

Abstract
FSAP (Factor VII-activating protease) is a new plasma-derived serine protease with putative dual functions in haemostasis, including activation of coagulation Factor VII and generation of urinary-type plasminogen activator (urokinase). The (auto-)activation of FSAP is facilitated by polyanionic glycosaminoglycans, such as heparin or dextran sulphate, whereas calcium ions stabilize the active form of FSAP. In the present study, extracellular RNA was identified and characterized as a novel FSAP cofactor. The conditioned medium derived from various cell types such as smooth muscle cells, endothelial cells, osteosarcoma cells or CHO (Chinese-hamster ovary) cells contained an acidic factor that initiated (auto-)activation of FSAP. RNase A, but not other hydrolytic enzymes (proteases, glycanases and DNase), abolished the FSAP cofactor activity, which was subsequently isolated by anion-exchange chromatography and unequivocally identified as RNA. In purified systems, as well as in plasma, different forms of natural RNA (rRNA, tRNA, viral RNA and artificial RNA) were able to (auto-)activate FSAP into the two-chain enzyme form. The specific binding of FSAP to RNA (but not to DNA) was shown by mobility-shift assays and UV crosslinking, thereby identifying FSAP as a new extracellular RNA-binding protein, the KD estimated to be 170–350 nM. Activation of FSAP occurred through an RNA-dependent template mechanism involving a nucleic acid size of at least 100 nt. In a purified system, natural RNA augmented the FSAP-dependent Factor VII activation several-fold (as shown by subsequent Factor Xa generation), as well as the FSAP-mediated generation of urokinase. Our results provide evidence for the first time that extracellular RNA, present at sites of cell damage or vascular injury, can serve an important as yet unrecognized cofactor function in haemostasis by inducing (auto-)activation of FSAP through a novel surface-dependent mechanism.
Keywords: blood coagulation, extracellular RNA, Factor VII-activating protease (FSAP), haemostasis, plasma hyaluronan-binding protease
Abbreviations: CHO, Chinese-hamster ovary; DMEM, Dulbecco's modified Eagle's medium; FSAP, Factor VII-activating protease; HBS, Hepes-buffered saline; HCV, hepatitis C virus; HUVEC, human umbilical vein endothelial cell; PPACK, Phe-Pro-Arg-CH2Cl
INTRODUCTION
Upon vascular injury, with the exposure of the subendothelium and deeper layers of the vessel wall, immediate adherence and aggregation of blood platelets prevent life-threatening blood loss and initiate the wound-healing process. Conceptually, vascular wall (extrinsic) components, as well as blood-borne (intrinsic) components, initiate two converging blood coagulation pathways, leading to the production of thrombin. The ‘extrinsic’ coagulation pathway is initiated in a tissue-factor-dependent manner, since this potent coagulation cofactor is not expressed on quiescent endothelium, but in deeper layers of the vessel or in association with activated platelets [1,2], and its contact with blood-borne Factor VII/VIIa is instrumental in the injury-dependent initiation of blood clotting. However, it still remains unclear how this crucial step in the induction of coagulation is initiated.
We have previously described a new haemostasis factor, designated FSAP (Factor VII-activating protease), which appears to be the most effective activator of Factor VII in vitro independent of tissue factor [3], but also promotes activation of (extravascular) pro-urokinase [4,5], as well as high-molecular-mass kininogen [6]. Furthermore, FSAP was found to exhibit Factor VIII-inhibitor bypassing activity [3], thus serving as a multifunctional haemostasis factor that had been previously ignored in this system [7]. Although the liver is the main organ of FSAP expression and production ([8], and H. Trusheim, V. Schmidt and K. T. Preissner, unpublished work), the protein is found at multiple sites in the organism, including the vessel wall (B. Knoblauch, J. Römisch, C. Kannemeier and K. T. Preissner, unpublished work), indicating that its broad distribution via the blood stream could be linked to its multiple functions in different tissues of the body. Moreover, a polymorphism in the protein [9] that is related to a 50% reduction in pro-urokinase-converting activity, without affecting the Factor VII-activating activity, renders this FSAP mutant a reasonable prognostic factor for late atherothrombotic complications [10]. However, information regarding the mode of (auto-)activation of FSAP or its functional properties in these situations is missing.
In order to unravel mechanisms of (auto-)activation of FSAP we took advantage of previous findings that identified FSAP as ‘plasma hyaluronan-binding protein’ [11], as well as previous observations from our laboratory showing that polyanionic components, such as heparin or dextran sulphate, together with calcium ions, were effective in promoting conversion of the proenzyme form of FSAP into the active two-chain serine protease [12]. As most of the initiation or activation reactions in haemostasis take place on (cellular) surfaces, we sought to study the influence of cellular factors on FSAP activation and function. Surprisingly, we identified cell-derived RNA species as potent cofactors for the (auto-)activation of FSAP and its function ex vivo to promote activation of respective proenzyme substrates, including Factor VII and pro-urokinase. These findings allow us to propose a new model for mechanisms of haemostasis enzyme formation following vascular injury or cell damage involving extracellular RNA.
