

Abstract
INTRODUCTION
Alzheimer's disease (AD) pathology is defined by β‐amyloid (Aβ) plaques and neurofibrillary tau, but Lewy bodies (LBs; 𝛼‐synuclein aggregates) are a common co‐pathology for which effective biomarkers are needed.
METHODS
A validated α‐synuclein Seed Amplification Assay (SAA) was used on recent cerebrospinal fluid (CSF) samples from 1638 Alzheimer's Disease Neuroimaging Initiative (ADNI) participants, 78 with LB‐pathology confirmation at autopsy. We compared SAA outcomes with neuropathology, Aβ and tau biomarkers, risk‐factors, genetics, and cognitive trajectories.
RESULTS
SAA showed 79% sensitivity and 97% specificity for LB pathology, with superior performance in identifying neocortical (100%) compared to limbic (57%) and amygdala‐predominant (60%) LB‐pathology. SAA+ rate was 22%, increasing with disease stage and age. Higher Aβ burden but lower CSF p‐tau181 associated with higher SAA+ rates, especially in dementia. SAA+ affected cognitive impairment in MCI and Early‐AD who were already AD biomarker positive.
DISCUSSION
SAA is a sensitive, specific marker for LB‐pathology. Its increase in prevalence with age and AD stages, and its association with AD biomarkers, highlights the clinical importance of α‐synuclein co‐pathology in understanding AD's nature and progression.
Highlights
SAA shows 79% sensitivity, 97% specificity for LB‐pathology detection in AD.
SAA positivity prevalence increases with disease stage and age.
Higher Aβ burden, lower CSF p‐tau181 linked with higher SAA+ rates in dementia.
SAA+ impacts cognitive impairment in early disease stages.
Study underpins need for wider LB‐pathology screening in AD treatment.
Keywords: Alzheimer's disease, co‐pathology, Lewy body, SAA
1. BACKGROUND
Biomarkers that accurately reflect underlying pathological features of Alzheimer's disease (AD) have significantly enhanced early diagnosis, subtyping, and the progress of clinical trials. In living individuals, imaging and fluid biomarkers are used for detecting the presence of β‐amyloid (Aβ) plaques and tau tangles, the two hallmark neuropathological changes in AD. However, many individuals diagnosed with dementia due to AD have co‐pathologies, which introduces heterogeneity to the pathogenesis and presentation of the symptoms and likely influences the disease progression. 1
A common co‐pathology of AD is Lewy bodies (LBs), composed of α‐synuclein (α‐syn) aggregates. 2 Seed Amplification Assays (SAAs) that detect aggregates of misfolded α‐syn in antemortem cerebrospinal fluid (CSF) have been shown to reliably predict LB‐pathology identified at autopsy. 3 SAA was first largely used in identifying α‐syn pathology in Parkinson's disease (PD), dementia with LB (DLB), and synucleinopathies. 4 , 5 More recent work showed α‐syn seeds detected by SAA (SAA‐positivity; SAA+) in asymptomatic individuals and individuals with cognitive impairment in the context of co‐pathologies. 5 , 6 , 7 , 8 Notably, Arnold et al. validated the effectiveness of SAA in detecting pathological α‐syn seeds pre‐ and post mortem CSF samples from an autopsy‐validated cohort with diverse neurodegenerative diseases, demonstrating high specificity (98.1%) and sensitivity (97.8%). The method proved adept at identifying limbic transitional and diffuse neocortical α‐syn inclusions in CSF samples collected ante‐mortem. 5 In two studies conducted in memory clinics, comprising cognitively unimpaired (CU) individuals and those with cognitive impairment from various causes, it was observed that individuals positive for both AD biomarker and SAA experiences a more rapid cognitive decline over 10 years to those positive for only one. The effects of SAA+ deteriorating attention, executive, visuospatial, and motor functions were noted to be independent of other factors. 7 , 8 These studies also found that SAA+ prevalence increased with age, particularly among CU individuals, and was more likely to occur alongside AD Aβ biomarker positivity, rather than AD tau biomarker positivity. 7 Furthermore, a smaller‐scale research cohort focusing on individuals with cognitive impairment and biomarker confirmation for AD‐related Aβ, tau pathologies, and neurodegeneration reported a significant presence of SAA+ (45%), with those positive for AD biomarkers and SAA+ more frequently exhibiting atypical dementia phenotypes. 6
In this study, the extent of LB co‐pathology within the Alzheimer's Disease Neuroimaging Initiative (ADNI) — a multi‐site, open‐access observational study — was evaluated using a clinically validated commercial Amprion SYNTap SAA. ADNI distinguishes itself as a comprehensive observational study that aimed at standardizing and validating the use of neuroimaging, fluid, and digital biomarkers for diagnosis and prognosis, as well as to inform the design of clinical trials for AD. It features standard study protocols and provides a wealth of accompanying clinical and biomarker data, including autopsy neuropathology reports in a subset. In addition to 577 CU individuals, this study incorporates the most extensive cohort of individuals with amnestic mild cognitive impairment (MCI; N = 654) and clinical diagnosis of mild‐to‐moderate AD dementia (N = 407), thus far studied with SAA, which are the primary populations targeted by recent disease‐modifying therapies for AD. The specific objectives of the study were to: (1) evaluate the detection capability of SAA for LB‐pathology in 78 cases with neuropathologic confirmation at autopsy; (2) investigate the prevalence of SAA+ in relation to well‐known AD‐related risk factors such as age, sex, race, ethnicity, APOE ε4 allele, and particular genetic variants, including Triggering receptor expressed on myeloid cells 2 (TREM2) and glucosylceramidase beta 1 (GBA1), alongside with polygenic risk scores (PRS) for AD and PD; (3) explore the prevalence of SAA+ in relation to continuous measures of AD biomarkers, including CSF Aβ42, p‐tau181, and the ratio of p‐tau181/Aβ42, and global cortical Aβ burden measured by positron emission tomography (PET) imaging; and (4) assess the differences in cognitive impairment and decline between individuals with and without SAA+. The working hypothesis posited that LB‐pathology would be associated with age and disease stage and that the prevalence of SAA+ would show a positive associated with the presence of AD pathology.
2. METHODS
2.1. Study design and participants
Data were obtained from the ADNI database (https://adni.loni.usc.edu/). The study employed a cross‐sectional analysis of biomarker, demographic, and diagnosis data and both cross‐sectional and longitudinal analyses of cognitive data. The analysis included all ADNI 1–3 participants with available CSF samples (N = 1638). We expected α‐syn pathology to be more prevalent with increasing age and disease progression; therefore, we selected the most recent CSF samples for examination to ensure the most accurate diagnostic information, detectable activity of the α‐syn seeding, and greatest relevance to post mortem neuropathological confirmation.
ADNI participants included CU individuals, individuals with MCI, and individuals with clinical diagnosis of dementia due to AD. Briefly, at the time of enrollment, ADNI participants were aged between 55 and 90 (inclusive) years, had a study partner able to provide an independent evaluation of functioning, and were proficient in English or Spanish. The enrollment criteria for CU participants included a Mini‐Mental State Examination (MMSE) score between 24 and 30 (inclusive), a Clinical Dementia Rating (CDR) of 0, no depression, no MCI, and no dementia. MCI participants were required to have MMSE scores between 24 and 30 (inclusive), a subjective memory complaint, objectively memory loss (adjusted for education) on the Wechsler Memory Scale Logical Memory II, a CDR of 0.5, no significant impairment in other cognitive domains, essentially preserved activities of daily living, and no dementia. Participants with dementia due to AD met the criteria with MMSE scores between 20 and 26 (inclusive), a CDR of 0.5 or 1.0, and meets NINCDS/ADRDA criteria for probable AD. Exclusion criteria at the time of ADNI study enrollment included significant neurological disease other than AD, any contraindications to neuroimaging or other ADNI protocols, neuroimaging evidence of infection, infarction, lacunes, or other focal lesions, psychiatric disorders, including psychotic features, alcohol abuse, significant systemic illness or unstable medical condition, laboratory abnormalities that could complicate the study, use of certain psychoactive medications, and participation in other clinical trials.
RESEARCH IN CONTEXT
Systematic review: We reviewed literature on Alzheimer's disease (AD) and Lewy body (LB)‐pathology biomarkers, searching through databases like PubMed until January 2024. We focused on studies about biomarker accuracy for α‐synuclein, noting α‐synuclein's role beyond Parkinson's disease (PD) and its importance in AD and cognition. The review stressed the need for precise biomarkers to clarify the complexity of co‐pathologies in AD.
Interpretation: Seed Amplification Assays (SAAs) accurately detect LB‐pathology. Our study shows a link between the presence of SAA markers and various factors such as the age, the disease stage, and the presence of AD biomarkers, offering new insights into how LB‐pathology interacts with AD characteristics and treatment strategies for patients with both AD and concomitant LB‐pathology.
