

Abstract
The presence of DPPII (dipeptidyl peptidase II; E.C. 3.4.14.2) has been demonstrated in various mammalian tissues. However, a profound molecular and catalytic characterization, including substrate selectivity, kinetics and pH-dependence, has not been conducted. In the present study, DPPII was purified from human seminal plasma to apparent homogeneity with a high yield (40%) purification scheme, including an inhibitor-based affinity chromatographic step. The inhibitor lysyl-piperidide (Ki~0.9 μM at pH 5.5) was chosen, as it provided a favourable affinity/recovery ratio. The human enzyme appeared as a 120 kDa homodimer. Mass spectrometric analysis after tryptic digestion together with a kinetic comparison indicate strongly its identity with QPP (quiescent cell proline dipeptidase), also called dipeptidyl peptidase 7. pH profiles of both kcat and kcat/Km clearly demonstrated that DPPII/QPP possesses an acidic and not a neutral optimum as was reported for QPP. Kinetic parameters of the human natural DPPII for dipeptide-derived chromogenic [pNA (p-nitroanilide)] and fluorogenic [4Me2NA (4-methoxy-2-naphthylamide)] substrates were determined under different assay conditions. DPPII preferred the chromogenic pNA-derived substrates over the fluorogenic 4Me2NA-derived substrates. Natural human DPPII showed high efficiency towards synthetic substrates containing proline at the P1 position and lysine at P2. The importance of the P1′ group for P2 and P1 selectivity was revealed, explaining many discrepancies in the literature. Furthermore, substrate preferences of human DPPII and dipeptidyl peptidase IV were compared based on their selectivity constants (kcat/Km). Lys-Pro-pNA (kcat/Km 4.1×106 s−1·M−1) and Ala-Pro-pNA (kcat/Km 2.6×106 s−1· M−1) were found to be the most sensitive chromogenic substrates for human DPPII, but were less selective than Lys-Ala-pNA (kcat/Km 0.4×106 s−1·M−1).
Keywords: dipeptidyl peptidase (DPP), kinetics, pH profile, proline, quiescent cell proline dipeptidase (QPP)
Abbreviations: Con A, concanavalin A; DPP, dipeptidyl peptidase; FAPα, fibroblast activation protein α; Lys-Pip, lysyl-piperidide; 4Me2NA, 4-methoxy-2-naphthylamide; NHS, N-hydroxysuccinimide; Nle, norleucine; pNA, p-nitroanilide; QPP, quiescent cell proline dipeptidase; Z, benzyloxycarbonyl
INTRODUCTION
Among the DPPs (dipeptidyl peptidases) able to cleave post-proline bonds, such as DPP IV, FAPα (fibroblast activation protein α), DPP8, DPP9 and DPPII [1–3], DPPIV has been studied most extensively [4]. In 1966, DPPII was first described in bovine anterior pituitary extracts in a report that dealt primarily with the properties of DPPI [5]. Based on the enzyme's ability to cleave dipeptides from β-naphthylamide derivatives with an unsubstituted N-terminus, but not from mono-amino acid arylamides, it was referred to as dipeptidyl aminopeptidase II [6,7]. Hydrolysis of Lys-Ala-derived chromogenic or fluorogenic substrates at acidic pH was routinely used to detect this enzyme. From then on, DPPII (E.C. 3.4.14.2) has been demonstrated in various mammalian tissues [6–17]. However, a profound enzymological and molecular characterization, especially of human natural DPPII is missing. For human DPPII, only the N-terminal sequence is available (GenBank® accession number S77568).
Human QPP (quiescent cell proline dipeptidase) was first described in 1999 when Chiravuri et al. [19] observed that inhibitors of post-proline-cleaving DPPs caused apoptosis in quiescent lymphocytes in a process independent of DPPIV. Araki et al. [20] suggested that DPPII was the rat orthologue of human QPP, based on a sequence identity of 78% and biochemical similarities.
In 2002, the Mammalian Gene Collection Program Team introduced the name DPP7 for a protein with the same sequence as QPP [21]. A comparison of the catalytic properties of partially purified human DPPII and QPP/DPP7 supported the hypothesis that QPP and DPPII are identical [22]. In the present paper, we reconcile two separate lines of enquiry on what have been thought to be different enzymes. If the hypothesis holds true, the same enzyme is discussed in the literature [22,23] under three different names: DPPII, QPP and DPP7.
Since DPPII is glycosylated [10,14,15,19], we preferred to use the natural enzyme for biochemical and enzymological characterization. In the present study, we introduce a protocol that can be used for high-yield purification of DPPII from human seminal plasma.
DPPII releases N-terminal Xaa-Pro and Xaa-Ala dipeptides from oligopeptides, especially tripeptides and from synthetic peptide substrates at acidic pH [7,12,14]. According to Underwood et al. [24], QPP cleaves substrates with equal efficiency at acidic and neutral pH. QPP is also suggested to be active towards larger peptides, such as chemokines with a penultimate proline residue [25,26]. In contrast, preliminary data in the literature indicate that DPPII only cleaves very short peptides [7,12,14]. The confusing results concerning the substrate selectivity of DPPII [27] and QPP, optimal pH, influence of ionic strength and fragmentary knowledge of kinetics substantiate the need for an unambiguous investigation of these parameters. In the past, chromogenic and fluorogenic substrates were used indiscriminately, and the influence of the leaving group on P2 and P1 preferences was never explored. In the present paper, we report the kinetic parameters (kcat and Km) of purified human natural DPPII for various chromogenic [pNA (p-nitroanilide)] and fluorogenic [4Me2NA (4-methoxy-2-naphthylamide)] dipeptide-derived substrates under different assay conditions. The leaving group on P1′ is revealed to have a great impact on the substrate selectivity.
The preference (kcat/Km) of natural human DPPII for particular residues in dipeptide-derived chromogenic substrates (pNA) was compared with the preference of natural human DPPIV.
MATERIALS AND METHODS
Materials
The DPPII inhibitors Lys-Pip (lysyl-piperidide) and N-(4-chlorobenzyl)-4-oxo-4-(1-piperidinyl)-1,3-(S)-butanediamine dihydrochloride (Figure 1) were synthesized as described previously [28].
Figure 1. Selective DPPII inhibitors.

1, Lys-Pip; 2, N-(4-chlorobenzyl)-4-oxo-4-(1-piperidinyl)-1,3-(S)-butane-diamine.
Seminal plasma was obtained from healthy donors. All chromatography media [Con A (concanavalin A)–Sepharose, NHS (N-hydroxysuccinimide)-activated Sepharose 4 Fast Flow, zinc-chelating Sepharose Fast Flow, HiTrap zinc-chelating HP (1 ml), HiTrap Q HP (1 ml) and Superdex 200 HR 10/30] were purchased from Amersham Biosciences. Lys-Ala-pNA, Ala-Pro-pNA, GlyPro-pNA, Lys-Pro-4Me2NA and Ala-Pro-4Me2NA were obtained from Bachem, and Lys-Ala-4Me2NA, Gly-Pro-4Me2NA, 4-methoxy-2-naphthylamine, BSA and cacodylic acid were from Sigma. Other dipeptide pNAs were synthesized as described in [29]. All other chemicals were obtained from ICN Biomedicals. Recombinant QPP was obtained by expression in Sf9 cells using the BACTO-BAC™ Baculovirus Expression System (Life Technologies) [22].
Enzyme assay
Routinely, DPPII activity was determined kinetically for 10 min at 37 °C by measuring the initial velocities of p-nitroaniline release (405 nm) from the substrate using a Spectramax plus microtitre plate reader (Molecular Devices).
One unit of DPPII activity was defined as the amount of enzyme that catalyses the release of 1 μmol of p-nitroaniline from the substrate per min under assay conditions. During its purification, DPPII enzymic activity was determined using Lys-Ala-pNA (1 mM) in 0.05 M cacodylic acid/NaOH buffer, pH 5.5, containing 10 mM EDTA and 14 μg/ml aprotinin, using a 10 μl enzyme sample in a final volume of 200 μl. During the characterization of purified DPPII, Ala-Pro-pNA (1 mM) was used, and aprotinin and EDTA were omitted from all buffers. Enzyme dilutions of the purified DPPII were made in the presence of 1 mg/ml BSA.
