


Abstract
CasX (also known as Cas12e), a Class 2 CRISPR-Cas system, shows promise in genome editing due to its smaller size compared to the widely used Cas9 and Cas12a. Although the structures of CasX–sgRNA–DNA ternary complexes have been resolved and uncover a distinctive NTSB domain, the dynamic behaviors of CasX are not well characterized. In this study, we employed single-molecule and biochemical assays to investigate the conformational dynamics of two CasX homologs, DpbCasX and PlmCasX, from DNA binding to target cleavage and fragment release. Our results indicate that CasX cleaves the non-target strand and the target strand sequentially with relative irreversible dynamics. The two CasX homologs exhibited different cleavage patterns and specificities. The dynamic characterization of CasX also reveals a PAM-proximal seed region, providing guidance for CasX-based effector design. Further studies elucidate the mechanistic basis for why modification of sgRNA and the NTSB domain can affect its activity. Interestingly, CasX has less effective target search efficiency than Cas9 and Cas12a, potentially accounting for its lower genome editing efficiency. This observation opens a new avenue for future protein engineering.
Graphical Abstract
Graphical Abstract.

Introduction
Clustered regularly interspaced short palindromic repeats-CRISPR associated (CRISPR-Cas) systems provide adaptive immunity against invading nucleic acids in bacteria and archaea (1–3). The immune response of the CRISPR-Cas system occurs in three stages: incorporation of foreign nucleic acid fragments into the CRISPR locus region, expression of CRISPR RNA (crRNA), and target interference. In the target interference step, crRNA assembles with Cas protein to form a ribonucleoprotein (RNP) interference complex that guides the Cas nuclease to its complementary target for cleavage. The RNA-guided DNA targeting function of CRISPR-Cas systems enables the development of various genome engineering tools. CRISPR-Cas systems can be divided into two classes based on the composition of effector proteins. Class 1 systems require multiple Cas proteins to achieve interference, whereas Class 2 systems only require a single multi-domain Cas protein (4,5). Cas9 and Cas12a, both Class 2 effectors, are extensively used in genome editing across various cell types and organisms (6–10).
CasX (Cas12e), identified by metagenomic analysis from groundwater bacteria, belongs to Class 2 systems (11). It specifically recognizes a 5′-TTCN PAM and has shown considerable genome editing efficacy when combined with single-guide RNA (sgRNA) in human cells. With a smaller size of approximately 980 amino acids compared to Cas9 and Cas12a, CasX offers enhanced convenience for AAV-mediated delivery (12). CasX has demonstrated substantial potential as an alternative genome editing tool alongside Cas9 and Cas12a (13–15). Despite using an RNA guide for DNA targeting, CasX shares minimal protein sequence similarity with Cas12a or Cas9, except for the presence of a RuvC nuclease domain. As evidenced by the reported cryo-electron microscopy (cryo-EM) structures of CasX–sgRNA–DNA ternary complexes (11), CasX employs the single RuvC domain to cleave both the target-strand (TS) and non-target-strand (NTS) DNA. This mechanism is analogous to that of Cas12a (16,17). Two unique CasX domains are crucial for DNA cleavage: the target-strand loading (TSL) domain adjacent to the TS, and the non-target-strand binding (NTSB) domain next to the NTS. The NTSB domain is essential for DNA unwinding, as validated by biochemical assays (11). Structural differences between CasX and other Cas proteins suggest distinct mechanisms utilized by CasX.
Two homologous proteins, CasX from Deltaproteobacteria (hereafter DpbCasX) and CasX from Planctomycetes (hereafter PlmCasX), are commonly used in research (18). They share 68% protein sequence identity and can utilize the same sgRNA, however, the cleavage activity of purified PlmCasX is significantly lower than that of DpbCasX in the presence of the original sgRNA, designated as sgRNAv1. Structural analysis of PlmCasX revealed that its Helical-II domain barely engages with the scaffold stem of sgRNA, while for DpbCasX, the scaffold stem tightly binds to its Helical-II domain. To address this issue, a new sgRNA, named sgRNAv2, was engineered with increased flexibility in its scaffold stem, resulting in marked enhancement of cleavage activities and editing efficiencies (15). Despite these advancements, CasX still exhibits limited editing efficiency in living cells (19).
Cryo-EM analysis and biochemical assays have significantly augmented our understanding of CasX. However, its comprehensive dynamic characteristics during targeting and cleavage remain elusive. Here, using single-molecule fluorescence resonance energy transfer (FRET) assays, we observed three distinct conformational states during the cleavage process of CasX: initial R-loop formation, NTS pre-cleavage, and TS pre-cleavage. These states transition unidirectionally from low-FRET to high-FRET states, indicating a sequential cleavage order of NTS and TS. Notably, DpbCasX and PlmCasX exhibit different dynamic behaviors during DNA cleavage, highlighting disparities in their NTS cleavage sites and target specificity.
Materials and methods
Plasmid construction, protein expression and purification
The point mutations of the CasX expression plasmids were introduced by QuikChange Site-Directed Mutagenesis with Q5 High-Fidelity DNA Polymerase (New England Biolabs) and confirmed by DNA sequencing. Escherichia coli strain Rosetta (DE3) cells were used to express the CasX proteins. CasX expression plasmids (100 ng) were transformed into competent Rosetta cells (100 μL), which were then incubated on ice for 30 min, following this, the cells were incubated at 42°C for 45 s. After 5 min on ice, 900 μL of Luria broth (LB) medium was added and after shaking at 37°C for 1 h, the recovered cells were then cultured on plate medium with 50 mg/ml ampicillin. After an overnight incubation at 37°C, 10 colonies were picked to the main culture containing Terrific broth (TB) and 50 mg/ml ampicillin, the culture was then kept shaking at 37°C to reach an OD600 of 0.6. The expression of proteins was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM and the subsequent incubation of the culture overnight at 16°C. The cells were harvested by centrifugation at 4000 rpm and resuspended in a lysis buffer (600 mM sodium chloride, 20 mM HEPES, pH 7.5, 10% glycerol, 50 mM imidazole, 1 mM TCEP) with 1 mM protease inhibitor PMSF (Sigma). The cells were lysed by sonication and the resulting debris was removed by centrifugation at 14 000 rpm for 45 min at 4°C. The supernatant was collected and incubated with Ni-NTA beads (GE Healthcare) for 2 h at 4°C. After washing with the lysis buffer, proteins were eluted with lysis buffer containing 350 mM imidazole, the elution was then mixed with TEV protease (protein:TEV = 20:1) and incubated overnight on ice to remove the MBP tag. When examining DNA cleavage activities of DpbCasX mutants (K187A, Q190A, R191A and K187A/Q190A/R191A), their MBP tag were not removed. The proteins were concentrated using a 30 kDa MWCO concentrator (Amicon) and mixed with a low salt heparin buffer (400 mM sodium chloride, 20 mM HEPES, pH 7.5, 10% glycerol, 1 mM TCEP), The proteins were then applied to a heparin column (GE Healthcare) on an Akta FPLC (GE Healthcare) and eluted by a sodium chloride gradient with high salt heparin buffer (2 M sodium chloride, 20 mM HEPES, pH 7.5, 10% glycerol, 1 mM TCEP). The purified proteins were verified by SDS-PAGE electrophoresis, concentrated, and flash frozen in liquid nitrogen before being stored at –80°C.
In vitro transcription and purification of RNA
All the sgRNAs were transcribed in vitro. Primers were procured from Sangon Biotech (Shanghai, China) and DNA templates were generated through PCR reactions using Q5 polymerase (New England Biolabs). The HiScribe T7 kit (NEB) was used for invitro transcription. sgRNAs were purified by gel electrophoresis on a 10% urea–PAGE gel. The bands containing sgRNAs were excised and incubated in a soaking buffer (1 mM EDTA, 100 mM NaOAc pH 5.2) at 50°C for 2 h. Samples were then precipitated by the addition of 100% ethanol, and the precipitated sgRNAs were pelleted via centrifugation and washed using 70% ethanol. The sgRNAs were then resuspended in nuclease-free water (Life tech) and stored at –80°C.
