

ABSTRACT
Introduction
Lung adenocarcinoma (LUAD) is one of the major histopathological types of non‐small cell lung cancer (NSCLC), including solid, acinar, lepidic, papillary and micropapillary subtypes. Increasing evidence has shown that micropapillary LUAD is positively associated with a higher percentage of driver gene mutations, a higher incidence of metastasis and a poorer prognosis, while lepidic LUAD has a relatively better prognosis. However, the novel genetic change and its underlying mechanism in the progression of micropapillary LUAD have not been exactly determined.
Methods
A total of 181 patients with LUAD who underwent surgery at the First Affiliated Hospital of Huzhou University from January 2020 to December 2022 were enrolled. Three predominant lepidic and three predominant micropapillary LUAD tissue samples were carried out using whole‐exome sequencing. Comprehensive analysis of genomic variations and the difference between lepidic and micropapillary LUAD was performed. In addition, the TMEM229A Q200del mutation was verified using our cohort and TCGA‐LUAD datasets. The correlations between the TMEM229A Q200del mutation and the clinicopathological characteristics of patients with LUAD were further analyzed. The functions and mechanisms of TMEM229A Q200del on NSCLC cell proliferation and migration were also determined.
Results
The frequency of genomic changes in patients with micropapillary LUAD was higher than that in patients with lepidic LUAD. Mutations in EGFR, ATXN2, C14orf180, MUC12, NOTCH1, and PKD1L2 were concomitantly detected in three predominant micropapillary and three predominant lepidic LUAD cases. The TMEM229A Q200del mutation was only mutated in lepidic LUAD. Additionally, the TMEM229A Q200del mutation had occurred in 16 (8.8%) patients, and not found TMEM229A R76H and M346T mutations in our cohort, while TMEM229A mutations (R76H, M346T, and Q200del) occurred only in 1.0% of the TCGA‐LUAD cohort. Further correlation analysis between the TMEM229A Q200del mutation and clinicopathological characteristics suggested that a lower frequency of the Q200del mutation was significantly associated with positive lymph node metastasis, advanced TNM stage, positive cancer thrombus, and pathological features. Finally, overexpression of TMEM229A Q200del suppressed NSCLC cell proliferation and migration in vitro. Mechanistically, overexpression of TMEM229A and TMEM229A Q200del both reduced the expression level of phosphorylated (p)‐ERK and p‐AKT (Ser473), and the reduced protein level of p‐ERK in the TMEM229A Q200del group was more pronounced compared to the TMEM229A group.
Conclusion
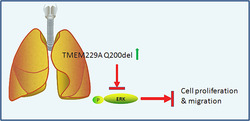
Our results demonstrated that the TMEM229A Q200del mutant may play a protective role in the progression of LUAD via inactivating ERK pathway, providing a potential therapeutic target in LUAD.
Keywords: lung adenocarcinoma, micropapillary component, mutation, transmembrane protein 229A, whole‐exome sequencing
The TMEM229A Q200del mutation had occurred in 16 (8.8%) patients and not found TMEM229A R76H and M346T mutations in our cohort. Further correlation analysis between the TMEM229A Q200del mutation and clinicopathological characteristics suggested that a lower frequency of the Q200del mutation was significantly associated with positive lymph node metastasis, advanced TNM stage, positive cancer thrombus, and pathological features. Overexpression of TMEM229A Q200del suppressed NSCLC cell proliferation and migration in vitro. Mechanistically, overexpression of TMEM229A and TMEM229A Q200del both reduced the expression level of phosphorylated (p)‐ERK and p‐AKT (Ser473), and the reduced protein level of p‐ERK in the TMEM229A Q200del group was more pronounced compared to the TMEM229A group.
Abbreviations
- ALK
anaplastic lymphoma kinase
- ARMS
amplification refractory mutation system
- ATS
American Thoracic Society
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
- DBS
double base substitution
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal–regulated kinase
- ERS
European Respiratory Society
- ExAC
the Exome Aggregation Consortium
- FFPE
formalin‐fixed paraffin‐embedded
- gnomAD
the Genome Aggregation Database
- IASLC
International Association for the Study of Lung Cancer
- InDel
small insertions and deletions
- LUAD
lung adenocarcinoma
- NSCLC
non‐small cell lung cancer
- ROS1
ROS proto‐oncogene 1 receptor tyrosine kinase
- RTCA
real‐time cellular analysis
- SBS
sequencing by synthesis
- SBS
single base substitution
- SNV
single nucleotide variant
- TCGA
The Cancer Genome Atlas Program
- TMEM229A
transmembrane protein 229A
- TNM
tumor‐node‐metastasis
- WHO
World Health Organization
1. Introduction
Lung adenocarcinoma (LUAD) is the most prevalent pathological subtype of lung cancer and exhibits diverse histological patterns and molecular characteristics, constituting approximately 60% of all lung cancer cases [1]. In 2015, the World Health Organization (WHO) primarily categorized LUAD into five histological classifications, namely solid, acinar, lepidic, papillary, and micropapillary pattern types [2]. Additionally, micropapillary LUAD cases are nonpredominant micropapillary adenocarcinomas. Previous study has shown that LUAD patients with micropapillary components often have lymphovascular invasion and pleural invasion, as well as lymph node or intrapulmonary metastasis features, and manifest more aggressive behavior and poorer outcomes than that other LUAD pattern types [3].
In consideration of the explanation for the mechanisms beyond tumorigenesis and malignancy discrepancy, several studies have been conducted to evaluate the molecular mechanisms and genetic changes of the LUAD subtype, particularly focusing on micropapillary LUAD [4, 5]. Recent advance have revealed that patients with micropapillary components exhibited disruption of the catenin‐cadherin complex, which contributed to its intracellular adherence [6, 7]. In addition, our previous study demonstrated that micropapillary LUAD has a significantly higher tumor mutation burden, such as EGFR mutations and ROS1 fusions. The incidence of coexisting EGFR mutations and ROS1 fusions is higher in this subtype compared to other subtypes of LUAD [8]. Warth et al. [9] also found that patients with micropapillary structure had a significantly higher tumor gene mutation burden and rearrangements of ROS1 or ALK than other histological subtypes of LUAD. Although specific genetic alterations associated with poorer prognosis of patients with micropapillary LUAD were identified, novel gene mutations and their related key mechanisms are not fully understood.
