

Abstract
The present study uses molecular docking and dynamic simulations to evaluate the inhibitory effect of flavonoid glycosides-based compounds on coronavirus Main protease (Mpro) and RNA polymerase. The Molegro Virtual Docker (MVD) software is utilized to simulate and calculate the binding parameters of compounds with coronavirus. The docking results show that the selected herbal compounds are more effective than those of chemical compounds. It is also revealed that five herbal ligands and two chemical ligands have the best docking scores. Furthermore, a Molecular Dynamics (MD) simulation was conducted for Hesperidin, confirming docking results. Analysis based on different parameters such as Root-mean-square deviation (RMSD), Root mean square fluctuation (RMSF), Radius of gyration (Rg), Solvent accessibility surface area (SASA), and the total number of hydrogen bonds suggests that Hesperidin formed a stable complex with Mpro. Absorption, Distribution, Metabolism, Excretion, And Toxicity (ADMET) analysis was performed to compare Hesperidin and Grazoprevir as potential antiviral medicines, evaluating both herbal and chemical ligand results. According to the study, herbal compounds could be effective on coronavirus and are admissible candidates for developing potential operative anti-viral medicines. Hesperidin was found to be the most acceptable interaction. Grazoprevir is an encouraging candidate for drug development and clinical trials, with the potential to become a highly effective Mpro inhibitor. Compared to RNA polymerase, Mpro showed a greater affinity for bonding with Hesperidin. van der Waals and electrostatic energies dominated, creating a stable Hesperidin-Mpro and Hesperidin-RNA polymerase complex.
Keywords: Main protease, RNA polymerase, SARS-CoV-2, Molecular simulation, ADMET analysis
Graphical abstract

Highlights
-
•
Insilico methods were used to investigate the inhibitory effect of flavonoid-based compounds on coronavirus Mpro and RNA polymerase.
-
•
The MVD software is utilized to simulate and calculate the binding parameters of compounds with coronavirus.
-
•
The ADMET analysis for Hesperidin and Grazoprevir were also compared as the best medicine against the virus in herbal and chemical ligand results.
-
•
The MD simulation was carried out with the GROMACS and revealed that the parameters such as RMSD, RMSF and Rg remained stable over time.
-
•
It was concluded that herbal compounds had more reasonable coronavirus Mpro and RNA polymerase inhibition.
1. Introduction
The ongoing global crisis caused by the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) has prompted extensive research efforts to identify effective methods to combat the virus. One promising approach is to develop vaccines that can prevent viral disease and create drug therapies to treat the illness [1,2]. The COVID-19 pandemic has led to significant advancements in antiviral therapies. These treatments can be administered early after diagnosis to prevent severe disease or used during later stages to improve patient outcomes. Direct-acting antivirals target the virus, while host-directed therapies aim to mitigate the inflammatory response in severe COVID-19 cases [[3], [4], [5]]. A significant factor in viral replication is the proteolytic cleavage of large precursor proteins, which microbial proteases facilitate [2,6]. Viral proteases can be targeted for drug design, and protease inhibitors can be tailored to the infection rate and treatment objectives [[7], [8], [9]]. The COVID-19 virus requires a complete inhibition of viral replication, and current antivirals such as Molnupiravir and Paxlovid are not significantly effective [7,10].
The 3C-like protease, known as Mpro, plays a crucial role in the post-translational processing of the replicase polyprotein, which is essential for viral replication. Using the crystal structure of SARS-CoV-2 Mpro, drugs with protease inhibitory characteristics can be developed to identify the mechanism of action of the protease RNA-dependent [8,11]. RNA polymerase is another critical enzyme for the replication and transcription of the viral genome and is a favorable target for antiviral drug investigation [[12], [13], [14]].
Recent studies have revealed insights into the mechanism of RNA polymerase's substrate RNA recognition. Additionally, the mechanism of inhibition of RNA polymerase by the nucleotide analog remdesivir has been clarified [12]. The findings suggest that glycosyl flavonoids could be potential agents against SARS-CoV-2 and other coronaviruses by inhibiting key targets involved in virus replication and reducing oxidative stress [15,16]. Following these discoveries, a multitude of research endeavors were initiated to investigate the potential inhibition of Mpro by glycosyl flavonoids. These studies involved the application of molecular docking technology and simulation studies to further analyze and understand the underlying mechanisms at a molecular level [[17], [18], [19], [20]]. It highlights the potential inhibitory activity of compounds like narcissoside, quercetin, myricetin, nicotiflorin and rutin through molecular docking and simulation studies providing crucial insights for the development of therapeutic agents against coronavirus infections. The study also emphasizes glycosylation's importance in enhancing these compounds' bioactive features [19,[21], [22], [23]]. Also, compounds like pectolinarin, rhoifolin, and baicalin have demonstrated inhibitory activity towards SARS-CoV-2 Mpro, which is attributed to the presence of sugar moieties that enhance their binding affinity to the active sites of the enzymes [20,24].
Different in silico studies have been conducted on FDA-approved drugs and small molecules against SARS-CoV-2 have been previously studied for their inhibitory effects on infectious organisms [[25], [26], [27]]. Furthermore, drugs used for the hepatitis C virus, such as Elbasvir, Grazoprevir, and Glecaprevir, show potential for inhibiting SARS-CoV-2 Mpro. Simeprevir, Paritaprevir, and Raltegravir also demonstrate strong interactions with the enzyme. These drugs could be promising candidates for further studies in managing COVID-19 [[28], [29], [30], [31]].
In our research, we carried out antiviral investigations using numerical modeling and simulation techniques, including insilico, molecular docking, and MD simulations. We focused on selecting FDA-approved small molecule drugs and natural compounds containing flavonoid glycosides, aiming to identify potential Mpro and RNA polymerase inhibitors from SARS-CoV-2. The findings suggest that both Hesperidin and Grazoprevir are potent inhibitors of Mpro and exhibit greater stability and interaction with the protease. Given that natural metabolites generally have lower cell toxicity and fewer side effects than synthetic drugs, Hesperidin may prove to be more effective than Grazoprevir for pharmaceutical applications, particularly in the treatment of infectious and inflammatory diseases. Additionally, the study explored the binding of Hesperidin to RNA polymerase using Docking and MD analysis. The results are expected to provide valuable insights into this interaction and may have implications for the development of RNA polymerase-targeted therapies.
2. Methods
2.1. Ligand preparation
In order to start the process, the first step was to obtain the chemical and herbal compounds from the PubChem Chemical Database [32]. These structures were then carefully adjusted using HyperChem software [33]. Two distinct methods were utilized to enhance and improve the structures - the molecular mechanics force field of MM+ and the semi-empirical approach of AM1 (Austin Model 1). After the structures were completed, they were saved in Mol2 format, which was used as the main input for further analysis using molecular docking software [34].
