

Abstract
Echistatin is a potent antagonist of the integrins αvβ3, α5β1 and αIIbβ3. Its full inhibitory activity depends on an RGD (Arg-Gly-Asp) motif expressed at the tip of the integrin-binding loop and on its C-terminal tail. Previous NMR structures of echistatin showed a poorly defined integrin-recognition sequence and an incomplete C-terminal tail, which left the molecular basis of the functional synergy between the RGD loop and the C-terminal region unresolved. We report a high-resolution structure of echistatin and an analysis of its internal motions by off-resonance ROESY (rotating-frame Overhauser enhancement spectroscopy). The full-length C-terminal polypeptide is visible as a β-hairpin running parallel to the RGD loop and exposing at the tip residues Pro43, His44 and Lys45. The side chains of the amino acids of the RGD motif have well-defined conformations. The integrin-binding loop displays an overall movement with maximal amplitude of 30°. Internal angular motions in the 100–300 ps timescale indicate increased flexibility for the backbone atoms at the base of the integrin-recognition loop. In addition, backbone atoms of the amino acids Ala23 (flanking the R24GD26 tripeptide) and Asp26 of the integrin-binding motif showed increased angular mobility, suggesting the existence of major and minor hinge effects at the base and the tip, respectively, of the RGD loop. A strong network of NOEs (nuclear Overhauser effects) between residues of the RGD loop and the C-terminal tail indicate concerted motions between these two functional regions. A full-length echistatin–αvβ3 docking model suggests that echistatin's C-terminal amino acids may contact αv-subunit residues and provides new insights to delineate structure–function correlations.
Keywords: RGD disintegrin, echistatin, integrin, NMR protein dynamics determination, off-resonance rotating-frame Overhauser enhancement spectroscopy (off-resonance ROESY)
Abbreviations: NOE, nuclear Overhauser effect; R.M.S.D., root mean square deviation; ROESY, rotating-frame Overhauser enhancement spectroscopy
INTRODUCTION
Venoms from Viperidae (vipers) and Crotalidae (rattlesnakes) snakes contain a number of different proteins that interfere with the coagulation cascade, the normal haemostatic system and tissue repair. Consequently, envenomations by these snakes generally results in persistent bleeding. Haemorrhage is the result of the synergistic action of certain metalloproteinases that degrade the extracellular matrix surrounding blood vessels and proteins that interfere with haemostasis. These proteins can be grouped into a few major protein families, including enzymes (serine proteinases, Zn2+-dependent metalloproteinases of the reprolysin family and group II phospholipase A2 isoenzymes) and proteins with no enzymic activity (C-type lectins and disintegrins) [1,2].
Snake venom haemorrhagic metalloproteinases have been classified into four classes, according to their domain structure [3]. PI metalloproteinases (20–30 kDa) are single-domain proteins with relatively weak haemorrhagic activity. The class PII metalloproteinases (30–60 kDa) contain a disintegrin domain at the C-terminus of a metalloproteinase domain structurally similar to that in the class PI. Haemorrhagins of the PIII class are large toxins (60–100 kDa) with the most potent activity, and comprise multidomain enzymes built up by an N-terminal metalloproteinase domain and C-terminal disintegrin-like and cysteine-rich domains. The metalloproteinases of the PIV group contain the mosaic structure of the PIII haemorrhagins and an additional C-type lectin module disulphide-bonded to the cysteine-rich domain.
Disintegrins are released in the venoms by proteolytic processing of PII metalloproteinases [4], and inhibit integrin–ligand interactions. According to their polypeptide length and the number of disulphide bonds, disintegrins have been classified into five groups, including monomeric (short, medium-sized, long and PIII) and dimeric proteins [5]. The structural diversity of disintegrins has been achieved during evolution through the selective loss of disulphide bonds [5]. The integrin inhibitory activity of disintegrins depends on the appropriate pairing of cysteine residues which determine the conformation of the inhibitory loop. In most single-chain disintegrins, the active sequence is the tripeptide RGD (Arg-Gly-Asp) [5,6]. RGD-containing disintegrins show different binding affinity and selectivity towards integrins, which recognize the RGD sequence in their ligands [7] (i.e. αIIbβ3, the platelet fibrinogen receptor; αvβ3, the vitronectin receptor; and α5β1, the fibronectin receptor). Structural studies of several short (echistatin) and medium (kistrin, flavoridin, albolabrin, salmosin and trimestatin) RGD-disintegrins revealed that the active tripeptide is located at the apex of a mobile loop protruding 14–17 Å (1 Å=0.1 nm) from the protein core [8–13].
Echistatin, a 49-residue RGD-containing peptide isolated from the venom of the saw scaled viper (Echis sochureki carinatus) [14], is the most thoroughly characterized disintegrin. Residues adjacent C-terminally to the primary integrin-binding motif of echistatin, R24GD26, contribute to the selective inhibition of αIIbβ3, αvβ3 and α5β1 integrins by echistatin [15]. Thus, whereas tryptophan favours the recognition of αIIbβ3 at position 27, aspartic acid appears to be critical for inhibiting integrins αvβ3 and α5β1. In addition, replacement of Met28 with asparagine completely abolished echistatin's ability to block each of these disintegrins, while substitution of leucine for Met28 selectively decreased the inhibitory activity of echistatin towards integrin α5β1 only. A number of studies also support a functional role for the C-terminal residues of echistatin in modulating the binding affinity of the disintegrin towards integrin αIIbβ3. Hence, an echistatin C-terminal synthetic peptide inhibited the binding of echistatin to integrin αIIbβ3, activated the integrin to bind immobilized ligands, induced the expression of activation-dependent conformational epitopes and increased fibrinogen binding to peptide-treated platelets [16]. In addition, deletion of the sequence P40RNP43 (Pro40-Arg-Asn-Pro43) and replacement of echistatin's C-terminal sequence [H44KGPAT49 (His44-Lys-Gly-Pro-Ala-Thr49)] with that of eristostatin [WNG (Trp-Asn-Gly)] [15–17] both decreased the inhibitory potential of the mutated echistatin on ADP-induced platelet aggregation. As a whole, these data indicate that the C-terminal tail may act in synergy with the integrin-binding loop to endow echistatin with high affinity and selectiveness for integrin receptors.
In medium-resolution NMR solution structures of echistatin reported previously [8,16–20] [PDB (Protein Data Bank) accession 2ECH], the N- and the C-terminal polypeptide stretches and the loop bearing the RGD recognition sequence show large dispersion, and from residue 42 onwards the protein is essentially disordered. These structural features, which have been interpreted as a realistic representation of the relative disorder of these regions in solution, precluded an in-depth understanding of the conformation and the relative orientation of the functional epitopes of echistatin. We have reported recently the solution structure [21] and internal backbone angular motions [22] of the short disintegrin obtustatin from the venom of Vipera lebetina obtusa (PDB code 1MPZ). The major finding from these studies was that the integrin-binding loop and the C-terminal tail displayed concerted motions in the 100–300 ps timescale, which can be interpreted by hinge movements articulated at residues located at the base of the integrin-binding loop (Trp20, His27, Tyr28) and in the C-terminal tail (Cys36 and Leu38). Since obtustatin and echistatin share identical disulphide connectivities and are highly homologous, a similar type of hinge motion may be expected. The aims of the present study were to refine the structure of echistatin, and to analyse in detail the internal motions of the backbone and side chain atoms of the disintegrin by homonuclear NMR using spectra recorded with triple-resonance cryogenic probes at 500 and 600 MHz. In contrast with previous models, our results show that the full-length C-terminal region and the active loop of echistatin have well-defined conformations. The C-terminal tail of echistatin folds as a β-hairpin running parallel to, and displaying concerted motions with, the RGD loop. These two functional regions of echistatin form a conformational epitope that is engaged in extensive interactions with target integrin receptors.
