

Key Points
-
•
Better clinical responses to teclistamab correlate with a more functional initial immune T-cell repertoire in the periphery and tumor site.
-
•
More durable responses to teclistamab associate with lower Tregs and lower expression of inhibitory receptors on T cells at baseline.
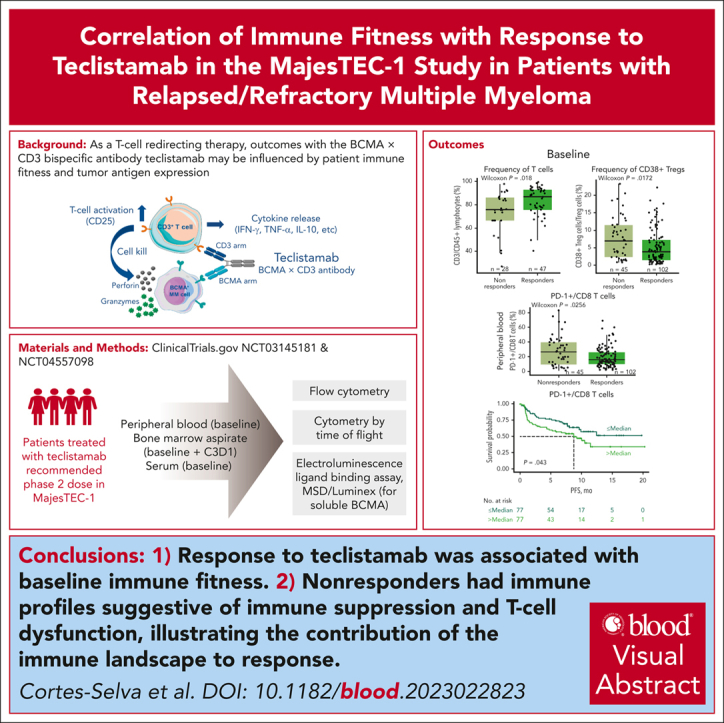
Visual Abstract
Abstract
Teclistamab, an off-the-shelf B-cell maturation antigen (BCMA) × CD3 bispecific antibody that mediates T-cell activation and subsequent lysis of BCMA-expressing myeloma cells, is approved for the treatment of patients with relapsed/refractory multiple myeloma (R/RMM). As a T-cell redirection therapy, clinical outcomes with teclistamab may be influenced by patient immune fitness and tumor antigen expression. We correlated tumor characteristics and baseline immune profiles with clinical response and disease burden in patients with R/RMM from the pivotal phase 1/2 MajesTEC-1 study, focusing on patients treated with 1.5 mg/kg of teclistamab (N = 165). Peripheral blood samples were collected at screening, and bone marrow samples were collected at screening and cycle 3. Better clinical outcomes to teclistamab correlated with higher baseline total T-cell counts in the periphery. In addition, responders (partial response or better) had a lower proportion of immunosuppressive regulatory T cells (Tregs), T cells expressing coinhibitory receptors (CD38, PD-1, and PD-1/TIM-3), and soluble BCMA and a T-cell profile suggestive of a more cytolytic potential, compared with nonresponders. Neither frequency of baseline bone marrow BCMA expression nor BCMA-receptor density was associated with clinical response to teclistamab. Improved progression-free survival was observed in patients with a lower frequency of T cells expressing exhaustion markers and immunosuppressive Tregs. Overall, response to teclistamab was associated with baseline immune fitness; nonresponders had immune profiles suggestive of immune suppression and T-cell dysfunction. These findings illustrate the importance of the contribution of the immune landscape to T-cell redirection therapy response. This trial was registered at www.ClinicalTrials.gov as #NCT03145181/NCT04557098.
Teclistamab, a bispecific antibody targeting the plasma cell antigen B-cell maturation antigen (BCMA) and stimulating T cells via CD3, is approved for relapsed/refractory multiple myeloma. Cortes-Selva and colleagues report that while the level of expression of BCMA was not predictive of responses seen in a phase 1/2 trial, analysis of the relationship between baseline immune status and responses was informative. The baseline function of the immune T-cell repertoire in blood and myeloma sites predicts clinical responses, with durable responses associated with fewer exhausted or immunosuppressive T cells.
Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by the accumulation of malignant plasma cells within the bone marrow (BM).1,2 Current treatment of MM often involves sequential lines of therapy with 3 major classes of agents (immunomodulatory agents [IMiDs], proteasome inhibitors, and anti-CD38 monoclonal antibodies).3, 4, 5, 6 Despite advances in therapy extending survival, MM remains largely incurable. Although initial and even sustained response to treatment and disease remission may occur, many patients with MM eventually relapse and become refractory to anti-MM therapy, leaving few remaining treatment options and resulting in poor prognoses and limited survival.
B-cell maturation antigen (BCMA) is preferentially expressed on the surface of normal and malignant plasma cells, and its activation promotes MM growth, making it an ideal therapeutic target for MM therapy.7,8 Teclistamab is the first approved BCMA×CD3 bispecific antibody for the treatment of triple-class exposed relapsed/refractory (R/R) MM (R/RMM), with weight-based dosing9,10 and the longest study follow-up of any bispecific antibody in MM. Teclistamab is an immunoglobulin G4 antibody that redirects CD3 T cells to BCMA-expressing cells, leading to T-cell activation and subsequent lysis of BCMA-expressing myeloma cells.11,12 The phase 1/2 multicohort, open-label MajesTEC-1 study investigated the safety and efficacy of teclistamab in patients with R/RMM who previously received at least 3 lines of therapy. In phase 1, the recommended phase 2 dose (RP2D) of teclistamab was identified as a weekly subcutaneous dose of 1.5 mg/kg preceded by step-up doses of 0.06 and 0.3 mg/kg.12 Results from patients (with no prior exposure to BCMA-targeted therapy) who were treated at the RP2D in the pivotal phase 1 and 2 portions of the study demonstrated that teclistamab was efficacious and well tolerated.13 For patients who received the RP2D across the dose escalation and expansion portions of the trial, the overall response rate was 63.0%, and the median duration of response was 18.4 months.13
Immune cell composition has been demonstrated to contribute to the response to immunotherapy,14 and effective T-cell redirection therapy is hypothesized to be dependent on immune fitness and tumor antigen expression.15 Understanding the relationships between baseline T-cell functional status or tumor BCMA levels and outcomes is therefore important to help optimize clinical response. T-cell exhaustion or dysfunction is frequently observed in patients with cancer after chronic antigen exposure and sustained T-cell receptor signaling.15, 16, 17, 18 Transient expression of inhibitory receptors on activated T cells plays a key role in maintaining immune homeostasis, finely regulating the balance between host-protective and autoimmune T-cell responses. However, in exhausted T cells, expression of these receptors remains high, and T-cell activity is reduced, thereby preventing optimal control of tumors. Recent data in other malignancies suggest that the level and type of T-cell dysfunction play a key role in determining tumor reactivity.14,19,20
Our report builds on emerging translational data15 by specifically demonstrating the impact of baseline immune cell state on response to teclistamab in R/RMM, and to our knowledge, it is the most comprehensive analysis of its kind, with a cohort of 165 patients, representing a major step forward in our understanding of immune fitness in response to therapy. Here, we show that therapy-responsive patients exhibit higher baseline total T-cell counts in the periphery, with lower expression of markers associated with T-cell exhaustion and a significantly higher expression of cytotoxic factors such as granzyme B and perforin. Moreover, a more durable response was defined by a less dysfunctional T-cell signature. Our data demonstrate the contribution of circulating and tumor microenvironment–associated T cells to the clinical response to teclistamab, independent of disease burden.