Future directions: The study emphasizes the clinical importance of SAA in detecting LB co‐pathology in AD, which may affect cognition and alter individual progression paths. It advocates for wider LB‐pathology screening in AD, aiding in tailored diagnostics and treatments.
2.2. CSF sample collection
CSF samples were initially collected into collection tubes supplied to each participating ADNI site, subsequently transferred to polypropylene transfer tubes, and then frozen on dry ice within 1 hour of collection. These samples were shipped overnight on dry ice to the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center. Upon arrival to the ADNI Biomarker Core laboratory, the CSF samples were thawed and aliquoted into 0.5 mL cryo for long term storage at −80˚C. Pristine CSF aliquots were provided for SAA analysis.
2.3. CSF α‐synuclein SAA processing
The α‐syn SAA testing was conducted by Amprion Clinical Laboratory (CLIA ID No. 05D2209417; CAP No. 8168002) using a clinically validated method in compliance with Clinical Laboratory Improvement Amendment (CLIA) standards. For the analysis, each CSF sample was tested in triplicate within a 96‐well plate using a reaction mixture comprised of 100 mM PIPES pH 6·5, 0·44 M NaCl, 0·1% sarkosyl, 10 µM ThT, 0·3 mg/mL recombinant αSyn, and 40 µL CSF, in a final volume of 100 µL. Each well contained two silicon nitride beads to enhance the consistency, with each plate incorporating both positive and negative assay quality control samples to assure assay accuracy. The plates were sealed with an optical adhesive film and inserted into a BMG LABTECH FLUOstar Ω Microplate Reader. They were incubated at 42°C, undergoing cycles of 1 minute of shaking followed by 14 min of rest, with fluorescence recorded after each shake (using an excitation wavelength 440 nm and emission wavelength of 490 nm). Following a total incubation time of 20 hours, the maximum fluorescence intensity for each well was logged, and an algorithm was applied to the triplicate reading to categorize the result.
CSF samples were classified as: “PD/DLB‐like Detected” if α‐syn aggregates were detected with an aggregation profile consistent with Type 1 seeds observed in PD and DLB; “MSA‐like Detected” if α‐syn aggregates matched Type 2 seeds typically seen in multiple system atrophy (MSA); or “Not Detected” if no α‐syn aggregates were detected. Samples that did not yield a definite result after two tests were classified as “Inconclusive.”
All CSF α‐syn SAA analyses were performed with the analysts blinded to the participants’ demographic details, clinical profiles, and AD biomarker data. The integrity of the blinding was maintained by utilizing unique specimen identifiers for randomly assigned sample shipments.
2.4. Post mortem neuropathological confirmation of LB pathology
ADNI study follows the National Institute on Aging‐Alzheimer's Association (NIA‐AA) guidelines for the neuropathologic examinations. The neuropathologic data are considered the “gold standard” against which the biomarker results such as CSF α‐syn SAA are compared.
Pathological lesions within the brain were assessed using established neuropathologic diagnostic criteria. Using the NIA‐AA protocol, an “ABC” score for AD neuropathologic change was generated which incorporates histopathologic assessments of Aβ deposits (A), staging of neurofibrillary tangles (B), and scoring of neuritic plaques (C). In addition, detailed methods for assessing commonly co‐morbid conditions such as Lewy body disease, vascular brain injury, hippocampal sclerosis, and TAR DNA binding protein (TDP) immunoreactive inclusions were included. The brain areas sampled for microscopic assessment included middle frontal gyrus (Block 1), superior and middle temporal gyri (Block 2), inferior parietal lobe (angular gyrus) (Block 3), occipital lobe to include the calcarine sulcus and parastriate cortex (Block 4), hippocampus and parahippocampal gyrus at the level of the lateral geniculate nucleus (Block 5), striatum (caudate nucleus and putamen) with olfactory cortex at the level of the nucleus accumbens (Block 6), thalamus and subthalamic nucleus (Block 8), midbrain with red nucleus (Block 9), pons with locus coeruleus (Block 11), medulla oblongata (Block 12), spinal cord (Block 13), cerebellum with dentate nucleus (Block 14), striatum and pallidum at the level of the anterior commissure to include nucleus basalis of Meynert, basal forebrain, and septum (Block 17), anterior cingulate gyrus at the level of the genu of the corpus callosum (Block 19), precentral gyrus (Block 21), amygdala and entorhinal cortex (Block 23), posterior cingulate gyrus and precuneus at the level of the splenium (Block 30).
Neuropathology data were captured using the Neuropathology Data Form Version 10 provided by the National Alzheimer's Coordinating Center (NACC). Neuropathological confirmation of the LB‐pathology was through α‐syn immunohistochemistry, adhering to the Consortium on DLB criteria. 9
2.5. Alzheimer's disease CSF biomarker assessments
Pristine aliquots of CSF were analyzed by the electrochemiluminescence immunoassays (ECLIA) Elecsys CSF Aβ42, CSF phospho‐tau181 (p‐tau181), and CSF total‐tau on a fully automated Elecsys cobas e 601 instrument and a single lot of reagents for each measured biomarker. The Roche Elecsys CSF immunoassays were used following a Roche Study Protocol at the ADNI Biomarker Laboratory, according to the kit manufacturer's instructions. Analyses were performed in a series of runs, each sample run one time (in singlicate) for each biomarker test, over the time period of November 17, 2016, through June 22, 2022, following a standard new lot rollover protocol from the manufacturer that involved repeated analyses of quality control samples. The analyte measuring ranges were lower technical limit to upper technical limit for each biomarker: 200 to 1700 pg/mL for the Elecsys CSF Aβ42 immunoassay, 8 to 120 pg/mL for the Elecsys CSF p‐tau181 immunoassay, and 80 to 1300 pg/mL for the Elecsys CSF total‐tau immunoassay. The results that are above the upper technical limit or below the lower technical limit are not included in the relevant analyses described below. Only CSF Aβ42 and CSF p‐tau181 were used in the relevant analyses described below.
The AD CSF biomarker positivity was defined as “Aβ42+” if CSF Aβ42 < 980 pg/mL and “p‐tau181+” if CSF p‐tau181 > 24 pg/mL. Overall AD CSF biomarker positivity “CSF_AD+” was defined by the ratio of p‐tau181/Aβ42 with a cutoff of > 0.025. These cutoff definitions were based on previously published methods 10 and the revised ADNI Biomarker Core protocol (UPENNBIOMK_ROCHE_ELECSYS_METHODS_20231109.pdf).
2.6. Alzheimer's disease positron emission tomography biomarker assessment
The radiochemical synthesis of florbetapir was overseen and regulated by Avid Radiopharmaceuticals and distributed to the qualifying ADNI sites. The radiochemical synthesis of florbetaben was overseen and regulated by Life Molecular Imaging and distributed to the qualifying ADNI sites. PET imaging was performed at each ADNI site according to standardized protocols. The florbetapir protocol entailed the injection of 370 MBq (10·0 mCi) ± 10% of tracer followed by an uptake phase of 50 minutes. At 50 minutes, subjects were positioned in the scanner and 4 × 5 minute frames of emission data were collected. The florbetaben protocol entailed the injection of 300 MBq (8.1 mCi) ± 10% of tracer followed by 20 minutes (4 × 5 minute frames) acquisition at 90–110 minutes post‐injection. PET/CT scans preceded these acquisitions with a CT scan for attenuation correction; PET‐only scanners performed a transmission scan following the emission scan. All PET scans underwent a rigorous quality control protocol and were processed to produce final images with standard orientation and voxel size of 2mm3. 11
A global standardized uptake value ratio (SUVR) was estimated across cortical summary regions (FreeSurfer v7.1 defined frontal, cingulate, parietal, and lateral temporal cortices) normalized to the whole cerebellum.
Direct SUVR‐to‐centiloid transformations provided by the ADNI PET Core were applied to florbetaben and florbetapir data for estimates of standardized global cortical Aβ burden in centiloid units. “Aβ‐PET+” was defined as centiloid > 20, which correspond to the recommended cross‐sectional thresholds for florbetapir (whole cerebellum‐normalized SUVR of 1.11 12 ) and florbetaben (whole cerebellum‐normalized SUVR of 1.08 13 ) PET images.
For participants with Aβ‐PET imaging data, Aβ‐PET images within 6 months of sample collection time of the CSF sample used for SAA analysis were selected.
2.7. Genetics data
Previously generated whole genome sequencing data were obtained from ADNI. We extracted known high‐risk variants in GBA1 (p·E356K, p·T408M, p·N409S, NM_000157·4), apolipoprotein E (APOE) ε4 haplotypes, and TREM2 (p·R47H, p·R62H, NM_018965·4) which are associated with PD, DLB, and AD. 14 , 15 , 16 PRS were calculated based on two recent AD genome‐wide association studies (GWAS) including APOE ε4 17 and excluding APOE ε4, 18 and a PD GWAS, 14 using the single nucleotide polymorphism (SNP) association weights from each GWAS using plink2.
