

Abstract
UPPS (undecaprenyl pyrophosphate synthase) catalyses consecutive condensation reactions of FPP (farnesyl pyrophosphate) with eight isopentenyl pyrophosphates to generate C55 UPP, which serves as a lipid carrier for bacterial peptidoglycan biosynthesis. We reported the co-crystal structure of Escherichia coli UPPS in complex with FPP. Its phosphate head-group is bound to positively charged arginine residues and the hydrocarbon moiety interacts with hydrophobic amino acids including L85, L88 and F89, located on the α3 helix of UPPS. We now show that the monophosphate analogue of FPP binds UPPS with an eight times lower affinity (Kd=4.4 μM) compared with the pyrophosphate analogue, a result of a larger dissociation rate constant (koff=192 s−1). Farnesol (1 mM) lacking the pyrophosphate does not inhibit the UPPS reaction. GGPP (geranylgeranyl pyrophosphate) containing a larger C20 hydrocarbon tail is an equally good substrate (Km=0.3 μM and kcat=2.1 s−1) compared with FPP. The shorter C10 GPP (geranyl pyrophosphate) displays a 90-fold larger Km value (36.0±0.1 μM) but similar kcat value (1.7±0.1 s−1) compared with FPP. Replacement of L85, L88 or F89 with Ala increases FPP and GGPP Km values by the same amount, indicating that these amino acids are important for substrate binding, but do not determine substrate specificity. With GGPP as a substrate, UPPS still catalyses eight isopentenyl pyrophosphate condensation reactions to synthesize C60 product. Computer modelling suggests that the upper portion of the active-site tunnel, where cis double bonds of the product reside, may be critical for determining the final product chain length.
Keywords: computer modelling, prenyltransferase, product specificity, site-directed mutagenesis, substrate specificity, undecaprenyl pyrophosphate synthase
Abbreviations: FPP, farnesyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate; GPP, geranyl pyrophosphate; IPP, isopentenyl pyrophosphate; LB, Luria–Bertani; OPPS, octaprenyl pyrophosphate synthase; TFMC-GP, 7-(2,6-dimethyl-8-phospho-2,6-octadienyloxy)-8-methyl-4-trifluoromethyl-chromen-2-one; TFMC-GPP, 7-(2,6-dimethyl-8-pyrophospho-2,6-octadienyloxy)-8-methyl-4-trifluoromethyl-chromen-2-one; UPP, undecaprenyl pyrophosphate; UPPS, UPP synthase; for brevity, the single-letter system for amino acids has been used: L85, for example means Leu-85
INTRODUCTION
A group of prenyltransferases catalyse chain elongation of C15 FPP (farnesyl pyrophosphate) (allylic substrate) to designated chain lengths through condensation reactions with specified numbers of C5 IPP (isopentenyl pyrophosphate) (homoallylic substrate) [1–3]. According to the stereochemistry of the double bonds formed from each IPP condensation, these prenyltransferases are classified as cis- or trans-type [4]. The trans-type prenyltransferases, such as OPPS (octaprenyl pyrophosphate synthase), bind FPP and IPP substrates presumably through two conserved Asp-rich DDXXD motifs, which chelate the Mg2+ ion to bind the pyrophosphate group [5–7]. In contrast, the cis-type UPPS (undecaprenyl pyrophosphate synthase) that catalyses consecutive condensation reactions of FPP with eight molecules of IPP to form the C55 product, binds its substrates through H-bonds, and through charged and hydrophobic interactions [8–10]. The enzyme product serves as a lipid carrier to transport lipid II (made in the cytoplasm) across the cell membrane for bacterial peptidoglycan biosynthesis [11]. UPPS is the first and the only cis-prenyltransferase whose three-dimensional structure has been solved and is being intensively studied as a model to understand the catalytic mechanism of cis-prenyltransferases [12]. Moreover, UPPS may serve as a new antibiotic target since it has been proven to be essential for bacterial survival [13].
We have solved the crystal structures of UPPS alone, the enzyme in complex with substrate and product analogues (ammonium sulphate and Triton X-100), and also the complex with the FPP substrate [9,10,12]. In this structure, FPP is bound at the top of the tunnel-shaped active site surrounded by the two α-helices (α2 and α3) and four β-strands (βA, βB, βD and βC) as shown in Figure 1(A) [9]. On the basis of the crystal structure [10], the pyrophosphate group of FPP is bound to the backbone NHs of G29 and R30 as well as to the side chains of N28, R30 and R39 through hydrogen bonds (Figure 1B). It was not determined how the pyrophosphate head group and the hydrocarbon tail of FPP contribute to the enzyme–substrate interaction. Previously, we synthesized an FPP analogue TFMC-GPP [7-(2,6-dimethyl-8 -pyrophospho-2,6-octadienyl-oxy)-8-methyl-4-trifluoromethyl-chromen-2-one], incorporating a fluorescent trifluoromethyl-chromen-2-one cross-linking to C10-GPP (geranyl pyrophosphate) [14]. In the present study, we have synthesized TFMC-GP [7-(2,6-dimethyl-8-phospho-2,6-octadienyloxy)-8-methyl-4-trifluoromethyl-chromen-2-one], containing a monophosphate group as a probe to evaluate its enzyme binding affinity, in comparison with the diphosphate substrate analogue and the phosphate-free farnesol.
Figure 1. Three-dimensional structure of UPPS in complex with FPP.