The transmembrane protein (TMEM) family genes are located in the different biological membranes of the cell and performed a wide variety of roles in physiological and pathological phenomena [10]. TMEM229A is a member of the TMEM family that plays an important role in tooth differentiation and development [11]. In addition, the single nucleotide variant (SNP) of TMEM229A rs7783359 was associated with sport performance [12, 13]. Our previous study also indicated that elevated TMEM229A suppressed NSCLC progression via inactivating ERK (extracellular signal–regulated kinase) pathway [14], but the function of TMEM229A mutants in LUAD is unknown.
In the present study, we aimed to investigate the evolutionary trajectory between LUAD histological subtypes and screen subtype‐specific genetic changes. In addition, we further studied the roles and mechanisms of TMEM229A Q200del mutant in the progression of LUAD, providing a novel therapeutic target for the treatment of LUAD, particularly in micropapillary LUAD.
2. Materials and Methods
2.1. Patient Samples
In the present study, all patients diagnosed with LUAD at the First Affiliated Hospital of Huzhou University from January 2020 to December 2022 were enrolled. Those patients diagnosed with LUAD were selected, but patients receiving presurgical therapy or combining other malignancies were excluded. The pathological diagnosis was confirmed using hematoxylin and eosin staining by two experienced pathologists (Qilin Shi and Hui Xia from the First Affiliated Hospital of Huzhou University). In the selection criteria, a total of 181 patients were enrolled. Among them, 54 cases harbored more than 5% of the micropapillary component, and the remaining cases were other LUAD subtypes according to the International Association for the Study of Lung Cancer (IASLC)/American Thoracic Society (ATS)/European Respiratory Society (ERS) [15], including 45 solid, 36 acinar, 28 lepidic, and 18 papillary subtypes. The clinicopathological characteristics collected, including sex, age, smoking history, tumor size, tumor differentiation, tumor‐node‐metastasis (TNM) stage, cancer thrombus, and lymph node metastasis. This study was approved by the Ethics Committee of the First Affiliated Hospital of Huzhou University (approved number: 2020KYLL049). Informed consent was obtained from all patients. The clinicopathological characteristics of all patients are shown in Table 1.
TABLE 1.
Association between TMEM229A Q200del mutation and clinicopathological features of patients with lung adenocarcinoma.
| Variables | TMEM229A | χ 2 | p | |
|---|---|---|---|---|
| WT (n = 165) | Q200del (n = 16) | |||
| Gender | 4.242 | 0.039* | ||
| Female | 75 (45.5%) | 3 (18.8%) | ||
| Male | 90 (54.5%) | 13 (81.2%) | ||
| Age | 2.103 | 0.147 | ||
| > 65 | 57 (34.5%) | 9 (56.3%) | ||
| ≤ 65 | 108 (65.5%) | 7 (43.7%) | ||
| Smoking history | 0.494 | 0.482 | ||
| Ever | 88 (53.3%) | 10 (62.5%) | ||
| Never | 77 (46.7%) | 6 (37.5%) | ||
| Tumor size (cm) | 0.155 | 0.693 | ||
| > 3.0 | 44 (26.7%) | 5 (31.3%) | ||
| ≤ 3.0 | 121 (73.3%) | 11 (68.7%) | ||
| Tumor differentiation | 0.155 | 0.693 | ||
| Well/moderate | 116 (70.3%) | 12 (75.0%) | ||
| Poor | 49 (29.7%) | 4 (25.0%) | ||
| Lymphatic invasion | 4.258 | 0.043* | ||
| Present | 50 (30.3%) | 2 (12.5%) | ||
| Absent | 115 (69.7%) | 14 (87.5%) | ||
| TNM stage | 4.459 | 0.035* | ||
| I + II | 131 (79.4%) | 9 (56.3%) | ||
| III + IV | 34 (20.6%) | 7 (43.7%) | ||
| Cancer thrombus | 4.174 | 0.044* | ||
| Present | 36 (21.8%) | 1 (6.3%) | ||
| Absent | 129 (78.2%) | 15 (93.7%) | ||
| Histological type | 9.152 | 0.002** | ||
| Micropapillary | 53 (32.1%) | 1 (6.3%) | ||
| Papillary | 14 (8.5%) | 4 (25.0%) | ||
| Solid | 43 (26.1%) | 2 (12.5%) | ||
| Acinar | 32(19.4%) | 4 (25.0%) | ||
| Lepidic | 23 (13.9%) | 5 (31.2%) | ||
p < 0.05,
p < 0.01.
2.2. DNA Extraction and Quantification
DNA extraction and purification from formalin‐fixed paraffin‐embedded (FFPE) tissues were performed using a commercial kit (cat. no. 56404, QIAamp DNA FFPE Tissue Kit, Qiagen, Germany) according to the manufacturer's protocol. Briefly, 5‐μm‐thick FFPE samples were dewaxed, and xylene was removed. After the samples were lysed, washed, and purified, the quantity and quality of DNA were eluted and measured using a NanoDrop 2000.
2.3. Whole‐Exome Sequencing
In all selected patients, three micropapillary LUAD (more than 50% of the micropapillary component, namely, served as predominant micropapillary LUAD) and three lepidic LUAD (more than 50% of the lepidic component, namely served as predominant lepidic LUAD) cases were selected, and whole‐exome sequencing was carried out using the Illumina HiSeq X Ten sequencing platform (Origingene of Biotechnology, Shanghai). Briefly, the sequencing libraries were further constructed by an Illumina Pair‐end (PE). Later, sequencing was performed based on sequencing by synthesis (SBS). Raw sequencing data were generated in a FASTQ file format and principally filtered on the total read volume, GC content, Q20 and Q30 percentage, and duplication rate. In addition, the sequencing data quality was assessed using the software FastQC (version 0.11.7), and the data were then compared with the human reference genome (hg19) using BWA software (https://ccb.jhu.edu/software/hisat2/ index.shtml). To ensure the reliability of the data, we clarified the criteria for the identifying and filtering on single nucleotide variants (SNVs), including (1) mutations with a number of supporting reads greater than 20 were saved; (2) mutations with non‐zero variant allele frequency (VAF) (> 0.1) were removed; (3) variants with low frequency (≤ 0.01) from the 1000 Genomes Project (https://www.internationalgenome.org/), the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org), and the Exome Aggregation Consortium (ExAC) were retained; and (4) only functional variants were presented. After sample coverage filtration, SNVs and small insertions and deletions (InDels) were identified by MuTect (version 1.14) from GATK (version 4.0) [16]. In addition, oncogenic driver genes were downloaded from the Cancer Gene Census in the COSMIC database (https://cancer.sanger.ac.uk/census), and then the mutational spectrum and absolute contribution of COSMIC v3 SBS (single base substitution) mutational characteristics were obtained by MutationalPatterns [17] on unfiltered somatic mutations, while the absolute exposures of COSMIC v3 DBS (double base substitution) InDel signatures were carried out by Sigminer [18].
