

Abstract
Ferrochelatase (EC 4.99.1.1), the terminal enzyme of the haem biosynthetic pathway, catalyses the chelation of Fe(II) into the protoporphyrin IX ring. The energetics of the binding between murine ferrochelatase and mesoporphyrin were determined using isothermal titration calorimetry, which revealed a stoichiometry of one molecule of mesoporphyrin bound per protein monomer. The binding is strongly exothermic, with a large intrinsic enthalpy (ΔH=−97.1 kJ · mol−1), and is associated with the uptake of two protons from the buffer. This proton transfer suggests that hydrogen bonding between ferrochelatase and mesoporphyrin is a key factor in the thermodynamics of the binding reaction. Differential scanning calorimetry thermograms indicated a co-operative two-state denaturation process with a single transition temperature of 56 °C for wild-type murine ferrochelatase. An increase in the thermal stability of ferrochelatase is dependent upon mesoporphyrin binding. Similarly, murine ferrochelatase variants, in which the active site Glu-289 was replaced by either glutamine or alanine and, when purified, contained specifically-bound protoporphyrin, exhibited enhanced protein stability when compared with wild-type ferrochelatase. However, in contrast with the wild-type enzyme, the thermal denaturation of ferrochelatase variants was best described as a non-co-operative denaturation process.
Keywords: differential scanning calorimetry, ferrochelatase, haem biosynthesis, isothermal titration calorimetry, porphyrin, thermal stability
Abbreviations: DSC, differential scanning calorimetry; ITC, isothermal titration calorimetry; E289Q etc., Glu-289→Gln etc
INTRODUCTION
Ferrochelatase (EC 4.99.1.1) catalyses the terminal step of the haem biosynthetic pathway, the insertion of ferrous iron into the protoporphyrin IX ring [1,2]. Mutations in the mammalian ferrochelatase gene cause erythropoietic protoporphryria, a disorder characterized by a decrease in ferrochelatase activity with accumulation of free protoporphyrin; the subsequent painful photosensitivity is the major clinical manifestation of the disorder [3]. Although ferrochelatases present in a group of Gram-positive bacteria appear to be water-soluble proteins [2], most of the isolated ferrochelatases are associated with the cytoplasmic membrane of prokaryotes and the mitochondrial inner membrane of non-photosynthetic eukaryotes [1,2].
The ferrochelatase molecule has a conserved overall fold, despite the variation in the oligomeric state among different species; Bacillus subtilis ferrochelatase is a monomer [4,5], and Saccharomyces cerevisiae [6] and human [7] ferrochelatases are homodimers. The X-ray crystallographic structures of B. subtilis [4,5], S. cerevisiae [6] and human [7] ferrochelatases revealed that each molecule contains two similar Rossmann-type domains delimiting the porphyrin-binding cleft. However, there are two major differences among the three structures. The first is centred at the C-terminus of the mammalian enzyme, which is involved in the co-ordination of the [2Fe–2S] cluster and homodimer stabilization [7]. The second is a N-terminal 12-residue loop, which resides at the entrance of the active site [7] and, due to its hydrophobic nature, has been postulated to be involved in membrane association [6–8]. In fact, [2Fe–2S] clusters have been identified in Schizosaccharomyces pombe [9], Caulobacter crescentus [10] and animal ferrochelatases [11,12], but not in the B. subtilis [4,5] and S. cerevisiae [6] enzymes, and the N-terminal 12-residue hydrophobic loop is absent in B. subtilis ferrochelatase [4,5], which is not a membrane-associated protein.
During the ferrochelatase-catalysed reaction, an out-of-plane distortion of the porphyrin macrocycle facilitates the insertion of the metal ion into the porphyrin ring [1,2]. This ferrochelatase-induced porphyrin distortion was demonstrated using resonance Raman spectroscopy [13–16], and the distortion was responsive to substitutions at the conserved active-site residues His-209 and Glu-289 in murine ferrochelatase [13]. Further, the structure of the N-methyl mesoporphyrin–ferrochelatase complex revealed that the enzyme had forced increased tilting of the pyrrole ring A [5]. Curiously, the substitution of murine ferrochelatase Glu-289 with either glutamine or alanine yielded ferrochelatase variants [E289Q (Glu-289→Gln) and E289A] with specifically-bound protoporphyrin IX [17]. Despite the significantly decreased catalytic activities of the E289Q and E289A variants, the endogenously bound protoporphyrin could serve as an amenable substrate for Zn2+ chelation [17]. The ability of the endogenously bound protoporphyrin to function as substrate [17] is consistent with the observed distortion of the porphyrin macrocycle in E289Q and E289A mutants [13].
In order to assess the effect of porphyrin binding on the thermodynamic stability of ferrochelatase, we used DSC (differential scanning calorimetry) to examine the thermal denaturation of murine wild-type ferrochelatase, E289Q and E289A variants in the absence or presence of the ligand. We found that porphyrin binding increased the thermal stability of ferrochelatase and variants, although the degree of enhancement varied. The thermodynamics of the porphyrin binding were investigated using ITC (isothermal titration calorimetry) to determine the binding stoichiometry, the enthalpy, entropy and free energy associated with the process. The binding of porphyrin to ferrochelatase occurred with a 1:1 stoichiometry and was coupled to a two-proton transfer.
EXPERIMENTAL
Materials
Mesoporphyrin was purchased from Frontier Scientific. All other chemicals were from Sigma or Riedel-de Haën and were of the highest purity available. Blue Sepharose CL-B6 and Superdex 200 chromatography resins were from Amersham Biosciences.