(A) The active site portion of UPPS surrounded by two α-helices (α2 and α3) and four β-strands (βA, βB, βD and βC) and bound FPP on the top of the tunnel is shown based on our published structure [10]. At the bottom of the tunnel, the large amino acid L137 acts as a seal to block further chain elongation and determine the chain length of the product [12]. The catalytically important amino acids including D26, H43, R30 and R39 near FPP are displayed. (B) The pyrophosphate of FPP is hydrogen-bonded to the backbone NH and side chain of R30, and backbone NH of G29 as well as the side-chains of R39 and N28 [10]. The hydrocarbon moiety of FPP is bound with hydrophobic amino acids including L85, L88 and F89, located on the α3 helix that moves towards the substrate during FPP binding. The distances are shown in Å (1 Å=0.1 nm).
For testing the enzyme–substrate interaction mediated by the hydrocarbon moiety of the allylic substrate, reaction kinetics parameters for using all-trans C10-GPP and C20-GGPP (genanylgeranyl pyrophosphate) as alternative allylic substrates were determined. The possibility of using FPP or GPP as a homoallylic substrate was also tested. The hydrocarbon moiety of FPP is bound to the hydrophobic amino acids including L85, L88 and F89, located on the α3 helix. These amino acids were mutated to probe the roles of L85, L88 and F89 in substrate recognition.
FPP is elongated by condensation with eight IPP molecules towards the bottom of the tunnel sealed by the large amino acid L137 (see Figure 1A), which blocks further chain elongation and determines the final chain length of the C55-product [12]. The reason why cis-prenyltransferases synthesize longer chain-length products than the trans-prenyltransferases has long been puzzling. As shown in the present study, the structure of C60-products resulting from the larger C20-GGPP substrates predicted by computer modelling is compared with that of the assumed C60-product synthesized from FPP and one extra IPP. On the basis of the results, we propose that the molecular shapes of both the active site of UPPS and its product are essential to determine the ultimate product chain length.
EXPERIMENTAL
Materials
Radiolabelled [14C]IPP (55 mCi/mmol) was purchased from Amersham Biosciences and FPP was obtained from Sigma. Taq DNA polymerase was obtained from Life Technologies. The plasmid mini-prep kit, the DNA gel extraction kit and the Ni2+-nitrilotriacetate resin were purchased from Qiagen. Potato acid phosphatase (2 units/mg) was purchased from Roche Molecular Biochemicals. FXa and the protein expression kit (including the pET32Xa/LIC vector and competent JM109 and BL21 cells) were obtained from Novagen. All commercial buffers and reagents were of the highest grade available.
Synthesis of TFMC-GP
The synthetic route for compound 2 (TFMC-GP) is shown in Scheme 1. To the solution containing 7-(8-bromo-2,6-dimethyl-2, 6-octadienyloxy)-8-methyl-1, 4-trifluoromethyl-chromen-2-one (compound 1) (230 mg, 0.5 mmol), which was synthesized according to our reported procedure [14], and CH3CN (2 ml), was added tetrabutylammonium dihydrogen phosphate (340 mg, 1.0 mmol) in 6 ml of CH3CN at 25 °C. The solution was stirred at room temperature (25 °C) for 6 h. After evaporation of solvent, the residue was dissolved in 3 ml of propan-2-ol/water (1:1) mixture and loaded on to an anion-exchange column of Dowex AG50X80 (NH4+ form) eluted with propan-2-ol/water (1:1) to give the crude product. The desired fractions were collected, the organic solvent was removed by a rotary evaporator and the remaining water was freeze-dried to yield a white solid. The solid was further purified by reversed-phase HPLC using a program of 20% B (80% A) for 5 min, followed by a linear gradient of 20% B (80% A) to 100% B (0% A) over 30 min with a flow rate of 2.5 ml/min [where A=25 mM aqueous ammonium bicarbonate (pH 8.0), B=acetonitrile]. The desired peak was collected, frozen and freeze-dried to afford the final product TFMC-GP (30%). 1H NMR (400 MHz, CD3OD) δ 1.68 (s, 3H), 1.73 (s, 3H), 2.07 (t, 2H, J=7.6 Hz), 2.21 (m, 2H), 2.27 (s, 3H), 4.39 (t, 2H, J=6.8 Hz), 4.57 (s, 2H), 5.39 (t, 1H, J=6.8 Hz), 5.58 (t, 1H, J=6.8 Hz), 6.69 (s, 1H), 7.07 (d, 1H, J=9.2 Hz), 7.58 (d, 1H, J=9.2 Hz); 13C NMR (100 MHz, CD3OD) δ 7.55, 13.10, 15.64, 25.99, 38.91, 62.05, 74.66, 107.05, 109.76, 111.69 (q, J=6.1 Hz), 114.73, 121.52 (q, J=8.3 Hz), 121.98 (q, J=273.1 Hz), 123.55, 128.81, 130.70, 139.39, 142.08 (q, J=31.9 Hz), 153.22, 160.74, 160.98; 31P NMR (162 MHz, CD3OD) δ 6.30; UV (H2O), λmax=336 nm, ε=10700 M−1·cm−1; IR: (KBr) 3449, 1738, 1610, 1101; FAB-MS: m/z 477.1 (M+H+), HRMS (FAB): m/z calculated for C21H25F3O7P (M+H+): 477.1290, found: 477.1288.
Scheme 1. Synthesis of compound 2 from previously prepared compound 1 [14].

Measurement of the inhibition constant for TFMC-GP
Measurement of the inhibition constant for TFMC-GP was performed in a reaction mixture containing 0.03 μM UPPS, 3 μM FPP and 50 μM [14C]IPP in a buffer of 100 mM Hepes/KOH (pH 7.5), 50 mM KCl, 0.5 mM MgCl2 and 0.1% Triton X-100 in the presence of various concentrations of the inhibitor. Triton X-100 (0.1%) was included in the reaction mixture so that product release did not limit the reaction, thus allowing the measurement of the IPP condensation rate at each inhibitor concentration [15]. Portions of the reaction mixture were periodically withdrawn and mixed with 10 mM EDTA to terminate the reaction and 1-butanol was utilized to extract the radiolabelled products for quantification using scintillation counting. The initial rates were obtained in the presence of 0–50 μM inhibitor and IC50 and Ki values of the inhibitor were determined by fitting the plot of reaction rates versus the inhibitor concentrations using the following equations [16]
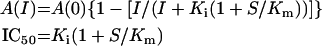 |
(1) |
In these equations, A(I) is the enzyme activity with the inhibitor concentration I, A(0) enzyme activity without inhibitor, I the inhibitor concentration, Ki the inhibition constant of the inhibitor, S the FPP concentration and Km the Michaelis constant for FPP.