RNA labeling
The RNA 3′ labeling procedure was based on selective periodate oxidation of RNA at its 3′ end, followed by the reaction of the oxidized product with hydrazide. The detailed procedure was previously described in detail (20). The concentrations of RNA and Cy3 were estimated by measuring the A260 and A549, respectively. The labeling efficiencies of the RNAs used in this study were ≥90%.
DNA labeling
Amine-derived DNA was procured from Sangon Biotech (Shanghai, China) and solubilized to 0.5 mM in 10 mM NaHCO3 (Sigma) buffer. NHS-fluorophores (Lumiprobe) were then added to a final concentration of 5 mM. Subsequently, the mixtures were incubated in the dark at 25°C for a minimum of 2 h. Samples were then precipitated by the addition of 100% ethanol, and the precipitated DNA was pelleted via centrifugation and washed using 70% ethanol. Labeled DNA was then dissolved in nuclease-free water (Life tech). The concentrations of DNA and Cy5 were estimated by measuring A260 and A649, respectively. The labeling efficiencies of the DNAs used in this study were equal to or greater than 90%.
In vitro cleavage assay
The DNA substrates were 5′ labeled by NHS-fluorophores. The CasX proteins were diluted to a concentration of 2 μM using the cleavage buffer (150 mM potassium chloride, 50 mM Tris–HCl, pH 7.5, 10 mM magnesium chloride, 1 mM DTT) containing 3 μM sgRNAs and incubated at 25°C for 1 h to reconstitute the RNP complex. The DNA cleavage reactions were initiated by mixing the CasX-sgRNA complex and the labeled DNA at 37°C with final concentrations of 500 and 10 nM, respectively. Sample aliquots were taken at designated time points and mixed with 2× urea loading buffer (8 M urea, 25 mM EDTA, 100 μg/ml heparin) and were incubated in 95°C heat blocks for 5 min. Following this, the samples were loaded into 15% urea–PAGE. To determine the cleavage sites of DpbCasX and PlmCasX, the samples were run on 20% urea-PAGE. The cleavage signals were then visualized using a gel scanner (Amersham Typhoon 5, GE Healthcare).
Single-molecule FRET assay
Single-molecule FRET experiments were conducted at 25°C or 37°C using a home-built objective-type TIRF microscope in the smFRET buffer (150 mM potassium chloride, 50 mM Tris–HCl, pH 7.5, 10 mM magnesium chloride) with an oxygen scavenging system containing 3 mg/ml glucose, 100μg/ml glucose oxidase (Sigma-Aldrich), 40 μg/ml catalase (Roche), 1 mM cyclooctatetraene (COT, Sigma-Aldrich), 1 mM 4-nitrobenzylalcohol (NBA, Sigma-Aldrich), 1.5 mM 6-hydroxy-2,5,7,8-tetramethyl-chromane-2-carboxylic acid (Trolox, Sigma-Aldrich). The details of the TIRF microscope have been previously described (21).
The CasX or CasX mutant was pre-incubated with sgRNA (protein:sgRNA = 1 μM:1.5 μM) in the cleavage buffer at 25°C for 60 min. The target DNA was immobilized on the cover slide via biotin-streptavidin interaction. Fluorescent signals were recorded at 500 ms/frame at 2 mW laser power or 2 s/frame at 0.6 mW laser power and started several seconds prior to the introduction of protein-sgRNA complexes. To capture the dynamic process of protein-sgRNA complexes binding to a DNA target, the pre-incubated protein-sgRNA complexes were diluted to working concentrations of 3 or 10 nM, respectively, depending on the binding affinity of CasX mutants to DNA targets.
For all our experiments, three repeats were performed. Usually, one field of view was measured in each repeat, from which 500–1000 single-molecule traces were recorded for further analysis.
Single-molecule FRET data analysis
Collected movies were analyzed using a custom-made software program. Fluorescence spots were fitted by a 2D Gaussian function within a 9-pixel by 9-pixel area, matching the donor and acceptor spots using a variant of the Hough transform (22). The background-subtracted total volume of the 2D Gaussian peak was used as the raw fluorescence intensity I. FRET traces exhibiting anti-correlation behaviors between donor and acceptor fluorescent signals were identified and subjected to further analysis via a Hidden Markov Model-based software HaMMy (23). Initial FRET efficiencies of FRET states were estimated by visual inspection of hundreds of traces and used as pre-set values for HaMMy to classify FRET states and transitions. The distributions of each FRET state and their dwell times were extracted. Further iterations of HaMMy analysis were performed if centers of the determined FRET distributions diverge significantly from the pre-set values.
To correct contribution of photobleaching from dwell time of binding events, the following equation was used:
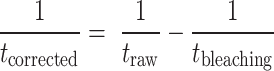 |
in which tcorrected is dwell time after correcting for photobleaching, traw is measured dwell time of transient binding events, and tbleaching is dwell time of stable binding events whose termination is caused by photobleaching.
The effective target search rates and the nonspecific binding rates were calculated as follows:
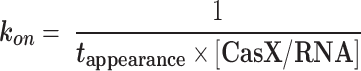 |
in which tappearance is the time from injection of CasX/sgRNA until appearance of CasX/sgRNA on immobilized dsDNA, [CasX/RNA] is the concentration of CasX/sgRNA binary complex. The effective target search efficiency was calculated as the ratio of the effective target search rate to the nonspecific binding rate.
Genome editing in fluorescent reporter human cells
GFP HEK293T reporter cells were seeded into 48-well plates and transfected 18–24 h later at 70–90% confluency according to the manufacturer's protocol with lipofectamine 3000 (Life Technologies). Approximately 300 ng of plasmid DNA encoding the sgRNA and wild-type or engineered CasX was added to each well. Twenty-four hours after transfection, GFP HEK293T reporter cells that had been successfully transfected were selected by the addition of 1.5 mg/ml puromycin to the cell culture media for 72 h. During this period, the cells were passaged regularly to maintain sub-confluent conditions. Once the cells in the control group, which had not been transfected with any plasmid, had died completely, the culture medium in the test group was replaced with fresh normal medium without puromycin. After a 2-day recovery, the cells were collected for analysis of the GFP signal strength via Flow Cytometer (BD LSRFortessa).
Results
Single-molecule FRET assay to capture conformational dynamics of DpbCasX-sgRNA-DNA
To directly visualize the conformational dynamics of DpbCasX during DNA cleavage following the formation of the DpbCasX-sgRNA-dsDNA ternary complex, we established a single-molecule FRET assay using a design similar to those for Cas12a proteins (16,17,24). FRET is a physical phenomenon that describes the energy transfer between two nearby fluorophores. The efficiency of FRET between two fluorophores is highly sensitive to their relative distance. By labeling molecules of interest with suitable fluorophores, single-molecule FRET is a powerful tool for capturing conformational changes of biomolecules in real time, thereby providing further mechanistic insights (25). Cy3 and Cy5 fluorophores, which are commonly used for FRET measurements, were attached to the 3′ end of sgRNA and the PAM distal end of the target strand (TS), respectively (Figure 1A, Supplementary Tables S1 and S2). The labeling of sgRNA did not affect CasX cleavage activities (Supplementary Figure S1A).
Figure 1.