Overproduction and purification of wild-type and mutant ferrochelatase forms
The wild-type murine ferrochelatase, E289Q and E289A variants were expressed under the Escherichia coli alkaline phosphatase promoter phoA [18], by growing the overproducing bacterial cells for 24 h at 21 °C in Mops medium containing 50 mg/l ampicillin, and were purified as described previously [17,18]. The purified protein was concentrated in a Diaflo stirred cell with an YM 30 membrane (Millipore), and subjected to gel-filtration chromatography on a Superdex 200 column (2.6 cm×45 cm). This gel-filtration step was necessary not only to remove most of the detergent from the Blue Sepharose elution buffer, which contained 1% sodium cholate [17,18], but also to exchange the protein buffer according to the requirements of further experiments. Thus the purified protein was eluted in any of the following three buffers: Bicine buffer (20 mM Bicine, pH 8, 0.5 M NaCl and 10% glycerol), Tris buffer (20 mM Tris/HCl, pH 8, 0.5 M NaCl and 10% glycerol) and phosphate buffer (20 mM potassium phosphate, pH 8, 0.5 M NaCl and 10% glycerol). Then the eluted purified protein was concentrated as described above and kept in liquid nitrogen until further use.
Porphyrin solutions
Porphyrin solutions were prepared following a slightly modified procedure from that previously described [17]. Typically, porphyrin stock solutions (approx. 3 mM) were prepared by dissolving the porphyrin powder (approx. 5 mg) in 2.3 ml of Tween 80 micelle solution (32 mM), keeping the solution in a ultrasonic bath for approx. 20 min, and subsequently diluting it 10-fold in the appropriate buffer. The final concentration of detergent was 3.2 mM, which is well above the critical micelle concentration of Tween 80 (i.e. 12 μM) [19].
SDS/PAGE, protein concentration determination and enzyme activity assay
Protein purity was estimated by SDS/PAGE [20] and was never less than 95%. Protein concentrations were determined by the bicinchoninic acid assay using BSA as standard. The enzymic activity was determined using the pyridine haemochromogen assay as described previously [21].
DSC: data acquisition and analysis
DSC measurements were performed on a MicroCal VP-DSC differential scanning calorimeter (MicroCal Inc.) with cell volumes of 0.517 ml, at temperatures ranging 15–100 °C and scan rates of 0.2, 0.5, 1.0, 1.2 and 1.5 °C/min. Before each run, at least two blank measurements were performed with the respective buffer in both compartments. The last of such runs was used as the baseline for the following measurement, with the protein solution in the sample compartment and the corresponding buffer in the reference compartment. Sample and reference solutions were properly degassed and carefully loaded into the calorimeter to eliminate bubbling effects. Each experiment was performed at least twice. Between experiments, both compartments were cleaned with water, followed by rinses with 0.1 M HCl and 0.1 M NaOH and lastly a thorough rinse with distilled water. Typically, the protein concentration was 15 μM and the experiments were performed in three different buffers: Bicine buffer (20 mM Bicine, pH 8, 0.5 M NaCl and 10% glycerol), Tris buffer (20 mM Tris/HCl, pH 8, 0.5 M NaCl and 10% glycerol) and phosphate buffer (20 mM potassium phosphate, pH 8, 0.5 M NaCl and 10% glycerol).
Raw calorimetric data were converted into heat capacity by subtracting the buffer baseline, determined under identical conditions, and dividing by the scan rate and the sample protein concentration. Thereafter the native baseline was subtracted from the original results in order to calculate excess heat capacities. The obtained data were subsequently analysed with the ORIGIN MicroCal software (MicroCal, LLC, Northampton, MA, U.S.A.). In all cases, the model used allowed the calculation of the denaturation temperature, Tm, the calorimetric enthalpy, ΔHcali, and the van't Hoff enthalpy, ΔHvHi. The ratio, r, between the calorimetric enthalpy and the van't Hoff enthalpy was calculated in all cases, in order to assess the validity of the two-state approximation [22,23].
ITC
Thermodynamic analysis of the porphyrin binding to ferrochelatase (or the variants E289Q or E289A) was performed using a titration calorimeter comprising a twin heat-conduction calorimeter (ThermoMetric AB, Järfälla, Sweden) with a 1 ml titration cell [24], a water bath and peripheral units (built at Lund University, Sweden). The system has been described in detail elsewhere [25]. A stirring speed of 70 rev./min was maintained with a gold stirrer [25] and the system was calibrated electrically, with a reproducibility of ±0.5%, by means of an insertion heater [26]. Ferrochelatase (15 or 20 μM final concentrations in 0.9 ml of any of the three buffers described above) previously loaded into the calorimetric cell, was titrated with the porphyrin solution by adding aliquots of 6.22–9.54 μl with a modified 500-μl gas-tight Hamilton syringe. The heats of dilution (of ferrochelatase and porphyrin) were determined in separate experiments. Each experiment consisted of 15–20 consecutive injections, and was repeated at least 3 times. The interval between injections was designed so that the voltage signal could return to the baseline, usually within 13 min. Data acquisition and syringe pump delivery were computer controlled, through the SIGMA software (Sven Hägg, Lund University, Lund, Sweden). The preparation and composition of any of the 3 buffers containing the protein and the porphyrin (i.e. mesoporphyrin) are described above, and the mesoporphyrin concentration was determined spectrophotometrically (i.e. at A399), so that the final mesoporphyrin concentration in the titrating solution was 0.3 mM. All experiments were performed at 298.15±0.01 K. The voltage-time peaks were integrated and the areas converted into the heat exchanged per injection by use of the calibration constant. The heat due to the binding reaction between porphyrin and enzyme was obtained as the difference between the heat of reaction and the corresponding heat of dilution. Thus the corrected heat for each injection can be transformed into the enthalpy of ligand–protein binding, ΔH, given per mol of added titrating agent. The data were analysed using a 1:1 binding stoichiometry. When ligand binding is coupled to a protonation/deprotonation event the measured enthalpy is buffer-dependent [27,28]. In order to verify whether porphyrin binding was coupled to a protonation or deprotonation reaction, the ligand–ferrochelatase binding process was studied in different buffers. If the binding reactions were to proceed with proton release or uptake, then the measured overall measured enthalpy will be different in different buffers, due to their different ionization enthalpies. Thus proton linkage contributions to the binding process could be determined by measuring the molar heat of binding in different buffers (i.e. Bicine, Tris and phosphate buffers). Furthermore, not only could the number of protons involved be calculated [see eqn (1)], but it also could be inferred whether they were released or taken up upon binding [from the sign of the slope of eqn (1)]. From the observed binding enthalpy in each buffer system, ΔHobs, we could therefore calculate the enthalpy change corrected for the ionization enthalpy of the buffer, ΔHo, i.e. the binding enthalpy that would be observed in a buffer with zero ionization enthalpy (ΔHbi=0), according to eqn (1) [27,28],
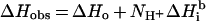 |
(1) |
as well as the number of protons (NH+) released (positive slope) from the buffer upon binding of the ligand to the protein.
