

SUMMARY
Ebola virus (EBOV), a major global health concern, causes severe, often fatal EBOV disease (EVD) in humans. Host genetic variation plays a critical role, yet the identity of host susceptibility loci in mammals remains unknown. Using genetic reference populations, we generate an F2 mapping cohort to identify host susceptibility loci that regulate EVD. While disease-resistant mice display minimal pathogenesis, susceptible mice display severe liver pathology consistent with EVD-like disease and transcriptional signatures associated with inflammatory and liver metabolic processes. A significant quantitative trait locus (QTL) for virus RNA load in blood is identified in chromosome (chr)8, and a severe clinical disease and mortality QTL is mapped to chr7, which includes the Trim5 locus. Using knockout mice, we validate the Trim5 locus as one potential driver of liver failure and mortality after infection. The identification of susceptibility loci provides insight into molecular genetic mechanisms regulating EVD progression and severity, potentially informing therapeutics and vaccination strategies.
Graphical abstract
In brief
Schäfer et al. use a genetic screening platform based on the collaborative cross (CC) to identify and elucidate the role of the Trim5 locus during Ebola virus (EBOV) infection and the development of EBOV disease (EVD) in mice.
INTRODUCTION
Infectious diseases exert considerable pressure on the sequence and genetic structure of mammalian genomes, and host genetic variation is known to influence disease susceptibility, progression, and severity.1 Despite this association, the specific genes and natural allele variants underlying the different disease outcomes to viral infections remain largely unknown in mammals, especially for sporadic emerging zoonotic viruses, such as Ebola virus (EBOV).
EBOV, first identified in 1976 in Zaire (today the Democratic Republic of the Congo [DRC]), has repeatedly re-emerged and caused severe human outbreaks of EBOV disease (EVD) in Sub-Saharan Africa.2 The 2013–2016 epidemic in West Africa was the most significant to date as the outbreak emerged in a population-dense area. Human EVD symptoms range from a mild array of non-specific symptoms to severe hemorrhagic multiorgan disease with high mortality, suggesting the potential for interhost genetic control.2 In the aftermath of the West African EVD epidemic, two vaccine modalities were approved by US and European authorities. The vesicular stomatitis virus (VSV)-based and adenovirus/modified vaccinia Ankara-based EBOV vaccines were approved in 2019 and 2020, respectively3–7 and used to protect targeted populations in the DRC against EVD during the largest EVD outbreak there in 2018/2019.8 In parallel, two different antibody therapies (REGN-EB3 and mAb144) have been approved to treat EVD.9–12 Consequently, the impact of recent EVD outbreaks in the DRC have been minimized using antibody therapies and vaccines.13 Despite these advances, further understanding of the host pathways and susceptibility loci that drive differential disease after infection could lead to improved therapeutics and diagnosis of at-risk individuals.
Non-human primates (NHPs) are considered the gold-standard model of EBOV pathogenesis as infection best recapitulates human EVD.14 However, NHPs often come with ethical concerns and are associated with high expense. As more practical animal models were needed, EBOV was mouse adapted (MA-EBOV) by serial passage in progressively older suckling mice, resulting in the MA-EBOV strain.15 Depending on the mouse strain, MA-EBOV infection can lead to a robust pro-inflammatory response, extensive and systemic organ damage, and widespread lymphocyte apoptosis. However, MA-EBOV infection does not reproduce other important hallmarks of EVD, including tissue fibrin deposition and coagulopathy in classic laboratory mouse strains.16
To overcome these challenges, we used the natural variation in the Collaborative Cross (CC), a recombinant inbred mouse genetic reference population, as a platform to identify pathogen susceptibility loci and genes that regulate EVD in rodents.17 The CC platform has successfully identified the conservation of a syntenous locus/genes that are encoded in human and mice, which regulate Sarbecovirus (severe acute respiratory syndrome coronavirus [SARS-CoV] and SARS-CoV-2) pathogenesis.18,19 As natural host genetic variation in the CC has previously been shown to regulate EVD susceptibility,20 we further identified CC recombinant inbred (RI) mouse models that not only replicated the range of human EVD disease phenotypes (e.g., asymptomatic to hemorrhagic disease/death) but provided models to identify polymorphic host loci that potentially regulate disease outcomes across diverse mammalian hosts.
In this study, we generated an intercross mapping population from two CC RI mouse strains and identified a significant susceptibility locus on chromosome 8 associated with viral RNA load in blood. Moreover, a second susceptibility locus was identified on chromosome 7 (chr7) that is significantly associated with EVD-like disease severity, weight loss, and mortality in mice. After narrowing the locus to a region that included the Trim5 paralogous gene cluster (six closely related Trim5 homologs: Trim12a and Trim12c, Trim30a, Trim30b, Trim30c, and Trim30d21–23), we deleted this Trim5 locus in the mouse. After infection, Trim5 locus-deficient mice (Trim−/−) developed less severe EVD-like disease with reduced mortality compared to wild-type littermates. Whole-genome expression and polymorphism analyses of these F2 animals identified the potential function of the wild-type Trim5 locus as a driver of exacerbated inflammatory (“cytokine storm”) and key liver metabolic responses, such as the activation of the nuclear factor κB (NF-κB) pathway, inflammation, bile acid metabolism, and fatty acid metabolism. Disruption of the Trim5 cluster was associated with less clinical disease and liver pathology and increased survival. Improved clinical outcomes were associated with significant reductions in the expression of genes associated with severe liver disease, such as bile acid metabolism, fatty acid metabolism, oxidative phosphorylation, and coagulation pathways. In addition to other genes and susceptibility loci, these experiments highlight a role for the Trim5 locus in EBOV pathogenesis, perhaps by regulating liver failure, while also emphasizing the utility of CC mouse models to decipher complex disease traits in mammals.
RESULTS
CC strains demonstrate different EVD-like disease after MA-EBOV infection
Natural genetic variation between mouse strains influences infection outcomes across an array of pathogens.18–20,24–26 Guided in part by earlier EBOV CC RI-F1 intercross outcomes,20 we characterized the impact of natural host genetic variation on susceptibility to MA-EBOV infection and EVD-like disease in CC RI parent strains. To identify EVD resistant and susceptible strains, we infected groups of female mice from eight different parental CC RI mouse strains (CC001/Unc, CC002/Unc, CC004/TauUnc, CC011/Unc, CC021/Unc, CC051/TauUnc, CC061GeniUnc, and CC074/Unc, hereafter referred to without suffixes) intraperitoneally (i.p.) with 100 focus-forming units (ffu) of MA-EBOV (Figure 1). We identified four CC mouse strains (CC011, CC021, CC051, and CC061) that developed mild to moderate EVD-like disease (Figures 1A and 1B). In addition, we identified four other CC mouse strains (CC01, CC002, CC004, and CC074) that demonstrated high susceptibility to EBOV infection and signs of EVD-like disease, as shown by rapid weight loss (~20% by 6 dpi) and uniform mortality (Figures 1C and 1D).
Figure 1. CC strains demonstrate different EVD-like disease after MA-EBOV infection.
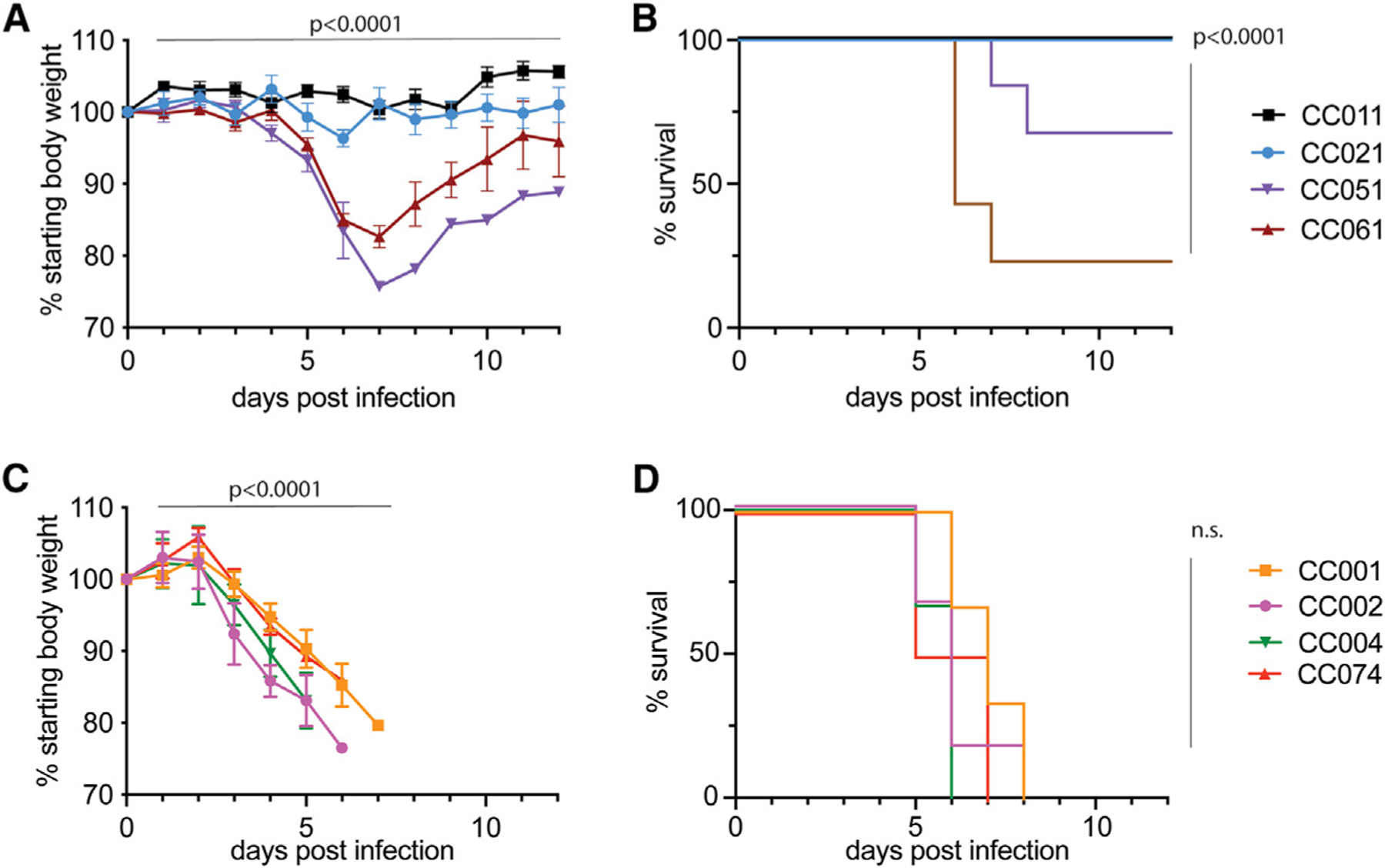
Age-matched female mice (n = 6 per group) of eight genetically diverse CC strains (CC001, CC02, CC004, CC011, CC021, CC051, CC061, and CC074) were infected i.p. with 100 ffu MA-EBOV and monitored daily for weight loss and mortality until day 12 post infection.
(A and B) (A) Weight loss and (B) survival curves for CC011, CC021, CC051, and CC061 mice.
(C and D) (C) Weight loss and (D) survival curves for CC001, CC002, CC004, and CC074 mice. Data were analyzed using Mann-Whitney test (weight loss) and log rank test (survival) and significance is indicated.
We then selected CC011 (resistant) and CC074 (susceptible) as the contrasting parental strains for an F2 mapping population with the goal to identify genetic loci that contribute to EVD-like disease during acute infection. During the generation of a large F2 mapping population, we conducted in-depth phenotyping of MA-EBOV infection in the parental CC011 and CC074 mice. We infected additional mice of both sexes, which were necropsied 6 days post infection (dpi) to investigate changes in weight loss (Figure 2A), survival (Figure 2B), liver EBOV ffu equivalents (Figure 2C), and gross pathology of the liver (Figure S1A).
Figure 2. Characterization of EBOV infection in the two parental strains, CC011 and CC074.