EXPERIMENTAL
Cell culture
HUVECs (human umbilical vein endothelial cells) were isolated and cultivated in endothelial cell basal medium containing 5% fetal calf serum, 10 ng/ml human basic fibroblast growth factor, 500 μg/ml gentamycin and 500 ng/ml amphocetin (Promocell, Heidelberg, Germany) on gelatin-coated dishes, as described previously [13]. HUVECs were used up to the fifth passage and subcultivated using trypsin in a split ratio of 1:5 every 5 days. Wild-type CHO (Chinese-hamster ovary) cells and a glycosaminoglycan-deficient cell line (CHO-745) were cultured in Ham's F-12 medium containing 10% fetal calf serum, 10 mg/ml glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin (Gibco, Paisley, Renfrewshire, Scotland, U.K.). Osteosarcoma cells (MG-63) were cultured in DMEM (Dulbecco's modified Eagle's medium) containing 10% fetal calf serum, 10 mg/ml glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin. Subcultivation of MG-63 and CHO cells was performed using trypsin and a split ratio of 1:5 every 3–5 days. All cell cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
Preparation of cell-conditioned medium
Cell-conditioned medium from different cell cultures mentioned above was prepared as exemplified for confluent HUVEC monolayers. Cells were incubated with serum-free medium for 1 h at 37 °C, washed with HBS (Hepes-buffered saline) at pH 7.4 and incubated with HBS at pH 7.4 or pH 9.0 for 10 min or 24 h respectively. The supernatant was collected, sterile-filtered and stored at −80 °C until used; cells were immediately included in the FSAP cofactor assay. Pretreatment of conditioned media was performed using alternatively 1 unit/ml RNase-free trypsin followed by the addition of 10-fold molar excess of soya-bean trypsin inhibitor or by addition of soya-bean trypsin inhibitor alone, 5 units/ml glycanase, 10 μg/ml DNase or RNase A, respectively, and then tested for the remaining FSAP cofactor activity as described below. Moreover, a butanol or chloroform/methanol (2:1, vol/vol) extract of conditioned medium was prepared, and after evaporation and dissolving in assay buffer, FSAP cofactor activity was measured.
Isolation of cellular RNA and ribosomes
Total RNA was isolated from confluent cell cultures of endothelial, smooth muscle, osteosarcoma and CHO cells, using the RNeasy Kit (Qiagen, Hilden, Germany) or Trizol LS reagent (Invitrogen, Karlsruhe, Germany) according to the manufacturer's instructions. For visual detection, isolated RNA was subjected to electrophoresis on a 1% agarose gel followed by ethidium bromide staining. Total RNA was quantified using a Gene Quant photometer (Amersham Pharmacia). For isolation of ribosomes, confluent dishes of CHO-745 cells were subjected to a ribosome-isolation procedure, as described previously [14]. In each case, two ribosomal preparations were performed in parallel, one in the presence of RNase A and the other in the presence of RNase A inhibitor (Amersham Pharmacia). The ribosome-containing fractions, as well as the isolated RNA thereof, were analysed in the FSAP cofactor assay (see below). RNA-containing samples were also analysed by agarose gel electrophoresis and visualized by ethidium bromide staining; RNA size markers (RNA ladder) were obtained from Fermentas (St. Leon-Rot, Germany). For chemical hydrolysis, 100 μg of total RNA (from CHO cells) was dissolved in 1 ml of water, 100 μl of pre-chilled 2 M NaOH was added and the mixture was incubated for up to 60 min on ice. At various time intervals, 110 μl samples were withdrawn, and the reaction was stopped by adding 10 μl of 2 M acetic acid, followed by ethanol precipitation. Each pellet was dissolved in water and tested for FSAP cofactor activity (see below). Parallel subsamples were subjected to agarose gel electrophoresis and visualized by ethidium bromide staining.
FSAP enzymic assays
FSAP was isolated from human plasma and characterized as described previously [12]. For analysing activation and enzymic activity of FSAP (cofactor assay) in cell culture supernatants or in isolated systems (using different RNA preparations or concentrations of different RNA species as indicated in the Figure legends), the hydrolysis of the chromogenic substrate S-2288 (H-D-Ile-L-Pro-L-Arg-p-nitroanilide dihydrochloride; Chromogenix, Mölndal, Sweden) at a final concentration of 0.3 mM in a final volume of 100 μl was followed over a period of 20 min at 37 °C at A405 in a microplate reader EL 808 (BioTek Instruments, Winooski, VT, U.S.A.). Prior to the assay, wells of a microtitre plate were blocked with 3% BSA, washed, and the test samples were included together with 18.75 nM single-chain FSAP in the reaction mixture at various concentrations dissolved in 20 mM Tris/150 mM NaCl, pH 8.2 [12]. To follow activation and enzymic activity of FSAP directly on cell surfaces, cultures of different cell types, as indicated above, were grown to confluence in wells of a microtitre plate, blocked with serum-free DMEM containing 3% BSA, and 18.75 nM single-chain FSAP was added and the reaction analysed as described above. Also, tumour necrosis factor-α (100 ng/ml) or PMA (100 ng/ml) were used to stimulate cell cultures from which the cell layer or the respective conditioned medium was analysed for FSAP cofactor activity. FSAP cofactor activity was also analysed in 50 μl aliquots of column fractions from affinity chromatography. FSAP activity in different reaction mixtures is expressed as substrate S-2288 turnover (μM/min) at maximal reaction velocity [12].
The formation of Factor Xa, induced by FSAP–rRNA, was measured in the presence of a coagulation factor concentrate (Beriplex P/N 500), including Factors VII, IX, X, and prothrombin (Aventis-Behring, Marburg, Germany), by the use of the chromogenic substrate S-2222 (N-benzoyl-L-Ile-L-Glu-Gly-L-Arg-p-nitroaniline hydrochloride; Chromogenix), as described previously [15]. The activation of pro-urokinase (Saruplase; Medac, Hamburg, Germany) by FSAP was followed over time as described previously [12], with the following modifications: 20 nM pro-urokinase diluted in a total volume of 100 μl TBS buffer, containing 1 mM CaCl2, was incubated with 2 nM single-chain FSAP in the absence or presence of 10 μg/ml dextran sulphate, 100 μg/ml rRNA or 100 μg/ml herring sperm DNA (Sigma), whereby the latter two additives were also pre-incubated with 10 μg/ml RNase A (Roche) at 37 °C for 1 h and tested separately. Generation of urokinase was followed in the presence of 0.2 mM of the chromogenic substrate S-2444 (pyro-L-Glu-Gly-L-Arg-p-nitroanilide hydrochloride; Chromogenix), which is a substrate for urokinase, at 37 °C in a microplate reader.
Anion-exchange chromatography
To isolate components with FSAP cofactor activity, 40 ml of conditioned medium (pH 9.0) from CHO-745 cells was applied on to an anion-exchange Mono-Q HR 5/5 column (1 cm×6 cm; Amersham Pharmacia), equilibrated with HBS at pH 9.0. After washing, bound substances were eluted with a salt gradient (0.1–1 M NaCl) in the same buffer, and 10-ml fractions were collected, desalted and concentrated by centrifugation in a 10 kDa cut-off spin column (Sartorius, Göttingen, Germany) at 3500 g. Protein concentration was determined by a protein assay kit (Pierce, Rockford, IL, U.S.A.). FSAP cofactor activity in the absence or presence of 10 μg/ml RNase A was analysed as described above.
