

Abstract
HIV-1 Vpr is a highly conserved accessory protein that is involved in many functions of the virus life cycle. Vpr facilitates the entry of the HIV pre-integration complex through the nuclear pore, induces G2 cell cycle arrest, regulates cell apoptosis, increases transcription from the long terminal repeat and enhances viral replication. Vpr contains a Leu/Ile-rich domain (amino acids 60–81) in its C-terminal part, which is critical for dimerization. The sequence comprising residues 52–96 is implicated in properties of the protein such as DNA interaction and apoptosis via interaction with the adenine nucleotide translocator. To understand the specific interactions of Vpr-(52–96), the ability of this peptide to dimerize via a leucine-zipper mechanism has been investigated, by NMR and fluorescence spectroscopy. In contrast with results from a study performed in the presence of trifluoroethanol, our results, obtained in 30% (v/v) [2H]acetonitrile, show that Vpr-(52–96) in solution still forms an α-helix spanning residues 53–75, but dimerizes in an antiparallel orientation, through hydrophobic interactions between leucine and isoleucine residues and stacking between His71 and Trp54. Moreover, to demonstrate the physiological relevance of the dimer structure, fluorescence spectroscopy experiments have been performed in a Mes buffer, which confirmed the formation of the dimer in aqueous solution and highlighted the spatial proximity between Trp54 and His71. Surprisingly, the leucine-zipper structure shown in the present work for Vpr-(52–96) mimics the structure of full-length Vpr-(1–96), and this could explain why some of the properties of Vpr-(52–96) and Vpr-(1–96) are identical, while some are even enhanced for Vpr-(52–96), particularly in the case of DNA transfection experiments.
Keywords: fluorescence, HIV, leucine zipper, NMR, Vpr
Abbreviations: ANT, adenine nucleotide translocator; HSQC, heteronuclear single-quantum coherence; NOE, nuclear Overhauser effect; r.m.s.d., root mean square deviation; TFE, trifluoroethanol
INTRODUCTION
HIV-1 contains several auxiliary proteins, including Vpr, a 14 kDa virion-associated protein containing 96 amino acids [1]. Vpr interferes with many functions in the viral life cycle, induces G2/M cell cycle arrest [2,3], controls apoptosis by permeabilizing the mitochondria membrane [4,5], increases transcription of the long terminal repeat [6], enhances viral replication [7] and helps the entry of the HIV-1 pre-integration complex containing the viral DNA [8]. Vpr shuttles between the nucleus and the cytoplasm. Moreover, Vpr concentrates in the nuclear envelope by interacting with components of the nuclear pore complex [9] and transfects various cell lines [10] very efficiently. Different fragments of Vpr have also been extensively studied. The N-terminal part (amino acids 1–51) was shown to play an important role in nuclear localization and in virion incorporation [11]. More precisely, Vpr-(13–33) is known to be essential for nuclear localization, whereas Vpr-(34–51) has been described to play an important role in Vpr oligomerization [12]. Lastly, the C-terminal domain (amino acids 52–96) of Vpr was shown to be involved in cell cycle arrest [13], to bind the nucleocapsid protein NCp7 [14], and to interact with HIV-1 RNA. Furthermore, this domain is thought to promote nuclear provirus transfer.
To gain insight into the structure–function relationships of Vpr, many structural studies have been performed on various domains {Vpr-(1–96) [15,16], Vpr-(1–51) [17] and Vpr-(52–96) [18]} and shorter fragments [12,19] of Vpr. Whatever the solvent and solution conditions employed, these fragments are characterized by long amphipathic α-helices spanning residues 13–33 for Vpr-(13–33), 34–51 for Vpr-(34–51), 17–29 and 35–46 for Vpr-(1–51) and 53–78 for Vpr-(52–96). Moreover, it was shown that Vpr-(52–96) dimerizes, and that residues Leu60 and Leu67, located on the hydrophobic face of the amphipathic α-helix, appear to be involved in this dimerization through a leucine-zipper arrangement, since their replacement by alanine [18] prevented dimerization. Since no long-range NOE (nuclear Overhauser effect) was observed in this previous work, in the presence of 30% (v/v) TFE (trifluoroethanol), a theoretical model of a coiled-coil Vpr-(52–96) dimer, suggested to have a parallel orientation, was proposed.
Furthermore, the three-dimensional structure of the entire Vpr protein (residues 1–96) has been determined in the presence of TFE or C2H3CN ([2H]acetonitrile) or in pure water by NMR [15,16]. The structure obtained in the presence of 30% (v/v) TFE revealed three well defined α-helices adopting a U-shaped form, while in the presence of 0–30% (v/v) C2H3CN, the protein shows the same well-defined α-helices folded around a hydrophobic core comprising Leu, Ile, Val and aromatic residues [16]. Within Vpr-(1–96), point mutational analysis of the Leu/Ile residues in the leucine-rich domain suggested that multiple Leu/Ile residues may be involved in maintaining an intramolecular leucine-zipper-like structure [20].
Taking into account the solvent effects on the structure of full-length Vpr-(1–96), study of the (52–96) domain in the presence of C2H3CN and at acidic pH was undertaken by NMR. In these conditions, it was possible to observe the formation of a leucine zipper through the antiparallel orientation of two α-helices connected via hydrophobic-residue interactions. These results were confirmed by fluorescence experiments.
MATERIALS AND METHODS
Sample preparation
Due to its cellular toxicity [21], Vpr cannot be easily obtained in large quantities by recombination. Thus, the (52–96) C-terminal domain of Vpr has been obtained by automated solid-phase synthesis using the Fmoc (fluoren-9-ylmethoxycarbonyl) strategy and then purified by reverse-phase HPLC, using procedures reported previously for the production of retroviral proteins [22]. The peptide was analysed by MS and found to have a purity greater than 98%. During protein synthesis, 17 labelled amino acids (95% 15N, 15% 13C) were introduced: Thr53, Thr55, Val57, Ala59, Leu61, Ile63, Leu64, Leu67, Leu68, Pro69, His71, Pro72, Gly75, Gly82, Thr89, Ala93 and Lys95. Since Vpr has the ability to arrest the cell cycle in G2 phase and has the capacity to form ion channels, it is highly toxic for cells generally used for expression, and Vpr or its fragments could not be obtained in any other way than by chemical synthesis.
