

Abstract
The aim of the present study was to explore interactions between surface-membrane DHPR (dihydropyridine receptor) Ca2+ channels and RyR (ryanodine receptor) Ca2+ channels in skeletal-muscle sarcoplasmic reticulum. The C region (725Phe-Pro742) of the linker between the 2nd and 3rd repeats (II–III loop) of the α1 subunit of skeletal DHPRs is essential for skeletal excitation–contraction coupling, which requires a physical interaction between the DHPR and RyR and is independent of external Ca2+. Little is known about the regulatory processes that might take place when the two Ca2+ channels interact. Indeed, interactions between C fragments of the DHPR (C peptides) and RyR have different reported effects on Ca2+ release from the sarcoplasmic reticulum and on RyR channels in lipid bilayers. To gain insight into functional interactions between the proteins and to explore different reported effects, we examined the actions of C peptides on RyR1 channels in lipid bilayers with three key RyR regulators, Ca2+, Mg2+ and ATP. We identified four discrete actions: two novel, low-affinity (>10 μM), rapidly reversible effects (fast inhibition and decreased sensitivity to Mg2+ inhibition) and two slowly reversible effects (high-affinity activation and a slow-onset, low-affinity inhibition). Fast inhibition and high-affinity activation were decreased by ATP. Therefore peptide activation in the presence of ATP and Mg2+, used with Ca2+ release assays, depends on a mechanism different from that seen when Ca2+ is the sole agonist. The relief of Mg2+ inhibition was particularly important since RyR activation during excitation–contraction coupling depends on a similar decrease in Mg2+ inhibition.
Keywords: DHPR–RyR interaction, dihydropyridine receptor (DHPR) II–III loop, excitation–contraction coupling, magnesium inhibition, peptide C, ryanodine receptor (RyR)
Abbreviations: RyR, ryanodine receptor; DHPR, dihydropyridine receptor; DIDS, di-isothiocyanostilbene-2,2′-disulphonic acid; EC, excitation–contraction; SR, sarcoplasmic reticulum; TES, N-tris[hydroxymethyl]methyl-2-aminoethanesulphonic acid
INTRODUCTION
During EC (excitation–contraction) coupling, the action potential in the transverse tubule membrane of striated muscle fibres leads to a release of Ca2+ from the SR (sarcoplasmic reticulum). Two key components of this process are the DHPR (dihydropyridine receptor; an L-type Ca2+ channel), which is the voltage sensor in the transverse tubule membrane [1–4], and the RyR (ryanodine receptor), which is the Ca2+ release channel in the SR. Molecular interactions between these two Ca2+ channels play a key role in EC coupling and are fundamentally different in skeletal and cardiac muscles [5]. In the heart, transverse tubule depolarization triggers an influx of Ca2+ through the DHPR, which activates RyRs and calcium release [6–8]. In skeletal muscles, the influx of Ca2+ is not required [5] and RyR activation depends on a physical link between the DHPR and RyR [9] that is dependent on cytoplasmic levels of Ca2+, ATP and Mg2+ [13–16].
A picture of how RyRs are regulated by the DHPR in skeletal muscles, in conjunction with intracellular Ca2+, Mg2+ and ATP is emerging. Isolated RyRs are inhibited by cytoplasmic Mg2+ (the half-inhibitory concentration of Mg2+, Ki, is ∼0.2 mM at physiological ionic strength [10]) and activated by Ca2+ (∼1 μM [11]) as well as ATP (∼0.3 mM, even in the absence of Ca2+; [12]). During transverse tubule depolarization and activation of the RyR by the DHPR, (i) the sensitivity of RyRs to inhibition by Mg2+ is markedly reduced [13,14], (ii) ATP is a key activator of calcium release from the SR [15] and (iii) although Ca2+ is not the major RyR activator in EC coupling, it augments RyR activation [16,17]. These findings support Lamb and Stephenson's proposition [13] that, during EC coupling, DHPRs somehow relieve Mg2+ inhibition and thus permit RyR activation by ATP and Ca2+. The regions of the DHPR that interact physically with the RyR have been examined in myocytes/myotubes as well as in cell-free systems. In the first case, cDNAs encoding chimaeric constructs of skeletal and cardiac DHPR α1 subunits injected into dysgenic myotubes (i.e. myotubes lacking DHPRs [1,3,18]) revealed that the cytoplasmic loop between repeats II and III of the skeletal DHPR α1 subunit (666Glu-Leu791) [2], and specifically residues 725Phe-Pro742, are the crucial determinants of skeletal EC coupling [19]. The recombinant II–III loop activates isolated skeletal but not cardiac RyRs [20]. Shorter synthetic sequences [21], AS (671Thr-Leu690) and CS (724Glu-Pro760), also interact with isolated RyRs [34–39]. AS activates skeletal RyRs [21–25] or inhibits them by blocking the ion-conduction pathway [23]. In spite of the importance of residues 725Phe-Pro742 for skeletal EC coupling [19], the regulation of RyRs by the corresponding synthetic peptide is not well understood. Ryanodine binding and Ca2+ release assays show that CS is a much weaker activator of RyRs than AS [21,22,26], whereas single-channel studies show that CS strongly activates RyRs [25,27].
In the present study, we have further defined the interactions between CS and skeletal RyRs (RyR1) in the presence of Ca2+, Mg2+ and ATP, which are key physiological regulators of the RyR. Importantly, Mg2+ and ATP were present in previous experiments examining Ca2+ release from SR [21,22,26], but were not present in single-channel studies [25,27]. To characterize further the interaction with the RyR, we have compared the effects of CS with those of related peptides, CC (the cardiac C sequence), since replacement of the crucial region in the α1C subunit with its skeletal counterpart restores skeletal EC coupling [19]. We also examined the two halves of CS, the CS1 (725Phe-Gly743) and CS2 (740Asp-Pro760). CS2 is more potent (Ki∼290 μM) compared with CS1 (Ki∼3 mM) in reversing the effects of AS (30 μM) on Ca2+ release from SR vesicles, [3H]ryanodine binding to RyRs and changes in MCA (4-methylcoumaryl-7-amide) fluorescence [22]. However, unlike CS2, only the CS1 region is crucial for skeletal-type EC coupling in myocytes [19].