Affinity chromatography
Single-chain FSAP (1 mg) was incubated with 10% 1 M sodium citrate solution, pH 6.0, for 30 min at 22 °C, followed by addition of 1 mM PPACK (D-Phe-Pro-Arg-CH2Cl; Calbiochem-Novabiochem GmbH, Bad Soden, Germany), incubation for 2 h at 22 °C, and dialysis against PBS. Subsequently, 0.25 g of CNBr-Sepharose (Amersham Pharmacia) was used to couple the inactivated two-chain FSAP according to the manufacturer's instructions. For isolation of FSAP cofactor, cell-conditioned medium, as well as isolated RNA from CHO-745 cells, was loaded on to the FSAP-coupled Sepharose, followed by washing with HBS. Bound substances were eluted with 2 M NaCl dissolved in HBS. All fractions were tested for FSAP cofactor activity as described above. Likewise, pro-urokinase or human serum albumin were immobilized to CNBr-Sepharose following the same protocol, and RNA binding was analysed as described before. In each case, eluted RNA fractions were subjected to agarose gel electrophoresis and revealed by ethidium bromide staining.
RNA–protein complexes
HCV (hepatitis C Virus) RNA (nt 1–402) was transcribed in vitro from plasmid pHCV-CUL, linearized with AatII, in the presence of [α-32P]UTP using T7 RNA polymerase. The resulting RNA contains the HCV internal ribosome entry site (nt 1–342) plus the first 60 nt of the HCV core-protein-coding sequence. For obtaining a control DNA fragment using PCR, two DNA oligonucleotides were used to generate an amplified fragment with exactly the same limits (HCV nt 1–402). This fragment was internally labelled using [α-32P]dCTP.
Binding and UV-cross-linking reactions, as described previously [14], were performed in the absence or presence of 10 pmol of FSAP (unless otherwise indicated) with 0.7 pmol of RNA in binding buffer (10 mM Tris/HCl, pH 7.5, containing 100 mM NaCl, 0.3 mM MgCl2, 2% glycerol and 1 mM dithiothreitol) in a 10-μl reaction volume. Protease inhibitors (50 units/ml aprotinin, 10 mM PMSF and 0.05 mM ZnCl2) were added as indicated. Reaction mixtures were incubated for 10 min at 30 °C, irradiated with UV light (254 nm) for 30 min at 0 °C and treated with 2 mg/ml RNase for 60 min at 37 °C. After addition of one-third volume of loading buffer (125 mM Tris/HCl, pH 6.8, containing 4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.02% Bromophenol Blue and 7 M urea), samples were boiled and subjected to SDS/PAGE (12% gel) followed by autoradiography. Alternatively, FSAP was UV cross-linked to [α-32P]UTP-labelled HCV RNA as described above, with the difference that samples were not treated with RNase A after UV cross-linking, but incubated in the absence or presence of proteinase K. After addition of loading buffer, samples were analysed by SDS/PAGE (6% gel) followed by autoradiography.
To analyse FSAP–RNA complexes by electrophoretic mobility-shift assay, binding reactions contained 0.15 pmol of [α-32P]UTP-labelled HCV RNA in 5 mM Hepes buffer, pH 7.4, containing 145 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 3.8% glycerol, 2 mM dithiothreitol and 0.1 mM EDTA, in a volume of 10 μl. Protease inhibitors (50 units/ml aprotinin, 10 mM PMSF and 0.05 mM ZnCl2) were added as indicated. Reaction mixtures were incubated for 20 min at 30 °C. After addition of 5 μl of 20% glycerol, samples were resolved on a non-denaturing 6% polyacrylamide gel [16]. In control experiments, a DNA fragment instead of RNA was used, which was generated by asymmetric PCR from the same sequence as the RNA transcript and labelled internally during the PCR reaction using [α-32P]dCTP.
To analyse FSAP–RNA complexes by filter-binding assays, nitrocellulose filters were soaked with washing buffer (20 mM Tris/HCl, pH 7.25, 2 mM MgCl2 and 1 mM dithiothreitol) and fixed into a slot-blot apparatus. The indicated FSAP concentrations were incubated on the filters in 100 μl of wash buffer for 15 min and then aspirated through the filters, followed by incubation with 3% BSA for 20 min. Binding buffer (100 μl; 10 mM Tris/HCl, pH 7.25, 145 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 3.8% glycerol, 2 mM dithiothreitol and 0.1 mM EDTA) containing 0.15 pmol of [α-32P]UTP-labelled HCV RNA was incubated with each filter well for 15 min, and 1 mM PPACK was present for binding assay of single-chain FSAP. The reaction mixtures were aspirated through the filters, followed by four wash cycles, and after drying filter pieces corresponding to the slots were excised, added to 2 ml of scintillation fluid and radioactivity measured in a scintillation counter. The values shown represent means±S.D. from 3–5 measurements. For estimation of the dissociation constant KD for the reaction
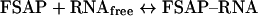 |
equation (1) was used:
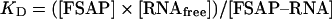 |
(1) |
Equation (2) is fulfilled if 50% binding of RNA to FSAP is achieved, thus obtaining KD=[FSAP].
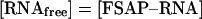 |
(2) |
The respective dissociation constants for single- and two-chain FSAP were derived from the sigmoidal binding curves at 50% RNA–FSAP complex formation (see Figure 5D).
Figure 5. Binding interactions between FSAP and RNA.