Methods
Study design and patient population
Full details of the study design of MajesTEC-1 (NCT03145181 and NCT04557098) have been previously published.12,13
Briefly, MajesTEC-1 is an ongoing, phase 1/2, open-label, multicenter clinical trial aimed at assessing the efficacy of teclistamab for the treatment of patients with R/RMM who have received at least 3 prior lines of therapy, including at least 1 proteasome inhibitor, 1 IMiD, and 1 anti-CD38 monoclonal antibody. Key eligibility criteria included age ≥18 years, documented MM diagnosis per International Myeloma Working Group criteria, measurable disease at screening, and an Eastern Cooperative Oncology Group performance score of 0 or 1.
The pivotal RP2D cohort comprised 165 patients analyzed per the clinical cutoff date of 16 March 2022, who received the RP2D of subcutaneous teclistamab 1.5 mg/kg weekly, following step-up doses (0.06 mg/kg and 0.3 mg/kg). An additional patient subset for cytometry by time of flight (CyTOF) in peripheral blood (PB) included 40 patients treated with active doses of teclistamab (0.72 mg/kg IV weekly with step-up doses or 0.72-6 mg/kg subcutaneously every other week with step-up doses). Treatment with teclistamab continued once weekly until disease progression, unacceptable toxicity, withdrawal of consent, or death, whichever occurred first. Responses were defined as a best clinical response of partial response or better per International Myeloma Working Group criteria, as assessed by an independent review committee.21,22 Exploratory end points included the assessment of soluble BCMA (sBCMA) and cytokines in serum, characterization of T-cell subsets and activation/exhaustion markers in PB and tumor samples, and membrane-bound BCMA on tumor plasma cells. Additional methods are described in the supplemental Appendix and supplemental Tables 1 and 2, available on the Blood website.
The study was conducted in accordance with the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Council for Harmonisation. The study protocol, amendments, and relevant documents were approved by the local independent ethics committee or institutional review board at each study site. All patients provided written informed consent.
Results
Peripheral immune profile of responders and nonresponders at baseline
Overall, 40 patients treated with teclistamab in the phase 1 part of the study and 125 patients from the phase 2 part were included (“pivotal RP2D” cohort; all treated at the RP2D). For the pivotal RP2D cohort, PB and tumor samples were taken at screening for flow cytometry analyses and profiled for immune composition, immune phenotype (Figure 1A), and other tumor characteristics. Samples from patients treated in the pivotal RP2D cohort were also profiled for cytokine production using the Meso Scale Discovery platform. CyTOF was performed with BM samples from the pivotal RP2D cohort, whereas CyTOF analyses of PB samples were carried out for an additional subset of 40 patients receiving active doses of teclistamab (as described in “Methods”). All patients for whom samples were available were included in the relevant analyses, as indicated by the number of patients in each figure. We used a comprehensive set of gating strategies for each of the target populations of interest for flow cytometry (supplemental Figure 1) and mass cytometry (supplemental Figure 2).
Figure 1.

Correlative immune factors of response to teclistamab monotherapy in the MajesTEC-1 study in the periphery at baseline. (A) Diagram of patients, samples, and analysis from the MajesTEC-1 study. (B) Frequency of T cells by response assessed by flow cytometry in the periphery at the RP2D dose, with representative staining shown on the right of the panel. (C) Absolute CD3 T-cell counts by flow cytometry relative to response. (D) Absolute CD8 T-cell counts assessed by flow cytometry (TBNK kit). (E) Percentage of Tregs from CD4 T cells at baseline relative to response at the RP2D dose, with representative staining shown on the right of the panel. (F) Frequency of CD38-expressing Tregs at baseline assessed by flow cytometry and relative to response, with representative staining shown on the right of the panel. (G) Frequency of naive CD8 T cells from the parent population of CD45+ T cells assessed in a subset of patients from the active dose cohorts assessed by CyTOF, with representative staining shown on the right of the panel. Statistical significance was evaluated using the Wilcoxon rank-sum test. CM, central memory; EM, effector memory; TBNK, T cells, B cells, and natural killer cells; TEMRA, terminally differentiated effector memory cells re-expressing CD45RA.
First, higher frequency and counts of peripheral CD3 T cells at baseline were observed in responders (partial response or better) than nonresponders (Figure 1B-C). Looking at T-cell subsets, we found higher cytotoxic CD8 T-absolute cell counts (Figure 1D) but no difference in CD4 T cells (supplemental Figure 3A) in responders than nonresponders. In addition, the proportion of peripheral immunosuppressive regulatory T cells (Tregs) at baseline was lower in responders than in nonresponders (Figure 1E). Moreover, a subset of Tregs expressing CD38, which has been associated with increased immunosuppression,23, 24, 25, 26 was also lower at baseline in responders than nonresponders (Figure 1F).