EXPERIMENTAL
Peptides
Peptides with the sequences shown in Figure 1 were synthesized as in [23].
Figure 1. Sequences of the skeletal and cardiac DHPR C regions and the synthetic DHPR C-region peptides used in the present study.

Note that several residues are given (double spaced) on either side of the skeletal C region used here to illustrate points made in the Discussion section.
Isolation of SR vesicles, lipid bilayers, solutions, single-channel recording and analysis
These techniques have been described previously in [28,29]. The cis (cytoplasmic) bath contained either 230 mM CsCH3O3S+20 mM CsCl+10 mM TES (N-tris[hydroxymethyl]methyl-2-aminoethanesulphonic acid; pH 7.4 adjusted with CsOH) (250 mM Cs+ solution) or 80 mM CsCH3O3S and 20 mM CsCl, 10 mM TES (100 mM Cs+ solution). The trans (luminal) solution contained either 30 mM CsCH3O3S+20 mM CsCl+10 mM TES (50 mM Cs+ solution) or 250 mM Cs+ solution+0.1–1.0 mM CaCl2. In experiments where vesicle fusion was performed with cis and trans baths containing 250 mM Cs+ solution, 500 mM mannitol was added to the cis bath to produce the osmotic gradient necessary for vesicle fusion. [Ca2+] was determined using a Ca2+ electrode (Fluka, Rolconkoma, NY, U.S.A.). [Mg2+] was estimated from the total amount of MgCl2 using published association constants [30] and the program ‘Bound and Determined’ [31]. Lipids were obtained from Avanti Polar Lipids (Alabaster, AL, U.S.A.), ATP, CsCH3O3S, CsCl and MgCl2 from Sigma, CaCl2 from BDH Chemicals (Poole, Dorset, U.K.) and TES, CsOH and DIDS (di-isothiocyanostilbene-2,2′-disulphonic acid) from MP Biomedicals (Seven Hills, NSW, Australia).
Positive current is defined as flow of positive charge from cytoplasm to lumen (cis to trans). Electrical potential is expressed as Vcytoplasm−Vlumen (i.e. Vcis−Vtrans). Channel activity was recorded at +40 mV. Channels were exposed to different conditions by flowing solutions over the bilayer using the local perfusion system with an exchange time of 1–2 s, depending on the flow rate [32].
DIDS modification
In one experiment, RyRs were modified by DIDS to slow the gating kinetics of the channels. The RyRs were almost fully activated by 30–90 s exposure to 500 μM DIDS at 1 mM cytoplasmic Ca2+ (P0±S.E.M.=0.94±0.01, n=11). Removal of DIDS decreased P0 to approx. 0.8 because the reversible component of DIDS activation was removed [32].
Statistics
The means were weighted with the number of channels in the bilayer. The S.E.M. was determined as:
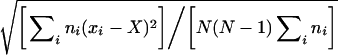 |
where N is the number of experiments, ni the number of RyRs in the bilayer in the ith experiment, xi the test value in the ith experiment and X the weighted mean of the test values xi, where
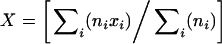 |
RESULTS
Activation of RyRs by 100 nM–1 μM DHPR C-region peptides
In the present study, RyRs were activated under control and test conditions either by cytoplasmic Ca2+ alone (100 μM) or by cytoplasmic ATP (2 mM) in the presence of free [Ca2+] between 20 and 200 μM. It has previously been shown that CS (≤10 μM) activates RyRs at cytoplasmic [Ca2+] over the range 1 nM–100 μM [27]. We show here that activation after adding peptide CS to the cytoplasmic side of the RyR depended on the presence of ATP (Figure 2). In the absence of ATP, the addition of 1 μM CS to the cytoplasmic side of the channels substantially increased RyR activity, whereas removal of peptide did not return P0 to control levels within the time frame of the experiments (Figure 2A). In contrast, in the presence of ATP, RyRs had a P0∼0.8 and were not stimulated further by the addition of 1 μM CS (Figure 2B).
Figure 2. RyRs are activated by the DHPR C-region peptide CS in the cytoplasmic bath.

(A) Current traces in a single experiment on two RyRs. The current baselines are denoted by the broken lines and channel openings are seen as upward current transitions. Channels were recorded (a) in cis/trans 250 mM/50 mM Cs+, 0.1 mM/1 mM Ca2+, (b) during exposure to 1 μM CS, (c) 15 s after removal of CS from the solution, (d) during a second exposure to 1 μM CS and (e) after removal again of the peptide. Reapplication of CS produced no additional increase in channel activity. (B) Two RyRs are activated by 2 mM ATP+0.5 mM Ca2+ (free [Ca2+]=200 μM) in the cytoplasmic bath (a) under control conditions and (b) during exposure to 1 μM CS.
RyR activation by the four peptides (CS, CS1, CS2 and CC) was measured during rapid application of 100 nM peptide to bilayers by local perfusion for 30–90 s. These exposures were brief enough and at sufficiently low concentrations that the inhibitory effects of the peptides (see below) were minor. Peptides were removed by flowing control solution over the bilayer for 30–120 s. Under control conditions, in the presence of 100 μM cytoplasmic Ca2+, the RyRs had P0 values of approx. 0.1. Application of each of the peptides increased channel activity with efficacies in the order CS>CS1∼CS2>CC (Figure 3A, black bars). Curiously, current levels marginally increased after the removal of peptides (Figure 3A, open bars).
Figure 3. RyR activation by the DHPR C-region peptides CS, CS1, CS2 and CC.

