

Abstract
The U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), and Pharmaceuticals and Medical Devices Agency (PMDA) guidances on small molecule drug-drug interactions (DDIs), with input from the International Transporter Consortium (ITC), recommend the evaluation of nine drug transporters. Although other clinically relevant drug uptake and efflux transporters have been discussed in ITC white papers, they have been excluded from further recommendation by the ITC and are not included in current regulatory guidances. These include the ubiquitously expressed equilibrative nucleoside transporters (ENT) 1 and ENT2, which have been recognized by the ITC for their potential role in clinically relevant nucleoside analog drug interactions for cancer patients. Although there is comparatively limited clinical evidence supporting their role in DDI risk or other adverse drug reactions (ADRs) compared to the nine highlighted transporters, several in vitro and in vivo studies have identified ENT interactions with non-nucleoside/-tide drugs, in addition to nucleoside/-tide analogs. Some noteworthy examples of compounds that interact with ENTs include cannabidiol and selected protein kinase inhibitors, as well as the nucleoside analogs remdesivir, EIDD-1931, gemcitabine, fialuridine. Consequently, DDIs involving the ENTs may be responsible for therapeutic inefficacy or off-target toxicity. Evidence suggests that ENT1 and ENT2 should be considered as transporters potentially involved in clinically relevant DDIs and ADRs, thereby warranting further investigation and regulatory consideration
Keywords: Adverse drug reaction, antineoplastic, antiviral, clinical efficacy, clinical trial, drug discovery, drug-drug interaction, drug transporter, European Medicines Agency, equilibrative nucleoside transporter (ENT), Food and Drug Administration, guidelines, inhibitor, International Transporter Consortium, localization, nucleoside analog drug, Pharmaceuticals and Medical Devices Agency, protein kinase inhibitor, substrate, toxicity
Introduction
Current guidances from the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Pharmaceutical and Medical Devices Agency (PMDA), with input from white papers written by the International Transporter Consortium (ITC)1–7, reference nine drug transporters for evaluation of small molecule drug-drug interactions (DDIs) in drug discovery and development programs, namely, organic anion transporter (OAT) 1, OAT3, organic cation transporter (OCT) 2, organic anion transporting polypeptide (OATP) 1B1, OATP1B3, P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), multidrug and toxin extrusion protein (MATE) 1, and MATE2-K. These transporters are mentioned specifically due to “clinical evidence suggesting their involvement in drug interactions”.1 Furthermore, alterations to the expression and function of these transporters are linked to adverse drug reactions (ADRs) because they play a critical role in the disposition of many pharmacologically active molecules.8 Therefore, investigating DDIs and ADRs early in the discovery process is important to inform any necessary clinical studies and understand the magnitude of the DDI and ADR risk. However, inclusion of transporters into the ITC recommendations and, consequently, regulatory guidances, must be supported by well-documented DDIs and robust evidence of their potential role in altering drug efficacy and safety.
Several additional drug uptake transporters, including OATP1A2, OATP2B1, OAT2, and OCT1, have been proposed and evaluated as transporters of emerging clinical importance in the literature, although current regulatory guidances and ITC recommendations omit some of these transporters. The ITC excludes some of these transporters due to insufficient clinical evidence in support of their importance in DDIs, lack of specific in vitro probe substrates, or the inadequate relative contribution of these transporters to the disposition of clinically relevant drugs. A previous white paper from the ITC also acknowledged the potential clinical importance of equilibrative nucleoside transporter (ENT) 1 and ENT2, however, the analysis was limited to the role of the ENTs in delivering nucleoside analog antineoplastics to target tissues.2 However, we suggest that the current literature indicates that ENTs can significantly influence the absorption, distribution, metabolism, and excretion characteristics of clinically relevant drugs. For example, the disposition of nucleoside analog drugs like remdesivir and gemcitabine may be affected by ENT downregulation/dysfunction or by co-administration of known ENT inhibitors (e.g., cannabidiol (CBD), Δ9-Tetrahydrocannabidiol (THC), and lorlatinib), which may lead to a reduction in drug elimination and consequently, increased exposure and likelihood of toxicity.9–13 Unfortunately, the lack of information on the ENTs compared to traditional transporters like OATP1B1 or P-gp potentially understates their role in drug disposition, which may have contributed to the comparative absence of studies characterizing their selectivity. The purpose of this review is to outline the general characteristics of the ENT family of transporters, the current knowledge of ENT-drug interactions in various in vitro models, and the important clinical implications involving ENTs to advocate for expanded ADR and DDI testing paradigms that include these transporters in drug discovery and development studies.
General Characteristics of ENT1 and ENT2
Nucleoside and nucleobase transporters play critically important roles in the flux of precursors for intracellular nucleotide synthesis. There are two major families of nucleoside and nucleobase transporter denoted as the (1) concentrative nucleoside transporters (CNTs) and (2) ENTs. The sodium-dependent CNTs are predominantly expressed in specific tissues such as the kidneys, intestines, and heart.14–16 Since less is currently known about the drug interactions with these transporters compared to ENTs, they will not be the focus of this review. In contrast, the ENTs are widely expressed in all tissues and cell types and consequently, are expected to exert greater impact on the distribution of their substrates. The ENT family of proteins encompasses four members, designated as ENT1–4 in humans, nonhuman primates, rodents, canines, and other species.14 Of the four members, ENT1, ENT2, and ENT3 are ubiquitously-expressed, sodium-independent uniporters that display distinct, but overlapping substrate and inhibitor selectivity.14–16 Excluding ENT4, the predominant physiological role for these transporters is to direct the movement of electroneutral nucleosides and nucleobases across membranes down their concentration gradients. Of the four ENTs, ENT1 and ENT2 are the most well-characterized family members and display relatively broad substrate selectivity. Targeted and untargeted transcriptomic and proteomic profiles for ENT1, ENT2, and other pharmacologically relevant drug transporters in various tissues and cell types has been reported.17–24 In general, ENT1 and ENT2 have ubiquitous but variable reported mRNA and protein expression throughout the human body. This is in contrast to the expression of some uptake transporters such as OATP1B1 or OATP1B3, which are highly expressed in certain tissues including the liver but remain undetectable in other tissues.19 Although basic, limited studies on the expression, selectivity, and roles of ENT3 and ENT4 have been performed, there remains a significant lack of information on these two transporters. Therefore, the rest of this review will focus on the roles of ENT1 and ENT2 in clinically relevant DDIs and ADRs.
Structural and Functional Characteristics of ENT1 and ENT2
Human ENT1 is a 456-residue protein with 11 transmembrane helices that shares 78% and 79% sequence identity with the 457-residue rat and 460-residue mouse orthologs, respectively.25–27 ENT1 primarily transports endogenous purine and pyrimidine nucleosides, some nucleobases, and nucleoside/-tide analog drugs.28, 29 Similarly, human ENT2 is a 456-residue protein with 11 transmembrane helices that shares 88% sequence identity with the 456-residue mouse and rat orthologs.25, 30 However, human ENT1 and ENT2 only share 47% sequence identity, suggesting they may play different physiological roles. As expected, ENT2 also transports purine and pyrimidine nucleosides, nucleobases, and nucleoside/-tide analog drugs with differing selectivity compared to ENT1.13, 28, 29, 31–33 Furthermore, ENT1 and ENT2 are predominantly expressed along the apical or basal membranes of polarized cells in many tissues, although expression on nuclear, mitochondrial, and lysosomal membranes has been reported.16, 34–37 These differences are important to consider when assessing the distribution of nucleoside/-tide analog drugs in various tissues. Notably, ENT1 is highly sensitive to the nontransported inhibitor, NBMPR, with a Ki in the nanomolar range. Although ENT2 is insensitive to low micromolar concentrations of nitrobenzylthioinosine (NBMPR), high micromolar concentrations of NBMPR exerts an observable inhibitory effect on ENT2-mediated transport.25–27
Recently, a crystal structure for human ENT1 bound to the inhibitors, NBMPR or dilazep, was reported in the literature.38 Interestingly, comparison of the binding sites for NBMPR and dilazep showed they were both shared and that several distinct amino acid residues that interacted with these molecules. Specifically, the Gln158 residue interacted with NBMPR and dilazep; however, Met33 and Phe307 only interacted with dilazep.38 The nearby Gly154 residue has been shown to be critical for NBMPR sensitivity in human ENT1 but the equivalent residue in ENT2 is a serine.38, 39 It has been suggested that the binding of NBMPR to the unique, central binding site in ENT1 prevents the protein from changing conformation from the outward-occluded position to the inward position. As a result, this is likely a substrate-independent mechanism because it prevents ENT1 from completing a transport cycle. While ENT2 lacks a Gly154 residue, NBMPR likely binds in the same location to prevent a substrate from being transported. Thus, the inhibitory potency of NBMPR is likely to be substrate-independent, given its unique hydrophobic binding site in ENT1 (and ENT2).
Other residues, such as Asn30, Met84, Phe334, and Asn338, are also important for the inhibitor sensitivity of ENT1.40–42 Through molecular docking simulations, Asp341 and Arg345 in ENT1 were found to be important in establishing interaction with the substrate, adenosine, but not with the inhibitor, dilazep.41 Further molecular docking studies, along with pharmacophore modeling of ENT1 substrates or inhibitors, should provide insight into the selectivity of this transporter.32, 33 While mutational analyses have been performed with human ENT2 to characterize its inhibitor sensitivity, a crystal structure for this transporter is still unavailable.43, 44 Because human ENT1 and ENT2 only share 47% sequence identity, only some amino acid residues are conserved between the two transporters, highlighting their distinct differences in substrate/inhibitor selectivity and specificity (Tables S1 and S2).42 However, importantly, many of these human ENT residues are conserved across various species, suggesting that compound selectivity across species may be similar.42 A summary of the key characteristics of human ENT1 and ENT2 compared to each other and with other common ADME uptake transporters (i.e., CNTs, OCTs, OATs, and OATPs) are presented in Table 1.