To verify the presence of other protease activity, other enzyme activities were determined using 1 mM Ala-pNA in 0.05 M Hepes buffer, pH 7.0 [membrane alanyl aminopeptidase (CD13)], 0.5 mM Gly-Pro-pNA in 0.05 M Tris/HCl buffer, pH 8.3 (DPPIV) [30], and 0.3 mM Z (benzyloxycarbonyl)-Gly-Pro-pNA in 0.1 M potassium phosphate buffer, pH 7.5, containing 1 mM dithiothreitol, 1 mM EDTA and 1 mM sodium azide (prolyl oligopeptidase) [31].
Protein content was determined according to the method of Bradford, with BSA as the standard [32].
Lys-Pip–Sepharose 4 Fast Flow affinity column preparation
The reversible competitive DPPII inhibitor Lys-Pip was synthesized by coupling di-Z-Lys to piperidine as described in [28].
The affinity gel used for the purification of DPPII from human seminal fluid was prepared by coupling a 4× molar excess of Lys-Pip to 20 ml of NHS-activated Sepharose 4 Fast Flow, using propan-2-ol as a coupling solvent for 24 h at room temperature (22 °C) with end-to-end agitation. After incubation, the gel was washed with coupling solvent, and the residual active groups on the medium were blocked by incubation with 2 column vol. of 0.2 M Tris/HCl buffer, pH 8.0, for 2 h. The matrix was treated with three cycles of alternating pH washes with 0.1 M sodium acetate, pH 4.0, and 0.05 M Tris/HCl, pH 8.5, each containing 0.5 M NaCl. After equilibration with 50 mM sodium acetate, pH 5.5, containing 14 μg/ml aprotinin and 0.15 M NaCl, the Lys-Pip affinity medium was ready for use.
Purification of DPPII
Human seminal plasma was separated from spermatozoa and prostasomes by centrifugation [33]. The resulting seminal fluid was loaded (1.2 ml/min) on to a 14 ml Con A–Sepharose column, equilibrated and washed (5 column vol.) with 20 mM Tris/HCl, pH 7.4, containing 1 mM CaCl2, 1 mM MnCl2, 14 μg/ml aprotinin and 0.5 M NaCl. Elution (1.2 ml/min) was performed with 0.5 M methyl-α-D-glucopyranoside (Sigma) in the same buffer. Fractions containing DPPII activity were loaded (0.3 ml/min) on to a 5 ml zinc-chelating Sepharose Fast Flow column equilibrated with 20 mM Tris/HCl, pH 7.4, containing 14 μg/ml aprotinin and 0.5 M NaCl. After washing with equilibration buffer, DPPII was eluted (1 ml/min) with 50 mM sodium acetate, pH 5.5, containing 14 μg/ml aprotinin. Active fractions (10 ml) were applied (0.3 ml/min) to a 6 ml Lys-Pip affinity column prepared and equilibrated as described above. The affinity column was washed with 5 column vol. of equilibration buffer and eluted (0.5 ml/min) with 0.75 M NaCl in the same buffer. Active fractions were concentrated on a Centriplus YM-10 centrifugal concentrator (Millipore). After dilution and adjusting the pH to 7.8, the sample was applied (1 ml/min) to a 1 ml HiTrap Q HP column equilibrated with 20 mM Tris/HCl, pH 7.8. Elution (1 ml/min) was performed with a linear 0–1 M NaCl gradient in 60 ml of equilibration buffer.
A final buffer exchange was accomplished by binding (0.4 ml/min) the active fractions on to a HiTrap zinc-chelating HP 1 ml column and elution (1 ml/min) of DPPII with 50 mM sodium acetate, pH 5.5. The purity of DPPII was determined by SDS/PAGE.
Purification of DPPIV
Soluble DPPIV used for substrate selectivity determinations was purified from human seminal plasma as described previously [33].
Estimation of molecular mass by SDS/PAGE
Purified DPPII was injected on to a Superdex 200 HR 10/30 gel-filtration column equilibrated with PBS, pH 7.2, at a flow rate of 0.5 ml/min. The native molecular mass of DPPII was determined by plotting the partition coefficient Kav against log (molecular mass) and using a calibration curve with ferritin (molecular mass, 440 kDa), catalase (molecular mass, 232 kDa), aldolase (molecular mass, 158 kDa), BSA (molecular mass, 67 kDa) and ovalbumin (molecular mass, 43 kDa).
SDS/PAGE (10%) was carried out in the presence of 2-mercaptoethanol essentially according to the Laemmli method in 25 mM Tris/HCl, 190 mM glycine, pH 8.3, and 0.5% (w/v) SDS using a Mini Protean II apparatus (Bio-Rad) [34]. The gels were standardized with broad-range molecular-mass-marker proteins from Bio-Rad [(myosin (200 kDa), β-galactosidase (116.3 kDa), phosphorylase b (97.4 kDa), BSA (66.2 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa), soybean trypsin inhibitor (21.5 kDa), lysozyme (14.4 kDa) and aprotinin (6.5 kDa)]. The gel was stained by a neutral silver-staining procedure modified after Heukeshoven and Dernick [35].
MS and protein identification
Purified DPPII was separated by SDS/10% PAGE, and the band was excised after staining with a MS-compatible silver-staining kit (SilverQuest, Invitrogen). The band was destained and cut into smaller pieces, before trypsin digestion according to the method of Jensen et al. [36]
The extracted peptides were desalted and concentrated by ZipTip C18 (Millipore), and eluted directly into home-made nano-electrospray needles. The needles for nano-electrospray MS were made with a micropipette puller (model P-2000; Sutter Instrument Co., Novato, CA, U.S.A.) from borosilicate glass capillaries GC100F-10, 1 mm o.d. (outer diameter)×0.58 mm i.d. (inner diameter) as described by Wilm and Mann [37]. They were gold-coated in batches of 12–14 in a vapour deposition instrument.
Electrospray mass spectra were acquired on a Q-TOF II quadrupole time-of-flight mass spectrometer (Micromass, Wythenshawe, U.K.) equipped with a dual-orthogonal sampling API source. Collision energy was manually tuned for each peptide for optimum MS/MS spectra.
Both MS and MS/MS spectra were used to search different public databases with the program Mascot (http://www.matrix-science.com) to identify the protein. All the MS/MS spectra were also inspected manually using the MassLynx 3.5 PepSeq software (Micromass).
The prediction of theoretical tryptic peptide sequences was performed using GPMAW 5.02 (Lighthouse Data, Odense, Denmark).
Stability
Thermostability studies were performed by incubation of purified DPPII (~10 μg/ml) at different temperatures (−80 °C, −20 °C, 4 °C and 37 °C) in 50 mM sodium acetate buffer, pH 5.5, in the presence of stabilizing agents (1 mg/ml BSA, 0.1% Tween 20 or 50% glycerol). At timed intervals, residual DPPII activity was assayed using Ala-Pro-pNA as described above. Stability at 37 °C was followed for up to 240 min. Stability at the other temperatures (−80 °C, −20 °C and 4 °C) was evaluated for up to 4 months.
The resistance to freeze/thaw cycles was also examined. Samples were stored at −80 °C and thawed on ice each cycle.
Kinetic properties
DPPII activity towards dipeptide-derived chromogenic substrates was assayed as described above. Hydrolysis of fluorogenic dipeptide-derived substrates was assayed by measuring the release of 4-methoxy-2-naphthylamine (excitation, 340 nm; emission 430 nm) at 37 °C for 10 min on a Tecan Spectrafluor Plus (Sercolab). The standard reaction mixture (200 μl) contained 15 ng of purified DPPII and one of at least eight different concentrations of substrate (0.01–11 mM) in 0.05 M cacodylic acid/NaOH buffer, pH 5.5, or in 0.05 M Hepes buffer, pH 7.0. The values for Km and Vmax were determined by plotting the initial velocities of product formation against the different substrate concentrations and fitting the data to the Michaelis–Menten equation by non-linear regression analysis using GraFit version 5 [38]. When using the fluorogenic substrates, the Michaelis–Menten equation was adapted to compensate for substrate inhibition (eqn 1) [39]:
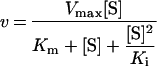 |
(1) |
where [S] is the substrate concentration and Ki is the substrate inhibition constant.