(A) Channel activity is expressed as P0. The degree of activation in response to peptide application (black bars) and peptide washout (open bars), normalized to control values before the addition of peptide. The four data groups on the left show the effect of 100 nM peptide in the presence of 100 μM Ca2+. The data on the right show the effect of 1 μM CS in the presence of 2 mM ATP+0.5 mM Ca2+ (free [Ca2+]=200 μM). Control values of P0 and the number of experiments are given in Table 1. (B) Time course of RyR activation. Ensemble traces were compiled from the RyR current summed from all experiments. Initially, RyRs were partially activated in the presence of 100 μM cytoplasmic Ca2+ with P0 values of approx. 0.1. The peptide was applied at zero time for 20–30 s. The exponential time constants, activation rates and number of experiments are given in Table 1.
The kinetics of RyR activation by 100 nM of each of the peptides was measured by compiling ensemble current traces from all experiments (Figure 3B). The exponential time constants for activation (τa) and their associated rate constants are given in Table 1. The activation time constant for CS2 was similar to the time of solution exchange and, thus, was a lower estimate of the actual activation rates. Activation by CS, CS1 and CC was significantly slower.
Table 1. Activation time constants (τa) for the DHPR C-region peptides.
Values of τa were obtained from exponential fits to the current-activation time courses shown in Figure 3. Pmin is the mean P0 before peptide application. N is the total number of bilayer experiments and n the total number of channels summed for each current time course.
| Pmin | N | n | τa (s) | kon (M−1·s−1) | |
|---|---|---|---|---|---|
| CS | 0.12 | 4 | 88 | 7.63±0.25 | (13.1±0.4)×105 |
| CC | 0.09 | 9 | 375 | 4.7±0.07 | (21±0.3)×105 |
| CS1 | 0.08 | 10 | 216 | 3.80±0.15 | (26±1)×105 |
| CS2 | 0.07 | 6 | 80 | 0.92±0.05 | (10.9±0.6)×106 |
In the presence of ATP, CS did not increase RyR activation (Figures 3A and 4). The results presented in Figure 4 show the concentration dependence of RyR activation by ATP in the presence and absence of CS. In the absence of CS (Figure 4, open circles), ATP (with 100 μM Ca2+) increased P0 from 0.19±0.07 to 0.63±0.14, whereas in the presence of CS (Figure 4, filled circles), RyRs had P0 of 0.4±0.1 and ATP could increase this to 0.75±0.11. Hill fits to the data showed that Ka for ATP activation was 400 μM both in the presence and absence of CS.
Figure 4. Activation of RyRs in the presence and absence of CS.

ATP (2 mM) alone (○, N=6), ATP+1 μM CS (●, N=4), ATP+75 μM CS (■, N=4). The curves show Hill fits (Hill coefficient=2) to each data set. The fitted Hill parameters are: (······), Ka=0.4 mM, Pmax=0.71, Pmin=0.13; (——), Ka=0.4 mM, Pmax=0.75 and Pmin=0.41; and (----), Ka=0.23 mM, Pmax=0.72 and Pmin=0.1. The baths contained (cis/trans) 250 mM/50 mM Cs+ and 0.1 mM/1 mM Ca2+ (free).
Fast inhibition of RyRs is most apparent with ≥10 μM DHPR C-region peptides
Fast inhibition of RyRs described in this section has not been reported previously. The existence of fast inhibition was indicated by several previous observations. First, we noticed that application of ≥10 μM CS to RyRs caused less initial activation when compared with that induced by 100 nM peptide (e.g. Figure 3). Secondly, RyR activity increased when 100 nM or 1 μM of the peptides was removed (e.g. Figures 2A and 3A), indicating that activity was decreased in the presence of the peptide. In seven other experiments, application of 10 μM CS increased P0 from 0.03±0.01 to 0.07±0.02 and then P0 increased further to 0.12±0.03 on washout of CS. These observations showed that RyRs were reversibly inhibited by CS and that the inhibition masked peptide activation. To measure the concentration dependence of inhibition, RyRs were permanently (within the time frame of the experiment) stimulated at the beginning of each experiment by a brief (30 s) application of 10 μM CS in the presence of 100 μM Ca2+. When CS was re-applied, P0 decreased in a concentration-dependent manner (Figure 5B, triangles) with Ki=28 mM and ni=1.5. When the same method was used in the presence of ATP, RyRs were inhibited by CS with a lower affinity, Ki=190 μM and ni=2.2 (Figure 5B, circles), in 100 or 250 mM Cs+ solutions. Figure 4 shows that, in the absence of ATP, RyRs are inhibited by 75 μM CS compared with 1 μM CS and that this inhibition is markedly alleviated by [ATP] as low as 0.2 mM.
Figure 5. Fast inhibition of RyRs by the DHPR C-region peptide CS.

(A) The gating of four RyRs are recorded at +40 mV. Channel activity during application and withdrawal of 100 μM CS reveals reversible peptide-induced inhibition. Before the control recording, the channels were exposed to 10 μM CS for 30 s so that the channels were activated by the peptide in the control and test situations. The broken line denotes the current baselines in each record. The baths contained (cis/trans) 250 mM/50 mM Cs+ and 0.1 mM/0.1 mM Ca2+. (B) Concentration dependences of RyR inhibition by CS, normalized to control values under three experimental conditions. Cytoplasmic solutions contained 250 mM Cs+ and 0.1 mM Ca2+ (△; P0 for control=0.24±0.05, N=5); 100 mM Cs+ and 2 mM ATP, 0.1 mM Ca2+ (free [Ca2+]=40 μM) (○; P0 for control=0.74±0.13, N=5); or 250 mM Cs+, 2 mM ATP, 0.1 mM Ca2+ (free [Ca2+]=20 μM) (●; P0 for control=0.42±0.11, N=3). Curves show Hill fits to the data with Ki=28 mM and ni=1.5 (······) and Ki=190 μM and ni=2.2 (——).