To investigate the immune profile and the functional status of peripheral T cells at a more granular level, CyTOF profiling was conducted in a subset of patients from the active dose cohort (n = 40) who had similar baseline characteristics to the RP2D cohort. We first used a manual gating approach for a hypothesis-driven analysis of T-cell subsets. Notably, the proportion of naive CD8 T cells was higher in patients with clinical response (Figure 1G). We then performed unsupervised clustering using Spanning-tree Progression Analysis of Density-normalized Events to identify immune cell phenotypes. Cells with closely related phenotypes were annotated into bubbles (supplemental Figure 4A). We used a tree blend plot to visualize trends toward higher frequencies of CD4 and CD8 T cells, with a lower proportion of natural killer cells, in responding patients (supplemental Figure 4A). However, these differences did not achieve significance at a cluster or bubble level between responders and nonresponders, likely due to small sample size and a stringent correction for multiple comparison (supplemental Figure 4B). Collectively, these results are in line with previous reports that have described a role for T-cell subsets in different malignancies and in response to immunotherapies, including T-cell redirecting agents.15,27,28
Correlation of BM microenvironment immunophenotype of patients with R/RMM with response to teclistamab
To characterize the immune composition and functionality in the BM microenvironment at baseline, a subset of patients from the RP2D cohort (n = 52; of whom 24 were responders and 28 nonresponders) was profiled by CyTOF to enable a more comprehensive evaluation using both extracellular and intracellular markers (supplemental Table 2). We found that responding patients had a higher proportion of BM CD8 T cells expressing granzyme B and perforin at baseline than that of nonresponders (Figure 2A-B). Furthermore, the frequency of Tregs in the BM was lower in responders than in nonresponders, consistent with the results in PB (Figure 2C). We then analyzed the population of non-Treg CD4 T cells and observed that the proportion of granzyme B–producing CD4 non-Tregs was higher in responders at baseline (Figure 2D). Next, significant differences in CD4 T cells between responders and nonresponders were observed in the proportion of naive and terminally differentiated effector memory (TEMRA) subsets among patients, in which responders exhibited a higher proportion of naive and TEMRA CD4 T cells (Figure 2E), with a similar trend in CD8 T cells (supplemental Figure 3B). Additionally, we did not observe significant differences at a cluster or bubble level between responders and nonresponders (supplemental Figure 5A). We next explored the difference in CD4 and CD8 T-cell composition within the BM of patients using the FreeViz algorithm and a supervised learning approach to identify any difference in cellular composition29,30 (supplemental Figure 5B). Cellular composition was visualized using contour plots, in which functional markers driving the separation based on response status are positioned distal from the center of the projection. In parallel, the phenotypes were confirmed using fan plots (supplemental Figure 5C). This polyfunctionality analysis revealed a shift toward granzyme B–positive and perforin–positive CD8 T cells and a naive CD4 T-cell phenotype in responders (supplemental Figure 5B-C). Consistent with the unsupervised analysis, we did not observe significant differences in the frequency of other immune populations in the BM at baseline (supplemental Figure 3C). These results suggest that at baseline, nonresponders show a less fit immune composition and function of their T-cell compartment in the BM microenvironment, which might lead to impaired teclistamab response.
Figure 2.

Higher proportions of granzyme B–producing and perforin-producing CD8 T cells, granzyme B–expressing CD4 non-Tregs, and naive CD4 T cells, as well as lower Treg proportion in the BM microenvironment at baseline were associated with lower response rates to teclistamab. (A) Frequency of CD8 T cells expressing granzyme B in a subset of 52 patients at the RP2D doses assessed at baseline by CyTOF. (B) Proportion of CD8 T cells expressing perforin relative to response assessed by CyTOF. (C) Percentage of CD4+CD27lowCD25hi Tregs in the BM microenvironment relative to response assessed at baseline by CyTOF. (D) Frequency of granzyme B–positive CD4 T cells (non-Tregs) assessed at baseline by CyTOF. (E) Proportion of the memory subset in the BM microenvironment at baseline assessed by CyTOF. Representative staining is shown on the right of each panel. Statistical significance was calculated using the Wilcoxon rank-sum test. CM, central memory; EM, effector memory; TEMRA, terminally differentiated effector memory cells re-expressing CD45RA.
An exhausted-like phenotype characterizes T cells in nonresponders at baseline in the periphery and tumor microenvironment
Next, we assessed the phenotype of baseline T cells in BM and PB to explore the expression of coinhibitory receptors involved in immune regulation in association with clinical response. Using flow cytometry, we characterized the frequency of T cells expressing markers such as PD-1, TIM-3, and CD38 that have been associated with a dysfunctional or exhausted phenotype (Figure 3A). We found a heterogeneous expression of CD38, PD-1, or TIM-3 on peripheral T cells, with a lower proportion of CD3 T cells coexpressing PD-1/TIM-3, CD8 T cells expressing PD-1, and CD4 T cells expressing CD38 in responders (Figure 3B). Similar observations were seen in the BM, where responders tended to exhibit a lower proportion of CD4 T cells expressing coinhibitory receptors (Figure 3C). Notably, previous reports suggest that after autologous stem cell transplant, T cells that recover maintain the expression of checkpoint inhibitors, such as PD-1.31 In our study, prior transplantation (autologous or allogeneic) did not affect the proportion of PB- or BM-derived CD8 T cells coexpressing PD-1 and TIM-3 or the proportion of immunosuppressive cells such as Tregs and CD38+ Tregs in responders compared with nonresponders (supplemental Figure 6). Together, these data suggest that the expression/coexpression of inhibitory receptors associated with T-cell dysfunction in both BM and PB at baseline influence the response to teclistamab therapy and are independent of prior transplant.
Figure 3.

Functional marker expression associated with exhaustion is higher in nonresponders in both the periphery and the BM microenvironments. (A) Staining of representative populations of interest (peripheral CD8, CD4, and CD3 T cells). (B) Frequency of PD-1+/CD8 T cells, CD38+/CD4 T cells, and PD-1+TIM-3+/CD3 T cells in the periphery at baseline in patients treated with the RP2D assessed by flow cytometry. (C) Frequency of PD-1+/CD8 T cells, CD38+/CD4 T cells, and PD-1+TIM-3+/CD3 T cells in the BM at baseline in patients treated with the RP2D assessed by flow cytometry. Groups were compared using the Wilcoxon rank-sum test.