Fluorescence titration experiments
The fluorescence spectrum of 1 μM TFMC-GP was collected before and after the addition of 1 μM UPPS in a buffer containing 100 mM Hepes/KOH (pH 7.5), 50 mM KCl and 0.5 mM MgCl2 at 25 °C. The spectra were also collected after the addition of 1, 2 and 3 μM FPP to outcompete TFMC-GP.
Measurements of kon and koff of the TFMC-GP using stopped-flow technology
The binding constant kon and release constant koff of the monophosphate FPP analogue (TFMC-GP) were measured using stopped-flow methods as described previously [14]. The compound's fluorescence above 530 nm (using a cut-off filter) was monitored using an excitation wavelength of 336 nm. In the measurements of kon, each concentration of UPPS (2.5, 3.75, 5 or 6.5 μM) was mixed with an equal volume of 0.5 μM TFMC-GP solution in a buffer containing 100 mM Hepes/KOH (pH 7.5), 0.5 mM MgCl2 and 50 mM KCl. The stopped-flow trace (average of four runs) monitored with time was fitted with the exponential equation shown below (eqn 3) (which was provided with the machine) to give the observed rate constant kobs for each enzyme concentration. The concentrations cited in parentheses and hereafter in the text are those after mixing. The slope of kobs versus enzyme concentration yielded the rate constant kon for the compound binding to UPPS. In the measurement of the dissociation rate constant koff, UPPS (0.5 μM) preincubated with TFMC-GP (7.5 μM) was mixed with an excess of competitor, FPP (25 μM). The stopped-flow trace was fitted with a single exponential equation (eqn 3) to obtain the koff for the compound. The Kd value for the compound binding to the UPPS was obtained from koff/kon.
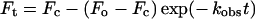 |
(3) |
In the above equation, Ft is the fluorescence at a given time point t, Fc the end-point fluorescence, Fo the initial fluorescence and kobs the observed rate constant.
Site-directed mutagenesis of UPPS
UPPS mutants were prepared by using PCR techniques in conjunction with the Escherichia coli Bos-12 UPPS gene template in the pET32Xa/Lic vector as described previously [17]. The mutagenic primers used were prepared by MDBio (Taipei, Taiwan). The mutagenic oligonucleotides for performing site-directed mutagenesis are as follows: 5′-AGTGCGGCAATGGAACTG-3′ for L85A (Leu85→Ala), 5′-ATGGAAGCGTTTGTGTGG-3′ for L88A and 5′-GAACTGGCTGTGTGGGCG-3′ for F89A. Subsequently, the forward primer 5′-GGTATTGAGGGTCGCATGTTGTCTGCT-3′ and reverse primer 5′-AGAGGAGAGTTAGAGCCATCAGGCTGT-3′ were used in combination with the PCR products obtained using the above mutagenic oligonucleotides to create the full-length mutant UPPS genes. The FXa cleavage site (IEGR) and the complementary sequences to the sticky ends of the linear vector pET-32Xa/LIC were included in these primers. Using a thermocycler (Applied Biosystems), 30 cycles of PCR were performed with the melting temperature at 95 °C for 2 min, annealing temperature at 42 °C for 1 min and polymerization temperature at 68 °C for 1 min. The PCR product was subjected to electrophoresis on 1.2% agarose gel in TAE buffer (40 mM Tris-acetate, pH 8, 1 mM EDTA), and the gel was then stained with ethidium bromide. The part of the gel containing the band of the correct size was excised, and the DNA was recovered using a DNA elution kit. The constructed gene of a mutant enzyme was ligated to the pET32Xa/LIC vector by incubation for 1 h at 22 °C. The recombinant UPPS plasmid was then used to transform E. coli JM109 competent cells that were streaked on an LB (Luria–Bertani) agar plate containing 100 μg/ml ampicillin. Ampicillin-resistant colonies were selected from the agar plate and grown in 5 ml of LB culture containing 100 μg/ml ampicillin overnight at 37 °C. The mutation was confirmed by sequencing the entire UPPS mutant gene of the plasmid obtained from the overnight culture. The correct construct was subsequently transformed to E. coli BL21 for protein expression. A 5 ml overnight culture of a single transformant was used to inoculate 500 ml of fresh LB medium containing 100 μg/ml ampicillin. The cells were grown to A600=0.6 and induced with 1 mM isopropyl β-D-thiogalactoside. After 4–5 h, the cells were harvested by centrifugation at 7000 g for 15 min. The purification of mutant UPPS was according to our reported procedure using Ni2+-nitrilotriacetate column chromatography and FXa for tag removal [17].