Table 1:
Summary of key features of human ENT1 and ENT2 compared to other human ADME uptake transporters.
| Characteristics | ENT1 | ENT2 | CNT1–3 | OCT1–3 | OAT1–3 | OATP1A-6A |
|---|---|---|---|---|---|---|
| Structure | 456 aa 11 transmembrane domains (47% identity with each other) |
649 – 691 aa 13 transmembrane domains |
554 – 556 aa 12 transmembrane domains |
542 – 563 aa 12 transmembrane domains |
643 – 848 aa 12 transmembrane domains |
|
| Tissue Expression | Ubiquitous (liver, kidney, intestine, brain, heart, testes, placenta, erythrocytes, etc.) | Kidney, intestine, heart, pancreas, skeletal muscle, etc. | Variable (liver [not OCT2], kidney, intestine, brain, heart, testes, etc.) | Variable (liver [OAT2], kidney, skeletal muscle, placenta, testes, etc.) | Variable (liver, kidney, intestine, brain, heart, testes, lung, etc.) | |
| Known Subcellular Localization | Plasma, nuclear, and mitochondrial membranes Endoplasmic reticulum |
Plasma and nuclear membranes | Plasma membrane | |||
| Transport Mechanism | Na+-independent uniport Bidirectional, facilitative diffusional transporter (down concentration gradients) |
Na+-dependent symporter Unidirectional, coupled transporter (up concentration gradients) |
Na+-independent uniporter Bidirectional, facilitative diffusional transporter |
Na+-independent antiporter Unidirectional, coupled transporter (evidence of bidirectional transport) |
Na+, K+, and Cl−-independent antiporter pH-sensitive Bidirectional, facilitative diffusional transporter |
|
| High affinity, low capacity | Low affinity, high capacity | |||||
| Common Substrates | Adenine, cytosine, guanine, thymine, uracil, adenosine, cytidine, guanosine, thymidine, uridine, inosine, gemcitabine, remdesivir, ribavirin, abacavir, fialuridine | Adenine, cytosine, guanine, thymine, uracil, adenosine, cytidine, guanosine, thymidine, uridine, inosine, hypoxanthine, gemcitabine, remdesivir, azathioprine, zidovudine | Cytosine, thymine, adenosine, cytidine, guanosine, thymidine, uridine, inosine, gemcitabine, ribavirin, zidovudine | Choline, acetylcholine, dopamine, norepinephrine, epinephrine, serotonin, histamine, MPP+, TEA+, metformin | Adefovir (OAT1), cGMP, cimetidine (OAT3), DHEAS, estrone 3-sulfate (OAT3), methotrexate (OAT3), para-aminohippurate (OAT1), prostaglandins, zidovudine | Variable (bile acids, DHEAS, hormones, prostaglandins, statins, bromsulphthalein, estrone 3-sulfate, estradiol-17β-glucuronide) |
| Common Inhibitors | NBMPR, dipyridamole, dilazep, draflazine, molnupiravir, imatinib, CBD, THC, verapamil, ticagrelor | NBMPR, dipyridamole, dilazep, draflazine, molnupiravir, imatinib, diazepam, lorazepam, tecadenoson | Decitabine, fludarabine, vidarabine | Atropine, cimetidine, clonidine, prazosin, trimethoprim, quinidine, quinine, verapamil | Benzylpenicillin, cyclothiazide, diclofenac, furosemide, indomethacin, ketoconazole, probenecid | Variable (antibiotics, antivirals, cyclosporin, diclofenac, indomethacin, MK-571, repaglinide, rifampicin, ritonavir) |
Cellular and Subcellular Localization of ENT1 and ENT2
The cellular and subcellular localization of the ENTs in different tissues is an important factor to consider when evaluating the disposition of endogenous nucleosides/-bases and xenobiotics. Although ENTs are ubiquitously expressed transporters, they can localize to different sides of polarized plasma membranes and/or on organelle membranes. For example, human ENT1 and ENT2 localize to the plasma membrane and nuclear membrane of BeWo cells.45 Furthermore, human ENT1 can localize to the mitochondrial membrane in heterologously expressing MDCK cells and human hepatocytes (Figure 1A).46, 47 Human ENT1 has also been detected in human liver and kidney microsomes, suggesting it may be present in the endoplasmic reticulum.48 ENT expression at both the plasma membrane and organelle membranes may provide a direct path to deliver therapeutics from the blood to the cytoplasm and then to certain organelles. For example, direct delivery of therapeutics to the mitochondria of cancer cells to disrupt mitochondrial metabolism via ENTs is a therapeutic strategy to treat cancers.
Figure 1: Tissue localization of human ENT1 and ENT2.
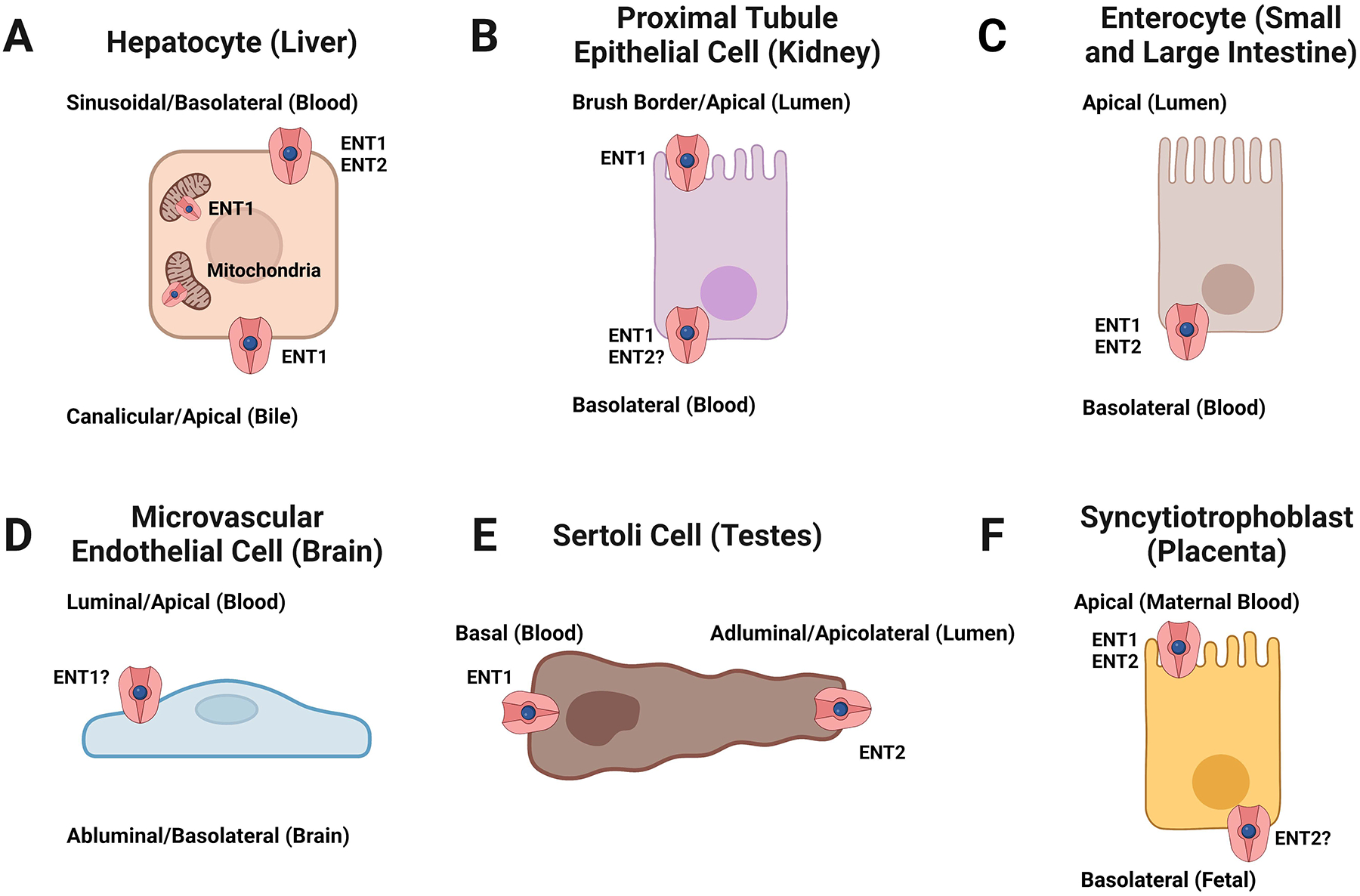
Localization of ENT1 and/or 2 in human (A) hepatocytes, (B) kidney proximal tubule epithelial cells, (C) enterocytes of the small and large intestine, (D) microvascular endothelial cells of the blood-brain barrier, (E) Sertoli cells of the blood-testis barrier, and (F) placental syncytiotrophoblasts. ENTs are bidirectional transporters that support nucleoside/-tide transport down their concentration gradients. ENT1/2 localization in brain microvascular endothelial cells and ENT2 localization in kidney proximal tubule epithelial cells or placental syncytiotrophoblasts remains unclear. Figure created with Biorender.com.
Apart from organelle expression, ENT1 and ENT2 are highly expressed at the plasma membranes in various cell types where they serve their primary function. Hepatocytes mediate the uptake of compounds from the portal blood across the sinusoidal/basolateral membrane into the cytoplasm, where they can be metabolized and excreted into the bile at the canalicular/apical membrane or back into the blood. In human hepatocyte sandwich cultures, ENT1 localized to both the canalicular/apical and sinusoidal/basolateral membranes, whereas ENT2 was only observed at the sinusoidal/basolateral membrane (Figure 1A).49 This is largely consistent with the observation that ENT1 and ENT2 are highly expressed in hepatocytes and in endothelial and Kupffer cells, although subcellular localization was indiscernible, using in situ hybridization.35 Nevertheless, ENT1 and ENT2 provide a transepithelial transport pathway for some compounds to cross through each side of a hepatocyte for recirculation or biliary elimination as an unmetabolized compound or metabolite.
Another major route for drug elimination is through the kidneys. In human kidneys, ENT1 localizes to the brush border/apical and basolateral membranes of proximal tubule epithelial cells of the nephron35, 50, which is consistent with earlier observations of brush border/apical and basolateral expression of heterologously expressed human ENT1 in polarized MDCK cells (Figure 1B).51 Intracellular and basolateral membrane expression of human ENT1 and ENT2 was also found in distal tubule epithelial cells but staining for ENT2 was not observed in proximal tubules35, although it would likely localize to the basolateral membrane if expressed.51 In the loop of Henle and collecting ducts, ENT1 is expressed at both the apical and basolateral membranes.50 Consequently, endogenous and exogenous ENT1 substrates can enter and exit through the blood or tubular fluid along the entire renal tubule based on their concentration gradients.
ENT1 expression in the human small intestine is predominantly concentrated to the crypt cells, with minimal expression at the brush border/apical membranes of enterocytes.35 This finding matches early functional studies in human intestinal epithelial cells that suggested ENT1 is absent from the brush border/apical membrane but may be present at the basolateral membrane, along with ENT2 (Figure 1C).52 These previous studies hypothesized that ENT1 and ENT2 are involved in nucleoside salvaging from the extracellular fluid at the basolateral membrane. In the colon, it has been suggested that ENT1 and ENT2 are functionally expressed at the basolateral membrane, based on observations in Caco-2 and T84 cells (Figure 1C).53–55 Thus, ENTs at the basolateral membrane are seemingly responsible for managing endogenous nucleoside homeostasis in the intestine but not nucleoside absorption from dietary intake. Nucleoside and nucleoside analog absorption from dietary sources is likely mediated by the CNTs expressed along the brush border/apical membranes of enterocytes.35 However, drugs that are ENT substrates can still access the intestinal epithelia through the circulatory system.