2.8. Clinical assessments
The global cognitive assessments including the Clinical Dementia Rating‐Sum of Boxes (CDR‐SB), the Alzheimer's Disease Assessment Scale‐cognitive subscale 13‐item (ADAS‐Cog13), the MMSE, based on a 30‐point questionnaire, and preclinical Alzheimer's composite score (PACC), and the domain‐specific cognitive assessments including the composite measures of memory, executive function, language, and visuospatial functioning 19 were analyzed as clinical outcome measures. The assessment of cognitive decline rates was confined to longitudinal data collected within 2 years of CSF sample collection. This restriction was imposed because the latest CSF samples were specifically chosen for SAA analysis, thereby constraining the prospective follow‐up clinical time points available for analysis.
Sleep disturbances and hallucinations are prominent neuropsychiatric symptoms associated with synucleinopathies. The extent of these symptoms often aligns with the severity and spread of α‐syn pathology. To assess the impact of presence of LB co‐pathology on sleep issues and hallucinations, specific questions from the Neuropsychiatric Inventory‐Questionnaire (NPI‐Q) are utilized. For sleep disturbances, the NPI‐Q inquires if the patient interrupts the caregiver's sleep, wakes up prematurely, or excessively naps. For hallucinations, it asks if the patient seems to hear voices or speaks with nonexistent individuals. Severity scoring for these items ranges from mild, indicating a noticeable but manageable change, to moderate, denoting a significant change, and severe, which reflects a very marked and dramatic change.
2.9. Statistical analysis
We conducted a retrospective analysis of neuropathological data derived from autopsy reports, wherein the presence of LB inclusions was established as the diagnostic gold standard, or “ground truth.” To evaluate the diagnostic performance of the SAA in detecting LB‐pathology, we calculated the SAA's sensitivity and specificity against the autopsy findings. The association between the SAA results and the gold standard was statistically examined using the chi‐squared test, and 95% confidence intervals (CIs) were computed to assess the precision of these estimates.
On the clinical ADNI cohort, descriptive statistics were computed on demographic, clinical, and biomarker measures collected at the time of CSF sample collection, categorized by clinical diagnosis and SAA results. Samples were designated as SAA− (“Not Detected”) if no α‐syn aggregates were present, and SAA+ (“PD/DLB‐like Detected”) if α‐syn aggregates conformed to Type 1 seeds, typically observed in PD and DLB. Cases with CSF α‐syn SAA results classified as “Inconclusive” or “MSA‐like Detected” were excluded from the principal analyses due to small sample size, as detailed in the Results section.
We performed separate logistic regression analyses to examine the relationship between AD biomarker positivity (categorized as CSF Aβ42+ or CSF Aβ42−, CSF p‐tau181+ or CSF p‐tau181−, CSF_AD+ or CSF_AD−, and Aβ‐PET+ or Aβ‐PET−) and SAA result, designated as SAA+ or SAA−. These analyses factored in age, sex, and APOE ε4 status to adjust for their known association with AD Aβ pathology. The model investigating CSF p‐tau181+/− status included an additional adjustment for CSF Aβ42 levels to account for the established interplay between AD Aβ and tau pathologies. Given the absence of Aβ‐PET data in 19% to 37% of our cohort, we used CSF AD biomarkers as the primary method for AD pathology assessment. However, we included Aβ‐PET analysis where available, acknowledging its status as the gold‐standard for assessing Aβ‐pathology.
To further assess the extent to which these pathological changes are associated with aging and nonlinearity of these associations, we constructed generalized additive models to analyze the relationship between age and positivity in SAA and AD biomarkers (CSF Aβ42, Aβ‐PET centiloid, CSF p‐tau181). CSF Aβ42 was included as a covariate in non‐linear age models for SAA‐positivity and CSF p‐tau181 positivity to model age effect independent of AD Aβ pathology. We employed the finite differences method to identify significant age intervals where there was a notable change in the prevalence of SAA or AD biomarker positivity. This approach involved discrete sampling across age and calculating the rate of change in the proportion of participants positive for SAA or AD biomarkers. We incorporated the uncertainty in estimates by computing 95% CIs around the derived rates of change, allowing us to determine the statistical significance of observed trends and delineate the age ranges most associated with these pathological states.
In addition to the initial analysis of the relationship between AD biomarker positivity and SAA result, we examined the association between SAA positivity and the continuous levels of AD biomarkers, to mitigate potential biases arising from binary categorization of AD biomarker positivity. These analyses factored in age to adjust for their known association with AD Aβ pathology. In our analysis of SAA positivity in relation to the continuous spectrum of CSF p‐tau181 levels, we additionally adjusted for CSF Aβ42 levels. Employing the finite differences method, as described above, we determined significant intervals within the AD biomarker levels where the prevalence of SAA positivity exhibited significant changes.
To assess the role of AD risk factor (such as sex, race, and ethnicity) and genetics (APOE ε4 allele status, and GBA1 and TREM2 variants) on LB‐pathology, we performed separate logistic regression analyses to evaluate their associations with SAA‐positivity. These analyses treated each AD risk factor as an independent categorical variable and the SAA results as the dependent variable. All models were adjusted for age and the other AD risk factors to assess their independent effects. Furthermore, we performed separate regression analyses to assess the association of SAA‐positivity with AD‐PRS and PD‐PRS, adjusting for biological sex, age, APOE ε4, and the first five genetic principal components.
To assess the impact of LB‐pathology on cognitive performance at the time of CSF sample collection, linear regression models were employed. These models were stratified by overall CSF AD biomarker positivity (CSF_AD+ vs. CSF_AD−) and SAA‐positivity (SAA+ vs. SAA−) and adjusted for age, sex, and APOE ε4 allele status. To assess the impact of LB‐pathology on the rates of cognitive decline, linear mixed‐effects models (LMM) were used, with cognitive variable of interest as the dependent variable and time from the CSF sample collection, a joint stratification for positivity in CSF_AD and SAA, and their interaction as independent variables. These models were adjusted for age, sex, and APOE ε4 status and included random intercepts in addition to random slope to address within‐subject correlations. The regression estimates for the CSF_AD and SAA stratified groups were compared using Tukey's honestly significant difference (HSD) test. The resulting p‐values and CIs were calculated based on the joint t‐distribution of z‐statistics and account for the probabilities in the context of multiple comparisons. Only the CSF_AD and SAA stratified groups with more than 20 participants were included in the analysis of cross‐sectional and longitudinal cognitive outcomes.
All analyses were performed within each clinical diagnosis (CU, MCI, Dementia) separately and were repeated for a subset who satisfied clinical criteria for eligibility to “Early‐AD” clinical trials, 20 that is, age of 50–85 years with a clinical diagnosis of MCI or Dementia, a CDR of 0.5 or 1, an MMSE score of 24–30, and biomarker evidence of AD Aβ‐pathology based on Aβ‐PET.
R version 4.3.1 was used for all statistical analyses and figures. Packages epiR, epitools, ggplot2, ggrepel, lme4, mgcv, mcr, multcomp, and tidyverse were used in data preparation and analysis.
3. RESULTS
3.1. Study cohort characteristics
The SAA analysis included 577 CU individuals, 654 with MCI, and 407 with Dementia enrolled in ADNI 1–3 studies; a detailed overview is presented in Table 1. One of the CU individuals failed screening for in‐clinic assessments and was excluded from further analysis.
TABLE 1.