Conformational dynamics of CasX cleavage captured by single-molecule FRET. (A) Diagram of the experimental design. The 3′ end of the sgRNA is labeled with Cy3 (green dots). The TS 34th nucleotide from the PAM is labeled with Cy5 (red dots). The process from adding CasX-sgRNA complex to the release of cleavage products is captured. (B) Representative single-molecule FRET trajectories of DpbCasX-sgRNAv2 on cognate DNA. The black curve represents the apparent FRET efficiency, while the orange lines represent Hidden Markov Modeling of FRET. The spontaneous appearance of Cy3 and FRET signals indicates the formation of CasX ternary complexes on immobilized dsDNAs (left blue arrow), and the disappearance of FRET indicates the dissociation of cleaved DNA fragments (right blue arrow). Three distinct FRET states are identified and assigned as S1, S2 and S3, in order of increasing values. (C) Time-dependent FRET probability density plots are synchronized at the appearance of FRET (t = 0, left panel) and at the disappearance of FRET (t = 0, right panel), enabling examining FRET evolution after ternary complex formation and before the release of cleaved DNA, respectively. N represents the number of events. Three independent experiments show consistent results. (D) Transition density plot displays the transition frequencies between different FRET states, initial and final FRET values for each transition event are accumulated into a 2D histogram. (E) FRET histograms of individual states were extracted from the original FRET trajectories after Hidden Markov Modeling classification. FRET values for each state are obtained through single Gaussian fitting. Each histogram is normalized by the total sum of all histograms.
In our assay, the Cy3-labeled DpbCasX-sgRNA binary complex was injected into a flow chamber containing immobilized Cy5-labeled target dsDNA (Figure 1A). Exposure time was 500 ms/frame unless otherwise indicated. The time from the injection of the binary complex to the occurrence of a stable binding event (Figure 1B, left blue arrow) was recorded as the appearance time. The emergence of Cy3 and Cy5 signals indicated the binding of DpbCasX-sgRNA to the target site, forming a ternary complex. The ternary complex then underwent a series of conformational changes, resulting in a gradual increase in the FRET signal until the abrupt disappearance of Cy5 and FRET signals (Figure 1B, right blue arrow). The disappearance of the FRET signal is likely attributed to the cleavage of DNA and the subsequent release of the labeled DNA fragment. The period during which the FRET signal persisted was recorded as the dwell time. It is notable that when the catalytically inactive DpbCasX (dDpbCasX, with D672A, E769A and D935A mutations) (11) was used in the same assay, its FRET dwell time was ∼8 times longer than that of DpbCasX (Supplementary Figure S1B). This result confirms that DNA cleavage is the main cause of the disappearance of Cy5 rather than photobleaching.
We examined a series of Cy5 labeling sites on the TS, ranging from 28 nt to 40 nt away from the PAM site, while maintaining the other Cy3 labeling site at the 3′ end of sgRNA. These designs consistently exhibited similar behavior, with an increase in FRET signals observed after ternary complex formation (Supplementary Figure S1C). Of those examined, the labeling site positioned 34 nt away from the PAM yielded the clearest FRET signals, manifesting in three discrete FRET states (Figures 1B and C) and was consequently used in the subsequent experiments. Analysis of single-molecule FRET trajectories employed hidden Markov model-based software (23) to identify FRET transitions among states (Figure 1D). Predominantly, transitions occurred from low-FRET to high-FRET states, while reverse reactions were less frequent.
To assign these three FRET states to different DpbCasX configurations (Figure 1E), the dynamics of dDpbCasX ternary complexes with a predesigned nick in the cleavage sites of either the TS or NTS were examined. When intact DNA or DNA with a nick in the TS was used, dDpbCasX could only sample the middle-FRET state (FRET efficiency E ∼ 0.5, defined as the S2 state) after transitioning through the low-FRET state (E ∼ 0.2, defined as the S1 state) (Supplementary Figure S1D). Only when DNA with a nick in the NTS was used, dDpbCasX eventually exhibited the high-FRET state (E ∼ 0.7, defined as the S3 state). These results indicate that S1 stands for the initial R-loop formation and the following S2 and S3 stand for the NTS pre-cleavage state and the TS pre-cleavage state, respectively. Furthermore, cleavage of the NTS is required for DpbCasX to sample the TS pre-cleavage state. Specifically, the high FRET efficiency of S3 suggests that the TS was bent or kinked to approach the catalytic center in the RuvC domain during cleavage, bringing Cy5 labeled at the TS close to Cy3 labeled at the 3′ end of the sgRNA. Thus, the NTS and TS are sequentially cleaved by CasX in a well-defined order, consistent with the previously proposed model (11) and similar to Cas12a (16,17).
Most of our single-molecule FRET experiments were performed at 37°C unless otherwise stated. Nevertheless, the DpbCasX ternary complex displayed similar FRET patterns at 25°C with all three FRET states, albeit with slower transition rates among them (Supplementary Figure S2).
Stability of the DpbCasX ternary complex modulated by PAM-proximal matches
Recognition of the target site and formation of a stable or transiently stable ternary complex are crucial steps for all Cas proteins to fulfill their enzymatic functions. Dead Cas proteins, such as dead Cas9 and dead Cas12a, can be fused with other effector proteins and repurposed for gene regulation, base editing, prime editing, and other applications due to their RNA-guided DNA-binding activities (26–28). Hence, the requirement for a minimum number of matches between the guide RNA and the target DNA for stable binding is essential for using Cas proteins in genome manipulation. For Cas9, ∼9 bp between the DNA target and the RNA spacer at the PAM-proximal end are required, while Cas12a requires ∼17 PAM-proximal matches (24,29). However, the effect of PAM-proximal matches on the stability of CasX on DNA is unknown.
To determine the minimal PAM-proximal matches required for stable binding of CasX to DNA, we examined single-molecule FRET signals of dDpbCasX on a range of DNA targets with varying numbers of PAM-proximal matches. In our assay, cognate DNA contained 20 matched bases to sgRNA, whereas 14m-, 16m-, 17m-, 18m- and 19m-DNA contained 14, 16, 17, 18 and 19 PAM-proximal matches to sgRNA, respectively (Figure 2A, Supplementary Figure S3A, Supplementary Table S2). The binding of dDpbCasX complexes to 18m-, 19m- and 20m-DNAs was remarkably stable, with the number of fluorescence dDpbCasX spots on DNAs barely changing after 1 h of incubation in the dark (Figure 2B). Thus, the disappearance of FRET signals of dDpbCasX on 20m-DNA under continuous laser excitation was caused by photobleaching (Supplementary Figure S3B), from which the photobleaching rate was quantified and applied to the estimated corrected dwell time of transient dDpbCasX binding on 14m-, 16m-, and 17m-DNAs (Figure 2B, Supplementary Figure S3B, details in Materials and methods). With 14m- and 16m-DNA, dDpbCasX formed a transiently stable complex on dsDNA with a dwell time of ∼5 s, mainly remaining in the S1 state (Figure 2B, Supplementary Figure S3A, B). When there were 17 or more PAM-proximal matches, dDpbCasX could transition to the S2 state with greatly extended dwell time. Additionally, the binding rates of dDpbCasX to DNA, corresponding to the effective target search rates, were enhanced with more matched bases (Figure 2C, Supplementary Figure S3C). Overall, our results showed that 18 or more PAM-proximal matches are required to form stable CasX ternary complexes with DNA. When PAM-proximal matches were fewer than 17 bp, dDpbCasX binds transiently to DNA, displaying repeatable binding and dissociation behaviors in single-molecule FRET trajectories (Supplementary Figure S3A).
Figure 2.