MATERIALS AND METHODS
Sample preparation
Echistatin was purified from Echis carinatus sochureki venom, purchased from Latoxan (Valence, France), by reversed-phase HPLC as described in [14]. Protein purity was assessed by SDS/PAGE, N-terminal sequencing (with an Applied Biosystems Procise 492 sequencer) and MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight) MS (using an Applied Biosystems DE-Pro spectrometer and α-hydroxycinammic acid saturated in 70% acetonitrile, containing 0.1% trifluoroacetic acid as the matrix). The complete amino acid sequence was determined in a single run, and it was identical with that of entry P17347 of the SwissProt database (http://us.expasy.org/sprot). The isotope-averaged molecular mass of echistatin was 5417 Da, and this value did not change upon incubation of the protein (5 mg/ml in 100 mM ammonium bicarbonate, pH 8.5, containing 5 M guanidinium chloride) with 50 mM iodoacetamide, indicating that the eight cysteine residues of echistatin were engaged in disulphide bonds. For NMR structure determination, echistatin was dissolved in H2O/2H2O (19:1) at a final concentration of 3 mM in a 5 mm Wilmad NMR tube. The solution was adjusted to pH 3.0 with HCl.
NMR spectroscopy
All two-dimensional spectra were recorded on Bruker Avance spectrometers working at proton frequencies of 500 and 600 MHz. Triple-resonance cryogenic NMR probes were used for all the measurements. Spectra were processed with XWIN-NMR, version 3.1, and analysed with SPARKY [23] software packages on a Pentium 4 workstation running the Linux operating system. Homonuclear two-dimensional spectra for structure determination were recorded at 298 K and included NOESY spectra at 500 and 600 MHz, with 100 and 200 ms of mixing time. In these experiments, solvent signal suppression was achieved by using the WATERGATE pulse sequence [24]. Recycling relaxation delay between transients was 2 s in all the NOESY spectra. Assignments of NOESY and ROESY (rotating-frame Overhauser enhancement spectroscopy) cross-peaks for structure determination and off-resonance ROESY relaxation analysis were assigned using echistatin resonances from the BioMagResBank (BMRB entry 2204 at http://www.bmrb.wisc.edu) with minor corrections.
Structure calculations
An initial set of unambiguous, non-overlapped NOE (nuclear Overhauser effect) cross-peaks was assigned manually. A preliminary set of structures was then obtained and used for further NOE cross-peak assignment by semi-automated analysis with the NOAH routine implemented in the DYANA software package [25]. Structural restraints used for NOAH semi-automated analysis included only the well-established disulphide bonds (Cys2–Cys11, Cys7–Cys32, Cys8–Cys37 and Cys20–Cys39) [26,27] and the manually assigned NOEs. These restraints were included in the calculation at the first cycle. The new set of extended structural restraints was then applied in further cycles of DYANA simulated annealing torsion angle dynamics for the generation of the final conformers. The final set of assigned NOE cross-peaks included a total of 977, of which 440 were structurally meaningful (Table 1). The 20 conformers with the best DYANA target function were refined further with one cycle of simulated annealing standard protocol in XPLOR [28]. All the calculation parameters concerning the simulated annealing protocol (i.e. NOE and van der Waals weighting, temperature, dielectric constants, and rates of heating and cooling) were set to the default values recommended by the user manual of the software.
Table 1. Summary of meaningful structural restraints.
| Type | Number |
|---|---|
| Total NOE-derived restraints | 440 |
| Intra-residual | 47 |
| Sequential | 101 |
| Backbone–backbone | 43 |
| Backbone–side chain | 11 |
| Side chain–side chain | 47 |
| Medium range (less than four residues apart) | 120 |
| Backbone–backbone | 32 |
| Backbone–side chain | 37 |
| Side chain–side chain | 51 |
| Long range | 172 |
| Disulphide bond constraints | 4 |
An overall amplitude of motion for the recognition loop was calculated based on the best 20 energy-refined DYANA models. In a first step, the disulphide cores of the molecules were superimposed. Then the disulphide core atom closest to the recognition loop (atom A) and the recognition loop atom furthest from the core (atom B) were identified. A triangle was built using the positions of atom A and the most distant positions of atom B (positions B1 and B2). The angle between the lines A–B1 and A–B2 was taken as the amplitude of motion of the loop.
Off-resonance ROESY relaxation analysis
Internal motions were analysed using the off-resonance ROESY method described by Schleucher and Wijmenga [29]. Briefly, cross-peak build-up rates in NOESY and ROESY spectra can be described as:
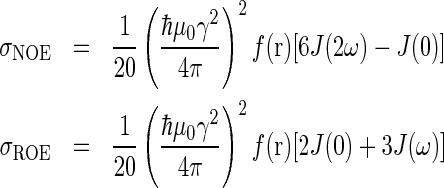 |
(1) |
where f(r) depends only on the radial component of internal motions, whereas J(ω), the spectral density function, depends only on the angular component of the internal motions. Thus the ratio of NOE and ROE (rotating-frame Overhauser effect) rates is considered to be independent of the radial component:
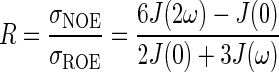 |
(3) |
In a ROESY experiment, the angle θi at which a spin i with a chemical shift Ωi will be locked by a spin lock of field strength γB1 and an offset Δ is defined by:
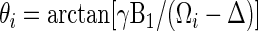 |
(4) |
Unmodified NOESY and ROESY spectra can be associated with values of the θ angle equal to 0° and 90° respectively. Therefore the angle at which the NOE and ROE effect contributions are equal, and thus the spectral signal is cancelled, provides a measure of the ratio R:
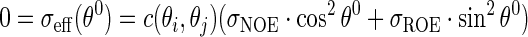 |
(5) |
where c(θi, θj) is a scaling factor that does not affect the θ0 value.
R can be determined measuring the value of θ at which the NOE and ROE contributions cancel. This θ value relates to angular internal motions:
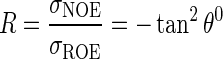 |
(6) |
Since the spectral density J(ω) depends only on the angular component of internal motions (eqn 3), low and high values of θ0 are inversely proportional to the degree of angular flexibility of a given vector.