Demographic, clinical, and biomarker characteristics of study participants at the time of CSF sample collection
| Parameter |
Cognitively unimpaired (N = 576) |
Mild cognitive impairment (N = 654) |
Dementia (N = 407) |
|---|---|---|---|
| Age (years) | 74·17 ± 7·19 [69·00, 79·00] | 74·15 ± 8·04 [68·00, 80·00] | 75·60 ± 7·82 [71·00, 81·00] |
| Male sex | 242 (42·0%) | 380 (58·1%) | 241 (59·2%) |
| Race (White/ Black or African American / Other or Unknown) | 515 (89·4%) / 38 (6·6%) / 23 (4·0%) | 608 (93·0%) / 23 (3·5%) / 23 (3·5%) | 391 (96·1%) / 8 (2·0%) / 8 (2·0%) |
| Ethnicity (Not Hispanic or Latino / Hispanic or Latino / Unknown) | 547 (95·0%) / 26 (4·5%) / 3 (0·5%) | 630 (96·3%) / 22 (3·4%) / 2 (0·3%) | 397 (97·5%) / 8 (2·0%) / 2 (0·5%) |
| Education (years) | 16·68 ± 2·44 [16·00, 18·00] | 16·14 ± 2·70 [14·00, 18·00] | 15·65 ± 2·73 [14·00, 18·00] |
| APOE ε4 carriers (heterozygote / homozygote) | 146 (25·4%) / 16 (2·8%) | 212 (32·5%) / 66 (10·1%) | 190 (46·7%) / 76 (18·7%) |
| APOE ε2 carriers | 80 (13·9%) | 52 (8·0%) | 21 (5·2%) |
| GBA1 variants carriers | 21 (6·1%) | 23 (4·7%) | 18 (5·6%) |
| TREM2 variants carriers | 11 (3·2%) | 20 (4·1%) | 12 (3·7%) |
| Bellenguez AD‐PRS | 0·03 ± 0·00 [0·03, 0·03] | 0·03 ± 0·00 [0·03, 0·03] | 0·03 ± 0·00 [0·03, 0·04] |
| Kunkle AD‐PRS | 0·01 ± 0·02 [0·00, 0·02] | 0·01 ± 0·02 [0·00, 0·03] | 0·02 ± 0·02 [0·01, 0·04] |
| Nalls PD‐PRS | −0·01 ± 0·00 [−0·01, −0·01] | −0·01 ± 0·00 [−0·01, −0·01] | −0·01 ± 0·00 [−0·01, −0·01] |
| Aβ PET (centiloid) | 20·83 ± 36·73 [−3·00, 31·00] | 38·95 ± 48·30 [−2·00, 76·75] | 81·19 ± 47·12 [54·50, 115·00] |
| Aβ PET+ (centiloid > 20) | 151 (32·5%) | 254 (51·0%) | 220 (86·3%) |
| CSF Aβ42 (pg/mL) | 1328·29 ± 635·29 [824·90, 1731·00] | 1044·65 ± 599·02 [609·90, 1349·00] | 667·17 ± 395·02 [451·25, 745·73] |
| CSF Aβ42+ (Aβ 42 < 980 pg/mL) | 194 (35·3%) | 360 (56·5%) | 362 (90·5%) |
| CSF p‐tau181 (pg/mL) | 21·96 ± 10·76 [14·93, 25·95] | 26·66 ± 14·70 [16·40, 32·55] | 35·94 ± 16·97 [24·58, 43·22] |
| CSF p‐tau181+ (p‐tau181 > 24 pg/mL) | 173 (31·5%) | 296 (46·5%) | 306 (76·5%) |
| CSF_AD+ (p‐tau181/Aβ42 > 0·025) | 140 (25·5%) | 329 (51·7%) | 361 (90·3%) |
| MMSE | 29·05 ± 1·18 [29·00, 30·00] | 27·72 ± 2·16 [26·25, 29·00] | 21·72 ± 4·46 [20·00, 25·00] |
| CDR‐SB | 0·10 ± 0·34 [0·00, 0·00] | 1·56 ± 1·08 [0·50, 2·00] | 5·60 ± 3·07 [3·50, 7·00] |
| ADAS‐Cog13 | 8·63 ± 4·42 [5·33, 11·00] | 15·76 ± 7·24 [10·67, 20·33] | 32·47 ± 10·56 [25·00, 38·00] |
| Memory composite score | 1·27 ± 0·76 [0·76, 1·73] | 0·28 ± 0·82 [−0·23, 0·76] | −1·15 ± 0·80 [−1·62, −0·61] |
| Executive function composite score | 0·97 ± 0·78 [0·45, 1·54] | 0·32 ± 0·87 [−0·23, 0·90] | −0·92 ± 1·03 [−1·65, −0·23] |
| Visual spatial composite score | 0·20 ± 0·66 [−0·08, 0·74] | 0·00 ± 0·76 [−0·55, 0·74] | −0·78 ± 1·08 [−1·49, −0·08] |
| Language composite score | 0·92 ± 0·71 [0·45, 1·43] | 0·26 ± 0·84 [−0·22, 0·85] | −0·89 ± 1·07 [−1·47, −0·10] |
| Sleep problems* (None / Mild / Moderate / Severe) | 267 (91·1%) / 17 (5·8%) / 8 (2·7%) / 1 (0·3%) | 308 (75·5%) / 64 (15·7%) / 28 (6·9%) / 8 (2·0%) | 207 (70·6%) / 50 (17·1%) / 25 (8·5%) / 11 (3·8%) |
| Hallucinations** (None / Mild / Moderate / Severe) | 293 (100%) / — / — / — | 404 (99·0%) / 3 (0·7%) / 1 (0·2%) / — | 271 (92·5%) / 12 (4·1%) / 6 (2·0%) / 4 (1·4%) |
| CSF aliquot storage duration (years) | 7.52 ± 4.46 4 , 11 | 9.67 ± 4.42 6 , 12 | 10.87 ± 4.14 9 , 15 |
Note: Continuous variables are reported in mean (standard deviation) [Q1, Q3] format. Categorical variables are reported in terms of count (percentage). Missing data counts and percentages for clinical and biomarker data are provided in Table S1.
Dementia participants were older than CU and MCI participants (Table 1). Participants with cognitive impairments (MCI and Dementia) had fewer years of education, were more likely to be male, APOE ε4 carriers, had greater AD biomarker burden (lower CSF Aβ42, and greater CSF p‐tau181 and Aβ‐PET centiloid), and had greater cognitive impairment than CU at the time of CSF sample collection.
CSF aliquots from participants with MCI and Dementia were stored for a longer duration before the CSF α‐syn SAA was performed, compared to those from CU participants (Table 1; p = 0.036 and p < 10−4, respectively).
3.2. Neuropathological confirmation of CSF 𝛼‐syn SAA results
Of the 1638 study participants, 78 had autopsy confirmation, with an ante‐mortem time of 4.3 ± 3.0 years (range 0 to 14 years) between CSF sample collection and time of death. The neuropathological analysis of the 78 individuals revealed LB‐pathology in the brains of 39 (50%) participants. Of the 39 with LB‐pathology, 16 showed diffuse neocortical, 7 transitional limbic, 11 amygdala‐predominant, 3 brainstem‐predominant, and 2 olfactory bulb LB‐pathology. A total of 31 out of 39 cases with LB‐pathology at autopsy also had intermediate to high AD neuropathological changes (ADNC) at autopsy, 7 with low ADNC, and 1 with no ADNC (Table S2). In contrast, 29 out of 39 cases without LB‐pathology at autopsy presented with intermediate to severe ADNC, 6 with low ADNC, and 4 with no ADNC.
Neuropathological examination served as the gold standard against which results of SAA were compared. Samples yielding positive SAA outcomes from individuals who exhibited LB pathological changes at autopsy were classified as true positives. Conversely, samples that tested negative for SAA from individuals devoid of LB pathology at autopsy were designated as true negatives. All but one of the 39 without LB‐pathology were SAA−; thus, the specificity for the CSF 𝛼‐syn SAA in this cohort was 97% (CI: 83%–99·9%). Of the 39 individuals with LB‐pathology at autopsy, 30 were PD/DLB‐like SAA+, 8 SAA−, and 1 inconclusive. Therefore, the overall sensitivity of the assay to detect LB‐pathology in any form was 79% (CI: 63%–90%), with varying sensitivity when stratified by LB‐pathology distribution: 100% in detecting diffuse neocortical LB‐pathology, 57% (CI: 18%–90%) in detecting transitional limbic LB‐pathology, and 60% (CI: 26%–88%) in detecting amygdala‐predominant LB‐pathology.
In comparison to the true positive cases (N = 30), the false negative cases (samples that yielded negative SAA results despite the presence of LB pathological changes identified during autopsy; N = 8) exhibited a longer average ante mortem time by 5.6 years (p < 0.0001). Additionally, false negative cases compared to true positive cases were associated with a higher severity of cerebral amyloid angiopathy (p = 0.047), a more frequent occurrence of aging‐related tau astrogliopathy (p = 0.0002), and a lesser degree of neuronal loss in the substantia nigra (p = 0.0004). The false negative and true positive cases did not differ in their levels of ADNC and TDP‐43 co‐pathology.
In a supplementary analysis, we further evaluated the performance of SAA within the autopsy‐confirmed cases to assess the potential effect of CSF aliquot age (i.e., time from CSF sample collection to CSF α‐syn SAA analysis) on the sensitivity of SAA in detecting LBD pathology. Our analysis revealed no significant association between the sensitivity of SAA and the CSF aliquot age (p = 0.132; Figure S1), suggesting that the SAA assay performance was robust to variances in CSF aliquot age.
3.3. CSF 𝛼‐syn SAA detection in clinical ADNI cohort
Nine out of 1637 participants in the clinical ADNI cohort (two CU, one MCI, six Dementia) had inconclusive SAA results and were excluded from the analysis. Overall, 366 out of 1628 participants (22.5%) were “PD/DLB‐like” SAA+. Three individuals (two CU and one MCI) had “MSA‐like” SAA results and excluded from the analysis because of small sample size in this group.
17 CSF specimens were visibly discolored, likely due to blood contamination. A sensitivity analysis, excluding these discolored specimens, revealed that the results were similar to those obtained in the main analyses (data not shown).
SAA had 100% reproducibility on 20 blind replica samples from 9 CU, 9 MCI, and 2 Dementia participants, of which 3 were SAA+ and 17 were SAA−.
