

Abstract
GalAGs (galactosaminoglycans) are one subset of the GAG (glycosaminoglycan) family of chemically heterogeneous polysaccharides that are involved in a wide range of biological processes. These complex biomacromolecules are believed to be responsible for the inhibition of nerve regeneration following injury to the central nervous system. The enzymic degradation of GAG chains in damaged nervous tissue by cABC I (chondroitinase ABC I), a broad-specificity lyase that degrades GalAGs, promotes neural recovery. In the present paper, we report the subcloning of cABC I from Proteus vulgaris, and discuss a simple methodology for the recombinant expression and purification of this enzyme. The originally expressed cABC I clone resulted in an enzyme with negligible activity against a variety of GalAG substrates. Sequencing of the cABC I clone revealed four point mutations at issue with the electron-density data of the cABC I crystal structure. Site-directed mutagenesis produced a clone with restored GalAG-degrading function. We have characterized this enzyme biochemically, including an analysis of its substrate specificity. By coupling structural inspections of cABC I and an evaluation of sequence homology against other GAG-degrading lyases, a set of amino acids was chosen for further study. Mutagenesis studies of these residues resulted in the first experimental evidence of cABC I's active site. This work will facilitate the structure–function characterization of biomedically relevant GalAGs and further the development of therapeutics for nerve regeneration.
Keywords: active site, chondroitinase ABC, chondroitin sulphate, dermatan sulphate, glycosaminoglycan, recombinant protein expression
Abbreviations: 4S, 4-O-sulphated; 6S, 6-O-sulphated; C4S, chondroitin 4-sulphate; C6S, chondroitin 6-sulphate; cABC, chondroitinase ABC; cAC, chondroitinase AC; cB, chondroitinase B; CE, capillary electrophoresis; DS, dermatan sulphate; ECM, extracellular matrix; GAG, glycosaminoglycan; GalAG, galactosaminoglycan; GalNAc, N-acetylgalactosamine; IdoA, iduronic acid; ΔUA, Δ4,5 unsaturated uronic acid
INTRODUCTION
GAGs (glycosaminoglycans) are linear acidic polysaccharides consisting of a disaccharide repeat unit of a hexosamine linked to an uronic acid. GAGs reside both in the ECM (extracellular matrix) and at the cell surface as constituents of proteoglycans. These sugars, apart from having important structural roles in the ECM, are also fundamental modulators of many biological processes, such as development, cell proliferation, signalling and inflammation [1–3]. The great chemical heterogeneity of these GAGs [4] is responsible for their wide-ranging biological influence. Each GAG disaccharide repeat unit can be customized through a variety of biosynthetic modifications that include epimerization of the uronic acid and variable sulphation. The establishment of a means to elucidate the fine structure of these complex polysaccharides is an essential biotechnological pursuit [5–8].
Two related enzymes with broad substrate specificity are produced by the bacterium Proteus vulgaris: cABC I (chondroitinase ABC I) and cABC II (chondroitinase ABC II) (EC 4.2.2.4) [9]. These enzymes depolymerize a variety of GAG substrates, including C4S (chondroitin 4-sulphate), DS (dermatan sulphate), C6S (chondroitin 6-sulphate) and hyaluronic acid. These enzymes have been purified and studied previously [9]. cABC I is a 997-amino-acid-residue endolytic enzyme that cleaves GAG substrates to tetrasaccharides and disaccharides. It degrades GalAGs (galactosaminoglycans) regardless of the C5 epimerization state of the uronic acid. This is particularly notable, as cABC I processes DS despite having little sequence or structural identity when compared with cB (chondroitinase B) (EC 4.2.2.4) [10]. The crystal structure of cABC I [11] reveals that it has three major domains and indicates that this enzyme shares considerable structural homology with Flavobacterium heparinum cAC (chondroitinase AC) (EC 4.2.2.5) [12], although there is little sequence identity between the enzymes.
Discrepancies in past cloned sequences of cABC I have augmented confusion in studying this enzyme [13,14]. Moreover, the cloning and expression of cABC I is challenging because of its size, stability and solubility. In addition to the difficulty of obtaining a pure recombinant protein, the broad substrate specificity complicates the elucidation of the mechanism of action. It is unclear how this GAG lyase is able to process such a broad range of substrates [4,9,11,15,16].
In the present study, we report the subcloning of cABC I from P. vulgaris, the recombinant expression of the enzyme in Escherichia coli and a one-step preparation of the pure protein. We offer both an activity assessment and a product profile determination of the recombinant enzyme against a spectrum of GAG substrates. Mutagenesis studies provide the first conclusive proof of residues that are critical for catalytic activity, paving the way for a more rigorous comprehension of the mode of action of cABC I.
EXPERIMENTAL
Subcloning of cABC I from P. vulgaris
Genomic DNA was isolated from cultures of P. vulgaris (ATCC number 6896) using a DNeasy purification kit from Qiagen. Primers were designed based on the previously published sequence of the gene (GenBank® accession number E08025) [13]. It has been reported previously that the active form of cABC I isolated from P. vulgaris is missing the N-terminal signal sequence [9]. Therefore two 5′ end primers were designed so as to generate a full-length clone and a truncated version of the gene by omitting 72 bases encoding the signal sequence. In order to facilitate cloning into a pET-28a vector (Novagen), the forward primer was constructed so as to incorporate an NdeI restriction site, and the reverse primer had a BamHI and a XhoI restriction site built in. The primers for cloning cABC I have the sequences: 5′-CATATGCCGATATTTCGTTTTACTGCA-3′ (forward primer for full-length gene), 5′-CATATGCCCACCAGCAATCCTGCATTTG-3′ (forward primer for truncated gene) and 5′-GGATCCTCGAGTCAAGGGAGTGGCGAGAGTTTG-3′ (reverse primer). PCR was run using P. vulgaris genomic DNA as the template, and a slightly longer extension time (3 min) was used to account for the length of the gene (2994 bp) being amplified.