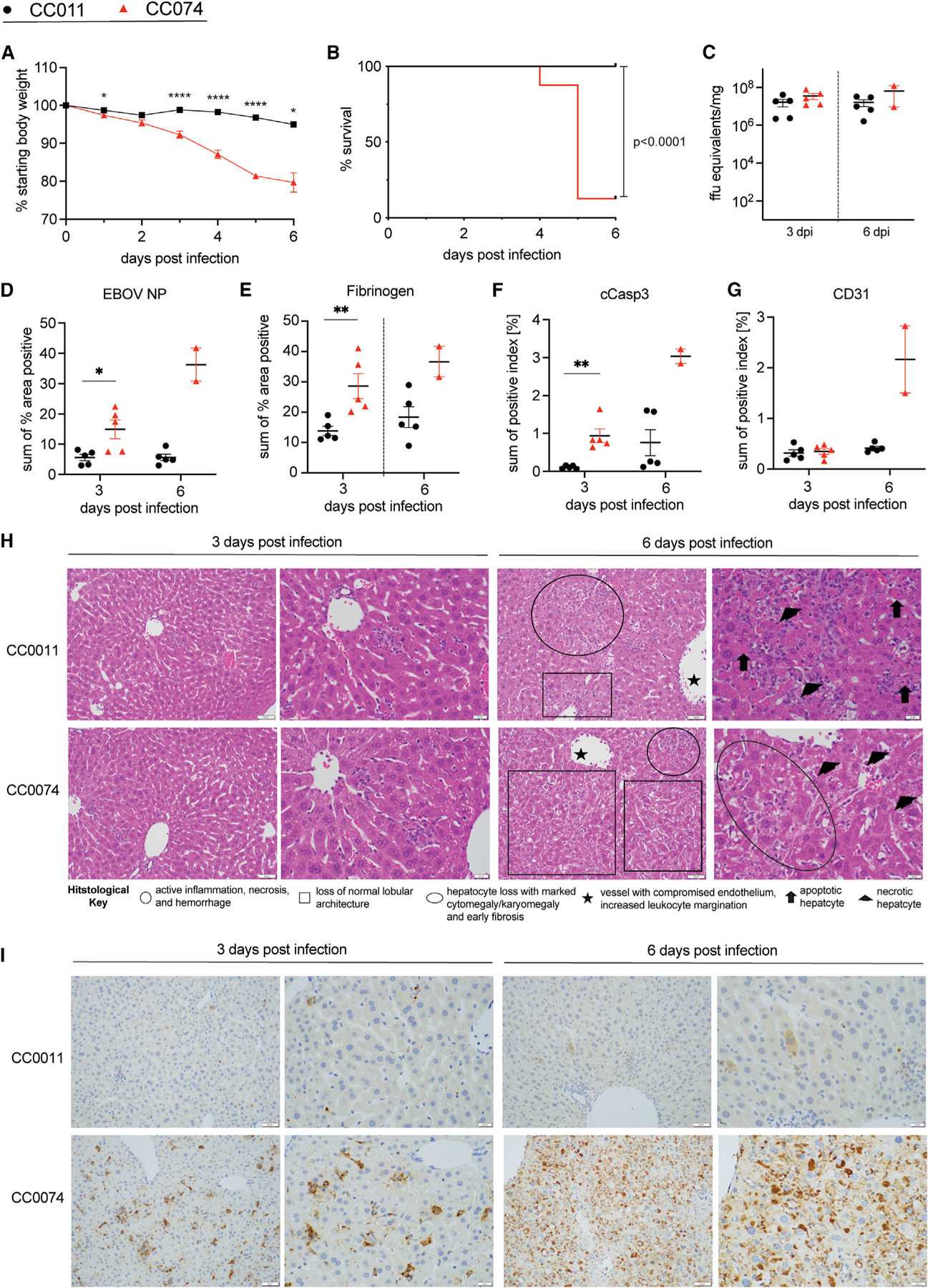
(A and B) Age-matched CC011 and CC074 mice (n = 16 each, 8–10 weeks, both sexes) were infected with 100 ffu MA-EBOV and mice were monitored daily for weight loss (A) and mortality (B).
(C–G) (C) Groups of CC011 and CC074 were used to evaluate the viral load in the liver (n = 5 female for all, except n = 2 males for CC074 on 6 dpi). Liver samples of matched mice from (C) were then used for quantification by IHC staining of (D) EBOV NP, (E) fibrinogen, (F) cleaved caspase 3, and (G) CD31 in the liver.
(H and I) Representative images (20× and 40×, respectively) of the (H) hematoxylin and eosin (H&E)-stained liver sections and (I) EBOV nucleoprotein-immunostained liver sections utilized for the histological assessment of differences between CC011 and CC074 at 3 and 6 dpi; scale bars, 50 μm. Data were analyzed using Mann-Whitney test (weight loss, liver titer, histological quantification) and log rank test (survival). Significance is indicated as *p < 0.05, **p < 0.005, and ****p < 0.0001.
Consistent with earlier assessments, neither male nor female CC011 mice showed clinical signs of disease, showing minimal weight loss and little to no evidence of gross pathology changes in the liver (Figures 1A and 1B; Figures 2A; S1A). In contrast, infection of CC074 mice resulted in signs of EVD-like disease with only a few survivors (~10%, only male mice) (Figures 1C and 1D; Figure 2B) that showed signs of organ damage, such as hepatic discoloration (Figure S1A). Importantly, EBOV replication, as measured by EBOV ffu/equivalents/mg of tissue, was not significantly different in either CC mouse strain on 3 or 6 dpi in the liver (Figure 2C).
However, histologically there was appreciably less immunostaining of the liver for EBOV nucleoprotein (NP) in the resistant CC011 as compared to the susceptible CC074 mice (Figures 2D and 2I; Table S1). Quantification of liver immunostaining identified a statistically significant difference between the two strains at 3 dpi. In both CC strains, Kupffer cells and leukocytes were the predominant cell types stained for EBOV nucleocapsid at 3 dpi. In CC074 at 3 dpi, hepatocytes were also frequently stained, while sporadic signal was noted in endothelial cells (Table S1). At 6 dpi, hepatocytes were the predominant cell type stained for EBOV nucleocapsid in both CC strains, which presented as either fine granular signals in CC011 hepatocytes or large aggregate staining in CC074 hepatocytes. The differences in immunostaining intensity and cellular tropism noted between the strains were similar to previous findings in susceptible and resistant CC F1 RIX mice evaluated using VP40, a viral matrix protein.20
Additional histological assessments for fibrinogen, cleaved caspase 3, and CD31 identified appreciably less immunostaining at 3 dpi as compared to 6 dpi for both CC011 and CC074 (Figures 2E–2G; Figure S1B; Table S1). Furthermore, there was appreciably less fibrinogen and cleaved caspase 3 immunostaining in CC011 as compared to CC074 livers at both 3 and 6 dpi. Quantification of fibrinogen and cleaved caspase 3 immunostaining identified a statistically significant difference between the two CC strains at 3 dpi (Figures 2E–2G). Further analysis of CD31 identified appreciably less signal in CC011 as compared to CC074 levels at 6 dpi but staining was equivalent between the two strains at 3 dpi (Figure 2G). Fibrinogen staining was most often associated with leukocytes (inflammation) and sinusoidal endothelium in both strains. Cleaved caspase 3 staining was most often associated with hepatocytes and leukocytes in nearly equal numbers except for the CC074 at 6 dpi where leukocyte staining predominated. An additional difference between the two CC strains was that cleaved caspase 3 staining of the vascular endothelium occurred less frequently in the CC011, as compared to CC074, livers on both 3 and 6 dpi. CD31 immunostaining was identified as excessive in the sinusoids of both CC strains at 6 dpi (Figure S1B).
Histological evaluation of hematoxylin and eosin (H&E)-stained liver sections identified several pathologic changes present in both CC strains to include neutrophilic inflammation, hepatocyte necrosis and apoptosis, mild hemorrhage, increased cytomegalic/karyomegalic cells, increased multinucleated cells, loss of lobular architecture, loss of vascular integrity, and increased leukocyte margination on the vascular endothelium (Figures 2H; Table S1). The pathologic changes were less severe at 3 dpi as compared to 6 dpi for both strains. Likewise, the pathologic changes were less severe in CC011, as compared to CC074, livers at both 3 and 6 dpi. In addition to having increased levels of inflammation at 6 dpi as compared to 3 dpi, the inflammation identified at 6 dpi for CC both strains also had increased numbers of histiocytic cells and often extended into the portal areas.
In summary, CC074 livers exhibited more pathologic changes as compared to CC011 livers, which were most apparent at 6 dpi. These changes included increased inflammation, hepatocellular necrosis and apoptosis, hepatocyte dysplasia (increased cytomegaly/karyomegaly and multinucleated cells), fibrinogen deposition, hemorrhage, and vascular leukocyte margination. The changes likewise included a loss of normal lobular architecture and diminished vessel integrity.
Disease phenotypes and identification of a severe EVD-like disease QTL on chr7
Next, we inoculated 6- to 8-week-old F2 mice (n = 236; 123 females and 113 males) i.p. with 100 ffu of MA-EBOV and monitored for weight loss and mortality. On 6 dpi, surviving mice were necropsied for determination of viral burden, differential gene expression, and histopathologic analysis. In this mapping population, there was a range of EVD-like disease with mice losing up to ~20% of their starting weight by 6 dpi (body weight changes at 6 dpi ranging from 82% to 105% of starting weight) across both male and female mice (Figure 3A; Figures S2A and S2B). The clinical disease spectrum expanded the range of clinical disease phenotypes observed with the two parental mouse strains (Figures 2A and 2B). Over the course of the infection, we also documented an overall 39% mortality in the F2 mice, starting on 4 dpi, with the highest number of mice succumbing on 6 dpi (Figure 3B). For all surviving F2 mice, viral replication, as measured by EBOV ffu equivalents/mg, was detected in the liver at 6 dpi by RT-qPCR (Figure S2C). Although EBOV RNA loads demonstrated an expanded range of titers as compared with the parent strains (Figure 2C), there was no strong correlation between ffu equivalents/mg in the liver and the weight loss in F2 mice (R2 = 0.037), further strengthening the observation that the disease phenotypes were unlikely due to degree of genome replication (Figure S2D).
Figure 3. Disease phenotypes and identification of a severe EVD-like disease QTL on chr7.
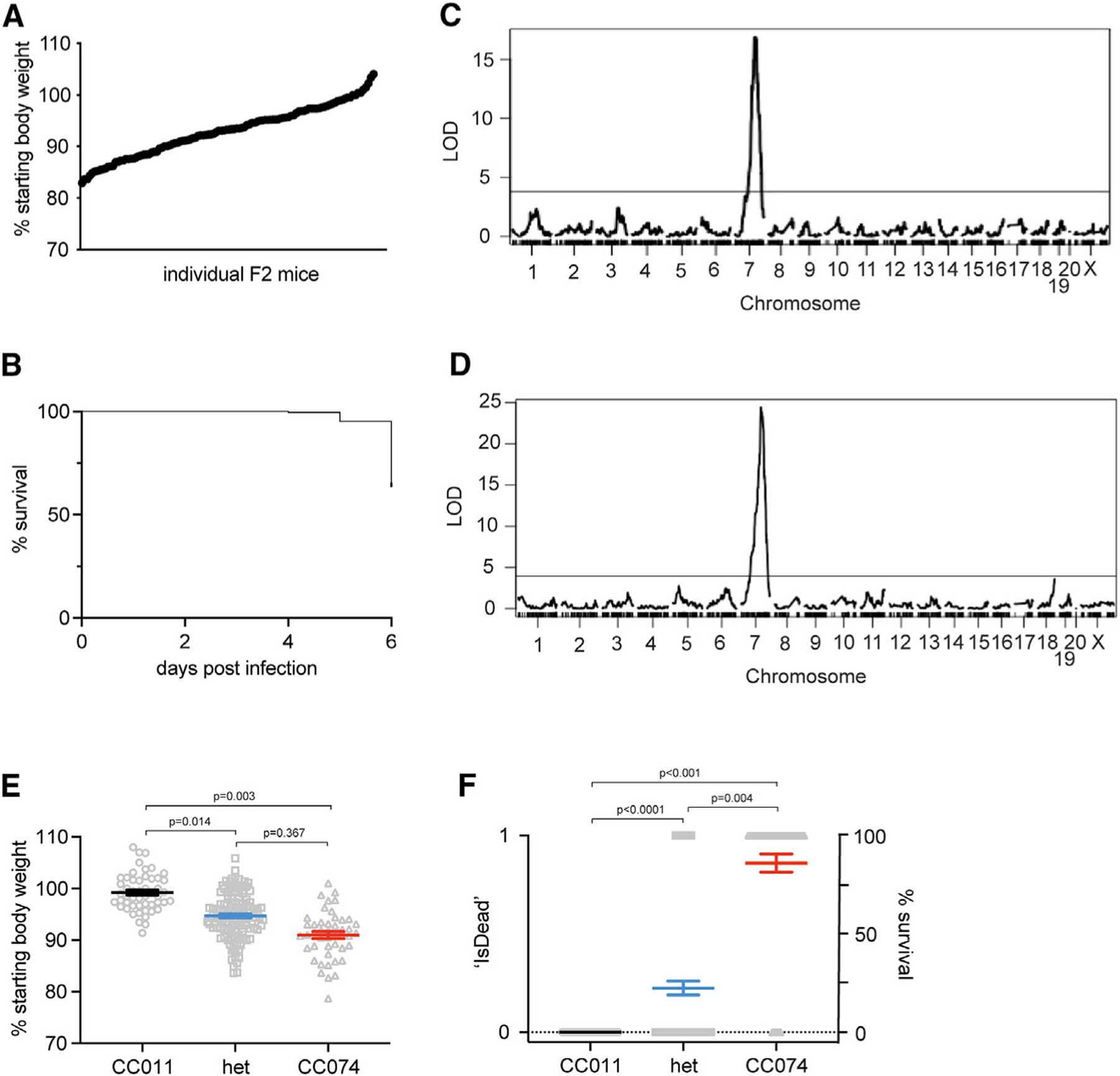
Eight- to 10-week-old CC011xCC074-F2 mice (n = 236; 123 females, 113 males) were generated and infected with 100 ffu MA-EBOV i.p. and followed for 6 days for weight loss and mortality.
(A) Weight loss on day 5 post infection of CC011xCC074-F2 mice.
(B) Survival of CC011xCC074-F2 mice over the time of infection.
(C) QTL map for the weight loss on day 5 post infection.
(D) QTL map for the overall survival rate over the time of infection. Phenotypes were grouped based on a homozygous CC011 genotype, a heterozygous genotype, and a homozygous CC074 genotype. (E) The allele plot for weight loss on 5 dpi, and (F) the allele plot for percentage survival over the time of infection; 0 = alive, 1 = dead. Data were analyzed using Mann-Whitney test (allele plots for weight loss and survival). LOD, log10 likelihood ratio; dotted line indicates the genome-wide p = 0.05 significance threshold.
Concurrent with the viral challenge, the F2 mice were genotyped with the MiniMUGA array. This genetic and the above-described phenotypic data were used for quantitative trait locus (QTL) mapping. We identified a single, highly significant (genome-wide p <0.001) genetic locus (QTL for EBOV susceptibility 1 [QES1]) on chr7 that was associated with body weight loss at 5 dpi and cumulative mortality at 6 dpi (Figures 3C and 3D). The confidence interval for the QTL (QES1) is located between 100 and 109 Mb on chr7 (peak marker position ~104.5 Mb). The resistant CC011 allele arose from both, A/J (proximal region until ~105 Mb) and WSB/EiJ (distal region), and this contrasted with the susceptible CC074 loci, which encoded a C57BL/6J haplotype (Figures 3E and 3F). As such, QES1 represented the major locus that drove differential EVD-like disease outcomes between CC011 and CC074 mice.