The catalytic-centre activity, kcat, was calculated using the molecular mass of 60 kDa per subunit of DPPII. Samples were analysed in duplicate and experiments were performed two to four times.
To examine the effect of ionic strength on the kinetic parameters, 150 or 750 mM NaCl was incorporated in the standard reaction mixture using Lys-Ala-pNA and Ala-Pro-pNA as the substrates.
pH profile
DPPII/QPP activity was measured over the pH range 3.0–8.5 (in increments of 0.25) using a buffer containing 0.025 M ethanoic (acetic) acid, 0.025 M cacodylic acid, 0.025 M Hepes and 0.1 M NaCl, titrated to the desired pH with NaOH or HCl. The change in ionic strength due to the pH titration was modest and had negligible effect on enzyme activity. The influence of pH on the hydrolysis rate was studied using the substrates Lys-Ala-pNA, Ala-Pro-pNA, Lys-Ala-4Me2NA and Ala-Pro-4Me2NA at two concentrations: a first equal to or lower than 0.5 Km, and a second far above the Km.
The reactions were initiated by the addition of enzyme. Data were fitted to a double-ionization equation (eqn 2) that resulted in a bell-shaped curve using GraFit version 5 [38]:
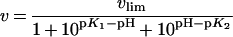 |
(2) |
The limiting value vlim stands for the maximum velocity of the active form of the enzyme and K1 and K2 are the acid dissociation constants of two enzymic groups whose state of ionization controls the velocity.
Inhibition measurements
Inhibition of DPPII/QPP activity was analysed by the spectrophotometric assay performed as described above, except that the DPPII inhibitor (dissolved and diluted in water) was pre-incubated with the enzyme for 15 min at 37 °C before the addition of substrate. IC50 values were obtained using substrate concentrations near the Km value, and at least ten different inhibitor concentrations (0–160 μM).
The Ki of the inhibitor for the active site was determined using seven different substrate concentrations (10–1000 μM Ala-Pro-pNA) and ten different inhibitor concentrations (0–160 μM).
Assuming competitive inhibition, the data were fitted to eqn (3) using GraFit version 5 [38]:
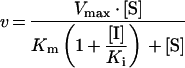 |
(3) |
RESULTS
Purification of human DPPII
DPPII was purified from human seminal plasma to apparent homogeneity by lectin-affinity chromatography, metal-chelating chromatography, a new affinity chromatography step and to final concentration by ion-exchange chromatography. This four-step purification procedure resulted in an approx. 7300-fold purification with a high yield of approx. 40%, as outlined in Table 1. Although the Con A–Sepharose chromatographic step removed 95% of the bulk protein, more than 100% of the Lys-Ala-pNA cleaving activity was recovered, suggesting the removal of an endogenous inhibitor or competing substrate that does not bind to the lectin. Introduction of the affinity column with the immobilized reversible DPPII inhibitor Lys-Pip was necessary for a final clean-up of the natural enzyme. The affinity constant of Lys-Pip for DPPII at pH 5.5 was ~0.9 μM. The IC50 values determined under similar conditions for DPPII and DPPIV (Ala-Pro-pNA at a concentration near the Km, at pH 7.0) were 0.1 μM and 166 μM respectively, indicating approx. 1000-fold selectivity for DPPII over DPPIV.
Table 1. Purification of DPPII from human seminal plasma.
| Step | Total protein (mg) | Total activity (units)* | Specific activity (units/mg) | Yield (%) | Purification (-fold) |
|---|---|---|---|---|---|
| Seminal fluid | 5458 | 54 | 0.0096 | 100 | 1 |
| Con A–Sepharose | 135 | 58 | 0.43 | 107 | 45 |
| Zinc-chelating Sepharose | 33 | 51 | 1.6 | 94 | 163 |
| Lys-Pip | 1.1 | 27 | 24 | 50 | 2536 |
| HiTrap Q | 0.3 | 24 | 70 | 44 | 7309 |
* One unit is defined as the amount of enzyme that cleaves 1 μmol of Lys-Ala-pNA per min under the assay conditions as described in the Material and methods section.
SDS/PAGE analysis of the purified DPPII protein (Figure 2) revealed the presence of a single protein band; when overloading the gel (1 μg of protein) a minor band appeared below DPPII. The purified enzyme showed no significant aminopeptidase, DPPIV or prolyl oligopeptidase activity.
Figure 2. SDS/PAGE analysis of purified human DPPII.

The purified enzyme was resolved by SDS/10% PAGE under reducing conditions. Lanes 1 and 2 contain 0.3 and 0.9 μg of purified DPPII respectively. Molecular-mass standards (sizes indicated in kDa) are shown in lane 3. Proteins were visualized using silver staining. The molecular mass estimated for DPPII was approx. 60 kDa.
Molecular mass of human DPPII
The molecular mass of purified human DPPII was estimated to be 60±2 kDa under denaturing conditions in the presence of 2-mercaptoethanol on SDS/PAGE (Figure 2). The enzyme was eluted from the Superdex 200 HR 10/30 gel-filtration column with an apparent molecular mass of 120±9 kDa. The native human DPPII therefore appears to exist as a homodimer composed of 60 kDa subunits.
Stability of the purified human DPPII
In order to prevent loss of enzyme activity due to storage or extremely low protein concentrations under assay conditions, the effect of three stabilizing agents (BSA, Tween 20 and glycerol) on the Ala-Pro-pNA-cleaving activity of purified DPPII was investigated at different temperatures.
Incubation of purified DPPII at 37 °C without the addition of a stabilizer resulted in a loss of approx. 30% of the initial activity after 2 h. In the presence of 1 mg/ml BSA or 0.1% Tween 20, 90% of the initial activity was maintained after 4 h. With 50% glycerol, there was some stabilization, but not to the same extent as with the other additives. Therefore the assays with purified DPPII were carried out in the presence of BSA.
Purified DPPII was stable for at least 4 months in the presence of 1 mg/ml BSA at 4 °C, −20 °C or −80 °C, and in the presence of 0.1% Tween 20 at −80 °C (95–105% residual activity).
In the presence of the stabilizing agents, the enzyme tolerated at least five subsequent freeze/thaw cycles. However, when no stabilizer was added, no residual DPPII activity could be detected following five subsequent freeze/thaw cycles (gradual loss).
Identification as QPP using MS
Nano-electrospray mass spectra of the tryptic peptides of the purified enzyme were acquired on an electrospray Q-TOF II mass spectrometer equipped with dual-orthogonal sampling API source (Figure 3A). A number of multiply charged peptides were selected for fragmentation. For example, electrospray MS/MS analysis of the doubly charged ion at m/z 596.26 produced the spectrum shown in Figure 3(B), which was interpreted to give the sequence QTSQAGFP(I/L).
Figure 3. MS analysis of silver-stained SDS/PAGE band.

(A) The nano-electrospray MS spectrum in the m/z region from 400 to 1000 of the peptides generated by tryptic digestion of the silver-stained band. Five multiple-charged peptides were chosen for fragmentation. These precursor ions are shown in the spectrum. The five multiple-charged peptides are 556.75(+2), 596.26(+2), 647.26(+2), 699.33(+2) and 732.86(+2). (B) The electrospray MS/MS spectrum for the double-charged ion at m/z 596.26 is shown in b and y series, and inspected amino acid sequence are indicated (one-letter amino acid codes are used).