NMR experiments
All NMR experiments were recorded on a sample at a concentration of 2 mM in water with 30% (v/v) C2H3CN, pH 3.0, at 293 K, 308 K and 313 K, on an Avance Bruker spectrometer operating at 600.14 MHz. Two-dimensional spectra were performed with 2048 real points in t2, a spectral width of 7062 Hz and 512 t1 increments. The transmitter frequency was set to the water signal. The solvent resonance was suppressed using a 3–9–19 pulse sequence [23] with gradients during the relaxation delay of 1.6 s between free induction decays. A mixing time of 200 ms has been used in NOESY experiments [24]. All heteronuclear spectra were recorded with a spectral width of 7936.5 Hz sampled over 2048 complex points in ω2 (1H) and a spectral width of 1200 Hz sampled over 128 complex points in ω1 (15N). Decoupling of the 15N nucleus during proton acquisition has been performed by application of a GARP sequence [25]. Experiments were recorded in the phase-sensitive mode using echo/antiecho gradient selection [26] and trim pulses in inept transfer. The initial analysis of Vpr-(52–96) dimerization was based on two-dimensional 1H/15N HSQC (heteronuclear single-quantum coherence) [27,28] spectra recorded at different concentrations ranging from 0.03 mM to 2.0 mM, pH 3.0, in a 30% (v/v) C2H3CN-containing solution of aqueous water. For all experiments, the temperature was controlled externally using a special temperature control system (BCU 05 Bruker).
Data analysis and structure calculation
All data were processed using XWINNMR software (Bruker). A π/6 phase-shifted sine bell window function was applied and data were zero filled once prior to Fourier transformation in both dimensions (t1 and t2). The final sizes of the frequency domains matrices were 2048 and 2048 real points in ω2 and ω1 respectively. For all experiments, 1H frequency scale was directly referenced to water. The data were then analysed with the Felix program (Accelrys, San Diego, CA, U.S.A.).
A set of distance restraints has been obtained from NOE cross-signal volume measured on a 200 ms mixing time NOESY at 303 K by integration of the peaks into distances by an R-6 dependency and a tolerance of 20% to take into account integration errors. The distances were calibrated using the distance between aromatic protons. No dihedral torsion angle restraints were included in the calculations. Standard protocols using distance geometry and simulated annealing were performed in X-PLOR 3.84 [29,30] on an SGI O2 R12000 computer.
Distance geometry and simulated annealing regularization Metric matrix distance geometry calculations were performed to embed and optimize initial structures. The experimentally obtained distance restraints were duplicated for the two monomers of Vpr-(52–96), during the embedding stage of the protocol. For optimization with simulated annealing (DGSA protocol) all distance restraints were specified with the ‘SUM’ averaging option in X-PLOR [30,31], which allows for ambiguity in the atom specification in input distance restraints. This feature is particularly useful, since many NOE cross-peaks arise from a combination of intramolecular and intermolecular proton pairs, and the inherent symmetry in the system prevents monomer-specific assignments of the cross-peaks.
Out of 100 attempts at embedding and optimization, the 10 best structures were selected based on the criteria of acceptable covalent geometry, low distance restraint violations, and favourable non-bonded energy. These structures were optimized further with restrained molecular dynamics simulations. Distance-restrained molecular dynamics were carried out in a vacuum with a distance-dependent dielectric constant. The dynamics were initiated at 5 K and the temperature was increased gradually to 1000 K in 5.0 ps and then equilibrated for 1.0 ps. The force constants for the distance restraints were kept at 8.4 kJ·mol−1·Å−2 (2.0 kcal·mol−1·Å−2) during these stages (1 Å=0.1 nm). Subsequently, the force constants for the distance restraints were scaled up to a final value of 126 kJ·mol−1·Å−2 (30 kcal·mol−1·Å−2) over 6.0 ps. The system was then allowed to evolve for 20.0 ps at 1000 K and then slow-cooled to 300 K in 14.0 ps and equilibrated for 10.0 ps. The co-ordinates saved every 0.5 ps in the last 4.0 ps were averaged. The resulting structure was subject to conjugate gradient minimization until a final gradient of 0.4 kJ·mol−1·Å−2 (0.1 kcal·mol−1·Å−2) was reached. All dynamics were carried out with a time step of 1.0 fs. Non-crystallographic symmetry restraints were maintained throughout. Quality of structures was evaluated with PROCHECK [32], and INSIGHT II (Accelrys) software was used to visualize structures.
The dimer dissociation constant was determined from fits to eqn (1), relating the observed resonance frequency, ωobs, to the population-weighted average of the monomer and the dimer resonance frequencies:
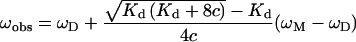 |
(1) |
where Kd is the dissociation constant of the monomer–dimer equilibrium, c is the total protein concentration, and ωM and ωD are the monomer and the dimer resonance frequencies respectively.
The Kd, ωM and ωD parameters were fitted to sets of data pairs consisting of ωobs and c, using non-linear least-squares optimization. Since the parameters are fitted all together to data for selected residues, the dissociation constant Kd can be determined with precision from a series of HSQC spectra recorded at different concentrations [33,34]. The fits were performed with a Levenberg–Marquardt non-linear least-squares optimization implemented in Gnuplot.
Absorbance and fluorescence spectroscopy
Absorption spectra were obtained using a Cary 400 spectrophotometer. Fluorescence emission spectra were recorded on a FluoroMax spectrofluorimeter (Jobin Yvon) equipped with a thermostatted cell compartment. Quantum yields were determined relative to Trp in water according to the method of Parker and Rees [35]. The quantum yield for Trp in water is taken as 0.14 [36]. At high concentrations of the peptide (>100 μM), the measurements were performed in a 50 μl microcell (optical length 0.3 mm). At lower peptide concentrations, conventional cells with a 0.4 mm optical length were used. The concentration dependence of the quantum yield of Vpr-(52–96) was fitted, using the Origin software, with an equation derived from eqn (1):
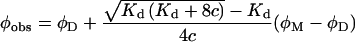 |
(2) |
where ϕM and ϕD are the quantum yields of the monomer and the dimer respectively. The pH dependence of the peptide fluorescence was determined by adding NaOH to the peptide in 25 mM Mes in the presence of 10 mM β-mercaptoethanol to avoid oxidation of the -SH groups. This experiment was done at peptide concentrations of 9.3 and 22 μM.