All infected CC011xCC074-F2 mice demonstrated high variation in the numbers of ffu RNA equivalents in blood (e.g., ~104 to >109 ffu equivalents/mL blood) (Figure S3A). Using these values, QTL mapping identified a significant QTL for ffu equivalents in the blood on chr8 (QES2) (Figure S3B; Table S2). The region spans from 115 to 129 Mb, with the resistant allele arising from 129/Sj and the susceptible allele either from PWK (~115–117 Mb) and NOD (~117–129 Mb). Again, no significant correlation was noted between the ffu equivalents in the blood and overall detectable weight loss (R2 = 0.04) (Figure S3C). In contrast, no significant QTL for ffu equivalents in the liver were detected (Figures S3D and S3E).
Validation of the Trim5 locus during MA-EBOV infection
QES1, regulating weight loss and mortality, is located on chr7 between 100 and 109 Mb, has high gene density (310 total annotated genes), including 192 olfactory receptor genes (a very large gene family in the mouse27), as well as the murine Trim5 locus and many other genes. Given this gene density, we applied a strict filtering of high-priority candidate variants (e.g., missense, frameshifts, and InDels affecting coding regions) that could be driving the different EVD-like disease outcomes between these strains. Based on whole-genome sequencing of the CC founder strains, we identified 418 missense mutations but no insertions or deletions in this region.28,29 Of genes containing a missense mutation, we found that many were members of the Trim gene family (Trim6, Trim12, Trim22, Trim30, and Trim34, Trim12a, Trim12c, Trim30a, Trim30b, Trim30c, and Trim30d) (Figure 4A; Table S2). In particular, half of those genes with missense mutations (e.g., Trim12a, 12c, 30a, 30b, 30c, and 30d) comprised the so-called Trim5 locus in mice, located at the peak of QES121 (Figure 4B). Comparing human and mouse Trim5 genes, Trim12c is considered the closest full-length mouse ortholog to human Trim5, based on amino acid sequence, function, and protein domain homology (Figure 4A).21,22 Importantly, a single missense SNP (H124Q) in Trim12c segregates between CC011 and CC074. This amino acid is part of a zinc-finger motif, and the substitution is predicted to have a damaging effect on the proper protein structure and, therefore, the adequate function of the protein (Figure S4A).30,31 Previous studies have shown that TRIM proteins are major regulators of the innate immune, inflammatory responses, and in some cases liver function.32–36
Figure 4. Characterization of EBOV infection in Trim+/+ and Trim−/− mice.
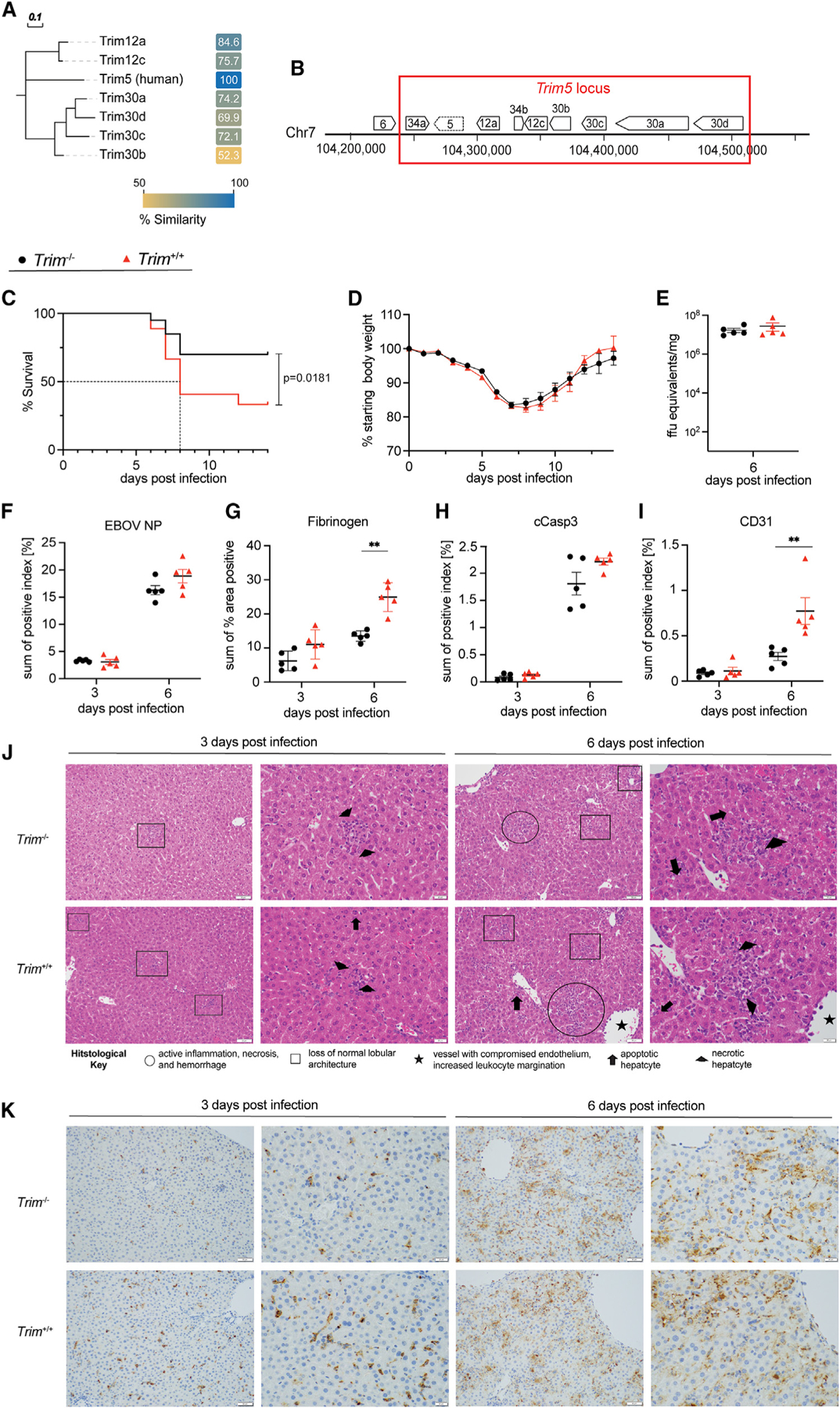
(A) A neighbor-joining phylogenetic tree was constructed from a multiple sequence alignment of the human TRIM5 protein and mouse orthologs (TRIM12a, TRIM12c, TRIM30a, TRIM30b, TRIM30c, and TRIM30d). Similarities (based on the Blosum62 similarity matrix) are included as a heatmap with similarity ranging from 50% (gold) to 100% (blue). The multiple sequence alignment was performed, the neighbor-joining tree was constructed in Geneious Prime, and the tree and distances were output as Newick and CSV files, respectively. The tree and heatmap were then rendered for publication in EvolView (https://www.evolgenius.info) and Adobe Illustrator CS.
(B) The Trim5 locus in wild-type C57BL/6J mice (indicated by red box). For validation of the Trim5 locus as a susceptibility region during MA-EBOV infection, age-matched Trim−/− (n = 20) and Trim+/+ littermates (n = 27) (all 8–10 weeks old, both sexes) were infected i.p. with 100 ffu MA-EBOV.
(C) Survival; dashed line indicates median survival.
(D–I) (D) Weight loss. A group of Trim+/+ and Trim−/− (n = 5 females for all at both time points) was then used to evaluate the (E) viral load in the liver (6 dpi only) and for quantification of (F) EBOV NP in the liver, (G) fibrinogen, (H) cleaved caspase 3, and (I) CD31 of Trim+/+ and Trim−/− mice by IHC staining.
(J and K) Representative images (20× and 40×, respectively) of the (J) H&E-stained liver sections and (K) EBOV nucleoprotein-immunostained liver sections utilized for the histological assessment of differences between Trim+/+ and Trim−/− mice at 3 and 6 dpi; scale bars, 50 μm. Circles are areas of active inflammation, necrosis, and hemorrhage. Squares are areas of active inflammation and necrosis. Stars denote vessels with compromised endothelium and increased leukocyte adherence/margination. Arrows denote apoptotic hepatocytes. Arrowheads denote necrotic hepatocytes. Data were analyzed using Mann-Whitney test (weight loss, liver titer, histological quantification) and log rank testing (survival). Significance is indicated as **p < 0.005.
To study the effect of the mapped Trim5 locus on EVD-like disease in mice, we used CRISPR-Cas9 genome editing to delete the entire Trim5 locus on the C57BL/6J genetic mouse background (Trim−/−; Figures 4B; S4B). Importantly, the Trim−/− mice were viable and robust, and genetic mapping studies revealed the appropriate deletion of the Trim5 locus (ΔTrim12a, 12c, 30a, 30b, 30c, and 30d) (Figures S4B–S4D). Next, we performed survival and weight-loss studies Trim−/− mice relative to wild-type littermates (Trim+/+) of both sexes to validate this locus as a susceptibility region for EVD-like disease after MA-EBOV infection. By 14 dpi, infected Trim−/− mice demonstrated an overall mortality of 25%, while wild-type littermates (Trim+/+) had a significantly higher mortality of 63% (log rank p <0.02) putting wild-type mice at a 2.63× (95% confidence interval [CI], 1.18–5.867) higher risk of death by MA-EBOV infection than Trim−/− mice (Figure 4C). Mice in these survival studies were also monitored daily for body weight changes, but we observed no significant differences in body weight between the Trim−/− mice and their wild-type littermates (Figure 4D).
Similar to the body weight findings, no differences were detected between the Trim−/− mice and their wild-type (Trim+/+) littermates with regard to viral genome equivalents in the liver on 6 dpi (Figure 4E). This observation was supported through histological assessments of the liver. Specifically, there were no readily appreciable differences in the staining intensity or cellular tropism of EBOV nucleocapsid immunostaining in the livers of the two Trim strains at 3 or 6 dpi (Figures 4F and 4K; Table S1). These findings contrast with what had previously been identified in the parental CC strains and suggest that loci other than the Trim5 locus likely regulate the viral tropism and viral protein expression as noted in the parental lines (Figures 2D and 2I).
Histological evaluation of H&E-stained liver sections identified the same types of pathologic changes in the Trim−/− and Trim+/+ mice as were previously described for the parental CC strains (Figures 4J; Table S1). These pathologic changes were less severe at 3 dpi as compared to 6 dpi for both Trim strains. Some pathologic changes (i.e., inflammation, hepatocellular necrosis and apoptosis) were identified to be slightly more severe in Trim+/+, as compared to Trim−/−, livers at 3 dpi, although no appreciable differences in pathology were identified between the Trim strains at 6 dpi (Figures 4J; 4S4G).
In addition to the pathologic changes identified through H&E histopathology, there were some immunostaining findings that differed between the two strains (Figures 4G–4I; Figure S4G; Table S1). For fibrinogen, there was appreciably less immunostaining for Trim−/− as compared to Trim+/+ livers at both 3 and 6 dpi (Figure 4G). For cleaved caspase 3 and CD31, there was less immunostaining for Trim−/− as compared to Trim+/+ livers at 6 dpi but not at 3 dpi (Figures 4H and 4I). Quantification of fibrinogen and CD31 immunostaining also identified a statistically significant difference between the two Trim strains at 6 dpi. Notably, these differences are primarily due to variations in staining intensity between the Trim−/− and Trim+/+ livers as the cellular tropism for fibrinogen, cleaved caspase 3, and CD31 immunostaining appear to be similar between the Trim strains at both 3 and 6 dpi. Specifically, for both Trim strains, fibrinogen staining was associated with leukocytes and sinusoids, cleaved caspase 3 staining was associated with hepatocytes and leukocytes at 3 dpi and included some vascular endothelium staining at 6 dpi, and CD31 staining was identified as excessive in the sinusoids at 6 dpi.
In summary, Trim+/+ livers exhibit more pathologic changes as compared to Trim−/− livers. These changes include increased inflammation, hepatocellular necrosis and apoptosis, fibrinogen deposition, hemorrhage, and diminished vessel integrity and are independent of viral replication as measured by viral genome equivalents and EBOV nucleoprotein immunostaining.
Altered inflammatory, cytokine, and liver metabolic signatures regulate severe EVD-like disease in the CC parents and CC011xCC074-F2 mice
To more precisely define host response patterns in the two parental strains, CC011 and CC074, after EBOV infection, we performed RNA sequencing (RNA-seq) on liver tissue at 3 and 6 dpi (Figure 5; Table S3). Pathway enrichment analysis on 3 dpi identified a significant upregulation of inflammatory responses, tumor necrosis factor alpha (TNFα), signaling, interleukin (IL)-6 pathway, and interferon pathways in both parental strains (Figures 5A; Table S4). The overall response was increased in the susceptible CC074 mice as compared with the resistant CC011 mice. Moreover, liver-associated metabolic pathways were strongly downregulated in CC074 mice (Figures 5A; Table S4). By 6 dpi, overall transcription signatures shift to liver disease/failure and breakdown of liver metabolic processes, such as fatty acid and bile acid metabolism, adipogenesis pathways in CC074-infected mice, which are significantly decreased in comparison to CC011 (Figures 5B; Table S4).
Figure 5. Differentially expressed gene signatures in the liver in the two parental strains, CC011 and CC074.