In an effort to define the kinetics of inhibition more clearly, we used channels whose gating kinetics were slowed by DIDS modification (see the Experimental section). DIDS-modified RyRs had a lower frequency of channel closures that made it easier to distinguish the closing events due to peptide inhibition (Figure 6A). The DIDS-modified RyRs were exposed to cycles of peptide application (10–60 s) and washout (20–90 s). Peptide inhibition occurred within 15–30 s of peptide application (Figures 6A and 6B). Peptide inhibition was fully reversed within 20–60 s of washout. Rates of RyR inhibition and recovery were determined from exponential fits to the current response. The apparent affinities for the DHPR C-region peptides, calculated from the on- and off-rates of inhibition, are given in Table 2. CS and CC (50 μM) each produced similar inhibition of DIDS-modified RyRs (Figure 6C). Peptides CS1 and CS2 at 50 μM were much weaker inhibitors than CS or CC. CS2 was a more potent inhibitor than CS1 at 150 μM.
Figure 6. Fast inhibition of DIDS-modified RyRs by DHPR C-region peptides.

(A) The gating of a single, DIDS-modified RyR during application and withdrawal of 50 μM CS under conditions described in Figure 5(A). Baselines are shown by the broken line. (B) Inhibition of seven DIDS-modified RyRs in one bilayer subjected to application and removal of 150 μM peptides CS1 and CS2. The solid line labelled ‘imax’ denotes the current when all channels are open. (C) DIDS-modified RyRs inhibited by DHPR C-region peptides. Data are normalized to P0 values in the absence of a peptide. The control values and number of experiments are given in Table 2.
Table 2. Time constants for inhibition (τi) and recovery (τr) and the associated reaction rates kon and koff for the DHPR C-region peptides on DIDS-modified RyRs.
Pmax is the value of P0 in the absence of peptide and N the total number of experiments and n the total number of channels over which the averages for τ were taken.
| Pmax | N | n | τi (s) | kon (M−1·s−1) | N | n | τr (s) | koff (s−1) | Ki (μM) (koff/kon) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 50 μM CS | 0.85±0.01 | 14 | 923 | 3.3±0.2 | (6.1±0.4)×103 | 13 | 809 | 12.9±0.7 | (7.8±0.1)×10−2 | 13 |
| 50 μM CC | 0.87±0.02 | 8 | 488 | 2.4±0.2 | (8.3±0.7)×103 | 8 | 488 | 23.4±2.2 | (4.3±0.4)×10−2 | 5.2 |
| 150 μM CS1 | 0.81±0.02 | 7 | 76 | 1.0±0.1 | (6.7±0.7)×103 | 4 | 48 | 3.6±0.3 | (2.8±0.2)×10−1 | 42 |
| 150 μM CS2 | 0.79±0.02 | 5 | 67 | 3.5±0.2 | (1.9±0.1)×103 | 4 | 60 | 10.2±0.7 | (9.8±0.7)×10−2 | 52 |
Slow inhibition by 50–150 μM DHPR C-region peptides
The C peptides also induced a very slow low-affinity inhibition. It was shown previously that when CS was applied to RyRs in bilayers at sufficiently high concentrations (50–150 μM) for several minutes, it produced channel closure that can take several minutes to reverse, if at all [27]. In the present study, we report that CS1, CS2 and CC at 50–150 μM induced a similar inhibition. CS1 inhibited 9/13 channels within 30–420 s, CS2 inhibited 3/9 channels within 60–260 s and CC inhibited 2/7 channels within approx. 40 s.
Ca2+/Mg2+ inhibition of RyRs is modified by 25–50 μM DHPR C-region peptides
Inhibition of RyRs by cytoplasmic Mg2+ is an important regulator of Ca2+ release in muscles. We have shown that Mg2+ inhibits RyRs by binding to either the Ca2+-activation sites (A-sites) or to low-affinity sites (I-sites) where Ca2+, Mg2+ and other bivalent cations can bind and inhibit the channel [33,34]. The effect of DHPR C-region peptides on Ca2+/Mg2+ inhibition of RyRs was measured to determine whether the peptides could mimic the change in the bivalent ion affinity of I-sites, which is so integral to EC coupling (see the Introduction section). To avoid effects of high-affinity peptide activation, RyRs were subjected at the start of each experiment to two cycles of exposure to peptides and washout to ensure that the channels were permanently activated by the peptide. To measure inhibition at the I-sites, RyRs were locally perfused with solutions containing various [Ca2+] or [Mg2+] in the presence of 2 mM ATP. Ca2+ and Mg2+ concentration dependences were obtained from the peptide-modified RyRs, both in the presence and absence of a peptide.
Figure 7 shows that application of CS rapidly and reversibly alleviates Ca2+ inhibition of RyRs in the presence of ATP. CS caused a small decrease in activity of channels that were not Ca2+-inhibited [i.e. when the cytoplasmic [Ca2+] was 40 μM (Figure 7A)]. Increasing cytoplasmic [Ca2+] to 1 mM resulted in a decrease in P0 due to Ca2+ inhibition at the I-sites (Figure 7B). Addition of CS to the inhibited channels caused a rapid increase in the current, which decreased as soon as the peptide was removed. The presence of CS (50 μM) decreased the sensitivity of RyRs to inhibition by Ca2+ under high (250 mM) and low (100 mM) ionic strength conditions (Figure 7C). The half-inhibitory concentration Ki and Hill coefficient ni for Ca2+ and Mg2+ are given in Table 3. The Ki for I-site inhibition is shifted to higher bivalent cation concentrations when the ionic strength is higher (Figure 7C and [35]). This ionic strength dependence was retained in the presence of CS.
Figure 7. Alleviation of Ca2+ inhibition by CS.