The time-resolved fluorescence measurements were carried out with the single-photon counting time-correlated technique, using the stable picosecond excitation pulses provided by a pulse-picked frequency tripled Ti-sapphire laser (Tsunami; Spectra Physics) pumped by a Millenia X laser (Spectra Physics) as described previously [37].
RESULTS
Determination of conditions for NMR spectroscopy
During the solid-phase synthesis of Vpr-(52–96), 17 labelled amino acids were introduced specifically at several positions in the (52–96) C-terminal sequence to facilitate the assignment of NMR spectra (Figure 1). C2H3CN at 30% (v/v) was added to the solvent to prevent the strong tendency of the domain peptide to oligomerize in pure water. During the study of Vpr-(1–96) structure, we showed that the use of TFE, a hydrophobic solvent, eliminated oligomerization, but forced the structure to adopt an open U-shaped form. The use of acetonitrile (C2H3CN), which is less hydrophobic than TFE, prevented Vpr oligomerization while still maintaining the tertiary structure of the protein due to hydrophobic interactions between the three helices. Thus acetonitrile made it possible to obtain good quality spectra that allowed investigation of dimer formation [16].
Figure 1. Primary sequence of the Vpr-(52–96) C-terminal domain.

The identified Leu/Ile-rich domain (residues 60–84) and the basic domain (residues 70–96) are specified. The 17 labelled amino acids incorporated during the chemical synthesis are represented in bold.
NMR analysis and intramolecular versus intermolecular NOE restraints
DQF (double-quantum-filtered)-COSY [38,39] and TOCSY [40] experiments were used to identify spin systems, and NOESY experiments [24,34] were used for sequential and long-range distance assignment [41]. The 17 labelled amino acids (95% 15N, 15% 13C) have been identified unambiguously from two-dimensional 15N HSQC [23,27,28], and confirmed the proton assignment (see Supplementary Table 1 at http://www.BiochemJ.org/bj/387/bj3870333add.htm). Although many overlaps occur in the aliphatic region, characteristic connectivities dαN(i,i+3) and dαβ(i,i+3) and the strong NOEs dNN(i,i+1) and dβN(i,i+1) have been identified from amino acids 53–75, indicating the presence of an α-helix spanning these residues. The C-terminal moiety (residues 76–96) of the peptide does not present any particular secondary structure element.
The antiparallel dimerization of the molecule through the formation of a leucine zipper is indicated by the correlation peaks observed between the aromatic protons of Trp54 and His71 and between the protons of Trp54 and Leu68 (Figure 2), and the unusual chemical shift of His71 protons. To obtain the structure of the homodimer with two-fold axis symmetry, it was necessary to discriminate between inter- and intra-molecular NOEs. The usual method to assign unambiguously the inter- and intra-molecular NOEs [42] by mixing equal amounts (1:1) of selectively labelled and unlabelled protein was difficult in our case, since only 17 amino acids were labelled.
Figure 2. Extracts of the NOESY spectrum recorded with a 100 ms mixing time for the Vpr-(52–96) domain.

Shown are the particular chemical shift for the 4H proton of His71 (b), its correlation with the aromatic protons of Trp54 (a, b, d), and correlations between Trp54 and Leu68 protons (c).
Structure calculation of the leucine-zipper-forming domain
The structure of the Vpr-(52–96) dimer was calculated by restrained simulated annealing, molecular dynamics and energy minimization implemented with a total of 573 NOE-derived distances. Of these distances, 275 are intraresidual, 128 are sequential and 148 are medium range (|i–j|≤4). Moreover, 22 intermolecular distances were identified and defined unambiguously, while all other restraints were defined ambiguously for structure calculation in X-PLOR [29,30]. The distinction between intramolecular and intermolecular correlations was straightforward concerning residues far from each other in the sequence, whereas this differentiation was impossible for residues close to each other both in the monomer and in the dimer. About 12 constraints per residue allowed a good definition of the structure to be obtained.
Out of 100 calculated structures, 10 were selected according to their low overall energy and their low number of distance restraint violations. None of the structure exhibited a NOE violation greater than 0.2 Å and all of them presented good covalent geometry, with no bond or angle violations. The distribution of the ϕ,ψ angles revealed that 98% of the residues were found in the allowed region, while 2% were located in non-favourable regions (Table 1). The superimposition of the 10 lowest-energy structures on their backbone atoms showed a good convergence of the structures (Figure 3). The pairwise r.m.s.d. (root mean square deviation) was calculated for the monomer and evaluated to 0.90±0.22 Å on the backbone atoms for residues 53–75, which form the best defined part of the structure. The pairwise r.m.s.d. calculated on the backbone atoms for the dimer was calculated as 1.40±0.30 Å for the same domain. Input data for structural calculations and statistics are listed in Table 1.
Table 1. Structural statistics for the structure of Vpr-(52–96).
1 kcal=4.184 kJ.
| Parameter | Value | |
|---|---|---|
| Restraints for calculation | ||
| Total NOE restraints | 573 | |
| Intraresidue | 275 | |
| Sequential (|i=j|=1) | 128 | |
| Medium range (|i–j|≤4) | 148 | |
| Intermolecular | 22 | |
| Structure statistics | ||
| R.m.s.d. | ||
| Bonds (Å) | (1.87–2.38)×10−3 | |
| Bond angles (°) | 0.48–0.54 | |
| Improper torsions (°) | 0.36–0.38 | |
| NOE restraints (Å) | (1.76–2.41)×10−2 | |
| Final energies (kcal/mol) | ||
| Total | 136.43–186.09 | |
| Bonds | 5.33–8.69 | |
| Angles | 97.94–120.01 | |
| Improper angles | 15.76–17.97 | |
| Van der Waals | 8.18–27.41 | |
| NOE | 9.19–17.30 | |
| Ramachandran plot | ||
| Residues in most favourables regions (%) | 60.00 | |
| Residues in additional allowed regions (%) | 32.82 | |
| Residues in generously allowed regions (%) | 5.13 | |
| Residues in disallowed regions (%) | 2.05 | |
| Atomic r.m.s.d. (Å) on the backbone atoms | Vpr-(52–75) monomer* | Vpr-(52–75) dimer† |
| Pairwise | 0.90±0.22 | 1.40±0.30 |
| To mean structure | 0.88±0.14 | 1.30±0.15 |
* The superimposition was performed for both monomers of Vpr-(52–96) separately; results were the same for the two monomers.