The PCR product was initially ligated into the pCR® 4-TOPO vector using the TOPO TA Cloning Kit (Invitrogen) and transformed into TOP10 E. coli cells. Plasmid DNA was isolated from positive colonies, and the cABC I gene was excised from the TOPO vector using the previously engineered NdeI and XhoI restriction sites. The excised gene was ligated into pET-28a that had been digested with the same restriction enzymes. The ligation products were then transformed into DH5α E. coli cells, and plasmid DNA that was isolated from the colonies was screened by restriction digestion for incorporation of the cABC I gene. Plasmid DNA isolated from the positive colonies was also sequenced to confirm incorporation of the gene.
Site-directed mutagenesis studies
The QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used with plasmid DNA template to produce mutants of cABC I. Briefly, mutants were produced by plasmid denaturation and annealing of complementary oligonucleotide primers containing the desired mutation. This was followed by extension of the primers with a temperature cycler and PfuTurbo DNA polymerase, resulting in a mutated plasmid with staggered nicks. Digestion of hemimethylated DNA of the parental template with DpnI endonuclease selects for the mutation-containing synthesized DNA. Primer sequences for mutagenesis studies are presented in Table 1. The mutated plasmids were transformed into XL1-Blue supercompetent cells. The plasmids were prepared using a miniprep kit (Qiagen). Each clone was sequenced to confirm the presence of the desired mutation. Plasmid DNA was used to transform BL21 (DE3) E. coli.
Table 1. Summary of primer sequences for site-directed mutagenesis studies.
Mutation codons are indicated in bold; bases modified in order to create the desired point mutations are underlined. Forward primers for each mutant are listed first.
| Mutant | Primer pair sequences |
|---|---|
| Thr154→Ala | 5′-ACTGGCTGGCGTGCTGTGGGAGTCTCT-3′ |
| 5′-AGAGACTCCCACAGCACGCCAGCCAGT-3′ | |
| Val309→Ile | 5′-GGAACGCAAGGCAGACATCTGATCACTGATAAACAAATC-3′ |
| 5′-GATTTGTTTATCAGTGATCAGATGTCTGCCTTGCGTTCC-3′ | |
| Pro322→Leu | 5′-CAACCAGAGAATCTTAACTCTCAAGATAAACAACTATTTG-3′ |
| 5′-CAAATAGTTGTTTATCTTGAGAGTTAAGATTCTCTGGTTG-3′ | |
| Pro694→Gln | 5′-GGTTGGGATTGGAATAGAATGCAAGGGGCAACCACT-3′ |
| 5′-AGTGGTTGCCCCTTGCATTCTATTCCAATCCCAACC-3′ | |
| His501→Ala | 5′-TGATGGTACAGCATGGCGAGCTGAAGGCAACTATCCGGGCTA-3′ |
| 5′-TAGCCCGGATAGTTGCCTTCAGCTCGCCATGCTGTACCATCA-3′ | |
| Tyr508→Ala | 5′-GGCAACTATCCGGGCGCCTCTTTCCCAGCC-3′ |
| 5′-GGCTGGGAAAGAGGCGCCCGGATAGTTGCC-3′ | |
| Arg560→Ala | 5′-CCGCTTGCAGGAGCACACCCTTTTAACTCACCTTCG-3′ |
| 5′-CGAAGGTGAGTTAAAAGGGTGTGCTCCTGCAAGCGG-3′ | |
| Glu653→Ala | 5′-CACCAATGTTTGGTCATCTGCAATTTATAACAAAGATAACCGT-3′ |
| 5′-ACGGTTATCTTTGTTATAAATTGCAGATGACCAAACATTGGTG-3′ | |
| His561→Ala | 5′-CCGCTTGCAGGAAGAGCCCCTTTTAACTCACCTTCG-3′ |
| 5′-CGAAGGTGAGTTAAAAGGGGCTCTTCCTGCAAGCGG-3′ | |
| His712→Ala | 5′-GACAGTCCTAAACCTGCTACCTTAATGCAACGTGGAGAG-3′ |
| 5′-CTCTCCACGTTGCATTAAGGTAGCAGGTTTAGGACTGTC-3′ | |
| Arg500→Ala | 5′-CCTGATGGTACAGCATGGGCACATGAAGGCAACTATCCGGGC-3′ |
| 5′-GCCCGGATAGTTGCCTTCATGTGCCCATGCTGTACCATCAGG-3′ |
Recombinant expression and protein purification of cABC I and mutants
Recombinant cABC I and the site-directed mutants were expressed in E. coli and purified essentially as described previously [8]. Cultures for expression contained 40 μg/ml kanamycin. The presence and purity of the proteins were assessed by SDS/PAGE analysis using pre-cast NuPAGE 12% Bistris gels, the XCell SureLock Mini-Cell and Simply Blue SafeStain (Invitrogen). Relative protein concentration was calculated using the Bradford assay (Bio-Rad protein assay kit) with BSA (Sigma) as a standard. The His6 tag was cleaved using the Thrombin Capture Kit (Novagen) as described previously [17].
Structural characterization
CD spectra were recorded at 25 °C on an Aviv 202 CD spectrophotometer using quartz cuvettes with optical path length of 1 mm. Scans were collected between 300 and 195 nm with a 1.0 nm bandwidth and a scan rate of 1 nm/min. Three scans were averaged for each protein. For melting experiments, spectra were collected at 5 °C intervals from 5 to 80 °C. Recombinant proteins were concentrated and buffer-exchanged into 50 mM sodium phosphate, pH 7.5, using Centricon 10 filters (Millipore). Protein content was quantified by standard methods using the Bio-Rad protein assay kit. All spectra were collected using a protein concentration of 0.2 mg/ml. The buffer contribution was accounted for in all spectra. The signal was normalized to molar ellipticity, [θ]M, in degrees·cm2·dmol−1.