SAA+ occurred with greater frequency in individuals with MCI (19%; 123 out of 651) and Dementia (38%; 152 out of 400) compared to in CU individuals (16%; 91 out of 572). The SAA+ rate of “early‐AD” participants was 24% (72 out of 304). After adjusting for differences in age, sex, APOE ε4, and CSF Aβ42 levels, participants with Dementia diagnosis had odds ratios (OR) of 1.99 [1.54–2.60] and 1.70 [1.38–2.11] for SAA+ compared to CU and MCI, respectively.
3.4. Demographic and biomarker characteristics of CSF 𝛼‐syn SAA‐positive participants
Characteristics of SAA+ participants and SAA− participants within each diagnostic group separately are summarized in Table 2. SAA+ CU and SAA+ MCI participants were significantly older than their SAA− counterparts. SAA+ Dementia participants differed significantly from their SAA− counterparts by exhibiting greater Aβ burden (i.e., lower CSF Aβ42 levels), after accounting for differences in age, sex, and APOE ε4 status, yet lower CSF p‐tau181 levels, after accounting for differences in age, sex, APOE ε4 status, and CSF Aβ42 levels. SAA+ Early‐AD participants were older and had greater Aβ burden (i.e., greater Aβ PET centiloid and lower CSF Aβ42 levels) compared to SAA− Early‐AD participants, after accounting for differences in age, sex, and APOE ε4 status.
TABLE 2.
Demographic and biomarker characteristics within each diagnostic group, stratified by CSF α‐syn SAA results
| Cognitively unimpaired | Mild cognitive impairment | Dementia | Early‐AD | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter |
SAA+ (N = 91) |
SAA‐ (N = 481) |
p‐value |
SAA+ (N = 123) |
SAA‐ (N = 528) |
p‐value |
SAA+ (N = 152) |
SAA‐ (N = 248) |
p‐value |
SAA+ (N = 72) |
SAA‐ (N = 232) |
p‐value |
| Age (years) | 76·7 (6·40) | 73·7 (7·18) | <0·0001 | 75·8 (7·67) | 73·7 (8·08) | 0·009 | 76·3 (7·42) | 75·1 (8·05) | 0·10 | 75·1 (6·39) | 73·3 (6·60) | 0·046 |
| Male sex | 46 (50·5%) | 194 (40·3%) | 0·09 | 74 (60·2%) | 304 (57·6%) | 0·67 | 93 (61·2%) | 143 (57·7%) | 0·56 | 42 (58·3%) | 125 (53·9%) | 0·60 |
| Race | ||||||||||||
| White | 83 (91·2%) | 428 (89·0%) | 0·28 | 117 (95·1%) | 488 (92·4%) | 0·18 | 147 (96·7%) | 237 (95·6%) | 0·26 | 69 (95·8%) | 222 (95·7%) | 0·88 |
| Black or African American | 7 (7·7%) | 31 (6·4%) | 1 (0·8%) | 22 (4·2%) | 1 (0·7%) | 7 (2·8%) | 1 (1·4%) | 5 (2·2%) | ||||
| Other or Unknown | 1 (1·1%) | 22 (4·6%) | 5 (4·1%) | 18 (3·4%) | 4 (2·6%) | 4 (1·6%) | 2 (2·8%) | 5 (2·2%) | ||||
| Ethnicity | ||||||||||||
| Not Hispanic or Latino | 88 (96·7%) | 455 (94·6%) | 0·61 | 118 (95·9%) | 509 (96·4%) | 0·53 | 150 (98·7%) | 240 (96·8%) | 0·31 | 68 (94·4%) | 226 (97·4%) | 0·82 |
| Hispanic or Latino | 3 (3·3%) | 23 (4·8%) | 4 (3·3%) | 18 (3·4%) | 1 (0·7% | 7 (2·8%) | 4 (5·6%) | 6 (2·6%) | ||||
| Unknown | — | 3 (0·6%) | 1 (0·8%) | 1 (0·2%) | 1 (0·7%) | 1 (0·4%) | — | — | ||||
| Education (years) | 16·9 (2·68) | 16·7 (2·39) | 0·52 | 16·2 (2·90) | 16·1 (2·65) | 0·66 | 15·8 (2·77) | 15·6 (2·72) | 0·37 | 16·3 (2·84) | 16·0 (2·62) | 0·54 |
| APOE ε4 carriers | ||||||||||||
| Heterozygote | 22 (24·2%) | 123 (25·7) | 0·92 | 39 (32·0%) | 173 (32·9%) | 0·10 | 75 (49·3%) | 113 (45·6%) | 0·76 | 36 (50·0%) | 113 (48·9%) | 0·45 |
| Homozygote | 3 (3·3%) | 13 (2·7%) | 19 (15·6%) | 47 (8·9%) | 27 (17·8%) | 46 (18·5%) | 17 (23·6%) | 42 (18·2%) | ||||
| APOE ε2 carriers | 16 (17·6%) | 64 (13·4%) | 0·37 | 15 (12·2%) | 36 (6·8%) | 0·07 | 5 (3·3%) | 16 (6·5%) | 0·25 | 3 (4·2%) | 7 (3·0%) | 0·93 |
| GBA1 variants carriers | 1 (1·6%) | 20 (7·2%) | 0·18 | 6 (6·3%) | 17 (4·3%) | 0·6 | 6 (5·3%) | 12 (6·0%) | 0·99 | 2 (3·5%) | 6 (3·3%) | 1 |
| TREM2 variants carriers | 0 (0%) | 9 (3·2%) | 0·32 | 3 (3·2%) | 17 (4·3%) | 0·82 | 4 (3·5%) | 8 (4·0%) | 1 | 1 (1·8%) | 9 (4·9%) | 0·52 |
| a Bellenguez AD‐PRS | 0·033 (0·0022) | 0·033 (0·0024) | 0·52 | 0·033 (0·0023) | 0·033 (0·0023) | 0·83 | 0·034 (0·0024) | 0·034 (0·0024) | 0·98 | 0·034 (0·0020) | 0·034 (0·0024) | 0·78 |
| a Kunkle AD‐PRS | 0·0091 (0·016) | 0·0062 (0·016) | 0·64 | 0·019 (0·022) | 0·013 (0·021) | 0·28 | 0·024 (0·021) | 0·025 (0·022) | 0·78 | 0·028 (0·021) | 0·024 (0·023) | 0·30 |
| a Nalls PD‐PRS | ‐0·011 (0·0034) | ‐0·011 (0·0035) | 0·82 | ‐0·011 (0·0036) | ‐0·011 (0·0036) | 0·91 | ‐0·011 (0·0036) | ‐0·011 (0·0035) | 0·34 | ‐0·011 (0·0041) | ‐0·011 (0·0037) | 0·44 |
| a Aβ PET (centiloid) | 21·6 (34·1) | 20·5 (36·9) | 0·89 | 49·3 (53·2) | 36·8 (46·9) | 0·20 | 84·9 (44·0) | 79·4 (48·7) | 0·34 | 93·8 (39·7) | 81·3 (35·6) | 0·02 |
| a CSF Aβ−42 (pg/mL) | 1370 (758) | 1330 (609) | 0·26 | 906 (561) | 1080 (604) | 0·07 | 614 (315) | 702 (436) | 0·02 | 610 (223) | 709 (267) | 0·01 |
| b CSF p‐tau181 (pg/mL) | 24·3 (13·4) | 21·4 (10·1) | 0·28 | 27·1 (12·9) | 26·6 (15·1) | 0·42 | 33·3 (16·4) | 37·6 (17·3) | 0·04 | 34·4 (18·3) | 34·1 (16·5) | 0·48 |
| a Aβ PET+ (centiloid > 20) | 26 (40·0%) | 123 (31·1%) | 0·30 | 51 (58·0%) | 202 (49·6%) | 0·85 | 85 (90·4%) | 133 (84·2%) | 0·12 | 72 (100%) | 232 (100%) | — |
| a CSF Aβ−42+ (Aβ42 < 980 pg/mL) | 34 (38·2%) | 156 (34·2%) | 0·86 | 79 (65·8%) | 280 (54·5%) | 0·21 | 138 (92·6%) | 218 (89·3%) | 0·25 | 64 (92·8%) | 202 (89·0%) | 0·44 |
| b CSF p‐tau181+ (p‐tau181 > 24 pg/mL) | 31 (34·8%) | 140 (30·6%) | 0·68 | 58 (48·3%) | 236 (46·0%) | 0·48 | 107 (71·8%) | 193 (79·1%) | 0·19 | 49 (71·0%) | 160 (70·5%) | 0·58 |
| a CSF AD+ (p‐tau181/Aβ42 > 0·025) | 27 (30·3%) | 111 (24·3%) | 0·78 | 73 (60·8%) | 255 (49·7%) | 0·31 | 139 (93·3%) | 215 (88·1%) | 0·07 | 64 (92·8%) | 201 (88·5%) | 0·45 |
| Sleep problems | ||||||||||||
| None | 49 (89·1%) | 216 (91·5%) | 0·64 | 67 (78·8%) | 240 (74·5%) | 0·51 | 74 (67·9%) | 127 (71·3%) | 0·76 | 34 (70·8%) | 104 (72·7%) | 0·97 |
| Mild | 5 (9·1%) | 12 (5·1%) | 9 (10·6%) | 55 (17·1%) | 19 (17·4%) | 31 (17·4%) | 9 (18·8%) | 27 (18·9%) | ||||
| Moderate | 1 (1·8%) | 7 (3·0%) | 7 (8·2%) | 21 (6·5%) | 12 (11·0%) | 13 (7·3%) | 4 (8·3%) | 9 (6·3%) | ||||
| Severe | — | 1 (0·4%) | 2 (2·4%) | 6 (1·9%) | 4 (3·7%) | 7 (3·9%) | 1 (2·1%) | 3 (2·1%) | ||||
| Hallucinations | ||||||||||||
| None | 55 (100%) | 236 (100%) | — | 85 (100%) | 318 (98·8%) | 0·59 | 97 (89·0%) | 169 (94·9%) | 0·18 | 47 (97·9%) | 138 (96·5%) | 0·57 |
| Mild | — | — | — | 3 (0·9%) | 5 (4·6%) | 6 (3·4%) | — | 3 (2·1%) | ||||
| Moderate | — | — | — | 1 (0·3%) | 4 (3·7%) | 2 (1·1%) | 1 (2·1%) | 2 (1·4%) | ||||
| Severe | — | — | — | — | 3 (2·8%) | 1 (0·6%) | — | — | ||||
Abbreviations: SAA−, α‐syn seeding aggregates not detected; SAA+, α‐synuclein aggregates detected with an aggregation profile consistent with the characteristic seeding seen in PD and DLB.