† The superimposition was performed for both monomers.
Figure 3. Structure of the Vpr-(52–96) dimer.

Superimposition of 10 NMR refined structures of Vpr-(52–96) showing the arrangement of the leucine-zipper structure. The helix spanning residues 53–75 is depicted in black and shows a good convergence with a low r.m.s.d. calculated on the backbone atoms. The domain encompassing residues 80–96 is shaded in grey and is disordered.
Each domain within the dimer structure displays the same characacteristics as the monomer structure calculated in the presence of 30% (v/v) TFE [18]. The structure is an amphipathic α-helix with a large hydrophobic face composed of residues Trp54, Gly56, Val57, Leu60, Ile61, Ile63, Leu64, Leu67, Leu68, Ile70, His71 and Ile74, and a less extended hydrophilic face constituted by residues Glu58, Arg62, Gln65, Gln66, Arg73 and Arg77. Leucine, valine and isoleucine residues constituting the hydrophobic interface of the dimer, with Trp54/His71 clamping the structure, are represented on Figure 4.
Figure 4. Residues implicated at the interface of the dimer.

View of the leucine-zipper structure showing the stacking of residues Trp54 and His71 (in dark grey) at each edge. Amino acids involved in the hydrophobic interface are shown in light grey. Only the domain comprising residues 53–75 is represented.
The relative orientation of the residues essential for formation of the leucine-zipper motif can be represented on a helical wheel (Figure 5). Many isoleucine and leucine residues are in the required position to interact to form the leucine zipper, and the stability of the dimer is ensured by hydrophobic interactions taking place at the interface. The dimeric structure is also maintained by the stacking of residues Trp54 and His71. A supplementary hydrophobic platform comprising residues Gly56, Leu60, Ile63, Leu67, Ile70 and Ile74 remains for additional interactions (Figure 5).
Figure 5. Helical wheel representation of the Vpr-(52–96) dimer.

The relative arrangement of the two α-helices forming the leucine-zipper dimer in antiparallel mode is shown. Leu/Ile residues are found at the interface of the dimer characterized by a hydrophobic face and a hydrophilic one. A second hydrophobic platform and a hydrophilic one seem to be available for supplementary interactions. Hydrophobic residues are underlined.
Characterization of the dimer interface and dissociation constant determination
Dimerization of the Vpr-(52–96) C-terminal domain and the dissociation constant for the monomer–dimer equilibrium were investigated by following the 1H and 15N chemical shift variations over a concentration range from 2.0 to 0.03 mM by heteronuclear NMR experiments. Figure 6 shows the superposition of the 15N HSQC spectra registered at the lowest and highest concentrations displaying the greatest chemical shift variations. Significant concentration-dependent 1H and 15N chemical shift variations were examined for a subset of residues involved at the interface of the dimer and are reported in Table 2. The largest chemical shift variations were observed for residues Thr55, Ile61, His71 and Phe72 in the 1H dimension, and Thr55, Val57, Ile61 and Phe72 in the 15N dimension. The absolute values of the 1H and 15N chemical shift deviations varied from 0.005 to 0.12 p.p.m. and from 0.02 to 0.76 p.p.m. respectively. As can be observed, the amino acids experiencing the most important chemical shift variations are located either directly at or close to the dimer interface.
Figure 6. Heteronuclear spectra of Vpr-(52–96).

Superimposition of the results from the echo/antiecho HSQC experiments performed at 600 MHz, 303 K and pH 2.6, at low concentration (0.03 mM) in black and at high concentration (2.0 mM) in red. The 17 labelled amino acids have been assigned and are identified on the spectrum. Arrows indicate residues with the most substantial 1H chemical shift variations: Thr55, Ile61, Leu67, Leu68, His71 and Phe72.
Table 2. Concentration-dependent amide proton and nitrogen chemical shift variations for Vpr-(52–96).
i and f indicate the chemical shift measured at 31×10−6 M and 2.0×10−3 M respectively.
| Chemical shift (p.p.m.) | ||||||
|---|---|---|---|---|---|---|
| Residue | δHi | δHf | ΔδH | δNi | δNf | ΔδN |
| Thr53 | 8.33 | 8.31 | 0.02 | 114.09 | 114.18 | 0.09 |
| Thr55 | 7.85 | 7.96 | 0.11 | 113.81 | 114.39 | 0.58 |
| Val57 | 7.79 | 7.78 | 0.01 | 120.58 | 121.23 | 0.65 |
| Ala59 | 7.83 | 7.81 | 0.02 | 120.48 | 120.65 | 0.17 |
| Ile61 | 7.92 | 8.04 | 0.12 | 118.48 | 119.25 | 0.77 |
| Ile63 | 7.71 | 7.76 | 0.05 | 119.67 | 119.84 | 0.17 |
| Leu64 | 8.22 | 8.24 | 0.02 | 120.21 | 120.11 | 0.10 |
| Leu67 | 8.14 | 8.10 | 0.04 | 118.93 | 119.03 | 0.10 |
| Leu68 | 8.07 | 8.05 | 0.02 | 119.44 | 119.25 | 0.19 |
| Phe69 | 8.02 | 8.03 | 0.01 | 118.51 | 118.46 | 0.05 |
| His71 | 8.03 | 7.96 | 0.07 | 118.01 | 117.29 | 0.72 |
| Phe72 | 8.07 | 7.95 | 0.12 | 117.25 | 117.08 | 0.17 |
| Gly75 | 8.01 | 7.99 | 0.02 | 109.98 | 110.09 | 0.11 |
| Gly82 | 8.12 | 8.09 | 0.03 | 111.68 | 111.68 | 0.00 |
| Thr89 | 7.93 | 7.91 | 0.02 | 114.30 | 114.35 | 0.05 |
| Ala93 | 7.96 | 7.93 | 0.03 | 122.90 | 122.93 | 0.03 |
| Lys95 | 8.10 | 8.09 | 0.01 | 122.44 | 122.57 | 0.13 |
Since the rate of exchange between the monomeric and dimeric states is much higher than the difference in chemical shifts between the two states, the observed resonance frequency, ωobs, may be related to the population-weighted average of the monomer and dimer resonance frequencies according to eqn (1). The mean Kd values calculated from the 1H and 15N data were (9±3)×10−4 M and (6±8)×10−4 M respectively, and are thus fully consistent. Due to larger shift changes relative to line widths and higher digital resolution in the 1H dimension, greater precision has been obtained for the 1H data set. Relative populations of monomer and dimer at different concentrations were calculated (Figure 7). The analysis was limited by the significant line broadening of the NMR spectrum attributable to increased transverse relaxation rates and by the increased population of dimers at a given sample concentration.