2.4. Polymerase Chain Reaction (PCR)
PCR assays were performed using PrimeSTAR Max DNA Polymerase (cat. no. R045A, Takara Biotechnology Co. Ltd.) according to the manufacturer's protocol. The thermocycling conditions used for the PCR were as follows: initial denaturation at 95°C for 5 min, followed by 35 cycles at 94°C for 30 s, 58°C for 30 s and 72°C for 30 s, and extension at 72°C for 5 min. To construct the human TMEM229A mutants (accession no. NM_001136002.2), the specific primer sequences used in the experiment were as follows: TMEM229A Q200del forward, 5′‐CCTTCGTCTTCAATTTCCTCC‐3′ and reverse, 5′‐GCTGTA GTGGAGGTGGAAGTAGAG‐3′. TMEM229A R76H forward, 5′‐GCTGTCCACTG CTGAAGCGC‐3′ and reverse, 5′‐CGCTGCTGCAGGTACACCTT‐3′. TMEM229A M346T forward, 5′‐GGTGCCCATCTACGTGATCT‐3′ and reverse, 5′‐TTAGTTAGCTGGTACGTACTGC‐3′.
2.5. DNA Electrophoresis and Identification
The size of the PCR product (Q200del, 554 bp; R76H, 222 bp; and M346T, 217 bp, respectively) was identified by DNA electrophoresis. Later, the PCR product was purified using a TIANgel Midi Purification Kit (cat. no. #DP209‐03, Tiangen Biochemical Technology Co. Ltd.). All purification products were sequenced and identified by Shanghai Sangon Biotech Co. Ltd. (Shanghai).
2.6. TCGA Databases
The function and clinical significance of TMEM229A mutations in LUAD were explored using TCGA databases (http://www.cbioportal.org). Fifteen studies and 6936 patients with LUAD were enrolled in the study.
2.7. Cell Culture
Two NSCLC cell lines (A549 and H23) were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, cat. no. L110KJ, BasalMedia, Shanghai) supplemented with 10% FBS (Gibco; HyClone; Cytiva), 100 μg/mL streptomycin and 100 IU/mL penicillin (Sigma‐Aldrich; Merck KGaA) in a humidified incubator under 5% CO2 at 37°C.
2.8. Plasmid Construction and Cell Transfection
To construct the full‐length human TMEM229A proteins (accession no. NM_001136002.2), the specific primer was designed, and the corresponding TMEM229A DNA was cloned and inserted into a pcDNA3.1‐myc/his vector (abbreviation: vector; cat. no. V800‐20; Invitrogen; Thermo Fisher Scientific Inc.). The recombinant plasmids were referred to as pcDNA3.1‐TMEM229A‐myc/his (abbreviation: OE‐TMEM229A). For pcDNA3.1‐TMEM229A‐Q200del‐myc/his mutant plasmid, we directly synthesized from Shanghai Sangon Biotech Co. Ltd. according to OE‐TMEM229A plasmid. Once cells reached approximately 80% confluency, transfections were carried out using Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific Inc.), according to the manufacturer's instructions. The primer sequence used in the experiment was as follows: TMEM229A forward, 5′‐CCCAAGCTTGCCACCATGGCGGGGAGCG‐3′ and reverse, 5′‐CGCGGATCCGGTTAGCTGGTACGTACTGCAC‐3′.
2.9. Real‐Time Cellular Analysis (RTCA)
The RTCA xCELLLigence system (ACEA Biosciences Inc.; Agilent Technologies Inc.) was widely used to monitor cell morphology, proliferation, and migration in a noninvasive procedure [19]. A cell index was served as indicate the cell number and cell adhesion. Upon cells adhered to the surface of the E‐16 plate or CIM plate, an electronic record was changed and converted into the cell index by the xCELLLigence system. After cells transfected with different plasmids, cell proliferation and migration were carried out using RTCA assay as previously described [20]. Briefly, for cell proliferation assays, 50‐μL culture medium was added to measure the background, and then 100‐μL culture medium containing 6 × 103 A549 cells and 1 × 104 H23 cells were seeded into the E‐16 plate. For cell migration assays, the CIM plate was consisted of the lower chamber (165‐μL culture medium was added) and the upper chamber (30‐μL serum‐free culture medium was added) and left to stand for 1 h in a humidified incubator and then measured the background. Subsequently, the cells (6–10× 104) were mixed with serum‐free culture medium and seeded into the CIM plate. The data were recorded and analyzed using xCELLLigence software 2.0 (ACEA Biosciences Inc.; Agilent Technologies Inc.) [19].
2.10. RNA Extraction and Reverse Transcription‐Quantitative PCR
Total RNA was isolated using FastPure Cell/Tissue Total RNA Isolation Kit (cat. no. RC112‐01, Nanjing Vazyme Biotech Co. Ltd.), according to the manufacturer's protocol. Briefly, cells were lysed in Buffer RL, and the genomic DNA was removed using FastPure gDNA‐Filter columns III. Subsequently, RNA was dissolved in ethanol and washed with Buffer RW1 and Buffer RW2. Finally, the purified RNA was dissolved in RNase‐free ddH2O. The RNA was reverse transcribed into cDNA using the PrimeScript RT reagent kit (Takara Biotechnology Co. Ltd.), as previously described [21]. Reverse transcription‐quantitative PCR was performed following the instructions of the UltraSYBR Green PCR Master mix (cat. no. CW0957H; CWBio) on an ABI 7500 system (Applied Biosystems; Thermo Fisher Scientific Inc.). 18S ribosomal RNA (18sRNA) was used an endogenous housekeeping gene for normalization, and the relative expression level was calculated using the 2−ΔΔCq method [22]. The primer sequences used in the experiment were as follows: TMEM229A forward, 5′‐CGACCTACCCCGCTTTCTTTT‐3′ and reverse, 5′‐GCTCCCACCGTAGATGAAGAT‐3′; and 18sRNA forward, 5′‐GTAACCCGTTGAACCCCATT‐3′ and reverse, 5′‐CCATC CAATCGGTAGTAGCG‐3′.