Variations of θ0 values are largest for motions in the timescale of 100–300 ps and therefore these motions are the main contributors to the internal dynamics detected in the present study. In this timescale (τe values of 100–300 ps), θ0 appears to exhibit a parallel behaviour to the maximum of the S2 Lipari–Szabo order parameter. Thus θ0 provides an estimation of the relative angular flexibility for a given NOE vector in a similar fashion to that extracted from relative differences in S2 for the 15N–1H or 13C–1H bond vectors in traditional heteronuclear relaxation studies.
For the determination of internal motions of echistatin, an independent set of off-resonance ROESY spectra was recorded on each spectrometer at 298 K as described in [22,29]. In general, experimental parameters were as similar as possible to those reported previously for obtustatin [22]. Relaxation delay was 3.0 s including pre-saturation of water resonance. All ROESY experiments were recorded with a mixing time of 100 ms, spectral width of 10 p.p.m. and transmitter offset at the water position equivalent to 4.69 p.p.m. at 25 °C. For each experiment, 512 t1 and 2048 t2 complex data points were acquired. All experiments had 32 scans. For detecting positive cross-peaks, as an indicative of chemical exchange processes, a regular ROESY (on-resonance) was recorded at each spectrometer with parameters identical with those of the off-resonance ROESY experiments. Values for γB1 were 7180 and 9259 for the 500 and 600 MHz experiments respectively, corresponding to approx. 50% of the used spectral width, as recommended in the literature [29]. Table 2 shows values of frequency offsets and equivalent θ values at the transmitter frequency (middle of the spectrum) used in all off-resonance ROESY experiments for evaluation of the optimum range of offset frequencies. ROESY cross-peaks were fitted to Lorentzian curve peak shapes and integrated using SPARKY internal commands. ROESY cross-peaks showing severe overlapping or very low signal-to-noise ratios were discounted from further analysis. Graphical representation, second-order polynomial fitting and zero-crossing identification for cross-peak intensity against θ dependence were carried out using home-made Gnuplot macros (http://www.gnuplot.info/) and UNIX shell scripts.
Table 2. Experimental parameters used for recording off-resonance ROESY spectra at 500 and 600 MHz proton frequencies.
| 500 MHz | 600 MHz | ||
|---|---|---|---|
| Offset (Hz) | θc* (°) | Offset (Hz) | θc* (°) |
| 21000 | 21.0 | 22200 | 25.5 |
| 18000 | 24.6 | 20000 | 28.0 |
| 16000 | 27.7 | 18200 | 31.0 |
| 15000 | 29.6 | 16500 | 34.0 |
| 13000 | 34.0 | 15000 | 37.0 |
| 11000 | 39.9 | 14200 | 39.1 |
| 10000 | 43.1 | 13100 | 42.0 |
| 12000 | 45.0 | ||
| 11100 | 48.2 | ||
* θc is the value of θ of the frequency at the middle of the spectrum.
Hydrodynamic calculations
For comparison with experimental data and for an initial estimation of the global correlation time τm, HydroNMR version 5.a software [30] was employed with the echistatin's NMR structure reported in the present study (PDB code 1RO3). Strong cross-linking by four disulphide bonds along with an R.M.S.D. (root mean square deviation) value of 1.2 Å for all residues among the 20 NMR conformers of echistatin (0.7 Å for the 32 best-defined residues) indicated that echistatin fulfils the level of overall structure globularity and rigidity required for this type of analysis. Temperature and solvent viscosity for calculations were set to 25 °C and 0.010 poises respectively, as recommended in the software manual. Theoretical rigid limit values of θ0 for each set of off-resonance ROESY experiments were estimated using the calculated value of τm.
Surface electrostatic potential
The surface electrostatic potential distribution of the best overall model of echistatin was calculated with MOLMOL [31], based on simple Coulomb interactions and implicit hydrogen atom positions. Solvent-accessible surface was calculated using the MOLMOL surfaces command with default parameters. Atom charges and the protonation state of the amino acids were derived with the “pdb_charge” MOLMOL macro.
Docking of echistatin into integrin αVβ3
A model for the integrin αVβ3–echistatin complex was constructed by simulation using the docking program HADDOCK [32] and the co-ordinates of the αVβ3 integrin extracellular domains (αV residues 1–439; β3 residues 55–434) in complex with a cyclic RGD pentapeptide (PDB code 1L5G). In a first stage, HADDOCK calculated 500 conformations using a rigid-body docking protocol. Next, the best 100 structures in terms of their inter-molecular energies were refined by simulated annealing in two stages. AIR (Ambiguous Interaction Restraints) [32] were derived from the known interactions of the RGD motif of the cyclic pentapeptide in the PDB structure 1L5G. Initial constraints were contacts between the arginine residue and αv residues Asp150, Tyr178, Thr212, Gln214 and Ala215, and between the aspartate residue of the RGD motif and β3 residues Arg216 and Ala218 and with the MIDAS Mn2+. Both, the side-chains and the backbone atoms at the interface (including residues 149–152, 176–181, 211–216 and 217–219 of αV, residues 120–124 and 212–221 of β3, and residues 23–27 and 44–46 of echistatin) were allowed to move in a semi-rigid-body docking protocol to search for conformational rearrangements. Using these parameters, the standard HADDOCK protocol for protein docking was applied with minor modifications. This protocol combines several stages of molecular dynamics calculations, including heating and cooling with a progressive increase of the flexibility at the binding interface. The resulting 50 molecules with the lowest intermolecular energy values were refined with explicit solvent molecules included. The structures were classified by clustering using the pairwise R.M.S.D. differences.
RESULTS AND DISCUSSION
NMR structure of echistatin
Two-dimensional NMR spectra of echistatin (∼3 mM, pH 3.0, 25 °C) showed good chemical shift dispersion in the HN–Hα region, which allowed the straightforward assignment of non-overlapped cross-peaks. Assignments of NOESY and ROESY cross-peaks for structure determination and off-resonance ROESY relaxation analysis were obtained on the basis of previously published data [20], as described in the Materials and methods section. Overall, chemical shifts observed in our spectra were in good agreement with those obtained from the BMRB database (BMRB 2204), although minor differences were detected for some cross-peaks. Statistics on echistatin NOE assignments and structure calculations are shown in Tables 1 and 3 respectively. The atomic co-ordinates of the final 20 lowest energy conformers of echistatin are accessible from the PDB under the code 1RO3. Figure 1 shows the superposition of the final 20 lowest energy conformers of echistatin (1RO3; Figure 1A) compared with the best eight models reported in previous structural studies on echistatin (2ECH; Figure 1B). The R.M.S.D. for the backbone atoms between the mean structures of 1RO3 and 2ECH was 4.1 Å, and this value was reduced to 2.6 Å if amino acid residues 3–19 and 29–47 only were taken into consideration. The structural elements that define the strongly disulphide-bonded core of the protein are mainly conserved in both structures, although it is evident that both echistatin models depart in structural features that are discussed below.