sBCMA in serum, but not membrane-bound BCMA in tumors, correlates with clinical response
The baseline expression of membrane-bound BCMA on BM plasma cells (BMPCs) was found to be prevalent and highly variable among patients (Figure 4A). However, neither BCMA expression nor BCMA-receptor density (Figure 4B) was associated with clinical responses to teclistamab. Interestingly, higher serum sBCMA levels at baseline were associated with significantly lower response rates13 (P < .001; Figure 4C) and critical high-risk disease characteristics such as the presence of soft tissue extramedullary plasmacytomas (Figure 4D), revised International Staging System (ISS) stage (Figure 4E), high tumor burden (BMPCs ≥ 60%; Figure 4F), and elevated levels of lactate dehydrogenase (LDH; Figure 4G; all P < .01), which indicate that sBCMA levels act as a surrogate for tumor disease burden, consistent with previous reports.32,33 Analysis of sBCMA at baseline in correlation with tumor burden (BMPC %) for 140 patients showed a positive albeit weak correlation (R = 0.28; P < .001; supplemental Figure 7), which might be partly explained by the measurement in different sites (sBCMA in blood vs BMPCs in BM) and other factors such as the patchy nature of MM in BM leading to underestimation or overestimation of BMPC percentage or the presence of sBCMA–producing extramedullary plasmacytomas. In addition, sBCMA is probably reflective not only of tumor burden but also the mean BCMA expression on tumor cells. Importantly, a binomial logistic regression examining baseline sBCMA and other indicators of high-risk disease (including BMPC %, albumin, LDH, beta-2 microglobulin, ISS stage III, revised ISS stage III, and presence of extramedullary plasmacytomas) showed that only sBCMA concentration was significantly associated with response (P < .0001), such that, as baseline sBCMA levels increase, the probability of achieving a clinical response decreases.
Figure 4.

Impact of BCMA expression and sBCMA on levels of response, and impact of high-risk disease characteristics (extramedullary plasmacytomas, revised ISS stage, tumor burden, and LDH levels) on serum sBCMA levels. (A) Frequency of BCMA+CD45lowCD38+CD138+ BMPCs relative to response in the RP2D cohort at baseline. (B) Density of BCMA expression in CD45lowCD38+CD138+ BMPCs depicted as log10 normalized values and relative to response assessed by flow cytometry. (C) sBCMA levels in serum at baseline from patients in the RP2D cohorts. (D) Quantification of sBCMA levels in serum associated with soft tissue extramedullary plasmacytoma. (E) Quantification of serum sBCMA levels relative to revised ISS staging (I, II, or III). (F) Quantification of serum sBCMA levels relative to tumor burden. (G) Quantification of serum sBCMA levels relative to baseline LDH level (low, normal, or high). Groups were compared using the Wilcoxon rank-sum test. Data for 1 patient with outlier values (sBCMA >1250 ng/mL) were included in the analyses but not included on the plots.
Higher proportion of exhausted T cells correlate with disease burden
Evaluating T-cell functional phenotype relative to disease burden, we found a higher frequency of baseline BM CD8 T cells expressing PD-1 and coexpressing PD-1 with TIM-3 or CD38 in patients who had high disease burden (BMPCs ≥ 60%; Figure 5A-C). Similarly, patients with high tumor burden had a higher proportion of CD4 T cells expressing CD25 and CD38 (Figure 5D-E). Similar trends were also observed in PB (supplemental Figure 8). Interestingly, when we assessed various disease factors, including BMPCs ≥80%, spike in serum M protein ≥5 g/dL, and serum free light chain ≥5000 mg/L, and generated a composite tumor burden score (as defined in the supplemental Appendix), we found that higher baseline frequency of CD8 T cells in BM coexpressing PD-1/TIM-3 and PD-1/CD38 (Figure 5F-G) and higher proportion of CD4 T cells coexpressing PD-1/TIM-3 and PD-1/CD38 (Figure 5H-I) were also associated with high composite tumor burden. Furthermore, we explored whether these T cells were induced shortly after treatment (from baseline to cycle 3 day 1) and found that teclistamab-mediated induction of BM PD-1+ CD38+ CD8 T cells was lower in patients with high levels of BMPCs (≥60%; Figure 5J), which is indicative of less robust immune activation shortly after treatment administration.
Figure 5.

Immunophenotypic analyses of T cells at baseline and at cycle 3 day 1 after teclistamab monotherapy in BM correlate with disease burden status. (A-C) Frequency of PD-1, PD-1/TIM-3, and PD-1/CD38 expression in the CD8 T-cell subset relative to percentage of BMPCs at baseline from the RP2D cohort assessed by flow cytometry. (D-E) Frequency of CD4 T cells expressing CD25 or CD38 at baseline relative to disease burden assessed by the percentage of BMPCs. (F-G) Frequency of PD-1/TIM-3 and PD-1/CD38 expression in CD8 T cells relative to composite tumor burden. (H-I) Frequency of CD4 T cells coexpressing PD-1/TIM-3 or PD-1/CD38 assessed by flow cytometry relative to tumor burden at baseline. (J) Maximum fold change from baseline to cycle 3 day 1 of CD8 T cells coexpressing PD-1 and CD38 relative to low (BMPCs <60%) or high (BMPCs ≥60%) tumor burden. Representative staining is shown on the right of panels B-E,H,I. Statistical analyses were performed using the Wilcoxon rank-sum test for panels A-E,J or Kruskal-Wallis test for panels F-I.
Finally, we assessed the production of cytokines in correlation with these immune cell populations expressing higher exhaustion-associated receptors and observed correlation with levels of serum inhibitory cytokines, such as interleukin-10, and promyeloma survival cytokines, such as interleukin-8 at baseline (supplemental Figure 9). We also observed a weak albeit significant correlation of PD-1+/TIM-3+ T cells with sBCMA (supplemental Figure 9F), supporting that higher exhaustion marker expression correlates with disease burden and impaired functionality in these cells.
Immune correlatives are associated with response independent of tumor burden
So far, our results indicate an association of a number of baseline immune factors in BM and blood from patients with teclistamab response. Furthermore, some of these immune correlatives were associated with tumor burden or a composite tumor burden score. To assess correlatives that are associated with response independently of tumor burden, we performed an exploratory analysis and compared the values for each biomarker of interest between response groups and tested for their correlation with tumor burden (Table 1). We included in the exploratory analysis the frequency of several T-cell subsets and biomarkers that are indicative of T-cell function, which may be key to inform mechanisms of action. Among the relevant correlatives, we found 20 biomarkers to be associated with clinical response (P < .05), including CD4 T cells expressing CD38 in BM (P = .004) or coexpressing PD-1 and TIM-3 in PB or BM (P = .029 and P = .032, respectively). From these, we found 7 biomarkers that were positively correlated with tumor burden, including BM CD4 T cells coexpressing PD-1 and CD38 (Rs = 0.324; P = .001). Overall, CD38 expression in tumor CD4 or CD8 T cells was found to be one of the main immune correlatives associated with disease burden. Furthermore, among the factors independent of disease burden (P > .05), we identified CD3 and CD8 T-cell counts and T-cell frequencies and CD3 T cells in the BM and blood coexpressing the inhibitory receptors PD-1 and TIM-3. Altogether, the identification of factors associated with response but not related to tumor burden may inform the use of teclistamab therapy.