(A, B) The gating of three RyRs recorded at +40 mV. The cytoplasmic solution contained 250 mM Cs+ and 2 mM ATP and the indicated free [Ca2+]. In these experiments, luminal [Ca2+]=1 mM. (A) Channel activity in 40 μM Ca2+ is slightly inhibited by application of 50 μM CS (note the increased frequency of short closures). (B) The presence of 1 mM Ca2+ has two effects on channel activity. It slightly decreased the channel conductance by partially blocking the Cs+ flow and decreased the open probability of channel opening (the latter being Ca2+ inhibition). During application of 50 μM CS in the presence of 1 mM Ca2+, channel activity reversibly increased as a result of decreased Ca2+ inhibition. The broken lines denote the current baselines in each record and the solid lines labelled ‘imax’ denote the current when all channels are open. (C) Ca2+ inhibition normalized to P0 at 10–40 μM Ca2+ in the presence of 250 mM Cs+ (○, ●) and 100 mM Cs+ (□, ■) and in the absence (○, □) or presence of 50 μM CS (●, ■). The data are the mean P0. The Hill fit parameters for all the experiments, including normalizing values at low [Ca2+] and [Mg2+], are given in Table 3.
Table 3. Summary of the parameters of Hill fits to the [Ca2+] and [Mg2+] dependences of P0 in the absence and presence of DHPR C-region peptides.
Pmax is the P0 at low bivalent-ion concentration, Ki the half-inhibitory concentration of X2+ and ni the Hill coefficient. N is the number of bilayers and n the number of channels measured.
| DHPR C-region peptide | Experimental conditions | X2+ | Pmax | Ki (μM) | ni | N | n |
|---|---|---|---|---|---|---|---|
| – | 250 mM Cs+, 2 mM ATP | Ca2+ | 0.86±0.03 | 980±055 | 3.0±0.6 | 5 | 51 |
| 50 μM CS | 250 mM Cs+, 2 mM ATP | Ca2+ | 0.84±0.06 | 2300±400 | 2.6±1.1 | 4 | 31 |
| – | 100 mM Cs+, 2 mM ATP | Ca2+ | 0.73±0.06 | 210±037 | 2.0±0.5 | 3 | 40 |
| 50 μM CS | 100 mM Cs+, 2 mM ATP | Ca2+ | 0.49±0.05 | 460±071 | 4.5±2.8 | 3 | 40 |
| – | 100 mM Cs+, 2 mM ATP | Mg2+ | 0.69±0.07 | 250±40 | 2.3±0.7 | 5 | 70 |
| 50 μM CS | 100 mM Cs+, 2 mM ATP | Mg2+ | 0.46±0.07 | 420±110 | 2.7±2.0 | 6 | 65 |
| 25 μM CS1 | 100 mM Cs+, 2 mM ATP | Mg2+ | 0.58±0.06 | 570±120 | 1.7±1.4 | 4 | 75 |
| 25 μM CS2 | 100 mM Cs+, 2 mM ATP | Mg2+ | 0.62±0.07 | 350±41 | 3.2±2.0 | 4 | 12 |
| *Native | 250 mM Cs+ | Ca2+ | 710±80 | 18 | |||
| *MHS | 250 mM Cs+ | Ca2+ | 1160±220 | 13 | |||
| *Native | 100 mM Cs+ | Ca2+ | 204±32 | 18 | |||
| *MHS | 100 mM Cs+ | Ca2+ | 957±250 | 9 |
* Native and MHS refer to normal and malignant hyperthermia-susceptible RyRs respectively from pig skeletal muscles [39].
Similarly, DHPR C-region peptides decreased RyR sensitivity to Mg2+ inhibition. Mg2+ inhibition was measured with 10–40 μM free cytoplasmic Ca2+ to saturate the A-sites and ensure that inhibition was mainly due to Mg2+ binding to the low-affinity I-sites [33]. Figure 8 shows the effect of DHPR C-region peptides on Mg2+ inhibition at low ionic strength. CS (50 μM) caused a decrease in RyR activity in the absence of Mg2+ (Figure 8A). In the presence of 0.3 mM Mg2+, the peptide addition had a stimulating effect (Figure 8B). Comparison of these records indicates that CS decreased the amount of Mg2+ inhibition in these channels (Figure 8C). Experiments using 25 μM CS1 and CS2 also showed that these peptides shifted Mg2+ inhibition to higher concentrations (Figure 8C and Table 3). After washout of these peptides, the Ki values for Ca2+ and Mg2+ inhibition at high and low ionic strengths were similar to those seen previously in native RyRs (Table 3), indicating that Ca2+/Mg2+ inhibition was not altered during high-affinity activation by CS.
Figure 8. Alleviation of Mg2+ inhibition by DHPR C-region peptides.

(A, B) The gating of five RyRs recorded at +40 mV. The cytoplasmic solution contained 100 mM Cs+ and 2 mM ATP and the indicated [Mg2+] and luminal [Ca2+]=1 mM. (A) Channel activity in 10 μM Ca2+ is partially inhibited in the presence of 50 μM CS. (B) In the presence of 0.3 mM Mg2+, 50 μM CS slightly increased channel activity. Comparison of the traces in (A, B) shows that Mg2+ produced less inhibition in the presence of CS. The broken lines denote the current baselines in each record and the solid lines labelled ‘imax’ denote the current when all channels are open. (C) Mg2+ inhibition in 100 mM Cs+ in the presence and absence of peptides. Data are normalized to P0 at 10 μM Ca2+ and zero Mg2+. In the presence of peptides (filled symbols), RyRs are less sensitive to bivalent ion inhibition. The broken and solid curves are Hill fits to ○ and ● respectively. The Hill fit parameters for all the experiments, including normalizing values at low [Ca2+] and [Mg2+], are given in Table 3.
DISCUSSION
In the present study, we find that regulation of RyRs by DHPR C-region peptides is strongly dependent on the cytoplasmic Ca2+, Mg2+ and ATP. This dependence can explain previous differences between the effects of the peptides on Ca2+ release from SR and on single-channel activity. A significant finding was that the DHPR C-region peptides decreased the apparent affinity of the RyR for Mg2+ inhibition. This is a particularly important result since the C region of the DHPR II–III loop is the determinate of skeletal-type EC coupling [2], a process that is proposed to activate RyRs by relieving their Mg2+ inhibition [13]. The experiments revealed other novel effects of these peptides on RyR activity. In the present study, the inhibition of RyRs by CS seen previously [27] is shown to be due to two mechanisms that are clearly distinguished by their kinetics. One mechanism produces inhibition with onset and recovery times of minutes after peptide application and removal. On the time scale of the bilayer experiments, this inhibition process is non-reversible. The other mechanism causes reversible inhibition with onset and recovery times of seconds. Fast inhibition occurs at the same time as, and partially masks, a slowly reversible activation process and both effects are substantially decreased in the presence of ATP.