High expression of ENT1 and ENT2 has also been observed in other tissues, including the brain, testes, and placenta.17–19, 22–24, 26, 34, 36, 56–63 Brain microvascular endothelial cells express ENT1 and ENT2; however, their exact subcellular localization is debatable, with some studies suggesting apical/luminal membrane expression based on functional experiments (Figure 1D).17, 22–24, 59, 63, 64 This would permit substrate access into the microvascular endothelial cells, although another nucleoside transporter is necessary to complete the transcellular transport pathway. It has been suggested that ENT2 is expressed at the basolateral/abluminal membrane in rats; however, there is no human data to confirm these observations.63 Within human brain tissue, ENT1 protein was found in the temporal and occipital cortex, thalamus, midbrain, and the caudate nucleus, putamen, and globus pallidus regions of the basal ganglia.57 On the other hand, ENT2 protein was primarily observed in the cerebellum, brainstem, pons, midbrain, medulla, and thalamus.57 Subcellular localization of ENT1 and ENT2 was not determined in that study and currently remains unknown. ENT1 and ENT2 are expressed in the lung58, 60–62 with one study suggesting apical membrane expression at the olfactory epithelium and weak expression in the respiratory epithelium in rats.65 However, further studies are necessary to determine their cellular and subcellular location in human respiratory tissues. In the testes, ENT1 is primarily localized to the basal membrane and ENT2 localized to the apicolateral membrane of Sertoli cells, which comprise the blood-testis barrier (Figure 1E).36 This testicular expression pattern may provide a transepithelial transport pathway for some compounds to access the seminiferous tubule lumen and affect spermatogenesis. Placental ENT1 and ENT2 expression has been shown at the apical/brush border membrane of syncytiotrophoblasts but are largely absent from the basolateral membranes (Figure 1F).34, 35, 56 However, a more recent study has shown that ENT2 is localized to the basolateral membrane of syncytiotrophoblasts.66 Collectively, drugs that are ENT substrates can be widely distributed throughout the body.
Modeling ENT1- and ENT2-Mediated Transport and Disposition
Nucleoside/-tide analog drugs have a long history of success in treating diseases, including bacterial and viral infections, select cancers, rheumatic disorders, as well as being used as platelet aggregation inhibitors. Most nucleoside/-tide analogs are used as antivirals or antineoplastics due to their ability to inhibit genomic replication of viruses or cancer cells. However, many of these drugs are hydrophilic and exhibit poor membrane permeability without assistance from transporters, and yet are prodrugs that must be intracellularly metabolized into their active metabolites. A selection of clinically-relevant nucleoside/-tide analog drugs, their respective pharmacological function, and their published interactions with human ENTs are listed in Table S1. There are conflicting reports on the interaction between nucleoside/-tide analogs or non-nucleoside/-tide drugs and the ENTs; however, it is possible that these differences are a consequence of evaluating ENT function in different model systems. One major limitation in some of these studies is that the effects of competitive ENT substrates and (nontransported) inhibitors on transport of a model substrate are generally indistinguishable. As a result, some studies may incorrectly report ENT substrates as inhibitors, depending on experimental design; substrates can act as competitive inhibitors and some inhibitors may be unreported substrates. In this review, drugs that are known to be transported by an uptake/efflux transporter are denoted as “substrates,” and drugs that are only known to inhibit transporter-mediated uptake as “inhibitors”, although they may still prove to be substrates.
An example of this is abacavir, which has been reported as an ENT1 substrate56 and a nontransported ENT1 and ENT2 inhibitor.31 Uptake of [3H]abacavir in the presence or absence of NBMPR was measured in both studies, although different cell models were used in each study. Interestingly, Cerveny et al. found that abacavir was an ENT1 substrate in BeWo cells and in microvillous plasma membrane vesicles isolated from human term placentas, but it was not a substrate in fresh human placental villous fragments based on the inhibitory capacity of NBMPR or a molar excess of uridine.56 As a positive control, ENT1 was shown to be involved in [3H]adenosine uptake in each cell model, eliminating the argument that ENT1 was simply not expressed in fresh human placental villous fragments. Furthermore, the effect of NBMPR was different in each model, indicating model-specific responses to [3H]adenosine and [3H]abacavir exposure instead of ENT-specific responses. This raises an important concern when interpreting conflicting observations on ENT-substrate/inhibitor interactions. Contrasting with these observations, a study conducted by our group using ENT-expressing HeLa cells demonstrated that uptake of [3H]abacavir was uninhibited by NBMPR, uridine, or unlabeled abacavir31, suggesting abacavir does not enter cells by a transporter-mediated mechanism, but by passive diffusion. Similarly, zidovudine was shown to be a substrate of ENT2 and 3 in several studies67, 68, an ENT1 and ENT2 inhibitor (but was transported by other unknown transporters) in another study31, or did not interact with ENT1 and ENT2 at all.69 Consequently, the differences between these studies indicate that standardized cell models and testing regimens are necessary to study ENT selectivity, which warrants further analysis. Because the ENTs are ubiquitously expressed, there are no standardized in vitro models to study ENT-mediated transport and inhibition.
Recently, novel ENT1 and ENT2 CRISPR/Cas9 knockout HeLa S3 cell lines were generated to study the selectivity profiles of these two transporters separately.33 These knockout models are especially useful since the contribution of the CNTs to nucleoside and nucleobase transport in HeLa S3 cells is negligible.11, 13, 31–33 Importantly, statistically significant inhibitory interactions between competitive substrates or inhibitors on ENT2-mediated uridine uptake were still clearly distinguishable from the background signal in those cells. Another recent study generated a pair of CRISPR/Cas9 knockout cell lines for ENT1 and ENT2 using HAP1 cells.70 This cell line exhibited the same lack of CNT-mediated transport of uridine as the HeLa S3 knockout cell lines described previously, which is ideal for studying ENT function. These ENT1 and ENT2 knockout models are especially useful for high-throughput drug-transporter interaction analyses for preclinical studies. Furthermore, HeLa cells exhibit low expression of endogenous transporters71, although the clonal HeLa S3 cell line may exhibit different genotypical and phenotypical characteristics. Some groups have opted to use ENT1 and ENT2 cDNA/cRNA-injected Xenopus laevis oocytes, but this model system is comparatively inconvenient and provides low throughput compared to conventional in vitro assays.26, 30, 72–74 A yeast overexpression system for human ENT1 and ENT2 has also been reported, although the 50% inhibitory concentration (IC50) data obtained using these cells was significantly different compared to other in vitro systems in the same study.10, 70, 75–77 Consequently, the data obtained from this yeast model to predict DDI risks or ADRs would be misleading. Another high-throughput approach is to overexpress ENT1 or 2 in nucleoside transporter-deficient cells like the PK15 cell lines designed by Ward et al.78 This approach could solve the throughput issue compared to injected oocytes; however, endogenous transporters may still interact with certain test compounds and obfuscate ENT-specific transport. Given the advancement of gene editing technologies like CRISPR/Cas9, it may be ideal to use in vitro models that selectively overexpress or knock down the ENT1, ENT2, and other nucleoside transporters (e.g., CNTs).
Nevertheless, once ENT-specific drug interactions have been identified, validation in primary and immortalized cell lines derived from tissues can follow because ENTs are ubiquitously expressed and are functionally essential. For example, Jouan et al. observed 89% of uridine uptake in human lung carcinoma A549 cells was mediated by ENT1 based on pharmacological inhibition by NBMPR, and the remainder was primarily attributed to ENT2 with negligible contribution by the CNTs or passive diffusion.70 Inhibitable ENT-mediated adenosine uptake has also been shown in the bronchial BEAS-2B cell line.79 These observations complement early studies conducted by Plagemann et al. with the then-unnamed ENTs.80, 81 In the testes, Sertoli cells functionally express both ENT1 and ENT2, although ENT1 is the predominant transporter.21, 36, 82 Also, placental microvillous membrane vesicles of syncytiotrophoblasts, fresh placental villous fragments, and the placental BeWo cell line can support ENT-mediated nucleoside uptake.45, 56, 69, 73, 74 The commonly used human hepatoma HepaRG cell line and primary human hepatocytes have been reported to functionally express ENT1 and ENT283, and as described earlier, ENT1 and ENT2 also mediate nucleoside uptake in colonic carcinoma-derived Caco-2 and T84 cells.37, 53, 55 Importantly, other primary cells or cell lines can be used, depending on preclinical study demands, but initial studies should be performed with a standardized cell model to identify ENT-specific interactions before moving onto tissue-specific cell types.
Because the ENTs can facilitate the disposition of antineoplastic and antiviral therapeutics, they can be used to deliver therapeutics for the treatment of viral infections (e.g., human immunodeficiency, Zika, Ebola, and SARS-CoV-2 viruses) and refractory or relapsing cancers (e.g., leukemias) in pharmacological and immunological sanctuary sites like the testes.84–88 Clofarabine is typically used to treat pediatric patients with refractory or relapsed acute lymphoid/myeloid leukemias82 and has the potential to cross blood-testis barrier through the ENT1 and ENT2 transepithelial transport pathway (Figure 1E).36 Clofarabine is a documented ENT substrate and inhibitor33, 89, and has been used as a probe ENT substrate to demonstrate the contribution of ENTs to the testicular disposition of clofarabine in Sertoli cells and in rats.82 In Miller et al., after observing a decrease in clofarabine uptake in the presence or absence of NBMPR using in vitro models, the in vivo disposition of that molecule across the rat blood-testis barrier was further explored.82 Rats were dosed NBMPR-P, the phosphorylated prodrug of NBMPR, via IP injection at 10 mg/kg in 7.5% DMSO in sterile saline or vehicle 20 minutes before clofarabine, lamivudine, or vehicle control dosing. This was followed by dosing with clofarabine at a dose that would achieve detectable levels of compound in plasma and testes or at a therapeutically relevant dose based on preliminary experiments.82 A second group dosed with lamivudine, which is known to not interact with the ENTs, was included as a control. The plasma concentrations of NBMPR and each probe drug were monitored in addition to testicular tissue concentrations. In whole, clofarabine is an example of an ENT substrate with clinical importance relating to therapeutic efficacy rather than having a potential DDI risk since Imax/IC50 values are less than 0.1 for ENT1, and the Imax/IC50 for ENT2 encompasses a wide range of values (Figure 2, Table S1). Nucleoside/-tide analog therapeutics such as clofarabine that can reach sanctuary sites may reduce the need for other cancer treatment options (e.g., orchiectomy and radiotherapy) or reduce the sexual transmission of some viruses. However, importantly, this strategy can be applied to other test compounds to evaluate their ability to cross the blood-testis barrier via the ENTs or be adapted to study ENT-mediated drug disposition to other tissues.
Figure 2: Predicted DDI risk of clinically relevant drugs for human ENT1 and ENT2.