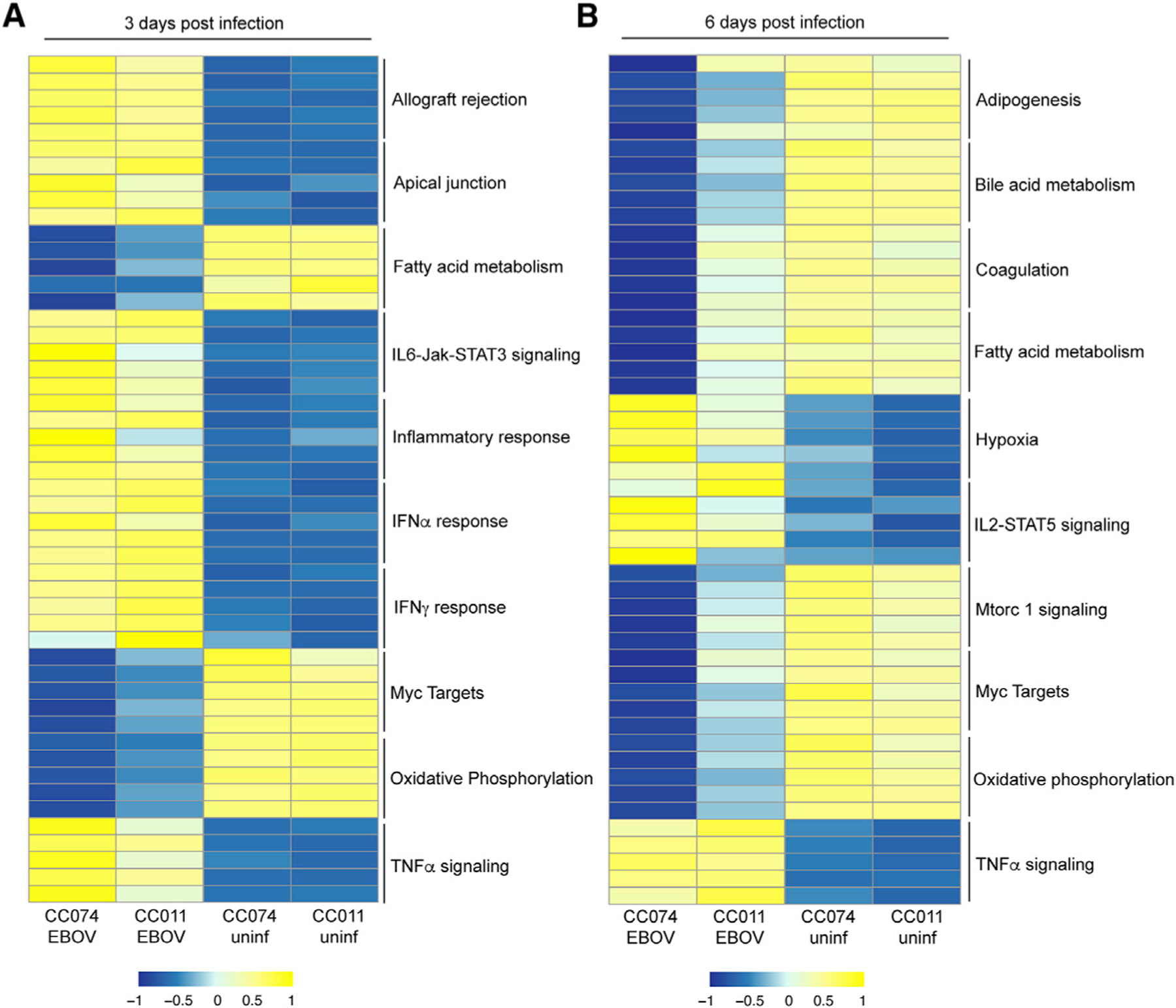
Heatmap of the top five ranked genes in the top 10 significantly enriched pathways in infected CC011 and CC074 mice compared to their respective mock-infected controls (<1.5 log2 fold over mock; p < 0.05); shown are the average expression level for each gene for each group on (A) 3 dpi and (B) 6 dpi, (n = 5 EBOV-infected females for all time points, except for n = 2 EBOV-infected males for CC074 on 6dpi, n = 3 mock-infected females for all time points).
For studying the transcriptome in the CC011xCC074-F2 mapping cross, CC011xCC074-F2 mice were stratified based on their body weight (weight loss at 6 dpi), and 15 mice at each of the extreme ends of bodyweight loss animals were selected for RNA-seq analysis (Figure S5; Table S3). Mice in the low-weight-loss (LWL) group had no to moderate bodyweight loss, while mice in the high-weight-loss (HWL) group lost significant amounts of their bodyweight over the course of infection. Nine mock-infected F2 mice (UI) were used as controls to identify infection-related responses (Figure S5A). The HWL mice not only showed slightly higher levels of viral load in the liver compared to the group LWL mice (Figure S5B) but also had more severe gross pathological changes in the liver (Figure S5C). Upregulated genes in the HWL mice were associated with activation of inflammatory response, TNFα signaling, apoptosis, and loss of vasculature integrity pathways. On the other hand, transcriptomic signatures for liver metabolic pathways, such as bile acid metabolic processes, were strongly downregulated, indicative of acute liver injury (Figure S5D; Table S5).
The Trim5 locus and liver failure
To further define the role of the Trim5 locus in EBOV infection and disease, we performed RNA-seq on the liver of Trim+/+ and Trim−/− mice on 3 and 6 dpi (n = 5 for each genetic background for each dpi, and n = 3 mock-infected mice). As previously reported, no significant difference in weight loss or viral load in the liver was detectable between these groups (Figures 4E; S4E). By 3 dpi, Trim5 locus gene expression was only evident in the Trim+/+, but not Trim−/−, mice, reflecting the deletion of this locus in the Trim−/− mice (Figure S4D).
Transcriptional expression analyses for 3 dpi only revealed significant expression changes of genes within the deleted Trim5 locus and no significant enrichment of specific pathways was reached (Table S3). Pathway enrichment analysis on 6 dpi, however, demonstrated the overall downregulation of genes belonging to key liver metabolic pathways. However, several of these pathways were more significantly downregulated in the Trim+/+ mice as compared to Trim−/− mice. Importantly, altered expression levels were noted in several crucial liver-associated metabolic pathways, including fatty acid, bile acid, adipogenesis, and xenobiotic metabolism in Trim+/+ mice. Additionally, genes responsible for coagulation regulation, a hallmark of EVD, were also significantly downregulated in Trim+/+ in comparison to Trim−/− mice (Figure 6A; Table S3; Table S6). In contrast genes involved in DNA repair, myc targets, and protein secretion were significantly upregulated in Trim−/− in comparison to Trim+/+ mice (Figures 6A; Table S6).
Figure 6. Differentially expressed gene signatures in the liver in Trim+/+ and Trim−/− mice.
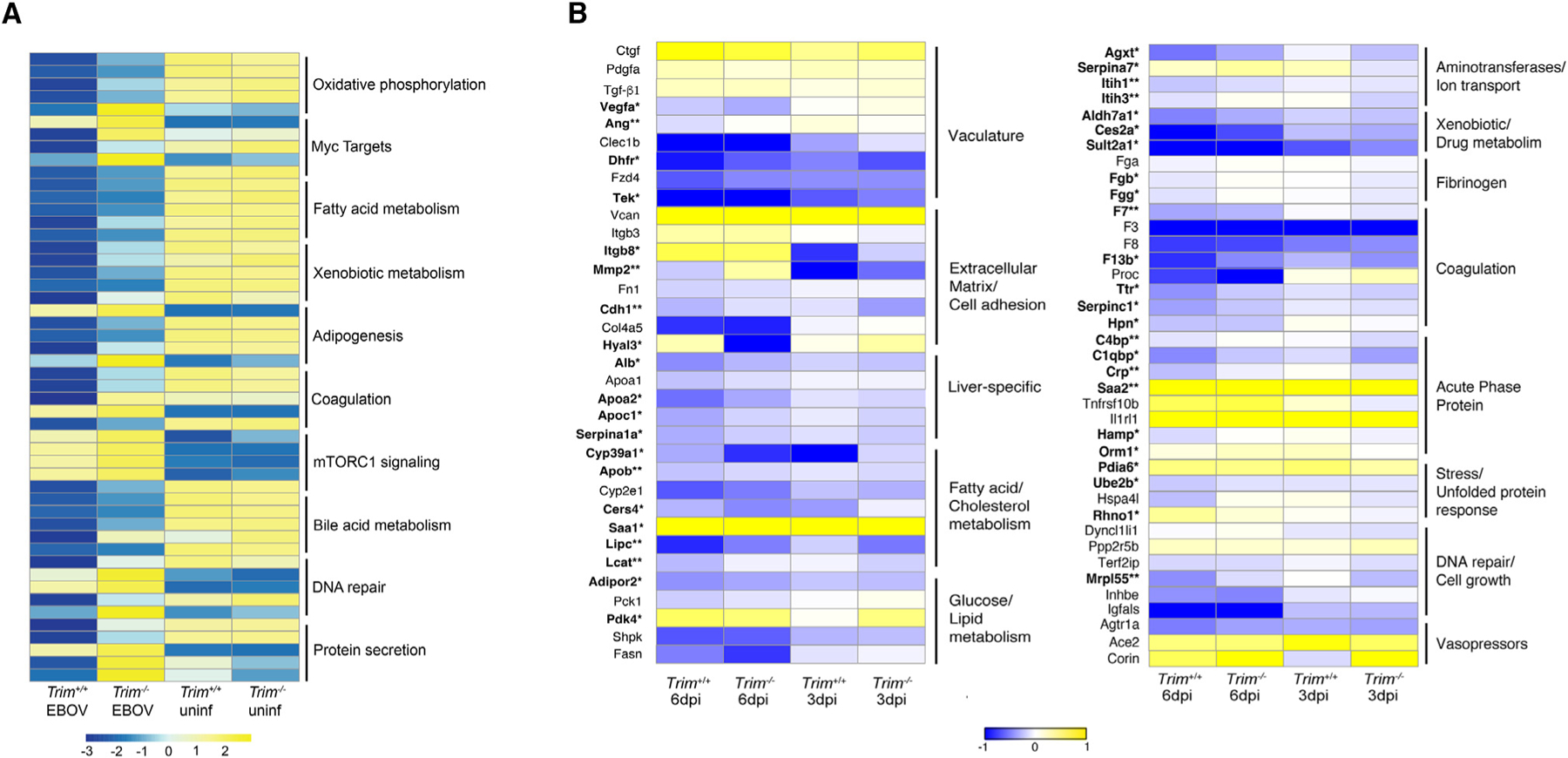
(A) Heatmap of the top five ranked genes in the top 10 significantly enriched pathways in infected Trim+/+ and Trim−/− mice compared to their respective mock-infected controls (<1.5 log2 fold over mock; p < 0.05); shown are the average expression levels for each gene for each group on 6 dpi (n = 5 females for all time points, n = 3 females for all time points for mocks).
(B) Heatmap of key genes being significantly affected during the development of EVD-like disease in the liver (in bold). For each gene, the average normalized transcript counts (log2 cpm.) are shown. Data were analyzed using Mann-Whitney test; significance is indicated as p* < 0.05, **p < 0.005 (n = 5 EBOV-infected females for all time points, n = 3 mock-infected females for all time points).
We next compared the changes in gene expression in key signature genes, which have been highlighted in EBOV-infected human liver organoids, several human liver cells lines, and in the liver of NHPs (Figures 6B; Table S7).20,37–39 Importantly, coordinated expression patterns were noted in Trim+/+ mice at 6 dpi, in the vasculature (Tek, Ang, Vegfa, Dhfr), extracellular matrix (Igtb8, Mmp2, Cdh1, Hyal3), fatty acid/cholesterol metabolism (Cyp39a1, Apob, Cers4, Saa1, Lipc, Lcat), glucose/lipid metabolism (Adipor2, Pdk4), aminotransferases/ion transport (Agxt, Serpina7, Itih1, Itih3), xenobiotic/drug metabolism (Aldh7a1, Ces2a, Sult2a1), other liver-specific genes (Alb, Apoa2, Apoc1, Serpina1a), fibrinogen (Fgb, Fgg), coagulation (F7, F13b, Ttr, Hpn, Serpinc1), acute phase proteins (C4bp, C1qbp, Crp, Saa2, Hamp, Orm1), stress/unfolded protein response (Pdia6, Ube2b, Rhno1), and DNA repair/cell growth (Mrpl55) (Figures 6B; Table S7).
DISCUSSION
The development of small rodent models for EBOV infection has been limited by restricted wild-type virus replication and the failure of mice to develop hallmarks of EVD.40,41 The current study, building on earlier work,20 showed that MA-EBOV-infected CC RI mice develop a spectrum of EVD-like disease syndromes ranging from mild asymptomatic infections (e.g., CC011) to a recapitulation of several hallmarks of EVD, including upregulation of inflammatory cytokines and chemokines, coagulopathy, and gross pathological damage in the liver that likely progresses to liver failure (CC074). The CC011 and CC074 infections are characterized by similar viral RNA loads at 3 and 6 dpi in the liver, yet paradoxically show dramatic alterations in viral nucleoprotein expression, tissue tropism, and pathology, including fibrinogen deposition, apoptotic cell death (cleaved caspase 3), and endothelial cell permeability (CD31). Real-time RT-qPCR is a common strategy to quantify EBOV RNA loads in humans and in animal models.42–45 Paradoxically, and despite similar virus RNA loads at 3 and 6 dpi, the tropism and intensity of EBOV NP staining is dramatic and extensive in CC074 but scattered and weak in CC011. Similar findings were previously reported in some CC F1 RIX mice, including altered tropism and significant reductions in live virus titers and genome equivalents.20
Despite this observation, QES2 was mapped in chr8 that regulated viral RNA loads in the blood, but not liver, and the phenotype appeared uncoupled from the Trim+/+ and Trim−/− locus in C57BL/6 mice (QES1). These data suggest a potential disconnect between RNA and viral protein production, an accelerated degradation of viral protein, or a possible defect in EBOV egress.46–48 Based on CRISPR-Cas9 screens and other empirical studies, a variety of host genes assist in EBOV egress, including several vacuolar protein sorting-associated proteins.49–52 Although speculative, chr8 QTL contains the Chmp1a gene, which is a component in the endosomal sorting complex required for transport III (ESCRT III).53 EBOV and other enveloped viruses use the ESCRT pathway during budding and release of progeny virions, providing a potential candidate gene.47,49,54,55 Consequently, the molecular mechanisms regulating this phenotype in select CC lines remains an important question that will be addressed in future studies.
