

Abstract
Introduction
Altered complement component 3 (C3) activation in patients with alpha-1 antitrypsin (AAT) deficiency (AATD) has been reported. To understand the potential impact on course of inflammation, the aim of this study was to investigate whether C3d, a cleavage-product of C3, triggers interleukin (IL)-1β secretion via activation of NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome. The objective was to explore the effect of AAT augmentation therapy in patients with AATD on the C3d/complement receptor 3 (CR3) signalling axis of monocytes and on circulating pro-inflammatory markers.
Methods
Inflammatory mediators were detected in blood from patients with AATD (n=28) and patients with AATD receiving augmentation therapy (n=19). Inflammasome activation and IL-1β secretion were measured in monocytes of patients with AATD, and following C3d stimulation in the presence or absence of CR3 or NLRP3 inhibitors.
Results
C3d acting via CR3 induces NLRP3 and pro-IL-1β production, and through induction of endoplasmic reticulum (ER) stress and calcium flux, triggers caspase-1 activation and IL-1β secretion. Treatment of individuals with AATD with AAT therapy results in decreased plasma levels of C3d (3.0±1.2 µg/mL vs 1.3±0.5 µg/mL respectively, p<0.0001) and IL-1β (115.4±30 pg/mL vs 73.3±20 pg/mL, respectively, p<0.0001), with a 2.0-fold decrease in monocyte NLRP3 protein expression (p=0.0303), despite continued ER stress activation.
Discussion
These results provide strong insight into the mechanism of complement-driven inflammation associated with AATD. Although the described variance in C3d and NLRP3 activation decreased post AAT augmentation therapy, results demonstrate persistent C3d and monocyte ER stress, with implications for new therapeutics and clinical practice.
Keywords: alpha1 antitrypsin deficiency, innate immunity, rare lung diseases
WHAT IS ALREADY KNOWN ON THIS TOPIC.
There are increased levels of the complement activation product C3d in the circulation and airways of individuals with alpha-1 antitrypsin (AAT) deficiency (AATD). Crucially however, there is a lack of knowledge on the impact of C3d on the course of inflammation in AATD.
WHAT THIS STUDY ADDS
Plasma deficiency of AAT leads to increased C3d, and cognate receptor CR3 on circulating monocytes, with subsequent production of endoplasmic reticulum (ER) stress and IL-1β governed by the NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome. Post AAT augmentation therapy, increased circulating levels of AAT corresponds with a reduction in C3d, leading to significantly reduced inflammasome activation and IL-1β production in vivo.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Results identify a positive impact of AAT therapy on monocytes, thereby modifying activities that trigger inflammation. However, persistent cellular ER-stress responses and NLRP3 activation highlight the need for developing multitargeted drug combinations for disease modification.
Introduction
Alpha-1 antitrypsin (AAT) is a 52 kDa glycosylated protein synthesised primarily by hepatocytes. It is the archetypal serine protease inhibitor, capable of exerting potent anti-inflammatory and immunomodulatory effects.1 AAT deficiency (AATD) is a rare hereditary disorder, accounting for 2%–3% of all instances of emphysema, and results from mutations in the SERPINA1 gene on the long arm of chromosome 14. The most pathogenic allele is Pi*Z which occurs in >95% of individuals with AATD, with homozygosity affecting severe plasma deficiency.2 In ZZ-AATD, the misfolded Z protein accumulates in the endoplasmic reticulum (ER) of hepatocytes,3 leading to a gain of function and predisposition to liver cirrhosis, but also a failure to secrete protective levels of AAT into the systemic circulation.4 The subsequent loss of function is characterised at the lung by insufficient protection of the alveoli from proteolytic damage, with lung disease presenting as early as the fourth or fifth decade of life.5 6 Intravenous augmentation therapy with plasma-purified AAT is an approved treatment available for AATD-associated emphysema, and the Randomised Trial of Augmentation Therapy in Alpha-1 Proteinase Inhibitor Deficiency (RAPID trial) and its open-label extension study RAPID-OLE, demonstrated a slowed progression of emphysema.7 8 Nevertheless, these latter clinical trials were unsuccessful in determining changes in lung function,7 8 and therefore additional research on the effectiveness and mechanisms of action are required.
More recently, a greater understanding of the pathways leading to lung inflammation has shed light on AATD as an inflammatory condition, in which proinflammatory mediators including interleukin-1β (IL-1β) and complement activation play a role.9 10 In this regard, C3d levels were elevated in ZZ-AATD individuals compared with non-AATD patients with COPD, and correlate with radiographic evidence of emphysema and decline in lung function.10 While C3d deposition is a recognised complication after lung transplantation, associated with early bronchiolitis obliterans syndrome,11 the role of C3d in driving inflammation in AATD is unknown. In contrast, the proinflammatory cytokine IL-1β is strongly implicated in pulmonary disease with levels recorded in stable COPD and during an exacerbation, and in epithelial lining before the onset of symptomatic lung damage in AATD.12 The predominant complex driving IL-1β production is the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, and markedly, NLRP3-deficient mice demonstrate a less severe COPD phenotype.13 A recent study reported NLRP3 expression in response to Alu RNA through TLR7 activation in AATD macrophages,14 however, C3d mediated signalling, linking complement activation to IL-1β production is unexplored. This study aimed to investigate IL-1β in AATD associated with NLRP3/caspase-1 inflammasome assembly uniquely responsive to C3d signalling, and to elucidate the impact of AAT augmentation therapy.
Materials and methods
Study design
Plasma samples donated by patients with AATD (Pi*ZZ phenotype) (n=38) were obtained from the Alpha-1 Foundation DNA and Tissue Bank at the University of Florida, USA and the National AATD Clinic in Beaumont Hospital, Ireland (n=9) (table 1) (n=47 total). Of the 47 patients with AATD, plasma was donated by 19 patients who were receiving AAT augmentation therapy administered intravenously at a dosage of 60 mg/kg body weight weekly. Healthy control (HC) volunteers (table 1) showed no evidence of any disease and had no respiratory symptoms; none were taking medication, all non-smokers and all proven MM phenotype with serum AAT concentrations within the normal range (23–28 µM). For the isolation of monocytes, patients with AATD were recruited from the National AATD Clinic in Beaumont Hospital, Ireland (table 2) or Icahn School of Medicine at Mount Sinai (table 3).
Table 1.
Characteristics of patients with AATD and those receiving AAT augmentation therapy from the Alpha-1 Foundation DNA and Tissue Bank at the University of Florida, USA and the National Alpha-1 Antitrypsin Deficiency Clinic in Beaumont Hospital, Ireland
| Healthy control donors | AATD without augmentation therapy | AATD with augmentation therapy | P value | |
| Number of participants | 16 | 28 | 19 | |
| Age, years (mean±SD) | 34.6±7.2 | 60.6±13.0 | 55.6±7.1 | 0.1546 |
| Gender (male/female) | 7/9 | 16/12 | 8/11 | – |
| AAT genotype (MM/ZZ) | 16/0 | 0/28 | 0/19 | – |
| Baseline plasma AAT (μM) | 25.0±3.0 | 5.2±1.8 | 20.0±7.0 | <0.0001 |
| Former smokers (%) | 0 | 71.42 | 63.15 | – |
| Emphysema (%) | 0 | 35.71 | 63.15 | – |
Genotyping and phenotyping of patients with AATD are as described in the online supplemental methodology section. Data are expressed as number or mean±SD. Comparisons between patients with AATD and those receiving AAT augmentation therapy was performed using non-parametric Kolmogorov-Smirnov t-test. Plasma from healthy donors and patients was used in experiments of figure 1.
AAT, alpha-1 antitrypsin; AATD, alpha-1 antitrypsin deficiency.
Table 2.
Clinical details of the healthy control donors and patients with AATD, who donated blood for monocyte isolation and were recruited from the National Alpha-1 Antitrypsin Deficiency Clinic in Beaumont Hospital, Ireland
| Healthy control donors | AATD without augmentation therapy | P value | |
| Number of participants | 24 | 6 | |
| Age, years (mean±SD) | 49.62±13.92 | 55.86±15.08 | 0.0763 |
| FEV1 (% predicted) | 98.60±11.17 | 45.16±22.88 | 0.0001 |
| FEV1/FVC (% predicted) | 59.15±20.72 | 54.30±15.00 | 0.9853 |
| DLCO % predicted (%) | – | 63.33±11.00 | – |
Data are expressed as number or mean±SD. Monocytes were isolated from blood of healthy control donors and individuals with AATD and used in experiments of figures 2–7A. Descriptive statistical comparisons by groups performed using non-parametric Kolmogorov-Smirnov t-test.