Group difference significance after accounting for differences in age, sex, and APOE ε4 status. Unadjusted results are provided in Table S3.
Group difference significance after accounting for differences in age, sex, APOE ε4 status, and CSF Aβ42 levels. Unadjusted results are provided in Table S3.
3.5. Prevalence of CSF 𝛼‐syn SAA positivity by AD biomarker positivity status
We tested the association of the prevalence of SAA positivity with AD biomarker positivity within each diagnostic group (CU, MCI, Dementia, and Early‐AD) separately (Figure 1).
FIGURE 1.
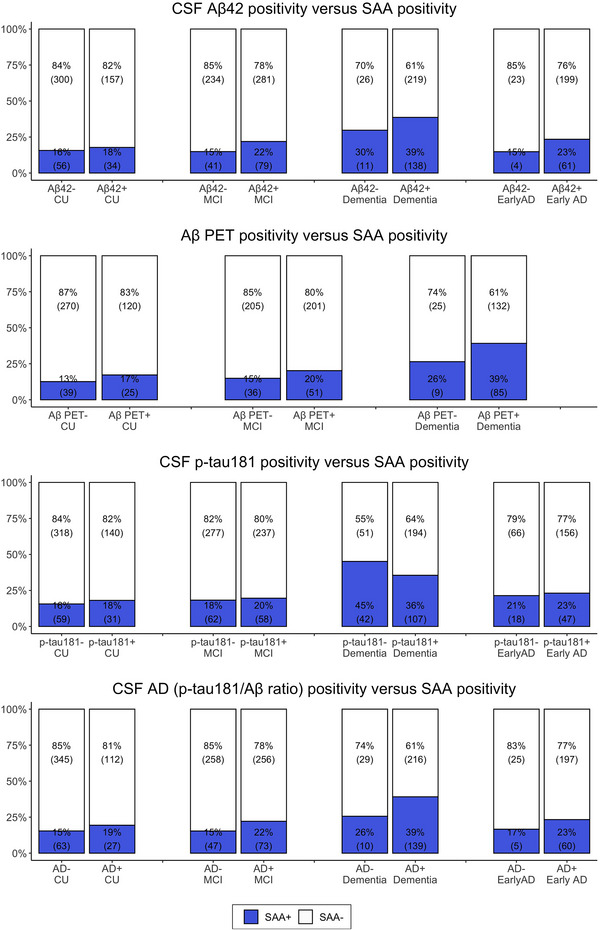
Prevalence of SAA positivity in relation to AD biomarker positivity within each diagnostic group (CU, MCI, Dementia, and Early‐AD) separately
When stratified for biomarker evidence for AD Aβ‐pathology (i.e., CSF Aβ42 < 980 pg/mL or Aβ‐PET centiloid > 20), SAA+ prevalence was higher in the Aβ+ groups compared to their Aβ− counterparts within each diagnosis. These differences were though not statistically significant after controlling for differences in age, sex, and APOE ε4 status. Among Aβ+ participants, Aβ+ Dementia participants had a 2‐fold greater OR for SAA+ compared to Aβ+ CU (CSF Aβ42+: OR 2.03 [1.50–2.79] and Aβ‐PET+: OR 2.3 [1.48–3.12]) and over 1.75‐fold greater OR for SAA+ compared to Aβ+ MCI (CSF Aβ42+: OR 1.75 [1.39–2.22] and Aβ‐PET+: OR 1.90 [1.42–2.56]), all adjusted for age, sex, and APOE ε4 status. SAA+ prevalence within Aβ+ MCI did not differ from those of Aβ+ CU. Among participants without Aβ deposition, the SAA+ rate within the Aβ− Dementia group was twice as high as that within the Aβ− non‐demented groups, without statistical significance after adjusting for differences in age, sex, and APOE ε4 status.
When stratified for CSF p‐tau181 biomarker status (CSF p‐tau181 > 24 pg/mL), SAA+ prevalence was lower in the p‐tau181+ compared to their p‐tau181− counterparts within Dementia (36% vs. 45%), without statistical significance after adjusting for differences in age, sex, APOE ε4 status, and CSF Aβ42 levels. In contrast, p‐tau181+ and p‐tau181− participants had similar SAA+ trends among CU, MCI, and Early‐AD groups (i.e., 18% vs. 16%, 20% vs. 18%, and 23% vs. 21%, respectively). Furthermore, among p‐tau181+ participants, p‐tau181+ Dementia participants had greater OR for SAA+ compared to p‐tau181+ CU (OR 1.80 [1.27–2.58]) and p‐tau181+ MCI (OR 1.63 [1.25–2.15]), accounting for differences in age, sex, APOE ε4 status, and CSF Aβ42 levels. Additionally, p‐tau181+ MCI participants had greater OR for SAA+ compared to p‐tau181+ CU (OR 1.35 [1.00–1.83]). Similar patterns of increase in OR for SAA+ with clinical severity was also observed among p‐tau181−, such that p‐tau181− Dementia had an OR of 2.06 [1.39–3.06] and 2.00 [1.34–2.93] for SAA+ compared to p‐tau181− CU and p‐tau181− MCI, respectively, and p‐tau181− MCI had an OR of 1.49 [1.10–2.05] for SAA+ compared to p‐tau181− CU, after accounting for differences in age, sex, APOE ε4 status, and CSF Aβ42 levels.
Finally, when stratified for CSF biomarker evidence for overall AD‐pathology (CSF_AD+; p‐tau181/Aβ42+ < 0.025), SAA+ prevalence was higher in the CSF_AD+ groups compared to their CSF_AD− counterparts within each diagnosis. These differences were statistically significant only in Dementia after controlling for differences in age, sex, and APOE ε4 status, with an OR of 2.21 [1.01–5.17] for SAA+. In the presence of AD pathology across clinical diagnosis groups, CSF_AD+ Dementia had a higher OR for SAA+ compared to CSF_AD+ CU (OR 2.02 [1.45–2.87]) and CSF_AD+ MCI (OR 1.78 [1.40–2.27]), accounting for differences in age, sex, and APOE ε4 status. In the absence of AD pathology, although CSF_AD– Dementia had a higher rate of SAA+ compared to non‐demented CSF_AD– (i.e., 26% vs. 15%), these differences were not statistically significant after controlling for differences in age, sex, and APOE ε4 status.
3.6. Non‐linear age models: Positivity in CSF 𝛼‐syn SAA and AD biomarkers
We modeled the prevalence of positivity in SAA and AD biomarkers as a nonlinear function of age (Figure 2). An increase in positivity for SAA and all AD biomarkers (CSF Aβ42, Aβ ‐PET centiloid, and CSF p‐tau181) was observed with a more pronounced trend for AD biomarkers within CU and MCI. Among Dementia, positivity in AD biomarkers decreased with age, while SAA+ remained relatively stable. In contrast, the Early‐AD participants exhibited a substantial increase in SAA+ with age, despite consistent rates of CSF p‐tau181+ over the age spectrum.
FIGURE 2.