Figure 7. Monomer–dimer equilibrium for Vpr-(52–96) in 30% (v/v) C2H3CN.

The population of monomers, pM, as a function of the peptide concentration was calculated according to the equation pM=(ωobs−ωD)/(ωM−ωD). The symbols (×) indicate the experimental data points included in the fit to eqn (1) in order to determine the Kd. The continuous and broken lines indicate the fitted populations of monomers and dimers respectively.
Comparison with the C-terminal domain of previously determined structures
The present structure established in the presence of 30% (v/v) C2H3CN and the same domain in the full-length protein Vpr-(1–96) [16] have been compared. The helix, spanning residues 53–75, is a little shorter than in the Vpr-(1–96) structure. The orientation of the hydrophobic residues is conserved and located on the same face of the helix, and many of these residues are involved at the interface of the dimer or in the formation of the hydrophobic core in the full protein. A superimposition of the 53–75 domain of one α-helix of the dimer and of the same domain in the whole protein gives an r.m.s.d. of 1.2 Å calculated on the backbone atoms. Interestingly, the second α-helix 53–75 of the dimer adopts the same position as the N-terminal α-helix 15–34 in the whole protein, in the opposite direction. These data show that the hydrophobic residues are protected from the solvent at the interface of the dimer [Vpr-(52–96)] as in the cluster in the whole protein [Vpr-(1–96)]. In all cases, the C-terminal domain spanning residues 80–96 is disordered (Figure 8).
Figure 8. Superimposition of Vpr-(1–96) with Vpr-(52–96).

The two averaged minimized structures of the dimeric leucine zipper formed by the C-terminal domain Vpr-(52–96) (red) and the full-length Vpr-(1–96) (yellow) protein are superimposed.
Fluorescence spectroscopy
To assess independently the dimer–monomer equilibrium of Vpr-(52–96), fluorescence experiments were performed by taking advantage of the presence of a single N-terminal Trp residue. The dependence of the quantum yield of Trp54 in Vpr-(52–96) on peptide concentration was investigated. The rationale of this approach is that the spatial proximity between Trp54 and His71 shown by NMR in the head-to-tail dimer should lead to strong quenching of the Trp54 fluorescence. Indeed, His has been shown to be an efficient quencher, but only in its protonated form [43,44]. The concentration dependence of the quantum yield of Vpr-(52–96) was first determined under the NMR conditions (30% acetonitrile, pH 3.0), where the His residues are expected to be fully protonated. In agreement with our hypothesis, we observed a dramatic decrease in the quantum yield when the peptide concentration was increased (Figure 9). This decrease was not associated with any change in the maximum emission wavelength, which remained at 448 nm, a wavelength typical of solvent-exposed Trp residues. The concentration dependence of the quantum yield of Vpr-(52–96) was fitted to eqn (2). We found a Kd value for the monomer–dimer equilibrium of (7±3)×10−4 M, in excellent agreement with the Kd values obtained by NMR. Moreover, the values of ϕM and ϕD were found to be 0.025 and 0 respectively. The negligible fluorescence of the dimers suggests that, due to their close proximity, the Trp54 and His71 residues may form ground-state interactions that totally quench Trp fluorescence. Such interactions have already been reported between 3-methylindole and N-acetylhistine at high concentrations (>0.2 M) [44]. In agreement with this, the time-resolved fluorescence parameters of Vpr-(52–96) at high concentrations (2 mM) differ only slightly from those at low concentration (1 μM), confirming that the dimers either are not emissive or emit with a lifetime lower than the resolution (20 ps) permitted by our device (results not shown).
Figure 9. Dependence of Vpr-(52–96) quantum yield on peptide concentration.

The single Trp residue at position 54 of Vpr-(52–96) was excited at 295 nm. The quantum yields were determined in 30% (v/v) CH3CN, pH 3.0 (●), and 25 mM Mes, pH 4.5 (■). Solid lines correspond to the fit of the experimental points with eqn (2).
To strengthen our conclusions further and exclude any artifacts due to acetonitrile, the concentration dependence of the quantum yield of Trp54 was investigated further in 25 mM Mes, pH 4.5. In line with the results obtained in 30% (v/v) 2H3CN, we found a steep decrease in the Vpr-(52–96) quantum yield when the peptide concentration was increased (Figure 9). Using eqn (2) to fit the data, a dissociation constant of (6±2)×10−5 M was obtained, indicating a shift with respect to the monomer–dimer equilibrium in 30% acetonitrile by around one order of magnitude. The fitted quantum yield of the dimer was again very small, suggesting that a head-to-tail dimer may also be observed in the Mes buffer. To confirm this, the pH dependence of the fluorescence of Trp54 was monitored in this buffer. In line with the fluorescence quenching properties of the protonated form of His and the usual pKa values of His residues, a sharp fluorescence change with a mid-transition at around pH 5.5 was observed (Figure 10). From the sigmoidal shape of the transition, it is further inferred that the protonation of the dimers may be considered as an ‘all-or-none’ reaction, where the two His residues in the dimer are protonated simultaneously. To check this hypothesis, we fitted this pH titration to the following equation:
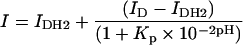 |
(3) |
where I is the fluorescence intensity at a given pH, and ID and IDH2 are the fluorescence intensities of the unprotonated and protonated dimer respectively. The protonation constant Kp describes the simultaneous protonation of the two His residues in the dimer. A very good fit was obtained with this model, giving a Kp value of 9.1×1010 M−2. In sharp contrast, no satisfactory fit could be obtained if either non-cooperative binding of the two protons or binding of a single proton is assumed (Figure 10, broken line). Notably, the non-zero value of the fluorescence intensity at acidic pH is probably related to the population of Vpr-(52–96) monomers existing at the peptide concentration (22 μM) of the experiment. It was confirmed at low peptide concentration (1 μM) that the fluorescence intensity of the monomers exhibited negligible sensitivity to pH, suggesting that the protonation of His71 does not significantly affect the fluorescence of Trp54 on the same monomer (results not shown).
Figure 10. pH dependence of Vpr-(52–96) quantum yield.