Measurements of Km and kcat values for wild-type and mutant UPPS
For the measurements of kinetic parameters, wild-type or mutant UPPS (0.01 μM wild-type, 0.3 μM L85A, 0.1 μM L88A or 0.1 μM F89A) was added to initiate the reaction of FPP or GGPP with [14C]IPP in 200 μl solutions. For Km and kcat determinations, a large range of substrate concentrations was used to estimate the Km for each substrate first. To measure accurately the IPP Km value, 5 μM FPP was utilized to saturate the enzyme, and IPP concentrations of 0.5–5-fold Km were employed. For FPP Km measurements, 0.2–20 μM FPP was used along with 20 μM [14C]IPP. For GGPP Km measurements, 0.2–20 μM GGPP was used along with 20 μM [14C]IPP. C10-GPP at 1–20 μM was also tested as an alternative substrate along with 20– 400 μM [14C]IPP for their Km measurements catalysed by the wild-type UPPS (0.01 μM). All reactions were performed in 100 mM KOH/Hepes buffer, pH 7.5, 50 mM KCl and 0.5 mM MgCl2 at 25 °C in the presence of 0.1% Triton X-100. To measure the initial rate, 40 μl portions of the reaction mixture were periodically withdrawn within 10% substrate depletion and mixed with 10 mM EDTA for reaction termination. The radiolabelled products were then extracted with butan-1-ol, and radioactivities associated with either aqueous or butanol phases were quantified separately by using a Beckman LS6500 scintillation counter to determine the initial reaction velocity. Initial velocity data were fitted to eqn (4) to obtain Km and kcat values by non-linear regression using the Kaleida-Graph computer program. The kcat was calculated from Vmax/[E].
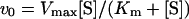 |
(4) |
where v0 is the initial velocity, [E] the enzyme concentration, [S] the substrate concentration, Vmax the maximum velocity and Km the Michaelis constant.
The final products of the UPPS reaction were prepared using 0.1 μM enzyme, 5 μM FPP, GGPP, or GPP and 50 μM [14C]IPP reaction for 6 h. The products were analysed by TLC as described previously [17].
Single-turnover reaction of UPPS using GGPP as a substrate
The single-turnover reaction of UPPS with GGPP was performed by a Kintek RFQ-3 Rapid Chemical Quench apparatus (Kintek Instruments, Austin, TX, U.S.A.) as described previously [15]. The reaction was initiated by mixing 15 μl of the enzyme (10 μM) preincubated with all-trans-GGPP (1 μM), and an equal volume of [14C]IPP (50 μM) solution in buffer containing 100 mM Hepes (pH 7.5), 0.5 mM MgCl2 and 50 mM KCl, at 25 °C in the presence of 0.1% Triton X-100. The enzyme reaction was terminated by quenching with 67 μl of 0.6 M NaOH after specified time periods of 0.2–50 s. The intermediates and products formed during the single-turnover reaction were extracted with butan-1-ol and converted into polyprenols by acid phosphatase and analysed on TLC.
Molecular structural modelling of C55-UPP product, C60 (eight cis double bonds) and assumed C60 (nine cis double bonds)
The construction of molecular models, structure optimization and conformational analysis were done using the Discover module of Insight II software using the CFF91 forcefield. Computer modelling to predict the three-dimensional structures of C60 (eight cis double bonds) and C60 (nine cis double bonds) were performed on a SGI R5000 workstation. By setting the known overall structure of two Triton X-100 molecules in the UPPS active site as in [9], the conformations of the C55 and C60 products were fully optimized by steepest descent first, and then by conjugate gradient algorithm (see http://www.molvis.indiana.edu/app_guide/InsighII/ for this commonly used modelling method) until root-mean-square derivative <0.05 kcal·mol−1·Å−1 (1 cal=4.184 J, 1 Å=0.1 nm), acceptable for molecular modelling.
RESULTS
Role of pyrophosphate of the allylic substrate in binding with UPPS
The diphosphate fluorescent analogue (TFMC-GPP), which was previously abbreviated as CPCF3, serves as an inhibitor (Ki=0.57 μM with respect to FPP) and alternative substrate (Km=0.69 μM and kcat=0.02 s−1) for E. coli UPPS [14]. The fluorescence intensity of this compound quenched by binding with UPPS enzyme can be recovered by adding FPP [14], indicating that the compound binds at the same position as the FPP does. Using stopped-flow technology, the association and dissociation rate constants were found to be kon=55.3 μM−1·s−1 and koff=31.6 s−1 respectively [14]. The Kd (0.57 μM) value derived from kon and koff, Ki (0.57 μM) and Km values (0.69 μM) of TFMC-GPP are all similar to the Km value (0.4 μM) of FPP. In the present study, the same fluorescent FPP analogue TFMC-GP, except with a monophosphate head group, also binds to UPPS in a competitive way with respect to FPP. Its fluorescence was reduced by adding UPPS and recovered by displacing with FPP (Figure 2A). However, this monophosphate analogue displays a 9-fold larger inhibition constant (Ki=5.0±0.3 μM) than that of the diphosphate TFMC-GPP (Figure 2B). On the other hand, farnesol, which lacks phosphate, failed to inhibit the UPPS reaction even at 1 mM (Figure 2B shows the data up to 200 μM). Farnesol was not able to displace TFMC-GP from the active site of UPPS in the stopped-flow experiment (results not shown). These results indicate the importance of the diphosphate group for the allylic substrate FPP to bind to the UPPS active site.
Figure 2. The fluorescent monophosphate analogue of FPP serves as an inhibitor for UPPS.

(A) The fluorescence of 1 μM TFMC-GP (■) was quenched by adding 1 μM UPPS (●) and gradually recovered by adding 1 (+), 3 (▲) and 5 (○) μM FPP to compete out the inhibitor, indicating that TFMC-GP binds at the same site as FPP does. (B) The reaction velocities of UPPS were measured at different concentrations of the TFMC-GP (□), in comparison with TFMC-GPP (○) and farnesol (×). The data for TFMC-GPP were adopted from our previous paper [14]. The Ki values of the compound in the presence of 0.03 μM UPPS, 3 μM FPP and 50 μM IPP were determined to be 5.0±0.3 μM. Farnesol does not inhibit the UPPS reaction at the concentrations indicated.