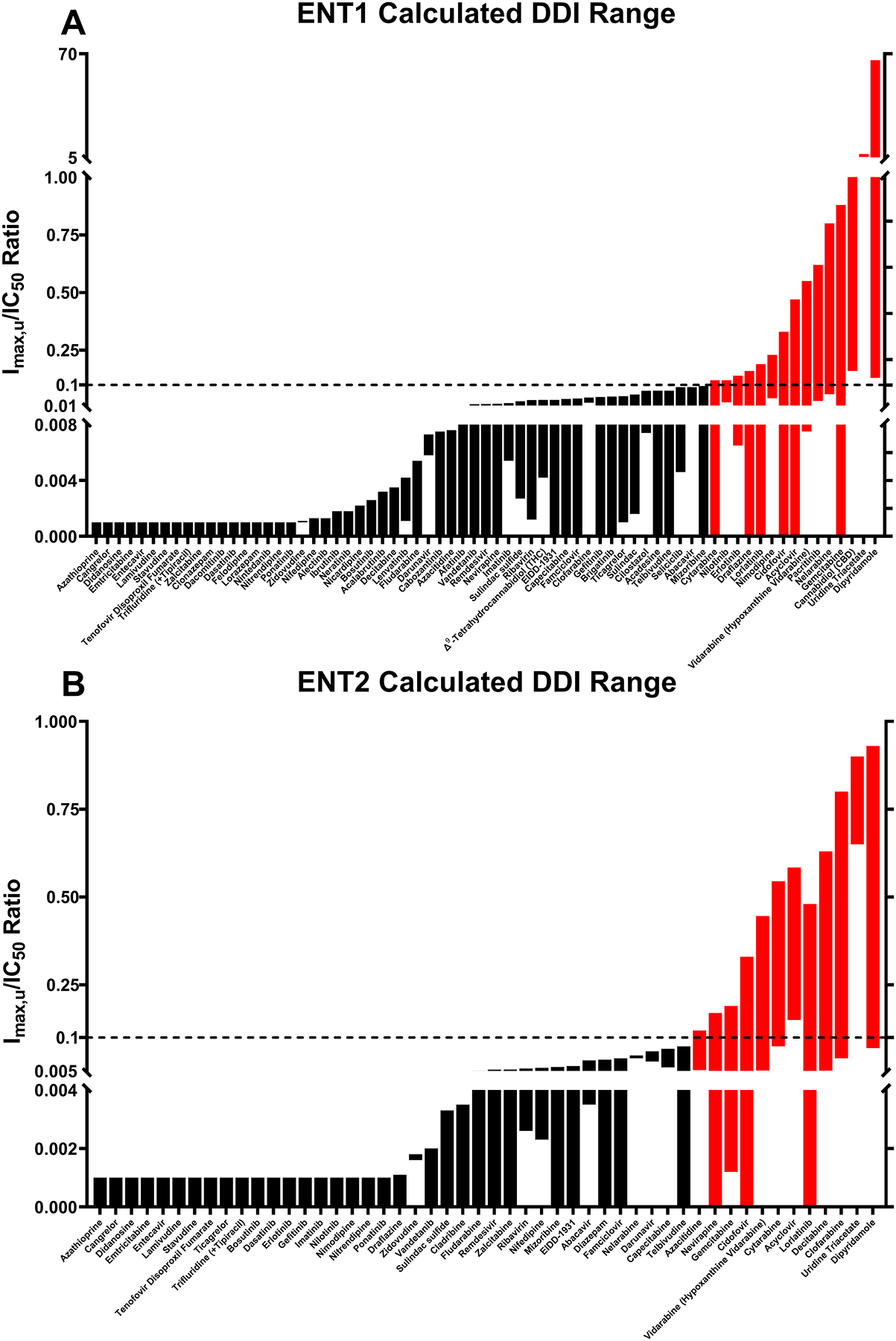
A range of calculated Imax,u/IC50 values for clinically relevant drugs with human (A) ENT1 or (B) ENT2 are graphically summarized based on available information. The dotted line indicates Imax,u/IC50 = 0.1 and drugs predicted to be at risk of in vivo DDIs (Imax,u/IC50 ≥ 0.1) are indicated in red. Refer to Tables S1 and S2 for full data sets and associated references.
Alternative approaches to evaluate in vivo drug disposition such as the use of ENT-knockout rodents is plausible64, 90–92, although knocking out ENT1 or ENT2 in mice results in an array of physical, physiological, and behavioral changes.58, 60, 64, 90, 93–97 Nevertheless, both ENT1- and ENT2-null mice remain viable and can be powerful tools to evaluate ENT-mediated drug disposition in vivo. In Paproski et al., wild-type mice dosed with [18F]3′-deoxy-3′-fluorothymidine ([18F]FLT, a positron emission tomography radiotracer and substrate for ENT1, ENT2, CNT1, and CNT3) exhibited widespread distribution of the compound into many tissues including the liver, kidney, heart, muscle, and lungs.64 Conversely, in wild-type mice pretreated with 15 mg/kg NBMPR-P or ENT1-null mice, the disposition of [18F]FLT to several tissues (i.e., liver, kidney, heart, etc.) exhibited a statistically significant decrease. Interestingly, there was a considerable increase in the disposition of [18F]FLT to the bone, bone marrow, and spleen, which was attributed to the expression of CNT1 and CNT3 in these tissues. It is also possible that ENT2 contributed to a portion of [18F]FLT uptake in some tissues because maximum plasma concentrations (Cmax) of NBMPR were likely high enough to partially inhibit its activity like in Miller et al.82 Similarly, in Endres et al., the tissue distribution of ribavirin was significantly altered in ENT1-null mice compared to their wild-type counterparts.92 This observation is in agreement with previous studies that have described ribavirin as a substrate of ENT1 (Table S1).32, 67, 68, 72, 74, 98, 99 These ENT1-knockout animal models can also be helpful in identifying other novel transport pathways that support the uptake of some drugs in vivo. For example, Anderson et al. observed that despite strong evidence that cytarabine is a substrate of ENT1 and ENT2 (Table S1)67, 91, 98–100, genetic or pharmacological removal (NBMPR inhibition) of the transporter in mice had no effect on systemic disposition of the drug.91 Instead, the authors hypothesized that there are one or more carrier-mediated mechanisms for cytarabine uptake that can be inhibited by NBMPR in mice and are not ENT1, OCTN1, OATP1B2, or MATE1. It is also unlikely that ENT2 may have played a role, given the administration of a 100 mg/kg oral dose of NBMPR prior to cytarabine dosing, although NBMPR Cmax values were not reported. At the present, there have been no reports of disposition studies performed in ENT2-null animal models, despite playing a major role in the disposition of many drugs (Table 1, Table S1).
Clinical Implications Involving ENT1 and ENT2
Efforts to identify the clinical relevance of ENTs have been largely limited to evaluating their interactions with nucleoside/-tide analog drugs; however, recent discoveries have pointed out that non-nucleoside/-tide drugs also interact with these transporters. A notable non-nucleoside/-tide inhibitor of ENT1 is CBD, which inhibits the transporter with a Ki < 250 nM and IC50 values for transport inhibition between 120 – 420 nM.9, 12 CBD is one of the primary active cannabinoids in cannabis plants, which has garnered widespread popularity as a recreational and medicinal drug in the United States and other countries. One study found that a ~19.2 mg smoke-inhaled dose of CBD reached a mean Cmax of 350 nM after 3 minutes with a mean half-life of about 31 hours in a five-patient cohort.101 Typical recreational and medicinal dosages of CBD are much higher due to its poor oral and inhalation bioavailability.102–104 A systematic review of CBD pharmacokinetic studies showed mean Cmax values ranging from sub-nanomolar to 2.00 μM, depending on the route of administration and dose102; however, most of the studies reported in this review used <20 mg single doses. Various clinical trials have evaluated the effectiveness and tolerability of single or consecutive daily doses of CBD (up to 1500 mg) for the treatment of various conditions.103, 104 In a cross-sectional study on 387 recreational CBD users, 29.5% reported using less than 24 mg per day with 60.2% using 25 mg or more per day.105 Collectively, these CBD dosages would achieve Cmax values that are expected to significantly exceed 2.00 μM and, consequently, the Ki/IC50 of ENT1. Assuming a Cmax of 2.00 μM (which is within the normal range of concentrations for medicinal and recreational use), the Imax,u/IC50 ratio for CBD (Figure 2, Table S2) indicate that it would be expected to cause a DDI when mainstream CBD products are concurrently used with therapies (e.g., antineoplastics and antivirals) that rely on ENT transport, because they may lower the therapeutic efficacy of these drugs. Interestingly, the primary active cannabinoid, THC, was also found to inhibit ENT1-mediated adenosine and thymidine transport with IC50 values ranging from 0.17 – 0.48 μM; however, it is unknown if it directly binds to and inhibits ENT1 like CBD.9 Based on a mean Cmax of 0.200 μM for THC (Figure 2, Table S2), the Imax,u/IC50 ratio suggests that it would not be a perpetrator in ENT DDIs. Additional preclinical and clinical studies are warranted to support these hypotheses, given the relatively potent IC50 values for CBD and THC, as well as the predicted DDI risk of CBD with ENTs.
In addition to CBD and THC, a variety of non-nucleoside/-tide drugs also inhibit ENT function. Many protein kinase inhibitors (PKIs) including imatinib, gefitinib, and vandetanib are potent inhibitors of ENTs (Table S2). Additional IC50 studies for PKIs and the ENTs are needed, and are particularly important as PKIs, including gefitinib and vandetanib, have been suggested for combination therapies with select nucleoside analogs. For example, gemcitabine is the standard therapy for many types of cancers and has been shown to be an in vitro and in vivo substrate of ENT1–3 with less than 10% bound to plasma proteins.67, 68, 100, 106–109 Notably, patients with low ENT1 expression in cancerous cells receiving gemcitabine treatment has been associated with significantly lower survival rates.110–112 Michaelis constants (Kt) for gemcitabine range from 0.500 – 329 μM for human ENT1 and 1.50 – 740 μM for ENT2 depending on experimental models.106, 108, 109 Although this is a wide range of Kt values, they encompass the mean Cmax values of up to 155 μM (Imax,u = 140 μM) for gemcitabine in humans113–115 and support the notion that the ENTs are relevant mediators of in vivo gemcitabine transport. Mean Cmax values at steady state for gefitinib are approximately 1.04 μM in healthy adults, but it can reach nearly 5 μM in pediatric patients based on independent studies and clinical trials (NCT00042991, NCT01610336, and NCT01982955).116–119 The mean Cmax for vandetanib is slightly higher at approximately 1.80 μM.120 Approximately 91% and 83 – 96% of gefitinib and vandetanib is bound to plasma proteins, respectively.119, 120 Reported IC50 values for gefitinib and vandetanib using various in vitro ENT1 models range from 2.00 – 14.0 and 11.0 – 37.0 μM, although there may be inconsistencies with these IC50 values due to the experimental model. (Table S2).10, 70, 75–77 Based on the Imax,u/IC50 ratios (values ≥ 0.1 are considered at risk for DDIs, Figure 2, Table S2) used to predict in vivo transporter inhibition referenced by regulatory guidances1, 6, 7, it is unlikely that these gefitinib-gemcitabine and vandetanib-gemcitabine combination therapies failed due to DDIs with the ENTs. Notably, there is a predicted risk of clinical DDIs for some PKIs, but not all, based on the Imax,u/IC50 ratios for erlotinib, lorlatinib, nilotinib, and pacritinib (Figure 2, Table S2). This observation suggests that there are unique structural features of these PKIs that allow them to act as ENT inhibitors, which are not present in other molecules.