Table 3. Structural statistics for the best 20 energy-refined DYANA NMR conformers of echistatin.
| Statistic | Value |
|---|---|
| Residual DYANA target function value (Å) | 1.12±0.21 |
| Residual NOE distance constraint violations | |
| >0.2 Å | 9±4 |
| Maximum (Å) | 0.22±0.09 |
| Residual disulphide bond constraint violations | |
| >0.2 Å | 0 |
| Maximum (Å) | 0 |
| Residual van der Waals close contacts violations | |
| >0.2 Å | 3±1 |
| Maximum (Å) | 0.22±0.04 |
| R.M.S.D. from ideal geometry | |
| Bond length (Å) | 0.0089±0.0005 |
| Bond angles (deg.) | 1.37±0.41 |
| R.M.S.D. N, Cα, C' (3–19, 29–47) to mean co-ordinates (Å) | 0.511±0.101 |
| R.M.S.D. N, Cα, C' (21–31) to mean co-ordinates (Å) | 0.621±0.142 |
| R.M.S.D. heavy atoms (3–19, 29–47) to mean co-ordinates (Å) | 1.051±0.291 |
| Residues distribution on Ramachandran plot | |
| In allowed regions | 714 (76.0%) |
| In unfavourable regions | 151 (16.1%) |
| In disallowed regions | 75 (8.0%) |
Figure 1. NMR confirmation and structural features of full-length echistatin.

(A) The best 20 three-dimensional structures of echistatin (PDB code 1RO3) superimposed over the backbone of well-defined residues (3–19 and 29–41). (B) The best eight models of the previously reported echistatin (PDB code 2ECH) superimposed over the backbone of well-defined residues (3–19 and 29–41). In (A) and (B) the residue backbone and side-chain atoms of the recognition R24GD26 motif have been plotted in red. (C) Representation of the surface electrostatic potential of echistatin (PDB code 1RO3) calculated using the MOLMOL program [31]. Positively and negatively charged areas are coloured blue and red respectively. The RGD motif and residues Pro43, His44 and Lys45 located at the tip of the integrin-binding loop and C-terminal loop respectively are circled. (D) Cartoon of the echistatin (PDB code 1RO3) mean structure in the ‘sausage’ representation and colour-coded according to the flexibility calculated from mean-normalized θ0 values. Values of θ0 for backbone Hα–HN NOEs have been mapped on N atoms, while θ0 values for sequential NOEs have been mapped on Cα and carbonyl atoms. θ0 values are associated with the following colours: red, θ0<0.75; orange, 0.75<θ0<0.85; green, 0.85<θ0<0.95; dark blue, θ0>0.95. Light blue indicates absence of data. Disulphide bonds are represented by yellow wires.
NOESY cross-peaks in the side chain proton region were of critical importance for obtaining a defined conformation for the C-terminal tail region (Figure 2). The detection of very weak long-distance NOEs between residues in the active loop and the C-terminal region was achieved using cryoprobes. Subsequent cycles of semi-automated NOE assignment process detected a not-previously reported strong network of NOEs between these two structural elements. Distribution of the final set of long-range NOE restraints along the whole amino acid sequence of echistatin is shown in Figure 3, where the network of NOEs between the C-terminal region and the rest of the protein can be appreciated.
Figure 2. Side-chain proton region of the NOESY spectra of echistatin.

The spectra were recorded at 600 MHz and at 25 °C with 200 ms of mixing time. Critical NOE cross-peaks for close contacts between the C-terminal tail and the rest of the protein have been labelled.
Figure 3. Plot of the number of NOE assigned cross-peaks used in the structure calculations.

Intra-residual (black), sequential (dark grey), short-range (less than four residues apart, light grey) and long-range (white) NOE assigned cross-peaks used in the structure calculations are shown, along with the amino acid sequence of echistatin.
A compact globular core maintained by a tight network of disulphide bonds, from which several loops protrude, defines the structure for echistatin depicted in Figure 1(D). An irregular loop similar to a β-hairpin harbours the recognition RGD tripeptide at its tip. Disulphide bonds linking residues Cys2–Cys11 and Cys20–Cys39 were confirmed by observation of Hβ–Hβ long-distance NOEs. The sequential and spatial proximity of Cys7 and Cys8 make it very difficult to confirm the remaining two disulphide bonds unambiguously by NMR. However, disulphide bridges have been determined previously by chemical methods [26,27] and therefore were also included as structural restraints. Although the above structural features have been described previously for the echistatin structure 2ECH, the refined NMR structure of echistatin in the present paper also significantly departs from the echistatin structure reported previously. A major distinct feature of the echistatin models 2ECH and 1RO3 is that, in the latter, the full-length C-terminal polypeptide is visible and folds as a β-hairpin running parallel to the RGD loop and exposing at the tip residues Pro43, His44 and Lys45. In addition, the side chains of residues in the active tripeptide (Arg24 and Asp26) are well-defined and are exposed to the solvent. Local R.M.S.D. values for backbone atoms suggest that mobility is conferred to the recognition site by a slight hinge effect located at the basis of the loop similar to that previously reported for obtustatin [22], a close homologue of echistatin. The amplitude of the motion of the active loop in the NMR structure of echistatin reported here is approx. 30°, shorter than that reported for obtustatin (35° [22]) and in marked contrast with the value of 60–70° previously described for the echistatin structure 2ECH [8]. A small network of hydrogen bonds keeps the conformation of the protruding active loop in a similar fashion to that observed in β-hairpin structures. In obtustatin, whose recognition loop is three residues shorter than in echistatin, and which does not display any regular secondary structure, a single hydrogen bond keeps the loop active conformation [22]. Although echistatin appeared to have an overall less flexible recognition loop than that observed in obtustatin, high R.M.S.D. values for the backbone atoms of residues Arg24, Gly25 and Asp26 suggested an increased flexibility for the active tripeptide, which may partially compensate for the shorter degree of overall motion of the active loop, providing further mobility to the integrin-recognition motif (see below).
An analysis of the conformation of the C-terminal region of echistatin showed details that were worth noticing. The length of the C-terminal region of echistatin is quite similar to that observed for the C-terminal tail of obtustatin, and residues Pro43 and Gly46 of echistatin occupy equivalent positions as Pro40 and Gly41 in obtustatin. However, unlike obtustatin, the R.M.S.D. values for backbone atoms of the C-terminal region of echistatin do not suggest the existence of a hinge motion within the C-terminal tail. Overall, the NMR structure of echistatin in the present paper shows a compact solvent-accessible surface (Figure 1C). Charges within the RGD loop of echistatin (C20KRARGDDMDDYC32) are not evenly distributed, with positive electrostatic potential upstream of Arg24 and negative potential downstream of Asp26. The net positive electrostatic character of the C-terminal tail of echistatin, running parallel to the positively charged stretch of the RGD loop, contributes further to this charge segregation (Figure 1C), which may have functional relevance.
Internal motions and θo values
Recently, new methods for the detection of internal motions by homonuclear NMR have been developed [29] that allow the study of motions in a wider spatial range than traditional 15N or 13C relaxation methods analysing the proton–heteronucleus bond. Also worthy of notice is that homonuclear NMR methods overcome the need for isotopic labelling of the samples, thus allowing the study of molecules isolated from natural sources. Although many dynamics studies on proteins using natural abundance 13C relaxation have been reported in the literature [33,34], the outcomes usually refer to very local dynamics (one-bond and methyl groups), and phenomena such as hinge effects or co-ordinated motions can hardly be detected. In the case of disintegrins, this kind of motion has been reported previously [22] and therefore a longer range of dynamics study over traditional one-bond relaxation measurements is preferred.
