR domain constructs.

293T cells were transiently transfected with caspase 9 (Casp-9) and FLAG-tagged BIR domain proteins or vector. After 40 h, cells were lysed in Nonidet P40 lysis buffer (120 mM Tris/HCl, 150 mM NaCl, 1% Nonidet P40, 1 mM DTT and protease inhibitor cocktail) and lysates immunoprecipitated (IP) with anti-FLAG antibody. Samples were then immunoblotted (W) with anti-caspase 9 and anti-FLAG antibodies. (A) Comparison of chimeric proteins MLXBIR3 and MLXBIR3SG with wild-type MLBIR and XIAP-BIR3. (B) Comparison of double and triple mutant MLBIR-Q constructs with wild-type MLBIR. (C) Comparison of XIAP-BIR3 triple mutant with wild-type XIAP-BIR3.
Anti-apoptotic effect of MLXBIR3SG
To determine if the MLXBIR3 and MLXBIR3SG chimeric proteins can block doxorubicin-induced apoptosis, the BIR domain proteins were transiently expressed in MCF-7 cells, and the cells were subsequently treated with the indicated cytotoxic agent. Assessment of apoptosis revealed that MLXBIR3SG was significantly more efficient in inhibiting doxorubicin-induced apoptosis than wild-type MLBIR or MLXBIR3 (Figure 3).
Figure 3. Anti-apoptotic effect of BIR domain constructs.

MCF7 cells were transiently transfected with the reporter plasmid pCMV-βgal and either vector control alone or BIR domain constructs. Following transfection, cells were exposed to doxorubicin (adriamycin; 0.5 μg/ml), stained with X-gal and apoptosis assessed by counting live and dead transfected cells. The percentage apoptosis represents the mean±S.D. for at least four sample points and is representative of three independent experiments. Asterisks designate that the difference between these two values is statistically significant (P<0.05). The relative transfection levels were evaluated by immunoblotting with anti-FLAG antibody.
Smac binding
The more efficient inhibition of apoptosis by MLXBIR3SG, relative to wild-type MLBIR, could be due to the improved binding and inhibition of caspase 9. Alternatively, since IAP antagonism is required for apoptosis to proceed [25,35,36], loss of the ability of the chimeric construct to bind to the natural IAP-antagonists, such as Smac, might also contribute to the observed increase in inhibition of apoptosis. However, co-expression of Smac with MLBIR, MLXBIR3, MLXBIR3SG or XIAP-BIR3 demonstrated that association of the four IAP constructs with the processed mature form of Smac occurred with similar efficiencies (Figure 4A).
Figure 4. Binding of ML-IAP, XIAP and their chimeric proteins to Smac.

(A) MLXBIR3 and MLXBIR3SG chimeric proteins interact with mature Smac. HEK-293T cells were transiently transfected with FLAG-tagged Smac and Myc-tagged BIR domain proteins or vector control. After 40 h, cells were lysed in Nonidet P40 lysis buffer (120 mM Tris/HCl, 150 mM NaCl, 1% Nonidet P40, 1 mM DTT and protease inhibitor cocktail) and lysates were immunoprecipitated (IP) with anti-Myc antibody. Samples were then immunoblotted (W) with anti-FLAG and anti-Myc antibodies. FL-SMAC designates full-length Smac protein, and SMAC designates the mature processed protein that is produced as a result of overexpression [18]. (B) Over-expression of ML-IAP diminishes the XIAP–Smac interaction. HEK-293T cells were transiently transfected with vector control (−) or increasing amounts of Myc–ML-IAP (1 μg and 4 μg) and treated 20 h later with 1.5 μg/ml of doxorubicin. Cells were lysed 40 h following transfection in Nonidet P40 lysis buffer and lysates were immunoprecipitated with control anti-FLAG (ct) or anti-XIAP (X) antibodies. Samples were then centrifuged, and the supernatant re-immunoprecipitated with anti-Myc antibody (2nd IP). Following extensive washing with lysis buffer, immunoprecipitates and portions of total cell lysates were subjected to SDS/PAGE and immunoblotted with anti-Smac, anti-XIAP and anti-Myc antibodies.