RESULTS AND DISCUSSION
DSC thermograms for the thermal denaturation of ferrochelatase: the scanning rate effect and determination of thermodynamic transition parameters
The thermograms for the thermal denaturation of wild-type ferrochelatase in Tris buffer exhibited a single transition temperature, but indicated that the process was irreversible both in the absence or presence of porphyrin, as there was a deep decrease in the heat capacity values starting very soon after the onset of denaturation, and no heat capacity peaks were detected in reheating scans (results not shown). In fact, aggregation was also immediately apparent in the protein samples removed from the calorimetric cells. Denaturation was then studied in Bicine and phosphate buffers. In these two buffer systems, aggregation was apparent only at higher temperatures (much higher than Tm), probably just slightly distorting the high temperature side of the calorimetric curve. The major goal of the DSC experiments was to measure the thermodynamic parameters associated with the temperature-induced denaturation of ferrochelatase, in the absence or presence of ligand. However, with irreversible processes, the analysis of DSC thermograms may be hindered, as irreversible denaturation of proteins is a kinetic process, and thus not amenable to treatment by equilibrium thermodynamics [29,30]. Nevertheless, in some cases the analysis of the effect of the scanning rate on the DSC transitions validates the equilibrium thermodynamics analysis of irreversible DSC measurements [31,32], and makes possible a thermodynamic study of the protein's denaturation process. Briefly, theoretical analyses of irreversible DSC measurements based on the model of Lumry and Eyring [33] and further described in [34–36], which assumes that reversible unfolding of a protein is followed by a rate-limiting irreversible step, indicate that, at high scanning rates, kinetic distortions of DSC transitions due to irreversible processes must become negligible [31], thus allowing a full equilibrium treatment of the reversible denaturation step. In this light, we examined the effect of the heating scanning rate on the transition temperature for the thermal denaturation of ferrochelatase in Bicine buffer (Figure 1).
Figure 1. DSC thermograms of wild-type ferrochelatase.

(A) Effect of scan rate on melting temperature (Tm). The thermal denaturation of ferrochelatase (15 μM) was monitored in Bicine buffer (see the Experimental section for details). DSC thermograms were obtained at different scan rates: (□), 0.2 K/min (Tm=49.8 °C); (○), 0.5 K/min (Tm=50.3 °C); (Δ), 1 K/min (Tm=56.0 °C); (▲), 1.2 K/min (Tm=56.7 °C); (+), 1.5 K/min (Tm=56.9 °C). The shown DSC curves result from baseline subtraction of the original thermograms (see the Experimental section). Inset: plot of Tm versus heating rate. (B) Analysis of the unfolding of ferrochelatase in bicine buffer. Protein concentration was 15 μM, and the scan rate 1 °C/min. Buffer corrected results were normalized to protein concentration, and thereafter the native baseline was subtracted, so as to calculate excess heat capacity. The experimental data are shown as open triangles and the continuous line represents the fitted line to the ‘non-two-state’ option, with one transition (see the Experimental and Results and discussion sections for further details of the analysis).
Although the temperature associated with the maximum Cp increases with the increase in the scanning rate (i.e. 49.8 °C for 0.2 °C/min and 50.5 °C for 0.5 °C/min), the transition temperature approached a plateau with increasing heating rates. The observed initial increase associated with the lower scanning rates could not be accounted for by the time constant of the instrument. In fact, for the 1.0–1.5 °C/min range, the transition temperature, Tm, remained practically constant, with a value of 56.5±0.5 °C (Figure 1A). The small temperature variation is not significant if we consider both the ‘within sample’ uncertainty in transition temperatures (±0.5 °C) and the instrumental response time. Therefore, the transition temperature was considered to be independent of the scanning rate for the 1.0–1.5 °C/min range (Figure 1A, inset), which allowed us to analyse the thermal denaturation of ferrochelatase by equilibrium thermodynamics. Our fitting of the DSC data covered a limited temperature range (from 40 °C up to a few degrees above the temperature of maximum heat capacity), since the high temperature side of the denaturation curve can be slightly distorted due to protein aggregation. We used the option ‘non-two-state’ in the ORIGIN MicroCal software, in order to get the calorimetric and van't Hoff enthalpies (one example of the obtained fit can be seen in Figure 1B). The thermodynamic parameters of the transition in two different buffers (bicine and phosphate) are summarized in Table 1. Both the calorimetric enthalpy (ΔHcali) and the van't Hoff enthalpy (ΔHvHi) for wild-type ferrochelatase have similar values of approx. 400 kJ·mol−1 in Bicine and phosphate buffers, and consequently the ratios r are approx. 1 (Table 1), indicative of a co-operative, two-state transition denaturation process. The transition temperature for the thermal denaturation of ferrochelatase (56.0 °C) is surprisingly similar to the value of 54 °C obtained for human haemopexin, a plasma haem transporter that binds haem with a binding constant of approx. 1×1012 M−1 [37]. The similarity in the Tm values for ferrochelatase and haemopexin is curious, and one is tempted to speculate that it might be inherent to the haem-binding pocket, as both proteins bind haem, although clearly with different affinities. However, there is not an obvious common protein fold between haemopexin and ferrochelatase and the similarity between the Tm values might just be coincidental.