Off-resonance ROESY data for echistatin were analysed with protocols and homemade scripts as described previously for the characterization of internal angular motions in the 100–300 ps time scale of the disintegrin obtustatin [22]. A total of 126 assigned cross-peaks from 16 off-resonance ROESY spectra recorded at two magnetic fields were analysed. Cross-peak intensities were fitted to a second-order polynomial function of the θ value, θ zero-crossing point was calculated, and θ0 values were averaged for each magnetic field and normalized with respect to the theoretical rigid limit. The theoretical global correlation time of echistatin 1RO3, calculated with the HydroNMR software, was 3.7 ns. This value is in good agreement with previously calculated correlation times reported in the literature (4.1±0.5 ns) [19].
Differences in the average normalized value of θ0 can be correlated with differences in S2 [22], and values of mean normalized θ0 for sequential and intra-residue HN–Hα ROE cross-peaks indicate the flexibility of ψ and Φ dihedral angles respectively. Higher values of θ0 indicate lower angular mobility. These values have been mapped on the three-dimensional structure and the amino acid sequence in Figures 1(D) and 4 respectively. On the other hand, analysis of intra-residue HN–Hβ, Hα–Hβ and Hα–Hγ ROE cross-peaks provides information on the flexibility of side chains. Values of θ0 for the most relevant side chains in echistatin are reported in Table 4. Several long-range ROE cross-peaks have been analysed to study relative motions of different structural elements. Values of θ0 for these cross-peaks are also shown in Table 4.
Figure 4. Internal angular motions of echistatin analysed by off-resonance ROESY.

(A) Plots of the θ0 values for intra (black line) and sequential (grey line) backbone Hα–HN cross-peaks measured at 500 (upper panel) and 600 (lower panel) MHz proton frequencies along the amino acid sequence of echistatin. (B) Mean normalized θ0 values for data at 500 and 600 MHz (upper panel) for intra (black) and sequential (grey) NOE Hα–HN cross-peaks with experimental errors, compared with backbone atom co-ordinate R.M.S.D. values (lower panel) from NMR solution structure (PDB code 1RO3), along with the amino acid sequence of echistatin.
Table 4. Mean-normalized θ0 values (averaged for data at 500 and 600 MHz) for cross-peaks involving side chains of well-defined residues (w), amino acids of the integrin-recognition site (r), C-terminal region residues (c) and residues in disulphide bridging (ss).
| NOE cross peak | Normalized-mean θ0 |
|---|---|
| Glu3 HN–Hβ (w) | 0.75±0.05 |
| Thr36 HN–Hβ (w) | 0.77±0.07 |
| Lys14 HN–Hβ (w) | 0.67±0.11 |
| Asn33 HN–Hβ (w) | 0.79±0.05 |
| Leu14 Hα–Hβ (w) | 0.71±0.04 |
| His44 HN–Hβ (c) | 0.71±0.05 |
| Lys45 HN–Hβ (c) | 0.68±0.04 |
| Arg41 HN–Hβ (c) | 0.73±0.06 |
| Arg41 Hβ–Arg22 HN (c) | 0.61±0.06 |
| Cys2 Hβ–Cys11 Hβ (ss) | 0.79±0.12 |
| Cys20 Hβ–Cys39 Hβ (ss) | 0.74±0.08 |
| Arg24 Hα–Hβ (r) | 0.71±0.08 |
| Asp26 Hα–Hβ (r) | 0.62±0.09 |
| Arg24 Hβ–Asp26 Hβ (r) | 0.48±0.29 |
In general, the NMR structural dispersion along the amino acid sequence is in good agreement with the flexibility values derived from θ0. Minor differences involve the RGD motif. Values of θ0 for this region are higher than at the base of the loop and lower than in the intervening loop stretches, invoking a minor hinge effect also suggested by local R.M.S.D. values. Other regions of echistatin showing increased flexibility include the residues Glu3, Ser4, Arg9 and Lys12 which are solvent-exposed as calculated with PROCHECK [35], and the backbone atoms of residues Gly17, Thr18 and Asp29, located at the base of the integrin binding loop (Figures 1D and 4). Residues in the tip of the recognition loop, including the RGD motif, also show some degree of flexibility. The increased backbone flexibility at the base of the RGD loop suggested that the mobility of the RGD loop might be due to a hinge effect articulated at the base of the loop, similar to that reported for obtustatin [22]. On the other hand, hydrogen-bonded core residues and cysteines that are engaged in disulphide bonds show, as expected, restricted mobility. Analysed long-range intra-loop ROE cross-peaks are listed in Table 4. Angular mobilities of these ROE vectors are partially restricted, reflecting their engagement in the hydrogen-bond network that maintains the recognition loop in its active conformation.
Internal motions in the C-terminal region of echistatin 1RO3 are significantly restricted. Hence, the backbone atoms of residues in the range 36–46 show values of θ0 higher than those of residues 47–49 and of residues located at the base of the integrin-recognition loop. These θ0 values, which indicate local restricted angular mobility, are in good agreement with the low structural dispersion in this region of the echistatin NMR structure. Unlike in the case of obtustatin, no hinge effect has been detected in the C-terminal region of echistatin. However, the short amplitude of the recognition loop overall motion, along with the network of long-range NOEs between residues of the RGD loop and the C-terminal tail (Figure 3), indicate the existence of concerted motions between the C-terminal and the RGD loops. It is also worth mentioning the relative low degree of angular flexibility of the side chains of amino acids His44 and Lys45 (θ0 for HN–Hβ ROE cross-peak of 0.71 and 0.68 respectively), which exhibit similar spatial orientation to the lateral chains of residues Arg24 and Asp26 of the integrin-recognition motif.
Dynamics of the RGD recognition motif
Analysis of the angular mobility showed that the backbone atoms of the amino acids of the R24GD26 motif are highly mobile in the time scale measured. Amino acids Ala23 and Asp26 exhibited equal or even higher levels of backbone angular mobility than residues Arg24 and Gly25 (Figures 1D and 4), indicating that the enhanced motion of the RGD motif at the tip of the integrin-binding loop may be articulated at Ala23 and Asp26. This conclusion is in good agreement with a previous 15N relaxation study of the active tripeptide of echistatin [19]. In this 15N study, values of S2 for Ala23, Gly25 and Asp26 were 0.29, 0.50 and 0.46 respectively, indicating that Ala23 represents a more flexible residue than those in the active site, therefore supporting the proposed small hinge effect for the active-site-flanking amino acids. However, in contrast with the aforementioned 15N relaxation study [19], our results do not support a large amount of chemical exchange for the amino acids in the active site. All analysed cross-peaks correspond to negative intensities (opposite to diagonal sign) in a regular ROESY spectrum, clearly indicating the absence of chemical exchange in the millisecond timescale. It must be mentioned that the values of Rex observed by 15N relaxation studies are in the range of 0.1 to 1 s, and that the values of τe for these residues measured by 15N relaxation analysis (1.2–2.7 ns) belong to a much longer timescale than that studied here (100–300 ps). Timescales of the chemical-exchange processes measured in the present study and in the 15N relaxation studies [19] are rather different, and therefore both analyses may be considered as complementary.