(A) Purified human single-chain FSAP (scFSAP) was incubated at 30 °C for 10 min in the absence or presence of [α-32P]UTP-labelled HCV RNA and in the absence or presence of protease inhibitors as indicated. Samples were irradiated with UV light (254 nm), treated with 2 mg/ml RNase A at 37 °C for 60 min, denatured and separated by denaturing SDS/PAGE (12% gel) which was stained with Coomassie Brilliant Blue (left-hand panel). For detection of protein bound to radioactive RNA, X-ray film was exposed overnight (right-hand panel). Size markers indicate positions of 14C-labelled marker proteins at the left margin. Binding of RNA to two-chain FSAP (B) or single-chain FSAP (C) was analysed in an electrophoretic mobility-shift assay. [α-32P]UTP-labelled HCV RNA (15 nM) was incubated in the absence (lanes 1) or presence of FSAP at the following concentrations: 0.03 μM (lanes 2), 0.1 μM (lanes 3), 0.2 μM (lanes 4), 0.3 μM (lanes 5), 1 μM (lanes 6), 2 μM (lanes 7), and RNA–two-chain FSAP (B) or RNA–single-chain FSAP complexes (C) were separated on a non-denaturing 6% polyacrylamide gel followed by exposure of the dried gel to visualize the RNA-containing bands. (D) Filter-binding assay with FSAP and HCV RNA in the presence (continuous curve) or absence (broken curve) of protease inhibitor (for details, see the Experimental section). The KD values obtained from the binding curves at 50% maximal binding are indicated (broken lines).
RESULTS
In order to test the influence of conditioned cell media from different cell cultures, as well as their influence itself on FSAP, the (auto-)activation of the isolated proenzyme was followed in the presence of these additives in a chromogenic substrate assay. As compared with medium alone, conditioned media from different cell types, including HUVEC, MG63, CHO or CHO-745 cells, increased FSAP activity several-fold (Figure 1), regardless whether the conditioned media were prepared at pH 7.4 or pH 9.0. The conditioned media-derived FSAP cofactor activity was retained after dialysis and was resistant to heat denaturation (Table 1). Likewise, in the presence of washed intact cell cultures, an equivalent elevation of FSAP activity was observed (Figure 1). These results indicate that, independent of the cell type, cell-derived conditioned media, as well as cellular surfaces, contain cofactor(s) that induce (auto-)activation of FSAP, reminiscent of the effect of heparin or dextran sulphate, as reported previously [12].Treatment of, for example, HUVEC cultures with PMA or tumour necrosis factor-α, which both increase the fraction of apoptotic cells, did not elevate the content of FSAP cofactor activity in the respective supernatants (results not shown).
Figure 1. FSAP cofactor activity in cell-conditioned media and on cell surfaces.

Isolated human single-chain FSAP was incubated with cell-conditioned media from different cell types as indicated (grey bars), as well as on washed cultures of the same cells (black bars), and FSAP protease activity as compared with buffer control was measured by amidolytic assay using the chromogenic substrate S-2288. The results are expressed as the means±S.D. (n=3), and a representative experiment out of four is shown.
Table 1. Identification of the FSAP cofactor from cell-conditioned medium.
Conditioned medium from CHO-745 cells was treated as indicated and subsequently tested for FSAP cofactor activity as compared with buffer control (representing 100%) containing each of the additives indicated. The results are representative of one experiment out of four.
| Biomolecule classification | Treatment of cell-conditioned medium | FSAP activity (% of control) |
|---|---|---|
| Protein | Protease inhibitors | 350 |
| Heat denaturation | 350 | |
| Trypsin, soya-bean trypsin inhibitor | 450 | |
| Carbohydrate | Glycosidase | 200 |
| Neuraminidase | 250 | |
| Lipid | Butanol extract | 200 |
| Methanol/chloroform extract | 200 | |
| Nucleic acid | Dnase | 450 |
| RNase A | 100 |
In order to identify the respective biomolecular nature of FSAP cofactor substance(s), conditioned medium from CHO-745 cells (that are devoid of heparinoid components) was subjected to various treatments to allow hydrolysis/loss of cofactor activity. Neither incubation with trypsin, protease inhibitors, such as benzamidine, PMSF, treatment with glycosidases (heparinase or heparitinase) or neuraminidase, extraction of lipids using butanol or methanol/chloroform nor treatment with DNase totally destroyed FSAP cofactor activity. Alone, treatment of conditioned medium with RNase A resulted in the loss of cofactor activity, indicating that extracellular RNA provides a new putative cofactor for (auto-)activation of FSAP (Table 1). These results were corroborated by comparing the FSAP cofactor activity of pH 7.4 conditioned medium from CHO (or endothelial) cells before and after treatment with RNase A (results not shown). In the latter case, no augmentation of FSAP activation was noted, indicating that the predominant cofactor activity is very likely to be derived from extracellular RNA. Moreover, addition of RNase A to different cell types shown above resulted in the complete prevention of cell surface-dependent elevation of FSAP activity, as seen in Figure 1.
To further isolate and characterize the respective cofactor(s) and to substantiate our primary findings, conditioned medium from CHO-745 cells was subjected to anion-exchange chromatography (Figure 2A), Following medium application to and the washing of column with physiological ionic strength buffer, the cofactor activity was eluted along a sodium chloride gradient at approx. 0.5 M NaCl in fraction 5. Both, the applied conditioned medium, as well as fraction 5, contained appreciable amounts of nucleic acid material, as revealed by ethidium bromide staining following agarose gel electrophoresis (molecular size range 100–2000 nt) (Figure 2A). In parallel, functional FSAP cofactor activity was determined as described above, and unfractionated medium, fraction 5, as well as authentic RNA isolated from CHO-745 cells, showed an equivalent potential for FSAP (auto-)activation when comparable amounts of RNA were applied. Despite the lower content of RNA seen in the conditioned medium, additional factors that promote FSAP activation may be present. Moreover, pre-treatment of conditioned medium, fraction 5 or isolated RNA with RNase A almost completely abolished this FSAP cofactor activity in all cases (Figure 2B). Together, these results strongly indicate that the new FSAP cofactor contains authentic RNA either present in cell conditioned media or on the surface of cell cultures.
Figure 2. Isolation of FSAP cofactor from conditioned medium of CHO-745 cells.