AATD, alpha-1 antitrypsin deficiency; DLCO, diffusing capacity of lung for carbon monoxide; FEV1, forced expiratory volume in 1 s (% predicted); FVC, forced vital capacity (% predicted).
Table 3.
Clinical details of healthy control subjects and patient with AATD cohorts either receiving or not receiving AAT augmentation therapy from Icahn School of Medicine at Mount Sinai
| Healthy controls | AATD without augmentation therapy | AATD with augmentation therapy | P value | |
| Number of samples | 8 | 6 | 6 | |
| Age, years (mean±SD) | 49.1±13.3 | 53.3±12.2 | 54.3±8.6 | 0.6798 |
| Gender (male/female) | 5/3 | 3/3 | 4/2 | 0.9845 |
| Baseline plasma AAT (μM) | 25.2±5.0 | 6.7±3.4 | 14.5±2.8 | <0.0001 |
| Former smokers (%) | 0 | 83.3 | 83.3 | 0.0022 |
| FEV1 % predicted (%) | – | 55.3±20.5 | 51.4±15.7 | 0.5655 |
| FVC % predicted (%) | – | 86.3±12.3 | 84.4±13.3 | 0.7218 |
| FEV1/FVC % (%) | – | 55.3±20.5 | 51.2±9.3 | 0.5203 |
| DLCO % predicted (%) | – | 61.3±16.9 | 57.3±15.2 | 0.6757 |
Six patients were receiving plasma purified AAT from CSL Behring (Zemaira). HC and patient samples were employed in figure 5B and figure 7B–F. For AATD five out of six samples were used for mRNA and three out of six samples were used for protein expression, determined by sample availability. Descriptive statistical comparisons by groups performed and p values shown.
DLCO, diffusing capacity of lung for carbon monoxide; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity (% predicted).
thorax-2023-221071supp001.pdf (588.5KB, pdf)
thorax-2023-221071supp002.pdf (119.2KB, pdf)
A detailed description of the methodology is provided in the methodology section of online supplemental file 2. In brief, purified monocytes were assessed for NLRP3 and IL-1β expression by Real-Time quantitative Polymerase Chain Reaction (qRT-PCR), sodium dodecyl sulfate polyacrylamide gel electrophoresis and western blotting. IL-1β, tumour necrosis factor-α (TNF-α) and C3d were measured by ELISA, and caspase-1 activity was detected by bioluminescence. Intracellular calcium (Ca2+) was measured by fluorometric assay. C3d and CR3 membrane expression was measured by flow cytometry. A complete list of inhibitors, primers and antibodies used are available in supplementary methodology (online supplemental tables 1–3, respectively).
Statistical analysis
Data were analysed using GraphPad Prism V.8.0 for PC. Appropriate descriptive statistics (eg, mean, SD, median, frequencies and counts) were used to describe patient characteristics and outcomes of interest. Statistical significance was calculated using a Student’s t-test when comparisons were made between two groups when normally distributed. Non-normal data were analysed by the two tailed non-parametric Mann-Whitney U test. Linear and multiple regression modelling was employed to examine the significance of any relationship within, or between, the clinical data. Adjustment was performed for covariates that included age and was reported using the standardised analysis of covariance (ANCOVA). Two tailed non-parametric Spearman correlation test was used to determine the positive and/or negative correlation between two continuous variables. One-way analysis of variance (ANOVA) was used to determine statistical significance when comparing three or more groups followed by Tukey’s posthoc multiple comparison test. A value of p<0.05 was considered statistically significant.
Results
AAT augmentation therapy impacts C3d and IL-1β plasma levels in patients with AATD
Plasma samples were isolated from patients with AATD who were receiving augmentation therapy (n=19), patients with AATD who had never received augmentation therapy (n=28) and also healthy individuals (n=16) (table 1). Significantly increased plasma levels of AAT were detected in patients receiving AAT therapy compared with patients not receiving weekly infusions (20±7.0 vs . 5.2±1.8 µM, p<0.0001) (figure 1A). By use of a specific ELISA for a neo-epitope exposed only on C3d, levels were quantified in patients with AATD. Plasma C3d was detected in significantly higher concentrations in samples of AATD compared with HC (range: 1.2–6.6 µg/mL vs 0.2–0.6 µg/mL, p<0.0001), and were significantly reduced in AATD plasma of patients receiving AAT augmentation therapy (range: 0.5–2.2 µg/mL, p<0.0001) (figure 1B). Moreover, mean plasma IL-1β levels were significantly higher in AATD samples compared with HC (115.4±30 pg/mL vs 17.5±6.0 pg/mL, p<0.0001), but significantly decreased levels were detected in samples of patients receiving AAT therapy (73.3±20 pg/mL, p<0.0001) (figure 1C). Of note, on comparing samples of AATD post augmentation therapy to HC, results revealed that neither C3d (range: 0.6–2.1 µg/mL vs 0.1–0.3 µg/mL, p=0.0095) nor IL-1β levels decreased to HC levels (range: 31.3–99.4 pg/mL vs 11.0–30.0 pg/mL, p<0001). Although an inverse correlation was detected between AAT and C3d (r=−0.7101, 95% CI: −0.8283 to −0.5313, p<0.0001) and AAT and IL-1β (r=−0.6489, 95% CI: −0.8182 to −0.4951, p<0.0001) (online supplemental figure 1A, B), in contrast, a significant positive correlation was recorded between plasma C3d and IL-1β (r=0.7160, 95% CI: 0.5399 to 0.832, p<0.0001) (figure 1D), which remained significant following correction for age (β=0.0059, p=0.0001). These results prompted an investigation into the mechanism, and impact, of C3d-induced IL-1β production.
Figure 1.
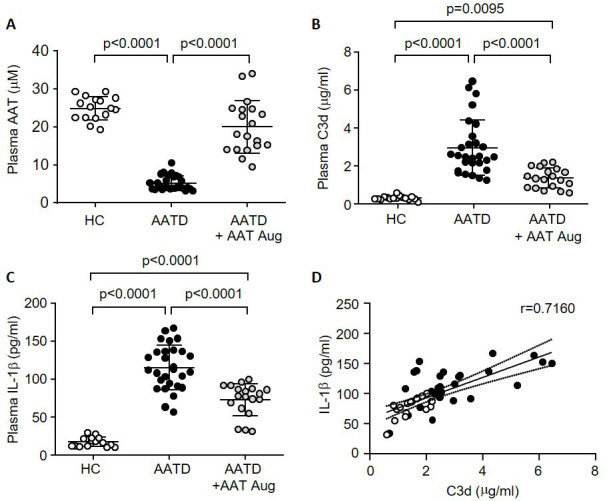
Increased plasma IL-1β and C3d levels in AATD are decreased post AAT augmentation therapy. Levels of AAT (A), C3d (B) or IL-1β (C) were determined in plasma of healthy control subjects (HC, n=16), patients with AATD who are not on AAT therapy (n=28) and patients with AATD receiving weekly AAT augmentation therapy (AATD+AAT Aug, n=19). Significance was tested by one-way ANOVA, followed by Tukey’s posthoc multiple comparison test. (D) Positive correlation between C3d and IL-1β plasma levels (n=47; p<0.0001, two-tailed non-parametric Spearman correlation test). AAT, alpha-1 antitrypsin; AATD, alpha-1 antitrypsin deficiency; HC, healthy control.
CR3 receptor inhibition blocks C3d-induced IL-1β expression in monocytes
Physiologically relevant concentrations of exogenous C3d (2.5 µg/mL) triggered release of IL-1β by monocytes (38.5±6.0 pg/mL; p=0.0002) and reached a plateau at approximately 10 µg/mL C3d (120±15.0 pg/mL, p<0.0001), a concentration used in further experiments (online supplemental figure 1C). CR3 can bind to the C3d fragment of C3,15 and in the current study the ability of monocyte CR3 to recognise and bind C3d was confirmed by flow cytometry (online supplemental figure 1D). In control experiments, the inclusion of a CR3 blocking antibody (clone mAb 107) known to target the CR3 ligand-binding domain, blocked C3d receptor engagement (~56% reduction, p=0.0006) (online supplemental figure 1E). Selectness for C3d was confirmed as other complement fragments, including C5, generated significantly reduced levels of IL-1β compared with C3d (14.2±3.0 pg/mL vs 120±17.0 pg/mL; p<0.0001) (figure 2A). Moreover, C3d is formed by the cleavage of C3a, and produced significantly increased levels of IL-1β compared with the larger C3a molecule (120±17.0 pg/mL vs 58.6±8.4 pg/mL; p<0.0001). A positive control for this latter experiment included lipopolysaccharide (LPS) in combination with adenosine-5′-triphosphate (figure 2A), known to trigger IL-1β secretion by monocytes via the purinergic receptor P2×7R.16
Figure 2.