Dynamics and cleavage of DpbCasX. (A) Schematic of mismatched DNA. (B) The number of dDpbCasX-sgRNAv2 complexes bound on immobilized cognate or mismatched dsDNAs before and after 1 hour of incubation in the dark (top) and the FRET dwell time of dDpbCasX on immobilized mismatched dsDNAs after correcting for photobleaching (bottom). The original dwell time distributions without correction are shown in Supplementary Figure S3B. (C) Effective target search rates of dDpbCasX or DpbCasX on dsDNA to form stable or transient stable complex (dwell time 1.5 s or longer). The distributions of appearance time are shown in Supplementary Figure S3C and S4E. The CasX RNP concentration was 3 nM. The search rate of DpbCasX on 14m-DNA is beyond our detection limit (< 0.3 μM−1s−1). (D) Cleavage activity of DpbCasX. The cleavage rates for 20m- (2.6 ± 0.1 min−1), 19m- (2.6 ± 0.3 min−1), 18m- (1.6 ± 0.2 min−1), 17m- (0.43 ± 0.03 min−1), 16m- (0.14 ± 0.02 min−1) and 15m-DNA (0.020 ± 0.002 min−1) were fitted by a single exponential decay curve. (E-F) FRET histograms (E) and dwell time (F) of individual states for different DNAs. (G) Effective target search rates of DpbCasX on 16m-DNA, 16m-Nick in TS DNA, and 16m-Nick in NTS DNA. The appearance time are shown in Supplementary Figure S5A. The CasX RNP concentration was 3 nM. (H) Cleavage activity of DpbCasX on 16m-DNA (0.15 ± 0.04 min−1) and 16m-Nick in NTS DNA (1.4 ± 0.1 min−1) using Cy5-labeled TS. Cleavage activity of DpbCasX on 16m-DNA (0.16 ± 0.06 min−1) and 16m-Nick in TS DNA (0.20 ± 0.05 min−1) using Cy5-labeled NTS. Three repeats were performed. Mean and SEM are shown where appropriate.
DNA cleavage of DpbCasX modulated by PAM-proximal matches
The minimal number of gRNA-DNA matches required for complete state transition, as observed by FRET assays, is supported by in vitro cleavage experiments. Cleavage of 18m- and 19m-DNA by DpbCasX exhibited comparable cleavage rates to the cognate 20m-DNA (∼2 min−1). In contrast, 17m-, 16m- and 15m-DNA exhibited gradually reduced cleavage rates (0.1–0.4 min−1), while 14m-DNA displayed no detectable cleavage (Figure 2D, Supplementary Figure S4A). Consistent with the cleavage assay, DpbCasX complexes containing 17 or more matched bases could transition sequentially from S1 to S2 and eventually to S3 states (Figures 2E, F; Supplementary Figure S4B, C), while reverse transitions occurred at much lower frequencies (Supplementary Figure S4D). When the number of matched bases decreased to 16, the DpbCasX complex remained mainly in the S1 and S2 states and barely sampled the S3 state under experimental conditions, corresponding to its greatly reduced cleavage rate (Figure 2D, F). 14 or fewer matched bases caused the transiently formed DpbCasX complex to remain only in the S1 state and exhibit no cleavage activity. There was almost no transient binding of the DpbCasX complex to 4m-DNA in our experiments, indicating that ∼8 PAM-proximal matches are needed to form the transient S1 state containing a partially formed R-loop. Hence, we proposed that DpbCasX has a 5–8 nucleotide long PAM-proximal seed region, which has also been identified in other Cas nucleases and RNA-based regulatory systems to aid in target search and recognition (30–33).
The effective search rates of DpbCasX on DNA were reduced when the number of matched bases decreased (Figure 2C, Supplementary Figure S4E), following the same trend as the rates captured with dDpbCasX. Interestingly, when a nick was introduced at the NTS cleavage site of the 16m-DNA, both binding and cleavage rates were greatly accelerated (Figures 2G, H; Supplementary Figure S5), whereas the influence of a nick at the TS cleavage site was minor. Therefore, the intact NTS likely serves as a checkpoint to modulate stable R-loop formation and subsequent DNA cleavage in the presence of mismatches. Consistently, once the NTS is cleaved, subsequent TS cleavage occurs quickly even with only 16 PAM-proximal matches (Figure 2H). Taken together, sufficient matches in the PAM-proximal region were pivotal for CasX to accomplish conformational changes and pass the checkpoint to permit NTS cleavage.
PlmCasX exhibits varied cleavage patterns and high mismatch sensitivity
Two CasX homologs, DpbCasX and PlmCasX, have been investigated in previous studies, Notably, DpbCasX exhibited higher activities in vitro, whereas PlmCasX demonstrated higher activities in vivo (11,15). To elucidate the differences between these two CasX proteins, we applied the same single-molecule FRET assay to capture the conformational dynamics of PlmCasX. The activities of PlmCasX guided by labeled sgRNAs were minimally affected (Supplementary Figure S6A). Unlike DpbCasX, PlmCasX exhibited only two major FRET states: a low-FRET state with E ∼ 0.2 and a middle-FRET state with E ∼ 0.5, defined as the S1 and S2 states, respectively (Figures 3A-B). The NTS pre-cleavage state mimicked by dead PlmCasX (dPlmCasX) with TS-nicked DNA, exhibits a middle FRET, similar to dPlmCasX with intact DNA (Supplementary Figure S6B). Interestingly, the FRET efficiency of the TS pre-cleavage state of dPlmCasX is highly dependent on the nick site on the NTS. Nicking the NTS between the 12th and 13th nucleotides resulted in a high-FRET S3 state for dPlmCasX, whereas nicking the NTS between the 14th and 15th nucleotides resulted in only a middle-FRET TS pre-cleavage state (Figure 3C). dDpbCasX exhibits identical behaviors to dPlmCasX, showing an NTS nick site-dependent FRET pattern. Therefore, the observation that DpbCasX displays the high-FRET S3 state during DNA cleavage, whereas PlmCasX does not (Figures 1C and 3B), could be attributed to their different cleavage patterns on NTS. Specifically, the FRET patterns suggested that the NTS cleavage site of DpbCasX is closer to the PAM than that of PlmCasX, resulting in a broader bending of the fluorophore-labeled TS to access the catalytic pocket. This hypothesis was confirmed by our biochemical assay (Figure 3D). DpbCasX cleaved NTS DNA mainly at 11–12 nt from PAM, while PlmCasX cleaved at 11, 12 and 14 nt from PAM. Meanwhile, DpbCasX cleaved TS DNA at 22–23 nt from PAM and PlmCasX cleaved TS DNA at 21–23 nt from PAM. Thus, DpbCasX and PlmCasX have different NTS and TS cleavage patterns, leading to different behaviors in FRET assays.
Figure 3.

Dynamics and cleavage of PlmCasX. (A) Representative single-molecule FRET trajectories of PlmCasX-sgRNAv2 complex on cognate DNA. Two distinct FRET states are indicated and assigned as S1 and S2 from low to high values. (B) Time-dependent FRET probability density plots of PlmCasX on cognate DNA. (C) Time-dependent FRET probability density plots of dPlmCasX and dDpbCasX on DNA with NTS-Nick at positions 12, 13 or NTS-Nick at positions 14, 15. (D) Cleavage sites of DpbCasX and PlmCasX on NTS and TS. Cleavage reaction was terminated 15 s after mixing CasX-sgRNA complex and target DNA. (E) The number of dPlmCasX-sgRNAv2 complexes bound on immobilized 20m- or 19m-dsDNAs before and after 1 h of incubation in the dark (top) and the FRET dwell time of dPlmCasX on immobilized dsDNAs after correcting for photobleaching (bottom). The original dwell time distributions without correction are shown in Supplementary Figure S6D, E. (F) Effective target search rates of dPlmCasX. The appearance time are shown in Supplementary Figure S6F. The CasX RNP concentration was 3 nM. (G) The cleavage activity of PlmCasX. Cleavage rates of 20m- (1.0 ± 0.1 min−1), 19m- (0.88 ± 0.09 min−1), 18m- (0.54 ± 0.09 min−1), 17m- (0.081 ± 0.004 min−1), 16m- (0.03 ± 0.01 min−1) and 15m-DNA (0.008 ± 0.001 min−1) were fitted by a single exponential decay curve. Three repeats were performed. Mean and SEM are shown where appropriate.