The peptide concentration was 22 μM in 25 mM Mes and 10 mM β-mercaptoethanol. Excitation and emission wavelengths were 295 and 350 nm respectively. The solid line corresponds to a fit of the experimental points with eqn (3). The broken line corresponds to a fit with an equation assuming either binding of a single proton or non-cooperative binding of two protons.
DISCUSSION
In the present work, we have confirmed that the structure adopted by the Vpr-(52–96) domain is a well defined α-helix, spanning residues 53–75, as determined in an earlier study [18] performed in 30% (v/v) TFE. However, in contrast with this previous study, we show herein that, in the presence of 30% (v/v) C2H3CN, Vpr-(52–96) was able to form a head-to-tail dimer through interaction between two leucine-zipper-like domains. Although such dimers have already been suggested from CD and gel filtration experiments [18], the present study constitutes the first direct structural evidence for the formation of a dimer involving the leucine-zipper-like motif within Vpr-(52–96).
The proposed antiparallel dimeric structure is strongly substantiated by the following evidence: (i) the large upfield shift undergone by the 4H proton of His71 resulting from the current cycle due to the stacking with Trp54, (ii) the strong fluorescence quenching of Trp54 by the protonated form of His71 in the opposite monomer and (iii) the co-operative protonation of the His71 residues in the dimer. In this dimeric structure, residues Trp54, Val57, Ile61, Leu64, Leu68, His71 and Gly75 constitute the hydrophobic interface. Another hydrophobic domain constituted by residues Gly56, Leu60, Ile63, Leu67, Ile70 and Ile74 remains accessible to interact with additional Vpr-(52–96) partners (Figure 5). A flexible region, encompassing residues 76–96, follows the helical region.
It appears that the two helices of the Vpr-(52–96) dimer mimic the first and third helices of the full-length Vpr-(1–96) structure (Figure 8). In both structures, the hydrophobic residues (Gly56, Leu60, Ile63, Leu67, Ile70 and Ile74) remain accessible for interaction. However, in Vpr-(52–96), two hydrophobic platforms exist, since two monomers are present. In the full-length protein, only one hydrophobic domain is available, while the second one is replaced by the hydrophilic region made of residues Arg12, Glu13, Asp17, Glu21, Glu24, Asn28 and Arg32 found in the first helix.
Vpr-(52–96) interacts with various partners such as proteins and nucleic acids and, among its most important properties, it is an apoptogenic peptide. It induces mitochondrial membrane permeabilization via a specific interaction with the ANT (adenine nucleotide translocator) located in the inner membrane of the mitochondria [45]. Vpr-(52–96) is also able to transfect various cell lines [10,46]. The dimeric structure reported in the present work could help to explain how Vpr-(52–96) is able to interact with host or viral effectors. Its resemblance to the entire protein structure could also explain the similarities between Vpr-(1–96) and Vpr-(52–96) with regard to their interactions, while the differences in structure might explain some of the variations in the interactions of Vpr-(52–96) and Vpr-(1–96), depending on the residues of Vpr implicated.
Recently, the effects of full-length Vpr and of the C-terminal peptide Vpr-(52–96) on the integration properties of integrase have been compared [47]. Both of them, at concentrations above 1.6 μM, inhibit the homologous integration activity of integrase. Conversely, at concentrations below 1.6 μM, Vpr-(1–96) is still an inhibitor, whereas Vpr-(52–96) stimulates integration. This phenomenon can be explained in the following way. At concentrations above 1.6 μM, Vpr-(52–96) is most probably a dimer, mimicking and exhibiting the same properties as Vpr-(1–96), i.e. inhibiting integration. In this case, the same hydrophobic platform remains accessible for interaction and prevents integrase activity. The domain comprising residues 71–82 is certainly not involved in this process, since it is as accessible in the dimer as in full-length Vpr. At lower concentrations, Vpr-(52–96) is in the monomeric form, and its effect will be different because its three-dimensional structure has changed. A second hydrophobic platform appears that was previously buried within the dimer, and is available for additional interactions, which inhibit integration activity. Thus the accessible residues are different from those in the Vpr-(52–96) dimer or in the full-length protein. This might explain the differences between the effects of this domain and of the full-length protein on integrase integration activity at low concentrations.
The capacity of Vpr-(1–96) and some of its fragments to interact with oligonucleotides has also been studied [10]. It was shown that the basic residues implicated in DNA interactions are located in the domain comprising residues 70–80 [48,49] containing an H(S/F)RIG motif. In transfection assays, Vpr-(52–96) is more efficient than Vpr-(1–96). These results are fully consistent with the present study. Since the Vpr-(52–96) domain may dimerize, it presents two basic domains located at the C-terminal end of each monomer. This will double the capacity of this peptide to interact with nucleic acids, compared with Vpr-(1–96), which possesses only one such domain. This might explain why Vpr-(52–96) is a better transfection agent than the full-length protein.
Vpr induces apoptosis [4] in cells via its interaction with ANT. The full-length protein or its 52–96 domain alone kills Jurkat lymphoma cells, while Vpr-(1–51) is unable to perform such an activity. Vpr-(52–96) conserves this property, even when the peptide is in the monomeric state. It has been shown that Vpr-(71–96) can also induce apoptosis, and that mutation of Arg73, Arg77 or Arg80 reduces the apoptogenic effect of Vpr-(52–96). These results are consistent with the structure determined in the present work whereby these three arginine residues, within the 71–82 domain, are not involved in the dimerization of Vpr-(52–96) and have the same accessibility whether the domain is a dimer or not.
The knowledge of the dimeric structure of Vpr-(52–96) could form the basis for the design of new therapeutic molecules related to the transfection properties of Vpr-(52–96) or to its apoptogenic abilities. These new agents could be new transfection factors or novel molecules showing antitumour activities.
Online data
Acknowledgments
We thank Etienne Piémont for his help in lifetime measurements, and Hugues de Rocquigny for advice and discussion. This work was supported by grants from the Agence Nationale de la Recherche sur le SIDA (ANRS) and SIDACTION. S.B. and H.B. were supported by a grant from Ministère de l'Enseignement Supérieur et de la Recherche (MESR).