Table 1.
Correlation of biomarkers with response and tumor burden by flow cytometry
| Biomarker | Biomarker ∼ clinical response |
Biomarker ∼ BMPCs, % |
||
|---|---|---|---|---|
| P value | Spearman correlation coefficient (Rs) | Spearman P value | Correlated with tumor burden | |
| BM CD38+/CD4+ T cells, % | .004 | 0.203 | .018 | Y |
| PB PD-1+TIM-3+/T cells, % | .005 | 0.100 | .226 | N |
| PB CD38+/CD3+CD4+CD8− T cells, % | .009 | 0.057 | .486 | N |
| PB CD45+CD3+, ×106/L | .012 | −0.106 | .359 | N |
| PB PD-1+/CD8+ T cells, % | .014 | 0.058 | .483 | N |
| BM CD38+/T cells, % | .014 | 0.207 | .016 | Y |
| PB TIM-3+/T cells, % | .017 | 0.033 | .687 | N |
| PB CD3+/CD45+ lymphocytes, % | .017 | 0.129 | .271 | N |
| PB PD-1+TIM-3+/CD8+ T cells, % | .018 | 0.129 | .116 | N |
| PB CD38+ Tregs, % | .019 | 0.004 | .964 | N |
| BM CD38+/CD8+ T cells, % | .020 | 0.276 | .001 | Y |
| PB CD45+CD3+CD8+, ×106/L | .024 | –0.135 | .243 | N |
| PB Tregs/CD4+ T cells, % | .029 | 0.164 | .046 | Y |
| BM PD-1+TIM-3+/CD4+ T cells, % | .029 | 0.149 | .084 | N |
| PB PD-1+/T cells, % | .032 | 0.016 | .842 | N |
| PB PD-1+TIM-3+/CD4+ T cells, % | .032 | 0.171 | .037 | Y |
| BM PD-1+TIM-3+/T cells, % | .033 | 0.135 | .116 | N |
| BM TIM-3+/CD8+ T cells, % | .041 | −0.052 | .547 | N |
| BM PD-1+CD38+/CD3+CD8−CD4+ cells, % | .042 | 0.324 | .001 | Y |
| BM PD-1+CD38+/CD3+ cells, % | .044 | 0.279 | .004 | Y |
Wilcoxon rank-sum test was used to identify differences in the specified biomarkers in responders vs nonresponders (biomarker ∼ clinical response P value). Association of each biomarker to tumor burden (biomarker ∼ BMPCs, %) was assessed using the Spearman correlation, and the correlation coefficient (Rs) and P value are reported. Biomarkers are flagged as correlated with tumor burden (Y) if Spearman P value <.05.
N, no; Y, yes.
Immune characteristics affect PFS
We also explored the contribution of immune status and T-cell subsets to progression-free survival (PFS). In MajesTEC-1, patients had a median PFS of 11.3 months.13 Importantly, we found that inferior PFS was associated with a higher proportion of peripheral CD8 T cells expressing PD-1 (P = .043; Figure 6A), a higher frequency of peripheral Tregs (P = .019; Figure 6B), and a higher proportion of BM CD4 T cells expressing CD25 (P = .00013; Figure 6C) and CD38 (P = .024; Figure 6D), all markers related to T-cell functionality. However, multivariate analyses using Cox regression analysis, including BM CD25+/CD4 T cells and CD38+/CD4 T cells as well as PB PD-1+/CD8 T cells and Tregs, showed that, when accounting for these flow cytometry parameters, only BM CD25+/CD4 T cells were associated with improved PFS (less than the median BM CD25+/CD4 T cells; hazard ratio, 0.39; 95% confidence interval, 0.22-0.70; P = .002; supplemental Figure 10). Altogether, these data indicate that patient immune state at baseline contributes to the response to teclistamab therapy and PFS after treatment.
Figure 6.

Higher proportions of Tregs and expression of PD-1, CD25, and CD38 in T-cell subsets were associated with inferior PFS at baseline. (A-B) Kaplan-Meier curves of PFS for patients from the RP2D cohort according to proportion of baseline peripheral PD-1+ CD8 T cells (A) and Tregs (B) in the periphery. (C) Kaplan-Meier curves of PFS according to proportion of BM CD25-expressing CD4 T cells at baseline. (D) Kaplan-Meier curves of PFS according to proportion of BM CD38-expressing CD4 T cells at baseline. Dark green lines represent patients with proportions below the median, and light green lines represent patients with proportions equal to or higher than the median PFS in MajesTEC-1. Statistical significance was calculated using the Cox proportional hazards model.
Discussion
Evidence from several cancer types suggests that the immune status of both the periphery and the tumor contributes to the response to immunotherapy.15,34,35 Patients with R/RMM vary in disease manifestation and time to progression, are difficult to treat, and exhibit substantial heterogeneity in their response to available treatment options.36,37 Moreover, the initial T-cell repertoire has been suggested to predetermine the response to CD3 bispecific antibodies.15 As such, gaining a deeper understanding of patient immunity at the start of treatment is crucial to fully realize the potential of immunotherapy and to use these therapies in the most clinically beneficial manner.