Determination of optimal biochemical conditions for recombinant cABC I activity
For these studies, C6S and DS were dissolved at a 1 mg/ml concentration in various buffers in an attempt to determine the relative effects of pH, temperature, ionic strength and sodium acetate concentration on enzyme activity (characterization with C4S was cost-prohibitive). The activity of a fixed amount of recombinant active cABC I (0.2 μg) was assessed based on the change in absorbance at 232 nm per min (ΔA232/min) as reaction conditions were varied. The effect of pH was investigated by using two different buffer systems: (1) 50 mM sodium phosphate buffer, pH 6.5, 7.0, 7.5, 8.0, 8.5 or 9.0, and (2) 50 mM Tris buffer, pH 7.5, 8.0, 8.5 or 9.0. Activity at various temperatures (25–45 °C) was also investigated. To determine the relative effect of ionic strength, the NaCl concentration was varied from 0 to 1.0 M in 50 mM Tris buffer (pH 8.0). Previous studies have suggested that addition of sodium acetate to the buffer enhances the activity of cABC I [9]. To confirm this, we varied the sodium acetate concentration from 0 to 0.5 M in 50 mM Tris buffer (pH 8.0).
The temperature study was carried out using a temperature-controlled UV spectrophotometer (DU 800, Beckman–Coulter) in a quartz cuvette at a 1 ml final reaction volume. The other optimization experiments were carried out on a SpectraMax 190 (Molecular Devices) using a 96-well quartz plate. Eight enzyme reactions (i.e. one column of the plate) could be initiated and monitored simultaneously using our set-up. This semi-high-throughput approach enabled us to sample multiple reaction conditions in an easily repeatable manner. The temperature on the SpectraMax was set to 37 °C for these experiments. Absorbance at 232 nm was monitored for 2–4 min, and activity was calculated based on the initial rate of product formation.
cABC I product profile and activity analysis
For product profile analysis, CE (capillary electrophoresis) was performed using similar conditions to those developed for the separation of heparan sulphate GAG disaccharides [18]. Substrates used included C6S from shark cartilage (Sigma), DS from porcine intestinal mucosa (Sigma) and C4S from sturgeon notochord (Seikagaku). For activity experiments, 2 μl of enzyme was placed in 248 μl of 50 mM Tris/HCl and 50 mM sodium acetate, pH 8.0, with 1 mg/ml substrate (0.25 mg/ml for hyaluronan) at 37 °C. Product formation was monitored as an increase in absorbance at 232 nm as a function of time in our semi-high-throughput format. Initial rates represent <10% substrate turnover. cABC (protease-free) was purchased from Seikagaku. Substrates used in these studies are described above and additionally include chondroitin from shark cartilage (Seikagaku), hyaluronan from human umbilical cord (Sigma), heparin (Celsus) and heparan sulphate (Celsus). The quantity of enzyme used in each reaction was measured using the Bio-Rad protein assay kit. A kinetic analysis of cABC I employed our semi-high-throughput spectrophotometric approach, and is essentially as described previously [19]. A volume of 1 μl of 0.2 μg/μl cABC I was added to 249 μl of a solution containing different concentrations of GalAG substrates (C4S, C6S and DS) in 50 mM Tris/HCl and 50 mM sodium acetate, pH 8.0. Each well contained different substrate concentrations ranging from 0.1 to 5 mg/ml. Product formation was monitored by measuring the absorbance at 232 nm every 2 s.
To evaluate the kinetic data, the initial reaction rate (vo) was first determined from the value of the slope from the plot of product formation as a function of time. The values of Vmax and Km were extracted from the slope and y-intercept of the Hanes plot generated by monitoring the product formation and using eqn (1):
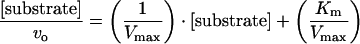 |
(1) |
where [substrate] represents the substrate concentration. The kcat was calculated by dividing Vmax by the concentration of enzyme in the reaction. A molar absorption coefficient (ε) for the product of the enzymic reaction of 3800 M−1·cm−1 was used [8]. The calculated value for the path length of the well using a 250 μl volume for the reaction was 0.904 cm. The analyses were performed in triplicate.
RESULTS
Cloning of the cABC I gene from the P. vulgaris genome
The gene for cABC I was cloned from P. vulgaris genomic DNA as a full-length version and as the mature enzyme, without its putative leader sequence. The PCR product of approx. 3 kb was subcloned into pET-28a, via an intermediate TOPO cloning step, to facilitate its incorporation and expression in E. coli. cABC I was expressed in E. coli as described above, with an N-terminal His6 tag. The histidine tag enabled quick purification of the enzyme over a charged Ni2+ column. Expression of the initial clone resulted in an enzyme with very low activity against GalAG substrates.
DNA sequencing analysis revealed a number of differences between our sequence and the previously published sequence of the gene (GenBank® accession number E08025). The major irregularity was observed in the resulting amino acid sequence between residues 494–530, which can be attributed to a pair of frame-shift errors in the published DNA sequence [13]. After position 1771, there should be an additional cytosine base (underlined) in the published sequence (CGC CCT G instead of CGC CTG), that would result in a proline residue instead of leucine at position 494. At position 1870, there is an additional thymidine base (underlined) which should be removed (TCA GTG GGT instead of CAG TTG GGT), thereby resulting in a better alignment between the published sequence and our cloned sequence. Other errors in the sequence of Sato et al. [13] produce differences in amino acids at positions 125 (proline instead of leucine), 369 (valine instead of methionine), 670 (glycine instead of alanine) and 865 (arginine instead of serine). These errors in the previously published sequence have also been observed by Huang et al. [11].