To provide a more quantitative measure of Smac binding to the various BIR constructs, a fluorescence polarization-based competition assay was developed using a 5-carboxyfluorescein-labelled peptide probe based on the N-terminus of the Drosophila IAP antagonist Hid (Hid-FAM). The binding affinities of the Hid-FAM probe to the chimeric BIR constructs (determined directly by fluorescence polarization) are similar to those determined for binding to the wild-type ML-IAP constructs (20–30 nM; Table 2), indicating that the chimeric substitutions have not perturbed significantly the peptide-binding site of ML-IAP-BIR. Similarly, the Ki values for MLXBIR3 and MLXBIR3SG are essentially the same as those for MLBIR or the slightly shorter MLBIR-Q, whether the competitor is mature Smac protein or a 9-mer peptide corresponding to the mature Smac N-terminus (AVPIAQKSE) (Table 2). These data indicate that the peptide-binding sites on the chimeric BIR domains are identical to that of wild-type ML-IAP. Therefore, the increased efficiency of inhibition of apoptosis observed for MLXBIR3SG relative to MLBIR can be attributed to its improved binding to and inhibition of caspase 9.
The affinities of the ML-IAP and chimeric constructs for mature Smac (Ki=0.026–0.059 μM) are significantly greater than the corresponding affinities for the Smac-based 9-mer peptide (Ki=0.20–0.35 μM) (Table 2). By contrast, previous reports have shown that XIAP-BIR3 binds the Smac-based peptide and mature Smac with similar affinities (Ki=0.4–0.8 μM) [8,37], consistent with the present study (Table 2). Significantly higher affinity binding to mature Smac was reported recently, however, for an XIAP construct containing both BIR2 and BIR3 domains [38]. In the fluorescence polarization-based competition assay the XIAP-BIR2-BIR3 construct binds mature Smac with essentially the same affinity as MLBIR (Table 2). In these cases, however, the reported Ki values should be considered as lower limits, because they are comparable with the Kd determined for the Hid-FAM probe. For this reason, the binding of MLBIR, MLXBIR3SG and XIAP-BIR2-BIR3 to mature Smac was investigated further using SPR methods.
The SPR measurements show that Smac binds to MLBIR, MLXBIR3SG and XIAP-BIR2-BIR3 with similar high affinities (Table 3). The Kd of 0.77 nM obtained for Smac binding to XIAP-BIR2-BIR3 is consistent with the previously reported affinity for this interaction [38]. The approx. 100-fold increase in the affinity of MLBIR and MLXBIR3SG for the mature Smac protein relative to the Smac-based 9-mer peptide suggests that additional protein–protein contact sites exist on the ML-IAP-BIR domain outside of the already characterized peptide-binding site.
Table 3. SPR kinetic parameters for Smac binding to selected BIR domains.
| Protein | kon (M−1·s−1) | koff (s−1) | Kd (nM) |
|---|---|---|---|
| MLBIR | 4.18×105 | 7.63×10−4 | 1.89 |
| MLXBIR3SG | 4.20×105 | 8.16×10−4 | 1.94 |
| XIAP-BIR2-BIR3 | 3.26×105 | 2.52×10−4 | 0.77 |
Since MLBIR and XIAP-BIR2-BIR3 bind Smac with similar affinities, the possibility that ML-IAP would compete with the XIAP–Smac interaction was investigated. To that end an ML-IAP-expressing plasmid was transiently transfected into HEK-293T cells, and the cells were treated with doxorubicin in order to activate apoptosis and consequently process endogenous full-length Smac to its mature form. In the absence of ML-IAP, a strong interaction between endogenous XIAP and mature Smac was observed (Figure 4B). However, with increasing expression of ML-IAP, the XIAP–Smac association was reduced significantly, and a new complex of ML-IAP and Smac was detected after a second round of immunoprecipitations (Figure 4B). Therefore, over-expression of ML-IAP can reduce the XIAP–Smac interaction by competing directly with XIAP binding to Smac.