MaxEnt3 was used to resolve the multiple charged peaks to singly charged peaks. The mono-isotopic masses were used for identification of the protein based on peptide mass fingerprinting. The Mascot search engine was used to match the SwissProt (Chordata) database. Human QPP/DPP7 (GenBank® accession number AAF12747) was the most significant hit (Mowse score 96) matching 11 individual peptides, covering 20% of the protein. The experimental mass of each of the 11 peptides was within 0.11 mass unit of the calculated value. The peptides were distributed over the entire sequence and were also found in regions not conserved in the QPP sequence of other mammalian species. One peptide overlapped with the N-terminal sequence of human DPPII from placenta (GenBank® accession number S77568) (Figure 4). For nine out of the 11 peptides, an exact match was only found for DPPII/QPP/DPP7.
Figure 4. Alignment of the amino acid sequences of QPP and DPPII, and sequence coverage of the identified tryptic peptides of the purified human DPPII.

(A) ClustalW multiple sequence alignment of the N-terminal sequences of human QPP (hQPP), and human and pig DPPII (hDPPII and pDPPII respectively). (B) ClustalW multiple sequence alignment of human and mouse QPP (hQPP and mQPP respectively), and rat DPPII (rDPPII). Residues most probably forming the catalytic triad are labelled with an asterisk. The consensus sequence for the active site serine residue of serine-type proteases (Gly-Xaa-Ser-Xaa-Gly) is underlined the leucine zipper motif is marked in bold. The tryptic peptide sequences of the purified human DPPII identified by MS are marked with grey boxes in the human QPP sequence. GenBank® accession numbers are as follows: hQPP, AAF12747; hDPPII, S77568; pDPPII, S70349; mQPP, AAG01154; rDPPII, Q9EPB1.
Comparison of natural human DPPII and recombinant QPP/DPP7
The kinetic parameters of DPPII purified from human seminal plasma and recombinant QPP/DPP7 (residues Ala26 to end, and containing an N-terminal FLAG tag) [22] were compared in Table 2. Human DPPII and QPP showed very similar Km values for the frequently used substrates and IC50 values for both inhibitors (Figure 1) tested. The small differences in the results are within the standard error of the experiments. The influence of ionic strength was investigated simultaneously for both enzyme preparations using the same reagent dilutions at pH 7.0 with Ala-Pro-pNA (typically used for QPP). The increase in NaCl concentration (0–150–750 mM) resulted in a similar increase of Km values and a decrease in Vmax for both the purified DPPII and the recombinant QPP. The combined pH profiles of DPPII and QPP of kcat for Lys-Ala-pNA and Ala-Pro-pNA are shown in Figure 5.
Table 2. Comparison of recombinant human QPP and purified human DPPII regarding Km and IC50 values observed with different substrates and inhibitors.
Each assay was carried out in 0.05 M cacodylic acid buffer, pH 5.5, and 0.05 M Hepes buffer, pH 7.0, at 37 °C with the substrates mentioned in the Table. The measurements with DPPII and QPP were carried out simultaneously using the same reagent dilutions for both enzymes. Values for Km and IC50 compound 1 are in μM. Values for IC50 compound 2 are in nM. Errors represent S.E.M. on the fit. IC50 values were obtained using substrate concentrations near the Km value. n.d., not determined.
| pH 5.5 | pH 7.0 | |||
|---|---|---|---|---|
| DPPII | QPP | DPPII | QPP | |
| Km – substrate analysis | ||||
| Lys-Ala-pNA | 570±40 | 560±30 | 40±5 | 35±3 |
| Ala-Pro-pNA | 40±4 | 30±3 | 9±2 | 7±3 |
| Gly-Pro-pNA | 210±30 | 180±30 | 70±6 | 60±5 |
| Nle-Nle-pNA | 20±3 | 15±1 | n.d. | n.d. |
| IC50 compound 1* | ||||
| Lys-Ala-pNA | 1.72±0.06 | 1.19±0.03 | 0.052±0.002 | 0.043±0.002 |
| Ala-Pro-pNA | 2.64±0.04 | 2.26±0.04 | 0.063±0.001 | 0.049±0.002 |
| IC50 compound 2† | ||||
| Lys-Ala-pNA | 0.95±0.04 | 1.10±0.02 | 1.00±0.04 | 0.95±0.01 |
| Ala-Pro-pNA | 1.45±0.03 | 1.30±0.02 | 1.05±0.02 | 1.00±0.02 |
Figure 5. pH profiles of purified DPPII and recombinant QPP with different substrates.

Measurements were carried out using the same reagent dilutions for both purified DPPII (△) and recombinant QPP (○) in 0.025 M ethanoic acid, 0.025 M cacodylic acid and 0.025 M Hepes buffer, containing 0.1 M NaCl (titrated with NaOH or HCl to the desired pH) at 37 °C as described under the Materials and methods section, with (A) Lys-Ala-pNA and (B) Ala-Pro-pNA. The initial rate was proportional to kcat. The experimental data were fitted to a double ionization equation (eqn 1) that results in a bell-shaped curve using GraFit software version 5 [38].
pH profile of human DPPII
The hydrolytic activity of human DPPII for Lys-Ala- and Ala-Pro- derived chromogenic (-pNA) and fluorogenic (-4Me2NA) substrates was assayed in the pH range 3.0–8.5 using a single composite buffer system containing NaCl to maintain comparable ionic strength and identical buffering substances over the entire pH range. The pH-dependency of kcat was obtained from initial rate measurements at [S] much greater than Km. To investigate the pH profile of kcat/Km, a substrate concentration much less than Km was used. Owing to substrate inhibition at high concentrations when using the -4Me2NA substrates, substrate concentrations to obtain initial rates proportional to kcat were carefully chosen.
With all substrates, the kcat profiles of human DPPII were optimal at pH values of approx. 5.0 while the kcat/Km profiles had a peak at pH ~5.5. At pH 8.0, no DPPII activity could be detected using this buffer system. All profiles were bell-shaped, indicating two ionization states contributing to substrate binding and enzyme turnover. The curve fitting yielded the pK1 and pK2 values listed in Table 3.
Table 3. pH-dependence of DPPII catalysis.
The reactions were performed at 37 °C in 0.025 M ethanoic acid, 0.025 M cacodylic acid and 0.025 M Hepes buffer, containing 0.1 M NaCl. The experimental data were fitted to a double-ionization equation (eqn 2), and the pK1 and pK2 values obtained are listed. Errors represent S.E.M. on the fit.
| Substrate | [S] (mM) | pK1 | pK2 | vlim‡ (μM/min) | Estimated pHopt |
|---|---|---|---|---|---|
| Lys-Ala-pNA | 0.029* | 4.57±0.04 | 6.62±0.03 | 0.46±0.01 | 5.6 |
| 9.5† | 4.00±0.03 | 5.58±0.03 | 26.1±0.6 | 4.8 | |
| Lys-Ala-4Me2NA | 0.029* | 4.52±0.04 | 6.53±0.04 | 0.133±0.004 | 5.5 |
| 0.95† | 4.18±0.02 | 5.98±0.02 | 1.97±0.04 | 5.1 | |
| Ala-Pro-pNA | 0.009* | 4.22±0.03 | 6.88±0.03 | 0.76±0.01 | 5.6 |
| 0.95† | 3.43±0.02 | 6.36±0.02 | 6.99±0.07 | 4.9 | |
| Ala-Pro-4Me2NA | 0.048* | 3.88±0.04 | 6.64±0.03 | 0.125±0.002 | 5.3 |
| 0.95† | 3.60±0.06 | 6.44±0.05 | 0.83±0.03 | 5.0 |
* Initial rate obtained is proportional to kcat/Km.
† Initial rate obtained is proportional to kcat.
‡ Values normalized to an identical enzyme concentration (1.3 nM).
Kinetic parameters of human DPPII
Kinetic parameters of natural human DPPII for the commercially available dipeptide-derived chromogenic (-pNA) and fluorogenic (-4Me2NA) substrates were determined under two different assay conditions. A first, at acidic pH (50 mM cacodylic acid, pH 5.5), said to be the optimum of DPPII [8,14], and a second at neutral pH (50 mM Hepes buffer, pH 7.0), typically used for QPP [24] (Table 4). The substrate selectivity of DPPII was determined by comparison of the selectivity constants kcat/Km for the different substrates. DPPII preferred the chromogenic pNA-derived substrates over the fluorogenic 4Me2NA-derived ones. All of the Michaelis–Menten curves for the 4Me2NA-derived substrates showed substrate inhibition. However, the fluorogenic leaving group 4-methoxy-2-naphthylamine (1–1000 μM) showed no inhibitory effect on DPPII activity. Purified human DPPIV showed a minor preference for the pNA-substrates (results not shown).