2.2. Target/receptor preparation
For the study, two essential targets of the coronavirus were focused on - Mpro protein (6M03) and RNA polymerase (7BTF), which were obtainable on the RCSB Protein Data Bank website (https://www.rcsb.org/) [13,35,36], shown in Fig. 1. The details of the proteins were downloaded and Discovery Studio software was used to remove any additional cofactors in the PDB files. However, water molecules and some cofactors within the protein PDB files were kept to create accurate molecular docking. This ensured the conditions were as close to their normal state as possible [37,38]. Fig. 1 displays the detailed information of the selected protein structures.
Fig. 1.

Molecular structures of A)6M03 and B)7BTF proteins (3D).
2.3. MVD process
The protein and compound structures were prepared for docking using MVD software [39]. The "preparation molecule for docking" module in the MVD package was utilized to identify potential protein cavities that could serve as receptors for ligand binding. Docking parameters were set, including a grid resolution of 30 Å, a maximum iteration of 1500, and a total population size of 50. Internal electrostatic interaction, sp2-sp2 torsions, and internal H-bond interactions were noted to assess the binding affinity and interactions of the compounds with Mpro. A simplex evolution at the maximum step 300, with a neighborhood distance factor of 1, was set, and ten sets of docking were conducted. Post-dock energy minimization was performed using the Nelder-Mead Simplex Minimization. The results were analyzed using Molegro Molecular Viewer 6.0 and Discovery Studio 2017 R2 Client, and the best-interacting compound from each dataset was selected [[40], [41], [42]]. Fig. 2 shows the cavities of the targets (6M03 and 7BTF) that have the most significant potential to bind to the ligands.
Fig. 2.

Area of the molecular structure of A) 6M03 and B) 7BTF proteins, the cavities of the targets that interact with the ligands and dock their green color.
2.4. MD simulation
An in-depth analysis of the binding between ligands and proteins was conducted using MD simulation. The simulation with the most optimal docking mode was chosen based on the energy of the interactions between the receptor and ligand observed during the docking study. Throughout the simulations, the GROMOS 54A7 force field was applied via the GROMACS program (version 2019.1) [43]. A neutral charge was achieved for the simulation box by adding a sufficient amount of Na+ and Cl− ions to a water solvent. The system's energy was minimized using the GROMOS 54A7 force field for a maximum of 50000 steps. During the NVT stage, the temperature was fixed at 300 K using the Berendsen thermostat, and the system was equilibrated for one ns in the canonical (NVT) ensemble. The Parrinello-Rahman pressure coupling method was used during the isothermal-isobaric (NPT) stage to maintain the system's pressure at 1 bar during the one ns run. Finally, the MD simulation was conducted for 100 ns using a time step of 2 fs, with the Particle-Mesh-Ewald method [44,45] being utilized to calculate long-range electrostatic interactions and the LINCS algorithm applied.
3. Results
3.1. Molecular docking study
3.1.1. MVD molecular docking studies related to Mpro
All compounds were classified based on the Mol dock score within the docking software, and compounds with better binding capability were selected for further evaluation. Hesperidin, Monoxerutin, Vitexin, Isoquercitrin, and Diosmin were among the herbal compounds that showed good binding ability, along with some chemical compounds such as Grazoprevir Primuline compositions, which had the most substantial binding ability to bind to 6M03. Based on the modelling results, Hesperidin and Grazoprevir were the most effective among the compounds explored in this research, and their molecular docking results, ligand map of structures, and the target 6M03 were shown in Fig. 3, Fig. 4. Fig. 3A displays the Pi-Pi (p-p) Stacks, Pi-Alkyl, Unfavorable Bump, Water Hydrogen, Carbon Hydrogen and Conventional Hydrogen bonds of Hesperidin molecule with amino acid residues of receptor 6M03, including His A41, Cys A145, HOH 492, HOH 427, HOH 463, HOH 465, Asn A142, Glu A166, Met A165, Gln A189, Thr A26, His A41, His A163, Leu A141and Phe A140. Grazoprevir, on the other hand, forms Alkyl or Pi (p) alkyl, Unfavorable bump, and Hydrogen atoms of water molecule bonds with amino acid residues based on Val A:104, Phe A:294, HOH434, and HOH482 of the 6M03 receptor, as displayed in Fig. 3B.
Fig. 3.

Schematics of the molecular docking between A) the Hesperidin, B) the Grazoprevir, and C) the Monoxerutin ligands and the 6M03 receptor along with the ligand maps.
Fig. 4.

Schematics of the molecular docking between A) the Primuline, B) the Vitexin, C) the Isoquercitrin and D) the Diosmin ligands and the 6M03 receptor along with the ligand map.
The amino acid residues of Phe A:294, Arg A:298, Ile A:152, HOH441, HOH482, and Asp A:295 of the 6M03 receptor form various types of bonds with monoxerutin, including Pi-pi T-shaped, Pi-pi-Alkyl, and Conventional Hydrogen, Water Hydrogen, and Carbon Hydrogen bonds. These bonds are illustrated in Fig. 3C.
Primuline interacts with amino acid residues (Pro A:108, Phe A:294, Gln A:110, Asp A:295, Thr A:111 and HOH428, HOH434) of the 6M03 receptor shown in Fig. 4A through Pi-Alkyl, Pi-Pi Stacked, Unfavorable Bump, Conventional Hydrogen and Water Hydrogen bonds.
Fig. 4B shows that Vitexin forms Pi-Pi Stacked, Conventional Hydrogen, and Water Hydrogen bonds with amino acid residues (Phe A:294, Gln A:110, Arg A:105, and HOH441, HOH434, HOH482) of the 6M03 receptor.
Isoquercitrin interacts with amino acid residues (Gln A:110, HOH441, Phe A:294, Asp A:295, and HOH434, HOH478, HOH482) of the 6M03 receptor (Fig. 4C) through Unfavorable Doner-Doner and Unfavorable Bump, Pi-Pi Stacked and Amide-Pi Stacked, Conventional Hydrogen and Water Hydrogen bonds.
Diosmin interacts with amino acid residues (Phe A:294, Arg A:105, Thr A:292, Gln A: 110 and HOH441, HOH434, HOH441, HOH428, HOH482, HOH466) of the 6M03 receptor by forming Pi-Alkyl, Carbon Hydrogen, Conventional Hydrogen, and Water Hydrogen bonds (Fig. 4D).
The study found that specific candidates can effectively inhibit virus protease by interacting with the active site during the catalytic process and inhibiting the amino acids of the virus. The docking software in this work identified five enzymatic flap-protected areas.