To identify biomarkers associated with clinical response and explore why some patients fail to respond to teclistamab, we evaluated baseline immune cell status and relevant tumor correlatives with clinical determinants in patients receiving teclistamab from the MajesTEC-1 study. This is, to our knowledge, the most comprehensive analysis of patient data elucidating determinants of response to a BCMAxCD3 bispecific antibody in patients with R/RMM. Through analysis of the peripheral immune compartment, we found that clinical response to teclistamab was associated with higher numbers of CD3 T cells at baseline, with responders exhibiting higher frequencies and numbers of total T cells and cytotoxic CD8 T cells than nonresponders. Expansion of phenotypically cytotoxic T cells has been hypothesized to be relevant to the antitumor response, which potentially explains why no such differences were observed for CD4 T cells.38 Transcriptional analysis of T-cell clones before and after T-cell engager (TCE) therapy recently showed expansion of an enriched CD8 effector cell population was found in clinical responders.15 Here, we show that in the BM microenvironment, CD8 T cells from responding patients showed a cytolytic profile and exhibited higher proportions of CD8 T cells expressing granzyme B and perforin. Furthermore, CyTOF analysis demonstrated that the proportion of naive CD8 T cells in the periphery at baseline was higher in responders than nonresponders. Circulating naive CD8 T cells may serve as a determinant of response due to the low level of exhaustion and enhanced replicative and differentiating potential that may enable them to become an effector cell type. Previous reports have suggested that naive-derived effector cells may have superior antitumor activity,39 may be relatively resistant to immunosuppressive cytokines such as transforming growth factor β,40 and may preferentially migrate to inflamed tissues.41 Our results suggest that an initial pool of cells with cytotoxic potential in the BM may be critical for the mechanism of action of teclistamab. This is in line with a previous report that found clonotypes responsive to TCEs exhibited a phenotype consistent with a cytotoxic-effector profile15 and is also supported by our findings that BM CD4 T-cell TEMRA are associated with response. Published reports suggest that more differentiated TEMRAs exhibit a higher cytotoxic profile, more pronounced cytokine production,42 and upregulation of cytotoxic molecules such as CD244, perforin, and granzyme B, and transcription factors Runx3, T-bet, and Hobit.43 Moreover, our findings of a role for naive T cells in response to teclistamab support a previous report in which therapy with bispecific TCEs led to a phenotypic shift of initially naive T-cell clones to effector and memory clusters, highlighting the role of naive CD8 T cells as an alternative route of immune response induced by TCE therapy.15
Our results highlight the role of baseline immunosuppressive Treg populations in clinical response to teclistamab: patients who responded to teclistamab treatment had lower frequencies of Tregs in both the tumor microenvironment and in circulation, and as such, likely exhibited less of a suppressive immune landscape than nonresponders. Additionally, Treg subsets were marked by higher expression of CD38 in nonresponders. Frequency of CD38 Tregs has been associated with less durable responses to anti-CD38 antibodies such as daratumumab and isatuximab,44 and higher levels of CD38 in Tregs correlated with a more suppressive state.26 Our data support that the accumulation of these cells as well as their functional characteristics are key correlatives to reduced response. In addition, the proportion of total, CD4, and CD8 T cells found to express or coexpress exhaustion markers CD38, TIM3, and PD-1 was lower in patients who achieved a clinically meaningful outcome with teclistamab, indicating that responders may display fitter immune status than nonresponders. Importantly, these data highlight the correlation between both PB and BM T cells at baseline in achieving better clinical outcomes, indicating that both PB and BM compartments may provide valuable information in patients treated with bispecifics. Previous studies have suggested that peripheral immune subsets can be predictive of intratumoral immune responses and may serve as a prognostic factor in patients with cancer.45 Moreover, this study expands on the understanding of T-cell responses in the BM, which is of critical value because sampling and evaluation of immune cells has been limited to PB in most other studies in humans.
Baseline sBCMA was associated with lower response rates, which can be explained by its association with high-risk features such as high BMPC infiltration, presence of extramedullary plasmacytomas, ISS stage III, high tumor burden, and elevated LDH levels.46, 47, 48 In a separate population pharmacokinetic analysis, baseline sBCMA was not a significant covariate affecting teclistamab pharmacokinetics, suggesting that sBCMA does not act as a sink for teclistamab.32 Importantly, our multivariate analysis identified baseline sBCMA as a predictive biomarker of response, with an inverse correlation between the 2 parameters.
Regarding tumor characteristics, the number of BCMA molecules expressed on BMPCs was not associated with clinical response to teclistamab. Similarly, the frequency of BCMA-expressing BMPCs was also not associated with response. The expression of BCMA was prevalent and heterogenous among patients with R/RMM, which suggests that, in the presence of BCMA expression, immune-mediated determinants are key to the response to teclistamab. However, although BCMA target loss is regarded as an infrequent occurrence driving the resistance to bispecific antibody therapy, other molecular alterations (such as missense mutations, in-frame deletions, and nontruncating mutations) have been identified as mediators of relapse after TCE therapy,49 and therefore, the detection of BCMA expression by flow cytometry does not preclude the presence of a mutation interfering with TCE binding and subsequent resistance to teclistamab. Hence, future studies evaluating the BCMA locus at the time of relapse will be of interest.
Higher tumor burden, as measured by the frequency of BMPCs, was also associated with an increased baseline proportion of T cells expressing markers of T-cell exhaustion in both BM and PB. Similar results were observed for patients with a high composite tumor burden score (BMPCs ≥ 80%; serum M-spike ≥ 5 g/dL; or serum free light chain ≥ 5000 mg/L), because these patients demonstrated an exhaustion-type phenotype represented by higher baseline frequencies of T cells in BM expressing PD-1/TIM-3 and PD-1/CD38 and Tregs. In addition, patients with BMPCs ≥60% did not induce T cells as effectively as those with BMPCs <60% after short-term treatment with teclistamab. Notably, our study identifies critical factors correlating with response independently of tumor burden, such as cytotoxic CD8 T-cell counts, CD3 T-cell frequencies and counts, and T cells expressing coinhibitory receptors (ie, CD8 T cells in blood or BM expressing TIM-3 or PD-1/TIM-3), which might inform the use of teclistamab alone and in combination with other immunotherapies.
Baseline T-cell characteristics were also associated with PFS. Exhausted T-cell phenotype, in terms of higher frequencies of T cells expressing PD-1 and CD8 and regulatory CD4 T cells in the periphery and of CD4 T cells expressing CD25 or CD38 in BM, correlated with decreased PFS, again highlighting the importance of immune status in achieving better clinical outcomes.
Despite high response rates (63%) with teclistamab in MajesTEC-1, it is critical to understand the characteristics of nonresponders to guide decisions regarding the potential application of combination therapy. Herein, the results showed that response to teclistamab was associated with immune fitness at baseline. Among nonresponders, baseline immune profiles were associated with T-cell dysfunction and immune suppression, with lower T-cell numbers, including those of naive T cells, and a higher frequency of immunosuppressive cells and T cells expressing markers of exhaustion. Additional studies are ongoing to evaluate these correlatives in patients treated earlier in their disease, when immune status may be more favorable. Moreover, from a mechanistic perspective, these findings support the clinical investigation of teclistamab in combination with agents that have immunomodulatory actions, such as daratumumab (currently being investigated in the phase 3 MajesTEC-3 study [NCT05083169]), checkpoint inhibitors (being investigated in the phase 1 TRIMM-3 study [NCT05338775]), IMiDs (being investigated in the phase 3 MajesTEC-4 study [NCT05243797]), and cereblon E3 ligase modulating drugs.