At the protein level, the sequence reported by Ryan et al. [14] is identical with the sequence by Huang et al. [11]. Therefore our clone is in close agreement to the sequence of Ryan et al. [14]. However, there were four point mutations present in our clone at variance with the sequence suggested by Ryan et al. [14]. Since expression of our initial clone resulted in a low-activity enzyme, we decided to ‘repair’ these point mutations using site-directed mutagenesis techniques. All four point mutations were corrected so as to conform precisely with the protein sequences reported in the crystal structure [11] and by Ryan et al. [14].
Recombinant expression and purification of cABC I
To establish the functionality of our ‘fixed’ cABC I clone, we set out to recombinantly express the protein in E. coli. Expression of the original full-length clone generated an enzyme almost wholly present in the insoluble fraction. The yield of soluble recombinant enzyme was greatly improved by the engineered removal of the hydrophobic N-terminal signal sequence. This result is consistent with most GAG-degrading enzymes that we have studied [8]. This sequence tag is most likely responsible for targeting to a specific location in the periplasm. cABC I purification generally yielded upwards of 35 mg of protein from 500 ml of culture (Table 2). SDS/PAGE analysis (Figure 1) revealed a highly pure band at approx. 110 kDa, in close agreement with previously reported masses of cABC I [9,13] and its theoretical mass of 112614 Da based solely on amino acid composition.
Table 2. Purification of recombinant cABC I.
| Fraction | Protein yield (mg) | Specific activity (1 mg/ml C6S) (units/mg) | x-Fold purification* |
|---|---|---|---|
| Crude lysate | 195 | 92.1 | |
| Elution from Ni2+ column | 35 | 197.9 | 2.1 |
| Thrombin cleavage† | 35 | 230.6 | 2.5 |
* The x-fold purification was determined relative to the specific activity measured for crude lysate.
† Cleavage of the His6 tag from the recombinant protein was confirmed by Western blot analysis using an anti-(His tag) antibody (Amersham Biosciences). No detectable difference in activity against GalAG substrates was observed between the recombinant cABC I and its His-tag-cleaved counterpart.
Figure 1. SDS/PAGE analysis of the purification of recombinant cABC I.

Summary of protein expression and purification following expression in BL21(DE3) as His6 N-terminal fusion proteins. Lane 1, cell pellet; lane 2, crude lysate; lane 3, Invitrogen SeeBlue Plus2 pre-stained standard; lane 4, inactive recombinant cABC I; lane 5, active recombinant cABC I (altered via site-directed mutagenesis). Sizes are indicated in kDa to the right of the gel.
A check for the absorbance at 232 nm, suggestive of the double bond formed in the degradation reaction, was performed spectrophotometrically with 5.0 μg of each recombinant enzyme (the original and the clone that underwent mutagenesis repair) and 1 mg/ml C6S or DS. Expression and purification of the ‘fixed’ truncated clone restored robust processing activity against a variety of GAG substrates. The original enzyme showed negligible activity against both of the GalAG substrates, whereas the ‘fixed’ version acted on both C6S and DS at healthy rates.
Biochemical conditions for optimal in vitro activity
Having established the broad substrate specificity of the recombinant cABC I, we next sought to optimize the reaction conditions so as to achieve maximal enzyme activity. These parameters included temperature, pH, ionic strength and dependence on sodium acetate. A Tris buffer system was chosen, as it resulted in a greater relative activity than phosphate buffer. The enzyme displayed maximal activity at pH 8.0 and was essentially inactive for both C6S and DS at pH 9.0 (Figure 2).
Figure 2. cABC I biochemical reaction conditions.

(A) pH profile. Inset, effect of buffer system on enzyme activity: (1) 50 mM Tris buffer, pH 8.0; (2) 50 mM sodium phosphate buffer, pH 8.0; open bars are DS; closed bars are C6S. (B) Effect of reaction temperature. (C) NaCl titration. (D) Sodium acetate titration. For all panels: ○, DS; ●, C6S.
We also examined the recombinant enzyme's activity against C6S and DS with regard to ionic strength. Recombinant cABC I was optimally active at 62.5 mM NaCl for C6S and 125 mM for DS. For C6S, 50% inhibition occurred at slightly more than 125 mM NaCl, with activity virtually ablated at approx. 400 mM NaCl. For DS, 50% inhibition occurred at ∼250 mM NaCl, and activity was essentially negligible at >500 mM NaCl.
In terms of temperature optima, with C6S as the substrate, the recombinant enzyme demonstrated maximal activity at 37 °C. At slightly over 40 °C, enzyme activity was 50% inhibited, and activity fell dramatically at 45 °C. For DS, a greater level of enzyme activity was evident over the range from 25 to 40 °C, with an optimum between 30 and 37 °C. At 45 °C, processing of DS by the recombinant cABC I was inhibited by over 60%. For both GalAG substrates, 37 °C was chosen as the optimal temperature for biochemical experiments.
It has been reported previously that acetate promotes cABC I activity [9]. Our investigation found that 50 mM sodium acetate provided optimal activity with C6S as the substrate, and 100 mM sodium acetate with DS as the substrate. An absence of sodium acetate in the reaction buffer inhibited enzyme activity against C6S by approx. 50%, and resulted in an almost complete decline in activity against DS.
cABC I activity analysis
The specific activity of recombinant cABC I acting on a variety of substrates was assessed (Table 3). These experiments are consistent with previously reported data for the cABC I enzyme purified from P. vulgaris and available commercially as “protease-free chondroitinase ABC I” from Seikagaku [9]. Kinetic parameters were determined for recombinant cABC I against C6S, DS and C4S substrates, and are summarized in Table 4. Our kinetic analysis corroborates our specific activity results, wherein cABC I seems to prefer C4S and C6S over DS (Figure 3).
Table 3. Specific activity of recombinant cABC I on GAG substrates.