It is important to note that if the reported IC50 values for gefitinib and vandetanib are much lower than observed for other PKIs, this ENT DDI hypothesis may partially explain why the gefitinib-gemcitabine or vandetanib-gemcitabine combination therapies did not demonstrate improved efficacy in patients with advanced non-small-cell lung cancer or locally advanced/metastatic pancreatic cancer, respectively, over gemcitabine alone.121, 122 For example, imatinib is reported to have an IC50 of 110 μM for human ENT1 and 230 μM for ENT2 using a yeast overexpression model, which is in stark contrast to significantly lower ENT1 IC50 (16.0 μM) using human lymphoblastic CEM cells in the same study77 and the unpublished findings in our group using HeLa S3 knockout cell lines (13.4 μM for ENT1 and 5.72 μM for ENT2). Furthermore, erlotinib is has an IC50 of 34.0 μM for human ENT1 in a yeast overexpression model, but ranges from 1.60 – 6.00 μM in mammalian cell lines.10 Consequently, these significantly different IC50 values and incomplete datasets makes it difficult to predict the risk of DDIs for certain PKIs and the ENTs at present. Nonetheless, it is clear that some PKIs may be involved in ENT-associated DDIs and further investigation into a wider array of PKIs is urged.
While clinical DDI studies with ENT perpetrator and victim drugs are limited, there is some evidence suggesting that ENT perpetrator drugs cause an increase in adenosine plasma levels. It is conceivable that these drugs are inhibiting ENT-mediated uptake of adenosine (and other endogenous nucleosides) in many tissues and cell types, including erythrocytes. For example, nimodipine is a potent inhibitor of ENT1 and is a drug at risk of causing a clinical DDI with ENT substrate drugs (Figure 2, Table S2). Nimodipine has been shown to increase plasma levels of adenosine in humans, which is speculated to be attributed to its inhibitory effect on nucleoside transporters (e.g., ENTs).123 While it is not a drug predicted to be at risk of causing a clinical DDI, ticagrelor also causes a significant increase in adenosine plasma levels in humans (Figure 2, Table S2).124 This is a signfiicant concern if current in vitro models for the ENTs are unable to predict clinical DDIs. Nevertheless, it is possible that the pharmacokinetics of an ENT victim drug administered concomitantly or consecutively could be negatively affected by perpetrator drugs like nimodipine or ticagrelor.
Nucleoside/-tide analog drugs are the predominant molecules studied in the context of ENT function. Due to the ubiquitous expression of the ENTs, nucleoside/-tide analog drugs can be widely distributed to many tissues. However, pathophysiological alterations to ENT expression and function can have severe effects on the absorption, distribution, metabolism, and excretion of these xenobiotics. Two of the most recent nucleoside/-tide analog drugs are remdesivir and molnupiravir, which were granted FDA emergency use authorization for the treatment of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory disease coronavirus 2 (SARS-CoV-2).125, 126 Remdesivir and molnupiravir are prodrugs that must undergo metabolic transformation into their active metabolites. Molnupiravir is not stable in plasma and is rapidly converted into EIDD-1931, which does not bind to plasma proteins.127 On the other hand, remdesivir is highly bound to plasma proteins (88–93.6%), whereas its metabolite GS-441524 is only 2% bound.128 Interestingly, remdesivir can passively diffuse across membranes but has also been shown to be a substrate of ENT1, ENT2, OATP1B1, and P-gp13, 129, as well as an inhibitor of OATP1A2, OATP1B3, OATP2B1, OCT1, and MATE1.129, 130 The study conducted by Nies et al. concluded that while remdesivir is a substrate for OATP1B1, the low uptake rates suggest that OATP1B1 is unlikely to play a major role in hepatocellular uptake.130
Remdesivir was a relatively potent inhibitor of ENT-mediated uridine transport with an IC50 of 39 μM for ENT1 and 77 μM for ENT2; however, it had an estimated Michaelis constant (Kt) for transport of 2.0 μM for ENT1 and 41 μM for ENT2.13 These values suggest that remdesivir is a better substrate and inhibitor of ENT1 than ENT2. Notably, the Cmax for a single 200 mg IV dose and multiple 100 mg IV doses of remdesivir is 7.27 μM and 3.70 μM, respectively.128, 131 Based on the calculated maximum unbound plasma concentration (Imax,u) for a single dose of remdesivir of 0.67 μM, and 0.030 μM for multiple doses, remdesivir may not be a clinically relevant substrate for ENT2 compared to ENT1. It is also possible that ENT2 has a high capacity for remdesivir transport (and, potentially, high intrinsic clearance), which could compensate for its lower affinity in a clinical setting. On the other hand, molnupiravir was a poor inhibitor of ENT1 and ENT2 with an IC50 of 701 μM for ENT1 and 851 μM for ENT2, but its primary active metabolite, EIDD-1931 was a slightly more potent inhibitor with an IC50 of 259 μM for ENT1 and 467 μM for ENT2.13 In the same study, EIDD-1931 was shown to be a substrate of ENT1 and ENT2 with an estimated Kt of 1.5 μM for ENT1 and 76 μM for ENT2, but molnupiravir was not.13 EIDD-1931 has a mean Cmax of 9.02 μM in healthy patients and 7.08 μM in COVID-19 patients after multiple 800 mg oral doses, which means that ENT1 may have significant interactions at clinically relevant concentrations.127 EIDD-1931 is also a substrate of CNT1, CNT2, and CNT3, whereas molnupiravir is a substrate for only CNT1 (and neither a substrate nor inhibitor of CNT2, CNT3, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, MATE2K, multidrug resistance-associated protein 2, P-gp, or BCRP).127, 132 Interestingly, “the lack of clinically significant CNT- or ENT-mediated DDIs reported in the literature” is cited as a reason for not anticipating clinical DDIs with molnupiravir and EIDD-1931 in the FDA emergency use authorization review.127 However, the lack of interest in evaluating the ENTs outside of nucleoside/-tide analog drugs may have contributed to this conclusion and thus, calls for further investigation into the ENT-associated DDI and ADR potential of xenobiotics.
While the tissue distribution of remdesivir and EIDD-1931 would be expected to be enhanced given their moderate passive permeability and the ubiquitous expression of ENT1 and ENT2, these therapeutics do not exhibit favorable clinical outcomes despite positive readouts in preclinical studies and early clinical trials.133–136 This phenomenon is potentially due to ENT downregulation58, 62, 137, 138 and increased secretion of the competitive substrate, adenosine58, 139, in the lungs of COVID-19 patients, which may adversely affect the disposition of remdesivir, molnupiravir, and their metabolites to the pulmonary cells of infected individuals.11 Furthermore, Acute lung injury and tissue hypoxia are typical complications in patients infected with SARS-CoV-2.137, 138 Interestingly, acute lung injury is known to increase NF-κB expression and extracellular adenosine levels as an anti-inflammatory response, where NF-κB represses ENT expression and adenosine competes for uptake.58, 139 During tissue hypoxia, HIF-1α levels are significantly elevated, which also leads to ENT1 repression.62 Further in vivo studies are warranted to link these two observations and establish the clinical importance of the ENTs in COVID-19 antiviral delivery to the lungs of SARS-CoV-2 patients. These complications can trigger hypoxic and inflammatory responses in the pulmonary epithelia.140 Together, these observations are highly suggestive that pathophysiological alterations to ENT expression and function can adversely affect the absorption, distribution, metabolism, and excretion of some compounds.
In addition to remdesivir and molnupiravir, there are many other nucleoside/-tide analogs in or approaching clinical trials such as the recent investigational new drug, galidesivir.141 Like remdesivir and molnupiravir, galidesivir was repurposed to treat COVID-19; however, clinical trials were unsuccessful, and the focus shifted towards targeting other infectious diseases including the Marburg virus.141 Potential interactions between galidesivir and the human ENTs have yet to be defined, although it is highly likely, given the findings from previous studies with structurally related analogs (Table S1). Interactions between other widely used nucleoside/-tide analog drugs include adefovir, sofosbuvir, and valaciclovir and the human ENTs are also currently unknown. However, this absence of evidence does not rule out the possibility that the ENTs may interact with these nucleoside/-tide analogs. Further evaluation of ENT interactions with these clinically relevant nucleoside/-tide analogs is necessary and may improve the development of future drug candidates. Many of the drugs listed in Tables 1 and 2 interact with one or more human ENTs as a substrate or an inhibitor, suggesting that the ENTs may also be involved in unsafe DDIs for the untested drugs or other non-nucleoside/-tide drugs. These findings support the notion that ENTs may be involved in clinically important drug interactions with remdesivir, molnupiravir (EIDD-1931), and other drugs, which requires further scrutiny in drug discovery and development.
While the intention of drug discovery and development programs is to design therapeutics that safely treat diseases or disorders, there is always a risk of off-target toxicity and other ADRs that need to be predictable. The most infamous example of a nucleoside analog which failed in clinical trials due to severe hepatotoxicity and myopathy that was not predicted by preclinical animal toxicity studies is fialuridine.142–144 Fialuridine was designed to treat chronic hepatitis B virus infection and demonstrated success in decreasing hepatitis B viral DNA levels in the blood of patients early into treatment during phase II clinical trials.143 However, severe side effects began to appear after 9 weeks of treatment, which ultimately led to the deaths of 5 out of 15 patients and an additional 2 patients requiring liver transplants.142, 143 It was found that fialuridine-induced hepatotoxicity and myopathy was associated with widespread mitochondrial damage143, which was later attributed to mitochondrial uptake by ENT1 and its incorporation into mitochondrial DNA via its 3’-hydroxyl moiety.46, 47 Kinetic parameters for ENT1 transport of fialuridine are unknown; however, it does inhibit ~45% of ENT1-mediated uridine transport at 50 μM, its uptake is completely inhibited by 10 μM NBMPR, and is described as an “excellent substrate” of ENT1.46 Although fialuridine is not an exceptionally potent inhibitor of ENT1-mediated transport, its uptake into cells and through mitochondria is only reliant on its concentration gradient. The mean Cmax for a single 5 mg dose of fialuridine in healthy adults is approximately 0.640 μM145, although the patients in phase II clinical trials received a 0.1 or 0.25 mg/kg (6 or 15 mg, assuming a body weight of 60 kg) dose of fialuridine per day.142, 143 Furthermore, initial estimates for the half-life of fialuridine in humans ranged from 1 – 4 hours but a later pharmacokinetic study revealed a more accurate elimination half-life of 28 – 30 hours.142, 145 Consequently, these patients likely had higher mean Cmax values over their treatment period, resulting in greater and longer exposure to this drug.