The herbal compounds form strong bonds with Arg A: 105 and A: 298, Ile A: 152 and A: 106, and Phe A: 294 amino acids and have two strong bonds with two enzymatic flaps.
In summary, this study shows that Diosmin can interact with particular amino acid residues that exist in the 6M03 receptor through different types of bonds. The compounds identified in this study can effectively inhibit virus protease by interacting with the active site and inhibiting the amino acids of the virus. The docking software identified five enzymatic flap-protected areas, and the herbal compounds formed strong bonds with specific amino acids and enzymatic flaps.
The strong interaction between certain compounds and enzymatically active regions has created special functional groups. These unique groups have demonstrated the potential to be highly effective with minimal side effects, generating significant interest in utilizing plant compositions as a potent strategy for treating coronavirus infections. This promising approach holds potential for addressing the challenges of infection treatment with greater specificity and reduced adverse effects.
Upon careful analysis, it was discovered that Hesperidin, a naturally occurring compound found in citrus fruits, and Grazoprevir, a synthetic chemical compound, exhibited significant interactions with Mpro, a key protein in a specific biological process (Table 1).
Table 1.
The results obtained parameters from the interaction between herbal and chemical compounds and Mpro of SARS-COV-2.
| Compound | Structure | Mol dock Score (Kcal/mol) | Rerank Score (Kcal/mol) | Hbond (Kcal/mol) |
|---|---|---|---|---|
| Hesperidin (herbal) | 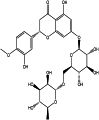 |
−156.107 | −109.347 | −10.6331 |
| Grazoprevir (chemical) | 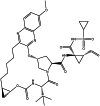 |
−143.818 | −76.1644 | 0 |
| Monoxerutin (herbal) | 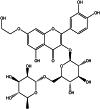 |
−139.471 | −66.9329 | −10.03 |
| Primuline (chemicl) | 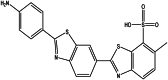 |
−113.146 | −70.443 | −10.12 |
| Vitexin (herbal) | 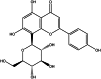 |
−105.07 | −73.450 | −8.44 |
| Isoquercitrin (herbal) | 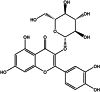 |
−103.249 | −63.2798 | −8.15 |
| Diosmin (herbal) | 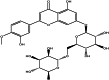 |
−101.588 | −40.0992 | −8.70 |
| Methyl 4-[[5-[[4-(2-amino-2-oxoethoxy)phenyl]methylidene]-3-methyl-4-oxo-1,3-thiazolidin-2-ylidene]amino]benzoate (chemical) | 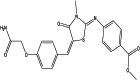 |
−89.1612 | −69.9767 | −2.86 |
| Troxerutin (herbal) |  |
−88.7576 | 174.365 | −9.42 |
| Proanthocyanidin (herbal) | 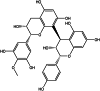 |
−81.6266 | −44.0925 | −0.57 |
| Icariin (herbal) |  |
−78.8867 | 2.2742 | −2.61 |
| Hidrosmin (herbal) | 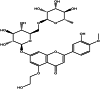 |
4.20513 | 536.421 | 0.16 |
| Methyl 4-[[(5E)-5-[[4-(2-amino-2-oxoethoxy)phenyl]methylidene]-3-methyl-4-oxo-1,3-thiazolidin-2-ylidene]amino]benzoate (chemical) | 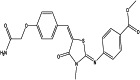 |
100.295 | −36.4086 | −0.59 |
| Crocin (herbal) | 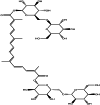 |
192.268 | 2102.37 | 1.28 |
| Ritonavir (chemical) |  |
285.506 | 2422.54 | 0.01 |
| Paritaprevir (chemical) |  |
332.37 | 2520.22 | 1.88 |
3.2. Prediction on ADMET
Many studies have stated that Hesperidin is helpful for prophylaxis and treatment of COVID-19 [[46], [47], [48], [49]]. Moreover, some researchers confirmed that Grazoprevir could be an effective therapeutic agent for COVID-19 treatment [[50], [51], [52]]. Therefore, we compared ADMET for both compounds. To evaluate this comparison, we used SWISSADMI server.Therefore, we compared ADMET for both compounds. ADMET refers to Chemical Absorption, Distribution, Metabolism, Excretion, and Toxicity analysis based on a comprehensive database for predicting compounds' pharmacokinetics before experimental procedures [53]. According to Lipinski's rule, if an orally active drug has one of the following features, it can be interpreted that this compound is of low permeation and poor absorption: with more than five hydrogen bond donors or with more than ten hydrogen bond acceptors, or with a molecular mass more than 500 Da, or with an octanol-water partition (log P) that does exceed 5 [54]. Like Hesperidin, Grazoprevir violates the number of H-bond acceptors and molecular weight (Table 2).
Table 2.
Predicted ADMET profile of hesperidin and Grazoprevir.
| Descriptor | Hesperidin | Grazoprevir |
|---|---|---|
| Molecular weight (g/mol) | 610.56 | 766.90 |
| Number of rotatable bonds | 7 | 9 |
| Number of H-bond donors | 8 | 3 |
| Number of H-bond acceptors | 15 | 12 |
| Log P | −0.72 | 4.142 |
| Gastrointestinal absorption | Low | Low |
| BBB permeability | No | No |
| Substrate of P-gp | Yes | Yes |
| CYP1A2 inhibitor | No | No |
| CYP2C19 inhibitor | No | No |
| CYP2C9 inhibitor | No | No |
| CYP2D6 inhibitor | No | No |
| CYP3A4 inhibitor | No | No |
| Bioavailability score | 0.17 | 0.17 |
| Log CLtot (mL/min/kg) | 0.211 | – |
| MRTD (mg/day) | 424 | 355 |
| Hepatotoxicity | No | No |
| Ames toxicity | No | No |
| hERG channel inhibition | Yes | Yes |
The inadequate water solubility of Grazoprevir, with a Log P value of 4.142, and the limited lipophilicity of Hesperidin, with a Log P value of −0.72, suggest that their oral bioavailability is low. This challenge can be addressed using intravenous administration or oral drug delivery systems. Distribution and permeability of these compounds into the blood-brain barrier (BBB) P-glycoprotein (P-gp) substrates analysis [55] implies that Hesperidin and Grazoprevir similarly cannot permeate through the BBB (Table 2). Also, it is noteworthy that the efflux of P-gp-mediated could limit Hesperidin and Grazoprevir distribution. Cytochrome P450 (CYP) superfamily carries out the drug Metabolism. According to the analysis in Table 2, Hesperidin and Grazoprevir are not likely to inhibit any types of CYP. Ames Test checks for mutagenicity, identifying potentially carcinogenic chemicals. Studies have shown Hesperidin dont have carcinogenic and hepatotoxic properties, while MRTD, which Maximum Recommended Therapeutic Dose, is more than Grazoprevir in Hesperidin.