Table 1. Thermodynamic transition parameters for wild-type ferrochelatase in two buffer systems.
Thermodynamic parameters were obtained from the analysis of the DSC curves with the ORIGIN MicroCal software (see the Experimental section for details). The model used allowed the calculation of the denaturation temperature, Tm, calorimetric enthalpy (ΔHcali) and van't Hoff enthalpy (ΔHvHi). r is the calorimetric to Van't Hoff enthalpy ratio. The heating rate was 1.0 °C/min, and in each case at least three measurements were performed. The uncertainties reported are the S.D. of the obtained values.
| Buffer | Tm (°C) | ΔHical (kJ·mol−1) | ΔHvHi (kJ·mol−1) | r |
|---|---|---|---|---|
| Bicine | 56.0±0.3 | 477±5 | 403±3 | 1.2 |
| Phosphate | 55.8±0.3 | 417±6 | 444±7 | 0.94 |
DSC thermograms for the thermal denaturation of E289Q and E289A
Binding of mesoporphyrin to ferrochelatase (1:1 molar ratio) results in an enhanced stability of the protein, which is reflected in a shift of Tm from 56.0 °C to 58.3 °C (Table 2). Similarly, the mutation of Glu-289 with glutamine caused an increase in protein stability (Tm=61.0 °C, Table 2). Previously, E289Q was demonstrated to be purified with endogenous protoporphyrin [17], specifically bound in the active site [13]. Conceivably, the substitution of the carboxylate side-chain of Glu-289 with the carboxamide side-chain of glutamine stabilized the binding of the endogenously-bound porphyrin to a greater extent in E289Q than in the wild-type ferrochelatase–mesoporphyrin complex, and, in so doing, contributed to the stabilization of the variant as a whole. Increased transition temperatures and thermal stabilities upon binding of ligands to proteins have been previously observed [37–39]. Indeed, the binding of haem to apo-haemopexin caused a 12.5 °C increase in the Tm of haem–haemopexin in relation to that of the apo-protein, which correlated with an increase in the molar enthalpy [37].
Table 2. Thermodynamic transition parameters for ferrochelatase, with or without porphyrin ligand, and Glu-289 variants.
The thermograms were obtained from protein samples in Bicine buffer. The heating rate was 1.0 °C/min, and in each case at least three measurements were performed. The uncertainties reported are the S.D. of the obtained values. Thermodynamic parameters were obtained from the analysis of the DSC curves with the ORIGIN MicroCal software (see the Experimental section for details). The model used allowed the calculation of the denaturation temperature, Tm, calorimetric enthalpy (ΔHcali) and van't Hoff Enthalpy (ΔHvHi). r is the calorimetric to van't Hoff enthalpy ratio. FC, wild-type ferrochelatase; MP, mesoporphyrin IX.
| Tm (°C) | ΔHcali (kJ·mol−1) | ΔHvHi (kJ·mol−1) | r | |
|---|---|---|---|---|
| FC | 56.0±0.3 | 477±5 | 403±3 | 1.2 |
| FC+MP* | 58.3±0.3 | 506±10 | 319±3 | 1.6 |
| E289A | 57.5±0.1 | 920±9 | 185±2 | 5.0 |
| E289Q | 61.0±0.2 | 565±6 | 194±2 | 2.9 |
* FC and MP were reacted in a 1:1 molar ratio.
In contrast to the co-operativity for the thermal denaturation of wild-type ferrochelatase, both E289Q and E289A variants exhibited r values (for the ratio between the calorimetric and the van't Hoff enthalpies) much greater than 1, namely 3 for E289Q and 5 for E289A (Table 2). These values reflect a non-co-operative denaturation, i.e. the defined co-operative unit is smaller than the protein [23] and the denaturation proceeds with significantly populated intermediate states. Often mutations that affect either the interaction between protein domains or localized structure can bring about protein destabilization and a change in the degree of co-operativity of the transition, as very small changes in structure can result in significant changes in thermodynamic parameters [40,41]. Carra et al. [41] reported that mutagenesis of residues involved in maintaining electrostatic interactions between the two subdomains defining the active site of Staphylococcal nuclease resulted in decreased co-operativity of the structure of the protein and a three-state mechanism model (instead of a two-state) to best describe the folding energetics [41]. Further, for the E289A variant, the stabilizing effect provided with porphyrin binding, as shown in the wild-type ferrochelatase–porphyrin complex, can be opposed by the destabilizing effect of the point mutation (Table 1). Perhaps, the stabilizing effect of the endogenously-bound porphyrin is not sufficiently large to compensate for the destabilization induced by the substitution of the charged glutamate residue with the smaller, non-polar alanine residue.