As mentioned above, the study performed by Rasmussen et al. observed a similar disconnect between viral RNA and viral protein in the susceptible CC line.20 However, several human studies used viral load as a measure of disease severity, reaffirming the importance of this metric to disease severity.56–60
Human EBOV outbreaks are sporadic and typically of insufficient size to provide opportunity for robust genome-wide association studies. CC RI mice, CC F1 RIX, or lines genetically modified at defined susceptibility loci provide for effective, cost-effective models for identifying candidate host susceptibility genes and studying virus-host interactions that regulate disease. The development of more authentic disease platforms for EBOV countermeasure development/testing are needed prior to expensive NHP experimentation.20,61
To better understand the genetic loci that regulate EVD and host susceptibility, we generated an F2 mapping population. We demonstrated that a highly significant QTL on chr7, QES1 (100–109 Mb), was associated with disease severity and mortality following MA-EBOV infection in these mice, while a second QTL on chr8, QES2 (115–129 Mb), was associated with genome equivalents in the blood. Focusing on genes near the QTL peak, we used sequence information, SNP comparisons, host expression signatures, and protein function predictions to identify the Trim5 gene cluster as a highly plausible candidate. These findings were further supported by the enrichment of highly expressed genes involved in NF-κB-activated pathways and the inflammatory response in highly susceptible F2 mice, as well as the role of TRIM5 in development of liver damage during HCV infection.62 The Trim5 cluster in the mouse encodes several paralogues, including Trim12a, Trim12c, Trim30a, Trim30b, Trim30c, and Trim30d, interspersed by Trim34a and Trim34b.21,22 Trim proteins are important players in the innate immune response, and human TRIM5 has been shown to be a strong activator of inflammatory responses.21,63,64 In humans, the Trim5 gene also regulates retrovirus cross-species transmission and functions as an innate immune sensor. Natural genetic variants in the Trim5 gene are associated with higher plasma levels of inflammation biomarkers and HCV clearance after antiviral therapy in HIV/HCV coinfected patients.62 In conjunction with the Trim34 gene, TRIM5 restricts HIV-1 uncoating by binding capsids and directing them into degradation machinery, while simultaneously enhancing recognition of infected cells by CD8 T cells.64–66 After mosquito-borne flavivirus infections, TRIM5a suppresses replication by binding to the viral protease NS2B/3 to promote ubiquitination and proteasomal degradation.36
In contrast to these earlier studies, we provide evidence for an alternative pathway for Trim5 regulation of disease severity and mortality. Although our Trim−/− mice lack the murine Trim5, Trim34, and other paralogues, viral genome equivalents and protein expression in the liver by real-time PCR and immunohistochemistry (IHC) are similar in vivo, suggesting that there is disconnect between viral load and weight loss/severity of disease and that other mechanisms might mediate altered mortality caused by MA-EBOV infection. These data are consistent with earlier findings that have shown that Trim6 ubiquitinates VP35 to promote EBOV replication.35 However, recent studies have also demonstrated that Trim12c is a strong activator of the NF-κB, inflammatory, and interferon pathways22 and that Trim34 expression prevents colon inflammation and functions in influenza A virus-activated programmed cell death.67,68 In support, several studies have shown that NF-κB activation and the inflammatory response in NHPs and in humans infected with EBOV are a critical indicator of severe EVD and death.69–73
Our liver transcriptome analysis of highly susceptible parental CC074 mice on 3 dpi and in susceptible F2 mice resulted in very similar expression and disease profiles previously reported from NHP studies, which showed a strong early activation of inflammatory response genes and a downregulation of liver metabolism genes.38 This study also demonstrated a strong upregulation of inflammatory, apoptotic, and innate immune responses in the liver, yet there were no significant differences in the virus tropism and early gene expression in either Trim+/+ or Trim−/− animals at these time points.
Rather, and as seen in susceptible CC074 parents at 6 dpi and in the highly susceptible F2, but less so in CC011 and resistant F2 cohorts, we detected the downregulation of several crucial liver-specific metabolic pathways, such as bile acid and fatty acid metabolism, cholesterol homeostasis, coagulation, fibrosis, and adipogenesis, in the susceptible Trim+/+ mice, suggesting the development of significant liver damage, potentially leading to liver failure.37,38,74 Supporting the dysregulation of liver metabolic pathways, oxidate phosphorylation and xenobiotic metabolic response pathways were also significantly downregulated in Trim+/+ but less so in Trim−/− mice, pathways that have been shown to be crucial for the liver function during oxidative stress responses and detoxification.38,75,76 Consistent with more severe gross liver pathology noted in the Trim+/+ mice, the expression of coagulation genes, a well-established clinical indicator of severe EBOV infection and the development of EVD, were significantly downregulated.37,38,77
Earlier studies have attributed a role in the development of liver disease, such as fibrosis, fatty liver, and cirrhosis, to Trim genes and, in conjunction with TRAF6 and TGF-b, Trim genes may promote liver fibrosis.78 Taken together, these data suggest that the imbalance and dysregulation of genes within pathways of key liver functions seen in the Trim+/+ mice interferes with the normal physiological function of the liver and so clearly demonstrate the crucial contribution of the liver in the severity of MA-EBOV infection and EVD-like disease development. Wild-type Trim5 (Trim+/+) clearly affects crucial metabolic liver pathways by downregulating the expression of key genes in several pathways, thereby causing the imbalance and dysfunction of the liver during EBOV infection.
Trim genes are highly polymorphic, evolving rapidly and encoding a variety of functional domains such as Ring domain, B-box domain, coiled domain, and some for a PRY-SPRY domain.79,80 As an important species-specific restriction factor for retroviruses, flaviviruses, and other virus pathogens, TRIM5 has likely been under sustained evolutionary selection pressure and encodes numerous allele variants.1 In rodents, the wild-type Trim5 cluster appears to drive severe liver failure. PolyPhen II predicts that a polymorphism in the B-box domain ablates Trim12c function preventing the protein to homodimerize and thus to inhibit its K63-ubiquitiylation function and attenuate liver disease during MA-EBOV infection.30 Taken together, we present strong support for the hypothesis that the Trim5 cluster participates in regulating EVD-like disease severity and mortality in infected mice. However, given the significance of QES1, and the fact that the Trim−/− mice do not uniformly replicate the clinical disease phenotypes seen in CC074 and vulnerable F2 mice (e.g., virus tropism, weight loss), these data suggest that other genes under this QTL likely contribute to disease severity as well. Such hypotheses are not unprecedented as syntenic regions that encode multiple genes reside on Chr3 in humans and Chr9 in mice, and at least two of the genes in this locus contribute to SARS-CoV-2 pathogenesis.18,19 Our data argue that the Trim5 gene cluster plays a significant role in liver disease, supporting the hypothesis that severe liver disease in humans may also be Trim5 cluster related. Future studies will need to investigate the precise molecular mechanism(s) by which Trim5 alters crucial liver metabolic pathways leading to liver dysfunction during EBOV infection and to identify the QTL and gene(s) that regulate virus tropism and viral protein expression differences in the liver of CC074 and CC011.
Zoonosis events often result in limited access to and/or numbers of human cases, hampering genome-wide association studies (GWASs). Our study highlights the power of genetic reference populations to map and elucidate the role of complex genetic traits and genetic variation on infectious diseases. The availability of contrasting mouse models of EVD-like disease severity that reflect human outcomes provides a mechanistic platform to identify novel gene candidates that regulate virus replication, tropism, and disease. In appropriately selected large population screens, highly penetrant genetic variants can be identified easily, as can their impacts on specific aspects of disease outcome. Moreover, targeted crosses between highly discordant strains can help to map and identify more complex genetic interaction networks, including emergent traits that are penetrant only in the context of specific genetic backgrounds, or epistatic interaction networks. Here, we identify two susceptibility loci that regulate filovirus infection and disease in mammals. While these studies implicated the Trim5 locus as an important candidate, causality will require the establishment of an allelic series by the introgression of targeted susceptibility alleles from the CC074 Trim5 locus into the resistant CC0011 genetic background by CRISPR-Cas9 gene editing. Unfortunately, the number of paralogous genes in the Trim5 cluster, coupled with highly conserved sequences, will complicate such approaches. Importantly, this approach will provide candidate genes and expression networks that complement the analysis of human datasets and provide opportunities for identifying common and unique loci that regulate EVD across mammals while narrowing the numbers of candidate genes under the QTL interval for downstream studies. While highlighting the broader utility of understanding the role of natural genetic variation in disease, these models should improve insight into the virus-host genetic and pathogenic mechanisms of EVD and provide new screening platforms for countermeasure development.
Limitations of the study
The gold-standard model for filovirus pathogenesis studies is performed in NHPs, which best recapitulate human EBOV disease phenotypes but are too expensive for complex trait mapping studies. While standard laboratory mice do not fully recapitulate the clinical disease spectrum seen in EBOV-infected human cases, MA-EBOV in the CC genetic reference population has been reported to closely approximate the human condition, strengthening the use of this resource.20 While our data support this earlier hypothesis, the use of two extreme-phenotype CC parental strains for F2 mapping studies does focus genetic loci mapping studies on a fraction of the genetic diversity that is encoded within the entire CC population. Consequently, important EBOV disease-driving loci may not be represented in this cross because genetic variation is lacking. Although a major disease-driving susceptibility loci was identified on chr7, validation with Trim5 locus knockout (KO) mice only partially rescued the clinical disease phenotypes, demonstrating that other loci likely exist under this QTL. Moreover, candidate deleterious alleles were predicted PolyPhen II analysis in the Trim5 locus, but validating a causal role for the allele variant in disease severity will require detailed biochemical studies coupled with the generation of an allelic series in the two parental CC mouse strains used in the cross. While the lack of availability human QTL mapping data limits our ability to determine if common disease driving loci and genes exist across species (e.g., mice and humans), our data do help prioritize candidate gene-association studies in human populations. Previous genetic mapping studies in humans and mice have identified common susceptibility loci and genes that are in synteny with regions identified in human GWAS studies,18,19 supporting a compelling hypothesis that justifies downstream studies. While we have mapped a novel locus that regulates viral load in the blood, earlier studies in the CC population identified lines that showed discordant measures between virus titer, RNA virus load, and viral antigen expression in the liver.20 As neither study identified mechanism, loci, or genes that regulate the disconnect between virus titer, RNA viral load, and viral protein expression in tissue, the highly reproducible findings across different laboratories support the need for critical downstream studies that are designed to identify the loci and underlying causal genes that drive this phenotypic disconnect. A strength of the study is that the candidate genes and expression networks identified in extreme-phenotype models of outbred populations has the potential to complement the analysis of human datasets that can identify the key disease-driving loci and genes that regulate EVD across mammals.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents should be directed and will be fulfilled by the Lead Contact Alexandra Schäfer (aschaefe@email.unc.edu).
Materials availability
Reagents specific to this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The raw data and normalized gene expression levels are available at the GEO database (https://www.ncbi.nlm.nih.gov/geo/),81,82 ID: GSE165142 and ID: GSE262041.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Ethics statement
All infectious work with EBOV was performed in the maximum containment laboratory at the Integrated Research Facility, Rocky Mountain Laboratories (RML), Division of Intramural Research (DIR), National Institute of Allergy and Infectious Disease (NIAID), National Institutes of Health (NIH) according to standard operating protocols (SOPs) approved by the RML Institutional Biosafety Committee (IBC). The animal work was approved by the RML Institutional Animal Care and Use Committee (IACUC) and performed according to the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care, International and the Office of Laboratory Animal Welfare. All procedures on animals were carried out by trained personnel following SOPs approved by the IBC and IACUC. Humane endpoint criteria in compliance with IACUC-approved scoring parameters (weight loss >25%, ataxia, lethargy (animal is reluctant to move), bloody discharge from nose, mouth, rectum or urogenital area, tachypnea, dyspnea or paralysis of the limbs) were used to determine when animals should be humanely euthanized.
Mouse studies and virus quantification
Collaborative Cross mice were purchased directly from the System Genetics Core Facility (SGCF) at UNC between 2016 and 2018. All mice in the SGCF are kept on a 12h light:dark cycle, and on standard chow. F1 and resultant F2 mice were bred directly at the SGCF. F2 mice were bred such that all grand-parental combinations were used, and mice were randomized at weaning such that each experimental cage contained non-siblings. Mice were shipped to RML at 6–7 weeks of age. CC mice were generated with institutional approval (IACUC ID 20–210).