After conducting detailed ADMET studies, it became evident that hesperidin displayed more favorable pharmacokinetic and safety profiles compared to Grazoprevir. Consequently, Hesperidin was selected for further in-depth research and development.
3.2.1. MVD molecular docking studies related to RNA polymerase
Since Hesperidin had the best performance and interaction with Mpro among the studied drugs, its interaction with RNA Polymerase was also investigated in this research work. MVD molecular docking conformation and analysis showed Hesperidin that interacted with RNA Polymerase. Free Total Energy or MolDock Score values were subject to negative energy values, indicating that the binding events of the complexes were spontaneous. Table 3 displays the docked configuration of the complexes with the related parameters. Schematic molecular docking results and ligand map of Hesperidin and the 7BTF targets are shown in Fig. 5. The MolDock Score value for Hesperidin was −104.691, where it was docked to RNA Polymerase receptor, respectively.
Table 3.
The obtained parameters from the interaction between Hesperidin and RNA Polymerase of SARS-COV-2.
| Compound | Mol dock Score (Kcal/mol) | Rerank Score (Kcal/mol) | Hbond (Kcal/mol) |
|---|---|---|---|
| Hesperidin (herbal) | −157.602 | −104.691 | −19.6493 |
Fig. 5.

Schematics of the molecular docking between the Hesperidin ligand, the 7BTF receptor, and the ligand map.
Hesperidin forms Conventional Hydrogen Bonds, van der Waals, Amide Pi-Stacked, Pi-Lone Pair, and Pi-Alkyl with amino acids of the RNA polymerase using Glu 796, Ala 797, Trp 800, Thr 801, Glu 802, Gly 808 and Glu811.
3.3. MD simulation of hesperidin with Mpro and RNA polymerase
To investigate the stability and conformations of the protein-ligand complexes, MD simulations of 100 ns were performed. Parameters of the model, including RMSD, RMSF, Rg, SASA, and the total number of hydrogen bonds, were analyzed to study the system's structural changes and stability of Mpro and RNA polymerase with Hesperidin.
The lowest average RMSD calculations can be applied to evaluate a protein's or other molecules' conformational changes over time. The lower the RMSD value, the more stable the protein is. As shown in Fig. 6A, in the first 20 ns, the RMSD value reaches its maximum at around 0.3 for free Mpro and Mpro in interaction with Hesperidin.
Fig. 6.

The RMSD value A) Hesperidin and Mpro and B) Hesperidin and RNA polymerase.
Moreover, after 30 ns, the RMSD increases and becomes stable at around 0.4 for Mpro alone and 0.3 for Mpro complex. While Hesperidin changes protein structure, the complex conformation remains stable at the end of simulation time. The RMSD study reveals that the complex of Mpro-Hesperidin is almost stable with RMSD values smaller than 0.5 in the simulation time [56].
For RNA polymerase (Fig. 6B) in the first ten ns, the RMSD value reaches its maximum at around 0.45 for free RNA polymerase and 0.63 for RNA polymerase in interaction with Hesperidin. It then becomes stable at these values. The lower RMSD value of the complexes compared to the proteins suggests that the complex is stabilized [57].
RMSF is calculated to investigate the stability of the complex structure further. Fig. 7 displays the variation of RMSF value for the backbone of Mpro and RNA polymerase.
Fig. 7.

The RMSF value A) Hesperidin and Mpro and B) Hesperidin and RNA polymerase.
The flexibility of the backbone structure is sketched, which shows fluctuation during the simulation time. The high RMSF value indicates high flexibility, whereas the low RMSF value indicates limited motions during simulation. The RMSF plot of Mpro-Hesperidin and RNA polymerase-Hesperidin in the active site pocket areas fluctuated around a small range of 0.1–0.4 nm and 0.1 to 0.6, nm respectively. RNA polymerase-Hesperidin had higher fluctuations than Mpro-Hesperidin. Overall, RMSF fluctuates at around 0.1 nm for most residues, confirming the stability of the protein-ligand interactions [58].
Rg is a parameter that represents the compactness of a structure. The Rg value indicates the conformational flexibility of a protein, based on the moment of inertia calculated for the carbon alpha atoms from its center of mass. Higher Rg values indicate greater conformational flexibility [59]. Before the MD simulation, the Mpro and Hesperidin's gyration radius was estimated to be 2.2 Å. Then, Rg decreases to just below 2.1 Å during 40 ns and remains stable at this value for the last 60 ns. In the case of the Mpro-Hesperidin complex, Rg reveals a different trend and falls to a minimum of around 2.1 Å at 60 ns. Then, the last 40 ns of simulation Rg of the Mpro-Hesperidin complex remains stable at 2.11 Å, similar to the free Mpro (Fig. 8A). Hesperidin binding with Mpro made the protein more compact, indicating the high stability of the complex, which is observed in the RMSD analysis. An initial fluctuation in the RMSDs of Mpro might be due to the entry of a significant ligand like Hesperidin into the hydrophobic cavity of Mpro. Therefore, Hesperidin does not induce any significant structural deformation and represents the equilibrated manner of the protein structurally [60].
Fig. 8.

The Radius of gyration value A) Hesperidin and Mpro and B) Hesperidin and RNA polymerase.
In the case of the RNA polymerase-Hesperidin complex, the value of the Rg starts from 2.9 at the beginning of the simulation. It reaches 2.6 for the RNA polymerase alone and 2.53 for the RNA polymerase- Hesperidin during a decreasing process (Fig. 8B).
Another critical parameter in protein-ligand binding analysis is the distance of the ligand from the protein in the simulation. The Hesperidin is located within 0.14 nm of Mpro and RNA polymerase. This distance was constant during the simulation, which is another approved ligand-bound with the protein at an appropriate distance of 0.14 nm; this binding remains stable over the whole simulation time.
3.3.1. Hydrogen bond interaction network at the interaction interface
The number of hydrogen bonds plays a vital role in studying interactions of proteins and drugs during simulation. The measurement of the intermolecular H-bond evaluates the stability and pattern of hydrogen bonds in our system. A hydrogen bond interaction network is formed between the Mpro with Hesperidin and RNA polymerase with Hesperidin during the 100 ns dynamics simulation. Generally, during the simulation procedure, there are at least two or three hydrogen bonds between Hesperidin and Mpro, which even increase to nine (Fig. 9A), and two or three hydrogen bonds between Hesperidin and RNA polymerase, which even increase to six (Fig. 9B). As shown, Mpro forms more hydrogen bonds with Hesperidin than RNA polymerase, confirming the docking results.
Fig. 9.

The hydrogen bond interaction between A) Hesperidin and Mpro and B) Hesperidin and RNA polymerase.