Importantly, preclinical toxicity studies in rodents were unable to predict this severe human hepatic mitotoxicity and myopathy because, unlike human ENT1, mouse and rat ENT1 are not trafficked to the mitochondrial membrane.46, 47 This is because the rodent orthologs lack the mitochondrial targeting sequence Pro71, Glu72, and Asn74 (PEXN motif) present in human ENT1.47 Instead, the PEXN motif is substituted with Pro71, Asp72, and Ser74 in rodents. Interestingly, the PEXN motif is present in canines and cynomolgus monkeys, but not in rabbits or pigs. Although 70-day fialuridine dosing regimen in F-344 rats did cause increased levels of apoptosis and nuclear atypia in the liver, clinical signs were not observed, consistent with a lack of a mitochondrial ENT1-mediated uptake mechanism.144 On the other hand, 90-day fialuridine dosing in beagle dogs and 30-day dosing in rhesus monkeys did not produce any hepatotoxic effects.144 It is possible that the lack of hepatotoxicity observed in beagle dogs and rhesus monkeys may be partially explained by the delayed toxic effect of fialuridine, because the first patient to suffer liver failure had been treated for over 10 weeks in a 24-week daily dosing clinical trial and the duration of the toxicity studies in some of these preclinical species was insufficient.142, 143 While beagle dogs were treated for 90 days, species differences in the rates of fialuridine uptake, phosphorylation into its active metabolite, and incorporation into replicating DNA (occurrence and rate of DNA synthesis varies) may provide additional explanation for these observations. Fialuridine is an interesting example of a clinically important molecule, which has a severe and unusual mechanism of toxicity that is reliant on ENT-mediated transport into mitochondria and not predictable in certain species like rodents. However, it is also important to note that severe clinical hepatotoxicity is not limited to fialuridine and is observed with other structurally similar molecules such as clevudine and clofarabine, as they are directly involved in mitochondrial dysfunction.146–150 In clinical trials for clofarabine, nine patients died due to multi-organ failure (associated with liver, kidney, small intestine, and colon toxicity), myopathy, and sepsis.146, 150 Thus, it stands to reason that the mechanism in which clevudine and clofarabine can cross the mitochondrial membrane to exert their pharmacological function and toxicity is through ENT1-mediated transport. Together, these findings highlight the unique and challenging issues when translating the findings from certain preclinical models to humans. Furthermore, these observations point to the clinical importance of the ENTs, which must be accounted for through additional ADR and DDI testing paradigms.
Conclusions
The FDA, EMA, and PMDA drug interaction guidances1, 6, 7 provide recommendations, based in part from input provided by ITC white paper recommendations, for assessing transporter-mediated DDIs and serve as the standard for drug discovery and development programs. As a result, many transport studies have an implicit bias towards assessing OAT1, OAT3, OCT2, OATP1B1, OATP1B3, P-gp, BCRP, MATE1, and MATE2-K. While these transporters are critical for the disposition of many molecules, we believe that the current data suggests that ENTs may also be involved in important ADRs and DDIs. Despite the ITC previously acknowledging ENT1 and ENT2 as transporters with potential clinical relevance2, there are fewer studies characterizing ENT function compared to many other transporters. ENT1 and ENT2 are involved in the transport of clinically relevant nucleoside/-tide analog drugs, such as remdesivir, EIDD-1931, fialuridine, and gemcitabine, that are used to treat a range of indications from viral infections to cancers (Table S1). Furthermore, ENT1 has known inhibitory interactions with non-nucleoside/-tide drugs including PKIs such as gefitinib, lorlatinib, and other commonly used drugs, like CBD, at clinically relevant concentrations (Table S2). Consequently, there could be a significant risk of clinical DDIs that increase toxicity and/or decrease overall therapeutic efficacy when certain inhibitors are taken in combination with ENT substrates. In summary, greater recognition of ENTs as transporters potentially implicated in clinical DDIs or ADRs will encourage further investigative studies into their selectivity and clinical relevance and thereby improve drug discovery and development programs.
Supplementary Material
Acknowledgements
We thank Siennah Greenfield and David Klein for their exceptional contributions to the direction of this field and the thoughtful discussions during the drafting of this manuscript. We would also like to thank David Rodrigues for his expertise and constructive review of our manuscript.
Funding:
This work was supported by funding from the National Institutes of General Medical Sciences [Grants R01GM123643 and R01GM129777] and National Institute of Environmental Health Sciences [Grant T32ES007091].
Footnotes
Conflicts of Interest: The authors declared no competing interests for this work.
SUPPORTING INFORMATION
Supplementary information accompanies this paper on the Clinical Pharmacology & Therapeutics website (www.cpt-journal.com).
Contributor Information
Raymond K. Hau, College of Pharmacy, Department of Pharmacology & Toxicology, The University of Arizona, Tucson, AZ, USA
Stephen H. Wright, College of Medicine, Department of Physiology, The University of Arizona, Tucson, AZ, USA
Nathan J. Cherrington, College of Pharmacy, Department of Pharmacology & Toxicology, The University of Arizona, Tucson, AZ, USA
References
- 1.In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. U.S. Food and Drug Administration. <https://www.fda.gov/regulatory-information/search-fda-guidance-documents/in-vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions> (2020). Accessed February 5, 2023.
- 2.Hillgren KM et al. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 94, 52–63 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Zamek-Gliszczynski MJ et al. Transporters in Drug Development: International Transporter Consortium Update on Emerging Transporters of Clinical Importance. Clin Pharmacol Ther 112, 485–500 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Zamek-Gliszczynski MJ et al. Transporters in Drug Development: 2018 ITC Recommendations for Transporters of Emerging Clinical Importance. Clin Pharmacol Ther 104, 890–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Transporter Consortium et al. Membrane transporters in drug development. Nat Rev Drug Discov 9, 215–36 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guideline on the investigation of drug interactions. European Medicines Agency. <https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf> (2013). Accessed February 5, 2023.
- 7.Guideline on drug interaction for drug development and appropriate provision of information. Pharmaceuticals and Medical Devices Agency. <https://www.pmda.go.jp/files/000228122.pdf> (2019). Accessed February 5, 2023.
- 8.Zolk O & Fromm MF Transporter-mediated drug uptake and efflux: important determinants of adverse drug reactions. Clin Pharmacol Ther 89, 798–805 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Carrier EJ, Auchampach JA & Hillard CJ Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci U S A 103, 7895–900 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damaraju VL et al. Erlotinib, gefitinib, and vandetanib inhibit human nucleoside transporters and protect cancer cells from gemcitabine cytotoxicity. Clin Cancer Res 20, 176–86 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Hau RK, Wright SH & Cherrington NJ PF-07321332 (Nirmatrelvir) does not interact with human ENT1 or ENT2: Implications for COVID-19 patients. Clin Transl Sci 15, 1599–605 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liou GI et al. Mediation of cannabidiol anti-inflammation in the retina by equilibrative nucleoside transporter and A2A adenosine receptor. Invest Ophthalmol Vis Sci 49, 5526–31 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller SR et al. Remdesivir and EIDD-1931 Interact with Human Equilibrative Nucleoside Transporters 1 and 2: Implications for Reaching SARS-CoV-2 Viral Sanctuary Sites. Mol Pharmacol 100, 548–57 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldwin SA et al. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch 447, 735–43 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Young JD The SLC28 (CNT) and SLC29 (ENT) nucleoside transporter families: a 30-year collaborative odyssey. Biochem Soc Trans 44, 869–76 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Young JD, Yao SY, Baldwin JM, Cass CE & Baldwin SA The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med 34, 529–47 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Al-Majdoub ZM et al. Proteomic Quantification of Human Blood-Brain Barrier SLC and ABC Transporters in Healthy Individuals and Dementia Patients. Mol Pharm 16, 1220–33 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Couto N et al. Quantification of Proteins Involved in Drug Metabolism and Disposition in the Human Liver Using Label-Free Global Proteomics. Mol Pharm 16, 632–47 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Nishimura M & Naito S Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet 20, 452–77 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Bleasby K et al. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: a resource for investigations into drug disposition. Xenobiotica 36, 963–88 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Hau RK, Miller SR, Wright SH & Cherrington NJ Generation of a hTERT-Immortalized Human Sertoli Cell Model to Study Transporter Dynamics at the Blood-Testis Barrier. Pharmaceutics 12, 1005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uchida Y et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem 117, 333–45 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Shawahna R et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm 8, 1332–41 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Ohtsuki S et al. Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol Pharm 10, 289–96 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Yao SY et al. Molecular cloning and functional characterization of nitrobenzylthioinosine (NBMPR)-sensitive (es) and NBMPR-insensitive (ei) equilibrative nucleoside transporter proteins (rENT1 and rENT2) from rat tissues. J Biol Chem 272, 28423–30 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Griffiths M et al. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat Med 3, 89–93 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Handa M et al. Cloning of a novel isoform of the mouse NBMPR-sensitive equilibrative nucleoside transporter (ENT1) lacking a putative phosphorylation site. Gene 262, 301–7 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Yao SY, Ng AM, Cass CE, Baldwin SA & Young JD Nucleobase transport by human equilibrative nucleoside transporter 1 (hENT1). J Biol Chem 286, 32552–62 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackey JR, Baldwin SA, Young JD & Cass CE Nucleoside transport and its significance for anticancer drug resistance. Drug Resist Updat 1, 310–24 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Griffiths M et al. Molecular cloning and characterization of a nitrobenzylthioinosine-insensitive (ei) equilibrative nucleoside transporter from human placenta. Biochem J 328 (Pt 3), 739–43 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller SR et al. Nucleoside Reverse Transcriptase Inhibitor Interaction with Human Equilibrative Nucleoside Transporters 1 and 2. Drug Metab Dispos 48, 603–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller SR et al. Multiple Computational Approaches for Predicting Drug Interactions with Human Equilibrative Nucleoside Transporter 1. Drug Metab Dispos 49, 479–89 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller SR et al. Predicting Drug Interactions with Human Equilibrative Nucleoside Transporters 1 and 2 Using Functional Knockout Cell Lines and Bayesian Modeling. Mol Pharmacol 99, 147–62 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barros LF et al. Immunolocalisation of nucleoside transporters in human placental trophoblast and endothelial cells: evidence for multiple transporter isoforms. Pflugers Arch 429, 394–9 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Govindarajan R et al. In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am J Physiol Regul Integr Comp Physiol 293, R1809–22 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Klein DM et al. Basolateral uptake of nucleosides by Sertoli cells is mediated primarily by equilibrative nucleoside transporter 1. J Pharmacol Exp Ther 346, 121–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Senyavina NV, Gerasimenko TN, Fomicheva KA, Tonevitskaya SA & Kaprin AD Localization and Expression of Nucleoside Transporters ENT1 and ENT2 in Polar Cells of Intestinal Epithelium. Bull Exp Biol Med 160, 771–4 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Wright NJ & Lee SY Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol 26, 599–606 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.SenGupta DJ & Unadkat JD Glycine 154 of the equilibrative nucleoside transporter, hENT1, is important for nucleoside transport and for conferring sensitivity to the inhibitors nitrobenzylthioinosine, dipyridamole, and dilazep. Biochem Pharmacol 67, 453–8 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Visser F et al. Residues 334 and 338 in transmembrane segment 8 of human equilibrative nucleoside transporter 1 are important determinants of inhibitor sensitivity, protein folding, and catalytic turnover. J Biol Chem 282, 14148–57 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Wu Z et al. Insight into the nucleoside transport and inhibition of human ENT1. Curr Res Struct Biol 4, 192–205 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rehan S, Shahid S, Salminen TA, Jaakola VP & Paavilainen VO Current Progress on Equilibrative Nucleoside Transporter Function and Inhibitor Design. SLAS Discov 24, 953–68 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Visser F, Baldwin SA, Isaac RE, Young JD & Cass CE Identification and mutational analysis of amino acid residues involved in dipyridamole interactions with human and Caenorhabditis elegans equilibrative nucleoside transporters. J Biol Chem 280, 11025–34 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Visser F et al. Mutation of residue 33 of human equilibrative nucleoside transporters 1 and 2 alters sensitivity to inhibition of transport by dilazep and dipyridamole. J Biol Chem 277, 395–401 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Mani RS et al. Demonstration of equilibrative nucleoside transporters (hENT1 and hENT2) in nuclear envelopes of cultured human choriocarcinoma (BeWo) cells by functional reconstitution in proteoliposomes. J Biol Chem 273, 30818–25 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Lai Y, Tse CM & Unadkat JD Mitochondrial expression of the human equilibrative nucleoside transporter 1 (hENT1) results in enhanced mitochondrial toxicity of antiviral drugs. J Biol Chem 279, 4490–7 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Lee EW, Lai Y, Zhang H & Unadkat JD Identification of the mitochondrial targeting signal of the human equilibrative nucleoside transporter 1 (hENT1): implications for interspecies differences in mitochondrial toxicity of fialuridine. J Biol Chem 281, 16700–6 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Nakamura K et al. Large-scale multiplex absolute protein quantification of drug-metabolizing enzymes and transporters in human intestine, liver, and kidney microsomes by SWATH-MS: Comparison with MRM/SRM and HR-MRM/PRM. Proteomics 16, 2106–17 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Govindarajan R et al. Expression and hepatobiliary transport characteristics of the concentrative and equilibrative nucleoside transporters in sandwich-cultured human hepatocytes. Am J Physiol Gastrointest Liver Physiol 295, G570–80 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Damaraju VL et al. Localization of broadly selective equilibrative and concentrative nucleoside transporters, hENT1 and hCNT3, in human kidney. Am J Physiol Renal Physiol 293, F200–11 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Mangravite LM, Xiao G & Giacomini KM Localization of human equilibrative nucleoside transporters, hENT1 and hENT2, in renal epithelial cells. Am J Physiol Renal Physiol 284, F902–10 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Patil SD & Unadkat JD Sodium-dependent nucleoside transport in the human intestinal brush-border membrane. Am J Physiol 272, G1314–20 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Mun EC, Tally KJ & Matthews JB Characterization and regulation of adenosine transport in T84 intestinal epithelial cells. Am J Physiol 274, G261–9 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Ward JL & Tse CM Nucleoside transport in human colonic epithelial cell lines: evidence for two Na+-independent transport systems in T84 and Caco-2 cells. Biochim Biophys Acta 1419, 15–22 (1999). [DOI] [PubMed] [Google Scholar]
- 55.Senyavina NV & Tonevitskaya SA Effect of Hypoxanthine on Functional Activity of Nucleoside Transporters ENT1 and ENT2 in Caco-2 Polar Epithelial Intestinal Cells. Bull Exp Biol Med 160, 160–4 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Cerveny L et al. Equilibrative Nucleoside Transporter 1 (ENT1, SLC29A1) Facilitates Transfer of the Antiretroviral Drug Abacavir across the Placenta. Drug Metab Dispos 46, 1817–26 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Jennings LL et al. Distinct regional distribution of human equilibrative nucleoside transporter proteins 1 and 2 (hENT1 and hENT2) in the central nervous system. Neuropharmacology 40, 722–31 (2001). [DOI] [PubMed] [Google Scholar]
- 58.Morote-Garcia JC et al. Repression of the equilibrative nucleoside transporters dampens inflammatory lung injury. Am J Respir Cell Mol Biol 49, 296–305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Billington S et al. Interindividual and Regional Variability in Drug Transporter Abundance at the Human Blood-Brain Barrier Measured by Quantitative Targeted Proteomics. Clin Pharmacol Ther 106, 228–37 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Eckle T et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J 27, 3078–89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi DS et al. Genomic organization and expression of the mouse equilibrative, nitrobenzylthioinosine-sensitive nucleoside transporter 1 (ENT1) gene. Biochem Biophys Res Commun 277, 200–8 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Eltzschig HK et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med 202, 1493–505 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Redzic ZB et al. Polarized distribution of nucleoside transporters in rat brain endothelial and choroid plexus epithelial cells. J Neurochem 94, 1420–6 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Paproski RJ et al. Biodistribution and uptake of 3’-deoxy-3’-fluorothymidine in ENT1-knockout mice and in an ENT1-knockdown tumor model. J Nucl Med 51, 1447–55 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Genter MB, Krishan M, Augustine LM & Cherrington NJ Drug transporter expression and localization in rat nasal respiratory and olfactory mucosa and olfactory bulb. Drug Metab Dispos 38, 1644–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Errasti-Murugarren E, Diaz P, Godoy V, Riquelme G & Pastor-Anglada M Expression and distribution of nucleoside transporter proteins in the human syncytiotrophoblast. Mol Pharmacol 80, 809–17 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Boswell-Casteel RC & Hays FA Equilibrative nucleoside transporters-A review. Nucleosides Nucleotides Nucleic Acids 36, 7–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Govindarajan R et al. Facilitated mitochondrial import of antiviral and anticancer nucleoside drugs by human equilibrative nucleoside transporter-3. Am J Physiol Gastrointest Liver Physiol 296, G910–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karbanova S et al. Role of nucleoside transporters in transplacental pharmacokinetics of nucleoside reverse transcriptase inhibitors zidovudine and emtricitabine. Placenta 60, 86–92 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Jouan E et al. Differential Inhibition of Equilibrative Nucleoside Transporter 1 (ENT1) Activity by Tyrosine Kinase Inhibitors. Eur J Drug Metab Pharmacokinet 46, 625–35 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ahlin G et al. Endogenous gene and protein expression of drug-transporting proteins in cell lines routinely used in drug discovery programs. Drug Metab Dispos 37, 2275–83 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Choi MK, Kim MH, Maeng HJ & Song IS Contribution of CNT1 and ENT1 to ribavirin uptake in human hepatocytes. Arch Pharm Res 38, 904–13 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Nishimura T et al. Quantification of ENT1 and ENT2 Proteins at the Placental Barrier and Contribution of These Transporters to Ribavirin Uptake. J Pharm Sci 108, 3917–22 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Yamamoto T et al. Ribavirin uptake by cultured human choriocarcinoma (BeWo) cells and Xenopus laevis oocytes expressing recombinant plasma membrane human nucleoside transporters. Eur J Pharmacol 557, 1–8 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Damaraju VL, Kuzma M, Mowles D, Cass CE & Sawyer MB Interactions of multitargeted kinase inhibitors and nucleoside drugs: Achilles heel of combination therapy? Mol Cancer Ther 14, 236–45 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Huang M, Wang Y, Cogut SB, Mitchell BS & Graves LM Inhibition of nucleoside transport by protein kinase inhibitors. J Pharmacol Exp Ther 304, 753–60 (2003). [DOI] [PubMed] [Google Scholar]
- 77.Damaraju VL, Weber D, Kuzma M, Cass CE & Sawyer MB Selective Inhibition of Human Equilibrative and Concentrative Nucleoside Transporters by BCR-ABL Kinase Inhibitors: IDENTIFICATION OF KEY hENT1 AMINO ACID RESIDUES FOR INTERACTION WITH BCR-ABL KINASE INHIBITORS. J Biol Chem 291, 18809–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ward JL, Sherali A, Mo ZP & Tse CM Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem 275, 8375–81 (2000). [DOI] [PubMed] [Google Scholar]
- 79.Allen-Gipson DS et al. Ethanol blocks adenosine uptake via inhibiting the nucleoside transport system in bronchial epithelial cells. Alcohol Clin Exp Res 33, 791–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wohlhueter RM, Brown WE & Plagemann PG Kinetic and thermodynamic studies on nitrobenzylthioinosine binding to the nucleoside transporter of Chinese hamster ovary cells. Biochim Biophys Acta 731, 168–76 (1983). [DOI] [PubMed] [Google Scholar]
- 81.Plagemann PG & Wohlhueter RM Nucleoside transport in cultured mammalian cells. Multiple forms with different sensitivity to inhibition by nitrobenzylthioinosine or hypoxanthine. Biochim Biophys Acta 773, 39–52 (1984). [DOI] [PubMed] [Google Scholar]
- 82.Miller SR et al. Testicular disposition of clofarabine in rats is dependent on equilibrative nucleoside transporters. Pharmacol Res Perspect 9, e00831 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mayati A et al. mRNA Expression and Activity of Nucleoside Transporters in Human Hepatoma HepaRG Cells. Pharmaceutics 10, 246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nesbit ME Jr. et al. Testicular relapse in childhood acute lymphoblastic leukemia: association with pretreatment patient characteristics and treatment. A report for Childrens Cancer Study Group. Cancer 45, 2009–16 (1980). [DOI] [PubMed] [Google Scholar]
- 85.Byrn RA & Kiessling AA Analysis of human immunodeficiency virus in semen: indications of a genetically distinct virus reservoir. J Reprod Immunol 41, 161–76 (1998). [DOI] [PubMed] [Google Scholar]
- 86.Kiessling AA et al. Human immunodeficiency virus in semen arises from a genetically distinct virus reservoir. AIDS Res Hum Retroviruses 14 Suppl 1, S33–41 (1998). [PubMed] [Google Scholar]
- 87.Ma X et al. Pathological and molecular examinations of postmortem testis biopsies reveal SARS-CoV-2 infection in the testis and spermatogenesis damage in COVID-19 patients. Cell Mol Immunol 18, 487–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Robinson CL et al. Male germ cells support long-term propagation of Zika virus. Nat Commun 9, 2090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.King KM et al. A comparison of the transportability, and its role in cytotoxicity, of clofarabine, cladribine, and fludarabine by recombinant human nucleoside transporters produced in three model expression systems. Mol Pharmacol 69, 346–53 (2006). [DOI] [PubMed] [Google Scholar]