Table 4. Comparison of the steady-state kinetic parameters for chromogenic (-pNA) and fluorogenic (-4Me2NA) substrate hydrolysis by DPPII.
Each kinetic experiment was carried out in 0.05 M cacodylic acid buffer, pH 5.5, and in 0.05 M Hepes buffer, pH 7.0, at 37 °C. The data represent the means±S.D. of two to four separate experiments. n.d., not determined.
| -pNA | -4Me2NA | ||||
|---|---|---|---|---|---|
| Substrate | Kinetic parameter | pH 5.5 | pH 7.0 | pH 5.5 | pH 7.0 |
| Lys-Ala- | Km (μM) | 570±80 | 50±10 | 500±50 | 38±4 |
| kcat (s−1) | 230±30 | 21±3 | 78±4 | 6.1±0.1 | |
| kcat/Km (×106 s−1·M−1) | 0.40±0.08 | 0.4±0.1 | 0.16±0.02 | 0.16±0.02 | |
| Lys-Pro-* | Km (μM) | 19±3 | n.d. | 33±1 | 3.1±0.3 |
| kcat (s−1) | 78±2 | n.d. | 35.0±0.5 | 3.0±0.1 | |
| kcat/Km (×106 s−1·M−1) | 4.1±0.7 | n.d. | 1.10±0.04 | 1.0±0.1 | |
| Ala-Pro- | Km (μM) | 40±1 | 16±1 | 260±30 | 101±7 |
| kcat (s−1) | 105±7 | 38±1 | 27±1 | 8.1±0.2 | |
| kcat/Km (×106 s−1·M−1) | 2.6±0.2 | 2.4±0.2 | 0.10±0.01 | 0.080±0.006 | |
| Gly-Pro- | Km (μM) | 230±10 | 93±1 | 610±80 | 240±20 |
| kcat (s−1) | 67±1 | 28±2 | 1.2±0.1 | 0.41±0.01 | |
| kcat/Km (×106 s−1·M−1) | 0.29±0.01 | 0.30±0.02 | 0.0020±0.0003 | 0.0017±0.0001 | |
* Data from one experiment±S.E.M. on the fit.
Among the commercially available substrates, DPPII exhibited highest efficiency towards the chromogenic Ala-Pro-pNA. Lys-Ala-4Me2NA was preferred over Ala-Pro-4Me2NA and Gly-Pro-4Me2NA.
Catalytic parameters of human DPPII for various chromogenic substrates are shown in Table 5. Among the substrates tested, Lys-Pro-pNA and Nle-Nle-pNA (Nle is norleucine) (chosen because of its high apparent affinity in a previous substrate specificity study of QPP [22]) showed the lowest Km values and therefore highest apparent affinity for human DPPII. Lys-Ala-pNA showed the highest activity (Vmax).
Table 5. Steady-state kinetic parameters for dipeptide-derived chromogenic (-pNA) substrate hydrolysis by natural human DPPII.
Each kinetic experiment was carried out in 0.05 M cacodylic acid buffer, pH 5.5, at 37 °C. Errors represent S.E.M. on the fit. Nle, norleucine.
| Substrate-pNA | Km (μM) | kcat (s−1) | kcat/Km (×106 s−1·M−1) |
|---|---|---|---|
| Nle-Nle-* | 19±2 | 18.4±0.1 | 0.968±0.102 |
| Lys-Ala-* | 567±79 | 229.3±27.1 | 0.404±0.074 |
| His-Ala- | 223±16 | 12.8±0.3 | 0.057±0.004 |
| Tyr-Ala- | 220±21 | 11.0±0.3 | 0.050±0.005 |
| Pro-Ala- | 1376±228 | 15.3±1.3 | 0.011±0.002 |
| Lys-Pro-* | 19±3 | 77.7±2.0 | 4.089±0.654 |
| Ala-Pro-* | 41±1 | 105.4±7.3 | 2.571±0.189 |
| Asn-Pro- | 101±10 | 184.6±4.8 | 1.828±0.187 |
| Arg-Pro-* | 40±3 | 65.8±8.2 | 1.645±0.239 |
| Phe-Pro- | 58±9 | 93.0±3.1 | 1.603±0.254 |
| Ser-Pro- | 80±7 | 116.9±2.6 | 1.461±0.132 |
| His-Pro- | 83±6 | 28.4±0.5 | 0.342±0.025 |
| Gly-Pro-* | 227±11 | 67.3±1.3 | 0.296±0.016 |
* Data represent the means±S.D. of two to four separate experiments.
Studying the influence of the assay conditions on the catalytic parameters revealed a decrease of both Km and kcat values upon the change from the acidic to the neutral assay. Since the decrease was proportional for both parameters, selectivity constants (kcat/Km) were similar for both conditions (at pH 5.5 and pH 7.0) for each substrate tested.
When the parameters were measured in the composite buffer (as used for the pH profile) at pH 5.5 and pH 7.0, the selectivity constants were significantly higher at pH 5.5. This acidic optimum was also seen in the pH profiles (Figure 5). These results clearly demonstrate the influence of assay conditions on catalytic parameters and may explain confusing reports in literature concerning the optimal pH of QPP and DPPII.
DPPII activity was markedly influenced by the ionic strength of the assay medium. As shown in Figure 6, Km values of Lys-Ala-pNA and Ala-Pro-pNA increased at both pH 5.5 and pH 7.0. The effect of NaCl on Km was most prominent with Lys-Ala-pNA at pH 7.0. The Km increased 24-fold (55 μM to 1330 μM) following the increase in ionic strength from 0 to 0.75 M NaCl. The Vmax values all decreased, except with Lys-Ala-pNA, at pH 5.5. NaCl concentrations up to 1 M in the enzyme sample (final NaCl concentration in assay 50 mM) had no significant influence on the DPPII activity measured under standard assay conditions.
Figure 6. Effects of increasing NaCl concentrations on the steady-state kinetic parameters.

Initial velocities of substrate cleavage were measured with different concentrations of substrate at pH 5.5 and pH 7.0 in buffer containing (○) 0 M NaCl, (□) 0.15 M NaCl or (△) 0.75 M NaCl. The Michaelis–Menten plot of the experimental data was generated using GraFit version 5 [38]. (A) Lys-Ala-pNA in 0.05 M cacodylic acid buffer, pH 5.5. (B) Lys-Ala-pNA in 0.05 M Hepes buffer, pH 7.0. (C) Ala-Pro-pNA in 0.05 M cacodylic acid buffer, pH 5.5. (D) Ala-Pro-pNA in 0.05 M Hepes buffer, pH 7.0.
Comparison of substrate preferences of DPPII and DPPIV
Substrate preferences of DPPII and DPPIV purified from human seminal plasma were compared based on the selectivity constants (kcat/Km) (Figure 7). Lys-Pro-pNA and Ala-Pro-pNA were found to be the most sensitive substrates for human DPPII, but they were less selective than Lys-Ala-pNA.
Figure 7. Comparison of the substrate preferences of human DPPII and human DPPIV.

Selectivity constants kcat/Km (×106 M−1·s−1) of human DPPII and human DPPIV were compared for substrates of the type P1-P2-pNA in 50 mM cacodylic acid, pH 5.5, and 50 mM Tris/HCl, pH 8.3, respectively.
DISCUSSION
In the present study, DPPII was purified from human seminal plasma to apparent homogeneity using a high-yield scheme including an inhibitor-based affinity chromatographic step. Lys-Pip was chosen as the affinity ligand over more selective DPPII inhibitors [40] in order to facilitate elution. With the above scheme, it was possible to obtain sufficient natural human DPPII for experiments such as the molecular identification and catalytic characterization of the enzyme. Previously, human DPPII has been partially purified from brain, kidney, seminal fluid and placenta [22,41–44], and to apparent homogeneity from kidney [45]. However, only an N-terminal sequence (33 amino acids) and partial catalytic data were reported.