The binding of UPPS with the monophosphate analogue was then monitored by using the stopped-flow spectrofluorimeter. A representative stopped-flow trace obtained by mixing 0.5 μM TFMC-GP with an equal volume of 5 μM UPPS is shown in Figure 3(A) (kobs=384±21 s−1). The binding rate constant kon obtained from the slope of the observed rate versus UPPS concentration (the inset in Figure 3A) was 43.5±7 μM−1·s−1, comparable with the kon (55.3±8 μM−1·s−1) of TFMC-GPP. However, the dissociation constant koff of TFMC-GP (192±14.7 s−1), determined from the stopped-flow competition experiments (Figure 3B) by mixing an equal volume of 0.5 μM enzyme preincubated with 7.5 μM TFMC-GP with 25 μM FPP, was six times faster than that (31.6 s−1) of TFMC-GPP. Thus, the Kd value derived from koff/kon of TFMC-GP is 4.4 μM, approx. 8-fold larger than that of TFMC-GPP. Thus, this difference in Kd value is mainly caused by the larger dissociation rate constant of TFMC-GP.
Figure 3. UPPS binding assay by stopped-flow spectrofluorimetry.

(A) Binding process of UPPS with TFMC-GP was monitored by using the stopped-flow spectrofluorimeter. A representative stopped-trace obtained by mixing 0.5 μM TFMC-GP with an equal volume of 5 μM UPPS is shown. The data were fitted with a single exponential equation to obtain kobs=384±21 s−1. The kon was determined from the slope of the plot of kobs values versus concentrations of the compound shown in the inset of this Figure. (B) The increase in fluorescence by mixing an equal volume of 0.5 μM enzyme preincubated with 7.5 μM TFMC-GP with 25 μM excessive FPP. By fitting the data with a single exponential equation, the rate constant for the dissociation of the compound from UPPS was determined to be 192±14.7 s−1.
Role of hydrocarbon moiety of allylic substrate in UPPS reaction
Next, we examined the contribution of the C15 hydrocarbon moiety of FPP in binding with UPPS. The C10-GPP was tested as an alternative allylic substrate but showed a 90-fold larger Km value (36±4 μM) and a 20-fold larger IPP Km value (83±13 μM) compared with the kinetic parameters of FPP (Km=0.4 μM) as an allylic substrate and IPP (Km=4 μM) as a homoallylic substrate. However, the kcat (1.7±0.1 s−1) of GPP is similar to that (2.5 s−1) for the reaction of FPP and IPP. When the shortest C5-IPP was used as both allylic and homoallylic substrates, only residual activity was detected (kcat=8.5×10−5 s−1). On the other hand, the all-trans C20-GGPP, a stereoisomer of the C20 intermediate of the UPPS reaction, can serve as a satisfactory allylic substrate (kcat=2.1 s−1, Km=0.3 μM) with comparable kinetic parameters to those of FPP (Tables 1 and 2).
Table 1. Kinetic parameters of wild-type and mutant E. coli UPPS using FPP and IPP as substrates.
| UPPs | kcat (s−1) (FPP) | Km (FPP) (μM) | Km (IPP) (μM) | Rel. kcat* |
|---|---|---|---|---|
| Wild-type† | 2.5±0.1 | 0.4±0.1 | 4.1±0.3 | 1 |
| L85A | 0.095±0.01 | 1.6±0.3 | 617±123 | 0.04 |
| L88A | 0.23±0.04 | 2.1±0.7 | 173±55 | 0.1 |
| F89A | 0.61±0.09 | 2.6±0.2 | 84±17 | 0.24 |
* kcat relative to that of wild-type.
† Kinetic parameters obtained from [15].
Table 2. Kinetic parameters of wild-type and mutant E. coli UPPS using GGPP and IPP as substrates.
| UPPs | kcat (s−1) (GGPP) | Km (GGPP) (μM) | Km (IPP) (μM) | Rel. kcat* |
|---|---|---|---|---|
| Wild-type† | 2.1±0.1 | 0.3±0.1 | 13±1 | 1 |
| L85A | 0.18±0.03 | 1.2±0.3 | 60±10 | 0.086 |
| L88A | 0.32±0.07 | 0.7±0.1 | 42±11 | 0.15 |
| F89A | 0.70±0.11 | 1.0±0.4 | 18±2 | 0.33 |
* kcat relative to that of wild-type.
† Kinetic parameters obtained from [15].
Since C5-IPP to C20-GGPP can all act as allylic substrates but with different activities, we also tested whether FPP and GPP can serve as a homoallylic substrate. After incubation for 24 h with a much higher (1 μM) enzyme concentration, still no product was formed from 5 μM [3H]FPP (allylic substrate) and 50 μM FPP (homoallylic substrate) or 5 μM [3H]FPP (allylic substrate) and 50 μM GPP (homoallylic substrate) (results not shown).
L85, L88 and F89 are essential in binding substrate but not important in distinguishing GGPP from FPP
On the basis of our previously solved crystal structure of UPPS in complex with the substrate, interactions between the C15 hydrocarbon tail of the FPP molecule and the active site residues of UPPS are mostly hydrophobic [10]. The side-chains of L85, M25, L88, A47, V50, I141, A69, A92 and F89 of UPPS may be involved in the hydrophobic interactions [10]. Three of these residues, L85, L88 and F89, are located on the α3 helix, and move towards the bound FPP during a change in protein conformation [10]. We performed site-directed mutagenesis studies to examine their role in substrate binding and catalysis. As shown in Table 2, the mutants L85A, L88A and F89A showed 4, 5 and 6 times larger FPP Km values. Apparently, the weaker hydrophobic interaction in the mutant UPPS leads to improper positioning of FPP and also causes a larger IPP Km and poor activity (IPP is bound after FPP as shown in [8]). The IPP Km values of the mutants L85A, L88A and F89A are respectively 154, 43 and 21 times larger, and the kcat values of the mutants are 25, 10 and 5 times smaller respectively when compared with the wild-type (Table 1). The L85 side-chain is closer to the electrophilic C1 of FPP, which is attacked by IPP, thus L85A shows a more remarkably reduced IPP affinity and kcat value.