References
- 1.Frankel A. D., Young J. A. HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Re F., Luban J. HIV-1 Vpr: G2 cell cycle arrest, macrophages and nuclear transport. Prog. Cell Cycle Res. 1997;3:21–27. doi: 10.1007/978-1-4615-5371-7_2. [DOI] [PubMed] [Google Scholar]
- 3.Jowett J. B., Planelles V., Poon B., Shah N. P., Chen M. L., Chen I. S. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2+M phase of the cell cycle. J. Virol. 1995;69:6304–6313. doi: 10.1128/jvi.69.10.6304-6313.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacotot E., Ravagnan L., Loeffler M., Ferri K. F., Vieira H. L., Zamzami N., Costantini P., Druillennec S., Hoebeke J., Briand J. P., et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 2000;191:33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart S. A., Poon B., Jowett J. B., Chen I. S. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 1997;71:5579–5592. doi: 10.1128/jvi.71.7.5579-5592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanitharani R., Mahalingam S., Rafaeli Y., Singh S. P., Srinivasan A., Weiner D. B., Ayyavoo V. HIV-1 Vpr transactivates LTR-directed expression through sequences present within -278 to -176 and increases virus replication in vitro. Virology. 2001;289:334–342. doi: 10.1006/viro.2001.1153. [DOI] [PubMed] [Google Scholar]
- 7.Ayyavoo V., Mahalingam S., Rafaeli Y., Kudchodkar S., Chang D., Nagashunmugam T., Williams W. V., Weiner D. B. HIV-1 viral protein R (Vpr) regulates viral replication and cellular proliferation in T cells and monocytoid cells in vitro. J. Leukocyte Biol. 1997;62:93–99. doi: 10.1002/jlb.62.1.93. [DOI] [PubMed] [Google Scholar]
- 8.Fouchier R. A., Malim M. H. Nuclear import of human immunodeficiency virus type-1 preintegration complexes. Adv. Virus Res. 1999;52:275–299. doi: 10.1016/s0065-3527(08)60302-4. [DOI] [PubMed] [Google Scholar]
- 9.Le Rouzic E. G., Mousnier A., Rustum C., Stutz F., Hallberg E., Dargemont C., Benichou S. Docking of HIV-1 VPR to the nuclear envelope is mediated by the interaction with the nucleoporin hCG1. J. Biol. Chem. 2002;277:45091–45098. doi: 10.1074/jbc.M207439200. [DOI] [PubMed] [Google Scholar]
- 10.Kichler A., Pages J. C., Leborgne C., Druillennec S., Lenoir C., Coulaud D., Delain E., Le Cam E., Roques B. P., Danos O. Efficient DNA transfection mediated by the C-terminal domain of human immunodeficiency virus type 1 viral protein R. J. Virol. 2000;74:5424–5431. doi: 10.1128/jvi.74.12.5424-5431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao X. J., Subbramanian R. A., Rougeau N., Boisvert F., Bergeron D., Cohen E. A. Mutagenic analysis of human immunodeficiency virus type 1 Vpr: role of a predicted N-terminal alpha-helical structure in Vpr nuclear localization and virion incorporation. J. Virol. 1995;69:7032–7044. doi: 10.1128/jvi.69.11.7032-7044.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engler A., Stangler T., Willbold D. Structure of human immunodeficiency virus type 1 Vpr(34–51) peptide in micelle containing aqueous solution. Eur. J. Biochem. 2002;269:3264–3269. doi: 10.1046/j.1432-1033.2002.03005.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y., Lu Y., Ratner L. Arginine residues in the C-terminus of HIV-1 Vpr are important for nuclear localization and cell cycle arrest. Virology. 1998;242:414–424. doi: 10.1006/viro.1998.9028. [DOI] [PubMed] [Google Scholar]
- 14.de Rocquigny H., Petitjean P., Tanchou V., Decimo D., Drouot L., Delaunay T., Darlix J. L., Roques B. P. The zinc fingers of HIV nucleocapsid protein NCp7 direct interactions with the viral regulatory protein Vpr. J. Biol. Chem. 1997;272:30753–30759. doi: 10.1074/jbc.272.49.30753. [DOI] [PubMed] [Google Scholar]
- 15.Wecker K., Morellet N., Bouaziz S., Roques B. P. NMR structure of the HIV-1 regulatory protein Vpr in H2O/trifluoroethanol. Comparison with the Vpr N-terminal (1-51) and C-terminal (52-96) domains. Eur. J. Biochem. 2002;269:3779–3788. doi: 10.1046/j.1432-1033.2002.03067.x. [DOI] [PubMed] [Google Scholar]
- 16.Morellet N., Bouaziz S., Petitjean P., Roques B. P. NMR structure of the HIV-1 regulatory protein VPR. J. Mol. Biol. 2003;327:215–227. doi: 10.1016/s0022-2836(03)00060-3. [DOI] [PubMed] [Google Scholar]
- 17.Wecker K., Roques B. P. NMR structure of the (1-51) N-terminal domain of the HIV-1 regulatory protein Vpr. Eur. J. Biochem. 1999;266:359–369. doi: 10.1046/j.1432-1327.1999.00858.x. [DOI] [PubMed] [Google Scholar]
- 18.Schuler W., Wecker K., de Rocquigny H., Baudat Y., Sire J., Roques B. P. NMR structure of the (52-96) C-terminal domain of the HIV-1 regulatory protein Vpr: molecular insights into its biological functions. J. Mol. Biol. 1999;285:2105–2117. doi: 10.1006/jmbi.1998.2381. [DOI] [PubMed] [Google Scholar]
- 19.Engler A., Stangler T., Willbold D. Solution structure of human immunodeficiency virus type 1 Vpr(13–33) peptide in micelles. Eur. J. Biochem. 2001;268:389–395. doi: 10.1046/j.1432-1033.2001.01895.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang L., Mukherjee S., Narayan O., Zhao L. J. Characterization of a leucine-zipper-like domain in Vpr protein of human immunodeficiency virus type 1. Gene. 1996;178:7–13. doi: 10.1016/0378-1119(96)00312-5. [DOI] [PubMed] [Google Scholar]
- 21.Piller S. C., Ewart G. D., Jans D. A., Gage P. W., Cox G. B. The amino-terminal region of Vpr from human immunodeficiency virus type 1 forms ion channels and kills neurons. J. Virol. 1999;73:4230–4238. doi: 10.1128/jvi.73.5.4230-4238.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cornille F., Wecker K., Loffet A., Genet R., Roques B. Efficient solid-phase synthesis of Vpr from HIV-1 using low quantities of uniformly 13C-, 15N-labeled amino acids for NMR structural studies. J. Pept. Res. 1999;54:427–435. doi: 10.1034/j.1399-3011.1999.00129.x. [DOI] [PubMed] [Google Scholar]