The natural DPPII from human seminal plasma appeared to be a homodimer of 120 kDa composed of 60 kDa subunits. These data are in good agreement with the molecular masses determined for DPPII from other sources [9,10,12,14,45] and for human QPP [24,26]. In contrast, Huang et al. [17] reported that the DPPII purified from porcine seminal plasma was composed of three identical subunits.
Since 11 tryptic peptides of the purified human DPPII, identified by MS, matched the QPP sequence, and since the purified and recombinant preparations show almost identical kinetic behaviour, DPPII is likely to be identical with QPP. The possibility that QPP and DPPII are encoded by two separate, but very closely related, loci is very unlikely, but cannot be ruled out completely. However, no very closely related human nucleotide or amino acid sequences have been reported so far. Using the mass spectrometric approach, it is not feasible to obtain 100% of coverage.
QPP (which we consider to be identical with DPPII) shows significant sequence similarities over the entire sequence with lysosomal Pro-Xaa carboxypeptidase (E.C. 3.4.16.2) (43% identity in 447-residue overlap) [46] and with human thymus-specific serine peptidase (28% identity in 467-residue overlap) [47]. These three peptidases are grouped in the S28 family of the clan SC [27,48] (see also the MEROPS peptidase database, http://merops.sanger.ac.uk/); unfortunately, no structures are available for family members. All three proteins possess N-glycosylation sites, but these are not conserved. DPPII/QPP contains a leucine zipper motif (amino acids 128–149) that is required for its dimerization and enzymic activity [26]. This motif is not present in lysosomal prolylcarboxypeptidase or human thymus-specific serine peptidase.
As a group, S28 peptidases do not show any significant homology with the S9 family, which includes prolyl oligopeptidase, oligopeptidase B, acylaminoacylpeptidase, FAPα and the DPPs IV, 8 and 9, nor with the S10 family of carboxypeptidases. However, around the putative catalytic Ser162 in DPPII/QPP, the sequence GGSYGGML corresponds to a consensus motif also found in the prolyloligopeptidase family (GXSXGGZZ, where X is any amino acid and Z represents a hydrophobic residue), suggesting an evolutionary relationship [50]. The putative catalytic aspartate (Asp418) and histidine (His443) are located far to the C-terminal end of the protein.
The recent comparison of the catalytic properties of recombinant QPP/DPP7 with those of partially purified human DPPII [a minor contamination with (an)other member(s) of the family of DPPs was suspected] suggested, but could not prove, their identity [22]. The purified human DPPII and recombinant QPP showed comparable Km and IC50 values (Table 2), and a similar effect of pH (Figure 5) and ionic strength.
Underwood et al. [24] reported that QPP and DPPII were different with respect to their pH profiles [24]. QPP was able to cleave substrate molecules as efficiently at acidic pH as at neutral pH. Fukasawa et al. [10] mentioned a similar high activity of rat kidney DPPII at neutral pH using Gly-Pro-β-naphthylamide as the substrate. DPPII from other sources only showed an acidic pH optimum for dipeptide-derived substrate cleavage [6–9,11–17,41]. We determined pH profiles using the same composite buffer over the entire pH range, containing NaCl in order to eliminate the influence of differences in ionic strength and buffering substances. In the pH profiles of both kcat and kcat/Km, only an acidic optimum was detectable with all substrates tested. As can be deduced from the kinetic parameters in Table 4, the affinity of the substrates for DPPII (Km) was better under our neutral assay conditions, but the turnover number (kcat) decreased in comparison with the values in the acidic assay conditions. At roughly physiological ionic strength (composite buffer), the decrease in kcat outweighs the decrease in Km, resulting in a smaller selectivity constant at pH 7.0 (results not shown).
The optimum around pH 5.0–5.5 and a complete loss of activity at pH 8.0 are in agreement with the vesicular localization of the enzyme. Although DPPII was previously located at the lysosomes [6,11,14,51], more recent studies locate QPP (or DPPII) to vesicles distinct from lysosomes [25].
In enzymology textbooks [52,53], the pH-dependence of enzymes is usually described in terms of ionizable groups on the enzyme and the substrate. The pH-dependence for both kcat and kcat/Km was bell-shaped for all the substrates tested. The kcat/Km parameter reflects ionizable groups on the free enzyme and the free substrate. The pKa of the α-NH2 is ~7.8 and the lysine ε-NH2 is ~10.4. Therefore we conclude that the two ionizations governing the kcat/Km pH profile are located on the free enzyme. The curve fitting yielded the pK1 and pK2 values, listed in Table 3, for a group that must be deprotonated and one that must be protonated. Binding of the substrate changes the charge distribution near the enzyme's catalytic site, and charged groups on the enzyme may alter the pKa of the bound substrate. These changes are reflected in the observed shift of pK1 and pK2 in the kcat pH profile. It is intriguing to observe that the pK1 and pK2 values and their relative shifts upon substrate binding vary with the type of substrate used (Table 3). The effect of the P1 residue on the pKa values was most striking in the kcat profile. The effect of the leaving group was dependent on the residues in P1 and P2.
The Michaelis–Menten curves in the presence of increasing ionic strength showed a dose-dependent inhibitory effect of NaCl on human DPPII (Figure 6). High ionic strength resulted in a decreased affinity of the substrate for the enzyme, and was most prominent in the assay at neutral pH with Lys-Ala-pNA. Salt ions electrostatically shield the charged surface residues of DPPII, possibly competing with the substrate for enzyme binding. Whereas the decrease in kcat/Km is easily interpreted for the reaction of two oppositely charged molecules, the decrease in kcat is more difficult to explain. The affinity of the enzyme for cationic groups is reflected in its preference for cleavage of basic substrates. The neutral optimum for QPP, as suggested by Underwood et al. [24], which we could not confirm, was probably due to the difference in ionic strength and composition of the two buffering systems used.
There are no systematic studies on substrate selectivity of DPPII, and various data in the literature are contradictory. Most of the conclusions are based on relative rates instead of catalytic parameters, and preferences of DPPII for synthetic substrates are determined without considering the impact of the chromogenic or fluorogenic leaving group used. We compared the selectivity constants (kcat/Km) of pNA- and 4Me2NA-derived dipeptides for human DPPII. As can be seen in Table 4, the 4Me2NA-derived substrates were all cleaved with lower efficiency than the pNA-derived substrates. In contrast, DPPIV showed only a minor preference for the pNA-derived over 4Me2NA-derived substrates.
Among the commercially available substrates, DPPII preferred Ala-Pro-pNA over Lys-Ala-pNA, while Lys-Ala-4Me2NA was preferred over Ala-Pro-4Me2NA. These results showed that the residue at P1′ had an impact on substrate selectivity. However, in both types of substrates, Lys-Pro-pNA (not commercially available) and Lys-Pro-4Me2NA performed best, pointing to an overriding P2 preference for lysine, independent of the P1′. The inversion of substrate selectivity emphasizes the importance of the leaving group at P1′. Discrepancies due to different leaving groups could also be detected in other studies [10,17,51]. The reported preference of rat brain DPPII for Gly-Pro-4-nitroanilide over Lys-Ala-2-naphthylamide may be due to the different group at P1′ [12]. The influence of the residue at P1′ may also be responsible for the lack of predictability in rate relationships between tripeptides and their corresponding naphthylamides, as seen with DPPII from the porcine ovary [14] and bovine dental pulp [8].