SASA monitors the folding process. A rise in SASA value denotes a relative expansion of protein volume, and a weak fluctuation is expected over the simulation time. The change of SASA of Mpro and RNA polymerase during the time is shown in Fig. 10.
Fig. 10.

The SASA versus time at 300K of A) Hesperidin and Mpro and B)Hesperidin and RNA polymerase.
The SASA of Mpro and RNA polymerase are stable with a slight drop in the presence of Hesperidin, suggesting a suitable compact structure of the Mpro - Hesperidin and RNA polymerase- Hesperidin complexes.
The potential energy of the Mpro and RNA polymerase are analyzed for 100 ns, showing a slight increase in the energy of proteins in the presence of Hesperidin. Potential energy remains stable with some fluctuations between −1.12 × 106 and −1.13 × 106 Kcal/mol for Mpro and between −0.562 × 106 and −0.568 × 106 Kcal/mol for Mpro and Hesperidin complex. In the case of the RNA polymerase, potential energy remains stable with some fluctuations between −2.498 × 106 and −2.481 × 106 Kcal/mol for both RNA polymerase and RNA polymerase- Hesperidin complex.
These values confirm the system's stability, whereas energy is at its lower bound and remains almost constant at this level [61].
An effective approach for computing the complete free energy (ΔGbind) between the receptor (R) and the substrate (S) in biomolecular research is the molecular mechanics/Poisson Boltzmann surface area (MM/PBSA) method. The final total energy (Kcal/mol) between Hesperidin and Mpro is the sum of contributions of van der Waals, electrostatic, and SASA energy components from MD simulation. The contribution of each of the mentioned components in the total energy of the Hesperidin is shown in Table 4.
Table 4.
The final total energy (Kcal/mol) between hesperidin and Mpr.o.
| Compound | Van der Waals | Electrostatic | SASA | Binding free energy |
|---|---|---|---|---|
| Hesperidin-Mpro | −69.55 ± 12 | −184.98 ± 30 | −14.02 ± 2.45 | −4.71 ± 0.58 |
| Hesperidin-RNA polymerase | −238.357 ± 36 | −210.602 ± 54 | −4.84 ± 0.35 | −4.15 ± 0.28 |
Moreover, Table 4 shows van der Waal and Electrostatic energies, which are predominantly responsible for forming a stable hesperidin-Mpro and hesperidin-RNA polymerase complex. The nonpolar energy is usually calculated using the SASA energy.
3.3.2. Study the structural visualization of the interactions of hesperidin with Mpro and RNA polymerase in MD simulation
For a more detailed study of the molecular dynamics of hesperidin with Mpro and RNA polymerases related to COVID-19, the stability of the interactions at 25–100 ns have been compared (Fig. 11, Fig. 12).
Fig. 11.

The structural visualization of the interactions of hesperidin and RNA polymerases in MD simulation study at A)25, B)50, C)75, and D)100 ns.
Fig. 12.

The structural visualization of the interactions of hesperidin and Mpro in the MD simulation study at A)25, B)50, C)75, and D)100 ns.
Investigate the interaction of the hesperidin with RNA polymerase in 25 ns provided that amino acid units Glu802, Thr801, Ile779, and Cys799 in 50 ns amino acid units Glu811, Gly808, Trp800, Pro809, Thr801, Ile779, Lys780 have interacted with the drug (Fig. 11).
A study of the 75 ns time indicates that the amino acid units of Thr801, Trp800, Thr806, Cys799, and Lys798 interact with the drug, that the Thr801, Trp800 units are repeated compared to the time of 50 ns and Thr801, Cys799, Trp800 units compared to the time of 25 ns. Finally, in 100 ns, Thr801, Cys799, and Trp800 amino acid units have interacted with the drug, compared to the previous time of 75 ns, three amino acid units have been repeated and are among the stable amino acid units in interaction with the drug. The amino acid unit Thr801 has a good bond with the drug at all times and can play a significant role in deactivating the intended target.
Likewise, the analysis of the molecular dynamics results of the interaction of hesperidin with Mpro in Fig. 12 shows that in 25 ns, amino acids of Met49, Thr25, His41, Thr26, Thr24, Asn119, Cys145 and in 50 ns, amino acids of Leu27, Phe140, Giu166, Thr24, Thr26, Asn119 interact with the drug hesperidin.
Examining the time of 75 ns shows that the amino acid units Met49, HIS41, Thr25, and Thr45 interact with the drug, and the His41, Met49, and Thr25 units are repeated compared to the previous times. Finally, at 100 ns, amino acid units of Ser46, Asn142, Gln19, Asn119, Met49, Thr25, and Phe140 interacted with the drug, compared to previous times, amino acid units of Thr25, Met49, Phe140, and Asn119 were repeated and are among the stable amino acid units in interaction with the drug.
Meanwhile, the amino acid unit Asn119 has a good bond with the drug at all times and can play a significant role in deactivating the intended target. Examining drug interactions with amino acid units in simulation times and based on RMSF and H-bond molecular dynamics data reveals that drug penetration has been done in the intended target. Also, the drug has a suitable orientation for interaction with the intended receptor, and the molecular structure of the intended ligand is the same from 25 to 100 ns, proving the same pharmacophore compared to the initial time.
4. Discussion
The science world faces the challenge of saving human health against COVID-19 during the coronavirus outbreak. The computational studies for identifying potential inhibitors of SARS-CoV-2 focus on the use of molecular docking to evaluate anti-coronaviral agents. The studies suggest that glycosyl flavonoids have the potential to form stable complexes with the Mpro of SARS-CoV-2, indicating their effectiveness as antiviral agents. Several specific flavonoids, such as quercetin, isoquercitrin, rutin, baicalin, naringin, pelargonidin, and hesperidin aglycone, showed promising results in inhibiting the interaction of the SARS-CoV-2 Mpro and RNA polymerase [22,23,57,[62], [63], [64]]. Additionally, various glycosyl flavonoids demonstrated high binding energy values against multiple SARS-CoV-2 protein targets, suggesting their potential as therapeutic agents against the virus.
Insilico drug methodology of our investigation processes used in this paper showed that seven compounds could be antiviral therapeutics for potentially curing and preventing COVID-19. Among these compounds, compounds derived from herbals are more suitable candidates due to their natural bases and fewer side effects [3,65]. Fortunately, according to the results obtained in this study, herbal compounds had more reasonable interactions with the virus. Although Grazoprevir was similar to Hesperidin in the ADMET index, Hesperidin has the most acceptable interaction with the virus. The most significant result belongs to Grazoprevir, a pleasing precursor for drug manufacturing and clinical testing. It could perform to be as a top-notch Mpro inhibitor.