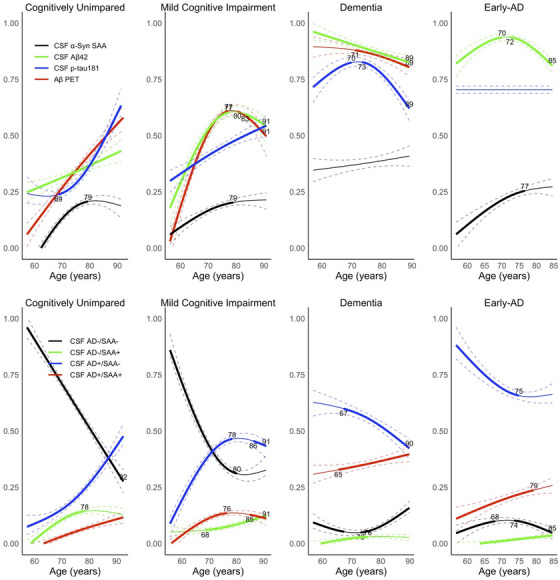
Top row: Proportions of biomarker positive groups with increasing age for AD biomarkers and SAA. Combined proportions may exceed one because individuals can test positive for more than one biomarker. Bottom row: Proportions of CSF_AD and SAA stratified groups with increasing age. CSF_AD positivity (AD+) refers to having CSF p‐tau181/Aβ42 > 0025. Early‐AD is a clinical subgroup of ADNI participants eligible for “Early‐AD” clinical trials (i.e., clinical diagnosis of MCI or Dementia; age range 50–85 years; CDR score 0·5 or 1·0; MMSE score 24–30; PET Aβ+)
When we repeated the non‐linear age model for prevalence in joint SAA and overall CSF AD biomarker positivity, the prevalence of participants positive for CSF_AD, SAA, or both (specifically, CSF_AD+/SAA−, CSF_AD−/SAA+, CSF_AD+/SAA+) increased with age, while the prevalence of CSF_AD−/SAA− participants decreased with age. This pattern remained consistent among both CU and MCI participants, and the increase in prevalence with age was particularly prominent for the CSF_AD+/SAA− group. However, within Dementia and Early‐AD participant groups, the prevalence of CSF_AD+/SAA− decreased with age, while the prevalence of CSF_AD+/SAA+ increased with age.
3.7. Non‐linear AD biomarker models for the prevalence of CSF 𝛼‐syn SAA positivity
Next, we modeled SAA+ prevalence as a function of continuous AD biomarker levels, while accounting for differences in age and CSF Aβ42 levels when applicable (Figure 3). There was an exponential increase in the proportion of SAA+ participants with decreasing CSF Aβ42 levels, particularly starting at 1117 pg/mL. Similarly, an increase in the proportion of SAA+ participants occurred between Aβ‐PET centiloid levels 12 and 100. There was a significant increase in the incidence of SAA+ with increasing CSF p‐tau181 levels up to 22 pg/mL, beyond which the SAA+ rate stabilized, presenting a trend of lower proportion of SAA+ with higher levels of CSF p‐tau181 starting at 26 pg/mL. When modeling against the CSF p‐tau181/Aβ42 ratio levels, we observed an increase in the incidence of SAA+ with greater CSF p‐tau181/Aβ42 ratio levels up to p‐tau181/Aβ42 = 0·089. However, for CSF p‐tau181/Aβ42 ratio levels greater than 0·108, there was a significant decline in the incidence of SAA+. When performing separate modeling within each diagnosis (Figure 3; Figure S2), Dementia participants displayed a decrease in the incidence of SAA+ with increasing Aβ‐PET burden beyond a centiloid of 106. This decrease was also observed with increasing CSF p‐tau181 levels consistently across the study range, and with increasing CSF p‐tau181/Aβ42 ratio values between 0·52 and 0·118.
FIGURE 3.
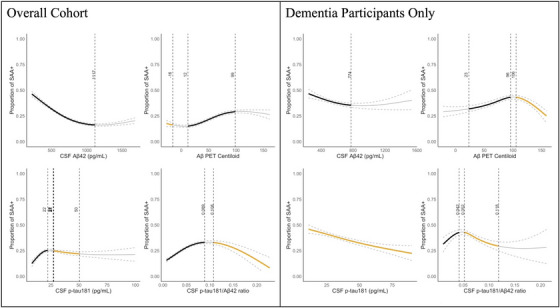
Proportions of SAA+ participants modeled as a function of continuous AD biomarker levels (CSF Aβ42, Aβ‐PET Centiloid, CSF p‐tau181, and CSF p‐tau181/Aβ42) for the overall study cohort and Dementia participants, separately. Models for CU, MCI, and Early‐AD cohorts are provided in Figure S2. The bold black and orange areas of the curve indicate statistically significant increasing and decreasing proportions of SAA+ participants with greater AD biomarker burden (i.e., decreasing CSF Aβ42 levels and increasing Aβ‐PET Centiloid, CSF p‐tau181, and CSF p‐tau181/Aβ42 levels), respectively. Models within each diagnosis separately are provided in Figure S2
3.8. Prevalence of CSF 𝛼‐syn SAA with respect to AD risk factors and genetics
We compared SAA+ prevalence in groups stratified based on AD risk factors, including biological sex, race, and ethnicity, and genetics (APOE ε4 genotype and GBA1 and TREM2 variants). There was a higher SAA+ prevalence in APOE ε4 homozygous compared to APOE ε4 non‐carrier or heterozygote among MCI and Early‐AD participants with an OR of 1.81 [1.16–2.80] and 2.02 [1.10–3.75], respectively. However, these differences did not reach statistical significance after adjusting for CSF Aβ42 levels. No significant associations with GBA1 and TREM2 variants were identified among diagnostic groups (CU, MCI, Dementia; Table 1) or between SAA+ and SAA− (Table 2).
SAA+ compared to their SAA− counterparts within each diagnosis did not differ in their PD‐PRS and Bellenguez AD‐PRS. SAA+ MCI, but not SAA+ CU or SAA+ Dementia, had greater Kunkle AD‐PRS than their SAA− counterparts; however, this difference was not significant after adjusting for APOE ε4 status, suggesting these differences are driven by APOE ε4.
3.9. Effects of CSF 𝛼‐syn SAA positivity on cross‐sectional cognitive impairment
We assessed the effects of SAA positivity on cognitive performance at the time of CSF sample collection within participants that were AD CSF biomarker positive.
CSF_AD+/SAA+ MCI participants demonstrated greater impairment in language, executive function, and memory and overall global cognition (ADAS Cog13 and PACC), compared to CSF_AD+/SAA− MCI participants (Figure 4; Figure S3‐S4). Similarly, CSF_AD+/SAA+ Early‐AD participants had greater impairment in global cognition (CDR‐SB and PACC) and executive function and language compared to CSF_AD+/SAA− Early‐AD participants. No significant differences in global cognition or specific cognitive domains were observed between CSF_AD+/SAA+ and CSF_AD+/SAA− within CU or Dementia.
The assessment of effects of SAA positivity on cognitive performance within participants without AD CSF biomarker positivity (i.e., CSF_AD− participants) was limited to CU and MCI due to the small sample size (N < 20) of CSF_AD−/SAA+ participants within Dementia and Early‐AD groups (Figure S3‐S5). No significant differences in global cognition or specific cognitive domain measures were found between CSF_AD−/SAA+ and CSF_AD−/SAA− participants in either CU or MCI.
3.10. Effects of CSF 𝛼‐syn SAA positivity on longitudinal cognitive decline
Next, we assessed the effects of SAA positivity on cognitive decline by modeling progression trajectories of longitudinal cognitive assessments within 2 years of CSF sample collection. In comparison to their CSF_AD+/SAA− counterparts, CSF_AD+/SAA+ CU participants exhibited a greater decline in global cognition measures (ADAS‐Cog13 and PACC; Figure 4 and Figure S3‐S4). However, there were no significant differences observed in global cognition or domain‐specific cognitive decline between CSF_AD+/SAA− and CSF_AD+/SAA+ participants within the MCI, Dementia, and Early‐AD groups. Similarly, there were no significant differences in global cognition and domain‐specific cognitive decline between CSF_AD−/SAA− and CSF_AD−/SAA+ participants within the CU and MCI groups.
FIGURE 4.

Within AD+ (CSF p‐tau181/Aβ42 > 0.025) participants in each diagnostic group separately, effect of SAA status (i.e., SAA+ vs. SAA−) on cognitive impairment and cognitive decline. The forest plots represent SAA+ effect estimates compared to SAA− based on linear regression models for the cross‐section cognitive impairment comparisons and linear mixed‐effects models for the longitudinal cognitive decline comparisons. The global cognitive assessments, including the CDR‐SB, the ADAS‐Cog13, the MMSE, based on a 30‐point questionnaire, and PACC, and the domain‐specific cognitive assessments including the composite measures of memory, executive function, language, and visuospatial functioning were analyzed as clinical outcome measures. The assessment of cognitive decline rates was confined to longitudinal data collected within a 2‐year timeframe of CSF sample collection. Results within AD− (CSF p‐tau181/Aβ42 ≤ 0.025) participants are provided in Figure S5.