MA-EBOV (passage 3) was propagated on Vero E6 cells, tittered on these cells and stored in liquid nitrogen.15 For infection studies, equal groups of mice of both sexes were infected at the age of 8–10 weeks. A virus dilution for injection of 100 ffu per mouse was prepared immediately before infection of mice with MA-EBOV by i.p. injection of 0.2 mL suspension in DMEM into two sites of the lower abdomen (0.1 mL per site) as previously described.20,83 All animals were monitored daily for body weight changes and at least once daily for clinical signs of disease. Animals were euthanized at >25% weight loss and/or signs of ataxia, extreme lethargy (unresponsive to touch), bloody discharge, tachypnea, dyspnea, or paralysis of limbs as approved by the IACUC. At the time of planned necropsy, mice were anesthetized, and changes in gross pathology of the liver (changes in color from burgundy red (score 0) to pale (score 4) and in integrity form firm (score 0) to soft (score 4)) were scored on a scale from 0 (no change) to 4 (100% changed). Blood was collected and liver and spleen samples were collected for histology, differential gene expression analysis and detection of viral load. Tissues were inactivated and removed from the BSL4 using validated and IBC-approved inactivation protocols.84,85 Total RNA was extracted from blood and tissue samples using Trizol (Invitrogen) according to manufacturer’s directions. Viral load was determined via RT-qPCR using a LightCycler 480II (Roche). The following conditions were used: 50°C for 10 min, 95°C for 5 min, followed by 40 cycles of 95°C for 10 s and 60°C for 45 s. The sequences of the EBOV NP primers and probe were as follows: forward primer: TCA TGG CAA TCC TGC AAC A, Reverse primer: TCG GTT GAA TCA TCC CAT TGT, probe: 6FAM – CAT CAG TGA ATG AGC ATG G -MGBNFQ. Samples were quantified against a standard curve of MA-EBOV RNA extracted from 10-fold serial dilutions of viral stocks with predetermined titers. Virus titers were shown as FFU equivalent of RNA copies in 1 mg of total tissue RNA.86
METHOD DETAILS
Histology and antigen staining
Formalin-fixed tissues were removed from BSL4 following validated and IBC-approved protocols. They were further processed on a Leica ASP 6025 tissue processer, embedded in paraffin (Leica Paraplast) and sectioned at 5um thickness. Sequential tissue sections were stained with hematoxylin and eosin (H&E, Richard Allan Scientific) or labeled for antigens using anti-cleaved Casp3 (9664S, Cell Signaling Technology) or anti-Fibrinogen (A008002-2, Agilent) on the Ventana Discovery automated staining platform (Roche) or using anti-CD31 (77699S, Cell Signaling Technology) or anti-EBOV NP (0301-012, IBT Bioservices) on the Bond III (Leica Biosystems) automated stainer.
Briefly, for labeling performed in the Discovery platform, antigen retrieval was accomplished using Ventana’s CC1 (pH 8.5), tissues were blocked, and the primary antibody was diluted as follows: anti-cC3 at 1:200 or anti-Fibrinogen antibody at 1:1000 using Discovery Casein Diluent (760–219, Roche). Ready-to use secondary antibodies Discovery OmniMap anti-Rabbit HRP (760–4311, Roche) was used, followed by Discovery Chromo Maps DAB (760–159, Roche) development and Hematoxylin II (790–2208, Roche) for nuclear staining.
For labeling performed on the Bond platform, slides were dewaxed in Bond Dewax solution (AR9222) and hydrated in Bond Wash solution (AR9590). Heat induced antigen retrieval was performed for 20 min at 100°C in Bond-Epitope Retrieval solution 1 pH-6.0 (AR9961). After pretreatment, slides were incubated with an anti-CD31 antibody at 1:100 dilution or with an anti-EBOV NP antibody at 1:2000 dilution for 1h followed by Novolink Polymer (RE7260-K) secondary, respectively. Antibody detection with 3,3′-diaminobenzidine (DAB) was performed using the Bond Intense R detection system (DS9263). Stained slides were dehydrated and cover slipped with Cytoseal 60 (23–244256, Thermo Fisher Scientific). A positive control was included for each run.
Automated quantification of cleaved Caspase-3, CD31, fibrinogen, and EBOV NP expression in mouse liver
The process of quantitative image analysis begins with the acquisition of high-resolution digital slides. FFPE sections of mouse liver and spleen were stained for Cleaved Caspase-3 (cC3), CD31, Fibrinogen, and EBOV NP with DAB detection on a Bond autostainer (Leica Biosystems), followed by scanning on a ScanScope AT2 slide scanner (Leica Biosystems) with a 40X power objective. The final 8-bit image per channel resolution was 0.2529 mmmicron per pixel (MPP). Images were uploaded to eSlide Manager as JPEG-compressed* Aperio SVS files and visualized with ImageScope version 12.4.4 (Leica Biosystems). Images were then imported to Definiens Architect XD 2.7 Build 60765 ×64 for analysis with Tissue Studio version 4.4.2.
Using the Tissue Studio portal within Definiens Architect, each tissue section was detected independently because each one represented a different subject (animal). Cellular (cC3) or Marker Area (CD31, Fibrinogen, and EBOV-NP) data were obtained for each whole tissue section. The program also calculated the total tissue area. To detect and segment individual cells, 20X apparent magnification was used. The hematoxylin (counterstain) and DAB (IHC marker) thresholds were set according to the stain control slides provided by the Pathology Services Core (PSC). The analysis output included all quantitative results as well as overlays representing cellular or marker area scores.
For cell analysis, a typical nucleus size of 61 μm2 and a hematoxylin threshold of 0.05 was used each nucleus was segmented and counted by Tissue Studio. Exceptionally small objects were excluded by filtering based on size. Then, each nucleus was scored according to the DAB intensity using an IHC threshold of 0.3. The positive intensity scores were counted on linear scale as Low, Medium, or High using Threshold Low/Medium = 0.6 and Threshold Medium/High = 0.9. A histological score was calculated as H-score = (3 × % High Positive) + (2 × % Medium Positive) + (1 × % Low Positive). The output also included total numbers of cells, percent positive and negative cells, and cell density (#/mm2).
For marker area analysis, Hematoxylin and marker (IHC) thresholds were then set according to control values. Exceptionally small objects were excluded by filtering based on size. Then, marker area was measured and scored on a linear scale as Low, Medium, or High using Threshold Low/Medium = 0.2–0.45 and Threshold Medium/High = 0.3–0.9. A histological score was calculated as H-score = (3 × % High Positive) + (2 × % Medium Positive) + (1 x % Low Positive). The output also included percent positive and negative areas, as well as unstained areas. The compression Quality was 70 and the compression ratio was 15–25.
Mouse genotyping and genetic mapping
Upon weaning, each F1 and F2 mouse was tail clipped. Genomic DNA was extracted from tail-clips using the Qiagen DNeasy blood and tissue kit Hilden, Germany). For processing and running on the MiniMUGA array, 1.5 mg of DNA were sent to Neogen (Neogen Inc, Lincoln).18,87 Upon receipt of genotype data from Neogen, we used all of our data (CC011 and CC074 genotypes, F1s and F2s) to filter these genotypes to informative markers in our study. Briefly, where CC011 and CC074 mice had alternate genotypes, where F1 animals had expected heterozygous (H) calls, and where expected mendelian ratios were seen in the genotypes of F2 animals. From this filtered set of 2750 markers, we conducted genetic mapping using the R/QTL package.88,89 Briefly, the scanone function was used to determine the strength of the regression of phenotypes on genotypes at each of the informative markers in the CC011xCC074-F2 cross. Rounds of 1,000 permutations were run to scramble the relationship between phenotypes and genotype to determine significance thresholds (LOD), providing an appropriate threshold of significance that is robust to the phenotype distribution and allele frequencies. It was ensured that long-range linkage disequilibrium was not driving these observations by fitting multifactor ANOVAs with single QTL and with sets of loci, for phenotypes where we identified multiple QTL only loci with statistically significant improvement in fit for the full model were considered.
RNA-sequencing data and bioinformatic analysis
Total RNA was extracted from liver samples using Trizol (Invitrogen) according to manufacturer’s directions. The initial quality and integrity of total RNA was evaluated using the Agilent Technologies Tapestation 2200 (Agilent Technologies). The RNA sequencing library was generated from 1000 ng total RNA using the KAPA Stranded RNA-Seq kit with RiboErase according to manufacturer’s directions (Roche Sequencing and Life Science). The pooled libraries were sequenced on 3 lanes Illumina NovaSeq 6000 using NovaSeq 6000 S4 XP Reagent Kit 100 Nu, paired end, with an average of 30,000,000 reads per RNA sample.
Analysis of raw reads was performed as previously described.90 Briefly, sequenced reads were mapped to the mouse reference transcriptome (Ensemble; Mus musculus version 108) using Kallisto (version 0.46.0). Transcript quantification data were normalized using the TMM method in EdgeR (version 3.38.4) and DEGs (p.Adj.val <0.01; logLC >2.0) were identified using linear modeling with limma (version 3.52.2) using R (version 4.2.0) in R Studio (version 2022-04-19). Gene ontology analysis was carried out using Gene Set Enrichment Analysis (GSEA; gsea-msigdb.org) to identify dysregulated pathways.
Generation of Trim−/− mice
Transgenic mice were generated with institutional approval (IACUC ID 18–314). The target region spanned approximately 300 kb encompassing the Trim5 group cluster, including Trim34a, Trim12a, Trim34b, Trim12c, Trim30b, Trim30c, Trim30a and Trim30d genes (Figures 4A and 4B). Cas9 guide RNAs flanking the target region were identified using Benchling software. Three guide RNAs at each end of the target region were selected for activity testing. Guide RNAs were cloned into a T7 promoter-gRNA scaffoldvector (UNC Animal Models Core) followed by T7 in vitro transcription (HiScribe T7 High Yield RNA Syntehsis Kit, New England BioLabs) and RNeasy spin column purification (Qiagen). Functional testing was performed by transfecting a mouse embryonic fibroblast cell line (UNC Animal Models Core) with in vitro transcribed guide RNA and recombinant Cas9 protein (UNC Animal Models Core). The guide RNA target site was amplified from transfected cells and analyzed by ICE (Synthego). Guide RNAs selected for genome editing in embryos were 5sg73B (protospacer sequence 5′- gTATAAGTTCCACGTGTT-3’; chr7:104,235,525bp) and 3sg66B (protospacer sequence 5′-gCTGGTAAAAGCCTAGTACGG-3’; chr7:104,535,557) (lower-case g indicates heterologous guanine added for in vitro transcription). A donor oligonucleotide was included to facilitate homologous recombination to produce a clean deletion event between the guide RNA cut sites (Trim5-983-DO-T1, sequence 5′-GCCCTTGTATGTAAAGTGTCACATCCTAGCTGTCAGCAT CAGGGTCCATCCTAACTAGGCTTTTACCAGAGTCCCTCAAGAGCAGAAATGTTCTCCAGACATGATACCTC −3′). However, editing events in the animals did not have the deletion junction corresponding to the oligonucleotide, indicating that the oligonucleotide was not a significant participant in the genome editing event (Figure S4B). C57BL/6J zygotes were electroporated with 1.2 mM Cas9 protein, 47 ng/ul each guide RNA and 400 ng/ul donor oligonucleotide and implanted in recipient pseudo-pregnant females. Resulting pups were screened by PCR with primers Trim5-5ScF1 and Trim5-5ScR1 (sequences below), which flank the deletion region such that a PCR product was only obtained if the ~300 kb deletion event had occurred in the animal. Four of 23 pups showed evidence of a deletion event by PCR. The deletion-specific PCR product from the 4 founder animals was sequenced to determine the precise junctions of the deletion events. Two male founders positive by PCR for the deletion were mated to wild-type C57BL/6J females for germline transmission of the deletion allele. The deletion-specific PCR was sequenced from positive offspring to verify the specific deletion endpoints. One founder transmitted two different deletion events to its offspring. The deletion events both encompass over 300 kb with endpoints near the guide RNA cut sites. Trim−/− animals were detected by PCR with primers Trim5–5ScF1 (5′- TGGATCTGCAGGTGACAAGTA −3′), Trim5–5ScR1 (5′- CGTAGAGTGTGAGAAATCCTTTG −3′) and Trim5–3ScR1 (5′- GGCGCAGATTGCTTTAAAC −3′). This assay gives a ~593 bp band for the Trim−/− allele and a 466 bp band for the Trim wild-type allele (Figures S4B and S4C).