Structure of MLXBIR3SG–peptide complex
To gain a more detailed understanding of the similarities and differences between wild-type and chimeric ML-IAP constructs, the MLXBIR3SG protein was crystallized in complex with the Smac 9-mer peptide. The two copies of the protein–peptide complex in the asymmetric unit of this crystal are very similar to one another, with an all-atom RMSD (root mean square deviation) of 1.4 Å (Cα RMSD 0.7 Å), excluding residues 100–101 that adopt different conformations in the two copies due to crystal packing. Residues 78–167 of MLXBIR3SG copy A and 78–172 of copy B are well ordered and visible in electron density maps; however, as observed for wild-type MLBIR [15], only the first four residues (AVPI) of the Smac-based 9-mer peptide are well ordered.
The structure of the MLXBIR3SG chimeric protein (Figure 5A) is essentially identical with that of wild-type MLBIR (Figure 5B), with a Cα RMSD of 0.6 Å excluding residues 100 –101. The peptide-binding region of MLBIR is unaffected by the chimera substitutions (Figure 5D): no atom of the chimeric residues is closer than 10 Å to the bound peptide. As would be expected, given the similarity of the peptide-binding site, the all-heavy-atom RMSD between the same peptide (AVPIAQKSE) bound to the two different proteins is less than 0.2 Å.
Figure 5. Crystal structure of the MLXBIR3SG–peptide complex.

(A) Schematic representation of the crystal structure of MLXBIR3SG in complex with the AVPIAQKSE peptide. The protein is shown in ribbon representation and the first four residues of the bound peptide are depicted in stick form, coloured by atom type; the bound zinc atom is shown as a sphere. Major secondary structure elements of the protein are labelled. The chimeric portions of the protein (Gly150 and helix-5) are coloured red. (B) The structure of wild-type MLBIR bound to the AVPIAQKSE peptide (PDB code 1OXQ; [15]) is shown in the same orientation as (A). (C) The structure of XIAP-BIR3 bound to Smac (PDB code 1G73; [14]) is shown in the same orientation as (A). Only the first four residues of Smac are depicted, in stick form. (D) Stereoview of the superposition of the peptide-binding sites of wild-type MLBIR and the chimeric protein MLXBIR3SG. The backbone of the MLXBIR3SG chimera is shown in a ribbon representation. Side chains of MLXBIR3SG and wild-type MLBIR that are within 3.8 Å of the peptide are shown in stick representation, as is the AVPI portion of the AVPIAQKSE peptide from both complexes. The MLXBIR3SG–AVPIAQKSE complex is coloured as in (A), whereas the wild-type MLBIR complex is coloured blue and depicted with thinner sticks.
Like wild-type MLBIR, the MLXBIR3SG chimera closely resembles other BIR domains, in particular XIAP-BIR3 (Figure 5C), which can be superposed on MLXBIR3SG with a Cα RMSD of 0.5 Å (chain C of PDB code 1G73 versus chain A of the MLXBIR3SG structure, again excluding residues 100–101). The chimeric XIAP residues in MLXBIR3SG adopt the same side chain conformations as they do in XIAP-BIR3. Three of these residues make up the majority of the interface with the rest of the BIR domain; the MLXBIR3SG chimeric substitutions for these residues (F162Y, V163I, V166I) add only three heavy atoms at the periphery of the interface, none of which affect packing of this helix. Thus helix-5 of MLXBIR3SG looks like the corresponding helix of XIAP-BIR3, but packs with the rest of the domain exactly like the equivalent helix in wild-type MLBIR.