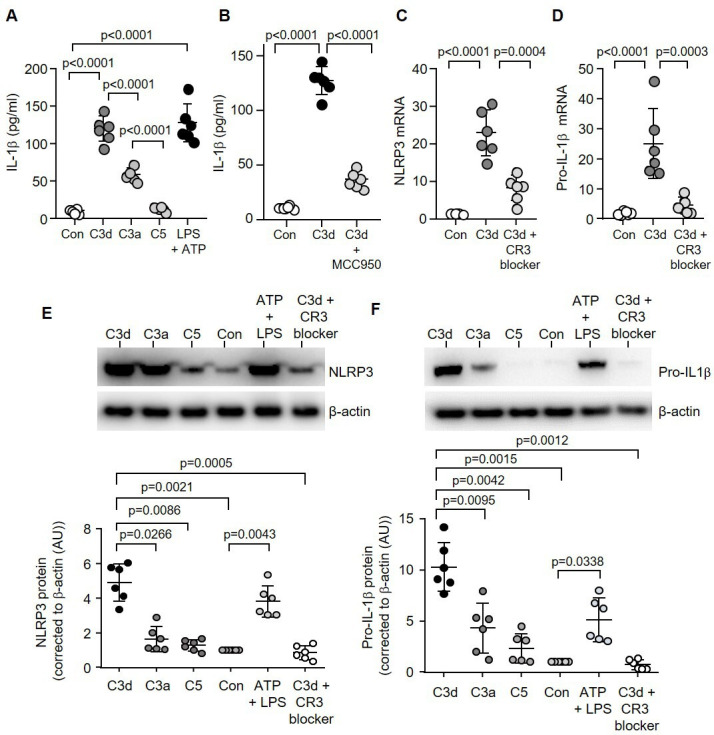
C3d signalling via CR3 triggers NLRP3 inflammasome activation and IL-1β release. Monocytes remained untreated (Con) or challenged with C3d, C3a, C5 (10 µg/mL) or ATP (3 mM) and LPS (50 ng/mL) (A), or MCC950 (1 µM) (B), and extracellular levels of IL-1β determined by ELISA (n=6 biological repeats, one-way ANOVA, followed by Tukey’s posthoc multiple comparison test). (C&D) NLRP3 (C) or pro-IL-1β (D) Real-Time quantitative Polymerase Chain Reaction of monocyte mRNA (fold change) post C3d treatment (10 µg/mL) with or without CR3 blocking antibody (C3d blocker, n=6 biological repeats, one-way ANOVA, followed by Tukey’s posthoc multiple comparison test). HC monocytes (1×105) were incubated with C3d, C3a, C5 or ATP and LPS. Cell lysates were collected and western blotted for NLRP3 (E) or pro-IL-1β (F). All results are expressed as relative densitometry units (DU), with representative western blots presented (n=6 biological repeats, one-way ANOVA, followed by Tukey’s posthoc multiple comparison test). HC, healthy controlIL-1β, interleukin-1β; LPS, lipopolysaccharide; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3.
Previous immune cell studies have identified NLRP3 as a key driver of IL-1β.16 In monocytes, IL-1β protein production in response to C3d was inhibited by the inclusion of MCC950 (37.0±8.0 pg/mL vs 127.5±13.0 pg/mL; p<0.0001), an inhibitor of the NLRP3 inflammasome (figure 2B).17 Moreover, C3d caused a significant 20-fold increase in NLRP3 (p<0.0001) and a 25-fold increase in pro-IL-1β (p<0.0001) mRNA expression, an effect averted by the CR3 blocker (p=0.0004 and p=0.0003, respectively) (figure 2C, D). Accordingly, by western blot analysis, a 5- fold increase in NLRP3 (p=0.0021) and a 10-fold increase in pro-IL-1β (p=0.0015) protein levels were detected in whole cell lysates of C3d challenged cells (figure 2E, F). Collectively, these results indicate that C3d-induced IL-1β production is CR3-NLRP3-dependent.
NLRP3 inflammasome is driven by C3d-induced NF-kβ signalling through the PI3K/AKT pathway in monocytes
Nuclear factor kappa β (NF-κβ) is a central mediator of the priming signal of NLRP3 inflammasome activation and acts by inducing the transcriptional expression of pro-IL-1β.18 Phosphorylation of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor-alpha (Ikβ-α) and subsequent degradation, releases the transcriptional activity of NF-κβ. In the current study, following exposure of monocytes to C3d, there was a significant 20-fold decrease in total Ikβ-α (p<0.0001) (online supplemental figure 2A). Preincubation with BAY 11–7082 (NF-κβ inhibitor) (p=0.0018), clone mAb 107 (CR3 blocker) (p=0.0085), LY-294002 (PI3 kinase inhibitor) (p=0.0050) or isobavachalcone (AKT inhibitor) (p=0.0079), blocked C3d-induced Ikβ-α degradation, by a minimum of 15-fold for each inhibitor (online supplemental figure 2A). Since NF-κβ can be activated through phosphatidylinositol 3-kinase (PI3K) and its major downstream kinase Akt,19 we investigated whether PI3K/Akt contributes to C3d-induced NF-κβ activation. Following the C3d challenge to monocytes, a significant 10-fold increase in phosphorylation of the p85 regulatory subunit of PI3K (p=0.0038) (online supplemental figure 2B) and Akt (p=0.0031) (figure 3A) was observed. As Akt phosphorylation was significantly inhibited by inclusion of Akt (p=0.0011), PI3K (p=0.0039) and NF-κβ inhibitors (p=0.0031) (figure 3A), this would suggest that C3d binding to CR3 triggers Akt activation by PI3K and that Akt is the upstream molecule of NF-κβ activity. In concurrence, C3d-induced mRNA expression of pro-IL-1β was significantly decreased by BAY 11-7082 (p<0.0001), clone mAb 107 (p<0.0001), LY-294002 (p<0.0001) and isobavachalcone (p<0.0001) (figure 3B), with a minimum 9-fold reduction observed for each inhibitor. This observation was further confirmed on the IL-1β protein level by ELISA of cell supernatants (40±5.0 pg/mL vs 120±17.0 pg/mL; p<0.0001) (figure 3C). Of note, the effect of C3d on NF-κβ activation also leads to production of TNF-α and in response to 2.5 µg/mL or 10 µg/mL C3d a significant increase in TNF-α was detected (38.5±6.0 and 121±15.0 pg/mL; p=0.0112 and p=0.0165, respectively) (online supplemental figure 2C). Collectively this block of experiments demonstrates that inhibition of NF-κβ activation abrogates the stimulatory effects of C3d on proinflammatory cytokines, including pro-IL-1β production.
Figure 3.