(A) Conditioned medium (CM) of CHO-745 cells was applied on to a HR5/5 Mono Q-Sepharose column, equilibrated in HBS, pH 9.0, and after the flow-through fraction (F) and the wash fraction (W) were eluted, the cofactor was desorbed with an NaCl gradient and enriched in fraction 5 as indicated. From the same volume of each sample, RNA was isolated and subjected to electrophoresis on 1% agarose gel and revealed by ethidium bromide staining. RNA size markers are shown along the left margin. (B) FSAP cofactor activity (similar amounts of RNA in all samples were applied) present in conditioned medium (CM) and the eluted fraction 5 was compared with authentic RNA isolated from CHO-745 cells in the absence (filled bars) or after pre-incubation with 10 μg/ml RNase A (hatched bars) using the chromogenic substrate assay. Results are expressed as the mean values of duplicate determinations and a representative experiment out of three is shown.
To confirm these findings we continued to isolate and characterize RNA from different sources in order to characterize its behaviour as FSAP cofactor, both from a structural and a functional point of view. As the starting material, either lysate or intact ribosomes from CHO-745 cells were taken, and when both fractions were tested for FSAP cofactor activity in the presence of an RNase inhibitor, an appreciable elevation of FSAP activity was demonstrated (Figure 3). Moreover, pre-incubation of both fractions with RNase A resulted in a complete loss of cofactor activity for the ribosomal fraction, whereas a partial, but significant, reduction in activity was seen for cell lysate (Figures 3A and 3C). Following isolation of RNA from both fractions, the RNA-dependent cofactor activity was retained in the presence of RNase inhibitor, whereas incubation with RNase A lead to complete loss of FSAP cofactor activity in both fractions, as expected (Figures 3B and 3D). These results indicate that RNA from intracellular sources can serve as cofactor for FSAP (auto-)-activation and that a certain size of RNA is required for this effect. In addition to natural rRNA from eukaryotic cells, tRNA from yeast, RNA from Escherichia coli (results not shown) or HCV, artificial RNA, such as poly(I-C) or poly(A-U), but not the DNA homologue poly(dI-dC), presented a similar and comparable cofactor activity for FSAP (see below).
Figure 3. RNA constitutes the FSAP cofactor activity present in cell lysate and ribosomes.

Ribosomes (A) and cell lysate (C) derived from CHO-745 cells in the presence of RNase inhibitor, as well as total RNA isolated from ribosomes (B) and cell lysate (D), were tested for FSAP cofactor activity in the absence (filled bars) or after pre-incubation with 10 μg/ml RNase A (hatched bars). In all cases, the respective RNA concentration was adjusted to 50 μg/ml. The results are expressed as the means±S.D. of three independent experiments. The isolated RNA preparations in both cases in the absence (lanes 1) or presence of RNase A (lanes 2) were analysed by electrophoresis on 1% agarose gels and revealed by ethidium bromide staining as indicated. RNA size markers, as well as the position of the 28 S and 18 S RNA, are indicated on the right-hand side.
To provide conclusive evidence that both FSAP and RNA can interact directly with each other, RNA isolated from CHO-745 cells was applied to immobilized FSAP, human serum albumin or urokinase respectively. For the latter two proteins, the majority of unbound RNA had already left each column together with the flow through and washing fractions, whereas RNA subjected to immobilized FSAP could only be eluted with 2 M NaCl (Figure 4A). To substantiate these findings, FSAP was allowed to bind to in vitro-synthesized 32P-labelled RNA of 402 nt from HCV and was then irradiated with UV light to induce covalent cross-links between RNA and protein. In contrast to the commonly used UV cross-linking assay (see below), excess RNA was not degraded with RNase after UV cross-linking, but the RNA and RNA–protein complexes were resolved on a low percentage SDS/polyacrylamide gel under reducing and denaturing conditions. In the absence of protein, free RNA migrated with an apparent molecular mass of approx. 115 kDa, i.e. with approx. 90% of its calculated mass of 128 kDa (Figure 4B). In the presence of FSAP, an additional band of approx. 175 kDa appeared, corresponding to an increase in molecular mass of approx. 60 kDa (representing the size of FSAP). Following treatment with proteinase K, this RNA–protein complex completely disappeared. When this UV cross-linking experiment was performed with a radiolabelled DNA fragment amplified from the mentioned HCV RNA template, no DNA–FSAP complex was seen (Figure 4B), indicating that only RNA, but not DNA, is able to interact directly with FSAP.
Figure 4. Specific binding of RNA to FSAP.

(A) RNA isolated from CHO-745 cells (lanes 1) was applied on to immobilized FSAP, human serum albumin (HSA) or pro-urokinase, and after elution of the flow-through (lanes 2) and wash fractions (lanes 3) each column was eluted with 2 M NaCl (lanes 4). RNA was isolated from the same volume of each fraction and analysed by electrophoresis on 1% agarose gels and revealed by ethidium bromide staining. RNA size markers, as well as the position of the 28 S and 18 S RNA, are indicated at the left- and right-hand sides respectively. (B) The formation of a distinct FSAP–RNA complex was analysed by incubation of [α-32P]UTP-labelled HCV RNA (lanes 1–3) or as control of [α-32P]CTP-labelled DNA (lanes 4–6) in the absence (lanes 1 and 4) or presence of FSAP (lanes 2 and 3, 5 and 6) followed by irradiation with UV light (254 nm) in the absence of RNase. In lanes 3 and 6, the reaction mixtures were treated with proteinase K after UV irradiation. Following denaturation, samples were separated by denaturing SDS/PAGE (8% gel), followed by autoradiography; radiolabelled size markers are indicated at the right-hand side.
To investigate the RNA-binding properties of FSAP in more detail, we performed a standard UV cross-linking assay, including RNase digestion (Figure 5). When FSAP was incubated in the absence of RNA, no FSAP cleavage occurred, whereas in the presence of RNA, single-chain FSAP was converted into the two-chain form. In the presence of protease inhibitors, however, cleavage of single-chain FSAP was largely inhibited, and RNA binding was detected mainly to uncleaved single-chain FSAP, but also to the heavy and light chains. In contrast, enzymically active two-chain FSAP only bound RNA via the heavy chain that was covalently marked by radiolabelled RNA in the UV cross-linking assay (Figure 5A). This indicates that within the uncleaved single-chain proenzyme, the heavy-chain and the light-chain moiety may have the potential to come into contact with RNA, whereas in a conformationally altered two-chain FSAP, the heavy chain of the molecule contains the dominant RNA-binding site. These considerations are consistent with a template mechanism in which two molecules of FSAP (proenzyme and enzyme) are brought together on the same RNA by differential binding (see below).