- 23.Piotto M., Saudek V., Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 24.Jeener J., Meier B. H., Bachmann P., Ernst R. R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979;71:4546–4553. [Google Scholar]
- 25.Shaka A., Barker P. B., Freeman R. Computer-optimized decoupling scheme for wideband applications and low-level operation. J. Magn. Reson. 1985;64:547–552. [Google Scholar]
- 26.Schleucher J., Schwendinger M., Sattler M., Schmidt P., Schedletzky O., Glaser S. J., Sorensen O. W., Griesinger C. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR. 1994;4:301–306. doi: 10.1007/BF00175254. [DOI] [PubMed] [Google Scholar]
- 27.Bax A., Ikura M., Kay L. E., Torchia D. A., Tschudin R. Comparison of different modes of two-dimensional reverse-correlation NMR for the study of proteins. J. Magn. Reson. 1990;86:304–318. [Google Scholar]
- 28.Norwood T. J., Boyd J. E., Heritage J. E., Soffe N., Campbell I. D. Comparison of techniques for 1H-detected heteronuclear 1H-15 N spectroscopy. J. Magn. Reson. 1990;87:488–501. [Google Scholar]
- 29.Nilges M., Clore G. M., Gronenborn A. M. Determination of three-dimensional structures of proteins from interproton distance data by dynamical simulated annealing from a random array of atoms. Circumventing problems associated with folding. FEBS Lett. 1988;239:129–136. doi: 10.1016/0014-5793(88)80559-3. [DOI] [PubMed] [Google Scholar]
- 30.Brünger A. T. New Haven, CT: Yale University Press; 1992. X-PLOR Software Manual, version 3.1. [Google Scholar]
- 31.Nilges M. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J. Mol. Biol. 1995;245:645–660. doi: 10.1006/jmbi.1994.0053. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 33.Akerud T., Thulin E., Van Etten R. L., Akke M. Intramolecular dynamics of low molecular weight protein tyrosine phosphatase in monomer-dimer equilibrium studied by NMR: a model for changes in dynamics upon target binding. J. Mol. Biol. 2002;322:137–152. doi: 10.1016/s0022-2836(02)00714-3. [DOI] [PubMed] [Google Scholar]
- 34.Kumar A., Ernst R. R., Wuthrich K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980;95:1–6. doi: 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- 35.Parker C. A., Rees W. T. Correction of fluorescence spectra and measurement of fluorescence quantum efficiency. Analyst. 1960;85:587–600. [Google Scholar]
- 36.Kirby E. P., Steiiner R. F. The influence of solvent and temperature upon the fluorescence of indole derivatives. J. Phys. Chem. 1970;74:4480–4490. [Google Scholar]
- 37.Ramos P., Coste T., Piemont E., Lessinger J. M., Bousquet J. A., Chapus C., Kerfelec B., Ferard G., Mely Y. Time-resolved fluorescence allows selective monitoring of Trp30 environmental changes in the seven-Trp-containing human pancreatic lipase. Biochemistry. 2003;42:12488–12496. doi: 10.1021/bi034900e. [DOI] [PubMed] [Google Scholar]
- 38.Rance M., Sorensen O. W., Bodenhausen G., Wagner G., Ernst R. R., Wuthrich K. Improved spectral resolution in cosy 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 1983;117:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 39.Marion D., Wüthrich K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 1983;113:967–974. doi: 10.1016/0006-291x(83)91093-8. [DOI] [PubMed] [Google Scholar]
- 40.Griesinger C., Otting G., Wüthrich K., Ernst R. R. Clean TOCSY for 1H spin system identification in macromolecules. J. Am. Chem. Soc. 1988;110:7870–7872. [Google Scholar]
- 41.Wüthrich K. Hoboken, NJ: Wiley Interscience; 1986. NMR of Proteins and Nucleic Acids. [Google Scholar]
- 42.Burgering M. J., Boelens R., Gilbert D. E., Breg J. N., Knight K. L., Sauer R. T., Kaptein R. Solution structure of dimeric Mnt repressor (1–76) Biochemistry. 1994;33:15036–15045. doi: 10.1021/bi00254a012. [DOI] [PubMed] [Google Scholar]
- 43.Shinitzky M., Goldman R. Fluorometric detection of histidine-tryptophan complexes in peptides and proteins. Eur. J. Biochem. 1967;3:139–144. doi: 10.1111/j.1432-1033.1967.tb19508.x. [DOI] [PubMed] [Google Scholar]
- 44.Chen Y., Barkley M. D. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 45.Jacotot E., Ferri K. F., El Hamel C., Brenner C., Druillennec S., Hoebeke J., Rustin P., Metivier D., Lenoir C., Geuskens M., et al. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J. Exp. Med. 2001;193:509–519. doi: 10.1084/jem.193.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coeytaux E., Coulaud D., Le Cam E., Danos O., Kichler A. The cationic amphipathic alpha-helix of HIV-1 viral protein R (Vpr) binds to nucleic acids, permeabilizes membranes, and efficiently transfects cells. J. Biol. Chem. 2003;278:18110–18116. doi: 10.1074/jbc.M300248200. [DOI] [PubMed] [Google Scholar]
- 47.Bischerour J., Tauc P., Leh H., de Rocquigny H., Roques B., Mouscadet J. F. The (52–96) C-terminal domain of Vpr stimulates HIV-1 IN-mediated homologous strand transfer of mini-viral DNA. Nucleic Acids Res. 2003;31:2694–2702. doi: 10.1093/nar/gkg364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Rocquigny H., Caneparo A., Delaunay T., Bischerour J., Mouscadet J. F., Roques B. P. Interactions of the C-terminus of viral protein R with nucleic acids are modulated by its N-terminus. Eur. J. Biochem. 2000;267:3654–3660. doi: 10.1046/j.1432-1327.2000.01397.x. [DOI] [PubMed] [Google Scholar]
- 49.Zhang S., Pointer D., Singer G., Feng Y., Park K., Zhao L. J. Direct binding to nucleic acids by Vpr of human immunodeficiency virus type 1. Gene. 1998;212:157–166. doi: 10.1016/s0378-1119(98)00178-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.