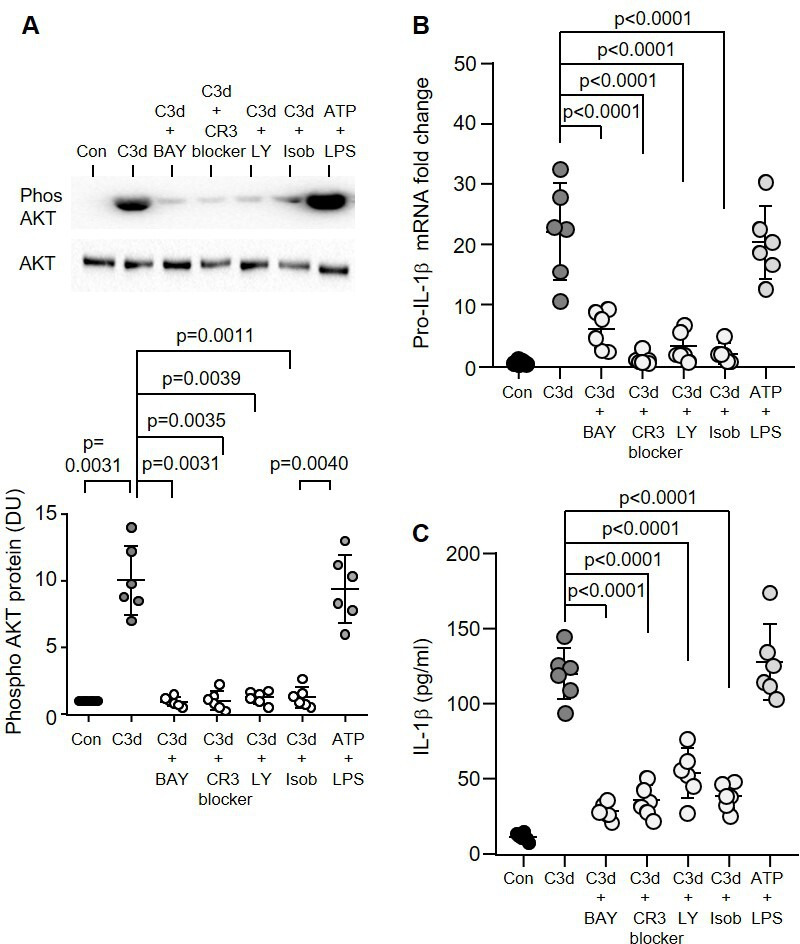
C3d activation of the PI3K/Akt–NF-κβ pathway. (A) Expression levels of AKT post C3d (10 µg/1×105 cells) treatment was measured by western blot analysis of monocyte whole cell lysates. Controls included preincubation with BAY 11-7082 (NF-κβ inhibitor, 5 µg/mL), CR3 blocker (clone mAb 107, 1 µg/mL), LY-294002 (LY) PI3 kinase inhibitor, 10 µM) or isobavachalcone (Isob) (AKT inhibitor, 5 mM) for 30 min prior to C3d challenge. Phosphorylation levels were normalised to respective total protein. Results are expressed as DU, with representative western blots presented (n=6 biological repeats, one-way ANOVA, followed by Tukey’s posthoc multiple comparison). (B) The effect of C3d (10 µg/mL) on pro-IL-1β mRNA levels by qPCR analysis and protein levels by ELISA (C) with or without the inclusion of CR3 and NF-κβ blockers (n=6 biological replicates, one-way ANOVA, followed by Tukey’s posthoc multiple comparison test). Data are expressed as mean±SD. DU, densitometry units; IL-1β, interleukin-1β; LPS, lipopolysaccharide.
Monocytes of patients with AATD demonstrate increased NF-kβ signalling and NLRP3 inflammasome activation
Next we investigated the C3d:CR3 proinflammatory signalling axes in AATD monocytes. By flow cytometry, C3d was found significantly increased by 2.27-fold on the outer membrane surface of circulating AATD monocytes compared with HC cells (9927±2033 vs 4355±1065 mean fluorescence intensity (MFI), p=0.0020) (figure 4A). AATD monocytes also expressed significantly higher levels of the cognate receptor, with ~2-fold increase in CR3 membrane expression (p=0.0022) (figure 4B). A potential consequence of increased C3d membrane expression was explored, and downstream NF-κβ activation revealed a significant 4-fold decrease in total Ikβ-α protein expression (p<0.0001), a corresponding significant 2-fold increase in p85 (p=0.0022) (online supplemental figure 3A, B, respectively) and 2.5-fold increase in Akt phosphorylation levels in AATD monocytes (p=0.0022) (figure 4C).
Figure 4.
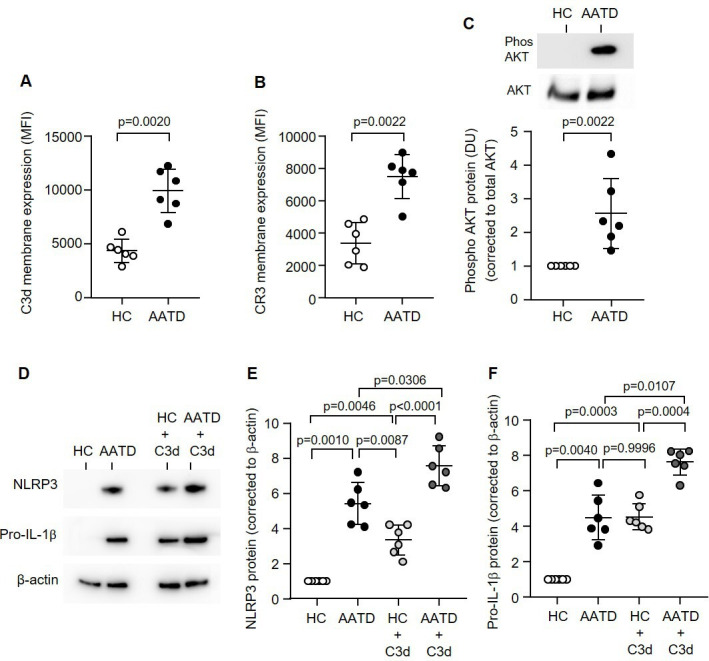
The CR3:C3d signalling pathway is increased in the monocytes of patients with AATD. C3d (A) or CR3 (B) levels were measured on the outer plasma membranes of monocytes isolated from AATD or HC by flow cytometry (n=6 subjects per group, non-parametric Mann-Whitney U test). (C) Expression levels of AKT in AATD or HC subjects measured by western blot analysis of monocyte whole cell lysates. Phosphorylation levels were normalised to respective total protein. Results are expressed as DU, with representative western blots presented (n=6 subjects per group, non-parametric Mann-Whitney U test). (D–F) NLRP3 (D and E) or pro-IL-1β (D and F) protein was detected by western blot analysis in HC or AATD monocytes either untreated or treated with C3d (10 µg/mL). Data are expressed as relative DU, with representative western blots presented (n=6 biological repeats, one-way ANOVA, followed by Tukey’s posthoc multiple comparison test). AATD, alpha-1 antitrypsin deficiency; DU, densitometry units; HC, healthy controls; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3.
In agreement with increased C3d and NF-κβ activation components, AATD monocytes demonstrated a 4.5-fold increase in levels of NLRP3 in comparison to HC cells (p=0.0010) (figure 4D, E). Experiments were expanded to investigate the impact of supplementing additional C3d in reactions. A significant difference was observed for both HC and AATD monocytes following C3d activation, with a 2.5-fold increase in NLRP3 protein expression levels recorded compared with untreated cells (p=0.0046 and p=0.0306, respectively) (figure 4D, E). Additionally, experiments explored pro-IL-1β levels in resting and C3d-challenged HC and AATD monocytes, with results mirroring those obtained for NLRP3. In comparison to HC cells, both resting and C3d-activated AATD monocytes demonstrated a 4-fold and 2.5-fold increase in pro-IL-1β protein levels (p=0.0040 and p=0.0107, respectively) (figure 4D,F). From this set of experiments, we conclude that disproportionate C3d-induced CR3 signalling in circulating AATD monocytes leads to increased intracellular levels of NLRP3 and pro-IL-1β.
C3d-induced ER stress and NLRP3 inflammasome activation
A range of signals may promote the inflammasome assembly including increased ER stress and intracellular Ca²+, leading to caspase-1 activity.20 We set out to determine whether C3d could induce this phenomenon. First, the protein expression of the ER stress-associated transcription factor ATF6 and the chaperone protein GRP78 in whole-cell lysates isolated from HC monocytes post C3d exposure was evaluated (10 µg/105 cells). Western blot results demonstrate a significant 4-fold increase in the active form of ATF6 (cleaved ATF6) (figure 5A) and 8-fold increase in GRP78 (online supplemental figure 4A) protein expression in monocytes in response to C3d (p=0.0003 and p=0.0053, respectively). Positive and negative controls for this experiment included the treatment of cells with the ER stress inducer thapsigargin (100 nM)21 or the inclusion of the CR3 blocker, respectively (figure 5A).
Figure 5.