Thermodynamic parameters of porphyrin binding to ferrochelatase
The thermodynamics of mesoporphyrin binding to ferrochelatase was investigated by ITC (Figure 2). Initially, experimental conditions were thoroughly scrutinized, given the poor solubility of porphyrin in aqueous solutions which implied the use of a detergent to stabilize the porphyrin in an aqueous milieu. In addition, ferrochelatase had to be maintained in a glycerol-containing buffer. Essentially, a series of prerequisites had to be fulfilled in order to examine the binding thermodynamics of a somewhat hydrophobic ligand (porphyrin) to a protein with membrane-associated properties (murine ferrochelatase). To avoid the interference of the heat of formation of detergent micelles, care was taken during the ITC experiments that the detergent (Tween 80) concentration was guaranteed to be above its critical micelle concentration. To ascertain that these requirements were met, several factors were tested and conclusions drawn. (i) In separate dilution experiments, the concentrated Tween 80 solution was titrated into the buffer, demonstrating a negligible heat effect and confirming that under our experimental conditions the micelles were only diluted and did not dissociate. (ii) Mesoporphyrin was stable as encapsulated in the micelles and no precipitate was ever found after titrations were completed, thus leading us to assume that mesoporphyrin only exchanged between the micelles and ferrochelatase. (iii) Although glycerol has a high exothermic heat of dilution in water [25], it exhibited an endothermic heat of dilution in 20 mM Tris, pH 8 (results not shown); thus to avoid potential calorimetric effects due to the differences in glycerol concentration in the buffers containing ferrochelatase and mesoporphyrin, both the ferrochelatase-containing solution added to the calorimetric cell and the mesoporphyrin solution used as titrating agent were prepared to contain the same glycerol concentration (i.e. 10%). (iv) Titration of buffer into the ferrochelatase-containing solution indicated that the heat of dilution of ferrochelatase was also negligible in the adopted experimental setup.
Figure 2. ITC of wild-type ferrochelatase with mesoporphyrin.

(A) Titration in Bicine buffer. Experiments were performed at 25 °C. The plot represents the integral heat for the titration in Bicine buffer of mesoporphyrin (336 μM, 8.6 μl/injection) into ferrochelatase (15 μM) versus the mesoporphyrin to ferrochelatase molar ratios (each point represents an injection). The continuous line is a guide for the eye and the broken lines show the crossing at a 1:1 binding stoichiometry. (B) Effect of buffer on the observed enthalpies. The observed enthalpy changes for the binding reaction of mesoporphirin to ferrochelatase (ΔHobs) in the three studied buffers are plotted as a function of the ionization enthalpy of the respective buffer as reported in [44]. The slope of the fitted curve yields the number of protons released by the buffer and the intercept yields the binding enthalpy (ΔHo) that would be observed in a buffer with ΔHionization=0 [see eqn (1) in the text].
The study was performed in the three buffer systems mentioned above: Tris, phosphate and Bicine. The calorimetric determination of KD for the binding of mesoporphyrin to ferrochelatase was not feasible due to too tight binding (KD=5 μM) [13] and therefore only the binding enthalpy could be obtained from the calorimetric experiments [42]. Figure 2(A) illustrates the heat evolved (integral heats) upon titration of wild-type ferrochelatase with mesoporphyrin in Bicine buffer. A binding stoichiometry of 1:1 for ferrochelatase–mesoporphyrin was obtained regardless of which of the three buffers titrations were performed.
In addition, a protonation/deprotonation event appears to be coupled to the binding of porphyrin to ferrochelatase, and thus the observed enthalpy change (ΔHobs) depends also on the enthalpy change associated with the ionization of the buffer (eqn 1). From the plot of the observed enthalpy change as a function of the values reported in the literature for the ionization enthalpy of the respective buffers [44], it could be inferred that the binding of mesoporphyrin to ferrochelatase is accompanied by the release of two protons from the buffer (Figure 2B). The binding of mesoporphyrin to ferrochelatase is an exothermic process, with a binding enthalpy of −97.1 kJ·mol−1 at 298.15 K (ΔHo, calculated according to eqn 1). This value is greater than those reported for the binding enthalpy of small inhibitor molecules to proteins with a similar binding constant to that of mesoporphyrin to ferrochelatase [43].
From the corrected binding enthalpy (−97.1 kJ·mol−1) and assuming a binding constant of Ka=2×105 M−1 (based on a dissociation constant, KD, of 5.0 μM [13]), the change in Gibbs energy for the binding reaction was calculated as ΔG°=−30.2 kJ·mol−1 at T=298.15 K and the entropic contribution as TΔS°=−66.9 kJ·mol−1, indicating that the binding of mesoporphyrin to ferrochelatase proceeds with a decrease in entropy. This unfavourable entropy change is nevertheless insufficient to prevail over the highly exothermic enthalpy, resulting in an overall favourable binding reaction (ΔG°=−30.2 kJ·mol−1). A decrease in entropy is, in principle, to be expected for a simple binding reaction. However, in many cases of binding of ligands to proteins the opposite is observed. This fact has been interpreted to be due to the hydrophobic effect as a major driving force in the stabilization of the protein–ligand complexes, since a part of the protein hydrophobic surface is buried upon binding [45,46]. It is important to emphasize the complexity of the system being studied, and caution prevents us from making an interpretation solely in terms of entropy changes. First, we assumed that the ligand (mesoporphyrin), which is hydrophobic and was encapsulated in micelles, was always transferred from the micelles to the protein-binding site; therefore the hydrophobic contribution to the binding process should not be apparent under our experimental set-up. Secondly, the binding seemed to occur with the uptake of two protons, which possibly could also contribute to an entropy decrease. Thus, although we refrain from a further interpretation for the observed entropy decrease, we would like to stress that the observed proton linkage with the binding of mesoporphyrin to wild-type ferrochelatase suggests that hydrogen bonding is an important factor in the definition of the free energy of the binding reaction.