Three substitutions are required to enhance ML-IAP affinity for caspase 9
Subsequent superposition of the structures of MLBIR and MLXBIR3SG on the structure of XIAP-BIR3 in complex with caspase 9 [26] indicated that improved MLBIR affinity for caspase 9 might be achieved with fewer amino acid substitutions (Figure 6). In particular, interactions observed between Gln399 of caspase 9 and XIAP residues Glu337 and Asn340 could be achieved adequately using the corresponding ML-IAP residues Asp161 and His164 respectively. By contrast, the interactions between the side chain of XIAP residue Leu344 and a hydrophobic pocket on caspase 9 could not be substituted effectively by the negatively charged ML-IAP residue, Glu168. As discussed above, ML-IAP residue Ser150 is a poor substitute for XIAP residue Gly326 due to steric clash; substitution of XIAP residue His343 with ML-IAP residue Gln167 might also be expected to disrupt the interface.
Figure 6. XIAP-BIR3–caspase 9 interface.

The structure of the XIAP-BIR3–caspase 9 complex (PDB code 1NW9; [26]) is shown; the backbone of XIAP-BIR3 is shown in ribbon representation (coloured gold) and selected side chains are shown in stick form (coloured by atom type, with gold carbons). Caspase 9 is shown as a molecular surface, except for the N-terminal peptide binding XIAP-BIR3; the first four residues of this peptide are shown in stick form (coloured by atom type, with pink carbons). Selected side chains from a superposed structure of wild-type MLBIR are shown in stick form (coloured by atom type, with blue carbons). Amino acid labels correspond to XIAP/ML-IAP residues. The side chain conformations of the ML-IAP residues Gln167 and Glu168 have been adjusted to mimic the conformations of the equivalent XIAP residues.
In order to test the hypothesis that ML-IAP might be converted to a high-affinity caspase 9 inhibitor with fewer substitutions than in the chimeric protein MLXBIR3SG, three double and one triple mutant of MLBIR-Q were constructed in which two or three of the residues Ser150, Gln167 or Glu168 were substituted with the corresponding XIAP residues, glycine, histidine or leucine respectively (i.e. four constructs containing the following mutations were made: S150G/Q167H, S150G/E168L, Q167H/E168L, S150G/Q167H/E168L). Apparent caspase 9 inhibition constants for these mutants (Table 2) are in good qualitative agreement with their relative caspase 9 binding efficiencies, as determined by co-immunoprecipitation (Figure 2B). As found for the chimeric protein MLXBIR3SG, the triple mutant, MLBIR-Q(S150G/Q167H/E168L), has an apparent Ki that is too low to measure accurately (≪10 nM), supporting the hypothesis that these three residues are responsible for ML-IAP being a significantly weaker inhibitor of caspase 9 than XIAP-BIR3. Of the double mutants, S150G/E168L shows the greatest improvement relative to wild-type ML-IAP (approx. 100-fold), suggesting that these two residues are more important than Gln167 in disrupting the MLBIR-Q–caspase 9 interface. If either of these residues are retained as wild-type, as in the double mutants S150G/Q167H or Q167H/E168L, the apparent affinity for caspase 9 is reduced by approx. 50-fold relative to the S150G/E168L double mutant, and by >200-fold relative to the triple mutant. Gln167, however, clearly also contributes negatively to the MLBIR-Q–caspase 9 interaction, since inclusion of the Q167H mutation in the triple mutant results in a significant improvement in binding relative to the S150G/E168L double mutant.
As might be expected based on the above results, mutation of the corresponding XIAP-BIR3 residues Gly326, His343 and Leu344 to their ML-IAP counterpart residues, serine, glutamate and glutamine respectively, significantly reduces caspase 9 binding efficiency of XIAP-BIR3 (Figure 2C).