2.11. Western Blotting
Total protein was extracted using radioimmunoprecipitation (RIPA) buffer containing protein and phosphatase inhibitors (Beyotime Institute of Biotechnology, Shanghai) according to our previous reports [14, 20]. An equal amount of protein was separated using 4%–20% SDS‐PAGE, and then the proteins were transferred onto 0.45‐μm PVDF membranes (EMD Millipore), followed by blocking with 5% bovine serum albumin (BSA) at room temperature for 1 h. The membranes were incubated with primary antibodies overnight at 4°C. After washing with PBS containing 0.1% Tween‐20, the membranes were incubated with the corresponding HRP‐conjugated secondary antibodies ([goat‐anti rabbit IgG (H + L) antibody; 1:2000; cat. no. A0208; Beyotime Institute of Biotechnology] or [goat‐anti mouse IgG (H + L) antibody; 1:2000; cat. no. A0216; Beyotime Institute of Biotechnology]) at room temperature for 1 h. The immunoblots were visualized using an ECL reagent (cat. no. P0018S; Beyotime Institute of Biotechnology) and detected by a Tanon 5200 system (Tanon Science & Technology Co. Ltd.). β‐actin was used an endogenous housekeeping gene for normalization, and the relative expression level of proteins was calculated using ImageJ v1.6.0 software (National Institutes of Health). The primary antibodies used in this experiment were as follows: anti‐TMEM229A (1:500; cat. no. ab107780; Abcam); anti‐ERK1/2 (1:2000; cat. no. 4695; Cell Signaling Technology Inc.); anti‐phosphorylated (p)‐ERK1/2 (1:2000; cat. no. 4370; Cell Signaling Technology Inc.); anti‐AKT (1:2000; cat. no. 4691; Cell Signaling Technology Inc.); anti‐p‐AKT (1:2000; Ser473; cat. no. 4060; Cell Signaling Technology Inc.); and anti‐β‐actin (1:5000; cat. no. M1210‐2; Hangzhou HuaAn Biotechnology Co. Ltd.).
2.12. Hematoxylin–Eosin (H&E) Staining
The LUAD tissues were preserved and immersed using 4% paraformaldehyde solution and then embedded in paraffin. The 5‐μm sections were stained using H&E staining solution (cat. no. C0105S; Beyotime Institute of Biotechnology). The images were visualized and analyzed under a light microscope at 100× magnification.
2.13. Statistical Analyses
The data were collected and analyzed using SPSS Statistics software (version 21.0; IBM Corp.). Data were presented as the mean ± standard error of mean of three independent experiments and were analyzed using an unpaired Student's t test or one‐way ANOVA followed by Tukey's post hoc test. Categorical variables were presented as frequencies and percentages, and variables among different groups were compared using chi‐square tests or Fisher's exact tests. p < 0.05 was considered statistically significant.
3. Results
3.1. Clinicopathological Characteristics of Patients
Among the 181 patients with LUAD, 54 (29.8%) had a micropapillary pattern, 45 (24.9%) had a solid pattern, 36 (19.9%) had acinar pattern, 28 (15.5%) had lepidic pattern, and 18 (9.9%) had papillary carcinoma. The clinicopathological characteristics of all patients are presented in Table 1.
3.2. Mutational Landscape of Micropapillary and Lepidic LUAD
According to a previous report, patients with micropapillary LUAD showed the worst prognosis, while lepidic LUAD had a protective role in prognosis [3]. The histological images of micropapillary and lepidic lesions are shown in Figure 1A. To explore the differences in novel gene mutations, three predominant micropapillary and three predominant lepidic LUAD patients were selected for whole‐exome sequencing. The results demonstrated that the lepidic LUAD group had 1402 gene snp and 274 gene indel mutations, but the micropapillary LUAD group had 2428 gene snp and 447 gene indel mutations, indicating that there are more gene mutations in micropapillary LUAD. In addition, EGFR mutations were identified as the most frequent driver gene, which was consistent with previous studies [3, 23]. Moreover, several other genes including ATXN2, C14orf180, MUC12, NOTCH1, and PKD1L2 were concomitantly mutated in the two subtypes (Figure 1B), suggesting that the PI3K‐AKT‐mTOR, Notch, MAPK, and GPCR signaling pathways were affected (Figure 1C,D). The PI3K‐AKT‐mTOR and Notch pathways play a role in cell growth, cell apoptosis, and metastasis, which may be associated with tumor progression and therapeutic resistance. Additionally, genome‐wide association studies had shown that loss‐of‐function mutations in ATXN2 gene may be associated with susceptibility to type I diabetes, obesity and hypertension, while NOTCH1 gene mutations were associated with aortic value disease, Adams‐Oliver syndrome, T‐cell acute lymphoblastic leukemia, chronic lymphocytic leukemia, and head and neck squamous cell carcinoma. These data imply that lepidic and micropapillary component from LUAD were shared the same several pathway‐specific mutations. We next explored SNV and Indel mutation data and identified the gene with an alternation frequency difference between the lepidic and micropapillary LUAD. The results indicated that the TMEM229A Q200del mutation was only present in lepidic LUAD cases, but not in micropapillary LUAD cases (Figure 1B). However, there was no other gene showed an alternation frequency difference between the two groups.
FIGURE 1.