Their MD simulations with the GROMACS revealed that RMSD, RMSF, and Rg remained stable over time. Moreover, the MD provided that Mpro forms more H-bonds with Hesperidin than RNA polymerase, confirming docking results. The SASA of both proteins are stable with a slight drop in Hesperidin, indicating compact Mpro-Hesperidin and RNA polymerase-Hesperidin complexes.
The potential energy of Mpro and RNA polymerase was stable, with some fluctuations observed. Hesperidin caused a slight increase in protein energy for Mpro. The energy remained stable for RNA polymerase, with fluctuations observed for the polymerase and polymerase-Hesperidin complex. van der Waals and Electrostatic energies dominate in forming a stable hesperidin-Mpro and hesperidin-RNA polymerase complex. SASA energy is used to calculate nonpolar energy.
After thorough analysis, it has been determined that Hesperidin can effectively interact with the intended receptor due to its suitable orientation. Furthermore, the molecular structure of the intended ligand remains unchanged in the MD simulation, providing clear evidence of the same pharmacophore as the initial time. However, further experimental investigation and in vitro and in vivo examinations are necessary regarding the drug's further development.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
CRediT authorship contribution statement
Elaheh Molaakbari: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Mohammad Reza Aallae: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Fereshteh Golestanifar: Writing – review & editing, Writing – original draft, Validation, Software, Methodology, Investigation, Formal analysis, Conceptualization. Zahra Garakani-Nejad: Writing – review & editing, Validation, Software, Methodology, Investigation. Ahmad Khosravi: Writing – review & editing, Validation, Investigation, Conceptualization. Mohsen Rezapour: Writing – review & editing, Validation, Investigation. Rahime Eshaghi Malekshah: Writing – review & editing, Writing – original draft, Validation, Methodology, Investigation. Mahsa Ghomi: Writing – review & editing. Guogang Ren: Writing – review & editing, Visualization, Validation.
Declaration of competing interest
The authors report there are no competing interests to declare.
Acknowledgments
The authors gratefully thank the founders of Kerman University, Mr. Alireza Afzalipour, and his wife, Mrs. Fakhereh Saba, for their support and kindness in training future generations of doctors, engineers, and scientists. Furthermore, the authors warmly thank Dr. Parviz Dabiri for supporting the research activities in the chemistry laboratories at Kerman University.
Contributor Information
Elaheh Molaakbari, Email: e.molaakbari@gmail.com.
Guogang Ren, Email: g.g.ren@herts.ac.uk.
References
- 1.Swanstrom R., Anderson J., Schiffer C., Lee S.K. Viral protease inhibitors. Handb. Exp. Pharmacol. 2009;189:85–110. doi: 10.1007/978-3-540-79086-0_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science. 2020;1979(368):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryu W.S. 2016. Molecular Virology of Human Pathogenic Viruses. [DOI] [Google Scholar]
- 4.COVID-19 Treatment Guidelines Panel . Disponible en. National Institute of Health; 2020. Coronavirus disease 2019 (COVID-19) treatment guidelines; p. 2019.https://covid19treatmentguidelines.nih.gov/ [Google Scholar]
- 5.Griffin D.O., Brennan-Rieder D., Ngo B., Kory P., Confalonieri M., Shapiro L., Iglesias J., Dube M., Nanda N., In G.K., Arkfeld D., Chaudhary P., Campese V.M., Hanna D.L., Sawcer D., Ehresmann G., Peng D., Smorgorzewski M., Amstrong A., Vinjevoll E.H., Dasgupta R., Sattler F.R., Mussini C., Mitjà O., Soriano V., Peschanski N., Hayem G., Piccirillo M.C., Lobo-Ferreira A., Rivero I.B., Hung I.F.H., Rendell M., Ditmore S., Varon J., Marik P. The importance of understanding the stages of covid-19 in treatment and trials. AIDS Rev. 2021;23 doi: 10.24875/AIDSRev.200001261. [DOI] [PubMed] [Google Scholar]
- 6.Adem S., Eyupoglu V., Sarfraz I., Rasul A., Ali M. Identification of potent COVID-19 main protease (Mpro) inhibitors from natural polyphenols: an in silico strategy unveils a hope against CORONA. 2020. [DOI] [PMC free article] [PubMed]
- 7.Swanstrom R., Anderson J., Schiffer C., Lee S.K. Viral protease inhibitors. Handb. Exp. Pharmacol. 2009;189:85–110. doi: 10.1007/978-3-540-79086-0_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prior A.M., Kim Y., Weerasekara S., Moroze M., Alliston K.R., Uy R.A.Z., Groutas W.C., Chang K.O., Hua D.H. Design, synthesis, and bioevaluation of viral 3C and 3C-like protease inhibitors. Bioorg. Med. Chem. Lett. 2013;23:6317–6320. doi: 10.1016/j.bmcl.2013.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Razzaq A., Shamsi S., Ali A., Ali Q., Sajjad M., Malik A., Ashraf M. Microbial proteases applications. Front. Bioeng. Biotechnol. 2019;7 doi: 10.3389/fbioe.2019.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lv Z., Chu Y., Wang Y. vol. 7. 2015. pp. 95–104. (HIV Protease Inhibitors: A Review of Molecular Selectivity and Toxicity, HIV/AIDS - Research and Palliative Care). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Y., Yin W., Xu H.E. RNA-dependent RNA polymerase: structure, mechanism, and drug discovery for COVID-19. Biochem. Biophys. Res. Commun. 2021;538 doi: 10.1016/j.bbrc.2020.08.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin W., Mao C., Luan X., Shen D.D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M., Chang S., Xie Y.C., Tian G., Jiang H.W., Tao S.C., Shen J., Jiang Y., Jiang H., Xu Y., Zhang S., Zhang Y., Xu H.E. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;1979:368. doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Zhang B., Yang X., Li J., Yang H., Liu Z., Xu W., Guddat L.W., Wang Q., Lou Z., Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;1979:368. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng Q., Peng R., Yuan B., Zhao J., Wang M., Wang X., Wang Q., Sun Y., Fan Z., Qi J., Gao G.F., Shi Y. Structural and biochemical characterization of the nsp12-nsp7-nsp8 core polymerase complex from SARS-CoV-2. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ngwa W., Kumar R., Thompson D., Lyerly W., Moore R., Reid T.E., Lowe H., Toyang N. Potential of flavonoid-inspired phytomedicines against COVID-19. Molecules. 2020;25 doi: 10.3390/molecules25112707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solnier J., Fladerer J.P. Flavonoids: a complementary approach to conventional therapy of COVID-19? Phytochemistry Rev. 2021;20 doi: 10.1007/s11101-020-09720-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godinho P.I.C., Soengas R.G., Silva V.L.M. Therapeutic potential of glycosyl flavonoids as anti-coronaviral agents. Pharmaceuticals. 