A DHPR II–III loop peptide relieves Ca2+/Mg2+ inhibition
The ability of the C-region peptide to decrease Mg2+ inhibition reveals a tantalizing parallel between the actions of the peptide and changes in RyR activity during EC coupling in skeletal muscles. It is surprising that the mere addition of the CS section of the DHPR II–III loop to the RyR in bilayers can mimic an effect of the DHPR on RyRs during EC coupling. The 2-fold decrease in RyR affinity for Mg2+ caused by CS is much smaller than the 10–100-fold decrease that occurs during EC coupling in skinned muscle fibres [36] and in triad preparations [37]. However, the effect of CS indicates that, in this very different isolated system, there is a loose interaction between the DHPR C-region peptides and the RyR, which can affect Ca2+/Mg2+ inhibition. Such an interaction is consistent with a recent finding by Gallant [38] that the Arg615→Cys mutation in pig skeletal RyRs not only decreases the apparent affinity of the RyR for Ca2+ and Mg2+ inhibition [39] but also decreases the activating effect of CS. Although the Arg615→Cys mutation and CS both relieve Ca2+/Mg2+ inhibition, the peptide and the mutation differ in their ionic strength dependences. The Arg615→Cys mutation has a larger effect on Ca2+/Mg2+ inhibition at low ionic strength (Table 3), whereas CS has the same effects both at high and low ionic strength.
Different peptide mechanisms are revealed by different experimental conditions
Effects of C-region peptides depend on the type of RyR agonists present. A slowly reversible activation by CS is seen when RyRs are activated by Ca2+ alone (without ATP; e.g. Figure 3). In the presence of ATP, this type of activation is absent. However, if Mg2+ is also present, CS can reversibly activate RyRs by decreasing their sensitivity to Mg2+ inhibition (e.g. Figure 8). These findings can reconcile disparate reports in the literature about the effect of these peptides on RyR1. This and other single-channel studies [25,27] show that RyR activation by peptide CS is much stronger and occurs with higher affinity than that revealed by measurements of Ca2+ release from SR vesicles where Ka>10 μM (C. S. Haarmann, A. F. Dulhunty and D. R. Laver, unpublished work; [21,22,26]). So far, single-channel studies have measured the effects of CS under conditions where Ca2+ is the sole agonist, whereas Ca2+ release experiments were performed in the presence of ATP and Mg2+ since these ligands are required for vesicle loading. Therefore CS-induced activation of SR Ca2+ release could be due to the relief of Mg2+ inhibition, which is a mechanism different from the high-affinity activation seen with Ca2+ alone. In the present study, we show that CS2 is a stronger and faster inhibitor of RyR than CS1, which may also explain why CS2 is more potent than CS1 in reversing the activation by AS [22].
CS has no effect on Ca2+ release in skinned fibres but decreases depolarization-induced Ca2+ release, suggesting that CS competes with the II–III loop for its activation site on the RyR [24]. It is unlikely that fast inhibition by CS could occur in these skinned fibre experiments due to the presence of ATP in the cytoplasmic bathing solution. It is intriguing that reversible inhibition is regulated by the physiological agonist ATP. Physiologically, reversible inhibition in the absence of ATP and its abolition in the presence of ATP could contribute to the decrease in Ca2+ release seen during fatigue where ATP levels are decreased.
Activation by CS (724Glu-Pro760) in the present study appears to differ from that seen with a similar peptide (720Leu-Glu765) [25]. CS activates with Ka<100 nM and is effectively non-reversible, whereas activation by residues 720Leu-Glu765 had Ka∼10 μM and is readily reversible. The additional amino acids at each end of CS (LKDV and PLAEL; Figure 1) appear to decrease significantly its binding affinity. This is not surprising given the frequency of charged amino acids near the ends of CS and in additional amino acids in 720Leu-Glu765. In a parallel situation, a significant loss of activity is seen when the predominantly positively charged peptide AS (671Thr-Leu690) is modified by the addition of residues 691–710, which contain several negative charges [40].
DHPR C region in EC coupling
Two requirements for changing cardiac to skeletal EC coupling in dysgenic myotubes expressing CaV1.2 are the localization of DHPRs opposite every other RyR and the activation of RyRs via an external Ca2+-independent interaction with the DHPR. Both these requirements are fulfilled when the C-region sequence is skeletal, in an otherwise cardiac context [2,19,41]. Some effects of C-region peptides are consistent with interactions between DHPRs and RyRs that may contribute to skeletal EC coupling. First, the localizing interaction is expected to be of high affinity and specific for the skeletal C region. Indeed, RyR activation by CS is greatest with the skeletal DHPR peptides that bind and activate with high affinity and have low off rates. Secondly, the activating interaction in EC coupling might be the desensitization of RyRs to Mg2+ inhibition [36]. Although the latter effect is not specific for the DHPR isoform, isoform specificity may depend on co-localization of DHPRs and RyRs (via the DHPR C region or by other regions that may, or may not, be conserved between CaV1.1 and CaV1.2). There is a further parallel between our results and EC coupling studies. We find that activation by CS is stronger than that by either CS1 or CS2. This is consistent with a stronger EC coupling if the whole C region is of skeletal sequence compared with the situation when CS1 is skeletal and CS2 is cardiac [19]. There appears to be a lack of C-region isoform specificity in fast inhibition, which indicates that both cardiac and skeletal DHPRs and RyRs could interact physically with each other if they were appropriately targeted. This appropriate targeting appears to be dependent on the CS1 region [19].