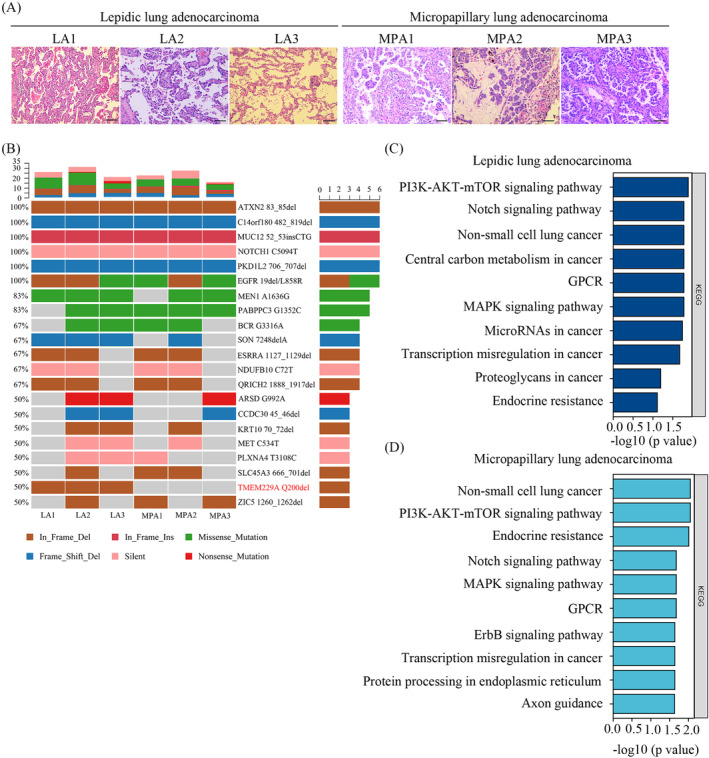
H&E‐stained slides and mutational landscape of micropapillary and lepidic lung adenocarcinoma. (A) The slides from three lepidic and three micropapillary lung adenocarcinoma patients were stained using hematoxylin–eosin staining (100× magnification). (B) Cancer‐associated genes with the top mutation in three lepidic and three micropapillary lung adenocarcinoma samples were analyzed using whole‐exome sequencing. (C) Pathways enriched in lepidic lung adenocarcinoma were analyzed by KEGG. (D) Pathways enriched in micropapillary lung adenocarcinoma were performed by KEGG.
3.3. Mutational Landscape of TMEM229A in LUAD
To explore the clinical significance of TMEM229A mutations in LUAD, the distribution and roles of TMEM229A mutants in TCGA databases patients with LUAD were first analyzed, and the results indicated that 15 associated studies were included, and 6936 LUAD patients were enrolled in this study. In all selected patients, the frequency of TMEM229A mutations was approximately 1% (Figure 2A), namely, R76H, Q200del, and M346T (Figure 2B). Additionally, Q200del was an inframe mutation, but R76H and M346T were missense mutations. Moreover, the alteration frequency of TMEM229A was analyzed in nine selected studies and showed that the frequency of TMEM229A mutations was low and mainly contained structural variants, mutations, and CNA data (Figure 2C). Finally, we deeply analyzed the RNA‐seq data and found that TMEM229A expression was not changed in different TMEM229A variants (Figure 2D). To verify the TMEM229A Q200del, R76H, and M346T mutations in LUAD, specific primers were designed and synthesized, and PCR was performed. The length of the PCR product of TMEM229A Q200del was 536 bp (Figure S1A,B). In addition, the PCR product was sequenced, and the results demonstrated that 16 of 181 LUAD patients had the TMEM229A Q200del mutation and not found R76H and M346T mutations (Table 1). Further correlation analysis between the TMEM229A Q200del mutation and clinicopathological characteristics suggested that a lower frequency of the Q200del mutation was associated with positive lymph node metastasis (p = 0.043), advanced TNM stage (p = 0.035), positive cancer thrombus (p = 0.044), and pathological features (p = 0.008) (Table 1). These data suggest that the TMEM229A Q200del mutation may play a protective role in the tumorigenesis of LUAD.
FIGURE 2.
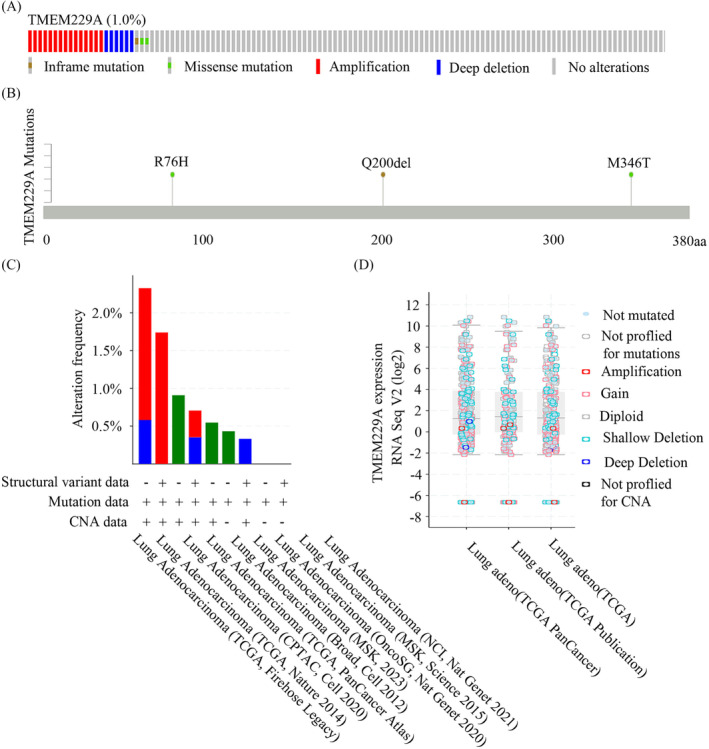
Mutational landscape of TMEM229A in lung adenocarcinoma. (A) The frequency of TMEM229A mutations in TCGA‐LUAD samples. (B) TMEM229A mutations (R76H, Q200del, and M346T) in TCGA‐LUAD samples. (C) The frequency of TMEM229A mutations in samples with Structural variant data, Mutation data, and CNA data. (D) The relationship between TMEM229A expression and TMEM229A mutations in TCGA‐LUAD samples.
3.4. TMEM229A Q200del Mutation Inhibits NSCLC Cell Proliferation and Migration via Inactivating ERK Pathway In Vitro
Our previous study demonstrated that TMEM229A was expressed at low levels in NSCLC and suppressed NSCLC progression by inactivating the ERK pathway, suggesting that TMEM229A is a suppressor gene in the development of NSCLC [14]. To further investigate the role of the TMEM229A Q200del mutation in LUAD, cell proliferation and migration assays were performed, and the results indicated that overexpression of TMEM229A Q200del mutant significantly upregulated TMEM229A expression and inhibited H23 and A549 (TMEM229A wild‐type) cell proliferation and migration in vitro (Figure 3A–D). In addition, the proliferative inhibition of TMEM229A Q200del mutant was more obvious than that of wild‐type TMEM229A (Figure 3C). Consistent with our previous report [14], overexpression of TMEM229A and its Q200del mutant resulted in a decrease in the expression levels of p‐ERK and p‐AKT (Ser473) (Figure 3E). Additionally, the TMEM229A Q200del group exhibited a significantly lower protein level of p‐ERK compared to the TMEM229A group (Figure 3E), indicating that the TMEM229A Q200del mutant inhibits LUAD progression via inactivating ERK pathway.