The stabilization conferred on the E289 variants by the specifically bound protoporphyrin (Table 2) is consistent with an impaired catalytic activity of the variants. Both Glu-289 variants are known to be less active than wild-type ferrochelatase, i.e. under turnover conditions, both possess activities in the order of 1% of that of the wild-type enzyme and both can convert the bound porphyrin to Zn-porphyrin, but are incapable of product release [17]. This impairment in enzymic activity correlates with the fact that if the porphyrin substrate is in a more stable environment, as in the case of the E289 variants, then the activation energy barrier to reach the transition state is greater and the conversion to product is less favoured. In Figure 3, the three-dimensional structure of B. subtilis ferrochelatase co-crystallized with N-methylmesoporphyrin exhibits the distortion brought on the ring A of the porphyrin macrocycle. The distortion present is of the saddled-ruffled type, the same type of distortion that the porphyrin substrate is thought to undergo during the ferrochelatase catalytic cycle [5,13,16]. Significantly, the side-chain oxygen atoms of Glu-264 (B. subtilis numbering, which corresponds to murine ferrochelatase Glu-289) are at a distance of approx. 4 Å from the tetrapyrrole ring A, which implies a close interaction between the porphyrin substrate and this active-site residue. Thus it is not surprising that its replacement would affect the stability and thermodynamics of porphyrin binding and consequently the catalytic process. Studies are underway to elucidate the role of conserved putative porphyrin binding residues on the thermodynamic stability of mammalian ferrochelatase.
Figure 3. Three-dimensional structure of B. subtilis ferrochelatase co-crystallized with N-methylmesoporphyrin.

The conserved Glu-264 (Glu-289 in murine ferrochelatase) and the bound porphyrin are highlighted. The image of the porphyrin–ferrochelatase complex (accession code 1C1H from the Protein DataBank) was generated using RasMol 2.7.2.1.
Acknowledgments
We acknowledge Professor Helena Santos and Tiago Faria (ITQB/UNL) for access to and assistance in using the MicroCal VP-DSC calorimeter, João Carita (ITQB/UNL) for growing E. coli cells and Isabel Pacheco (ITQB/UNL) for assistance with gel filtration chromatography. M.B. acknowledges Dr Arne Schön (Department of Biology, The Johns Hopkins University, Baltimore, MA, U.S.A.) for invaluable comments and discussions regarding the DSC results. This work was supported by Fundação para a Ciência e Tecnologia to CIQ(UP), Unidade de Investigação 81 (to M.B., G.B. and F.A.), POCTI/34973/BME/2000 (to R.F. and V.P.) and the American Cancer Society (grant number RSG-96-05106-TBE to G.C.F.). F.A. is recipient of a predoctoral fellowship (PRAXIS XXI/BD/18523/98) and G.B. is recipient of a post-doctoral fellowship (SFRH/BPD/5668/2001).
References
- 1.Franco R., Lloyd S. G., Moura J. J. G., Moura I., Huynh B. H., Ferreira G. C. Ferrochelatase: a new iron-sulfur center-containing enzyme. In: Ferreira G. C., Franco R., Moura J. J. G., editors. Iron Metabolism: Inorganic Biochemistry and Regulatory Mechanisms. Weinheim: Wiley-VCH; 1999. pp. 35–50. [Google Scholar]
- 2.Dailey H. A., Dailey T. A., Wu C. K., Medlock A. E., Wang K. F., Rose J. P., Wang B. C. Ferrochelatase at the millennium: structures, mechanisms and [2Fe–2S] clusters. Cell. Mol. Life Sci. 2000;57:1909–1926. doi: 10.1007/PL00000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deybach J. C. Porphyria. Painful photosensitivity. Lancet. 2001;358(suppl.):S49. doi: 10.1016/s0140-6736(01)07061-1. [DOI] [PubMed] [Google Scholar]
- 4.Al-Karadaghi S., Hansson M., Nikonov S., Jönsson B., Hederstedt L. Crystal structure of ferrochelatase: the terminal enzyme in heme biosynthesis. Structure (London) 1997;5:1501–1510. doi: 10.1016/s0969-2126(97)00299-2. [DOI] [PubMed] [Google Scholar]
- 5.Lecerof D., Fodje M., Hansson A., Hansson M., Al-Karadaghi S. Structural and mechanistic basis of porphyrin metallation by ferrochelatase. J. Mol. Biol. 2000;297:221–232. doi: 10.1006/jmbi.2000.3569. [DOI] [PubMed] [Google Scholar]
- 6.Karlberg T., Lecerof D., Gora M., Silvegren G., Labbe-Bois R., Hansson M., Al-Karadaghi S. Metal binding to Saccharomyces cerevisiae ferrochelatase. Biochemistry. 2002;41:13499–13506. doi: 10.1021/bi0260785. [DOI] [PubMed] [Google Scholar]
- 7.Wu C.-K., Dailey H. A., Rose J. P., Burden A., Sellers V. M., Wang B.-C. The 2.0 Å structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 2001;8:156–160. doi: 10.1038/84152. [DOI] [PubMed] [Google Scholar]