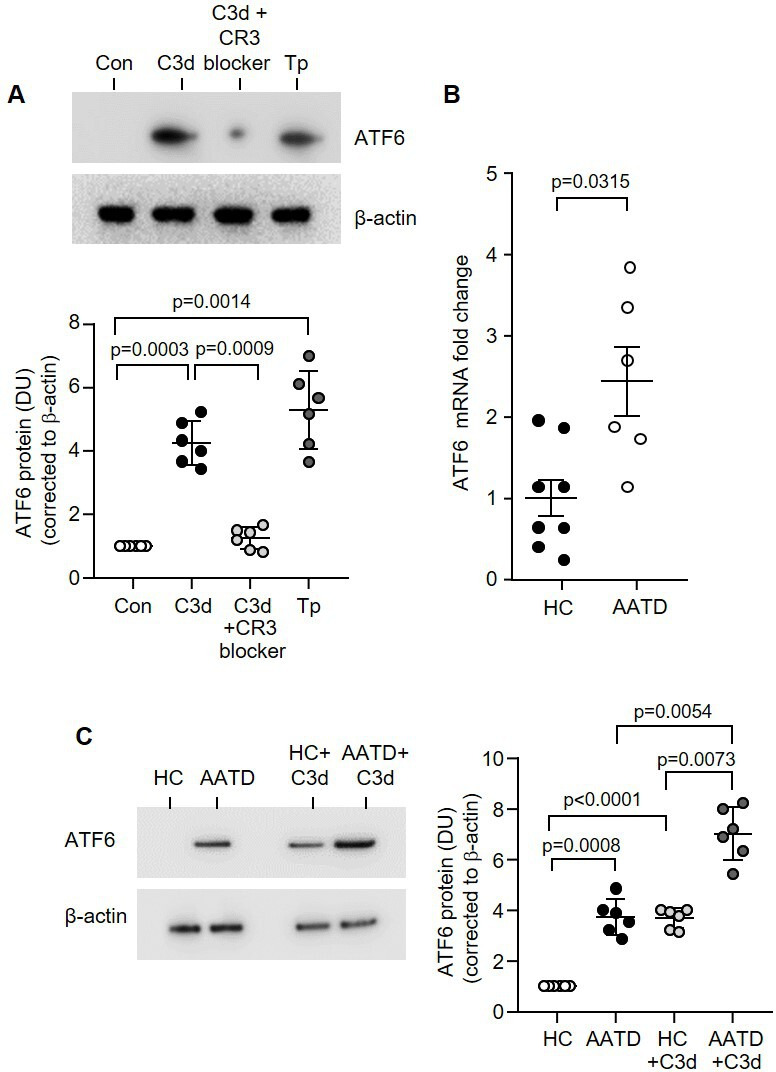
CR3 inhibition modulates C3d-induced ER stress in AATD monocytes. (A) Western blot and densitometry analysis of cleaved ATF6 expression in control (Con) untreated monocytes from HC or following treatment with C3d (10 µg/mL) for 16 hours. Negative and positive controls included preincubation with CR3 blocker (clone mAb 107, 1 µg/mL) and treatment with the ER stress inducer thapsigargin (Tp) (100 nM), respectively (n=6 biological repeats per group, one-way ANOVA). (B) ATF6 qRT-PCR of HC (n=8) or AATD (n=6) monocyte mRNA revealed significantly increased expression in AATD (non-parametric Mann-Whitney U test). (C) ATF6 expression in resting HC monocytes compared with AATD cells, and following C3d challenge (n=6 subjects per group, one-way ANOVA, followed by Tukey’s posthoc multiple comparison test). AATD, alpha-1 antitrypsin deficiency; ER, endoplasmic reticulum; HC, healthy individuals.
Next we investigated basal and C3d engendered ER stress in AATD cells, as polymers of AAT protein are implicated in ER stress.22 By use of a specific monoclonal antibody (2C1) that recognises AAT polymers,23 high concentrations of circulating polymers were recorded in AATD plasma (range: 240.55–1239.17 µg/mL), although, no relationship was detected between the plasma C3d level or IL-1β and the plasma polymer level in these patients (online supplemental figure 4B, C, respectively). Additionally, a signal for AAT polymers was detected in whole cell lysates of AATD monocytes (4.12±1.6 vs 0.25±0.05 ng/mg, p=0.0045), with significantly increased levels detected in C3d challenged AATD cells compared with untreated monocytes (7.12±2.5 vs 4.12±1.6 ng/mg, p=0.0250) (online supplemental figure 4D). In keeping with increased cellular polymers, gene expression of ER stress markers ATF6 and the second marker, GRP78, were significantly increased in AATD monocytes (figure 5B and online supplemental figure 4E, respectively) (p=0.0315 and p=0.0003, respectively). To corroborate this phenomenon, we used a Serpina1a-e knockout mouse model that lacks AAT expression and polymers.24 Results demonstrate that monocytes of Serpina1a-e−/− mice lack ER stress and possess similar gene expression levels of GRP78 and ATF6, compared with wild-type mice (p=0.4206 and p=0.6905, respectively) (online supplemental figure 5A, B). Serpina1a-e−/− mice also express similar levels of NLRP3 and IL-1β (p=0.5476 and p=0.2222, respectively) (online supplemental figure 5C, D). Moreover, AATD human monocytes not only exhibited higher protein expression of the ER stress markers ATF6 in resting non-stimulated cells compared with HC cells (p=0.0008) (figure 5C), but also in response to C3d. AATD monocytes showed a distinct sensitivity to C3d with a significant 1.8-fold increased ATF6 expression recorded compared with untreated AATD monocytes (p=0.0054) or C3d-treated HC cells (p<0.0001) (figure 5C), with similar results recorded for GRP78 (online supplemental figure 6).
Figure 6.
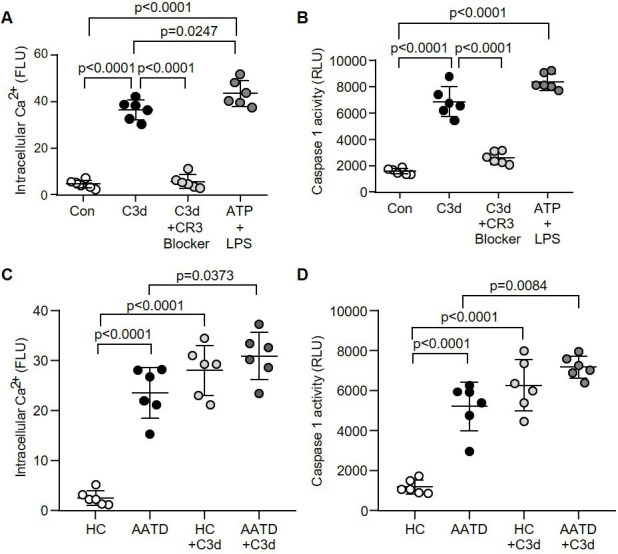
Increased caspase-1 activity in AATD monocytes in response to C3d. (A and B) Monocytes remained untreated (Con) or challenged with C3d (10 µg/mL), C3d in the presence of the CR3 blocker (clone mAb 107) or ATP (3 mM) and LPS (50 ng/mL) (positive control). Intracellular Ca2+ (A) and caspase-1 activity (B) were assessed by luminescence. RLU (n=6 biological repeats, one-way ANOVA followed by Tukey’s posthoc multiple comparison test). (C and D) Intracellular Ca2+ levels and caspase-1 activity in resting HC monocytes compared with AATD cells, and following C3d challenge (n=6 subjects per group, one-way ANOVA, followed by Tukey’s posthoc multiple comparison test). AATD, alpha-1 antitrypsin deficiency; HC, healthy control; LPS, lipopolysaccharide; FLU, fluorescence units; RLU, relative luminescence units.
As ER stress can lead to alterations in Ca2+ homeostasis,25 subsequent studies investigated the impact of C3d on cytosolic Ca2+ levels. To this end, a significant 8-fold increase in Ca2+ in cytosols of monocytes treated with C3d was recorded (p<0.0001), although lower than the ATP/LPS-induced increase in intracellular Ca2+ (p=0.0247) (figure 6A). Additionally, C3d challenged monocytes showed a 4.3-fold increase in caspase‐1 activity (p<0.0001), that in vitro was attenuated with the C3d blocker (p<0.0001) (figure 6B). Findings were further corroborated in AATD monocytes, which in comparison to HC cells, demonstrated a significant increase in cytosolic Ca2+ levels (p<0.0001) (figure 6C) and ~4.5 fold increase in caspase-1 activity (p<0.0001) (figure 6D). Moreover, in the presence of C3d, AATD monocytes expressed a further significant 1.3-fold increase in intracellular Ca2+ levels and 1.4-fold increase in caspase-1 activity compared with counterpart untreated cells (p=0.0373 and p=0.0084, respectively) (figure 6C, D). Taken together, the above results indicate that ER stress in circulating AATD monocytes leads to increased cytosolic Ca2+ and caspase-1 activity, a phenomenon further exacerbated by C3d:CR3 signalling.