Another possible explanation is that the TS cleavage of PlmCasX is extremely fast and beyond our temporal detection limit, leading to the absence of the high-FRET S3 state. However, even after increasing the acquisition rate of the camera by a factor of 10, we were still unable to capture the high-FRET S3 state during the DNA cleavage of PlmCasX. Therefore, this possibility remains unconfirmed.
Although dPlmCasX formed a stable complex on the cognate 20m-DNA target, even a single PAM-distal mismatch greatly reduced its stability and accelerated the dissociation of dPlmCasX (Figure 3E, Supplementary Figure S6C–E). PAM-distal mismatches also moderately decreased the binding rate of dPlmCasX to the target site (Figure 3F, Supplementary Figure S6F). Like DpbCasX, PAM-distal mismatches also prevented PlmCasX from transitioning to the cleavage states, resulting in the disappearance of the S2 state and greatly reduced cleavage activity (Figure 3G, Supplementary Figure S6C and S6G). Similar to DpbCasX, ∼ 8 PAM-proximal matches are needed to form the transient S1 state containing a partially formed R-loop, supporting that PlmCasX also has a 5–8 nucleotide long PAM-proximal seed region. In general, PlmCasX shows better specificity than DpbCasX for discriminating DNAs containing mismatches, likely due to the reduced stability of PlmCasX complexes on mismatch-containing DNAs (Figures 2B and 3E).
Modified sgRNA enhances the stability of PlmCasX on DNA
In a previous study, the structure of the Plm-sgRNAv1-DNA ternary complex was resolved, revealing that the scaffold stem of sgRNAv1 did not engage significantly with the Helical-II domain, unlike in DpbCasX. However, the transition from the NTS-loading state to the TS-loading state was facilitated by the interaction between the Helical-II domain and the sgRNA scaffold stem. To strengthen this interaction, an engineered sgRNA (sgRNAv2) was designed with a change of only three nucleotides, significantly enhancing the cleavage activity of CasX compared to the original sgRNA (sgRNAv1) (15). Therefore, sgRNAv2 was primarily used in this study. To elucidate the mechanism underlying the enhanced activity of sgRNAv2, we compared the conformational dynamics of PlmCasX guided by sgRNAv1 and sgRNAv2. sgRNAv2 substantially stabilized the dPlmCasX-sgRNA-DNA complex and accelerated the binding of dPlmCasX to target DNA (Figures 4A–C; Supplementary Figure S7A). For the cognate 20m-DNA, ∼80% of the dPlmCasX-sgRNAv1 binding events were transient with a dwell time of ∼5 s, resulting in a reduced overall dwell time (Figure 4A). When using wild-type PlmCasX, both sgRNAv1 and sgRNAv2 displayed similar single-molecule FRET patterns, including an initial transient S1 state followed by an S2 state (Figure 4D). Nevertheless, after 16 min, only ∼20% of Cy5-labeled DNA was cleaved by PlmCasX-sgRNAv1, compared to ∼70% by PlmCasX-sgRNAv2 (Figure 4D). Thus, PlmCasX-sgRNAv1 mainly forms a transient unstable binding to its target lasting ∼5 s (Supplementary Figure S7B), as observed with dPlmCasX, rarely leading to DNA cleavage. In contrast, sgRNAv2 greatly accelerates and stabilizes PlmCasX binding to DNA, facilitating cleavage.
Figure 4.

The conformational dynamics of PlmCasX affected by sgRNA and the nonspecific binding of CasX to DNA. (A) Distributions of dwell time of dPlmCasX-sgRNAv1 on cognate DNA with an exposure time of 2 s. The dwell time was fitted by a double exponential decay curve. (B) Dwell times of dPlmCasX-sgRNAv2 and dPlmCasX-sgRNAv1 on cognate DNA. (C) Effective target search rates of dPlmCasX-sgRNAv1 and dPlmCasX-sgRNAv2 on cognate DNA. The appearance time are shown in Supplementary Figure S7A. The CasX RNP concentration was 3 nM. (D) Time-dependent FRET probability density plots of PlmCasX-sgRNAv1 and dPlmCasX-sgRNAv2 on cognate DNA and corresponding DNA cleavage. The disappearance of fluorescent spots indicates Cy5-labeled DNA cleavage after CasX addition. (E) Schematic of CasX nonspecific binding and a representative single-molecule fluorescence trace. CasX-sgRNA complex was injected into a chamber containing dsDNA without a PAM sequence. The sudden appearance of Cy3 signal indicated the transient binding of CasX-sgRNA to DNA. The time from injection to Cy3 signal appearance was recorded as appearance time, and the duration of Cy3 signal was recorded as dwell time. (F) Distributions of appearance time of DpbCasX-sgRNAv2 and PlmCasX-sgRNAv2 on DNA without a PAM sequence. The exposure time was 2 ms/frame, and the CasX RNP concentration was 10 nM. (G) Nonspecific binding rates and effective target search rates of different Cas proteins (details in Methods). Data for CasX and correlated rates for Cas9 and Cas12a are shown in Supplementary Table S3 (29,35,36). (H) Effective target search efficiency of different Cas proteins defined as the ratio of the effective target search rate to the nonspecific binding rate shown in (G). Three repeats were performed. Mean and SEM are shown where appropriate.
CasX displays lower target search efficiency than Cas9 and Cas12a
Although the engineered sgRNAv2 enhances CasX activity, its overall genome editing efficiency remains significantly lower than that of Cas9 or Cas12a. Single-molecule assays indicate that, following target recognition, both CasX homologs need ∼10 s to complete DNA cleavage and fragment release (Figures 1B and 3A). Previous studies employing similar assays have identified that Cas12a needs 30–90 s to complete DNA cleavage and fragment release (16,17). Following DNA binding, SpCas9 needs ∼10 s to reach its DNA cleavage state (34). Together, for CasX, Cas12a and Cas9, DNA cleavage is fast and likely occurs within 10–100 s after the formation of a stable protein-RNA-DNA ternary complex. Thus, the differences in editing efficiencies are likely dominated by the efficiencies of these nucleases in target search and ternary complex formation.
With a high exposure rate (2 ms per frame), we measured the appearance time and dwell time of CasX on nonspecific dsDNA containing no PAM sequence and quantified the corresponding binding and dissociation rates (Figure 4E, F; Supplementary Figure S7C and Supplementary Table S3). We confirmed that almost no nonspecific transient binding events occurred in the absence of DNA (Supplementary Figure S7D). The nonspecific binding rates of both DpbCasX and PlmCasX on dsDNA were ∼10-fold faster than SpCas9 and 2–4-fold slower than Cas12a (Figure 4G, Supplementary Table S3) (29,35,36). The dissociation rates of DpbCasX and PlmCasX from nonspecific dsDNA were 2–20-fold slower than those of SpCas9 and Cas12a (Supplementary Table S3), indicating that CasX has more time to search for target sites during each transient nonspecific binding event. However, the effective search rates of DpbCasX and PlmCasX to locate correct target sites were 2.6 ± 0.3 and 2.1 ± 0.2 μM−1s−1, respectively (Supplementary Table S3), which are similar to SpCas9 and FnCas12a but significantly slower than AsCas12a and LbCas12a. We defined the effective target search efficiency of Cas proteins as the proportion of nonspecific binding events that lead to sequential target recognition and stable binding events. The effective target search efficiency of CasX, calculated as the ratio of the effective target search rate to the nonspecific binding rate, is significantly lower than that of Cas9, AsCas12a and LbCas12a (Figure 4H). This suggests that multiple encounters are required for CasX to recognize its target sites. Interestingly, FnCas12a also has a very low effective target search efficiency, similar to CasX, which correlates with its poor gene editing efficiency in cells compared to other Cas12a homologs (37,38). Therefore, improving the effective target search efficiency may be a viable strategy to enhance the editing efficiency of CasX and other Cas nucleases. Effective target search relies on effective target recognition, which includes recognition of the PAM and subsequent R-loop propagation. A recent study showed that an engineered Cas9 variant with greatly enhanced genome editing activity demonstrated increased DNA unwinding capacity (39).