FIGURE 3.
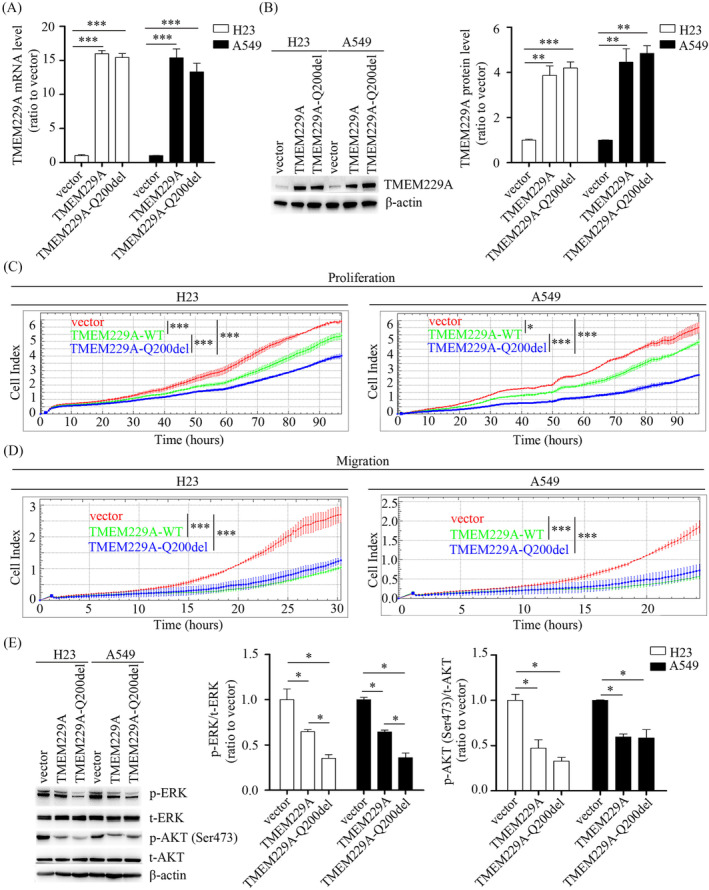
Overexpression of TMEM229A Q200del suppressed NSCLC cell proliferation and migration via inactivating ERK pathway. (A) H23 and A549 cells were transfected with vector, TMEM229A or TMEM229A‐Q200del for 24 h. The mRNA level of TMEM229A was measured in H23 and A549 using reverse transcription‐quantitative PCR. (B) H23 and A549 cells were transfected with vector, TMEM229A or TMEM229A‐Q200del for 48 h. The protein level of TMEM229A was detected by western blotting. β‐actin was used as a loading control. (C) After cells transfected with different plasmids, cell proliferation was assessed using RTCA assay. (D) After cells transfected with different plasmids, cell migration was performed using RTCA assay. (E) H23 and A549 cells were transfected with different plasmids. p‐ERK, t‐ERK p‐AKT Ser473, and t‐AKT were detected by western blotting. β‐actin was used as a loading control. Data represent the mean ± SEM from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 were determined by one‐way ANOVA with Tukey's post hoc analysis. OE, overexpression; Q200del, TMEM229A Q200del; p, phosphorylated; t, total.
4. Discussion and Conclusions
LUAD is a morphologically heterogeneous cancer of the lung, which possesses a unique histological, radiological, epidemiological, and clinical features [24]. In 2015, the WHO mainly classified LUAD into five histological categories, including solid, acinar, lepidic, papillary and micropapillary pattern types [2]. Recent studies have demonstrated that patients with the micropapillary or solid subtype of LUAD are associated with worse outcomes due to genomic diversity [25], a higher incidence of metastasis [7], the spread of tumors through air spaces [26], and other factors [5, 27]. In addition, our previous study discovered that patients with micropapillary LUAD had a higher prevalence of EGFR mutations, ROS1 rearrangement and combined mutations of EGFR, ROS1 and EML4‐ALK using the amplification refractory mutation system (ARMS) [8], which was consistent with other previous report [28]. Based on these observations, this study intends to screen new micropapillary LUAD‐associated genes using whole‐exome sequencing and further investigate their role in the tumorigenesis of LUAD.
In the present study, the most commonly mutated driver genes were EGFR, ATXN2, C14orf180, MUC12, NOTCH1, and PKD1L2 in patients with a micropapillary component and the lepidic subtype of LUAD. Previous report indicated that EGFR mutations are strongly associated with patients with the micropapillary subtype of LUAD [29]. In addition, most of the reported driver genes included KARS, PIK3CA, TP53, and ALK rearrangements in micropapillary LUAD [30]. Our results also found that one lepidic (1/3) and one micropapillary LUAD sample (1/3) had TP53 mutations, but KRAS and PIK3CA mutations and ALK rearrangements were not found, which may be attributed to the small sample size. As reported previously [4], several novel gene mutations were identified in micropapillary LUAD, such as ZNF469, TTN, TENM4, APOBEC, KEAP1, NOTCH4, PTP4A3, NAPRT, and RECQL4. Further studies discovered that these novel genes were regarded as oncogenes or tumor suppressor genes and played a pivotal role in the progression and prognosis of LUAD. Interestingly, we observed that the TMEM229A Q200del mutation existed only in lepidic LUAD, but not in micropapillary LUAD, which was firstly identified. According to the UniProt databases (https://www.uniprot.org/), TMEM229A is localized to the plasma membrane and is a seven‐TMEM with poorly defined biology. A previous report showed that downregulated expression of TMEM229A contributed to the progression from deciduous to permanent teeth [11]. In addition, our previous study confirmed that TMEM229A was lowly expressed in NSCLC and partly suppressed NSCLC progression through inactivating the ERK pathway, suggesting TMEM229A was a suppressor gene in the development and progression of NSCLC [14]. A recent whole‐genome sequencing study proved that the TMEM229A rs7783359 polymorphism was linked explicitly with reaction time in wrestlers.12 13 This study showed that the TMEM229A Q200del mutation was associated with lymph node metastasis, TNM stage, cancer thrombus and pathological pattern. Overexpression of TMEM229A Q200del mutant significantly inhibited NSCLC cell proliferation and migration. Moreover, the inhibition of TMEM229A Q200del mutant was more obvious than that of wild‐type TMEM229A. Mechanistically, overexpression of TMEM229A and TMEM229A Q200del mutant both reduced the expression levels of p‐ERK and p‐AKT (Ser473), and the reduced expression level of p‐ERK/t‐ERK in TMEM229A Q200del group was more obvious than that in TMEM229A group, suggesting the TMEM229A Q200del mutant inhibits LUAD progression via inactivating ERK pathway.