However, using C20-GGPP as a substrate, the IPP Km values of three mutant enzymes are larger than that of the wild-type, but significantly smaller than that using FPP as a substrate (Table 2). Furthermore, GGPP Km values of these mutants are slightly smaller than FPP Km values. These results suggest that a mutation in which a large amino acid L85, L88 or F89 is replaced with the smaller Ala may give more space in the FPP site to accommodate larger GGPP substrates. However, this effect is not obvious since the wild-type UPPS can still utilize GGPP as a satisfactory substrate as shown above.
Products of C20-GGPP and IPP under steady-state and single-turnover conditions
Interestingly, the enzyme reaction with C20-GGPP and IPP led to a C60 final product as judged from TLC (lane 2, Figure 4A), with one extra IPP compared with the C55 product synthesized from FPP (lane 1, Figure 4A) under steady-state conditions, where 5 μM GGPP and 50 μM [14C]IPP were used as substrates. Surprisingly, the longer GGPP still underwent eight IPP condensation reactions catalysed by UPPS. To ensure that the formation of C60 from GGPP was not due to the extended reaction time, but was immediately formed from C55, a single-turnover reaction where [UPPS]>[GGPP] was performed to monitor the GGPP chain elongation process in the active site. The reaction was terminated in a series of short periods of time using a rapid-quench apparatus. As shown in Figure 4(B), the intermediates and C60 product were formed during the single-turnover of UPPS (10 μM) with GGPP (1 μM), and the C60 product was readily formed without any delay from the C55. The radiolabelled [14C]IPP provided the radioactivity of the intermediates and product so that they could be visualized using a phosphoimager by exposing the TLC plate to a film. From the top to the bottom of the TLC, C25 to C60 products were observed. In contrast, using FPP as the substrate, the C20–C55 but not the C60 products were formed in the single-turnover reaction catalysed by UPPS [15].
Figure 4. Products generated by the wild-type and the mutant UPPS using FPP, GGPP or GPP as a substrate.

The reactions were performed with 1 μM wild-type, 5 μM FPP (lane 1), GGPP (lane 2) or GPP (lane 3), and 50 μM [14C]IPP in a buffer of 100 mM Hepes (pH 7.5), 0.1 mM Mg2+, and 50 mM KCl, in the presence of 0.1% Triton X-100. (A) The major products shown in lanes 1, 2 and 3 are C55, C60 and C50, synthesized from FPP, GGPP and GPP respectively. C55 was also formed from GPP, but it was still a minor product after an extended reaction time (6 h). (B) The formation of intermediates (C25–C55) and product (C60) of UPPS reaction using GGPP as a substrate. The C60 formation immediately follows the C55 production without delay. This indicates that a UPPS active site can readily accommodate a C60 product that contains eight cis double bonds.
Similar to FPP and GGPP, the shorter GPP also reacted with eight IPPs to form C50, but C55 was also formed after an extended reaction time (6 h) (see lane 3, Figure 4A).
Computer prediction of the three-dimensional structures of C55-UPP, C60 (eight cis double bonds) and assumed C60 (nine cis double bonds)
We previously proposed that a large residue, L137, serves as a ‘floor’ at the bottom of the active site tunnel to block further chain elongation of the C55 product so that only a C55 product is formed from the reaction of FPP with eight IPPs [12]. The fact that UPPS creates the C60 product containing eight cis double bonds from GGPP seems to argue against the proposed model. To explain this, we performed computer modelling to derive the most stable structures for both C60 products containing eight and nine cis double bonds respectively. As shown in Figure 5(A), the energy-minimized structure of C55-UPP (left panel) matches with the overall shape of the two Triton X-100 molecules (right panel) in the active site [9], indicating that the computer-predicted structure of the C55 product is truly its structure in the active site. As shown in the left panel of Figure 5(B), the C60 product containing eight cis double bonds adopts a similar structure to that of the C55 product, except with a longer C5 tail. In contrast, if UPPS catalysed nine IPP condensation reactions using FPP as a substrate, the assumed C60 containing nine cis double bonds adopts a more enlarged, circular shape (Figure 5B, right panel), which makes the formation of C60 (nine cis double bonds) unfavourable in the UPPS active site. From the data presented here, product chain length seems to be determined not only by the L137 at the bottom of the tunnel as shown in Figure 1, but it also seems to be regulated by the space in the upper portion of the active site tunnel, which the cis double bonds of the product occupy.
Figure 5. Computer structural modelling of the C55 and C60 products.

(A) The molecular shape of C55 UPP obtained by energy minimization using the computer program Discover (left panel). This predicted structure of UPP product matches with that of the two Triton X-100 molecules bound in the active site of UPPS (right panel) [9]. (B) Molecular shape of C60 containing eight cis double bonds elongated from all-trans-GGPP (left panel) and the assumed C60 containing nine cis double bonds (right panel). The former adopts a similar shape to that of UPP, whereas the latter has a different shape. The carbon atoms are shown in grey and oxygen atoms in black.
Consistent with this notion, GPP formed C50 product to maintain its overall structure of the eight cis double bonds. However, the space left between the end of C50-product and L137 is apparently sufficient for C50 to twist slightly for condensing with one more IPP to form the C55-product.
DISCUSSION
As demonstrated in the present study, the pyrophosphate head group is important for allylic substrate binding to UPPS. The affinity of the monophosphate FPP analogue was decreased 8-fold mainly due to its faster dissociation rate compared with the diphosphate analogue. Therefore, the loss of the distal phosphate, that forms hydrogen bonds with the side chain and backbone NHs of R30 as well as the side chain NH of R39, accounts for the 8-fold weaker binding. Furthermore, farnesol (an FPP analogue completely lacking the pyrophosphate group) does not interfere with the UPPS reaction, even at the high concentration of 1 mM.