- 8.Gora M., Rytka J., Labbe-Bois R. Activity and cellular location in Saccharomyces cerevisiae of chimeric mouse/yeast and Bacillus subtilis/yeast ferrochelatases. Arch. Biochem. Biophys. 1999;361:231–240. doi: 10.1006/abbi.1998.0990. [DOI] [PubMed] [Google Scholar]
- 9.Medlock A. E., Dailey H. A. Examination of the activity of carboxyl-terminal chimeric constructs of human and yeast ferrochelatases. Biochemistry. 2000;39:7461–7467. doi: 10.1021/bi000134p. [DOI] [PubMed] [Google Scholar]
- 10.Dailey T. A., Dailey H. A. Identification of [2Fe–2S] clusters in microbial ferrochelatases. J. Bacteriol. 2002;184:2460–2464. doi: 10.1128/JB.184.9.2460-2464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreira G. C., Franco R., Lloyd S. G., Pereira A. S., Moura I., Moura J. J. G., Huynh B. H. Mammalian ferrochelatase, a new addition to the metalloenzyme family. J. Biol. Chem. 1994;269:7062–7065. [PubMed] [Google Scholar]
- 12.Dailey H. A., Finnegan M. G., Johnson M. K. Human ferrochelatase is an iron-sulfur protein. Biochemistry. 1994;33:403–407. doi: 10.1021/bi00168a003. [DOI] [PubMed] [Google Scholar]
- 13.Franco R., Ma J.-G., Lu Y., Ferreira G. C., Shelnutt J. A. Porphyrin interactions with wild-type and mutant mouse ferrochelatase. Biochemistry. 2000;39:2517–2529. doi: 10.1021/bi991346t. [DOI] [PubMed] [Google Scholar]
- 14.Blackwood M. E., Rush T. S., Medlock A., Dailey H. A., Spiro T. G. Resonance Raman spectra of ferrochelatase reveal porphyrin distortion upon metal binding. J. Am. Chem. Soc. 1997;119:12170–12174. [Google Scholar]
- 15.Blackwood M. E., Rush T. S., Rosenberg F., Schultz P. G., Spiro T. G. Alternative modes of substrate distortion in enzyme and antibody catalyzed ferrochelation reactions. Biochemistry. 1998;37:779–782. doi: 10.1021/bi972616f. [DOI] [PubMed] [Google Scholar]
- 16.Lu Y., Sousa A., Franco R., Mangravita A., Ferreira G. C., Moura I., Shelnutt J. A. Binding of protoporphyrin IX and metal derivatives to the active site of wild-type mouse ferrochelatase at low porphyrin-to-protein ratios. Biochemistry. 2002;41:8253–8262. doi: 10.1021/bi025569m. [DOI] [PubMed] [Google Scholar]
- 17.Franco R., Pereira A. S., Tavares P., Mangravita A., Barber M. J., Moura I., Ferreira G. C. Substitution of murine ferrochelatase glutamate-287 with glutamine or alanine leads to porphyrin-bound variants. Biochem. J. 2001;356:217–222. doi: 10.1042/0264-6021:3560217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira G. C. Mammalian ferrochelatase, overexpression in Escherichia coli as a soluble protein, purification and characterization. J. Biol. Chem. 1994;269:4396–4400. [PubMed] [Google Scholar]
- 19.Bhairi S. M. Calbiochem–Novabiochem. U.S.A.: San Diego; 2001. A Guide to the Properties and Uses of Detergents in Biological Systems. [Google Scholar]
- 20.Laemmli U. K. Cleavage of structural proteins during assembly of head of bacteriophage-T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Franco R., Moura J. J. G., Moura I., Lloyd S. G., Huynh B. H., Forbes W. S., Ferreira G. C. Characterization of the iron-binding site in mammalian ferrochelatase by kinetic and Mössbauer methods. J. Biol. Chem. 1995;270:26352–26357. doi: 10.1074/jbc.270.44.26352. [DOI] [PubMed] [Google Scholar]
- 22.Lumry R., Biltonen R. Validity of the ‘two-state’ hypothesis for conformational transitions of proteins. Biopolymers. 1966;4:917–944. doi: 10.1002/bip.1966.360040808. [DOI] [PubMed] [Google Scholar]
- 23.Freire E. Thermal denaturation methods in the study of protein unfolding. Methods Enzymol. 1995;259:144–168. doi: 10.1016/0076-6879(95)59042-0. [DOI] [PubMed] [Google Scholar]
- 24.Bäckman P., Bastos M., Hallén D., Lönnbro P., Wadsö I. Heat conduction calorimeters: time constants, sensitivity and fast titration experiments. J. Biochem. Biophys. Meth. 1994;28:85–100. doi: 10.1016/0165-022x(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 25.Bastos M., Afonso M., Caçote M. H. M., Ramos M. J. Interactions in the model system α-cyclodextrin-glycerol. Experimental and theoretical study. J. Chem. Soc. Faraday Trans. 1997;93:2061–2067. [Google Scholar]
- 26.Briggner L-E., Wadsö I. Test and calibration processes for microcalorimeters, with special reference to heat conduction instruments used with aqueous systems. J. Biochem. Biophys. Meth. 1991;22:101–118. doi: 10.1016/0165-022x(91)90023-p. [DOI] [PubMed] [Google Scholar]
- 27.Eftink M., Biltonen R. Thermodynamics of interacting biological systems. In: Beezer A. E., editor. Biological Microcalorimetry. San Diego: Academic Press; 1980. pp. 343–412. [Google Scholar]