Overall, our results demonstrate that the mutation burden in micropapillary LUAD was greater than that in lepidic LUAD. In addition, the TMEM229A Q200del mutation appeared in lepidic LUAD but not in micropapillary LUAD. Moreover, overexpression of the TMEM229A Q200del mutant significantly suppressed NSCLC cell proliferation and migration via inactivating ERK pathway, providing a novel therapeutic target and a promising translational marker for the treatment of LUAD, particularly in micropapillary LUAD. However, the present study had several limitations. First, the cohort only selected three micropapillary components and three lepidic subtypes of LUAD for whole‐exome sequencing. Future studies with a larger number of samples will be conducted. Additionally, the investigation of TMEM229A Q200del's function in NSCLC cell lines by plasmid overexpression does not truly represent the function of the endogenous mutation. The function of endogenous TMEM229A Q200del mutant should be further analyzed via Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR). Finally, the mechanism of TMEM229A Q200del mutant in LUAD progression was not fully determined. Thus, we will further investigate the underlying mechanism of TMEM229A Q200del and TMEM229A in the development and progression of LUAD.
Author Contributions
Xilin Zhang and Zhaohui Dong designed and conceived the study. Yixian Liang, Yanping Xie, Huanming Yu, Wenjuan Zhu, and Chengyi Yin performed these experiments. Yixian Liang, Zhaohui Dong, and Xilin Zhang analyzed the data. Xilin Zhang and Yixian Liang wrote the manuscript. All authors have read the final manuscript.
Ethics Statement
This work was approved by the Ethics Committee of the First People's Hospital of Huzhou (approved number: 2020KYLL049), and informed consent was obtained from all patients.
Consent
Informed consent was obtained from all patients.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1 TMEM229A Q200del genotype was identified. (A) TMEM229A Q200del mutation of selected 10 cases was identified using DNA electrophoresis; (B) TMEM229A genotype was further performed using DNA sequencing.
Acknowledgments
The authors have nothing to report.
Funding: This research was kindly supported by Zhejiang Provincial Natural Science Foundation of China under Grant Nos. LY22H160026 to X.L.Z. and LTGY23H160004 to Z.H.D., The Scientific Technology Projects of Health and Medicine of Zhejiang Province under Grant no. 2023KY1180 to X.L.Z., and Huzhou Science and Technology Fund under Grant no. 2021GZB03 to X.L.Z.
Yi‐Xian Liang and Yan‐Ping Xie contributed equally to this work.
Contributor Information
Zhao‐Hui Dong, Email: 793701557@qq.com.
Xi‐Lin Zhang, Email: zhangxilin16@126.com.
Data Availability Statement
The corresponding datasets have been submitted to Sequence Read Archive (SRA).
database (BioProject ID: PRJNA1044823). The persistent URL directly linked with https://dataview.ncbi.nlm.nih.gov/object/PRJNA1044823?reviewer=ofkt4k3518m7mevjjb5mti2mjn. All datasets used and/or analyzed in this article are available from the corresponding authors upon reasonable request.
References
- 1. Siegel R. L., Miller K. D., Wagle N. S., and Jemal A., “Cancer Statistics, 2023,” CA: A Cancer Journal for Clinicians 73, no. 1 (2023): 17–48, 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 2. Travis W. D., Brambilla E., Nicholson A. G., et al., “The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification,” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 10, no. 9 (2015): 1243–1260, 10.1097/JTO.0000000000000630. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y., Wang R., Cai D., et al., “A Comprehensive Investigation of Molecular Features and Prognosis of Lung Adenocarcinoma With Micropapillary Component,” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 9, no. 12 (2014): 1772–1778, 10.1097/JTO.0000000000000341. [DOI] [PubMed] [Google Scholar]
- 4. Meng F., Zhang Y., Wang S., et al., “Whole‐Exome Sequencing Reveals the Genomic Features of the Micropapillary Component in Ground‐Glass Opacities,” Cancers (Basel) 14, no. 17 (2022): 4165, 10.3390/cancers14174165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jeon Y. J., Lee J., Shin S., et al., “Prognostic Impact of Micropapillary and Solid Histological Subtype on Patients Undergoing Curative Resection for Stage I Lung Adenocarcinoma According to the Extent of Pulmonary Resection and Lymph Node Assessment,” Lung Cancer 168 (2022): 21–29, 10.1016/j.lungcan.2022.04.005. [DOI] [PubMed] [Google Scholar]
- 6. Sun S., Li S., Li J., et al., “Prognosis of the Second Predominant Subtype in Lung Adenocarcinoma: A Retrospective Single‐Center Cohort Study,” Journal of Thoracic Disease 14, no. 12 (2022): 4846–4864, 10.21037/jtd-22-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Y., Byun A. J., Choe J. K., et al., “Micropapillary and Solid Histologic Patterns in N1 and N2 Lymph Node Metastases Are Independent Factors of Poor Prognosis in Patients With Stages II to III Lung Adenocarcinoma,” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 18, no. 5 (2023): 608–619, 10.1016/j.jtho.2023.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang X., Jiang Y., Yu H., Xia H., and Wang X., “A Comprehensive Study on the Oncogenic Mutation and Molecular Pathology in Chinese Lung Adenocarcinoma Patients,” World Journal of Surgical Oncology 18, no. 1 (2020): 172, 10.1186/s12957-020-01947-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warth A., Penzel R., Lindenmaier H., et al., “EGFR, KRAS, BRAF and ALK Gene Alterations in Lung Adenocarcinomas: Patient Outcome, Interplay With Morphology and Immunophenotype,” The European Respiratory Journal 43, no. 3 (2014): 872–883, 10.1183/09031936.00018013. [DOI] [PubMed] [Google Scholar]
- 10. Marx S., Dal Maso T., Chen J. W., et al., “Transmembrane (TMEM) Protein Family Members: Poorly Characterized Even if Essential for the Metastatic Process,” Seminars in Cancer Biology 60 (2020): 96–106, 10.1016/j.semcancer.2019.08.018. [DOI] [PubMed] [Google Scholar]