Conflict-of-interest disclosure: D.C.-S., D.V., S. Skerget, T.P., S. Stein, R.B., O.S.L., K.N., C.D., J.P., A.B., T.S., C.U., R.K., L.P., D.T., and S.X.W.L. are employees of Janssen and may have stock/other ownership interests in Janssen. L.S.W., J.G., and S.G. were employees of Janssen at the time the work was carried out and may have stock/other ownership interests in Janssen. R.I.V. was an employee of Janssen at the time the work was carried out and may have stock/other ownership interests in Janssen. P.M. has served in consulting/advisory roles and received honoraria from AbbVie, Amgen, Celgene, GlaxoSmithKline, Janssen, Oncopeptides, and Sanofi. S.Z.U. has served in a consulting or advisory role for AbbVie, Amgen, Bristol Myers Squibb/Celgene, Celgene, Genentech, Gilead Sciences, GlaxoSmithKline, Janssen, Karyopharm Therapeutics, Merck, and Takeda; and has received research funding from Amgen, Array BioPharma, Bristol Myers Squibb, Celgene, GlaxoSmithKline, Janssen, Merck, Pharmacyclics, Sanofi, Seattle Genetics, and Skyline Diagnostics. N.J.B. has served in consulting/advisory roles for Amgen, Celgene, Janssen, Karyopharm Therapeutics, Pfizer, Sanofi, and Takeda; received honoraria from AbbVie, Amgen, Celgene, Genentech/Roche, GlaxoSmithKline, Janssen, Karyopharm Therapeutics, Sanofi, and Takeda; and has received research funding from Celgene and Janssen. N.W.C.J.v.d.D. has served in consulting/advisory roles for AbbVie, Adaptive Biotechnologies, Amgen, Bayer, Bristol Myers Squibb, Celgene, Janssen, Novartis, Pfizer, Merck, Roche, Servier, and Takeda; and has received research funding from Amgen, BMS, Celgene, Cellectis, Janssen, and Novartis.
The current affiliation for R.I.V. is AbbVie Inc, North Chicago, IL.
Acknowledgments
The authors acknowledge Yann Abraham, Elena Ramos, and Aniko Meijer for their valuable contributions to mass cytometry analyses. Editorial and medical writing support for this article was provided by John Bilbruck and Corey Eagan of Eloquent Scientific Solutions and was funded by Janssen Global Services, LLC.
The MajesTEC-1 trial was funded by Janssen Research & Development, LLC.
Authorship
Contribution: D.C.-S. drafted the manuscript, performed experiments, contributed to data generation, and analyzed and interpreted data; D.V., C.D., and L.S.W. contributed to data generation and analyzed and interpreted data; S. Stein, T.P., R.B., O.L., K.C.-N., and J.P. performed experiments and contributed to data generation; S. Skerget, L.P., and D.T. contributed to data generation; A.B., T.S., C.U., R.K., and J.G. contributed to the study design, managed the clinical study and provided clinical oversight, and analyzed and interpreted data; S.G. contributed to the study design and analyzed and interpreted data; S.X.W.L. analyzed and interpreted data; P.M., S.Z.U., N.J.B., and N.W.C.J.v.d.D. were clinical investigators in the study; R.I.V. conceived and contributed to the project design, analyzed and interpreted data, and provided oversight of the work; and all authors reviewed and revised the manuscript, approved the final version, and agreed to submit the manuscript for publication.
Footnotes
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Cowan AJ, Green DJ, Kwok M, et al. Diagnosis and management of multiple myeloma: a review. JAMA. 2022;327(5):464–477. doi: 10.1001/jama.2022.0003. [DOI] [PubMed] [Google Scholar]
- 2.Bird SA, Boyd K. Multiple myeloma: an overview of management. Palliat Care Soc Pract. 2019;13 doi: 10.1177/1178224219868235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimopoulos MA, Chen C, Spencer A, et al. Long-term follow-up on overall survival from the MM-009 and MM-010 phase III trials of lenalidomide plus dexamethasone in patients with relapsed or refractory multiple myeloma. Leukemia. 2009;23(11):2147–2152. doi: 10.1038/leu.2009.147. [DOI] [PubMed] [Google Scholar]
- 4.Orlowski RZ, Nagler A, Sonneveld P, et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol. 2007;25(25):3892–3901. doi: 10.1200/JCO.2006.10.5460. [DOI] [PubMed] [Google Scholar]
- 5.Miguel JS, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–1066. doi: 10.1016/S1470-2045(13)70380-2. [DOI] [PubMed] [Google Scholar]
- 6.Usmani SZ, Nahi H, Plesner T, et al. Daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma: final results from the phase 2 GEN501 and SIRIUS trials. Lancet Haematol. 2020;7(6):e447–e455. doi: 10.1016/S2352-3026(20)30081-8. [DOI] [PubMed] [Google Scholar]
- 7.Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia. 2020;34(4):985–1005. doi: 10.1038/s41375-020-0734-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verkleij CPM, Broekmans MEC, van Duin M, et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv. 2021;5(8):2196–2215. doi: 10.1182/bloodadvances.2020003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.TECVAYLI . Janssen Biotech, Inc; 2022. Prescribing information.https://www.teclistamabhcp.com [Google Scholar]
- 10.TECVAYLI . Janssen Biologics BV; 2022. Summary of product characteristics. [Google Scholar]
- 11.Pillarisetti K, Powers G, Luistro L, et al. Teclistamab is an active T cell-redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020;4(18):4538–4549. doi: 10.1182/bloodadvances.2020002393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Usmani SZ, Garfall AL, van de Donk N, et al. Teclistamab, a B-cell maturation antigen × CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): a multicentre, open-label, single-arm, phase 1 study. Lancet. 2021;398(10301):665–674. doi: 10.1016/S0140-6736(21)01338-6. [DOI] [PubMed] [Google Scholar]
- 13.Moreau P, Garfall AL, van de Donk N, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495–505. doi: 10.1056/NEJMoa2203478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, van der Leun AM, Yofe I, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176(4):775–789.e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedrich MJ, Neri P, Kehl N, et al. The pre-existing T cell landscape determines the response to bispecific T cell engagers in multiple myeloma patients. Cancer Cell. 2023;41(4):711–725.e6. doi: 10.1016/j.ccell.2023.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Xia A, Zhang Y, Xu J, Yin T, Lu XJ. T cell dysfunction in cancer immunity and immunotherapy. Front Immunol. 2019;10:1719. doi: 10.3389/fimmu.2019.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T cell dysfunction and exhaustion in cancer. Front Cell Dev Biol. 2020;8:17. doi: 10.3389/fcell.2020.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller BC, Sen DR, Al Abosy R, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sivakumar S, Abu-Shah E, Ahern DJ, et al. Activated regulatory T-cells, dysfunctional and senescent T-cells hinder the immunity in pancreatic cancer. Cancers (Basel) 2021;13(8):1776. doi: 10.3390/cancers13081776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajkumar SV, Harousseau JL, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18):4691–4695. doi: 10.1182/blood-2010-10-299487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328–e346. doi: 10.1016/S1470-2045(16)30206-6. [DOI] [PubMed] [Google Scholar]