The hydrophobic moiety also contributes significantly to the UPPS–substrate interaction. Whereas UPPS utilizes GGPP with kcat and Km values similar to that for FPP, the Km value for GPP as an allylic substrate is remarkably larger. The homoallylic IPP Km value is also larger when using GPP as an allylic substrate. The larger Km values of GPP and IPP in this case cannot be due to the competition for the allylic and homoallyic binding sites between GPP and IPP, since GPP could not serve as a homoallylic substrate and IPP serves as a poor allylic substrate with extremely low activity, as shown by the data presented here. This indicates that the allylic substrate that is 5 or 10 carbons shorter than FPP causes remarkably lower affinity and reactivity, but if it is 5 carbons larger (GGPP) it serves as an equally good allylic substrate.
However, using all-trans C20-GGPP as an alternative substrate results in the production of the longer C60 compared with the normal product C55. In vivo, GGPP could lead to C60 product, which may not be as satisfactory a lipid carrier as UPP. This may become an important issue since more and more attempts have been made to produce useful isoprenoids using E. coli as a factory by over-producing GGPP, which serves as a precursor for many isoprenoid compounds [18–20]. Indeed, it has been observed that high-level expression of the plasmid-encoded crtE gene (a GGPP synthase) causes cytotoxicity of the E. coli host cells [21]. We also observed the same effect when E. coli expressed GGPP synthase (results not shown). This could be due to the incorporation of the C60 polyprenol, which is not an efficient lipid carrier, and/or IPP pools being depleted due to other primary metabolic biosyntheses. When other genes are introduced into the engineered E. coli, which can transform GGPP into the desired products, E. coli should still grow well.
The hydrophobic residues, including L85, L88 and F89, interact with the hydrocarbon moiety of FPP in the UPPS active site. These residues ensure the correct position and orientation of the allylic substrate FPP for an efficient nucleophilic attack by IPP. The substitution of L85, L88 and F89 with Ala lowered the substrate affinity and kcat values (Tables 1 and 2). It is notable that these three amino acids located on the α3 helix interact with the substrate only when FPP is bound and the closed conformation is formed [10,22]. It has been shown that the flexible loop (amino acids 72–83) preceding the α3 helix may pull this helix closer to the bound FPP substrate in the closed conformation [22]. As shown in the present study, amino acids such as L85, L88 and F89 on the α3 helix directly participate in substrate binding and catalysis.
On the basis of our results, the product chain length determination of UPPS may not only be dependent on the bottom part of the active site tunnel as shown in Figure 1(A), but may also be regulated by the space in the upper portion of the active site tunnel of UPPS. In general, cis-type linear prenyltransferases synthesize much longer chain-length products than trans-type enzymes. Rubber prenyltransferase (cis-type) can catalyse the incorporation of thousands of IPP units into FPP initiator [23], but trans-type enzymes identified thus far only generate up to a C50 product [24]. From the three-dimensional structures of cis-type UPPS and trans-type OPPS that we solved, we found that the UPPS is broader at the top portion of the tunnel (funnel-like) compared with the trans-type (cylinder-like) OPPS, which makes the all-trans C40 product to have a linear shape. The molecular shapes of the active sites of these cis- and trans-type prenyltarnsferases are apparently important for controlling the chain lengths of the products. For the cis-type rubber prenyltransferases, which form extremely large polymers [25], we predict that they must have a spacious top portion of the tunnel-like active site crevice plus a bottom and/or side opening to allow the continuation of chain elongation. This is supported by our computer-generated structural model for the rubber prenyltransferase (results not shown), but yet to be verified by the crystal structure.
References
- 1.Liang P. H., Ko T. P., Wang A. H.-J. Structure, mechanism and function of prenyltransferases. Eur. J. Biochem. 2002;269:3339–3354. doi: 10.1046/j.1432-1033.2002.03014.x. [DOI] [PubMed] [Google Scholar]
- 2.Ogura K., Koyama T., Sagami H. Polyprenyl diphosphate synthases. Subcellular Biochem. 1997;28:57–87. doi: 10.1007/978-1-4615-5901-6_3. [DOI] [PubMed] [Google Scholar]
- 3.Poulter C. D., Rilling H. C. Prenyl transferases and isomerase. In: Porter J. W., Spurgeon S. L., editors. Biosynthesis of Isoprenoid Compounds, vol. 1, Chapter 4. New York: John Wiley & Sons; 1982. pp. 161–224. [Google Scholar]
- 4.Ogura K., Koyama T. Enzymatic aspects of isoprenoid chain elongation. Chem. Rev. 1998;98:1263–1276. doi: 10.1021/cr9600464. [DOI] [PubMed] [Google Scholar]