In conclusion, the action of these peptides critically depends on the presence of the key physiological regulators, Ca2+, Mg2+ and ATP, so that peptide effects on isolated RyRs might not equate to their action in muscle but rather provide pointers to mechanisms underlying EC coupling. The manifold effects of the peptides do suggest that the C region of the DHPR is capable of a complex influence over RyR activity in resting muscles and during EC coupling.
Acknowledgments
We are grateful to S. Pace and J. Stivala for assistance with SR vesicle preparation and to P. Johnson for assistance with the peptide-inhibition experiments. D.R.L. was supported by Professorship from the Australian Research Council.
References
- 1.Knudson C. M., Chaudhari N., Sharp A. H., Powell J. A., Beam K. G., Campbell K. P. Specific absence of the alpha 1 subunit of the dihydropyridine receptor in mice with muscular dysgenesis. J. Biol. Chem. 1989;264:1345–1348. [PubMed] [Google Scholar]
- 2.Tanabe T., Beam K. G., Adams B. A., Niidome T., Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature (London) 1990;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- 3.Tanabe T., Beam K. G., Powell J. A., Numa S. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature (London) 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- 4.Adams B. A., Beam K. G. Muscular dysgenesis in mice: a model system for studying excitation-contraction coupling. FASEB J. 1990;4:2809–2816. doi: 10.1096/fasebj.4.10.2165014. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong C. M., Bezanilla F. M., Horowicz P. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N′-tetracetic acid. Biochim. Biophys. Acta. 1972;267:605–608. doi: 10.1016/0005-2728(72)90194-6. [DOI] [PubMed] [Google Scholar]
- 6.Fabiato A. Simulated calcium current can both cause calcium loading in and trigger calcium release from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 1985;85:291–320. doi: 10.1085/jgp.85.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nabauer M., Callewaert G., Cleemann L., Morad M. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. 1989;244:800–803. doi: 10.1126/science.2543067. [DOI] [PubMed] [Google Scholar]
- 8.Beuckelmann D. J., Wier W. G. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J. Physiol. (Cambridge, U.K.) 1988;405:233–255. doi: 10.1113/jphysiol.1988.sp017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider M. F., Chandler W. K. Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature (London) 1973;242:244–246. doi: 10.1038/242244a0. [DOI] [PubMed] [Google Scholar]
- 10.Lamb G. D. Voltage-sensor control of Ca2+ release in skeletal muscle: insights from skinned fibers. Front. Biosci. 2002;7:d834–d842. doi: 10.2741/A815. [DOI] [PubMed] [Google Scholar]
- 11.Smith J. S., Coronado R., Meissner G. Single channel measurements of the calcium release channel from skeletal muscle sarcoplasmic reticulum. Activation by Ca2+ and ATP and modulation by Mg2+ J. Gen. Physiol. 1986;88:573–588. doi: 10.1085/jgp.88.5.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laver D. R., Lenz G. K., Lamb G. D. Regulation of the calcium release channel from rabbit skeletal muscle by the nucleotides ATP, AMP, IMP and adenosine. J. Physiol. (Lond.) 2001;537:763–778. doi: 10.1111/j.1469-7793.2001.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamb G. D., Stephenson D. G. Importance of Mg2+ in excitation-contraction coupling in skeletal muscle. News Physiol. Sci. 1992;7:270–274. [Google Scholar]
- 14.Lamb G. D., Recupero E., Stephenson D. G. Effect of myoplasmic pH on excitation-contraction coupling in skeletal muscle fibres of the toad. J. Physiol. (Lond.) 1992;448:211–224. doi: 10.1113/jphysiol.1992.sp019037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blazev R., Lamb G. D. Low [ATP] and elevated [Mg2+] reduce depolarization-induced Ca2+ release in rat skinned skeletal muscle fibres. J. Physiol. (Lond.) 1999;520:203–215. doi: 10.1111/j.1469-7793.1999.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson K., Meissner G. T-tubule depolarization-induced SR Ca2+ release is controlled by dihydropyridine receptor- and Ca2+-dependent mechanisms in cell homogenates from rabbit skeletal muscle. J. Gen. Physiol. 1995;105:363–383. doi: 10.1085/jgp.105.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamb G. D., Junankar P. R., Stephenson D. G. Raised intracellular [Ca2+] abolishes excitation-contraction coupling in skeletal muscle fibres of rat and toad. J. Physiol. (Lond.) 1995;498:349–362. doi: 10.1113/jphysiol.1995.sp021056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaudhari N. A single nucleotide deletion in the skeletal muscle-specific calcium channel transcript of muscular dysgenesis (mdg) mice. J. Biol. Chem. 1992;267:25636–25639. [PubMed] [Google Scholar]
- 19.Nakai J., Tanabe T., Konno T., Adams B., Beam K. G. Localization in the II-III loop of the dihydropyridine receptor of a sequence critical for excitation-contraction coupling. J. Biol. Chem. 1998;273:24983–24986. doi: 10.1074/jbc.273.39.24983. [DOI] [PubMed] [Google Scholar]
- 20.Lu X., Xu L., Meissner G. Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. J. Biol. Chem. 1994;269:6511–6516. [PubMed] [Google Scholar]
- 21.El-Hayek R., Antoniu B., Wang J., Hamilton S. L., Ikemoto N. Identification of calcium release-triggering and blocking regions of the II-III loop of the skeletal muscle dihydropyridine receptor. J. Biol. Chem. 1995;270:22116–22118. doi: 10.1074/jbc.270.38.22116. [DOI] [PubMed] [Google Scholar]