2021;14 doi: 10.3390/ph14060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma A., Goyal S., Yadav A.K., Kumar P., Gupta L. In-silico screening of plant-derived antivirals against main protease, 3CLpro and endoribonuclease, NSP15 proteins of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022;40 doi: 10.1080/07391102.2020.1808077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.da Silva F.M.A., da Silva K.P.A., de Oliveira L.P.M., Costa E.V., Koolen H.H.F., Pinheiro M.L.B., de Souza A.Q.L., de Souza A.D.L. Flavonoid glycosides and their putative human metabolites as potential inhibitors of the sars-cov-2 main protease (Mpro) and rna-dependent rna polymerase (rdrp) Mem. Inst. Oswaldo Cruz. 2020;115 doi: 10.1590/0074-02760200207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherrak S.A., Merzouk H., Mokhtari-Soulimane N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: a molecular docking and simulation studies. PLoS One. 2020;15 doi: 10.1371/journal.pone.0240653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubey K., Dubey R. Computation screening of narcissoside a glycosyloxyflavone for potential novel coronavirus 2019 (COVID-19) inhibitor. Biomed. J. 2020;43 doi: 10.1016/j.bj.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abian O., Ortega-Alarcon D., Jimenez-Alesanco A., Ceballos-Laita L., Vega S., Reyburn H.T., Rizzuti B., Velazquez-Campoy A. Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol. 2020;164 doi: 10.1016/j.ijbiomac.2020.07.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salarizadeh N., Aallaei M.R., Zarei A., Malekshah R.E., Molaakbari E., Farajnezhadi A. Docking and molecular dynamics simulations of flavonoids as inhibitors of infectious agents: rutin as a coronavirus protease inhibitor. ChemistrySelect. 2022;7 doi: 10.1002/slct.202202043. [DOI] [Google Scholar]
- 24.Jo S., Kim S., Shin D.H., Kim M.S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2020;35 doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Savi C., Hughes D.L., Kvaerno L. Quest for a COVID-19 cure by repurposing small-molecule drugs: mechanism of action, clinical development, synthesis at scale, and outlook for supply. Org. Process Res. Dev. 2020;24:940–976. doi: 10.1021/acs.oprd.0c00233. [DOI] [PubMed] [Google Scholar]
- 26.Panoutsopoulos A.A. Known drugs and small molecules in the battle for COVID-19 treatment. Genes Dis. 2020 doi: 10.1016/j.gendis.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma A., Tiwari V., Sowdhamini R. Computational search for potential COVID-19 drugs from FDA-approved drugs and small molecules of natural origin identifies several anti-virals and plant products. J. Bio. Sci. 2020;45 doi: 10.1007/s12038-020-00069-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahman M.M., Saha T., Islam K.J., Suman R.H., Biswas S., Rahat E.U., Hossen M.R., Islam R., Hossain M.N., Al Mamun A., Khan M., Ali M.A., Halim M.A. Virtual screening, molecular dynamics and structure–activity relationship studies to identify potent approved drugs for Covid-19 treatment. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1794974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F., Han K., Blair R., Kenst K., Qin Z., Upcin B., Wörsdörfer P., Midkiff C.C., Mudd J., Belyaeva E., Milligan N.S., Rorison T.D., Wagner N., Bodem J., Dölken L., Aktas B.H., Vander Heide R.S., Yin X.M., Kolls J.K., Roy C.J., Rappaport J., Ergün S., Qin X. SARS-CoV-2 infects endothelial cells in vivo and in vitro. Front. Cell. Infect. Microbiol. 2021;11 doi: 10.3389/fcimb.2021.701278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bahadur Gurung A., Ajmal Ali M., Lee J., Abul Farah M., Mashay Al-Anazi K. Structure-based virtual screening of phytochemicals and repurposing of FDA approved antiviral drugs unravels lead molecules as potential inhibitors of coronavirus 3C-like protease enzyme. J. King Saud Univ. Sci. 2020;32 doi: 10.1016/j.jksus.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhowmik D., Nandi R., Jagadeesan R., Kumar N., Prakash A., Kumar D. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect. Genet. Evol. 2020;84 doi: 10.1016/j.meegid.2020.104451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolton E.E., Chen J., Kim S., Han L., He S., Shi W., Simonyan V., Sun Y., Thiessen P.A., Wang J., Yu B., Zhang J., Bryant S.H. PubChem3D: a new resource for scientists. J. Cheminf. 2011;3 doi: 10.1186/1758-2946-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Froimowitz M. HyperChem(TM): a software package for computational chemistry and molecular modeling. Biotechniques. 1993;14 [PubMed] [Google Scholar]
- 34.Aallaei M., Molaakbari E., Mostafavi P., Salarizadeh N., Maleksah R.E., Afzali D. Investigation of Cu metal nanoparticles with different morphologies to inhibit SARS-CoV-2 main protease and spike glycoprotein using Molecular Docking and Dynamics Simulation. J. Mol. Struct. 2022;1253 doi: 10.1016/j.molstruc.2021.132301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y., Zhu Y., Liu X., Jin Z., Duan Y., Zhang Q., Wu C., Feng L., Du X., Zhao J., Shao M., Zhang B., Yang X., Wu L., Ji X., Guddat L.W., Yang K., Rao Z., Yangl H. Structural basis for replicase polyprotein cleavage and substrate specificity of main protease from SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2022;119 doi: 10.1073/pnas.2117142119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein Data Bank. Nucleic Acids Res. 2000;28 doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.BIOVIA, Dassault Systèmes BIOVIA . Dassault Systèmes; San Diego: 2017. Discovery Studio Modeling Environment, Release 2017. [Google Scholar]
- 38.Emon N.U., Alam MdM., Akter I., Akhter S., Sneha A.A., Irtiza Md, Afroj M., Munni A., Chowdhury M.H., Hossain S. Virtual screenings of the bioactive constituents of tea, prickly chaff, catechu, lemon, black pepper, and synthetic compounds with the main protease (Mpro) and human angiotensin-converting enzyme 2 (ACE 2) of SARS-CoV-2. Futur J Pharm Sci. 2021;7 doi: 10.1186/s43094-021-00275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kusumaningrum S., Budianto E., Kosela S., Sumaryono W., Juniarti F. The molecular docking of 1, 4-naphthoquinone derivatives as inhibitors of Polo-like kinase 1 using Molegro Virtual Docker. J. Appl. Pharmaceut. Sci. 2014;4:47–53. [Google Scholar]