Conclusion
ML-IAP is a potent anti-apoptotic protein that is up-regulated in most melanoma cell lines tested [9,39–41] and appears to contribute to the chemoresistance of this disease [42]. The mechanism by which ML-IAP regulates apoptosis is not clear, although direct inhibition of caspases 3 and 9 has been reported [9,40]. ML-IAP inhibition of caspase 3 was reported to be comparable with that by c-IAP1 (cellular IAP1) (Ki, approx. 100 nM) [9,43], and is thus approx. 10 –100-fold weaker than inhibition by XIAP [3–5]. In the present study ML-IAP is found to be an even less potent inhibitor of caspase 9 (Ki=3–5 μM; approx. 300-fold weaker than XIAP), suggesting that endogenous levels of ML-IAP will have little direct effect on caspase 9 activity.
Construction of chimeric and mutant proteins has established that three amino acids in the single BIR domain of ML-IAP (Ser150, Gln167 and Glu168) are responsible for its reduced caspase 9 affinity and inhibition relative to XIAP-BIR3. The corresponding residues of XIAP-BIR3 (Gly326, His343 and Leu344) were identified previously as being critical for the interaction between XIAP and the homodimerization interface of caspase 9, and are consequently important for XIAP-mediated inhibition of caspase 9 [26]. Interestingly, the chimeric protein, MLXBIR3SG, and the triple mutant, MLBIR-Q(S150G/Q167H/E168L), both inhibit caspase 9 more efficiently than native XIAP-BIR3; reasons for this observation are not immediately apparent.
Given that inhibition of caspases is unlikely to be the primary mechanism by which ML-IAP regulates apoptosis, the binding of ML-IAP-BIR to mature Smac was investigated. The single BIR domain of ML-IAP was found to bind mature Smac considerably tighter than expected based on its affinity for the Smac-based N-terminal 9-mer peptide (Tables 2 and 3), suggesting that there are Smac contacts on the BIR domain outside the peptide-binding site. By contrast, XIAP requires both its second and third BIR domains to achieve comparable high-affinity binding to mature Smac [38] (Tables 2 and 3). ML-IAP binding to Smac competes directly with XIAP binding, since both IAP proteins have critical interactions with the N-terminal four residues of the processed IAP protein antagonist [8,14,15].
When ML-IAP is up-regulated in melanoma and other cancer cells its high-affinity interaction with Smac will effectively compete with the XIAP–Smac interaction, thereby precluding Smac from antagonizing XIAP-mediated inhibition of caspases. In a similar fashion, baculoviral Op-IAP (where Op is Orgyia pseudotsugata nuclear polyhedrosis virus) does not inhibit Drosophila caspase activity, but rather binds the Drosophila IAP protein antagonists, Reaper, Hid and Grim, and prevents these pro-apoptotic proteins from blocking the suppression of caspase activity by the insect cellular IAP proteins [44–48]; more recently, Op-IAP was also found to bind efficiently to Smac/DIABLO, thereby preventing Smac-mediated inhibition of mammalian IAPs [49]. By sequestering Smac, ML-IAP can block apoptosis despite it being a relatively weak caspase inhibitor. Thus ML-IAP expression in cancer cells leads to increased resistance to chemotherapeutic agents, because it contributes to caspase inhibition, primarily by blocking the ability of Smac to disrupt XIAP–caspase interactions.
Acknowledgments
We are grateful to Yigong Shi for sharing of results prior to publication. We also thank Corrine E. Olesen and Dina Wassafs (Applied Biosystems, Bedford, MA, U.S.A.) for help with collecting SPR data. This work is based in part upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation under award DMR 97-13424, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award RR-01646 from the National Institutes of Health, through its National Center for Research Resources.