To obtain more quantitative information about the binding interactions between FSAP and RNA, an electrophoretic mobility-shift assay was employed (Figures 5B and 5C). To avoid proteolysis of single-chain FSAP during the assay, protease inhibitors together with zinc ions were included in the reaction mixture, the latter being a potent inhibitor of FSAP (auto-)degradation (F. Nakazawa, C. Kannemeier, J. Römisch and K. T. Preissner, unpublished work). At plasma concentrations of FSAP of 0.2 μM [12], all RNA was bound within FSAP–RNA complexes, indicating that FSAP may bind RNA at a threshold close to the FSAP plasma concentration. At higher FSAP concentrations, migration of the FSAP–RNA complexes was even more retarded (Figures 5B and Figures 5C; lanes 5 to 7), indicating that more than one molecule of FSAP may bind to a single RNA template. The dissociation contant KD of the binding of RNA to single-chain FSAP (performed in the presence of protease inhibitors to prevent activation) (Figure 5D, continuous curve) amounted to 170 nM, whereas the KD for the two-chain FSAP–RNA interaction is 350 nM (Figure 5, broken curve). These values correspond well with the FSAP concentrations required for binding of RNA in the shift assays and are compatible with a template mechanism of activation as well.
On the basis of the findings described above that RNA may provide a template for FSAP (auto-)activation and that complete degradation of RNA resulted in loss of FSAP cofactor activity, additional experiments were performed to corroborate these implications. NaOH hydrolysis of RNA resulted in a time-dependent decrease in size of the nucleic acid with a concomitant loss of its cofactor activity. An RNA fragment size below 100 nt may not serve as an effective cofactor anymore (Figures 6A and 6B). Furthermore, the bell-shaped curve that was obtained for the RNA-concentration-dependent cofactor activity (using total RNA from yeast or CHO cells) supported the previous assumption that a template mechanism is operative for RNA-triggered FSAP (auto-)activation (Figure 6C). As the previous data already implicated an RNA-augmented conversion of FSAP proenzyme single-chain form into the two-chain active molecule, the time-dependent activation/fragmentation of FSAP by RNA was followed over time in the presence of calcium ions to stabilize the active form of the enzyme. The activation profile induced by tRNA was comparable with the one obtained with heparin [12], with a maximum activity between 10 and 30 min followed by a partial decline (Figure 6D). The related time-dependent biphasic fragmentation pattern of FSAP induced by RNA (auto-activation/degradation) was in agreement with the activity kinetics (results not shown).
Figure 6. FSAP activation by an RNA-dependent template mechanism.

RNA was chemically hydrolysed by NaOH, and aliquots of the reaction mixture were withdrawn at the indicated time points and analysed by 1% agarose gel electrophoresis (A), followed by staining with ethidium bromide. RNA size markers, as well as 28 S and 18 S RNA, are indicated at the left margin. (B) In parallel, aliquots of the reaction mixture were analysed for FSAP cofactor activity as described. For comparison, FSAP activation was carried out by dextran sulphate (DXS). The results are expressed as the means±S.D. (n=3), and a representative experiment out of three is shown. (C) The (auto-)activation of single-chain FSAP (18.75 nM) was followed in the absence (0) or presence of the indicated concentrations of CHO cell total RNA (open bars) or yeast total RNA (closed bars), as described in the Experimental section. Note the bell-shaped concentration dependence, characteristic of a template mechanism. (D) The activation of single-chain FSAP (18.75 nM) by 100 μg/ml tRNA (△) or 5 units/ml heparin (■) in the presence of 1 mM CaCl2 was followed over time by a chromogenic substrate assay using S-2288. Samples were taken at the indicated time points and the values (corrected for FSAP activity in the absence of additives) are expressed as the means±S.D. (n=3), and a representative experiment out of three is shown.
Finally, the ability of RNA to serve as novel cofactor for FSAP function was tested in two proenzyme activation assays. In a purified system containing the coagulation proenzymes Factors VII, IX, X and prothrombin, FSAP alone induced only a moderate generation of Factor Xa, possibly due to some active two-chain FSAP present in the reaction mixture (Figure 7A). Moreover, in the presence of rRNA (or artificial RNA as well, results not shown), a significant elevation of Factor VII activiation, reflected by an increase in Factor Xa generation, was observed. The cofactor activity of RNA was prevented by pre-incubation of the RNA with RNase. This system is solely dependent on the property of FSAP to activate Factor VII, since Factors IX, X or prothrombin do not serve as protein substrates for FSAP [3,7]. Likewise, in a time-dependent biphasic manner, FSAP, together with rRNA (but not DNA or degraded rRNA), promoted the generation of urokinase, comparable with the cofactor function of dextran sulphate [12] (Figure 7B), and this effect was significantly higher as compared with FSAP alone. Based on these findings we propose a function of extracellular RNA as a novel cofactor in the (auto-)activation of FSAP with subsequent functional consequences for macromolecular FSAP substrates.
Figure 7. FSAP activation by RNA and functional consequences.

(A) The influence of FSAP (18.75 nM) alone or together with rRNA (50 μg/ml, isolated from CHO-745 cells) for Factor Xa formation in vitro was analysed with coagulation factor concentrate (containing Factors VII, IX, X and prothrombin), dependent on Factor VIIa generation. Where indicated, the rRNA was pre-incubated for 30 min with RNase (100 μg/ml) prior to the assay procedure. The results are expressed as the means±S.D. (n=3), and a representative experiment out of five is shown. (B) The ability of 2 nM FSAP to activate 20 nM pro-urokinase in the absence of additives (◆) or in the presence of 10 μg/ml dextran sulphate (▲), 100 μg/ml rRNA (■), 100 μg/ml herring sperm DNA (○) or 100 μg/ml RNA pre-treated with 10 μg/ml RNaseA (●) was followed over time in a chromogenic substrate S-2444 assay. Values are corrected for FSAP-induced urokinase activity in the presence of RNase A alone. The results are expressed as the means+S.D. (n=3) of a typical experiment out of three.