Specific activity was determined by monitoring the increase in A232 for 5 min. The initial rate of increase in A232 was determined for each substrate. The enzyme activity in units (1 unit=1 μmol of product formed/min) was calculated from the initial rate using ε=3800 M−1·cm−1 for reaction products at pH 8.0. n.d., not detected.
| Substrate | Specific activity (units/mg of protein) |
|---|---|
| C4S | 290.8 |
| C6S | 174.6 |
| DS | 122.3 |
| Chondroitin | 69.8 |
| Chondroitin sulphate D | 54.2 |
| Chondroitin sulphate E | 34.8 |
| Hyaluronan | 14.8 |
| Heparin/heparan sulphate | n.d. |
Table 4. Kinetic analysis of cABC I with various substrates.
Results are the means±S.D. for at least three experiments.
| Substrate | Km (μM) | kcat (min−1) | kcat/Km (μM−1·min−1) |
|---|---|---|---|
| C6S | 1.2±0.6 | 37000±6500 | 31000 |
| DS | 2.5±0.5 | 27000±2500 | 11000 |
| C4S | 1.5±0.1 | 52000±1300 | 35000 |
Figure 3. Kinetic analysis of cABC I on GalAG substrates.

Hanes representations of recombinant cABC I on C4S (●), C6S (○) and DS (▼). Inset, a comparison of enzyme activity on 1 mg/ml C6S substrate in 50 mM Tris buffer, pH 8.0, with 50 mM sodium acetate. Expression of the original clone (▲) results in a protein with negligible activity; expression of the clone manipulated to conform to the desired sequence (△) results in robust enzyme activity.
We also compared the specific activity of our recombinant active cABC I with the commercially available purified “protease-free chondroitinase ABC I” from Seikagaku. Rate of product formation (ΔA232/min) was measured for 2–4 min at 37 °C using a 1 mg/ml C6S solution in 50 mM Tris buffer (pH 8.0) containing 50 mM sodium acetate. The Bradford assay was used to calculate the amount of protein present in each sample. Based on the results, our recombinant enzyme gave us a specific activity value of 164.2 m-units/μg, whereas the Seikagaku enzyme had a specific activity of 20.2 m-units/μg. The difference in specific activity could be the result of enzyme stability or storage conditions.
The activity of cABC I on these three substrates was also analysed by CE. This study represented an end-point assay for activity, and allowed for a characterization of the final products of cABC I digestion on all of the substrates after an 18 h incubation at 37 °C. For C4S and C6S, the product profile shows predominantly disaccharide products, with minor tetrasaccharide products also detected (Figure 4). In both of these cases, the respective monosulphated disaccharide [i.e. ΔUA-GalNAc4S or ΔUA-GalNAc6S, where ΔUA is Δ4,5 unsaturated uronic acid and GalNAc is N-acetylgalactosamine either 4-O-sulphated (4S) or 6-O-sulphated (6S)] represents the major product. In the case of DS, we also observed a mixture of disaccharides and tetrasaccharides as the final products of digestion. However, one of the tetrasaccharide peaks is much larger than those observed for the C4S and C6S substrates. This suggests that there may be some resistance by tetrasaccharide fragments within DS to cleavage by cABC I. The structure of the resistant tetrasaccharide was determined to be ΔUA-GalNAc4S-IdoA-GalNAc4S (IdoA is iduronic acid) based on co-elution (on CE) with a previously isolated pure dermatan tetrasaccharide having the same structure. In order to confirm that this tetrasaccharide is indeed resistant to cABC I action, we incubated the pure tetrasaccharide with enzyme at 37 °C and analysed the resulting products by CE. The CE trace showed that there is no breakdown of the tetrasaccharide, thereby confirming that cABC I cannot degrade this tetrasaccharide fraction in DS. The diminished ability of the recombinant cABC I to cleave DS tetrasaccharides is consistent with previous reports on cABC I action pattern [9].
Figure 4. CE analysis of recombinant active cABC I.

Product profiles for cABC I acting on (A) C4S, (B) C6S and (C) DS. ΔDi4S, ΔUA-GalNAc4S; ΔDi6S, ΔUA-GalNAc6S; ΔDi2S6S, ΔUA2S-GalNAc6S; ΔDi4S6S, ΔUA-GalNAc4S6S; Tetra, tetrasaccharide. Impurities in commercial substrate preparations result in the ΔDi6S peak in electrophoretogram (A) and the ΔDi4S peak in electrophoretogram (B). mAU, milli-absorbance units.
Mutagenesis studies
A comparison between the crystal structures of F. heparinum cAC and P. vulgaris cABC I revealed a similar linear arrangement of domains that are superficially similar in terms of overall structure. On a closer inspection of the catalytic domain of cAC with the middle domain of cABC I, one can observe a paucity of sequence identity. On comparison of these domains between the two enzymes, the lack of sequence identity persists throughout, except for this group of amino acids, which have been implicated previously as active-site players in cAC [20] and that seem to have counterparts in cABC I. Using this framework of potential conserved catalytic active site residues, we undertook site-directed mutagenesis studies to probe the importance of several residues in enzyme activity (Table 5). His501, Tyr508, Glu653 and Arg560 were all mutated to alanine and characterized in activity experiments. These residues were suggested previously by Huang et al. [11], and could constitute the active site, whereby the proton acceptance and donation mechanism could take place [21]. Since a general base, such as histidine, is believed to be a key component for this catalysis, we also mutated histidine residues (His561 and His712). These residues are not conserved between the two enzymes, but are in close proximity to the proposed active site.
Table 5. Activity analysis of cABC I mutants.