References
- 1.Salvesen G. S., Duckett C. S. IAP proteins: blocking the road to death's door. Nat. Rev. Mol. Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 2.Deveraux Q. L., Reed J. C. IAP family proteins – suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi R., Deveraux Q., Tamm I., Welsh K., Assa-Munt N., Salvesen G. S., Reed J. C. A single BIR domain of XIAP sufficient for inhibiting caspases. J. Biol. Chem. 1998;273:7787–7790. doi: 10.1074/jbc.273.14.7787. [DOI] [PubMed] [Google Scholar]
- 4.Sun C., Cai M., Gunasekera A. H., Meadows R. P., Wang H., Chen J., Zhang H., Wu W., Xu N., Ng S. C., Fesik S. W. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature (London) 1999;401:818–822. doi: 10.1038/44617. [DOI] [PubMed] [Google Scholar]
- 5.Riedl S. J., Renatus M., Schwarzenbacher R., Zhou Q., Sun C., Fesik S. W., Liddington R. C., Salvesen G. S. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 6.Deveraux Q. L., Leo E., Stennicke H. R., Welsh K., Salvesen G. S., Reed J. C. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun C., Cai M., Meadows R. P., Xu N., Gunasekera A. H., Herrmann J., Wu J. C., Fesik S. W. NMR structure and mutagenesis of the third Bir domain of the inhibitor of apoptosis protein XIAP. J. Biol. Chem. 2000;275:33777–33781. doi: 10.1074/jbc.M006226200. [DOI] [PubMed] [Google Scholar]
- 8.Liu Z., Sun C., Olejniczak E. T., Meadows R. P., Betz S. F., Oost T., Herrmann J., Wu J. C., Fesik S. W. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature (London) 2000;408:1004–1008. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 9.Vucic D., Stennicke H. R., Pisabarro M. T., Salvesen G. S., Dixit V. M. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr. Biol. 2000;10:1359–1366. doi: 10.1016/s0960-9822(00)00781-8. [DOI] [PubMed] [Google Scholar]
- 10.Du C., Fang M., Li Y., Li L., Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 11.Verhagen A. M., Ekert P. G., Pakusch M., Silke J., Connolly L. M., Reid G. E., Moritz R. L., Simpson R. J., Vaux D. L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 12.Chai J., Du C., Wu J. W., Kyin S., Wang X., Shi Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature (London) 2000;406:855–862. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 13.Srinivasula S. M., Hegde R., Saleh A., Datta P., Shiozaki E., Chai J., Lee R. A., Robbins P. D., Fernandes-Alnemri T., Shi Y., Alnemri E. S. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature (London) 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 14.Wu G., Chai J., Suber T. L., Wu J. W., Du C., Wang X., Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature (London) 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 15.Franklin M. C., Kadkhodayan S., Ackerly H., Alexandru D., Distefano M. D., Elliot L. O., Flygare J. A., Mausisa G., Okawa D. C., Ong D., et al. Structure and function analysis of peptide antagonists of melanoma inhibitor of apoptosis (ML-IAP) Biochemistry. 2003;42:8223–8231. doi: 10.1021/bi034227t. [DOI] [PubMed] [Google Scholar]
- 16.Wu J. W., Cocina A. E., Chai J., Hay B. A., Shi Y. Structural analysis of a functional DIAP1 fragment bound to grim and hid peptides. Mol. Cell. 2001;8:95–104. doi: 10.1016/s1097-2765(01)00282-9. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasula S. M., Datta P., Kobayashi M., Wu J. W., Fujioka M., Hegde R., Zhang Z., Mukattash R., Fernandes-Alnemri T., Shi Y., et al. Sickle, a novel Drosophila death gene in the reaper/hid/grim region, encodes an IAP-inhibitory protein. Curr. Biol. 2002;12:125–130. doi: 10.1016/s0960-9822(01)00657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vucic D., Deshayes K., Ackerly H., Pisabarro M. T., Kadkhodayan S., Fairbrother W. J., Dixit V. M. SMAC negatively regulates the anti-apoptotic activity of melanoma inhibitor of apoptosis (ML-IAP) J. Biol. Chem. 2002;277:12275–12279. doi: 10.1074/jbc.M112045200. [DOI] [PubMed] [Google Scholar]