NTSB domain participates in DNA unwinding and heteroduplex formation
The structures of CasX reveal a unique NTSB (non-target strand binding) domain adjacent to the NTS, which interact with both the NTS and the TS (Figure 5A) (11,15). Previous biochemical assays demonstrated that the DpbCasX mutant lacking the NTSB domain (CasXΔNTSB, lacking residues 101–191) is impaired in dsDNA cleavage but can still cleave unwound dsDNA and single-stranded DNA (ssDNA) targets (11). Our single-molecule FRET assay showed that DpbCasXΔNTSB on the cognate dsDNA target exhibited only a single transient middle-FRET state (E ∼ 0.5) and almost no cleavage activity (Figures 5B, C; Supplementary Figures S8A, B). The disappearance of the S1 state, which corresponds to the initial R-loop formation, indicates that CasXΔNTSB is unable to unwind DNA and perform subsequent cleavage.
Figure 5.

NTSB domain modulates DNA unwinding, cleavage and specificity. (A) Structure of DpbCasX–sgRNA–dsDNA (PDB: 6NY2) with the NTSB domain shown in red. (B) Cleavage activity of DpbCasXΔNTSB on cognate DNA, bulged DNA and bulged DNA with mismatches compared to DpbCasX on cognate DNA. (C–E) Time-dependent FRET probability density plots of DpbCasXΔNTSB-sgRNAv2 on cognate DNA (C), Bulge 3–6 DNA (D), and Bulge 11–20 DNA (E). (F) Cleavage activity of DpbCasX and DpbCasXΔNTSB on ssDNA. (G) Effective target search rates of DpbCasXΔNTSB-sgRNAv2 and DpbCasX-sgRNAv2 on ssDNA. The appearance time are shown in Supplementary Figure S9B. The CasX RNP concentration was 10 nM. (H) Left, three residues within the NTSB domain interact closely with the TS in both the NTS-cleavage and TS-cleavage states of DpbCasX. Right, the cleavage activity of DpbCasX mutants on DNAs with varying matches to sgRNA. (I) Diagram of GFP disruption assay in cells (left). Normalized GFP disruption using WT PlmCasX or PlmCasX-K188A, PlmCasX-Q191A targeting site 1–5 within GFP (right). GFP disruption percentage was normalized to their corresponding values of cognate target. Three repeats were performed. Mean and SEM are shown where appropriate.
When several internal DNA mismatches were introduced at the PAM-proximal end (bulge 3–6), DpbCasXΔNTSB could form the transient S1 state and transition to S2 and S3 states like WT DpbCasX, leading to significant DNA cleavage (Figures 5B, D; Supplementary Figure S8A). Similar results were observed with bulge 3–10 DNA (Supplementary Figure S8C). Conversely, when internal DNA mismatches were introduced at the PAM-distal end (bulge 11–20 and bulge 13–20), although DpbCasXΔNTSB could still cleave these DNAs, it predominantly displayed a single middle-FRET state (E ∼ 0.5) without the low-FRET S1 state, unlike WT DpbCasX on the bulged DNA (Figure 5E, Supplementary Figure S8D, E). This suggests that the RNA-DNA heteroduplex might form at the PAM-distal end and further extend to the PAM-proximal end, allowing DNA cleavage by DpbCasXΔNTSB at a much slower rate than WT DpbCasX. To test this hypothesis, we further introduced PAM-proximal mismatches into DNA containing PAM-distal bulge (bulge 11–20_Mismatch 1–10). The PAM-proximal mismatches completely abolished cleavage activity (Figure 5B), while having almost no influence on single-molecule FRET signals (Figure 5E, Supplementary Figure S8F), strongly supporting our hypothesis.
Furthermore, even when ssDNA complementary to the spacer of the sgRNA was used as a target, DpbCasXΔNTSB displayed lower activity than DpbCasX in biochemical assays (Figure 5F, Supplementary Figure S9A), correlating with its significantly reduced CasX–sgRNA–ssDNA complex formation rate (Figure 5G, Supplementary Figure S9B, ∼7-fold difference). In conclusion, the NTSB domain is crucial not only for DNA unwinding but also for RNA-DNA heteroduplex formation.
Previous studies have shown that mutations in residues interacting with the NTS and RNA/DNA heteroduplexes can modulate Cas9 specificity (40,41). Accordingly, we selected three residues within the NTSB domain: K187, Q190 and R191 (Figure 5H), because they interact with the TS in both NTS- and TS- cleavage states of DpbCasX (11). Mutating all three residues (K187A/Q190A/R191A) almost abolished cleavage activity (Figure 5H, Supplementary Figure S10A), confirming the essential role of these residues in target cleavage. When only one residue was mutated, DpbCasX-Q190A had similar activity to WT DpbCasX, whereas DpbCasX-K187A and DpbCasX-R191A showed reduced activity. Since PlmCasX exhibits higher editing efficiency than DpbCasX in living cells (11,15) and these three residues are highly conserved in both CasX homologs (Supplementary Figure S10B), we generated corresponding PlmCasX mutants to investigate their specificities in genome editing. In our GFP disruption assay, PlmCasX-R192A and PlmCasX-K188A/Q191A/R192A had almost no activity when targeting two sites within GFP (Supplementary Figure S10C), whereas PlmCasX-K188A exhibited reduced editing efficiency comparing to WT PlmCasX when targeting site 1–3 containing two distal mismatches within GFP (18m, Figure 5I, Supplementary Figure S10C, D, Supplementary Tables S4 and S5). Overall, weakening the interactions between the NTSB domain and nucleic acids reduces cleavage activity and can affect discrimination against off-target sites under certain conditions.
Discussion
By combining multiple single-molecule fluorescence assays, we provide a comprehensive characterization of CasX dynamics from nonspecific DNA binding to target recognition and DNA cleavage (Figure 6). In our simple purified system, DpbCasX and PlmCasX exhibit significantly lower target search efficiencies, defined as the ratio of the effective target search rate to nonspecific DNA binding rate, compared to Cas9 and Cas12a (Figure 4H). In a cellular environment, CasX search efficiency may be further attenuated by DNA-binding proteins and other competing components, contributing to its relatively low editing efficiency in cells (16,42–44). After recognizing its target site, CasX initiates R-loop formation, NTS cleavage, TS cleavage, and DNA fragment release sequentially, similar to Cas12a (16,17). The irreversible conformational dynamics of CasX resemble those of AsCas12a (16) but differ from the reversible transitions of FnCas12a (45) and LbCas12a (17) revealed by single-molecule FRET assays. In our experiments, 8 PAM-proximal matches are needed to initiate R-loop formation, whereas 4 PAM-proximal matches are not sufficient. Previous studies have shown that mismatches in the seed region of Cas9 abolish R-loop formation (29). Thus, our dynamic measurements suggested that the PAM-proximal seed region of CasX contains 5 – 8 nucleotides. PAM-distal mismatches not only reduce DNA cleavage but also promote dissociation of the CasX-sgRNA complex before cleavage. These factors collectively enhance CasX specificity.
Figure 6.

Proposed model for CasX binding and cleavage of DNA. After nonspecific binding to DNA, CasX’s efficiency in recognizing PAM and initiating R-loop formation is 5–8 times lower than that of SpCas9, AsCas12a and LbCas12a. After PAM recognition, sufficient PAM-proximal matches are required to sequentially drive R-loop formation, NTS cleavage and TS cleavage. The NTSB domain plays an important role in DNA unwinding and R-loop formation via its interactions with sgRNA and DNA. Insufficient matches between sgRNA and the spacer hinder NTS cleavage and promote the disassembly of the NTS pre-cleavage state. Thus, NTS cleavage acts as a critical checkpoint, after which TS cleavage and DNA fragment release occur rapidly.