DISCUSSION
In the present study a novel non-protein enzyme cofactor was discovered, identified and characterized as extracellular RNA that serves as negatively charged surface to promote the (auto-)-activation of FSAP, with putative dual functions in the haemostatic system [7]. This is the first report describing a formerly intracellular component possibly involved in the extracellular initiation or regulation of haemostasis. In particular, the affinity of FSAP for negatively charged components, such as hyaluronic acid, heparin or dextran sulphate, led us to search for additional factors that may promote FSAP activation in the presence of cells or cell-derived material. Here, we made the surprising observation that conditioned media from different cell types, as well as cellular surfaces themselves, were able to significantly promote (auto-)-activation of FSAP, indicative of a cell-derived factor whose nature was subsequently defined as RNA by several methodological approaches.
In order to corroborate these observations, total RNA from CHO-745 cells that lack the expression of heparan sulphate moiety (and thus may not interfere with FSAP cofactor function) [17] was isolated and tested together with ribosomes from the same cells for FSAP cofactor function. In both cases RNase-sensitive material was identified to be the principle cofactor in the (auto-)-activation of FSAP. In addition to rRNA, which constitutes the majority of total RNA isolated from eukaryotic cells, tRNA from yeast or bacterial RNA from E. coli, as well as viral RNA from HCV and artificial RNA [such as poly(I-C)], but not DNA, were active as FSAP cofactor. Collectively, these data strongly indicate that certain common structural requirements of different RNA species, such as nucleotide composition/sequence or secondary structure (including single- and double-stranded regions), appear to be necessary to confer cofactor function.
To characterize further the direct interaction between RNA and FSAP, specific binding of the proenzyme (but not of the structurally related pro-urokinase or albumin respectively) could be documented by affinity adsorption, as well as by mobility-shift assays following UV cross-linking. Moreover, the estimated value of the dissociation constant for the FSAP–RNA interaction was found to be within the range of the plasma concentration of the proenzyme, indicating that binding of RNA to FSAP could very well occur in vivo. Based on cross-linking experiments, single-chain FSAP provides multiple contacts with RNA, whereas the active two-chain enzyme binds to RNA exclusively via its heavy-chain (with approx. 50% affinity as compared with the single-chain protein), indicative of the fact that both forms of FSAP can interact with RNA in a complex that leads to (auto-)-activation of the proenzyme. This mechanistic interpretation is supported by the RNA dose–response and RNA hydrolysis data, whereby the (auto-)activation of FSAP occurs on an RNA template with a minimal size of about 100 nt. Work is in progress to characterize the RNA-binding domain(s) in FSAP, however, the protein does not contain typical sequence motifs found in intracellular RNA-binding proteins [18].
The ability of RNA to promote the FSAP-dependent activation of its macromolecular substrates Factor VII or pro-urokinase provides further evidence and underlines the contention that (auto-)-activation of FSAP by extracellular RNA may very well contribute to mechanisms of proenzyme conversion in relation to induction of haemostatic reactions. Here, cell-derived RNA that may become exposed to or released into the extracellular environment during vascular injury, cell damage or during apoptosis could serve as a natural cofactor for FSAP, particularly at tissue sites that are devoid of (plasma) RNases or where a transient excess of RNA may be available. Interestingly, observations from our laboratory indicate that appreciable amounts of extracellular RNA can be detected in plasma of patients suffering from acute myocardial infarction or sepsis (H. Trusheim, V. Schmidt, A. Shibamiya and K. T. Preissner, unpublished work), and different mRNA species have been amplified by PCR in plasma of patients with tumours [19,20], indicative of increased cellular turnover or tissue damage. Yet, we were unable so far to relate the content of extracellular RNA in these plasma samples with an increased potential for FSAP activation, possibly due to the excess of RNases or the presence of several as yet unidentified RNA-binding proteins. Interestingly, endothelial cells, in particular, were recently shown to produce and secrete RNase A homologue (RNase 1) in appreciable amounts, whereas a series of other cell types in culture do not [21]. Although speculative at the moment, the endothelial cell-specific expression and secretion of RNase may very well contribute to vascular homoeostasis in general, and be a strong counter-player for the putative cofactor role of cell-derived extracellular RNA in haemostatic reactions described here.
As to the physiological relevance of our observations about RNA as a novel FSAP cofactor in comparison with dextran sulphate [12], we propose that specific polyanionic substances in close proximity either to intact (such as dextran sulphate) or to damaged (such as RNA) cells serve as potent cofactors in the induction of FSAP-mediated proenzyme activation. It remains to be shown in which way FSAP contributes to the generation of intravascular Factor VIIa and/or the extravascular production of urokinase in vivo.
Acknowledgments
We are grateful to the excellent and skilful technical assistance of Veronika Schmidt, Thomas Schmidt-Wöll, Berid Altmann, Arno Stöhr and Annette Feussner. We also thank Dr Jürgen Römisch, Dr Thomas Weimer and Dr Sandip M. Kanse for many helpful suggestions, and we kindly acknowledge the continuous support of Professor Ryuichi Kamiyama (Tokyo, Japan). This project was supported in part by grants (SFB 535, SFB 547, GRK 370, GRK 534) from the Deutsche Forschungsgemeinschaft (Bonn, Germany) and by Aventis Behring (Marburg, Germany). F. N. is a recipient of an Ishidu Shun Memorial Scholarship.