Kinetic parameters are reported in μM (Km) and min−1 (kcat). n.d., activity was too low to be detected.+refers to an exhaustive digestion of the substrate; − indicates that no products were detected.
| C6S kinetics | DS kinetics | Activity on CE | |||||
|---|---|---|---|---|---|---|---|
| cABC I mutant | Km | kcat | Km | kcat | C6S | C4S | DS |
| His501→Ala | n.d. | n.d. | n.d. | n.d. | − | − | − |
| His561→Ala | 15.2 | 39100 | 6.90 | 3970 | + | + | + |
| His712→Ala | 8.60 | 1140 | 5.10 | 613 | + | + | + |
| Tyr508→Ala | n.d. | n.d. | n.d. | n.d. | − | − | − |
| Arg560→Ala | n.d. | n.d. | n.d. | n.d. | − | − | −* |
| Arg500→Ala | 19.9 | 419 | 35.7 | 162 | + | + | + |
| Glu653→Ala | n.d. | n.d. | n.d. | n.d. | − | − | − |
* The Arg560→Ala mutant did display some residual activity on DS.
Examination of the mutant enzymes in an end-point assay by CE demonstrated a number of residues that seem to be absolutely critical to catalysis. His501→Ala produced no products on overnight digestion with C6S, DS or C4S as the substrate (Figure 5). This is in sharp contrast with His561→Ala and His712→Ala, which both produced a product profile comparable with recombinant active cABC I against all three of these GalAGs after an overnight digestion. Tyr508→Ala, Glu653→Ala and Arg560→Ala were all unable to yield products in an exhaustive digestion with any of these substrates. These observations provide direct evidence that His501, Tyr508, Glu653 and Arg560 are crucial for the activity of our recombinant cABC I. The relative positions between these critical residues within the active-site cleft of cABC I lends further credence to the notion of this amino acid synergy as the enzyme active site.
Figure 5. Representative CE profiles of cABC I mutants.

Products of C6S degradation following digestion by (A) His501→Ala and (B) His561→Ala cABC I mutants. Tables 4 and 5 provide further information regarding the substrate specificity of recombinant cABC I and its mutants. ΔDi4S, ΔUA-GalNAc4S; ΔDi6S, ΔUA-GalNAc6S; ΔDi2S6S, ΔUA2S-GalNAc6S. mAU, milliabsorbance units.
CD spectroscopy was used to analyse the overall secondary structure of the proteins. The resulting spectra displayed a high α-helix and β-sheet content (Figure 6). These results are in agreement with the crystal structure of cABC I [11]. CD analysis was also used to confirm that the loss of activity displayed by the inactive mutants (His501→Ala, Tyr508→Ala, Glu653→Ala and Arg560→Ala) was not due to an alteration of the overall secondary structure of the protein. As shown in Figure 6, no significant differences on the CD spectra between the mutants and the recombinant cABC I were observed. To address further the effect of mutations on protein stability, heat-denaturation studies were used to compare the recombinant enzyme with its mutants. All proteins displayed melting transitions of 45±5 °C (Figure 6, inset). Although these results do not exclude the possibility of minor structural perturbations in the local environment, they do suggest that the overall structure and stability of the protein are not compromised when these particular residues are mutated to alanine.
Figure 6. CD spectra of cABC I and the inactive mutants.

The recombinant cABC I (——) and the mutants His501→Ala (····), Tyr508→Ala (– –), Glu653→Ala (–··–) and Arg560→Ala (— —) were concentrated and buffer-exchanged into 50 mM sodium phosphate buffer, pH 8.0. Proteins were analysed in a quartz cell with a 1 mm path length. All spectra were collected using a protein concentration of 0.2 mg/ml in sodium phosphate, pH 7.0. For melting experiments (inset), spectra were collected in 5 °C intervals from 5 to 80 °C. The slight deviations in spectra intensity can be attributed to errors inherent in protein quantification.
DISCUSSION
In the present paper, we report the subcloning of the cABC I gene from P. vulgaris, and its recombinant expression in E. coli. We also examine this recombinant cABC I biochemically, providing the first conclusive evidence of the residues that constitute the enzyme active site. The establishment of a well-characterized enzyme with defined GalAG substrate specificity provides insight into important structure–function relationships in biology.
Purification of cABC I directly from cultures of P. vulgaris resulted in preparations with low yields and considerable protease contamination [22,23]. These conditions spurred investigators toward recombinant production approaches. Early attempts to recombinantly express cABC I in E. coli were laden with difficulty. Expression of a soluble protein was hampered by both the size of the gene and the signal sequence. Random proteolysis also proved to be a nuisance to achieving the recombinant protein, and this complication has been observed previously [13]. Our one-step purification process allowed for the preparation of an abundance of soluble protein. It was, however, discovered that this protein lacked the ability to effectively degrade GalAG substrates. Sequence anomalies in the original clone suggest that there may have been underlying structural changes responsible for this eradication of enzyme activity.
The sequential repair via site-directed mutagenesis of the original truncated clone was accompanied by a restoration of enzyme activity. Indeed, the ‘repaired’ recombinant enzyme was able to process C4S, DS and C6S at robust rates. It was also able to degrade a variety of other GalAG substrates and hyaluronan at lesser rates. It is clear that our recombinant active cABC I possessed a specific activity that far exceeded that of the commercially available enzyme. Therefore it is evident that our cloning, expression and purification system does not at all compromise the activity of our cloned enzyme. It is possible that the disparity in specific activities between our recombinant enzyme and the commercially available cABC I is the result of different purification and storage practices rather than intrinsic enzymic properties. It is also feasible that an overestimation of active protein content for the commercial enzyme led to a dramatically diminished observed rate, as some portion of quantified protein may have been distorted in the isolation process.