Plasma-purified human AAT in vitro and in vivo is associated with reduced C3d-induced IL-1β
IL-1β is recognised to play an important role in emphysema and in the current study a negative correlation between the diffusing capacity of the lungs for carbon monoxide (DLCO) and levels of IL-1β was recorded (95% CI: -0.9564 to -0.2931, r=−0.7113, p=0.0371) (online supplemental figure 7A). Consequently, the decrease in circulating levels of IL-1β in response to AAT augmentation therapy required further consideration (figure 1C). In the current study, we explored a mechanism by which augmented AAT may impact C3d signalling and examined whether exogenous AAT may block C3d monocyte engagement. Monocytes were incubated with C3d in the presence of physiological concentrations of AAT (20 µM), followed by a fluorescein isothiocyanate (FITC)-labelled antibody against C3d to confirm C3d binding by flow cytometry (online supplemental figure 7B). Results demonstrated a significant 2.2-fold increase in C3d binding to monocytes, which was blocked by the inclusion of AAT (p<0.0001) (online supplemental figure 7C). Previous studies have indicated the importance of AAT glycans for exerting immune-regulatory capacity.1 To explore this in the current study, successful deglycosylation of AAT protein by PNGase enzyme was confirmed on Coomassie Blue stained SDS gels, separating at 45 kDa compared with 52 kDa fully glycosylated AAT (online supplemental figure 7D). By flow cytometry, deglycosylated AAT (20 µM) failed to inhibit C3d membrane interaction (online supplemental figure 7E). Additionally, while fully glycosylated AAT significantly decreased IL-1β secretion compared with untreated cells (42.0±18.0 pg/mL vs 127±11.0 pg/mL, p<0.0001), in contrast, neither deglycosylated AAT, nor an alternative plasma protein, human serum albumin (HSA), impacted IL-1β secretion (figure 7A). Collectively, these results highlight the importance of AAT glycans for controlling the C3d-induced inflammatory response and indicated that weekly AAT infusions may modulate NLRP3 inflammasome and pro-IL-1β activation in circulating monocytes of patients with AATD, which was next explored.
Figure 7.
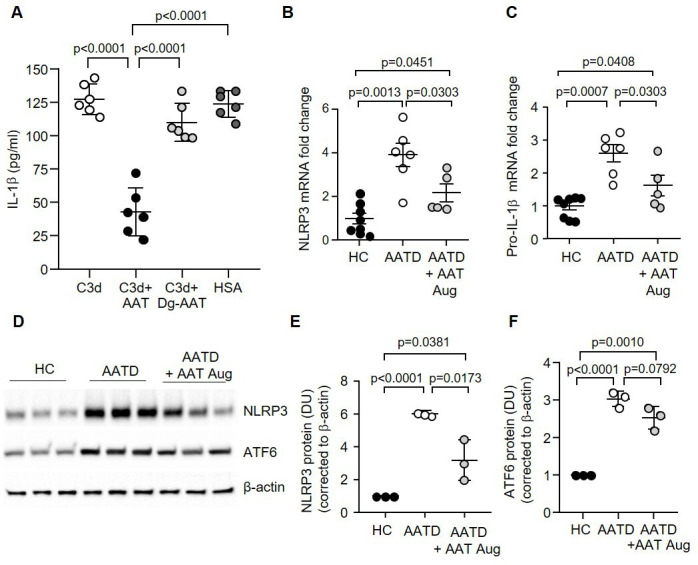
Monocyte NLRP3 is decreased, and levels of IL-1β expression are reduced after AAT augmentation therapy in vivo. (A) The effect of C3d (10 µg/mL) on IL-1β protein production determined by ELISA of cell supernatants from HC monocytes, with or without AAT, deglycosylated AAT (Dg-AAT) or control plasma protein HSA (1 mg/mL of exogenous protein) (n=6 biological replicates, one-way ANOVA). Data are expressed as mean±SD. (B and C) Monocytes were isolated from HC, patients with AATD not receiving augmentation therapy, or patients with AATD receiving weekly intravenous AAT therapy (AATD+AAT Aug). qPCR analysis for gene expression of NLRP3 (B) and IL-1β (C) demonstrated that augmentation therapy significantly decreased NLRP3 and IL-1β expression in patients with AATD when compared with patients not receiving therapy (n=5 and n=6 patients per group, respectively, one-way ANOVA). (D–F) Expression levels of NLRP3 (D and E) and ATF6 (D and F) in HC, patients with AATD not receiving AAT weekly infusions or AATD receiving intravenous AAT therapy measured by western blot analysis of monocyte whole cell lysates. NLRP3 levels were significantly different between the patients with AATD receiving therapy (n=3 subjects per group, one-way ANOVA). AAT, alpha-1 antitrypsin; AATD, alpha-1 antitrypsin deficiency; HC, healthy control; HSA, human serum albumin; IL-1β, interleukin-1β; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3.
Monocytes were isolated from patients with AATD who were on augmentation therapy, patients with AATD who had never received augmentation therapy and HC donors. Analysis by qRT-PCR revealed a 1.5-fold significant decrease in expression levels of NLRP3 and pro-IL-1β in AATD monocytes of patients receiving AAT augmentation therapy compared with those not on therapy (p=0.0303) (figure 7B, C). Of note, however, gene expression levels of NLRP3 and pro-Il-1β in monocytes of patients receiving AAT therapy remained significantly higher than HC cells (p=0.0451 and p=0.0408, respectively) (figure 7B, C). In concurrence, densitometry analysis of proteins on western blots of whole cell lysates revealed a twofold significant reduction in NLRP3 protein expression in monocytes of patients receiving augmentation therapy compared with patients not receiving intravenous AAT (p=0.0173) (figure 7D, E), but levels remained significantly higher than HC (p=0.0381). Subsequently, protein levels of ATF6 were assessed. Although increased ATF6 (p<0.0001) were recorded in AATD cells compared with HC, a small but non-significant trend towards reduced ATF6 protein expression was observed in monocytes of patients with AATD receiving AAT therapy compared with untreated patients (p=0.0792) (figure 7D, F). In summary, in agreement with reduced circulating C3d levels post AAT augmentation therapy, NLRP3 and pro-IL-1β protein levels are reduced in response to treatment with AAT. However, results also demonstrate continued ER stress-associated inflammation and NLRP3 activation in monocytes of patients with AATD receiving therapy, with levels remaining significantly higher than HC cells.
Discussion
This study demonstrates that circulating IL-1β levels are elevated in patients with AATD, and by use of in vitro and ex vivo human and murine models, we demonstrate that the effect of C3d on NLRP3 inflammasome activation involves NF-κβ gene regulation, in combination with ER stress-induced-caspase-1 activation for pro-IL-1β processing. Moreover, in patients with AATD receiving AAT augmentation therapy, circulating levels of C3d and monocyte NLRP3 inflammasome activation were significantly reduced, along with decreased plasma IL-1β levels, demonstrating an effect of AAT augmentation therapy on overall inflammation in AATD. These results are supported by a recent observational study that explored the consequences of discontinuation of AAT augmentation therapy in AATD.26 In this latter study, an increase in respiratory exacerbations emerged in association with systemic inflammation with a significant rise in IL-1β detected postdiscontinuation of therapy.26 The immune-regulatory effects of AAT were also reported in a double-dose AAT augmentation therapy study, demonstrating a greater decrease in proinflammatory cytokine levels when compared with standard dosing.27 Additionally, the current study revealed a strong correlation between IL-1β levels and C3d, a finding that remained statistically significant following correction for age, with C3d previously reported a cleavage-product of unchecked serine protease activity in AATD.10 Consequently, the observed reduction in IL-1β post AAT augmentation therapy could either be a result of increased protection against dysregulated neutrophil elastase-mediated C3 activation, or alternatively, the ability of infused glycosylated AAT to target and modulate C3d–CR3 engagement, as demonstrated here.