- 90.Bone DB, Antic M, Quinonez D & Hammond JR Hypoxanthine uptake by skeletal muscle microvascular endothelial cells from equilibrative nucleoside transporter 1 (ENT1)-null mice: effect of oxidative stress. Microvasc Res 98, 16–22 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Anderson JT, Hu S, Fu Q, Baker SD & Sparreboom A Role of equilibrative nucleoside transporter 1 (ENT1) in the disposition of cytarabine in mice. Pharmacol Res Perspect 7, e00534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Endres CJ et al. The role of the equilibrative nucleoside transporter 1 (ENT1) in transport and metabolism of ribavirin by human and wild-type or Ent1−/− mouse erythrocytes. J Pharmacol Exp Ther 329, 387–98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Warraich S et al. Loss of equilibrative nucleoside transporter 1 in mice leads to progressive ectopic mineralization of spinal tissues resembling diffuse idiopathic skeletal hyperostosis in humans. J Bone Miner Res 28, 1135–49 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Hinton DJ et al. Aberrant bone density in aging mice lacking the adenosine transporter ENT1. PLoS One 9, e88818 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen J et al. Altered glutamatergic neurotransmission in the striatum regulates ethanol sensitivity and intake in mice lacking ENT1. Behav Brain Res 208, 636–42 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu KC, Lee CY, Chou FY, Chern Y & Lin CJ Deletion of equilibrative nucleoside transporter-2 protects against lipopolysaccharide-induced neuroinflammation and blood-brain barrier dysfunction in mice. Brain Behav Immun 84, 59–71 (2020). [DOI] [PubMed] [Google Scholar]
- 97.Aherne CM et al. Coordination of ENT2-dependent adenosine transport and signaling dampens mucosal inflammation. JCI Insight 3, e121521 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mangravite LM, Badagnani I & Giacomini KM Nucleoside transporters in the disposition and targeting of nucleoside analogs in the kidney. Eur J Pharmacol 479, 269–81 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Osato DH et al. Functional characterization in yeast of genetic variants in the human equilibrative nucleoside transporter, ENT1. Pharmacogenetics 13, 297–301 (2003). [DOI] [PubMed] [Google Scholar]
- 100.Santini D et al. Prognostic role of human equilibrative transporter 1 (hENT1) in patients with resected gastric cancer. J Cell Physiol 223, 384–8 (2010). [DOI] [PubMed] [Google Scholar]
- 101.Ohlsson A et al. Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed Environ Mass Spectrom 13, 77–83 (1986). [DOI] [PubMed] [Google Scholar]
- 102.Millar SA, Stone NL, Yates AS & O’Sullivan SE A Systematic Review on the Pharmacokinetics of Cannabidiol in Humans. Front Pharmacol 9, 1365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Larsen C & Shahinas J Dosage, Efficacy and Safety of Cannabidiol Administration in Adults: A Systematic Review of Human Trials. J Clin Med Res 12, 129–41 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Millar SA et al. A systematic review of cannabidiol dosing in clinical populations. Br J Clin Pharmacol 85, 1888–900 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moltke J & Hindocha C Reasons for cannabidiol use: a cross-sectional study of CBD users, focusing on self-perceived stress, anxiety, and sleep problems. J Cannabis Res 3, 5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mackey JR et al. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst 91, 1876–81 (1999). [DOI] [PubMed] [Google Scholar]
- 107.Hammond JR, Lee S & Ferguson PJ [3H]gemcitabine uptake by nucleoside transporters in a human head and neck squamous carcinoma cell line. J Pharmacol Exp Ther 288, 1185–91 (1999). [PubMed] [Google Scholar]
- 108.Hioki M et al. Contribution of equilibrative nucleoside transporters 1 and 2 to gemcitabine uptake in pancreatic cancer cells. Biopharm Drug Dispos 39, 256–64 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Mackey JR et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 58, 4349–57 (1998). [PubMed] [Google Scholar]
- 110.Farrell JJ et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology 136, 187–95 (2009). [DOI] [PubMed] [Google Scholar]
- 111.Oguri T et al. The absence of human equilibrative nucleoside transporter 1 expression predicts nonresponse to gemcitabine-containing chemotherapy in non-small cell lung cancer. Cancer Lett 256, 112–9 (2007). [DOI] [PubMed] [Google Scholar]
- 112.Spratlin J et al. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res 10, 6956–61 (2004). [DOI] [PubMed] [Google Scholar]
- 113.Fogli S et al. Drug distribution and pharmacokinetic/pharmacodynamic relationship of paclitaxel and gemcitabine in patients with non-small-cell lung cancer. Ann Oncol 12, 1553–9 (2001). [DOI] [PubMed] [Google Scholar]
- 114.Abbruzzese JL et al. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol 9, 491–8 (1991). [DOI] [PubMed] [Google Scholar]
- 115.Grunewald R, Abbruzzese JL, Tarassoff P & Plunkett W Saturation of 2’,2’-difluorodeoxycytidine 5’-triphosphate accumulation by mononuclear cells during a phase I trial of gemcitabine. Cancer Chemother Pharmacol 27, 258–62 (1991). [DOI] [PubMed] [Google Scholar]
- 116.Hirano S et al. The pharmacokinetics and long-term therapeutic effects of gefitinib in patients with lung adenocarcinoma harboring the epidermal growth factor receptor(EGFR)mutation. Gan To Kagaku Ryoho 39, 1501–6 (2012). [PubMed] [Google Scholar]
- 117.Ranson M & Wardell S Gefitinib, a novel, orally administered agent for the treatment of cancer. J Clin Pharm Ther 29, 95–103 (2004). [DOI] [PubMed] [Google Scholar]
- 118.Swaisland HC et al. Single-dose clinical pharmacokinetic studies of gefitinib. Clin Pharmacokinet 44, 1165–77 (2005). [DOI] [PubMed] [Google Scholar]
- 119.Iressa (Gefitinib) Clinical Pharmacology and BioPharmaceutics Review(s). U.S. Food and Drug Administration. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206995orig1s000clinpharmr.pdf> (2014). Accessed February 5, 2023.
- 120.Caprelsa (Vandetanib) Clinical Pharmacology and BioPharmaceutics Review(s). U.S. Food and Drug Administration. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022405Orig1s000ClinPharmR.pdf> (2010). Accessed February 5, 2023.
- 121.Giaccone G et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J Clin Oncol 22, 777–84 (2004). [DOI] [PubMed] [Google Scholar]
- 122.Middleton G et al. Vandetanib plus gemcitabine versus placebo plus gemcitabine in locally advanced or metastatic pancreatic carcinoma (ViP): a prospective, randomised, double-blind, multicentre phase 2 trial. Lancet Oncol 18, 486–99 (2017). [DOI] [PubMed] [Google Scholar]
- 123.Blardi P et al. Nimodipine: drug pharmacokinetics and plasma adenosine levels in patients affected by cerebral ischemia. Clin Pharmacol Ther 72, 556–61 (2002). [DOI] [PubMed] [Google Scholar]
- 124.Bonello L et al. Ticagrelor increases adenosine plasma concentration in patients with an acute coronary syndrome. J Am Coll Cardiol 63, 872–7 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of COVID-19. U.S. Food and Drug Administration. <https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19> (2021). Accessed February 5, 2023.
- 126.Coronavirus (COVID-19) Update: FDA Authorizes Additional Oral Antiviral for Treatment of COVID-19 in Certain Adults. U.S. Food and Drug Administration. <https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-oral-antiviral-treatment-covid-19-certain> (2021). Accessed February 5, 2023.
- 127.Lagevrio (Molnupiravir) Emergency Use Authorization Review. U.S. Food and Drug Administration. <https://www.fda.gov/media/155241/download> (2021). Accessed February 5, 2023.
- 128.Veklury (Remdesivir) Summary Review. U.S. Food and Drug Administration. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/214787Orig1s000Sumr.pdf> (2020). Accessed February 5, 2023.
- 129.Veklury (Remdesivir) EUA Fact Sheet for Healthcare Providers. U.S. Food and Drug Administration. <https://www.fda.gov/media/137566/download> (2022). Accessed February 5, 2023.
- 130.Nies AT et al. Interaction of Remdesivir with Clinically Relevant Hepatic Drug Uptake Transporters. Pharmaceutics 13, 369 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Humeniuk R et al. Pharmacokinetic, Pharmacodynamic, and Drug-Interaction Profile of Remdesivir, a SARS-CoV-2 Replication Inhibitor. Clin Pharmacokinet 60, 569–83 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lagevrio (Molnupiravir) EUA Fact Sheet for Healthcare Providers. U.S. Food and Drug Administration. <https://www.fda.gov/media/155054/download> (2022). Accessed February 5, 2023.
- 133.Jayk Bernal A et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N Engl J Med 386, 509–20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Beigel JH et al. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med 383, 1813–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang Y et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 395, 1569–78 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Consortium WHOST et al. Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial Results. N Engl J Med 384, 497–511 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Leist SR et al. A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell 183, 1070–85 e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Taniguchi-Ponciano K et al. Increased expression of hypoxia-induced factor 1alpha mRNA and its related genes in myeloid blood cells from critically ill COVID-19 patients. Ann Med 53, 197–207 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Eckle T, Koeppen M & Eltzschig HK Role of extracellular adenosine in acute lung injury. Physiology (Bethesda) 24, 298–306 (2009). [DOI] [PubMed] [Google Scholar]
- 140.Bhattacharya S, Agarwal S, Shrimali NM & Guchhait P Interplay between hypoxia and inflammation contributes to the progression and severity of respiratory viral diseases. Mol Aspects Med 81, 101000 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Julander JG et al. An update on the progress of galidesivir (BCX4430), a broad-spectrum antiviral. Antiviral Res 195, 105180 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Institute of Medicine (US) Committee to Review the Fialuridine (FIAU/FIAC) Clinical Trials. Review of the Fialuridine (FIAU) Clinical Trials. (eds. Manning FJ and Swartz M) (The National Academies Press, Washington, DC, 1995). [PubMed] [Google Scholar]
- 143.McKenzie R et al. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med 333, 1099–105 (1995). [DOI] [PubMed] [Google Scholar]
- 144.Richardson FC, Engelhardt JA & Bowsher RR Fialuridine accumulates in DNA of dogs, monkeys, and rats following long-term oral administration. Proc Natl Acad Sci U S A 91, 12003–7 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bowsher RR et al. Sensitive and specific radioimmunoassay for fialuridine: initial assessment of pharmacokinetics after single oral doses to healthy volunteers. Antimicrob Agents Chemother 38, 2134–42 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Clolar (Clofarabine) FDA Package Insert. U.S. Food and Drug Administration. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021673s023lbl.pdf> (2004). Accessed February 5, 2023.
- 147.Seok JI et al. Long-term therapy with clevudine for chronic hepatitis B can be associated with myopathy characterized by depletion of mitochondrial DNA. Hepatology 49, 2080–6 (2009). [DOI] [PubMed] [Google Scholar]
- 148.Park SH et al. Clevudine Induced Mitochondrial Myopathy. J Korean Med Sci 32, 1857–60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Krawczyk J et al. Clofarabine in the treatment of poor risk acute myeloid leukaemia. Hematol Oncol 28, 118–23 (2010). [DOI] [PubMed] [Google Scholar]
- 150.Faderl S et al. A randomized study of clofarabine versus clofarabine plus low-dose cytarabine as front-line therapy for patients aged 60 years and older with acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 112, 1638–45 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.