- 19.Guo F., Nimmanapalli R., Paranawithana S., Wittman S., Griffin D., Bali P., O'Bryan E., Fumero C., Wang H. G., Bhalla K. Ectopic overexpression of second mitochondria-derived activator of caspases (Smac/DIABLO) or cotreatment with N-terminus of Smac/DIABLO peptide potentiates epothilone B derivative-(BMS 247550) and Apo-2L/TRAIL-induced apoptosis. Blood. 2002;99:3419–3426. doi: 10.1182/blood.v99.9.3419. [DOI] [PubMed] [Google Scholar]
- 20.Arnt C. R., Chiorean M. V., Heldebrant M. P., Gores G. J., Kaufmann S. H. Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J. Biol. Chem. 2002;277:44236–44243. doi: 10.1074/jbc.M207578200. [DOI] [PubMed] [Google Scholar]
- 21.Fulda S., Wick W., Weller M., Debatin K. M. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 2002;8:808–815. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 22.Yang L., Mashima T., Sato S., Mochizuki M., Sakamoto H., Yamori T., Oh-Hara T., Tsuruo T. Predominant suppression of apoptosome by inhibitor of apoptosis protein in non-small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine-conjugated Smac peptide. Cancer Res. 2003;63:831–837. [PubMed] [Google Scholar]
- 23.Chai J., Shiozaki E., Srinivasula S. M., Wu Q., Datta P., Alnemri E. S., Shi Y., Dataa P. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y., Park Y. C., Rich R. L., Segal D., Myszka D. G., Wu H. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- 25.Silke J., Hawkins C. J., Ekert P. G., Chew J., Day C. L., Pakusch M., Verhagen A. M., Vaux D. L. The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase 3- and caspase 9-interacting sites. J. Cell Biol. 2002;157:115–124. doi: 10.1083/jcb.200108085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiozaki E. N., Chai J., Rigotti D. J., Riedl S. J., Li P., Srinivasula S. M., Alnemri E. S., Fairman R., Shi Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell. 2003;11:519–527. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 27.Renatus M., Stennicke H. R., Scott F. L., Liddington R. C., Salvesen G. S. Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14250–14255. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boatright K. M., Renatus M., Scott F. L., Sperandio S., Shin H., Pedersen I. M., Ricci J.-E., Edris W. A., Sutherlin D. P., Green D. R., Salvesen G. S. A unified model for apical caspase activation. Mol. Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 29.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 30.Srinivasula S. M., Datta P., Fan X. J., Fernandes-Alnemri T., Huang Z., Alnemri E. S. Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J. Biol. Chem. 2000;275:36152–36157. doi: 10.1074/jbc.C000533200. [DOI] [PubMed] [Google Scholar]
- 31.Salvesen G. S., Nagase H. Inhibition of proteolytic enzymes. In: Beynon R. J., Bond J. S., editors. Proteolytic Enzymes: A Practical Approach. Oxford: IRL Press; 1989. pp. 83–104. [Google Scholar]
- 32.Keating S. M., Marsters J., Beresini M., Ladner C., Zioncheck K., Clark K., Arellano F., Bodary S. Putting the pieces together: Contribution of fluorescence polarization assays to small molecule lead optimization. In: Cohn G. E., editor. Proceedings of SPIE: In-Vitro Diagnostic Instrumentation, vol. 3913; 2000. pp. 128–137. [Google Scholar]