The optimal conditions for activity of our recombinant cABC I are similar to those obtained for the purified enzyme. However, we also observed that different buffer systems affect the processing activity on different substrates. For enzyme activity on C6S in 50 mM sodium phosphate buffer, the optimal pH was determined to be pH 7.0; however, in 50 mM Tris buffer, optimal activity was observed at pH 8.0. This discrepancy was not observed with DS as substrate, where the activity maximum was at pH 8.0 regardless of the buffer system. In both cases, however, the enzyme showed more activity in Tris buffer than in phosphate buffer. It was also observed that there was a slight difference in the concentration of NaCl and sodium acetate required for maximum activity on C6S as compared with DS. For DS, a higher concentration (approximately double) of both NaCl and sodium acetate in the buffer showed the highest activity in terms of product formation. Another interesting observation is the absolute importance of the presence of salt, i.e. either NaCl or sodium acetate, in the buffer system for activity of recombinant cABC I on DS. From Figures 2(C) and 2(D), we can see that, in the absence of any salt in the buffer, the enzyme activity on DS is approx. 5% of the observed maximum activity. However, with C6S as substrate, even when there is no salt in the buffer, we observe approx. 50–60% of the maximum activity.
We hypothesize that the salt requirement for DS is important in abolishing non-specific interactions of this substrate with the enzyme. DS has considerable intrinsic flexibility due to the presence of IdoA in its structure. Therefore it can potentially display a wider range of interactions than C6S, and may bind to positively charged patches on the surface of the enzyme, rather than in the active site. In the presence of salt, these non-specific interactions would be reduced markedly.
Our results with CE indicated that cABC I was unable to cleave a tetrasaccharide fragment within DS, and we identified this fragment to be ΔUA-GalNAc4S-IdoA-GalNAc4S. This is in contrast with the product profile obtained on treating DS with cB, where the major products are predominantly disaccharides [19]. This is not surprising since cABC I and cB have totally different structures and therefore may bind to and process DS very differently.
The crystal structure of cABC I [11] revealed a three-domain protein. The middle domain contains the catalytic site in a wide-open cleft. Despite very limited sequence identity with the catalytic domain of F. heparinum cAC, this middle domain of cABC I did contain a conserved grouping of residues that were implicated in catalysis in cAC [20]. These cABC I residues were His501, Tyr508, Arg560 and Glu653. Manipulation of these residues via mutagenesis to alanine resulted in knockout proteins: enzymes with a complete inability to degrade GAG substrates. Thus this tetrad of residues is crucial for enzyme activity (Figure 7). With regard to the β-elimination mechanism suggested previously for GAG lyases, it seems that this group of residues is potentially capable of performing the stabilization and proton shuffling responsibilities required for GAG degradation. The present study provides the first experimental evidence that this grouping of amino acids comprises the cABC I active site.
Figure 7. Active-site tetrad of cABC I.

Structural representation of cABC I. Inset, the active site residues of cABC I are shown in full.
It was demonstrated that His501 was in fact the histidine residue critical for catalysis. Two other histidine residues were examined, His561 and His712. Alanine mutants of these residues demonstrated that these histidine residues were not critical for GAG degradation. In fact, on an exhaustive digestion with GalAG substrate, both of these mutants provided a full product profile.
Previously, it was suggested that Arg500 was essential for cABC I's ability to process both CS and DS [11]. Arg500's side chain was predicted to be positioned toward the uronate carboxy group of either substrate, serving some role in charge neutralization. However, our product profile analysis and specific activity determinations with the Arg500→Ala mutant suggest that this residue is not actually critical for catalysis. In fact, with C6S, C4S and DS, an exhaustive digestion with Arg500→Ala resulted in a product profile virtually indistinguishable from those generated with our recombinant cABC I.
cABC I's broad substrate specificity complicates fine understanding of its GalAG-degradation mechanism. The present study outlines the cloning and expression of cABC I, provides a biochemical characterization of this enzyme and offers the first conclusive proof of the active site. This will allow for the further biochemical characterization of the enzyme, an essential element in advancing GAG sequencing biotechnology [16,17].
Acknowledgments
We thank Dr James Myette for helpful discussions, Dr Shiladitya Sengupta and Dr Sujan Rafiul Kabir for technical assistance, and the Multiuser Facility for the Study of Complex Macromolecular Systems (NSF-0070319 and NIH-GM68762) for access to instrumentation. This work was supported by National Institutes of Health (NIH) Grant GM57073. V.P. was funded through the NIH Biotechnology Training Grant (5-T32-GM08334) and a Dupont Fellowship (Massachusetts Institute of Technology). C.J.B. was funded through the NIH/National Institute of Environmental Health Sciences (NIEHS) Training Grant in Environmental Toxicology (5-T32-ES0720).