- 5.Chen A., Kroon P. A., Poulter C. D. Isoprenyl diphosphate synthases: protein sequence comparisons, a phylogenetic tree, and predictions of secondary structure. Protein Sci. 1994;3:600–607. doi: 10.1002/pro.5560030408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarshis L. C., Proteau P. J., Kellogg B. A., Sacchettini J. C., Poulter C. D. Regulation of product chain length by isoprenyl diphosphate synthases. Proc. Natl. Acad. Sci. U.S.A. 1996;93:15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo R. T., Kuo C. J., Chou C. C., Ko T. P., Shr H. L., Liang P. H., Wang A. H.-J. Crystal structure of octaprenyl pyrophosphate synthase from hyperthermophilic Thermotoga maritima and mechanism of product chain length determination. J. Biol. Chem. 2004;279:4903–4912. doi: 10.1074/jbc.M310161200. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y. H., Chen A. P.-C., Chen C.-T., Wang A. H.-J., Liang P. H. Probing the conformational change of Escherichia coli undecaprenyl pyrophosphate synthase during catalysis using an inhibitor and tryptophan mutants. J. Biol. Chem. 2002;277:7369–7376. doi: 10.1074/jbc.M110014200. [DOI] [PubMed] [Google Scholar]
- 9.Chang S. Y., Ko T. P., Liang P. H., Wang A. H.-J. Catalytic mechanism revealed by the crystal structure of undecaprenyl pyrophosphate synthase in complex with sulfate, magnesium, and Triton. J. Biol. Chem. 2003;278:29298–29307. doi: 10.1074/jbc.M302687200. [DOI] [PubMed] [Google Scholar]
- 10.Chang S. Y., Ko T. P., Chen A. P.-C., Wang A. H.-J., Liang P. H. Substrate binding mode and reaction mechanism of undecaprenyl pyrophosphate synthase deduced from crystallographic studies. Protein Sci. 2004;13:971–978. doi: 10.1110/ps.03519904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varki A., Cummings R., Esko J., Freeze H., Hart G., Marth J. Essentials of Glycobiology, Chapter 21. Plainview, NY: Cold Spring Harbor Laboratory Press; 1999. Bacterial polysaccharides; pp. 322–325. [Google Scholar]
- 12.Ko T. P., Chen Y. K., Robinson H., Tsai P. C., Gao Y.-G., Chen A. P.-C., Wang A. H.-J., Liang P. H. Mechanism of product chain length determination and the role of a flexible loop in Escherichia coli undecaprenyl-pyrophosphate synthase catalysis. J. Biol. Chem. 2001;276:47474–47482. doi: 10.1074/jbc.M106747200. [DOI] [PubMed] [Google Scholar]
- 13.Apfel C. M., Takacs B., Fountoulakis M., Stieger M., Keck W. Use of genomics to identify bacterial undecaprenyl pyrophosphate synthetase: cloning, expression, and characterization of the essential uppS gene. J. Bacteriol. 1999;181:483–492. doi: 10.1128/jb.181.2.483-492.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen A. P.-C., Chen Y. H., Liu H. P., Li Y. C., Chen C.-T., Liang P. H. Synthesis and application of a fluorescent substrate analogue to study ligand interactions for undecaprenyl pyrophosphate synthase. J. Am. Chem. Soc. 2002;124:15217–15224. doi: 10.1021/ja020937v. [DOI] [PubMed] [Google Scholar]
- 15.Pan J. J., Chiou S. T., Liang P. H. Product distribution and pre-steady-state kinetic analysis of Escherichia coli undecaprenyl pyrophosphate synthase reaction. Biochemistry. 2000;39:10936–10942. doi: 10.1021/bi000992l. [DOI] [PubMed] [Google Scholar]
- 16.Segel I. H. Wiley Classics Library edn. New York: John Wiley and Sons; 1993. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems; pp. 100–108. [Google Scholar]
- 17.Pan J. J., Yang L. W., Liang P. H. Effect of site-directed mutagenesis of the conserved aspartate and glutamate on E. coli undecaprenyl pyrophosphate synthase catalysis. Biochemistry. 2000;39:13856–13861. doi: 10.1021/bi001226h. [DOI] [PubMed] [Google Scholar]
- 18.Wang C. W., Oh M. K., Liao J. C. Engineered isoprenoid pathway enhances astaxanthin production in Escherichia coli. Biotechnol. Bioeng. 1999;62:235–241. doi: 10.1002/(sici)1097-0290(19990120)62:2<235::aid-bit14>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 19.Huang Q., Roessner C. A., Croteau R., Scott A. I. Engineering Escherichia coli for the synthesis of taxadiene, a key intermediate in the biosynthesis of taxol. Bioorg. Med. Chem. 2001;9:2237–2242. doi: 10.1016/s0968-0896(01)00072-4. [DOI] [PubMed] [Google Scholar]
- 20.Ravanello M. P., Ke D., Alvarez J., Huang B., Shewmaker C. K. Coordinate expression of multiple bacterial carotenoid genes in canola leading to altered carotenoid production. Metab. Eng. 2003;5:255–263. doi: 10.1016/j.ymben.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Math S. K., Hearst J. E., Poulter C. D. The crtE gene in Erwinia herbicola encodes geranylgeranyl diphosphate synthase. Proc. Natl. Acad. Sci. U.S.A. 1992;89:6761–6764. doi: 10.1073/pnas.89.15.6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang S. Y., Chen Y. K., Wang A. H.-J., Liang P. H. Identification of the active conformation and the importance of length of the flexible loop 72-83 in regulating the conformational change of undecaprenyl pyrophosphate synthase. Biochemistry. 2003;42:14452–14459. doi: 10.1021/bi035283x. [DOI] [PubMed] [Google Scholar]
- 23.Cornish K. Similarities and differences in rubber biochemistry among plant species. Phytochemistry. 2001;57:1123–1134. doi: 10.1016/s0031-9422(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 24.Koyama T. Molecular analysis of prenyl chain elongating enzymes. Biosci. Biotechnol. Biochem. 1999;63:1671–1676. doi: 10.1271/bbb.63.1671. [DOI] [PubMed] [Google Scholar]
- 25.Asawatreratanakul K., Zhang Y. W., Wititsuwannakul D., Wititsuwannakul R., Takahashi S., Rattanapittayaporn A., Koyama T. Molecular cloning, expression and characterization of cDNA encoding cis-prenyltransferases from Hevea brasiliensis. A key factor participating in natural rubber biosynthesis. Eur. J. Biochem. 2003;270:4671–4680. doi: 10.1046/j.1432-1033.2003.03863.x. [DOI] [PubMed] [Google Scholar]