On the other hand, the side-chain atoms of the echistatin's integrin-recognition tripeptide have limited mobility in comparison with other side chains of the protein (Table 4). Hence, ROE cross-peaks Hα–Hβ for Arg24 and Asp26 display high values of θ0, 0.71 and 0.62 respectively, indicating a well-defined side-chain conformation. However, the ROE cross-peak Arg24 Hβ–Asp26 Hα showed a very low value of θ0 and a poor model fitting, which is indicative of a mobile ROE vector and complex motions. The mobility of the ROE vector linking side chains of the recognition site clearly indicated the existence of complex mechanisms for the flexibility of these residues. The proposed hinge effect articulated at the active-site-flanking residues superimposed to the overall motion of the whole integrin-binding loop may contribute to the enhanced flexibility of the RGD motif side chains.
Docking model of αvβ3–echistatin complex
To gain structural insights into the inhibitory mechanism of echistatin, a putative model for a echistatin–integrin αVβ3 complex was built using HADDOCK 3.1 software and based on the crystal structure of integrin αVβ3 complexed with a cyclic RGD pentapeptide (PDB code 1L5G). HADDOCK calculations provided a set of convergent structures for the disintegrin–integrin complex. Specifically, four clusters of structures were obtained by fitting them over the interacting RGD residues with an average R.M.S.D. value for the backbone atoms of all the amino acids in all echistatin–integrin complexes of less than 1 Å. The energies of the four clusters were of the same order of magnitude, although the mean energy of the cluster selected for calculating the docking model was 2-fold lower than the corresponding value of the maximal mean energy cluster, and 25% lower than the nearest cluster energy. Figure 5 displays the lowest energy structure cluster of echistatin docked into the crevice between the α and the β subunits of integrin αVβ3. The average buried surface area of the complex interface is 1394 Å2. Table 5 shows the intermolecular contacts of the proposed echistatin–integrin αVβ3 complex. Among the residues of the integrin-binding motif, Asp26 contacts only the β3 subunit through interactions with Ser123 and the MIDAS cation, whereas Arg24 participates in an extensive network of interactions with αV residues. Noteworthy, echistatin residues flanking the RGD motif at its C- and N-terminal sites also interact selectively with the αv (Arg22) or the β3 (Asp27, Met28, Asp29) subunit (Table 5 and Figure 5). These residues may endow echistatin with its reported integrin-recognition specificity and affinity. Moreover, with the exception of the α1β1 inhibitory disintegrin obtustatin, the active tripeptides of all known disintegrins are flanked by positively (N-terminus) and negatively (C-terminus) charged residues [6]. Such a ‘plug’-based integrin-blocking mechanism supports the view that the conserved aspartate residue of the integrin-binding motifs of disintegrins (RGD, KGD, MGD, VGD, WGD, MLD, with the bold underlined residue indicating the conserved aspartate residue of the integrin-binding motifs) might be responsible for the binding of disintegrins to integrin receptors that share a β-subunit, whereas the N-terminal residue of the integrin-binding motif may dictate the integrin-inhibitory specificity through interactions with the α-subunit propeller domain [6]. On the other hand, residues located at the C-terminal side of the RGD motif determine the affinity of disintegrins to integrins [15,36,37]. The observation that replacement of Met28 with asparagine completely abolished echistatin's ability to block integrins αIIbβ3, αvβ3 and α5β1, while leucine at position 28 only decreased the inhibitory activity of echistatin towards integrin α5β1 [15], can be structurally explained with the docking model shown in Figure 5, according to which Met28 interacts with β-subunit residues (Table 5). On the other hand, the report that, compared with wild-type, the synthetic mutant Asp27→Trp of echistatin showed increased inhibitory activity against platelet aggregation and decreased potency to block vitronectin [38] has been interpreted on the basis of a trimestatin–αvβ3 docking model in terms of a specific contact of the corresponding Phe52 residue of trimestatin with residues of the propeller domain of αv [13]. However, comparison of the possible contacts between trimestatin and αvβ3 (Table 1 of [13]), and between echistatin and the same integrin (Table 5), shows that, as expected, they are conserved for conserved residues in the two disintegrins (the arginine and the aspartate of the RGD motif), but depart for those residues of both the RGD loop and the C-terminal region, with different side chains in trimestatin and echistatin. In particular, in the NMR structure of echistatin reported in the present paper, the side chain of Asp27 points in the opposite direction to its topologically equivalent Phe52 in the trimestatin structure, and makes contact solely with the β3 Mn2+ ion (Figures 5B and 5C). Thus a drastic change in the spatial orientation of the side chain of Trp27 in the echistatin mutant must be invoked. Alternatively, the presence of tryptophan at position 27 may force a local reorientation of the disintegrin molecule in the integrin RGD-binding crevice, with consequences for the interaction of the mutated echistatin with the α-subunit β-propeller domain.
Figure 5. Docking model of αvβ3–echistatin complex.

(A) Ribbon representation of a putative model of echistatin (blue) docked into the RGD-binding crevice of the globular head of integrin αvβ3 (PDB code 1L5G). The αV subunit β-propeller domain has been coloured orange and the β3 subunit A- and thigh domains are in yellow. (B) Detail of the interaction region showing the molecules in the same colour and orientation as in (A). The side chains of echistatin amino acids engaged in intermolecular contacts are plotted in the ball-and-stick representation and are labelled using the one-letter amino acid code. The β3 MIDAS Mn2+ ion is depicted as a violet sphere. (C) A close-up stereo view of the modelled αvβ3–echistatin interaction region displayed in the same orientation as in (A) and (B). Interacting residues (Table 5) are shown in the ball-and-stick representation, and the interactions, other than hydrophobic contacts, are displayed by broken lines.
Table 5. Possible intermolecular contacts between echistatin and αVβ3.
| Echistatin | αV | β3 |
|---|---|---|
| Lys15 | Met180, Lys181, Thr182 | |
| Arg22 | Tyr178 | |
| Arg24 | Asp150, Tyr178, Gln180, Asp218, Asp219 | |
| Asp26 | Mn2+, Ser123 | |
| Asp27 | Mn2+ | |
| Met28 | Lys125, Tyr122 | |
| Asp29 | Lys125 | |
| His44 | Asp150, Ala215, Ile216, Asp218 | |
| Lys45 | Gln214, Ile216 |
Noteworthy, despite the high degree of mobility allowed for the binding loop residues in the HADDOCK calculations, the backbone, as well as the side-chain atoms of the echistatin's integrin-recognition site amino acids, show low local dispersion in all the modelled disintegrin–integrin complexes. This strongly suggests an induced-fitting mechanism for the binding of echistatin to its target integrin receptor.