Since most bioactive peptides exert their function at a very low concentration, we used the selectivity constant kcat/Km to determine the efficiency of DPPII cleavage of various pNA-derived dipeptides (Table 5). Based on the Km, human DPPII showed a greater affinity for Xaa-Pro-type than for Xaa-Ala-type pNA substrates. This preference was in agreement with DPPII/QPP/DPP7 from other sources [16,19,22,45]. Mentlein and Struckhoff [12] could not detect a significant difference between the Xaa-Pro and the Xaa-Ala substrate types. The preferred N-terminal residues, as determined from the kcat/Km value, are basic (lysine, arginine), hydrophobic (alanine, phenylalanine) or uncharged (asparagine, serine) amino acids, as described previously [12,40]. In contrast with the results obtained by using a positional scanning synthetic combinatorial dipeptide-AMC substrate library [22], our human DPPII did accept aromatic residues (phenylalanine, histidine) at P2 for the pNA-substrates. Dipeptide-derived inhibitors for DPPII show comparable preferences for basic and neutral amino acids at P2 [40].
Comparison with the selectivity constants of DPPIV purified from human seminal plasma (Figure 7) showed that, although Lys-Pro-pNA and Ala-Pro-pNA were cleaved most efficiently by DPPII, Lys-Ala-pNA was more selective for DPPII determinations in crude samples due to its minor hydrolysis by DPPIV.
The knowledge of the molecular and catalytic properties of purified human natural DPPII, and the availability of a high yield purification protocol, should be helpful in the search for natural substrates of DPPII. Finally, the sequence identification of human DPPII is an essential step towards the unification of the nomenclature for the enzyme DPPII (E.C. 3.4.14.2).
Acknowledgments
This work was supported by the Research Foundation – Flanders (Belgium) [F. W. O. (Fonds voor Wetenschappelijk Onderzoek) – Vlaanderen]. M.-B. M. is a research assistant of the F. W. O.-Vlaanderen. We are grateful to Mrs Nicole Lamoen for her excellent technical assistance. We also thank KellyAnn D. Pryor and Frank Marsilio for expression and purification of QPP. Our thanks also go to the anonymous reviewers for their constructive comments.
References
- 1.Park J. E., Lenter M. C., Zimmermann R. N., Garin-Chesa P., Old L. J., Rettig W. J. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 1999;274:36505–36512. doi: 10.1074/jbc.274.51.36505. [DOI] [PubMed] [Google Scholar]
- 2.Abbott C. A., Yu D. M., Woollatt E., Sutherland G. R., McCaughan G. W., Gorrell M. D. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur. J. Biochem. 2000;267:6140–6150. doi: 10.1046/j.1432-1327.2000.01617.x. [DOI] [PubMed] [Google Scholar]
- 3.Olsen C., Wagtmann N. Identification and characterization of human DPP9, a novel homologue of dipeptidyl peptidase IV. Gene. 2002;299:185–193. doi: 10.1016/s0378-1119(02)01059-4. [DOI] [PubMed] [Google Scholar]
- 4.Lambeir A. M., Durinx C., Scharpe S., De Meester I. Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003;40:209–294. doi: 10.1080/713609354. [DOI] [PubMed] [Google Scholar]
- 5.McDonald J. K., Ellis S., Reilly T. J. Properties of dipeptidyl arylamidase I of the pituitary. J. Biol. Chem. 1966;241:1494–1501. [PubMed] [Google Scholar]
- 6.McDonald J. K., Reilly T. J., Zeitman B. B., Ellis S. Dipeptidyl arylamidase II of the pituitary. Properties of lysylalanyl-β-naphthylamide hydrolysis: inhibition by cations, distribution in tissues, and subcellular localization. J. Biol. Chem. 1968;243:2028–2037. [PubMed] [Google Scholar]
- 7.McDonald J. K., Leibach F. H., Grindeland R. E., Ellis S. Purification of dipeptidyl aminopeptidase II (dipeptidyl arylamidase II) of the anterior pituitary gland: peptidase and dipeptide esterase activities. J. Biol. Chem. 1968;243:4143–4150. [PubMed] [Google Scholar]
- 8.McDonald J. K., Schwabe C. Dipeptidyl peptidase II of bovine dental pulp: initial demonstration and characterization as a fibroblastic, lysosomal peptidase of the serine class active on collagen-related peptides. Biochim. Biophys. Acta. 1980;616:68–81. doi: 10.1016/0005-2744(80)90264-8. [DOI] [PubMed] [Google Scholar]
- 9.Fukasawa K., Fukasawa K. M., Hiraoka B. Y., Harada M. Purification and properties of dipeptidyl peptidase II from rat kidney. Biochim. Biophys. Acta. 1983;745:6–11. doi: 10.1016/0167-4838(83)90163-2. [DOI] [PubMed] [Google Scholar]
- 10.Fukasawa K. M., Fukasawa K., Higaki K., Shiina N., Ohno M., Ito S., Otogoto J., Ota N. Cloning and functional expression of rat kidney dipeptidyl peptidase II. Biochem. J. 2001;353:283–290. doi: 10.1042/0264-6021:3530283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imai K., Hama T., Kato T. Purification and properties of rat brain dipeptidyl aminopeptidase. J. Biochem. (Tokyo) 1983;93:431–437. doi: 10.1093/oxfordjournals.jbchem.a134197. [DOI] [PubMed] [Google Scholar]
- 12.Mentlein R., Struckhoff G. Purification of two dipeptidyl aminopeptidases II from rat brain and their action on proline-containing neuropeptides. J. Neurochem. 1989;52:1284–1293. doi: 10.1111/j.1471-4159.1989.tb01877.x. [DOI] [PubMed] [Google Scholar]
- 13.Hopsu-Havu V. K., Jansen C. T., Jarvinen M. Partial purification and characterization of an acid dipeptide naphthylamidase (carboxytripeptidase) of the rat skin. Arch. Klin. Exp. Dermatol. 1970;236:282–296. doi: 10.1007/BF00508328. [DOI] [PubMed] [Google Scholar]
- 14.Eisenhauer D. A., McDonald J. K. A novel dipeptidyl peptidase II from the porcine ovary: purification and characterization of a lysosomal serine protease showing enhanced specificity for prolyl bonds. J. Biol. Chem. 1986;261:8859–8865. [PubMed] [Google Scholar]
- 15.Lynn K. R. The isolation and some properties of dipeptidyl peptidases II and III from porcine spleen. Int. J. Biochem. 1991;23:47–50. doi: 10.1016/0020-711x(91)90007-a. [DOI] [PubMed] [Google Scholar]
- 16.Sentandreu M. A., Toldrá F. Partial purification and characterisation of dipeptidyl peptidase II from porcine skeletal muscle. Meat Sci. 2001;57:93–103. doi: 10.1016/s0309-1740(00)00082-6. [DOI] [PubMed] [Google Scholar]
- 17.Huang K., Takagaki M., Kani K., Ohkubo I. Dipeptidyl peptidase II from porcine seminal plasma: purification, characterization, and its homology to granzymes, cytotoxic cell proteinases (CCP 1–4) Biochim. Biophys. Acta. 1996;1290:149–156. doi: 10.1016/0304-4165(96)00013-x. [DOI] [PubMed] [Google Scholar]
- 18. Reference deleted.