References
- 1.Engelmann B., Luther T., Müller I. Intravascular tissue factor pathway – a model for rapid initiation of coagulation within the blood vessel. Thromb. Haemostasis. 2003;89:3–8. [PubMed] [Google Scholar]
- 2.Neuenschwander P. F., Fiore M. M., Morrissey J. H. Factor VII autoactivation proceeds via interaction of distinct protease–cofactor and zymogen–cofactor complexes. Implications of a two-dimensional enzyme kinetic mechanism. J. Biol. Chem. 1993;268:21489–21492. [PubMed] [Google Scholar]
- 3.Römisch J., Feussner A., Vermohlen S., Stöhr H. A. A protease isolated from human plasma activating factor VII independent of tissue factor. Blood Coagul. Fibrinolysis. 1999;10:471–479. [PubMed] [Google Scholar]
- 4.Etscheid M., Hunfeld A., Konig H., Seitz R., Dodt J. Activation of proPHBSP, the zymogen of a plasma hyaluronan binding serine protease, by an intermolecular autocatalytic mechanism. Biol. Chem. 2000;381:1223–1231. doi: 10.1515/BC.2000.150. [DOI] [PubMed] [Google Scholar]
- 5.Römisch J., Vermohlen S., Feussner A., Stöhr H. The FVII activating protease cleaves single-chain plasminogen activators. Haemostasis. 1999;29:292–299. doi: 10.1159/000022515. [DOI] [PubMed] [Google Scholar]
- 6.Etscheid M., Beer N., Fink E., Seitz R., Johannes D. The hyaluronan-binding serine protease from human plasma cleaves HMW and LMW kininogen and releases bradykinin. Biol. Chem. 2002;383:1633–1643. doi: 10.1515/BC.2002.184. [DOI] [PubMed] [Google Scholar]
- 7.Römisch J. Factor VII activating protease (FSAP): a novel protease in hemostasis. Biol. Chem. 2002;383:1119–1124. doi: 10.1515/BC.2002.121. [DOI] [PubMed] [Google Scholar]
- 8.Choi-Miura N. H., Otsuyama K., Sano Y., Saito K., Takahashi K., Tomita M. Hepatic injury-specific conversion of mouse plasma hyaluronan binding protein to the active hetero-dimer form. Biol. Pharm. Bull. 2001;24:892–896. doi: 10.1248/bpb.24.892. [DOI] [PubMed] [Google Scholar]
- 9.Römisch J., Feussner A., Nerlich C., Stoehr H. A., Weimer T. The frequent Marburg I polymorphism impairs the pro-urokinase activating potency of the factor VII activating protease (FSAP) Blood Coagul. Fibrinolysis. 2002;13:433–441. doi: 10.1097/00001721-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Willeit J., Kiechl S., Weimer T., Mair A., Santer P., Wiedermann C. J., Römisch J. Marburg I polymorphism of factor VII-activating protease: a prominent risk predictor of carotid stenosis. Circulation. 2003;107:667–670. doi: 10.1161/01.cir.0000055189.18831.b1. [DOI] [PubMed] [Google Scholar]
- 11.Choi-Miura N. H., Tobe T., Sumiya J., Nakano Y., Sano Y., Mazda T., Tomita M. Purification and characterization of a novel hyaluronan-binding protein (PHBP) from human plasma: it has three EGF, a kringle and a serine protease domain, similar to hepatocyte growth factor activator. J. Biochem. (Tokyo) 1996;119:1157–1165. doi: 10.1093/oxfordjournals.jbchem.a021362. [DOI] [PubMed] [Google Scholar]
- 12.Kannemeier C., Feussner A., Stöhr H. A., Weisse J., Preissner K. T., Römisch J. Factor VII and single-chain plasminogen activator-activating protease: activation and autoactivation of the proenzyme. Eur. J. Biochem. 2001;268:3789–3796. doi: 10.1046/j.1432-1327.2001.02285.x. [DOI] [PubMed] [Google Scholar]
- 13.Kanse S. M., Benzakour O., Kanthou C., Kost C., Lijnen H. R., Preissner K. T. Induction of vascular SMC proliferation by urokinase indicates a novel mechanism of action in vasoproliferative disorders. Arterioscler. Thromb. Vasc. Biol. 1997;17:2848–2854. doi: 10.1161/01.atv.17.11.2848. [DOI] [PubMed] [Google Scholar]
- 14.Ochs K., Zeller A., Saleh L., Bassili G., Song Y., Sonntag A., Niepmann M. Impaired binding of standard initiation factors mediates poliovirus translation attenuation. J. Virol. 2003;77:115–122. doi: 10.1128/JVI.77.1.115-122.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müller I., Klocke A., Alex M., Kotzsch M., Luther T., Morgenstern E., Zieseniss S., Zahler S., Preissner K., Engelmann B. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003;17:476–478. doi: 10.1096/fj.02-0574fje. [DOI] [PubMed] [Google Scholar]
- 16.Konarska M. M., Sharp P. A. Electrophoretic separation of complexes involved in the splicing of precursors to mRNAs. Cell. 1986;46:845–855. doi: 10.1016/0092-8674(86)90066-8. [DOI] [PubMed] [Google Scholar]
- 17.Murphy-Ullrich J. E., Westrick L. G., Esko J. D., Mosher D. F. Altered metabolism of thrombospondin by Chinese hamster ovary cells defective in glycosaminoglycan synthesis. J. Biol. Chem. 1988;263:6400–6406. [PubMed] [Google Scholar]
- 18.Burd C. G., Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 19.Tsui N. B., Ng E. K., Lo Y. M. Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin. Chem. 2002;48:1647–1653. [PubMed] [Google Scholar]
- 20.Ng E. K., Tsui N. B., Lam N. Y., Chiu R. W., Yu S. C., Wong S. C., Lo E. S., Rainer T. H., Johnson P. J., Lo Y. M. Presence of filterable and nonfilterable mRNA in the plasma of cancer patients and healthy individuals. Clin. Chem. 2002;48:1212–1217. [PubMed] [Google Scholar]
- 21.Landre J. B., Hewett P. W., Olivot J. M., Friedl P., Ko Y., Sachinidis A., Moenner M. Human endothelial cells selectively express large amounts of pancreatic-type ribonuclease (RNase 1) J. Cell. Biochem. 2002;86:540–552. doi: 10.1002/jcb.10234. [DOI] [PubMed] [Google Scholar]