- 33.Murshudov G. N., Vagin A. A., Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Perrakis A., Harkiolaki M., Wilson K. S., Lamzin V. S. ARP/wARP and molecular replacement. Acta Cryst. 2001;D57:1445–1450. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X. D., Zhang X. Y., Gray C. P., Nguyen T., Hersey P. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of human melanoma is regulated by smac/DIABLO release from mitochondria. Cancer Res. 2001;61:7339–7348. [PubMed] [Google Scholar]
- 36.Deng Y., Lin Y., Wu X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002;16:33–45. doi: 10.1101/gad.949602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kipp R. A., Case M. A., Wist A. D., Cresson C. M., Carrell M., Griner E., Wiita A., Albiniak P. A., Chai J., Shi Y., Semmelhack M. F., McLendon G. L. Molecular targeting of inhibitor of apoptosis proteins based on small molecule mimics of natural binding partners. Biochemistry. 2002;41:7344–7349. doi: 10.1021/bi0121454. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y., Rich R. L., Myszka D. G., Wu H. Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by Smac. J. Biol. Chem. 2003;278:49517–49522. doi: 10.1074/jbc.M310061200. [DOI] [PubMed] [Google Scholar]
- 39.Lin J.-H., Deng G., Huang Q., Morser J. KIAP, a novel member of the inhibitor of apoptosis protein family. Biochem. Biophys. Res. Commun. 2000;279:820–831. doi: 10.1006/bbrc.2000.4027. [DOI] [PubMed] [Google Scholar]
- 40.Kasof G. M., Gomes B. C. Livin, a novel inhibitor of apoptosis protein family member. J. Biol. Chem. 2001;276:3238–3246. doi: 10.1074/jbc.M003670200. [DOI] [PubMed] [Google Scholar]
- 41.Ashhab Y., Alian A., Polliack A., Panet A., Ben Yehuda D. Two splicing variants of a new inhibitor of apoptosis gene with different biological properties and tissue distribution pattern. FEBS Lett. 2001;495:56–60. doi: 10.1016/s0014-5793(01)02366-3. [DOI] [PubMed] [Google Scholar]
- 42.Nachmias B., Ashhab Y., Bucholtz V., Drize O., Kadouri L., Lotem M., Peretz T., Mandelboim O., Ben-Yehuda D. Caspase-mediated cleavage converts Livin from an antiapoptotic to a proapoptotic factor: implications for drug-resistant melanoma. Cancer Res. 2003;63:6340–6349. [PubMed] [Google Scholar]
- 43.Roy N., Deveraux Q. L., Takahashi R., Salvesen G. S., Reed J. C. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vucic D., Kaiser W. J., Harvey A. J., Miller L. K. Inhibition of reaper-induced apoptosis by interaction with inhibitor of apoptosis proteins (IAPs) Proc. Natl. Acad. Sci. U.S.A. 1997;94:10183–10188. doi: 10.1073/pnas.94.19.10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vucic D., Kaiser W. J., Miller L. K. Inhibitor of apoptosis proteins physically interact with and block apoptosis induced by Drosophila proteins HID and GRIM. Mol. Cell. Biol. 1998;18:3300–3309. doi: 10.1128/mcb.18.6.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaiser W. J., Vucic D., Miller L. K. The Drosophila inhibitor of apoptosis D-IAP1 suppresses cell death induced by the caspase drICE. FEBS Lett. 1998;440:243–248. doi: 10.1016/s0014-5793(98)01465-3. [DOI] [PubMed] [Google Scholar]
- 47.Seshagiri S., Vucic D., Lee J., Dixit V. M. Baculovirus-based genetic screen for antiapoptotic genes identifies a novel IAP. J. Biol. Chem. 1999;274:36769–36773. doi: 10.1074/jbc.274.51.36769. [DOI] [PubMed] [Google Scholar]
- 48.Wang S. L., Hawkins C. J., Yoo S. J., Muller H. A., Hay B. A. The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell. 1999;98:453–463. doi: 10.1016/s0092-8674(00)81974-1. [DOI] [PubMed] [Google Scholar]
- 49.Wilkinson J. C., Wilkinson A. S., Scott F. L., Csomos R. A., Salvesen G. S., Duckett C. S. Neutralization of SMAC/DIABLO by IAPs: a caspase-independent mechanism for apoptotic inhibition. J. Biol. Chem. 2004 doi: 10.1074/jbc.M408655200. DOI M408655200. [DOI] [PubMed]