- 28.Baker B. M., Murphy K. P. Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry. Biophys. J. 1996;71:2049–2055. doi: 10.1016/S0006-3495(96)79403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Ruiz J. M. Theoretical analysis of Lumry-Eyring models in differential scanning calorimetry. Biophys. J. 1992;61:921–935. doi: 10.1016/S0006-3495(92)81899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conejero-Lara F., Mateo P. L., Aviles F. X., Sanchez-Ruiz J. M. Effect of Zn2+ on the thermal denaturation of carboxypeptidase B. Biochemistry. 1991;30:2067–2072. doi: 10.1021/bi00222a010. [DOI] [PubMed] [Google Scholar]
- 31.Thórólfsoon M., Ibarra-Molero B., Fojan P., Petersen S. B., Sanchez-Ruiz J. M., Martínez A. L-Phenylalanine binding and domain organization in human phenylalanine hydroxylase: a differential scanning calorimetry study. Biochemistry. 2002;41:7573–7585. doi: 10.1021/bi0160720. [DOI] [PubMed] [Google Scholar]
- 32.Vogl T., Jatzke C., Hinz H.-J. Thermodynamic stability of Annexin V E17G: equilibrium parameters from an irreversible unfolding reaction. Biochemistry. 1997;36:1657–1668. doi: 10.1021/bi962163z. [DOI] [PubMed] [Google Scholar]
- 33.Lumry R., Eyring H. Conformation changes of proteins. J. Phys. Chem. 1954;58:110–120. [Google Scholar]
- 34.Galisteo M. L., Mateo P. L., Sanchez-Ruiz J. M. Kinetic study on the irreversible thermal denaturation of yeast phosphoglycerate kinase. Biochemistry. 1991;30:2061–2066. doi: 10.1021/bi00222a009. [DOI] [PubMed] [Google Scholar]
- 35.Freire E., van Osdol W. W., Mayorga O. L., Sanchez-Ruiz J. M. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Annu. Rev. Biophys. Biophys. Chem. 1990;19:159–188. doi: 10.1146/annurev.bb.19.060190.001111. [DOI] [PubMed] [Google Scholar]
- 36.Hernandez-Arana A., Rojo-Dominguez A., Altamirano M. M., Calcagno M. L. Differential scanning calorimetry of the irreversible denaturation of Escherichia coli glucosamine-6-phosphate deaminase. denaturation of Escherichia coli glucosamine-6-phosphate deaminase. Biochemistry. 1993;32:3644–3648. doi: 10.1021/bi00065a017. [DOI] [PubMed] [Google Scholar]
- 37.Wu M. L., Morgan W. T. Characterization of hemopexin and its interaction with heme by differential scanning calorimetry and circular dichroism. Biochemistry. 1993;32:7216–7222. doi: 10.1021/bi00079a018. [DOI] [PubMed] [Google Scholar]
- 38.Waldron T. T., Murphy K. P. Stabilization of proteins by ligand binding: application to drug screening and determination of unfolding energetics. Biochemistry. 2003;42:5058–5064. doi: 10.1021/bi034212v. [DOI] [PubMed] [Google Scholar]
- 39.Srinivas V. R., Singha N. C., Schwarz F. P., Surolia A. Differential scanning calorimetric studies of the glycoprotein, winged bean acidic lectin, isolated from the seeds of Psophocarpus tetrogonolobus. FEBS Lett. 1998;425:57–60. doi: 10.1016/s0014-5793(98)00197-5. [DOI] [PubMed] [Google Scholar]
- 40.Privalov G. P., Privalov P. L. Problems and prospects in microcalorimetry of biological macromolecules. Methods Enzymol. 2000;323:31–63. doi: 10.1016/s0076-6879(00)23360-0. [DOI] [PubMed] [Google Scholar]
- 41.Carra J. H., Anderson E. A., Privalov P. L. Three-state thermodynamic analysis of the denaturation of staphylococcal nuclease mutants. Biochemistry. 1994;33:10842–10850. [Google Scholar]
- 42.Freire E., Mayorga O. L., Straume M. Isothermal titration calorimetry. Anal. Chem. 1990;62:A950–A959. [Google Scholar]
- 43.Menze M. A., Hellmann N., Decker H., Grieshaber M. K. Binding of urate and caffeine to hemocyanin of the lobster Homarus vulgaris (E.) as studied by isothermal titration calorimetry. Biochemistry. 2000;39:10806–10811. doi: 10.1021/bi000008l. [DOI] [PubMed] [Google Scholar]
- 44.Goldberg R. N., Kishore N., Lennen R. M. Thermodynamic quantities for the ionization reactions of buffers. J. Phys. Chem. Ref. Data. 2002;31:231–370. [Google Scholar]
- 45.Baker B. M., Murphy K. P. Dissecting the energetics of a protein–protein interaction: the binding of ovomucoid third domain to elastase. J. Mol. Biol. 1997;268:557–569. doi: 10.1006/jmbi.1997.0977. [DOI] [PubMed] [Google Scholar]
- 46.Guzmán-Casado M., Sánchez-Ruiz J. M., El Harrous M., Giménez-Gallego G., Parrody-Morreale A. Energetics of myo-inositol hexasulphate binding to human acidic fibroblast growth factor. Effect of ionic strength and temperature. Eur. J. Biochem. 2000;267:3477–3486. doi: 10.1046/j.1432-1327.2000.01371.x. [DOI] [PubMed] [Google Scholar]
- 47.Shi Z., Ferreira G. C. Probing the active site loop motif of murine ferrochelatase by random mutagenesis. J. Biol. Chem. 2004;279:19977–19986. doi: 10.1074/jbc.M313821200. [DOI] [PubMed] [Google Scholar]