- 11. Kang J., Bai R., Liu K., Ji X. P., Li Y., and Han J. Y., “Identification of Significantly Different Modules Between Permanent and Deciduous Teeth by Network and Pathway Analyses,” Genetics and Molecular Research 15, no. 4 (2016): 7959, 10.4238/gmr15047959. [DOI] [PubMed] [Google Scholar]
- 12. Boulygina E. A., Borisov O. V., Valeeva E. V., et al., “Whole Genome Sequencing of Elite Athletes,” Biology of Sport 37, no. 3 (2020): 295–304, 10.5114/biolsport.2020.96272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Youn B. Y., Ko S. G., and Kim J. Y., “Genetic Basis of Elite Combat Sports Athletes: A Systematic Review,” Biology of Sport 38, no. 4 (2021): 667–675, 10.5114/biolsport.2022.102864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X., He Y., Jiang Y., et al., “TMEM229A Suppresses Non‐Small Cell Lung Cancer Progression via Inactivating the ERK Pathway,” Oncology Reports 46, no. 2 (2021): 176, 10.3892/or.2021.8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Travis W. D., Brambilla E., Noguchi M., et al., “International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma,” Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer 6, no. 2 (2011): 244–285, 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cibulskis K., Lawrence M. S., Carter S. L., et al., “Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples,” Nature Biotechnology 31, no. 3 (2013): 213–219, 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blokzijl F., Janssen R., van Boxtel R., and Cuppen E., “MutationalPatterns: Comprehensive Genome‐Wide Analysis of Mutational Processes,” Genome Medicine 10, no. 1 (2018): 33, 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang S., Tao Z., Wu T., and Liu X. S., “Sigflow: An Automated and Comprehensive Pipeline for Cancer Genome Mutational Signature Analysis,” Bioinformatics 37, no. 11 (2021): 1590–1592, 10.1093/bioinformatics/btaa895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan G., Du Q., Wei X., et al., “Application of Real‐Time Cell Electronic Analysis System in Modern Pharmaceutical Evaluation and Analysis,” Molecules 23, no. 12 (2018): 3280, 10.3390/molecules23123280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X., Chen Q., He Y., et al., “STRIP2 Motivates Non‐Small Cell Lung Cancer Progression by Modulating the TMBIM6 Stability Through IGF2BP3 Dependent,” Journal of Experimental & Clinical Cancer Research 42, no. 1 (2023): 19, 10.1186/s13046-022-02573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X., Guo H., Bao Y., Yu H., Xie D., and Wang X., “Exosomal Long Non‐Coding RNA DLX6‐AS1 as a Potential Diagnostic Biomarker for Non‐Small Cell Lung Cancer,” Oncology Letters 18, no. 5 (2019): 5197–5204, 10.3892/ol.2019.10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Livak K. J. and Schmittgen T. D., “Analysis of Relative Gene Expression Data Using Real‐Time Quantitative PCR and the 2(‐Delta Delta C(T)) Method,” Methods 25, no. 4 (2001): 402–408, 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23. Li C., Shen Y., Hu F., et al., “Micropapillary Pattern Is Associated With the Development of Brain Metastases and the Reduction of Survival Time in EGFR‐Mutation Lung Adenocarcinoma Patients With Surgery,” Lung Cancer 141 (2020): 72–77, 10.1016/j.lungcan.2020.01.007. [DOI] [PubMed] [Google Scholar]
- 24. Luca S., Zannini G., Morgillo F., et al., “The Prognostic Value of Histopathology in Invasive Lung Adenocarcinoma: A Comparative Review of the Main Proposed Grading Systems,” Expert Review of Anticancer Therapy 23, no. 3 (2023): 265–277, 10.1080/14737140.2023.2179990. [DOI] [PubMed] [Google Scholar]
- 25. Wang L. L., Ding L., Zhao P., et al., “Clinicopathological, Radiological, and Molecular Features of Primary Lung Adenocarcinoma With Morule‐Like Components,” Disease Markers 2021 (2021): 9186056, 10.1155/2021/9186056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khalil H. A., Shi W., Mazzola E., et al., “Analysis of Recurrence in Lung Adenocarcinoma With Spread Through Air Spaces,” Journal of Thoracic and Cardiovascular Surgery 166 (2023): 1317–1328.e4, 10.1016/j.jtcvs.2023.01.030. [DOI] [PubMed] [Google Scholar]
- 27. Shigenobu T., Takahashi Y., Masugi Y., et al., “Micropapillary Predominance Is a Risk Factor for Brain Metastasis in Resected Lung Adenocarcinoma,” Clinical Lung Cancer 22, no. 6 (2021): e820–e828, 10.1016/j.cllc.2021.04.001. [DOI] [PubMed] [Google Scholar]
- 28. Cancer Genome Atlas Research N , “Comprehensive Molecular Profiling of Lung Adenocarcinoma,” Nature 511, no. 7511 (2014): 543–550, 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li S., Li L., Zhu Y., et al., “Coexistence of EGFR With KRAS, or BRAF, or PIK3CA Somatic Mutations in Lung Cancer: A Comprehensive Mutation Profiling From 5125 Chinese Cohorts,” British Journal of Cancer 110, no. 11 (2014): 2812–2820, 10.1038/bjc.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang L., Mino‐Kenudson M., Roden A. C., et al., “Association Between the Novel Classification of Lung Adenocarcinoma Subtypes and EGFR/KRAS Mutation Status: A Systematic Literature Review and Pooled‐Data Analysis,” European Journal of Surgical Oncology: The Journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology 45, no. 5 (2019): 870–876, 10.1016/j.ejso.2019.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 TMEM229A Q200del genotype was identified. (A) TMEM229A Q200del mutation of selected 10 cases was identified using DNA electrophoresis; (B) TMEM229A genotype was further performed using DNA sequencing.
Data Availability Statement
The corresponding datasets have been submitted to Sequence Read Archive (SRA).
database (BioProject ID: PRJNA1044823). The persistent URL directly linked with https://dataview.ncbi.nlm.nih.gov/object/PRJNA1044823?reviewer=ofkt4k3518m7mevjjb5mti2mjn. All datasets used and/or analyzed in this article are available from the corresponding authors upon reasonable request.