It has previously been shown that sublytic levels of membrane attack complex of complement formation trigger NLRP3 inflammasome activation in lung epithelial cells.28 Building on the capacity of complement components as regulators of immune cell signalling, in the current study, C3d, a terminal product of complement activation, triggered a dual response in circulating HC monocytes, the first involving activation of the PI3K/Akt/NF-κβ signalling pathway, and the second, compromising ER homeostasis leading to increased ER stress. While, NF-κβ positively regulates gene expression of NLRP3 and pro-IL-1β, ER stress was accompanied by alterations in Ca2+ homeostasis resulting in increased cytosolic Ca2+ levels, as previously described following thapsigargin treatment.25 Both pathways converge on activation of caspase-1, which is ultimately responsible for cleaving proinflammatory pro-IL-1β to an active secreted molecule in monocytes. Of note, while this study focused on inflammasome activation of monocytes via the PI3K/AKT pathway, other mitogen-activated protein kinase (MAPK) pathways may play a part, and for example, it has been shown that the ERK/MEK pathway is involved in NF-κΒ activation in bronchial epithelial cells of patients with AATD.29 Moreover, in regards to the ability of C3d to trigger ER stress, this may in part be due to increased Z-AAT polymer formation in AATD cells, or alternatively, the proinflammatory influence of C3d. Indeed, studies have reported on the role of inflammatory-induced ER stress in a number of cell types and diseases including epithelial cells in inflammatory bowel disease30 and neutrophils in cystic fibrosis and also AATD.31 Akin to the effects of CXCL8, TNFα and CXCL7 on ER stress,31 results of the current study suggest C3d as an additional trigger of ER stress affecting the properties of both HC and AATD monocytes. Ultimately however, studies have revealed an essential role for NF-κβ activation in the production of pro-IL-1β.32 33 In the current study, we demonstrate that inclusion of the NF-κβ inhibitor Bay11-7082 significantly inhibited IL-1β expression in monocytes, whereas the addition of MCC950, a specific NLRP3 inhibitor, significantly decreased IL-1β secretion, indicating that C3d-induced IL-1β in monocytes is dependent on NLRP3 activation through caspase-1 protease activity. Of importance, there is a growing body of evidence supporting a role for NLRP3 in pulmonary conditions including lung ischaemia-reperfusion injury, asthma,34 allergic rhinitis35 and sarcoidosis,36 and in many cases, the prophylactic or therapeutic effects of MCC950 show promise.
Results of this study revealed that augmentation therapy significantly decreased expression levels of NLRP3 and pro-IL-1β in circulating monocytes of patients receiving AAT infusions, although levels remained significantly higher than HC samples, and this latter result is worthy of further discussion. Weekly infusions of AAT augmentation therapy result in a peak-and-trough effect, resulting in initially normal levels of AAT in the circulation for several days after infusion, but then levels fall to near or below the protective threshold.37 This may emphasise the need for further studies evaluating dosing regimens in AAT augmentation therapy in order to maintain daily raised levels of AAT throughout treatment, thereby preventing dysregulated C3d production. Of equal importance, AAT infusions would fail to negate formation of Z-AAT polymers. In this context, basal levels of ER stress in circulating innate immune cells including neutrophils and monocytes has been attributed to accumulation of Z-AAT within the ER compartment of AATD cells.22 38 In line with this concept, AAT polymers were detected in whole cell lysates of AATD monocytes, with increased levels recorded in C3d challenged cells. Of note, AAT polymers in monocytes were not previously identified,23 a variance perhaps explained by the use of alternate cell lysis buffer and detergents for protein and polymer extraction in the current study. Of interest, earlier investigations aimed at increasing Z-AAT secretion and potentially reducing cell stress include the use of chemical chaperones for improved protein folding, small peptides to block Z-AAT polymerisation or RNAi and CRISPR/Cas9 to decrease Z-AAT abnormal protein production.39 However, these later studies focused on hepatic AAT production to target the intrinsic liver disease found in AATD, and therefore much research is required to address ER stress in circulating monocytes as a result of Z-AAT accumulation.
Our study is not without limitations, including low patient numbers and a lack of longitudinal plasma and cell samples over the course of airways disease. Moreover, this study has focused solely on C3d, yet a further key component of the complement system to consider is C5. Of interest, a common C5 polymorphism (rs17611, 2404G>A) renders this protein more susceptible to proteolysis, producing a biologically active C5a-like molecule, which is implicated in inflammatory arthritis,40 until now, a phenomenon unexplored in AATD. Regardless of these shortcomings however, this study provides evidence of the positive immune-regulatory impact of AAT therapy on C3d induced NLRP3 and IL-1β activation of circulating monocytes.
In summary, elevated circulating levels of C3d in people with AATD with the most severe Pi*Z pathogenic allele, triggers NLRP3 inflammasome activation for increased expression of IL-1β. Although treatment of patients with AATD with AAT augmentation therapy significantly suppressed C3d and NLRP3 inflammasome activation and reduced plasma levels of the proinflammatory cytokine IL-1β, the positive impact on circulating monocytes should be considered carefully as both ER stress and NLRP3 levels of circulating AATD monocytes persists. This may have implications for combination therapeutics with concerted pharmacological interventions consisting of AAT augmentation therapy to block serine protease C3d production, in combination with specific NLRP3 inhibitors, or alternatively, therapeutic strategies to block Z-AAT polymerisation and ER stress in circulating immune cells.
Acknowledgments
We gratefully acknowledge the Alpha-1 Foundation DNA and Tissue Bank at the University of Florida for providing plasma samples from patients with AATD. We thank patients and healthy volunteers who graciously participated in this study.
Footnotes
@muellerlab
Contributors: DG and EPR conceived and planned the study and designed experiments. MB, EE, NGM and MC consented and recruited healthy volunteers and patients with AATD for sample analysis. DG, HY, RB, AY, LF, CM, EO and PG performed experiments, analysed and, or, interpreted the data. DG and EPR performed quality assurance and wrote the manuscript and EPR is guarantor.
Funding: In support of this work, EPR and PG acknowledge funding from the US Alpha-1 Foundation (Grant # 615848 and Grant # 614218, respectively). EPR acknowledges funding from the Medical Research Charities Group/Health Research Board Ireland (MRCG-2018-04).
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data that support the findings in this study are available from the corresponding author upon request.
Ethics statements
Patient consent for publication
Consent obtained directly from patient(s)
Ethics approval
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the IRB at Icahn School of Medicine at Mount Sinai and Beaumont Hospital Ethics Committee (protocol code Rec#18/52). Informed consent was obtained from all subjects involved in the study. Participating patients cannot be identified.