- 23.Uckun FM. Overcoming the immunosuppressive tumor microenvironment in multiple myeloma. Cancers (Basel) 2021;13(9) doi: 10.3390/cancers13092018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van de Donk N. Immunomodulatory effects of CD38-targeting antibodies. Immunol Lett. 2018;199:16–22. doi: 10.1016/j.imlet.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 25.van de Donk N, Usmani SZ. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol. 2018;9:2134. doi: 10.3389/fimmu.2018.02134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng X, Zhang L, Acharya C, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. 2017;23(15):4290–4300. doi: 10.1158/1078-0432.CCR-16-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sponaas AM, Waage A, Vandsemb EN, et al. Bystander memory T cells and IMiD/checkpoint therapy in multiple myeloma: a dangerous tango? Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.636375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zelle-Rieser C, Thangavadivel S, Biedermann R, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol. 2016;9(1):116. doi: 10.1186/s13045-016-0345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verkleij CPM, Frerichs KA, Broekmans MEC, et al. NK cell phenotype is associated with response and resistance to daratumumab in relapsed/refractory multiple myeloma. HemaSphere. 2023;7(5):e881. doi: 10.1097/HS9.0000000000000881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qing M, Zhou T, Perova T, et al. Immune profiling of patients with extranodal natural killer/T cell lymphoma treated with daratumumab. Ann Hematol. 2024;103(6):1989–2001. doi: 10.1007/s00277-023-05603-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lucas F, Pennell M, Huang Y, et al. T cell transcriptional profiling and immunophenotyping uncover LAG3 as a potential significant target of immune modulation in multiple myeloma. Biol Blood Marrow Transplant. 2020;26(1):7–15. doi: 10.1016/j.bbmt.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Girgis S, Wang Lin SX, Pillarisetti K, et al. Effects of teclistamab and talquetamab on soluble BCMA levels in patients with relapsed/refractory multiple myeloma. Blood Adv. 2023;7(4):644–648. doi: 10.1182/bloodadvances.2022007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stauffer A, Ray C, Hall M. A flexible multiplatform bioanalytical strategy for measurement of total circulating shed target receptors: application to soluble B cell maturation antigen levels in the presence of a bispecific antibody drug. Assay Drug Dev Technol. 2021;19(1):17–26. doi: 10.1089/adt.2020.1024. [DOI] [PubMed] [Google Scholar]
- 34.Sinha M, Zhang L, Subudhi S, et al. Pre-existing immune status associated with response to combination of sipuleucel-T and ipilimumab in patients with metastatic castration-resistant prostate cancer. J Immunother Cancer. 2021;9(5) doi: 10.1136/jitc-2020-002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, Li W, Yang X, et al. An immune landscape based prognostic signature predicts the immune status and immunotherapeutic responses of patients with colorectal cancer. Life Sci. 2020;261 doi: 10.1016/j.lfs.2020.118368. [DOI] [PubMed] [Google Scholar]
- 36.Gandhi UH, Cornell RF, Lakshman A, et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia. 2019;33(9):2266–2275. doi: 10.1038/s41375-019-0435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varughese P, Smith R, Xue M, et al. Real-world treatment patterns and outcomes of triple-class treated patients with multiple myeloma in the United States. Expert Rev Hematol. 2022;16(1):65–74. doi: 10.1080/17474086.2023.2154648. [DOI] [PubMed] [Google Scholar]
- 38.Raitakari M, Brown RD, Sze D, et al. T-cell expansions in patients with multiple myeloma have a phenotype of cytotoxic T cells. Br J Haematol. 2000;110(1):203–209. doi: 10.1046/j.1365-2141.2000.02131.x. [DOI] [PubMed] [Google Scholar]
- 39.Hinrichs CS, Borman ZA, Cassard L, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A. 2009;106(41):17469–17474. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen HH, Kim T, Song SY, et al. Naïve CD8(+) T cell derived tumor-specific cytotoxic effectors as a potential remedy for overcoming TGF-β immunosuppression in the tumor microenvironment. Sci Rep. 2016;6 doi: 10.1038/srep28208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2(4):251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 42.Larbi A, Fulop T. From “truly naïve” to “exhausted senescent” T cells: when markers predict functionality. Cytometry A. 2014;85(1):25–35. doi: 10.1002/cyto.a.22351. [DOI] [PubMed] [Google Scholar]
- 43.Tian Y, Babor M, Lane J, et al. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nat Commun. 2017;8(1):1473. doi: 10.1038/s41467-017-01728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitadate A, Kobayashi H, Abe Y, et al. Pre-treatment CD38-positive regulatory T cells affect the durable response to daratumumab in relapsed/refractory multiple myeloma patients. Haematologica. 2020;105(1):e37–e40. doi: 10.3324/haematol.2019.219683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niccolai E, Cappello P, Taddei A, et al. Peripheral ENO1-specific T cells mirror the intratumoral immune response and their presence is a potential prognostic factor for pancreatic adenocarcinoma. Int J Oncol. 2016;49(1):393–401. doi: 10.3892/ijo.2016.3524. [DOI] [PubMed] [Google Scholar]
- 46.Usmani SZ, Heuck C, Mitchell A, et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica. 2012;97(11):1761–1767. doi: 10.3324/haematol.2012.065698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al Saleh AS, Parmar HV, Visram A, et al. Increased bone marrow plasma-cell percentage predicts outcomes in newly diagnosed multiple myeloma patients. Clin Lymphoma Myeloma Leuk. 2020;20(9):596–601. doi: 10.1016/j.clml.2020.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23(15):3412–3420. doi: 10.1200/JCO.2005.04.242. [DOI] [PubMed] [Google Scholar]
- 49.Lee H, Ahn S, Maity R, et al. Mechanisms of antigen escape from BCMA- or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med. 2023;29(9):2295–2306. doi: 10.1038/s41591-023-02491-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.