References
- 1.Bernfield M., Gotte M., Park P. W., Reizes O., Fitzgerald M. L., Lincecum J., Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 2.Sugahara K., Mikami T., Uyama T., Mizuguchi S., Nomura K., Kitagawa H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 2003;13:612–620. doi: 10.1016/j.sbi.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Bao X., Nishimura S., Mikami T., Yamada S., Itoh N., Sugahara K. Chondroitin sulfate/dermatan sulfate hybrid chains from embryonic pig brain, which contain a higher proportion of L-iduronic acid than those from adult pig brain, exhibit neuritogenic and growth factor binding activities. J. Biol. Chem. 2004;279:9765–9776. doi: 10.1074/jbc.M310877200. [DOI] [PubMed] [Google Scholar]
- 4.Ernst S., Langer R., Cooney C. L., Sasisekharan R. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 1995;30:387–444. doi: 10.3109/10409239509083490. [DOI] [PubMed] [Google Scholar]
- 5.Venkataraman G., Shriver Z., Raman R., Sasisekharan R. Sequencing complex polysaccharides. Science. 1999;286:537–542. doi: 10.1126/science.286.5439.537. [DOI] [PubMed] [Google Scholar]
- 6.Sasisekharan R., Bulmer M., Moremen K. W., Cooney C. L., Langer R. Cloning and expression of heparinase I gene from Flavobacterium heparinum. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3660–3664. doi: 10.1073/pnas.90.8.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godavarti R., Davis M., Venkataraman G., Cooney C., Langer R., Sasisekharan R. Heparinase III from Flavobacterium heparinum: cloning and recombinant expression in Escherichia coli. Biochem. Biophys. Res. Commun. 1996;225:751–758. doi: 10.1006/bbrc.1996.1246. [DOI] [PubMed] [Google Scholar]
- 8.Pojasek K., Shriver Z., Kiley P., Venkataraman G., Sasisekharan R. Recombinant expression, purification, and kinetic characterization of chondroitinase AC and chondroitinase B from Flavobacterium heparinum. Biochem. Biophys. Res. Commun. 2001;286:343–351. doi: 10.1006/bbrc.2001.5380. [DOI] [PubMed] [Google Scholar]
- 9.Hamai A., Hashimoto N., Mochizuki H., Kato F., Makiguchi Y., Horie K., Suzuki S. Two distinct chondroitin sulfate ABC lyases: an endoeliminase yielding tetrasaccharides and an exoeliminase preferentially acting on oligosaccharides. J. Biol. Chem. 1997;272:9123–9130. doi: 10.1074/jbc.272.14.9123. [DOI] [PubMed] [Google Scholar]
- 10.Huang W., Matte A., Li Y., Kim Y. S., Linhardt R. J., Su H., Cygler M. Crystal structure of chondroitinase B from Flavobacterium heparinum and its complex with a disaccharide product at 1.7 Å resolution. J. Mol. Biol. 1999;294:1257–1269. doi: 10.1006/jmbi.1999.3292. [DOI] [PubMed] [Google Scholar]
- 11.Huang W., Lunin V. V., Li Y., Suzuki S., Sugiura N., Miyazono H., Cygler M. Crystal structure of Proteus vulgaris chondroitin sulfate ABC lyase I at 1.9 Å resolution. J. Mol. Biol. 2003;328:623–634. doi: 10.1016/s0022-2836(03)00345-0. [DOI] [PubMed] [Google Scholar]
- 12.Fethiere J., Eggimann B., Cygler M. Crystal structure of chondroitin AC lyase, a representative of a family of glycosaminoglycan degrading enzymes. J. Mol. Biol. 1999;288:635–647. doi: 10.1006/jmbi.1999.2698. [DOI] [PubMed] [Google Scholar]
- 13.Sato N., Shimada M., Nakajima H., Oda H., Kimura S. Cloning and expression in Escherichia coli of the gene encoding the Proteus vulgaris chondroitin ABC lyase. Appl. Microbiol. Biotechnol. 1994;41:39–46. doi: 10.1007/BF00166079. [DOI] [PubMed] [Google Scholar]
- 14.Ryan M. J., Khandke K. M., Tilley B. C., Lotvin J. A. Cloning and expression of the chondroitinase I and II genes from Proteus vulgaris. Pat. WO 94/25567. 1994
- 15.Bradbury E. J., Moon L. D., Popat R. J., King V. R., Bennett G. S., Patel P. N., Fawcett J. W., McMahon S. B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature (London) 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- 16.Morgenstern D. A., Asher R. A., Fawcett J. W. Chondroitin sulphate proteoglycans in the CNS injury response. Prog. Brain Res. 2002;137:313–332. doi: 10.1016/s0079-6123(02)37024-9. [DOI] [PubMed] [Google Scholar]
- 17.Myette J. R., Shriver Z., Kiziltepe T., McLean M. W., Venkataraman G., Sasisekharan R. Molecular cloning of the heparin/heparan sulfate Δ4,5 unsaturated glycuronidase from Flavobacterium heparinum, its recombinant expression in Escherichia coli, and biochemical determination of its unique substrate specificity. Biochemistry. 2002;41:7424–7434. doi: 10.1021/bi012147o. [DOI] [PubMed] [Google Scholar]
- 18.Rhomberg A. J., Ernst S., Sasisekharan R., Biemann K. Mass spectrometric and capillary electrophoretic investigation of the enzymatic degradation of heparin-like glycosaminoglycans. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4176–4181. doi: 10.1073/pnas.95.8.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pojasek K., Raman R., Kiley P., Venkataraman G., Sasisekharan R. Biochemical characterization of the chondroitinase B active site. J. Biol. Chem. 2002;277:31179–31186. doi: 10.1074/jbc.M201552200. [DOI] [PubMed] [Google Scholar]
- 20.Huang W., Boju L., Tkalec L., Su H., Yang H. O., Gunay N. S., Linhardt R. J., Kim Y. S., Matte A., Cygler M. Active site of chondroitin AC lyase revealed by the structure of enzyme–oligosaccharide complexes and mutagenesis. Biochemistry. 2001;40:2359–2372. doi: 10.1021/bi0024254. [DOI] [PubMed] [Google Scholar]
- 21.Jedrzejas M. J. Structural and functional comparison of polysaccharide-degrading enzymes. Crit. Rev. Biochem. Mol. Biol. 2000;35:221–251. doi: 10.1080/10409230091169195. [DOI] [PubMed] [Google Scholar]
- 22.Oike Y., Kimata K., Shinomura T., Suzuki S. Proteinase activity in chondroitin lyase (chondroitinase) and endo-β-D-galactosidase (keratanase) preparations and a method to abolish their proteolytic effect on proteoglycan. Biochem. J. 1980;191:203–207. doi: 10.1042/bj1910203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrisson F., van Hoof J., Vanroelen C. On the presence of proteolytic activity in glycosaminoglycan-degrading enzyme preparations. J. Histochem. Cytochem. 1986;34:1231–1235. doi: 10.1177/34.9.3525669. [DOI] [PubMed] [Google Scholar]