- 22.Saiki Y., El-Hayek R., Ikemoto N. Involvement of the Glu724-Pro760 region of the dihydropyridine receptor II-III loop in skeletal muscle-type excitation-contraction coupling. J. Biol. Chem. 1999;274:7825–7832. doi: 10.1074/jbc.274.12.7825. [DOI] [PubMed] [Google Scholar]
- 23.Dulhunty A. F., Laver D. R., Gallant E. M., Casarotto M. G., Pace S. M., Curtis S. Activation and inhibition of skeletal RyR channels by a part of the skeletal DHPR II-III loop: effects of DHPR Ser687 and FKBP12. Biophys. J. 1999;77:189–203. doi: 10.1016/S0006-3495(99)76881-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamb G. D., El-Hayek R., Ikemoto N., Stephenson D. G. Effects of dihydropyridine receptor II-III loop peptides on Ca2+ release in skinned skeletal muscle fibers. Am. J. Physiol. 2000;279:C891–C905. doi: 10.1152/ajpcell.2000.279.4.C891. [DOI] [PubMed] [Google Scholar]
- 25.Stange M., Tripathy A., Meissner G. Two domains in dihydropyridine receptor activate the skeletal muscle Ca2+ release channel. Biophys. J. 2001;81:1419–1429. doi: 10.1016/S0006-3495(01)75797-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto T., Rodriguez J., Ikemoto N. Ca2+-dependent dual functions of peptide C. The peptide corresponding to the Glu724-Pro760 region (the so-called determinant of excitation-contraction coupling) of the dihydropyridine receptor alpha 1 subunit II-III loop. J. Biol. Chem. 2002;277:993–1001. doi: 10.1074/jbc.M105837200. [DOI] [PubMed] [Google Scholar]
- 27.Haarmann C. S., Green D., Casarotto M. G., Laver D. R., Dulhunty A. F. The random-coil ‘C’ fragment of the dihydropyridine receptor II-III loop can activate or inhibit native skeletal ryanodine receptors. Biochem. J. 2003;372:305–316. doi: 10.1042/BJ20021763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahern G. P., Junankar P. R., Dulhunty A. F. Single channel activity of the ryanodine receptor calcium release channel is modulated by FK-506. FEBS Lett. 1994;352:369–374. doi: 10.1016/0014-5793(94)01001-3. [DOI] [PubMed] [Google Scholar]
- 29.Laver D. R., Roden L. D., Ahern G. P., Eager K. R., Junankar P. R., Dulhunty A. F. Cytoplasmic Ca2+ inhibits the ryanodine receptor from cardiac muscle. J. Membr. Biol. 1995;147:7–22. doi: 10.1007/BF00235394. [DOI] [PubMed] [Google Scholar]
- 30.Marks P. W., Maxfield F. R. Preparation of solutions with free calcium concentration in the nanomolar range using 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid. Anal. Biochem. 1991;193:61–71. doi: 10.1016/0003-2697(91)90044-t. [DOI] [PubMed] [Google Scholar]
- 31.Brooks S. P., Storey K. B. Bound and determined: a computer program for making buffers of defined ion concentrations. Anal. Biochem. 1992;201:119–126. doi: 10.1016/0003-2697(92)90183-8. [DOI] [PubMed] [Google Scholar]
- 32.O'Neill E. R., Sakowska M. M., Laver D. R. Regulation of the calcium release channel from skeletal muscle by Suramin and the disulfonated Stilbene derivatives DIDS, DBDS, and DNDS. Biophys. J. 2003;84:1674–1689. doi: 10.1016/S0006-3495(03)74976-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laver D. R., Baynes T. M., Dulhunty A. F. Magnesium inhibition of ryanodine-receptor calcium channels: evidence for two independent mechanisms. J. Membr. Biol. 1997;156:213–229. doi: 10.1007/s002329900202. [DOI] [PubMed] [Google Scholar]
- 34.Meissner G., Darling E., Eveleth J. Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry. 1986;25:236–244. doi: 10.1021/bi00349a033. [DOI] [PubMed] [Google Scholar]
- 35.Shomer N. H., Louis C. F., Fill M., Litterer L. A., Mickelson J. R. Reconstitution of abnormalities in the malignant hyperthermia-susceptible pig ryanodine receptor. Am. J. Physiol. 1993;264:C125–C135. doi: 10.1152/ajpcell.1993.264.1.C125. [DOI] [PubMed] [Google Scholar]
- 36.Lamb G. D., Stephenson D. G. Effect of Mg2+ on the control of Ca2+ release in skeletal muscle fibres of the toad. J. Physiol. (Cambridge, U.K.) 1991;434:507–528. doi: 10.1113/jphysiol.1991.sp018483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ritucci N. A., Corbett A. M. Effect of Mg2+ and ATP on depolarization-induced Ca2+ release in isolated triads. Am. J. Physiol. 1995;269:C85–C95. doi: 10.1152/ajpcell.1995.269.1.C85. [DOI] [PubMed] [Google Scholar]
- 38.Gallant E. M., Hart J., Eager K., Curtis S., Dulhunty A. F. Caffeine sensitivity of native RyR channels from normal and malignant hyperthermic pigs: effects of a DHPR II-III loop peptide. Am. J. Physiol. 2004;286:C821–C830. doi: 10.1152/ajpcell.00311.2003. [DOI] [PubMed] [Google Scholar]
- 39.Laver D. R., Owen V. J., Junankar P. R., Taske N. L., Dulhunty A. F., Lamb G. D. Reduced inhibitory effect of Mg2+ on ryanodine receptor-Ca2+ release channels in malignant hyperthermia. Biophys. J. 1997;73:1913–1924. doi: 10.1016/S0006-3495(97)78222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casarotto M. G., Green D., Pace S., Young J., Dulhunty A. F. Activating the ryanodine receptor with dihydropyridine receptor II-III loop segments: size and charge do matter. Front. Biosci. 2004;9:2860–2872. doi: 10.2741/1443. [DOI] [PubMed] [Google Scholar]
- 41.Grabner M., Dirksen R. T., Suda N., Beam K. G. The II-III loop of the skeletal muscle dihydropyridine receptor is responsible for the bi-directional coupling with the ryanodine receptor. J. Biol. Chem. 1999;274:21913–21919. doi: 10.1074/jbc.274.31.21913. [DOI] [PubMed] [Google Scholar]