Notably, the stability of the PlmCasX–sgRNA–DNA complex before cleavage is more sensitive to TS-sgRNA mismatches than DpbCasX, explaining PlmCasX’s higher specificity. PlmCasX and DpbCasX also show different cleavage patterns. In the NTS cleavage step, PlmCasX exhibits more promiscuous cleavage sites compared to DpbCasX, resulting in differences in TS cleavage sites and distinct TS pre-cleavage conformations observed in single-molecule FRET assays. Consequently, PlmCasX and DpbCasX may generate different editing profiles in cells, warranting further characterization for future cellular applications.
Enormous efforts have been made to improve the specificity of CRISPR-Cas nucleases (46–50), as off-target effects pose major challenges in gene editing (10). Methods to improve specificity fall into two categories: weakening non-essential interactions in Cas–RNA–DNA complexes to increase the energy barrier for DNA cleavage and increasing the stringency of checkpoints or adding new ones to improve specificity. Techniques such as truncated gRNAs (Tru-gRNAs) (51), high fidelity Cas9 (Cas9-HF1) (40), enhanced specificity Cas9 (eSpCas9) (41), and hyper-accurate Cas9 (HypaCas9) (52) belong to the first category, balancing specificity and activity by sacrificing some on-target activity (49,50,53–55). The second category includes SpCas9 (D1135E) with stricter PAM recognition (56), paired Cas9 nickase (57), and dCas9-FokI fusion nucleases (58), which increase the stringency of specific checkpoints or introduce an additional checkpoint to enhance overall specificity. Our study focused on key residues in the NTSB domain of CasX, which facilitate DNA unwinding and RNA-DNA heteroduplex formation. In-vitro and cellular assays showed that mutants with weakened NTSB domain interactions had reduced activity and potentially increased specificity at certain loci, consistent with the first category. Introducing additional checkpoints and/or modifying the stringency of existing checkpoints are potential routes to further improve CasX specificity.
In addition to specificity, editing efficiency is crucial for Cas nuclease applications (55). Optimizing sgRNA has enhanced CasX cleavage activity and editing efficiency (15) by stabilizing the PlmCasX–sgRNA complex at its target site (Figure 4B). However, the editing efficiency of CasX remains lower than that of Cas9 and Cas12a in cells. To address this, we introduced here a new parameter, effective search efficiency, representing the recognition probability of Cas nucleases upon target encounter. The effective search efficiencies of DpbCasX and PlmCasX (∼0.1) are significantly lower than those of SpCas9, AsCas12a and LbCas12a (0.5–0.8), suggesting that CasX’s low recognition probability limits its cellular efficiency. Enhancing effective search efficiency through mutations affecting PAM recognition and DNA unwinding is a feasible strategy, as demonstrated in Cas9 (39). Our mechanistic studies provide insights for CasX engineering and foundation for developing new variants through directed evolution.
Supplementary Material
Acknowledgements
Author contributions: W.X., W.W., J.J.G.L. and C.C. designed the experiments; W.X. prepared materials and performed biochemical and single-molecule experiments, D.L. performed biochemical and editing experiments in HEK293T cells, and W.X. and C.C. wrote the paper with input from D.L., W.W. and J.J.G.L.
Contributor Information
Wenjing Xing, State Key Laboratory of Membrane Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Danyuan Li, State Key Laboratory of Membrane Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing 100084, China.
Wenjuan Wang, Technology Center for Protein Sciences, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Jun-Jie Gogo Liu, State Key Laboratory of Membrane Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing 100084, China.
Chunlai Chen, State Key Laboratory of Membrane Biology, Beijing Frontier Research Center for Biological Structure, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Data availability
The data that support the findings of this study are in the manuscript and Supplementary Information.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
National Natural Science Foundation of China [21877069, 22277063, 22061160466 to C.C., 32150018 to J.J.G.L., 22007054, 32370391 to W.W.]; Ministry of Agriculture and Rural Affairs of China to J.J.G.L.; National Key Research and Development Program [2022YFF1002801 to J.J.G.L.]; Beijing Frontier Research Center for Biological Structure (to C.C. and J.J.G.L.); Tsinghua-Peking Joint Center for Life Sciences (to J.J.G.L.). Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
References
- 1. Horvath P., Barrangou R.. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010; 327:167–170. [DOI] [PubMed] [Google Scholar]
- 2. Marraffini L.A. CRISPR-Cas immunity in prokaryotes. Nature. 2015; 526:55–61. [DOI] [PubMed] [Google Scholar]
- 3. van Beljouw S.P.B., Sanders J., Rodríguez-Molina A., Brouns S.J.J.. RNA-targeting CRISPR-Cas systems. Nat. Rev. Microbiol. 2023; 21:21–34. [DOI] [PubMed] [Google Scholar]
- 4. Wright A.V., Nunez J.K., Doudna J.A.. Biology and applications of CRISPR systems: harnessing Nature's toolbox for genome engineering. Cell. 2016; 164:29–44. [DOI] [PubMed] [Google Scholar]
- 5. Makarova K.S., Wolf Y.I., Iranzo J., Shmakov S.A., Alkhnbashi O.S., Brouns S.J.J., Charpentier E., Cheng D., Haft D.H., Horvath P.et al.. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020; 18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hsu P.D., Lander E.S., Zhang F.. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014; 157:1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S.et al.. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015; 520:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Toth E., Weinhardt N., Bencsura P., Huszar K., Kulcsar P.I., Talas A., Fodor E., Welker E.. Cpf1 nucleases demonstrate robust activity to induce DNA modification by exploiting homology directed repair pathways in mammalian cells. Biol. Direct. 2016; 11:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tu M., Lin L., Cheng Y., He X., Sun H., Xie H., Fu J., Liu C., Li J., Chen D.et al.. A ‘new lease of life’: fnCpf1 possesses DNA cleavage activity for genome editing in human cells. Nucleic Acids Res. 2017; 45:11295–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang J.Y., Doudna J.A.. CRISPR technology: a decade of genome editing is only the beginning. Science. 2023; 379:eadd8643. [DOI] [PubMed] [Google Scholar]
- 11. Liu J.J., Orlova N., Oakes B.L., Ma E.B., Spinner H.B., Baney K.L.M., Chuck J., Tan D., Knott G.J., Harrington L.B.et al.. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature. 2019; 568:E8–E10. [DOI] [PubMed] [Google Scholar]
- 12. Han H., Yang Y., Jiao Y., Qi H., Han Z., Wang L., Dong L., Tian J., Vanhaesebroeck B., Li X.et al.. Leverage of nuclease-deficient CasX for preventing pathological angiogenesis. Mol. Ther. Nucleic Acids. 2023; 33:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karpov D.S., Demidova N.A., Kulagin K.A., Shuvalova A.I., Kovalev M.A., Simonov R.A., Karpov V.L., Snezhkina A.V., Kudryavtseva A.V., Klimova R.R.et al.. Complete and prolonged inhibition of Herpes simplex virus type 1 infection In vitro by CRISPR/Cas9 and CRISPR/CasX systems. Int. J. Mol. Sci. 2022; 23:14847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roberson E.D.O. A catalog of CasX genome editing sites in common model organisms. BMC Genomics [Electronic Resource]. 2019; 20:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuchida C.A., Zhang S., Doost M.S., Zhao Y., Wang J., O’Brien E., Fang H., Li C.P., Li D., Hai Z.Y.et al.. Chimeric CRISPR-CasX enzymes and guide RNAs for improved genome editing activity. Mol. Cell. 2022; 82:1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jeon Y., Choi Y.H., Jang Y., Yu J., Goo J., Lee G., Jeong Y.K., Lee S.H., Kim I.S., Kim J.S.et al.. Direct observation of DNA target searching and cleavage by CRISPR-Cas12a. Nat. Commun. 2018; 9:2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L.J., Sun R.R., Yang M.Y., Peng S.J., Cheng Y.X., Chen C.L.. Conformational dynamics and cleavage sites of Cas12a are modulated by complementarity between crRNA and DNA. iScience. 2019; 19:492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burstein D., Harrington L.B., Strutt S.C., Probst A.J., Anantharaman K., Thomas B.C., Doudna J.A., Banfield J.F.. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017; 542:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xin C., Yin J., Yuan S., Ou L., Liu M., Zhang W., Hu J.. Comprehensive assessment of miniature CRISPR-Cas12f nucleases for gene disruption. Nat. Commun. 2022; 13:5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang M.Y., Peng S.J., Sun R.R., Lin J.D., Wang N., Chen C.L.. The conformational dynamics of Cas9 governing DNA cleavage are revealed by single-molecule FRET. Cell Rep. 2018; 22:372–382. [DOI] [PubMed] [Google Scholar]