- 40.Byrnes J., Baskerville E., Caron B., Neylon C., Tenopir C., Schildhauer M., Budden A., Aarssen L., Lortie C. The four pillars of scholarly publishing: the future and a foundation. Ideas in Ecology and Evolution. 2014;7 doi: 10.7287/peerj.preprints. [DOI] [Google Scholar]
- 41.Vieira T.F., Sousa S.F. Comparing AutoDock and Vina in ligand/decoy discrimination for virtual screening. Appl. Sci. 2019;9 doi: 10.3390/app9214538. [DOI] [Google Scholar]
- 42.Pagadala N.S., Syed K., Tuszynski J. Software for molecular docking: a review. Biophys Rev. 2017;9:91–102. doi: 10.1007/s12551-016-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindah E. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015:1–2. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 44.Eshaghi Malekshah R., Fahimirad B., Aallaei M., Khaleghian A. Synthesis and toxicity assessment of Fe3O4 NPs grafted by ∼ NH2-Schiff base as anticancer drug: modeling and proposed molecular mechanism through docking and molecular dynamic simulation. Drug Deliv. 2020;27:1201–1217. doi: 10.1080/10717544.2020.1801890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pelalak R., Soltani R., Heidari Z., Malekshah R.E., Aallaei M., Marjani A., Rezakazemi M., Shirazian S. Synthesis, molecular dynamics simulation and adsorption study of different pollutants on functionalized mesosilica. Sci. Rep. 2021;11 doi: 10.1038/s41598-020-80566-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haggag Y.A., El-Ashmawy N.E., Okasha K.M. Is hesperidin essential for prophylaxis and treatment of COVID-19 Infection? Med. Hypotheses. 2020;144 doi: 10.1016/j.mehy.2020.109957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Attia G.H., Moemen Y.S., Youns M., Ibrahim A.M., Abdou R., El Raey M.A. Antiviral zinc oxide nanoparticles mediated by hesperidin and in silico comparison study between antiviral phenolics as anti-SARS-CoV-2. Colloids Surf. B Biointerfaces. 2021;203 doi: 10.1016/j.colsurfb.2021.111724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bellavite P., Donzelli A. Hesperidin and SARS-CoV-2: new light on the healthy function of citrus fruits. Antioxidants. 2020;9:742. doi: 10.3390/antiox9080742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harwansh R.K., Bahadur S. Herbal medicine in fighting against COVID-19: new battle with an old weapon. Curr. Pharmaceut. Biotechnol. 2022;23:235. doi: 10.2174/1389201022666210322124348. [DOI] [PubMed] [Google Scholar]
- 50.Behera S.K., Vhora N., Contractor D., Shard A., Kumar D., Kalia K., Jain A. Computational drug repurposing study elucidating simultaneous inhibition of entry and replication of novel corona virus by Grazoprevir. Sci. Rep. 2021;11:1–11. doi: 10.1038/s41598-021-86712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang M.M., Wang J., Dong L., Teng Y., Liu P., Fan J.J., Yu X.H. Lack of association of C3 gene with uveitis: additional insights into the genetic profile of uveitis regarding complement pathway genes. Sci. Rep. 2017;7:1–8. doi: 10.1038/s41598-017-00833-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balasubramaniam M., Reis R.J.S. Computational target-based drug repurposing of elbasvir, an antiviral drug predicted to bind multiple SARS-CoV-2 proteins. Chem. 2020 doi: 10.26434/chemrxiv.12084822.v2. [DOI] [Google Scholar]
- 53.Guan L., Yang H., Cai Y., Sun L., Di P., Li W., Liu G., Tang Y. ADMET-score–a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm. 2019;10:148–157. doi: 10.1039/c8md00472b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benet L.Z., Hosey C.M., Ursu O., Oprea T.I. BDDCS, the rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016;101:89–98. doi: 10.1016/j.addr.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wessler J.D., Grip L.T., Mendell J., Giugliano R.P. The P-glycoprotein transport system and cardiovascular drugs. J. Am. Coll. Cardiol. 2013;61:2495–2502. doi: 10.1016/j.jacc.2013.02.058. [DOI] [PubMed] [Google Scholar]
- 56.da Silva T.U., Pougy K. de C., Albuquerque M.G., da Silva Lima C.H., Machado S. de P. Development of parameters compatible with the CHARMM36 force field for [Fe4S4] 2+ clusters and molecular dynamics simulations of adenosine-5’-phosphosulfate reductase in GROMACS 2019. J. Biomol. Struct. Dyn. 2020:1–11. doi: 10.1080/07391102.2020.1847687. [DOI] [PubMed] [Google Scholar]
- 57.Yang S., Kar S. Protracted molecular dynamics and secondary structure introspection to identify dual-target inhibitors of Nipah virus exerting approved small molecules repurposing. Sci. Rep. 2024;14 doi: 10.1038/s41598-024-54281-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peng J., Li Y., Zhou Y., Zhang L., Liu X., Zuo Z. Pharmacophore modeling, molecular docking and molecular dynamics studies on natural products database to discover novel skeleton as non-purine xanthine oxidase inhibitors. J. Recept. Signal Transduction. 2018;38:246–255. doi: 10.1080/10799893.2018.1476544. [DOI] [PubMed] [Google Scholar]
- 59.Islam R., Parves M.R., Paul A.S., Uddin N., Rahman M.S., Al Mamun A., Hossain M.N., Ali M.A., Halim M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021;39:3213–3224. doi: 10.1080/07391102.2020.1761883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Al-Shabib N.A., Khan J.M., Malik A., Alsenaidy M.A., Rehman M.T., AlAjmi M.F., Alsenaidy A.M., Husain F.M., Khan R.H. Molecular insight into binding behavior of polyphenol (rutin) with beta lactoglobulin: spectroscopic, molecular docking and MD simulation studies. J. Mol. Liq. 2018;269:511–520. [Google Scholar]
- 61.Ahmad S.S., Sinha M., Ahmad K., Khalid M., Choi I. Study of Caspase 8 inhibition for the management of Alzheimer's disease: a molecular docking and dynamics simulation. Molecules. 2020;25:2071. doi: 10.3390/molecules25092071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu C., Zhou Q., Li Y., Garner L.V., Watkins S.P., Carter L.J., Smoot J., Gregg A.C., Daniels A.D., Jervey S., Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020;6 doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pillaiyar T., Meenakshisundaram S., Manickam M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today. 2020;25 doi: 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sacco M.D., Ma C., Lagarias P., Gao A., Townsend J.A., Meng X., Dube P., Zhang X., Hu Y., Kitamura N., Hurst B., Tarbet B., Marty M.T., Kolocouris A., Xiang Y., Chen Y., Wang J. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chaachouay N., Zidane L. Plant-derived natural products: a source for drug discovery and development. Drugs and Drug Candidates. 2024;3 doi: 10.3390/ddc3010011. [DOI] [Google Scholar]