References
- 1. Bergin DA, Hurley K, McElvaney NG, et al. Alpha-1 Antitrypsin: a potent anti-inflammatory and potential novel therapeutic agent. Arch Immunol Ther Exp (Warsz) 2012;60:81–97. 10.1007/s00005-012-0162-5 [DOI] [PubMed] [Google Scholar]
- 2. Greene CM, Marciniak SJ, Teckman J, et al. Alpha1-Antitrypsin deficiency. Nat Rev Dis Primers 2016;2. 10.1038/nrdp.2016.51 [DOI] [PubMed] [Google Scholar]
- 3. Lomas DA, Evans DL, Finch JT, et al. The mechanism of Z alpha 1-Antitrypsin accumulation in the liver. Nature 1992;357:605–7. 10.1038/357605a0 [DOI] [PubMed] [Google Scholar]
- 4. Carrell RW, Lomas DA. Alpha1-Antitrypsin deficiency--a model for conformational diseases. N Engl J Med 2002;346:45–53. 10.1056/NEJMra010772 [DOI] [PubMed] [Google Scholar]
- 5. Silverman EK, Sandhaus RA. Clinical practice. Alpha1-Antitrypsin deficiency. N Engl J Med 2009;360:2749–57. 10.1056/NEJMcp0900449 [DOI] [PubMed] [Google Scholar]
- 6. Stoller JK, Aboussouan LS. A review of Alpha1-Antitrypsin deficiency. Am J Respir Crit Care Med 2012;185:246–59. 10.1164/rccm.201108-1428CI [DOI] [PubMed] [Google Scholar]
- 7. Chapman KR, Burdon JGW, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe Alpha1 Antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet 2015;386:360–8. 10.1016/S0140-6736(15)60860-1 [DOI] [PubMed] [Google Scholar]
- 8. McElvaney NG, Burdon J, Holmes M, et al. Long-term efficacy and safety of Alpha1 Proteinase inhibitor treatment for emphysema caused by severe Alpha1 Antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med 2017;5:51–60. 10.1016/S2213-2600(16)30430-1 [DOI] [PubMed] [Google Scholar]
- 9. Serban KA, Mikosz A, Strange C, et al. Lectin complement pathway in emphysema. Am J Respir Crit Care Med 2019;199:659–61. 10.1164/rccm.201807-1380LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O’Brien ME, Fee L, Browne N, et al. Activation of complement component 3 is associated with Airways disease and pulmonary emphysema in Alpha-1 Antitrypsin deficiency. Thorax 2020;75:321–30. 10.1136/thoraxjnl-2019-214076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Westall GP, Snell GI, McLean C, et al. C3D and C4D deposition early after lung transplantation. J Heart Lung Transplant 2008;27:722–8. 10.1016/j.healun.2008.03.018 [DOI] [PubMed] [Google Scholar]
- 12. Kokturk N, Khodayari N, Lascano J, et al. Lung inflammation in Alpha-1-Antitrypsin deficient individuals with normal lung function. Respir Res 2023;24:40. 10.1186/s12931-023-02343-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang W, Ni H, Wang H, et al. Nlrp3 Inflammasome is essential for the development of chronic obstructive pulmonary disease. Int J Clin Exp Pathol 2015;8:13209–16. [PMC free article] [PubMed] [Google Scholar]
- 14. Lee J, Mohammad N, Lu Y, et al. Alu RNA induces Nlrp3 expression through Tlr7 activation in Α-1-Antitrypsin–deficient Macrophages. JCI Insight 2022;7:e158791. 10.1172/jci.insight.158791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vorup-Jensen T, Jensen RK. Structural Immunology of complement receptors 3 and 4. Front Immunol 2018;9:2716. 10.3389/fimmu.2018.02716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gabillard-Lefort C, Casey M, Glasgow AMA, et al. Trikafta Rescues CFTR and LOWERS monocyte P2X7R-induced Inflammasome activation in cystic fibrosis. Am J Respir Crit Care Med 2022;205:783–94. 10.1164/rccm.202106-1426OC [DOI] [PubMed] [Google Scholar]
- 17. McElvaney OJ, Zaslona Z, Becker-Flegler K, et al. Specific inhibition of the Nlrp3 Inflammasome as an antiinflammatory strategy in cystic fibrosis. Am J Respir Crit Care Med 2019;200:1381–91. 10.1164/rccm.201905-1013OC [DOI] [PubMed] [Google Scholar]
- 18. Liu T, Zhang L, Joo D, et al. NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2017;2:17023. 10.1038/sigtrans.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grandage VL, Gale RE, Linch DC, et al. Pi3-kinase/AKT is Constitutively active in primary acute myeloid leukaemia cells and regulates survival and Chemoresistance via NF-kB, Mapkinase and P53 pathways. Leukemia 2005;19:586–94. 10.1038/sj.leu.2403653 [DOI] [PubMed] [Google Scholar]
- 20. Paik S, Kim JK, Silwal P, et al. An update on the regulatory mechanisms of Nlrp3 Inflammasome activation. Cell Mol Immunol 2021;18:1141–60. 10.1038/s41423-021-00670-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thastrup O, Cullen PJ, Drøbak BK, et al. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the Endoplasmic Reticulum Ca2 (+)-Atpase. Proc Natl Acad Sci U S A 1990;87:2466–70. 10.1073/pnas.87.7.2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carroll TP, Greene CM, O’Connor CA, et al. Evidence for unfolded protein response activation in monocytes from individuals with Alpha-1 Antitrypsin deficiency. J Immunol 2010;184:4538–46. 10.4049/jimmunol.0802864 [DOI] [PubMed] [Google Scholar]
- 23. Van’t Wout EFA, van Schadewijk A, Lomas DA, et al. Function of monocytes and monocyte-derived Macrophages in Alpha1-Antitrypsin deficiency. Eur Respir J 2015;45:365–76. 10.1183/09031936.00046114 [DOI] [PubMed] [Google Scholar]
- 24. Borel F, Sun H, Zieger M, et al. Editing out five Serpina1 Paralogs to create a mouse model of genetic emphysema. Proc Natl Acad Sci U S A 2018;115:2788–93. 10.1073/pnas.1713689115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thastrup O, Cullen PJ, Drøbak BK, et al. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the Endoplasmic Reticulum Ca2(+)-Atpase. Proc Natl Acad Sci U S A 1990;87:2466–70. 10.1073/pnas.87.7.2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McElvaney OJ, Carroll TP, Franciosi AN, et al. Consequences of abrupt cessation of Alpha1-Antitrypsin replacement therapy. N Engl J Med 2020;382:1478–80. 10.1056/NEJMc1915484 [DOI] [PubMed] [Google Scholar]
- 27. Campos MA, Geraghty P, Holt G, et al. The biological effects of double-dose Alpha-1 Antitrypsin augmentation therapy. Am J Respir Crit Care Med 2019;200:318–26. 10.1164/rccm.201901-0010OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Triantafilou K, Hughes TR, Triantafilou M, et al. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to Nlrp3 Inflammasome activation. J Cell Sci 2013;126:2903–13. 10.1242/jcs.124388 [DOI] [PubMed] [Google Scholar]
- 29. van ’t Wout EFA, Dickens JA, van Schadewijk A, et al. Increased ERK signalling promotes inflammatory signalling in primary airway epithelial cells expressing Z Alpha1-Antitrypsin. hum Mol Genet. Hum Mol Genet 2014;23:929–41. 10.1093/hmg/ddt487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chotikatum S, Naim HY, El-Najjar N. Inflammation induced ER stress affects absorptive intestinal epithelial cells function and integrity. Int Immunopharmacol 2018;55:336–44. 10.1016/j.intimp.2017.12.016 [DOI] [PubMed] [Google Scholar]
- 31. McEnery T, White MM, Gogoi D, et al. Alpha-1 Antitrypsin therapy modifies neutrophil adhesion in patients with obstructive lung disease. Am J Respir Cell Mol Biol 2022;67:76–88. 10.1165/rcmb.2021-0433OC [DOI] [PubMed] [Google Scholar]
- 32. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-ΚB activating pattern recognition and cytokine receptors license Nlrp3 Inflammasome activation by regulating Nlrp3 expression. J Immunol 2009;183:787–91. 10.4049/jimmunol.0901363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kahlenberg JM, Lundberg KC, Kertesy SB, et al. Potentiation of Caspase-1 activation by the P2X7 receptor is dependent on TLR signals and requires NF-ΚB-driven protein synthesis. J Immunol 2005;175:7611–22. 10.4049/jimmunol.175.11.7611 [DOI] [PubMed] [Google Scholar]
- 34. Jeong JS, Lee KB, Kim SR, et al. Airway epithelial Phosphoinositide 3-kinase-Delta contributes to the modulation of fungi-induced innate immune response. Thorax 2018;73:758–68. 10.1136/thoraxjnl-2017-210326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang W, Ba G, Tang R, et al. Ameliorative effect of selective Nlrp3 Inflammasome inhibitor Mcc950 in an Ovalbumin-induced allergic rhinitis murine model. Int Immunopharmacol 2020;83:S1567-5769(19)33085-1. 10.1016/j.intimp.2020.106394 [DOI] [PubMed] [Google Scholar]
- 36. Huppertz C, Jäger B, Wieczorek G, et al. The Nlrp3 Inflammasome pathway is activated in Sarcoidosis and involved in Granuloma formation. Eur Respir J 2020;55:1900119. 10.1183/13993003.00119-2019 [DOI] [PubMed] [Google Scholar]
- 37. Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-Antitrypsin deficiency associated with emphysema. N Engl J Med 1987;316:1055–62. 10.1056/NEJM198704233161704 [DOI] [PubMed] [Google Scholar]
- 38. Hurley K, Lacey N, O’Dwyer CA, et al. Alpha-1 Antitrypsin augmentation therapy corrects accelerated neutrophil apoptosis in deficient individuals. J Immunol 2014;193:3978–91. 10.4049/jimmunol.1400132 [DOI] [PubMed] [Google Scholar]
- 39. McElvaney OF, Fraughen DD, McElvaney OJ, et al. Alpha-1 Antitrypsin deficiency: Current therapy and emerging targets. Expert Rev Respir Med 2023;17:191–202. 10.1080/17476348.2023.2174973 [DOI] [PubMed] [Google Scholar]
- 40. Giles JL, Choy E, van den Berg C, et al. Functional analysis of a complement polymorphism (Rs17611) associated with rheumatoid arthritis. J Immunol 2015;194:3029–34. 10.4049/jimmunol.1402956 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
thorax-2023-221071supp001.pdf (588.5KB, pdf)
thorax-2023-221071supp002.pdf (119.2KB, pdf)
Data Availability Statement
All data that support the findings in this study are available from the corresponding author upon request.