- 19.Chiravuri M., Schmitz T., Underwood R., Yardley K., Dayal Y., Huber B. T. A novel apoptotic pathway in quiescent lymphocytes identified by inhibition of a post-proline cleaving aminodipeptidase: a candidate target protease, quiescent cell proline dipeptidase. J. Immunol. 1999;163:3092–3099. [PubMed] [Google Scholar]
- 20.Araki H., Li Y., Yamamoto Y., Haneda M., Nishi K., Kikkawa R., Ohkubo I. Purification, molecular cloning, and immunohistochemical localization of dipeptidyl peptidase II from the rat kidney and its identity with quiescent cell proline dipeptidase. J. Biochem. (Tokyo) 2001;129:279–288. doi: 10.1093/oxfordjournals.jbchem.a002855. [DOI] [PubMed] [Google Scholar]
- 21.Mammalian Gene Collection Program Team. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. U.S.A. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leiting B., Pryor K. D., Wu J. K., Marsilio F., Patel R. A., Craik C. S., Ellman J. A., Cummings R. T., Thornberry N. A. Catalytic properties and inhibition of proline-specific dipeptidyl peptidases II, IV and VII. Biochem. J. 2003;371:525–532. doi: 10.1042/BJ20021643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenblum J. S., Kozarich J. W. Prolyl peptidases: a serine protease subfamily with high potential for drug discovery. Curr. Opin. Chem. Biol. 2003;7:496–504. doi: 10.1016/s1367-5931(03)00084-x. [DOI] [PubMed] [Google Scholar]
- 24.Underwood R., Chiravuri M., Lee H., Schmitz T., Kabcenell A. K., Yardley K., Huber B. T. Sequence, purification, and cloning of an intracellular serine protease, quiescent cell proline dipeptidase. J. Biol. Chem. 1999;274:34053–34058. doi: 10.1074/jbc.274.48.34053. [DOI] [PubMed] [Google Scholar]
- 25.Chiravuri M., Agarraberes F., Mathieu S. L., Lee H., Huber B. T. Vesicular localization and characterization of a novel post-proline-cleaving aminodipeptidase, quiescent cell proline dipeptidase. J. Immunol. 2000;165:5695–5702. doi: 10.4049/jimmunol.165.10.5695. [DOI] [PubMed] [Google Scholar]
- 26.Chiravuri M., Lee H., Mathieu S. L., Huber B. T. Homodimerization via a leucine zipper motif is required for enzymatic activity of quiescent cell proline dipeptidase. J. Biol. Chem. 2000;275:26994–26999. doi: 10.1074/jbc.M005445200. [DOI] [PubMed] [Google Scholar]
- 27.McDonald J. K. Dipeptidyl peptidase II. In: Barrett A. J., Rawlings N. D., Woessner J. F., editors. Handbook of Proteolytic Enzymes. London/San Diego: Academic Press; 1998. pp. 408–411. [Google Scholar]
- 28.Senten K., Van der Veken P., De Meester I., Lambeir A. M., Scharpe S., Haemers A., Augustyns K. Design, synthesis, and SAR of potent and selective dipeptide-derived inhibitors for dipeptidyl peptidases. J. Med. Chem. 2003;46:5005–5014. doi: 10.1021/jm0308803. [DOI] [PubMed] [Google Scholar]
- 29.Senten K., Van der Veken P., Bal G., Haemers A., Augustyns K. Polymer-assisted solution-phase parallel synthesis of dipeptide p-nitroanilides and dipeptide diphenyl phosphonates. Tetrahedron Lett. 2001;42:9135–9138. [Google Scholar]
- 30.Scharpé S., De Meester I., Vanhoof G., Hendriks D., Van Sande M., Van Camp K., Yaron A. Assay of dipeptidyl peptidase IV in serum by fluorometry of 4-methoxy-2-naphthylamine. Clin. Chem. 1988;34:2299–2301. [PubMed] [Google Scholar]
- 31.Goossens F., De Meester I., Vanhoof G., Scharpé S. A sensitive method for the assay of serum prolyl endopeptidase. Eur. J. Clin. Chem. Clin. Biochem. 1992;30:235–238. [PubMed] [Google Scholar]
- 32.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 33.De Meester I., Vanhoof G., Lambeir A. M., Scharpe S. Use of immobilized adenosine deaminase (EC 3.5.4.4) for the rapid purification of native human CD26/dipeptidyl peptidase IV (EC 3.4.14.5) J. Immunol. Methods. 1996;189:99–105. doi: 10.1016/0022-1759(95)00239-1. [DOI] [PubMed] [Google Scholar]
- 34.Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 35.Heukeshoven J., Dernick R. Simplified method for silver staining of proteins in polyacrylamide gels and the mechanism of silver staining. Electrophoresis. 1985;6:103–112. [Google Scholar]
- 36.Jensen O. N., Wilm M., Shevchenko A., Mann M. Sample preparation methods for mass spectrometric peptide mapping directly from 2-DE gels. Methods Mol. Biol. 1999;112:513–525. doi: 10.1385/1-59259-584-7:513. [DOI] [PubMed] [Google Scholar]
- 37.Wilm M., Mann M. Analytical properties of the nanoelectrospray ion source. Anal. Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 38.Leatherbarrow R. J. Horley, UK: Erithacus Software Ltd; 2001. GraFit Version 5. [Google Scholar]
- 39.Tipton K. F. Principles of enzyme assay and kinetic studies. In: Eisenthal R., Danson M. J., editors. Enzyme Assays. A Practical Approach. Oxford: IRL Press; 1992. pp. 26–27. [Google Scholar]
- 40.Senten K., Van der Veken P., Bal G., De Meester I., Lambeir A. M., Scharpe S., Bauvois B., Haemers A., Augustyns K. Development of potent and selective dipeptidyl peptidase II inhibitors. Bioorg. Med. Chem. Lett. 2002;12:2825–2828. doi: 10.1016/s0960-894x(02)00603-0. [DOI] [PubMed] [Google Scholar]
- 41.Kato T., Hama T., Nagatsu T. Separation of two dipeptidyl aminopeptidases in the human brain. J. Neurochem. 1980;34:602–608. doi: 10.1111/j.1471-4159.1980.tb11186.x. [DOI] [PubMed] [Google Scholar]
- 42.Mantle D. Characterization of dipeptidyl and tripeptidyl aminopeptidases in human kidney soluble fraction. Clin. Chim. Acta. 1991;196:135–142. doi: 10.1016/0009-8981(91)90066-l. [DOI] [PubMed] [Google Scholar]
- 43.Vanha-Perttula T. Studies on alanine aminopeptidase, dipeptidyl aminopeptidase I and II of the human seminal fluid and prostasomes. Sel. Top. Clin. Enzymol. 1984;2:545–564. [Google Scholar]
- 44.Lampelo S., Lalu K., Vanha-Perttula T. Biochemical studies on dipeptidyl peptidases I to IV of the human placenta. Placenta. 1987;8:389–398. doi: 10.1016/0143-4004(87)90066-x. [DOI] [PubMed] [Google Scholar]
- 45.Sakai T., Kojima K., Nagatsu T. Rapid chromatographic purification of dipeptidyl-aminopeptidase II from human kidney. J. Chromatogr. 1987;416:131–137. doi: 10.1016/0378-4347(87)80493-0. [DOI] [PubMed] [Google Scholar]
- 46.Rawlings N. D., Barrett A. J. Dipeptidyl-peptidase II is related to lysosomal Pro-X carboxypeptidase. Biochim. Biophys. Acta. 1996;1298:1–3. doi: 10.1016/s0167-4838(96)00153-7. [DOI] [PubMed] [Google Scholar]
- 47.Bowlus C. L., Ahn J., Chu T., Gruen J. R. Cloning of a novel MHC-encoded serine peptidase highly expressed by cortical epithelial cells of the thymus. Cell Immunol. 1999;196:80–86. doi: 10.1006/cimm.1999.1543. [DOI] [PubMed] [Google Scholar]
- 48.Tan F., Erdös E. G. Lysosomal Pro-X carboxypeptidase. In: Barrett A. J., Rawlings N. D., Woessner J. F., editors. Handbook of Proteolytic Enzymes. London/San Diego: Academic Press; 1998. pp. 405–407. [Google Scholar]
- 49. Reference deleted.
- 50.Tan F., Morris P. W., Skidgel R. A., Erdös E. G. Sequencing and cloning of human prolylcarboxypeptidase (angiotensinase C): similarity to both serine carboxypeptidase and prolylendopeptidase families. J. Biol. Chem. 1993;268:16631–16638. [PubMed] [Google Scholar]
- 51.Gossrau R., Lojda Z. Study on dipeptidylpeptidase II (DPP II) Histochemistry. 1980;70:53–76. doi: 10.1007/BF00508846. [DOI] [PubMed] [Google Scholar]
- 52.Fersht A. New York: W. H. Freeman and Company; 1999. Structure and Mechanism in Protein Science: a Guide to Enzyme Catalysis and Protein Folding. [Google Scholar]
- 53.Cornish-Bowden A. London: Portland Press Ltd; 1995. Fundamentals of Enzyme Kinetics. [Google Scholar]