The high-resolution structure of echistatin reported in the present paper, and particularly the fact that, in contrast with a previous echistatin NMR structure, its full-length C-terminal polypeptide is visible and folds as a β-hairpin running parallel to, and displaying concerted motions with, the RGD loop, prompted us to use the disintegrin–integrin model to investigate the structural grounds for the biological role of the C-terminal tail of echistatin. His44 and Lys45 at the tip of the β-hairpin appear to be engaged in a network of contacts with residues of the propeller domain of the αV subunit. This is consistent with biochemical data showing that deletion of the C-terminal region of echistatin reduced the ability of the disintegrin to inhibit platelet aggregation [7,16], and supports the view that the C-terminal tail contains determinants of integrin-blocking specificity.
Our docking model does not support the conclusion derived from cross-linking studies that the regions encompassing residues 109–118 and 209–220 represent contact sites in αvβ3 for echistatin analogues, with a photoreactive group in positions 28 and 45 respectively [39,40]. Residues 109–118 of β3 encompassing the proposed cross-linking site for p-benzoylphenylalanyl–(28)echistatin (where 28 indicates the position of the cross-linker attachment) correspond to the βA strand of the β3 A-domain [41], and are completely buried in the domain core. However, in our docking model (Figure 5), echistatin Met28 interacts with the β3 loop residues Tyr122 and Lys125 (Table 5). On the other hand, in our proposed echistatin–αvβ3 complex model, the closest distance between echistatin's Lys45 and any residue within the β3 209–220 region is approx. 20 Å. Hence, a large change in the relative orientation of the C-terminal region of echistatin upon binding to integrin αvβ3 might be invoked to reconcile the docking and the cross-linking models. However, the structure of the synthetic and chemically modified echistatin molecule has not been reported, and thus it is not clear whether it possess the same disulphide-bonding pattern and three-dimensional conformation as the 1RO3 NMR structure reported in the present paper.
Concluding remarks
The homonuclear NMR study reported in the present paper provides a refined structure and new insights into the dynamics and the conformation of biologically relevant epitopes of echistatin. Major structural features that were not apparent in previous echistatin structures are the overall hinge motion of the integrin-binding loop and the superimposed local enhanced flexibility of the RGD motif, and the conformation of the full-length C-terminal tail, which is now seen to fold over to form a hairpin that runs parallel to the integrin-recognition loop. The RGD loop and the C-terminal region of echistatin exhibit concerted motions and form a continuous patch involved in extensive interactions with both subunits of integrin receptors, providing the molecular basis for our understanding of the functional synergy between the RGD loop and the C-terminal sequence of echistatin.
Acknowledgments
This study has been partly financed by grants BCM2001-3337 from the Ministerio de Ciencia y Tecnología, and BFU2004-01432/BMC from the Ministerio de Educación y Ciencia, Madrid, Spain (to J.J.C.). We also thank Bruker España S.A. for financial support. Thanks are also due to the SCSIE of the University of Valencia for providing access to the NMR facility and to high-performance computing facilities.
References
- 1.Markland F. S. Snake venoms and the hemostatic system. Toxicon. 1998;36:1749–1800. doi: 10.1016/s0041-0101(98)00126-3. [DOI] [PubMed] [Google Scholar]
- 2.Ménez A. Chichester: John Wiley & Sons; 2002. Perspectives in Molecular Toxinology. [Google Scholar]
- 3.Jia L.-G., Shimokawa K.-i., Bjarnason J. B., Fox J. W. Snake venom metalloproteinases: structure, function and relationship to the ADAMs family of proteins. Toxicon. 1996;34:1269–1276. doi: 10.1016/s0041-0101(96)00108-0. [DOI] [PubMed] [Google Scholar]
- 4.Kini R. M., Evans H. J. Structural domains in venom proteins: evidence that metalloproteinases and nonenzymatic platelet aggregation inhibitors (disintegrins) from snake venoms are derived by proteolysis from a common precursor. Toxicon. 1992;30:265–293. doi: 10.1016/0041-0101(92)90869-7. [DOI] [PubMed] [Google Scholar]
- 5.McLane M. A., Marcinkiewicz C., Vijay-Kumar S., Wierzbicka-Patynowski I., Niewiarowski S. Viper venom disintegrins and related molecules. Proc. Soc. Exp. Biol. Med. 1998;219:109–119. doi: 10.3181/00379727-219-44322. [DOI] [PubMed] [Google Scholar]
- 6.Calvete J. J., Moreno-Murciano M. P., Theakston R. D. G., Kisiel D. G., Marcinkiewicz C. Snake venom disintegrins: novel dimeric disintegrins and structural diversification by disulphide bond engineering. Biochem. J. 2003;372:725–734. doi: 10.1042/BJ20021739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcinkiewicz C., Vijay-Kumar S., McLane M. A., Niewiarowski S. Significance of the RGD loop and C-terminal domain of echistatin for recognition of αIIbβ3 and αvβ3 integrins and expression of ligand-induced binding sites. Blood. 1997;90:1565–1575. [PubMed] [Google Scholar]
- 8.Saudek V., Atkinson R. A., Pelton J. T. Three-dimensional structure of echistatin, the smallest active RGD protein. Biochemistry. 1991;30:7369–7372. doi: 10.1021/bi00244a003. [DOI] [PubMed] [Google Scholar]
- 9.Adler M., Lazarus R. A., Dennis M. S., Wagner G. Solution structure of kistrin, a potent platelet aggregation inhibitor and GPIIb-IIIa antagonist. Science. 1991;253:445–448. doi: 10.1126/science.1862345. [DOI] [PubMed] [Google Scholar]
- 10.Senn H., Klaus W. The nuclear magnetic resonance solution structure of flavoridin, an antagonist of the platelet GPIIb-IIIa receptor. J. Mol. Biol. 1993;232:907–925. doi: 10.1006/jmbi.1993.1439. [DOI] [PubMed] [Google Scholar]
- 11.Smith K. J., Jaseja M., Lu X., Williams J. A., Hyde E. I., Trayer I. P. Three-dimensional structure of the RGD-containing snake toxin albolabrin in solution, based on 1H NMR spectroscopy and simulated annealing calculations. Int. J. Pept. Protein Res. 1996;48:220–228. doi: 10.1111/j.1399-3011.1996.tb00835.x. [DOI] [PubMed] [Google Scholar]
- 12.Shin J., Hong S.-Y., Chung K., Kang I., Jang Y., Kim D.-s., Lee W. Solution structure of a novel disintegrin, salmosin, from Agkistrodon halys venom. Biochemistry. 2003;42:14408–14415. doi: 10.1021/bi0300276. [DOI] [PubMed] [Google Scholar]
- 13.Fujii Y., Okuda D., Fujimoto Z., Horii K., Morita T., Mizuno H. Crystal structure of trimestatin, a disintegrin containing a cell adhesion recognition motif RGD. J. Mol. Biol. 2003;332:1115–1122. doi: 10.1016/s0022-2836(03)00991-4. [DOI] [PubMed] [Google Scholar]