QUANTIFICATION AND STATISTICAL ANALYSIS
Mann-Whitney tests were used to compare weight loss, viral load in liver tissue liver titer, and histological quantification. Log rank tests was used for survival analyses. Statistical analyses were performed in GraphPad Prism 10.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit-Anti-Cleaved-Caspase3 | CST | Cat#9664S |
| Polyclonal-Rabbit-Anti-Fibrinogen | Agilent | Cat#A008002-2 |
| Rabbit-Anti-CD31 | CST | Cat#77699S |
| Polyclonal-Anti-EBOV-NP | IBT Bioservices | Cat#0301-012 |
| OmniMap anti-Rabbit HRP | Roche | Cat#760-4311 |
| Novolink Polymer (RE7260-K) secondary | Roche | Cat#760-159 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| MA-EBOV | Bray et al.15 | N/A |
|
| ||
| Biological samples | ||
|
| ||
| blood and liver from mice | This paper | |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Trizol | Invitrogen | Cat#15596026 |
| Formalin, 10% | Fisher | Cat#SF100-4 |
| Hemotoxylin | Epredia | Cat#7211 |
| Eosin | Epredia | Cat#7111 |
| Clarifier 2 | Epredia | Cat#7402 |
| Discovery Omnimaps anti-RB HRP | Roche | Cat#5269679001 |
| Discovery Chromo Maps DAB | Roche | Cat#5266645001 |
| CC1 | Roche | Cat#6414575001 |
| Ventana Antibody Diluent with Casein | Roche | Cat#06440002001 |
| Bond Dewax | Leica Biosystems | Cat#AR9222 |
| Bond Wash | Leica Biosystems | Cat#AR9590 |
| Bond Epitope Retrieval solution A | Leica Biosystems | Cat#AR9961 |
| Novolink Polymer | Leica Biosystems | Cat#RE7260-K |
| Bond Intense R detection system | Leica Biosystems | Cat#DS9263 |
|
| ||
| Critical commercial assays | ||
|
| ||
| QuantiNova Probe PCR Kit | Qiagen | Cat#208254 |
| DNEasy blood and tissue kit | Qiagen | Cat#69506 |
| KAPA Stranded RNA-Seq Kit with RiboErase (HMR) library Prep kit | Roche | Cat#7962240001 |
| TruSeq UD Indexes | Illumina | Cat#20023785 |
| Novaseq6000, S4 flowcell | Illumina | Cat#20028313 |
|
| ||
| Deposited data | ||
|
| ||
| GSE165142 | GEO | https://www.ncbi.nlm.nih.gov/geo/ |
| GSE262041 | GEO | https://www.ncbi.nlm.nih.gov/geo/ |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Vero E6 | ATCC | Cat#CRL-1586 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Collaborative Cross mice (CC001/Unc, CC002/Unc, CC004/TauUnc, CC011/Unc, CC021/Unc, CC051/TauUnc, CC061GeniUnc, and CC074/Unc) | UNC System Genetics Core | https://csbio.unc.edu/CCstatus/index.py |
| C57BL/6J | The Jackson Laboratory | Cat#000664 |
| Trim−/− and Trim+/+ transgenic mice | This paper | |
|
| ||
| Oligonucleotides | ||
|
| ||
| EBOV-NP-f TCA TGG CAA TCC TGC AAC A | IDT | https://www.idtdna.com/pages |
| EBOV-NP-r TCG GTT GAA TCA TCC CAT TGT | IDT | https://www.idtdna.com/pages |
| EBOV-probe 6FAM – CAT CAG TGA ATG AGC ATG G -MGBNFQ. | IDT | https://www.idtdna.com/pages |
| 5sg73B - gTATAAGTTCCACGTGTT | IDT | https://www.idtdna.com/pages |
| 3sg66B - gCTGGTAAAAGCCTAGTACGG | IDT | https://www.idtdna.com/pages |
| Trim5-983-DO-T1 - GCCCTTGTATGTAAAGTGTCA CATCCTAGCTGTCAGCATCAGGGTCCATCCTAAC TAGGCTTTTACCAGAGTCCCTCAAGAGCAGAAA TGTTCTCCAGACATGATACCTC | IDT | https://www.idtdna.com/pages |
| Trim5-5ScF1 - TGGATCTGCAGGTGACAAGTA | IDT | https://www.idtdna.com/pages |
| Trim5-5ScR1 - CGTAGAGTGTGAGAAATCCTTTG | IDT | https://www.idtdna.com/pages |
| Trim5-3ScR1 - GGCGCAGATTGCTTTAAAC | IDT | https://www.idtdna.com/pages |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad v10 | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| R v4.2.0 or 4.2.2. | The R Project for Statistical Computing | https://www.r-project.org/ |
| Adobe Illustrator 2024 | Adobe | N/A |
| PyMOL v2.0 | Schrödinger, LLC | https://www.pymol.org/ |
| Kallisto v0.46.0 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html |
| EdgeR v3.38.4 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Limma v3.52.2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html |
| R Studio v2022-04-19 | The R Project for Statistical Computing | https://www.r-project.org/ |
| Phyre2 Protein Fold Recognition Server | ||
| Benchling | Benchling | https://www.benchling.com |
| ImageScope version 12.4.4 | Leica Biosystems | https://www.leicabiosystems.com/us/digital-pathology/manage/aperio-imagescope/ |
Highlights.
CC genetic reference population identifies genetic loci regulating EBOV pathogenesis in mice
An F2 population from two CC lines is either highly resistant or vulnerable to EBOV infection
A major locus on chromosome 7, encoding Trim5, drives severe EVD-like disease in mice
Gene expression signatures of liver damage mirror severe EVD in humans
ACKNOWLEDGMENTS
This study was supported by grants from the National Institute of Allergy and Infectious Diseases, NIH (U19AI100625, AI151797, and AI149644 to R.S.B. or to R.S.B., M.T.F., and F.P.M.d.V.; AI171292 to R.S.B.), the Intramural Research Program NIAID, NIH (to A.M. and H.F.), a Burroughs Wellcome Fund Postdoctoral Enrichment Program Award, and a Hannah H. Gray Fellowship from the Howard Hughes Medical Institute (to D.R.M.).
We would like to acknowledge the excellent technical support of Animal Models Core staff and their funding by the NCI Center Core Support Grant (P30 CA016086). We are grateful to the animal care staff at the NIAID Rocky Mountain Veterinary Branch for their assistance with conducting the mouse infection studies. We thank the Systems Genetics Core Facility (UNC) for maintaining and distributing CC mice and the Pathology Services Core for expert technical assistance with histopathology. The PSC is supported in part by an NCI Center Core Support Grant (5P30CA016080-42).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114127.
DECLARATION OF INTERESTS
D.O.C. is employed by, has equity ownership in, and serves on the board of directors of TransViragen, the company that has been contracted by UNC-Chapel Hill to manage its Animal Models Core Facility. R.S.B. is a member of advisory boards for VaxArt, Takeda, and Invivyd, focused on unrelated projects.
REFERENCES
- 1.Sawyer SL, Emerman M, and Malik HS (2007). Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLoS Pathog 3, e197. 10.1371/journal.ppat.0030197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacob ST, Crozier I, Fischer WA 2nd, Hewlett A, Kraft CS, Vega M.A.d.L., Soka MJ, Wahl V, Griffiths A, Bollinger L, and Kuhn JH (2020). Ebola virus disease. Nat. Rev. Dis. Prim 6, 13. 10.1038/s41572-020-0147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marzi A, Halfmann P, Hill-Batorski L, Feldmann F, Shupert WL, Neumann G, Feldmann H, and Kawaoka Y (2015). Vaccines. An Ebola whole-virus vaccine is protective in nonhuman primates. Science 348, 439–442. 10.1126/science.aaa4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geisbert TW, and Feldmann H (2011). Recombinant vesicular stomatitis virus-based vaccines against Ebola and Marburg virus infections. J. Infect. Dis 204, S1075–S1081. 10.1093/infdis/jir349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones SM, Feldmann H, Ströher U, Geisbert JB, Fernando L, Grolla A, Klenk HD, Sullivan NJ, Volchkov VE, Fritz EA, et al. (2005). Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med 11, 786–790. 10.1038/nm1258. [DOI] [PubMed] [Google Scholar]
- 6.Stanley DA, Honko AN, Asiedu C, Trefry JC, Lau-Kilby AW, Johnson JC, Hensley L, Ammendola V, Abbate A, Grazioli F, et al. (2014). Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat. Med 20, 1126–1129. 10.1038/nm.3702. [DOI] [PubMed] [Google Scholar]
- 7.Henao-Restrepo AM, Camacho A, Longini IM, Watson CH, Edmunds WJ, Egger M, Carroll MW, Dean NE, Diatta I, Doumbia M, et al. (2017). Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet 389, 505–518. 10.1016/S0140-6736(16)32621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wells CR, Pandey A, Parpia AS, Fitzpatrick MC, Meyers LA, Singer BH, and Galvani AP (2019). Ebola vaccination in the Democratic Republic of the Congo. Proc. Natl. Acad. Sci. USA 116, 10178–10183. 10.1073/pnas.1817329116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, Ploquin A, Doria-Rose NA, Staupe RP, Bailey M, et al. (2016). Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 351, 1339–1342. 10.1126/science.aad5224. [DOI] [PubMed] [Google Scholar]
- 10.Mulangu S, Dodd LE, Davey RT Jr., Tshiani Mbaya O, Proschan M, Mukadi D, Lusakibanza Manzo M, Nzolo D, Tshomba Oloma A, Ibanda A, et al. (2019). A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med 381, 2293–2303. 10.1056/NEJMoa1910993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pascal KE, Dudgeon D, Trefry JC, Anantpadma M, Sakurai Y, Murin CD, Turner HL, Fairhurst J, Torres M, Rafique A, et al. (2018). Development of Clinical-Stage Human Monoclonal Antibodies That Treat Advanced Ebola Virus Disease in Nonhuman Primates. J. Infect. Dis 218, S612–S626. 10.1093/infdis/jiy285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther-Bovendo H, Wei H, Aviles J, Hiatt E, et al. (2014). Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 514, 47–53. 10.1038/nature13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matz KM, Marzi A, and Feldmann H (2019). Ebola vaccine trials: progress in vaccine safety and immunogenicity. Expert Rev. Vaccines 18, 1229–1242. 10.1080/14760584.2019.1698952. [DOI] [PubMed] [Google Scholar]
- 14.St Claire MC, Ragland DR, Bollinger L, and Jahrling PB (2017). Animal Models of Ebolavirus Infection. Comp. Med 67, 253–262. [PMC free article] [PubMed] [Google Scholar]
- 15.Bray M, Davis K, Geisbert T, Schmaljohn C, and Huggins J (1998). A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis 178, 651–661. 10.1086/515386. [DOI] [PubMed] [Google Scholar]
- 16.Yamaoka S, Banadyga L, Bray M, and Ebihara H (2017). Small Animal Models for Studying Filovirus Pathogenesis. Curr. Top. Microbiol. Immunol 411, 195–227. 10.1007/82_2017_9. [DOI] [PubMed] [Google Scholar]
- 17.Ferris MT, Aylor DL, Bottomly D, Whitmore AC, Aicher LD, Bell TA, Bradel-Tretheway B, Bryan JT, Buus RJ, Gralinski LE, et al. (2013). Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog 9, e1003196. 10.1371/journal.ppat.1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schäfer A, Leist SR, Gralinski LE, Martinez DR, Winkler ES, Okuda K, Hawkins PE, Gully KL, Graham RL, Scobey DT, et al. (2022). A Multitrait Locus Regulates Sarbecovirus Pathogenesis. mBio 13, e0145422. 10.1128/mbio.01454-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schäfer A, et al. (2024). Genetic Loci reguate Sarbecovirus pathogenesis: A comparison across mice and humans. Virus Res 344, 199357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasmussen AL, Okumura A, Ferris MT, Green R, Feldmann F, Kelly SM, Scott DP, Safronetz D, Haddock E, LaCasse R, et al. (2014). Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science 346, 987–991. 10.1126/science.1259595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tareen SU, Sawyer SL, Malik HS, and Emerman M (2009). An expanded clade of rodent Trim5 genes. Virology 385, 473–483. 10.1016/j.virol.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang TH, Yoshimi R, and Ozato K (2015). Tripartite Motif (TRIM) 12c, a Mouse Homolog of TRIM5, Is a Ubiquitin Ligase That Stimulates Type I IFN and NF-kappaB Pathways along with TNFR-Associated Factor 6. J. Immunol 195, 5367–5379. 10.4049/jimmunol.1402064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lascano J, Uchil PD, Mothes W, and Luban J (2016). TRIM5 Retroviral Restriction Activity Correlates with the Ability To Induce Innate Immune Signaling. J. Virol 90, 308–316. 10.1128/JVI.02496-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noll KE, Whitmore AC, West A, McCarthy MK, Morrison CR, Plante KS, Hampton BK, Kollmus H, Pilzner C, Leist SR, et al. (2020). Complex Genetic Architecture Underlies Regulation of Influenza-A-Virus-Specific Antibody Responses in the Collaborative Cross. Cell Rep 31, 107587. 10.1016/j.celrep.2020.107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham JB, Swarts JL, Wilkins C, Thomas S, Green R, Sekine A, Voss KM, Ireton RC, Mooney M, Choonoo G, et al. (2016). A Mouse Model of Chronic West Nile Virus Disease. PLoS Pathog 12, e1005996. 10.1371/journal.ppat.1005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cartwright HN, Barbeau DJ, Doyle JD, Klein E, Heise MT, Ferris MT, and McElroy AK (2022). Genetic diversity of collaborative cross mice enables identification of novel rift valley fever virus encephalitis model. PLoS Pathog 18, e1010649. 10.1371/journal.ppat.1010649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ibarra-Soria X, Levitin MO, Saraiva LR, and Logan DW (2014). The olfactory transcriptomes of mice. PLoS Genet 10, e1004593. 10.1371/journal.pgen.1004593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava A, Morgan AP, Najarian ML, Sarsani VK, Sigmon JS, Shorter JR, Kashfeen A, McMullan RC, Williams LH, Giusti-Rodríguez P, et al. (2017). Genomes of the Mouse Collaborative Cross. Genetics 206, 537–556. 10.1534/genetics.116.198838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Institute, W.S. Mouse Genomes Project-querySNPs, indels or SVs http://www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel_1505. Accessed on 07/05/2021.