- 21. Peng S.J., Sun R.R., Wang W.J., Chen C.L.. Single-molecule photoactivation FRET: a general and easy-to-implement approach to break the concentration barrier. Angew Chem. Int. Edit. 2017; 56:6882–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Illingworth J., Kittler J.. A survey of the Hough transform. Comput. Vision Graph. 1988; 44:87–116. [Google Scholar]
- 23. McKinney S.A., Joo C., Ha T.. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006; 91:1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Singh D., Mallon J., Poddar A., Wang Y., Tippana R., Yang O., Bailey S., Ha T.. Real-time observation of DNA target interrogation and product release by the RNA-guided endonuclease CRISPR Cpf1 (Cas12a). Proc. Natl. Acad. Sci. U.S.A. 2018; 115:5444–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ha T., Fei J., Schmid S., Lee N.K., Gonzalez R.L., Paul S., Yeou S.. Fluorescence resonance energy transfer at the single-molecule level. Nat. Rev. Methods Primers. 2024; 4:21. [Google Scholar]
- 26. Anzalone A.V., Koblan L.W., Liu D.R.. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020; 38:824–844. [DOI] [PubMed] [Google Scholar]
- 27. Chen P.J., Liu D.R.. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 2023; 24:161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Knott G.J., Doudna J.A.. CRISPR-Cas guides the future of genetic engineering. Science. 2018; 361:866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh D., Sternberg S.H., Fei J., Doudna J.A., Ha T.. Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nat. Commun. 2016; 7:12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anders C., Niewoehner O., Duerst A., Jinek M.. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014; 513:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gorski S.A., Vogel J., Doudna J.A.. RNA-based recognition and targeting: sowing the seeds of specificity. Nat. Rev. Mol. Cell Bio. 2017; 18:215–228. [DOI] [PubMed] [Google Scholar]
- 32. Stella S., Alcón P., Montoya G.. Class 2 CRISPR-Cas RNA-guided endonucleases: swiss Army knives of genome editing. Nat. Struct. Mol. Biol. 2017; 24:882–892. [DOI] [PubMed] [Google Scholar]
- 33. Zeng Y., Cui Y., Zhang Y., Zhang Y.R., Liang M., Chen H., Lan J., Song G.T., Lou J.Z.. The initiation, propagation and dynamics of CRISPR-SpyCas9 R-loop complex. Nucleic Acids Res. 2018; 46:350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y.B., Mallon J., Wang H.B., Singh D., Jo M.H., Hua B.Y., Bailey S., Ha T.. Real-time observation of Cas9 postcatalytic domain motions. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2010650118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang M.Y., Sun R.R., Deng P.J., Yang Y.Z., Wang W.J., Liu J.J.G., Chen C.L.. Nonspecific interactions between SpCas9 and dsDNA sites located downstream of the PAM mediate facilitated diffusion to accelerate target search. Chem. Sci. 2021; 12:12776–12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun R.R., Zhao Y.Q., Wang W.J., Liu J.J.G., Chen C.L.. Nonspecific interactions between Cas12a and dsDNA located downstream of the PAM mediate target search and assist AsCas12a for DNA cleavage. Chem. Sci. 2023; 14:3839–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A.et al.. Cpf1 Is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015; 163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tóth E., Czene B.C., Kulcsár P.I., Krausz S.L., Tálas A., Nyeste A., Varga É., Huszár K., Weinhardt N., Ligeti Z.et al.. Mb- and FnCpf1 nucleases are active in mammalian cells: activities and PAM preferences of four wild-type Cpf1 nucleases and of their altered PAM specificity variants. Nucleic Acids Res. 2018; 46:10272–10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eggers A.R., Chen K., Soczek K.M., Tuck O.T., Doherty E.E., Xu B., Trinidad M.I., Thornton B.W., Yoon P.H., Doudna J.A.. Rapid DNA unwinding accelerates genome editing by engineered CRISPR-Cas9. Cell. 2024; 187:3249–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z.L., Joung J.K.. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016; 529:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Slaymaker I.M., Gao L.Y., Zetsche B., Scott D.A., Yan W.X., Zhang F.. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016; 351:84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Globyte V., Lee S.H., Bae T., Kim J.S., Joo C.. CRISPR/Cas9 searches for a protospacer adjacent motif by lateral diffusion. EMBO J. 2019; 38:e99466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rostain W., Grebert T., Vyhovskyi D., Pizarro P.T., Bellingen G.T.V., Cui L., Bikard D.. Cas9 off-target binding to the promoter of bacterial genes leads to silencing and toxicity. Nucleic Acids Res. 2023; 51:3485–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones D.L., Leroy P., Unoson C., Fange D., Curic V., Lawson M.J., Elf J.. Kinetics of dCas9 target search in Escherichia coli. Science. 2017; 357:1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stella S., Mesa P., Thomsen J., Paul B., Alcon P., Jensen S.B., Saligram B., Moses M.E., Hatzakis N.S., Montoya G.. Conformational activation promotes CRISPR-Cas12a catalysis and resetting of the endonuclease activity. Cell. 2018; 175:1856–1871. [DOI] [PubMed] [Google Scholar]
- 46. Tsai S.Q., Joung J.K.. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016; 17:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tycko J., Myer V.E., Hsu P.D.. Methods for optimizing CRISPR-Cas9 genome editing specificity. Mol. Cell. 2016; 63:355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Komor A.C., Badran A.H., Liu D.R.. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017; 168:20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu W.Y., Lebbink J.H.G., Kanaar R., Geijsen N., van der Oost J.. Genome editing by natural and engineered CRISPR-associated nucleases. Nat. Chem. Biol. 2018; 14:642–651. [DOI] [PubMed] [Google Scholar]
- 50. Anzalone A.V., Koblan L.W., Liu D.R.. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020; 38:824–844. [DOI] [PubMed] [Google Scholar]
- 51. Fu Y.F., Sander J.D., Reyon D., Cascio V.M., Joung J.K.. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014; 32:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A.. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017; 550:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim D., Luk K., Wolfe S.A., Kim J.S.. Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu. Rev. Biochem. 2019; 88:191–220. [DOI] [PubMed] [Google Scholar]
- 54. Kim N., Kim H.K., Lee S., Seo J.H., Choi J.W., Park J., Min S., Yoon S., Cho S.R., Kim H.H.. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 2020; 38:1328–.1336. [DOI] [PubMed] [Google Scholar]
- 55. Liu G.W., Lin Q.P., Jin S., Gao C.X.. The CRISPR-Cas toolbox and gene editing technologies. Mol. Cell. 2022; 82:333–347. [DOI] [PubMed] [Google Scholar]
- 56. Kleinstiver B.P., Prew M.S., Tsai S.Q., Topkar V.V., Nguyen N.T., Zheng Z.L., Gonzales A.P.W., Li Z.Y., Peterson R.T., Yeh J.R.J.et al.. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015; 523:481–U249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y.et al.. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013; 154:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guilinger J.P., Thompson D.B., Liu D.R.. Fusion of catalytically inactive Cas9 to fokl nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014; 32:577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are in the manuscript and Supplementary Information.