- 14.Gan Z. R., Gould R. J., Jacobs J. W., Friedman P. A., Polokoff M. A. Echistatin: a potent platelet aggregation inhibitor from the venom of the viper, Echis carinatus. J. Biol. Chem. 1988;263:19827–19832. [PubMed] [Google Scholar]
- 15.Wierzbicka-Patynowski I., Niewiarowski S., Marcinkiewicz C., Calvete J. J., Marcinkiewicz M. M., McLane M. A. Structural requirements of echistatin for the recognition of αvβ3 and α5β1 integrins. J. Biol. Chem. 1999;274:37809–37814. doi: 10.1074/jbc.274.53.37809. [DOI] [PubMed] [Google Scholar]
- 16.Wright P. S., Saudek V., Owen T. J., Harbeson S. L., Bitonti A. J. An echistatin C-terminal peptide activates GPIIbIIIa binding to fibrinogen, fibronectin, vitronectin and collagen type I and type IV. Biochem. J. 1993;293:263–267. doi: 10.1042/bj2930263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gould R. J., Polokoff M. A., Friedman P. A., Huang T.-F., Holt J. C., Cook J. J., Niewiarowski S. Disintegrins: a family of integrin inhibitory proteins from viper venoms. Proc. Soc. Exp. Biol. Med. 1990;195:168–171. doi: 10.3181/00379727-195-43129b. [DOI] [PubMed] [Google Scholar]
- 18.Cooke R. M., Carter B. G., Murray-Rust P., Hartshorn M. J., Herzyk P., Hubbard R. E. The solution structure of echistatin: evidence for disulphide bond rearrangement in homologous snake toxins. Protein Eng. 1992;5:473–477. doi: 10.1093/protein/5.6.473. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y., Suri A. K., Kominos D., Sanyal G., Naylor A. M., Pitzenberger S. M., Garsky V. M., Levy R. M., Baum J. Three-dimensional structure of echistatin and dynamics of the active site. J. Biomol. NMR. 1994;4:307–324. doi: 10.1007/BF00179342. [DOI] [PubMed] [Google Scholar]
- 20.Atkinson R. A., Saudek V., Pelton J. T. Echistatin: refined structure of a disintegrin in solution by 1H NMR and restrained molecular dynamics. Int. J. Pept. Protein Res. 1994;43:563–572. doi: 10.1111/j.1399-3011.1994.tb00558.x. [DOI] [PubMed] [Google Scholar]
- 21.Moreno-Murciano M. P., Monleón D., Marcinkiewicz C., Calvete J. J., Celda B. NMR solution structure of the non-RGD disintegrin obtustatin. J. Mol. Biol. 2003;329:135–145. doi: 10.1016/s0022-2836(03)00371-1. [DOI] [PubMed] [Google Scholar]
- 22.Monleón D., Moreno-Murciano M. P., Kovacs H., Marcinkiewicz C., Calvete J. J., Celda B. Concerted motions of the integrin-binding loop and the C-terminal tail of the non-RGD disintegrin obtustatin. J. Biol. Chem. 2003;278:45570–45576. doi: 10.1074/jbc.M307030200. [DOI] [PubMed] [Google Scholar]
- 23.Goddard T. D., Kneller D. G. San Francisco: University of California; 1999. SPARKY 3. [Google Scholar]
- 24.Piotto M., Sandek V., Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–670. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 25.Guntert P., Mummenthaler C., Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 26.Calvete J. J., Wang Y., Mann K., Schäfer W., Niewiarowski S., Stewart G. J. The disulfide bridge pattern of snake venom disintegrins, flavoridin and echistatin. FEBS Lett. 1992;309:316–320. doi: 10.1016/0014-5793(92)80797-k. [DOI] [PubMed] [Google Scholar]
- 27.Bauer M., Sun Y., Degenhardt C., Kozikowski B. Assignment of all four disulfide bridges in echistatin. J. Protein Chem. 1993;12:759–764. doi: 10.1007/BF01024934. [DOI] [PubMed] [Google Scholar]
- 28.Brunger A. T., Clore G. M., Gronenborn A. M., Karplus M. Three-dimensional structures of proteins determined by molecular dynamics with NOE interproton distance restraints. Proc. Natl. Acad. Sci. U.S.A. 1986;83:3801–3805. doi: 10.1073/pnas.83.11.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schleucher J., Wijmenga S. S. How to detect internal motion by homonuclear NMR. J. Am. Chem. Soc. 2002;124:5881–5889. doi: 10.1021/ja012040o. [DOI] [PubMed] [Google Scholar]
- 30.Garcia de la Torre J., Huertas M. L., Carrasco B. HYDRONMR: prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn. Reson. 2000;147:138–146. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- 31.Koradi R., Billeter M., Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 32.Dominguez C., Boelenes R., Bombin A. M. J. J. HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 33.Ma L., Hass M. A., Vierick N., Kristensen S. M., Ulstrup J., Led J. J. Backbone dynamics of reduced plastocyanin from the cyanobacterium Anabaena variabilis: regions involved in electron transfer have enhanced mobility. Biochemistry. 2003;42:320–330. doi: 10.1021/bi020553h. [DOI] [PubMed] [Google Scholar]
- 34.Palmer A. G., III, Wright P. E., Rance M. Measurement of relaxation rate constants for methyl groups by proton-detected heteronuclear NMR spectroscopy. Chem. Phys. Lett. 1991;185:41–46. [Google Scholar]
- 35.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 36.Scarborough R. M., Rose J. W., Naughton M. A., Phillips D. R., Nannizzi L., Arfsten A., Campbell A. M., Charo I. F. Characterization of the integrin specificities of disintegrins isolated from American pit viper venoms. J. Biol. Chem. 1993;268:1058–1065. [PubMed] [Google Scholar]
- 37.Lu X., Rahman S., Kakkar V. V., Authi K. S. Substitutions of proline 42 to alanine and methionine 46 to asparagine around the RGD domain of the neurotoxin dendroaspin alter its preferential antagonism to that resembling the disintegrin elegantin. J. Biol. Chem. 1996;271:289–294. doi: 10.1074/jbc.271.1.289. [DOI] [PubMed] [Google Scholar]
- 38.McLane M. A., Vijay-Kumar S., Marcinkiewicz C., Calvete J. J., Niewiarowski S. Importance of the structure of the RGD-containing loop in the disintegrins echistatin and eristostatin for recognition of αIIbβ3 and αvβ3 integrins. FEBS Lett. 1996;391:139–143. doi: 10.1016/0014-5793(96)00716-8. [DOI] [PubMed] [Google Scholar]
- 39.Yahalom D., Wittelsberger A., Mierke D. F., Rosenblatt M., Alexander J. M., Chorev M. Identification of the principal binding site for RGD-containing ligands in the αvβ3 integrin: a photoaffinity cross-linking study. Biochemistry. 2002;41:8321–8331. doi: 10.1021/bi025690t. [DOI] [PubMed] [Google Scholar]
- 40.Scheibler L., Mierke D. F., Bitanm G., Rosenblatt M., Chorev M. Identification of a contact domain between echistatin and the integrin αvβ3 by photoaffinity cross-linking. Biochemistry. 2001;40:15117–15126. doi: 10.1021/bi0109156. [DOI] [PubMed] [Google Scholar]
- 41.Xiong J. P., Stehle T., Zhang R., Joachimiak A., Frech M., Goodman S. L., Arnaout M. A. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science. 2001;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]