- 30.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Javanbakht H, An P, Gold B, Petersen DC, O’Huigin C, Nelson GW, O’Brien SJ, Kirk GD, Detels R, Buchbinder S, et al. (2006). Effects of human TRIM5alpha polymorphisms on antiretroviral function and susceptibility to human immunodeficiency virus infection. Virology 354, 15–27. 10.1016/j.virol.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 32.Sayah DM, Sokolskaja E, Berthoux L, and Luban J (2004). Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430, 569–573. 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 33.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, and Jung JU (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 34.Gralinski LE, Ferris MT, Aylor DL, Whitmore AC, Green R, Frieman MB, Deming D, Menachery VD, Miller DR, Buus RJ, et al. (2015). Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross. PLoS Genet 11, e1005504. 10.1371/journal.pgen.1005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bharaj P, Atkins C, Luthra P, Giraldo MI, Dawes BE, Miorin L, Johnson JR, Krogan NJ, Basler CF, Freiberg AN, and Rajsbaum R (2017). The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication. J. Virol 91, e00833–17. 10.1128/JVI.00833-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiramel AI, Meyerson NR, McNally KL, Broeckel RM, Montoya VR, Mendez-Solis O, Robertson SJ, Sturdevant GL, Lubick KJ, Nair V, et al. (2019). TRIM5alpha Restricts Flavivirus Replication by Targeting the Viral Protease for Proteasomal Degradation. Cell Rep 27, 3269–3283. 10.1016/j.celrep.2019.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scoon WA, Mancio-Silva L, Suder EL, Villacorta-Martin C, Lindstrom-Vautrin J, Bernbaum JG, Mazur S, Johnson RF, Olejnik J, Flores EY, et al. (2022). Ebola virus infection induces a delayed type I IFN response in bystander cells and the shutdown of key liver genes in human iPSC-derived hepatocytes. Stem Cell Rep 17, 2286–2302. 10.1016/j.stemcr.2022.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jankeel A, Menicucci AR, Woolsey C, Fenton KA, Mendoza N, Versteeg K, Cross RW, Geisbert TW, and Messaoudi I (2020). Early Transcriptional Changes within Liver, Adrenal Gland, and Lymphoid Tissues Significantly Contribute to Ebola Virus Pathogenesis in Cynomolgus Macaques. J. Virol 94, e00250–20. 10.1128/JVI.00250-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wynne JW, Todd S, Boyd V, Tachedjian M, Klein R, Shiell B, Dearnley M, McAuley AJ, Woon AP, Purcell AW, et al. (2017). Comparative Transcriptomics Highlights the Role of the Activator Protein 1 Transcription Factor in the Host Response to Ebolavirus. J. Virol 91, e01174–17. 10.1128/JVI.01174-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banadyga L, Wong G, and Qiu X (2018). Small Animal Models for Evaluating Filovirus Countermeasures. ACS Infect. Dis 4, 673–685. 10.1021/acsinfecdis.7b00266. [DOI] [PubMed] [Google Scholar]
- 41.Cross RW, Fenton KA, and Geisbert TW (2018). Small animal models of filovirus disease: recent advances and future directions. Expet Opin. Drug Discov 13, 1027–1040. 10.1080/17460441.2018.1527827. [DOI] [PubMed] [Google Scholar]
- 42.de La Vega MA, Caleo G, Audet J, Qiu X, Kozak RA, Brooks JI, Kern S, Wolz A, Sprecher A, Greig J, et al. (2015). Ebola viral load at diagnosis associates with patient outcome and outbreak evolution. J. Clin. Invest 125, 4421–4428. 10.1172/JCI83162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ward MD, Kenny T, Bruggeman E, Kane CD, Morrell CL, Kane MM, Bixler S, Grady SL, Quizon RS, Astatke M, and Cazares LH (2020). Early detection of Ebola virus proteins in peripheral blood mononuclear cells from infected mice. Clin. Proteonomics 17, 11. 10.1186/s12014-020-09273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeremiah Matson M, Ricotta E, Feldmann F, Massaquoi M, Sprecher A, Giuliani R, Edwards JK, Rosenke K, de Wit E, Feldmann H, et al. (2022). Evaluation of viral load in patients with Ebola virus disease in Liberia: a retrospective observational study. Lancet. Microbe 3, e533–e542. 10.1016/S2666-5247(22)00065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piorkowski G, Jacquot F, Quérat G, Carbonnelle C, Pannetier D, Mentré F, Raoul H, and de Lamballerie X (2017). Implementation of a non-human primate model of Ebola disease: Infection of Mauritian cynomolgus macaques and analysis of virus populations. Antivir. Res 140, 95–105. 10.1016/j.antiviral.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 46.Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol 32, 513–545. 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt O, and Teis D (2012). The ESCRT machinery. Curr. Biol 22, R116–R120. 10.1016/j.cub.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghosh S, and Marsh ENG (2020). Viperin: An ancient radical SAM enzyme finds its place in modern cellular metabolism and innate immunity. J. Biol. Chem 295, 11513–11528. 10.1074/jbc.REV120.012784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon TB, Hayward JA, Marsh GA, Baker ML, and Tachedjian G (2019). Host and Viral Proteins Modulating Ebola and Marburg Virus Egress. Viruses 11, 25. 10.3390/v11010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez MA, Vartanian JP, and Wain-Hobson S (1994). Hypermutagenesis of RNA using human immunodeficiency virus type 1 reverse transcriptase and biased dNTP concentrations. Proc. Natl. Acad. Sci. USA 91, 11787–11791. 10.1073/pnas.91.25.11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simmonds P, and Ansari MA (2021). Extensive C->U transition biases in the genomes of a wide range of mammalian RNA viruses; potential associations with transcriptional mutations, damage- or host-mediated editing of viral RNA. PLoS Pathog 17, e1009596. 10.1371/journal.ppat.1009596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flint M, Chatterjee P, Lin DL, McMullan LK, Shrivastava-Ranjan P, Bergeron É, Lo MK, Welch SR, Nichol ST, Tai AW, and Spiropoulou CF (2019). A genome-wide CRISPR screen identifies N-acetylglucosamine-1-phosphate transferase as a potential antiviral target for Ebola virus. Nat. Commun 10, 285. 10.1038/s41467-018-08135-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coulter ME, Dorobantu CM, Lodewijk GA, Delalande F, Cianferani S, Ganesh VS, Smith RS, Lim ET, Xu CS, Pang S, et al. (2018). The ESCRT-III Protein CHMP1A Mediates Secretion of Sonic Hedgehog on a Distinctive Subtype of Extracellular Vesicles. Cell Rep 24, 973–986.e8. 10.1016/j.celrep.2018.06.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Licata JM, Simpson-Holley M, Wright NT, Han Z, Paragas J, and Harty RN (2003). Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol 77, 1812–1819. 10.1128/jvi.77.3.1812-1819.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stuchell-Brereton MD, Skalicky JJ, Kieffer C, Karren MA, Ghaffarian S, and Sundquist WI (2007). ESCRT-III recognition by VPS4 ATPases. Nature 449, 740–744. 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- 56.Hartley MA, Young A, Tran AM, Okoni-Williams HH, Suma M, Mancuso B, Al-Dikhari A, and Faouzi M (2017). Predicting Ebola Severity: A Clinical Prioritization Score for Ebola Virus Disease. PLoS Neglected Trop. Dis 11, e0005265. 10.1371/journal.pntd.0005265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McElroy AK, and Spiropoulou CF (2014). Biomarkers for understanding Ebola virus disease. Biomarkers Med 8, 1053–1056. 10.2217/bmm.14.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Duan HJ, Chen HY, Ji YJ, Zhang X, Rong YH, Xu Z, Sun LJ, Zhang JY, Liu LM, et al. (2016). Age and Ebola viral load correlate with mortality and survival time in 288 Ebola virus disease patients. Int. J. Infect. Dis 42, 34–39. 10.1016/j.ijid.2015.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McElroy AK, Erickson BR, Flietstra TD, Rollin PE, Nichol ST, Towner JS, and Spiropoulou CF (2014). Biomarker correlates of survival in pediatric patients with Ebola virus disease. Emerg. Infect. Dis 20, 1683–1690. 10.3201/eid2010.140430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leligdowicz A, Fischer WA 2nd, Uyeki TM, Fletcher TE, Adhikari NKJ, Portella G, Lamontagne F, Clement C, Jacob ST, Rubinson L, et al. (2016). Ebola virus disease and critical illness. Crit. Care 20, 217. 10.1186/s13054-016-1325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Price A, Okumura A, Haddock E, Feldmann F, Meade-White K, Sharma P, Artami M, Lipkin WI, Threadgill DW, Feldmann H, and Rasmussen AL (2020). Transcriptional Correlates of Tolerance and Lethality in Mice Predict Ebola Virus Disease Patient Outcomes. Cell Rep 30, 1702–1713.e6. 10.1016/j.celrep.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Medrano LM, Rallón N, Berenguer J, Jiménez-Sousa MA, Soriano V, Aldámiz-Echevarria T, Fernández-Rodríguez A, García M, Tejerina F, Martínez I, et al. (2016). Relationship of TRIM5 and TRIM22 polymorphisms with liver disease and HCV clearance after antiviral therapy in HIV/HCV coinfected patients. J. Transl. Med 14, 257. 10.1186/s12967-016-1005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ozato K, Shin DM, Chang TH, and Morse HC 3rd. (2008). TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol 8, 849–860. 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pertel T, Hausmann S, Morger D, Züger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, et al. (2011). TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365. 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ganser-Pornillos BK, and Pornillos O (2019). Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol 17, 546–556. 10.1038/s41579-019-0225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohainle M, Kim K, Komurlu Keceli S, Felton A, Campbell E, Luban J, and Emerman M (2020). TRIM34 restricts HIV-1 and SIV capsids in a TRIM5alpha-dependent manner. PLoS Pathog 16, e1008507. 10.1371/journal.ppat.1008507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lian Q, Yan S, Yin Q, Yan C, Zheng W, Gu W, Zhao X, Fan W, Li X, Ma L, et al. (2021). TRIM34 attenuates colon inflammation and tumorigenesis by sustaining barrier integrity. Cell. Mol. Immunol 18, 350–362. 10.1038/s41423-020-0366-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X, Xiong J, Zhou D, Zhang S, Wang L, Tian Q, Li C, Liu J, Wu Y, Li J, and Wang J (2022). TRIM34 modulates influenza virus-activated programmed cell death by targeting Z-DNA-binding protein 1 for K63-linked polyubiquitination. J. Biol. Chem 298, 101611. 10.1016/j.jbc.2022.101611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wauquier N, Becquart P, Padilla C, Baize S, and Leroy EM (2010). Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Neglected Trop. Dis 4, e837. 10.1371/journal.pntd.0000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baize S, Leroy EM, Georges AJ, Georges-Courbot MC, Capron M, Bedjabaga I, Lansoud-Soukate J, and Mavoungou E (2002). Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol 128, 163–168. 10.1046/j.1365-2249.2002.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Villinger F, Rollin PE, Brar SS, Chikkala NF, Winter J, Sundstrom JB, Zaki SR, Swanepoel R, Ansari AA, and Peters CJ (1999). Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J. Infect. Dis 179, S188–S191. 10.1086/514283. [DOI] [PubMed] [Google Scholar]
- 72.Kerber R, Krumkamp R, Korva M, Rieger T, Wurr S, Duraffour S, Oestereich L, Gabriel M, Sissoko D, Anglaret X, et al. (2018). Kinetics of Soluble Mediators of the Host Response in Ebola Virus Disease. J. Infect. Dis 218, S496–S503. 10.1093/infdis/jiy429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McElroy AK, Harmon JR, Flietstra TD, Campbell S, Mehta AK, Kraft CS, Lyon MG, Varkey JB, Ribner BS, Kratochvil CJ, et al. (2016). Kinetic Analysis of Biomarkers in a Cohort of US Patients With Ebola Virus Disease. Clin. Infect. Dis 63, 460–467. 10.1093/cid/ciw334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kyle JE, Burnum-Johnson KE, Wendler JP, Eisfeld AJ, Halfmann PJ, Watanabe T, Sahr F, Smith RD, Kawaoka Y, Waters KM, and Metz TO (2019). Plasma lipidome reveals critical illness and recovery from human Ebola virus disease. Proc. Natl. Acad. Sci. USA 116, 3919–3928. 10.1073/pnas.1815356116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cichoż-Lach H, and Michalak A (2014). Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol 20, 8082–8091. 10.3748/wjg.v20.i25.8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramachandran A, and Jaeschke H (2018). Oxidative Stress and Acute Hepatic Injury. Curr. Opin. Toxicol 7, 17–21. 10.1016/j.cotox.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, and Hensley LE (2003). Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis 188, 1618–1629. 10.1086/379724. [DOI] [PubMed] [Google Scholar]
- 78.Qian H, and Chen L (2021). TRIM proteins in fibrosis. Biomed. Pharmacother 144, 112340. 10.1016/j.biopha.2021.112340. [DOI] [PubMed] [Google Scholar]
- 79.Johnson WE, and Sawyer SL (2009). Molecular evolution of the antiretroviral TRIM5 gene. Immunogenetics 61, 163–176. 10.1007/s00251-009-0358-y. [DOI] [PubMed] [Google Scholar]
- 80.Newman RM, Hall L, Connole M, Chen GL, Sato S, Yuste E, Diehl W, Hunter E, Kaur A, Miller GM, and Johnson WE (2006). Balancing selection and the evolution of functional polymorphism in Old World monkey TRIM5alpha. Proc. Natl. Acad. Sci. USA 103, 19134–19139. 10.1073/pnas.0605838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. (2013). NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41, D991–D995. 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210. 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haddock E, Feldmann H, and Marzi A (2018). Ebola Virus Infection in Commonly Used Laboratory Mouse Strains. J. Infect. Dis 218, S453–S457. 10.1093/infdis/jiy208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Haddock E, and Feldmann F (2017). Validating the Inactivation Effectiveness of Chemicals on Ebola Virus. Methods Mol. Biol 1628, 251–257. 10.1007/978-1-4939-7116-9_20. [DOI] [PubMed] [Google Scholar]
- 85.Haddock E, Feldmann F, and Feldmann H (2016). Effective Chemical Inactivation of Ebola Virus. Emerg. Infect. Dis 22, 1292–1294. 10.3201/eid2207.160233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsuda Y, Safronetz D, Brown K, LaCasse R, Marzi A, Ebihara H, and Feldmann H (2011). Protective efficacy of a bivalent recombinant vesicular stomatitis virus vaccine in the Syrian hamster model of lethal Ebola virus infection. J. Infect. Dis 204, S1090–S1097. 10.1093/infdis/jir379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sigmon JS, Blanchard MW, Baric RS, Bell TA, Brennan J, Brockmann GA, Burks AW, Calabrese JM, Caron KM, Cheney RE, et al. (2020). Content and Performance of the MiniMUGA Genotyping Array: A New Tool To Improve Rigor and Reproducibility in Mouse Research. Genetics 216, 905–930. 10.1534/genetics.120.303596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Broman KW, Wu H, Sen S, and Churchill GA (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890. 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- 89.Gatti DM, Svenson KL, Shabalin A, Wu LY, Valdar W, Simecek P, Goodwin N, Cheng R, Pomp D, Palmer A, et al. (2014). Quantitative trait locus mapping methods for diversity outbred mice. G3 (Bethesda) 4, 1623–1633. 10.1534/g3.114.013748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Amorim CF, Novais FO, Nguyen BT, Misic AM, Carvalho LP, Carvalho EM, Beiting DP, and Scott P (2019). Variable gene expression and parasite load predict treatment outcome in cutaneous leishmaniasis. Sci. Transl. Med 11, eaax4204. 10.1126/scitranslmed.aax4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data and normalized gene expression levels are available at the GEO database (https://www.ncbi.nlm.nih.gov/geo/),81,82 ID: GSE165142 and ID: GSE262041.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.