4. DISCUSSION
We performed CSF‐based α‐syn SAA on 1638 ADNI participants including a large sample of participants with amnestic MCI and dementia due to AD, of whom 78 had autopsy‐confirmation for LB‐pathology. The major findings of this study were: (1) SAA demonstrates 79% sensitivity and 97% specificity in detecting LB‐pathology in comparison to gold‐standard autopsy, with notably superior performance in identifying neocortical compared to limbic and amygdala‐predominant LB‐pathology. (2) SAA+ was 22% in the clinical ADNI cohort and increased with disease stage from CU (16%) to MCI (19%) to Dementia (38%). (3) SAA+ increased with age with positivity starting as early as 55 years. Furthermore, SAA+ comorbid with AD biomarker positivity increased with age while prevalence of AD biomarker positivity without comorbid SAA‐positivity declined, specifically in Early‐AD and Dementia individuals. (4) Increased SAA+ prevalence was associated with the greater Aβ‐pathology burden. In contrast, there was an inverse relationship between SAA+ prevalence and CSF p‐tau181 levels. (5) SAA positivity had an impact on cognitive impairment predominantly on those who were CSF AD‐biomarker positive with MCI and Early‐AD. Taken together, the observed increase in SAA+ with age and cognitive impairment stages and its association with AD biomarker levels underscores the clinical significance of LB co‐pathology in individuals with AD‐pathology, emphasizing its relevance in understanding disease etiology and progression.
In comparison to autopsy findings of LB‐pathology (N = 78), SAA demonstrated a sensitivity of 79% and specificity of 97%. Notably, the assay exhibited superior performance in detecting neocortical (100%) compared to limbic (57%) and amygdala‐predominant (60%) LB‐pathology. These findings in the ADNI cohort further validated the performance of the SYNTap SAA method used to detect 𝛼‐syn seeding, establishing its robustness and reliability as an assay for LB‐pathology, particularly at advanced stages which are more likely to contribute to dementia. Our results are comparable to neuropathology data analyzed with SAA in another study, 5 identifying pathological α‐syn seeds in pre‐ and post mortem CSF samples from individuals with a variety of neurodegenerative diseases in the context of co‐pathologies and revealing a notably higher sensitivity of SAA for limbic/transitional and diffuse neocortical LB‐pathology (97.8%) compared to those with amygdala‐predominant LB‐pathology (14.3%). The higher sensitivity of SAA in detecting amygdala‐predominant LB‐pathology (60% vs. 14%) in ADNI autopsy sub‐cohort is likely due to small sample sizes, differences in ante mortem time, differences in ante‐mortem clinical characteristics and, perhaps, different spread and amount of LB‐pathology among individuals within this diagnostic category. It is noteworthy that the sensitivity relative to autopsy is lower than sensitivity relative to clinical diagnosis reported for non‐genetic cases in cohorts with PD and DLB 3 , 4 , 21 ; therefore, our results should be interpreted in the context of AD co‐pathologies. We also acknowledge that, while there is an accumulating body of autopsy confirmation of SAA approaches, additional clinical‐pathological correlation studies on diverse cohorts are needed to fully understand the performance and limitations of SAA approaches especially in the presence of AD‐pathology.
We observed SAA‐positivity in 22% of the clinical ADNI cohort, with the highest SAA+ rate detected in the Dementia (16% CU, 19% MCI, and 38% Dementia). The stepwise increase in SAA+ prevalence across disease stages highlights a potential association between SAA‐positivity and the progression of neurodegenerative processes. SAA has consistently identified α‐syn co‐pathology in several studies involving elderly individuals and those with cognitive impairment. Our SAA+ rate, ranging from 19% to 38%, aligns with previously reported SAA+ rates of 23% to 45% in cognitively impaired populations. 6 , 8 In contrast, our observed SAA+ rate of 16% within CU ADNI participants exceeds the 4%–8% reported in previous studies involving CU individuals. 4 , 6 , 7 Notably, in one of these studies, CU participants were screened for dopamine transporter negativity likely contributing to lower SAA+ rates. 4 Additionally, the participants in these comparative studies were notably younger than ADNI CU participants (e.g., 63‐years vs. 71‐years) and exhibited lower rates of Aβ‐positivity (e.g., 14% vs. 32%). Both Aβ burden and advanced age are significant factors that could contribute to the observed disparity in reported SAA+ rates. We acknowledge that the prevalence of SAA+ in our study is relevant to older individuals of age greater than 55 years, such as those at risk for AD, and that additional study in populations with greater age variation is needed to understand the relationship between the prevalence of SAA+ and age in the general population. Another crucial consideration that warrants further investigation is the standardization of α‐syn SAA methods used across these studies to enhance the reliability and consistency of findings in multi‐cohort studies. Nevertheless, these ante mortem findings confirm autopsy results that up to 23% of elderly CU individuals without clinical evidence of PD/DLB or other neurodegenerative diseases present with incidental LB‐pathology at autopsy 22 and up to 45% of AD cases show co‐occurring LB‐pathology at autopsy. 23 The robust association between AD and LB‐pathology is not confined to sporadic AD cases; it extends to various forms of the disease. Specifically, LB‐pathology has been reported in 50% of autopsies performed by the Dominantly Inherited Alzheimer Network, 24 63% of autosomal dominant PSEN1 and PSEN2 carrier AD cases, 25 and 50% of AD cases with Down's syndrome. 26 This convergence of LB‐pathology with AD across diverse genetic and sporadic forms underscores the clinical relevance of SAA.
SAA+ prevalence increased with age independent of AD‐pathology, emphasizing an association between LB‐pathology and aging. The co‐occurrence of SAA+ and CSF AD‐biomarker positivity also increased with age, particularly in Early‐AD and Dementia. Conversely, prevalence of CSF AD‐biomarker positivity without comorbid SAA‐positivity decreased with age, especially in Early‐AD and Dementia. This suggests an age‐associated relationship between LB‐pathology, AD‐pathology, and disease presentation. In models accounting for age, we showed that SAA‐positivity increased with Aβ‐burden, aligning with previous research, where total 𝛼‐syn levels increase along with Aβ‐burden in CSF of AD patients. 27 The observed increase in SAA+ may be attributed to potential mechanisms, such as co‐seeding with Aβ or interference with normal 𝛼‐syn clearance due to Aβ accumulation. However, it remains speculative, and other factors like general brain degradation or inflammation could also contribute to the heightened susceptibility to aggregate accumulation. Nevertheless, this finding suggests a relationship between the aggregation of α‐syn and Aβ, the complex timing of which is hard to disentangle in our cross‐sectional study design. On the other hand, the higher SAA‐positivity in Dementia with lower CSF p‐tau181 levels raises questions about the biology of co‐pathologies in the AD spectrum, consistent with autopsy studies reporting significantly lower levels of tau pathology in participants with AD and LB co‐pathologies compared to those with pure AD pathology. 28 One clinical interpretation is that, in dementia patients with Aβ+ and SAA+ but low AD tau‐pathology, LB‐pathology contributes significantly to cognitive impairment. 29 Clinicopathological studies of patients with AD neuropathological changes have demonstrated that each major co‐pathology creates distinct, typically accelerated, trajectories of clinical progression compared to AD alone. 30 Evidence from similar clinicopathologic studies suggest that particularly comorbid AD and LB pathologies are important determinants of person‐specific trajectories of cognitive decline. 31 Furthermore, these studies suggest that the presence of co‐pathologies lowers the threshold for meeting clinical criteria for a diagnosis of dementia. 32 Therefore, the greater SAA‐positivity rate in the absence of tau pathology or in individuals with low tau‐pathology may be indicative of earlier presentation of symptoms in low tau‐burden individuals with mixed pathology etiology. At the same time, it is important to acknowledge methodological limitation such that in vitro and in vivo studies have shown interactions between Aβ, tau, and α‐syn, 33 , 34 suggesting that heterogeneous aggregates may have obscured detectable levels of each protein.
Individuals classified as CSF_AD+/SAA+ exhibited more pronounced global and domain‐specific cognitive impairments compared to their CSF_AD+/SAA− counterparts. This finding aligns with previous studies on 𝛼‐syn, AD, and cognitive decline. 35 Although not significant across all measures, SAA‐positivity had the highest impact on CSF_AD+ MCI and Early‐AD individuals, suggesting a nuanced influence of SAA‐positivity on cognitive impairment, specific to the disease stage and primarily affecting those with CSF_AD+.
The limitations of this study should be acknowledged. Despite the geographical diversity of the ADNI, the absence of racial and ethnic diversity among ADNI participants is a notable limitation. The analysis of cognitive decline trajectories may not be sufficiently powered due to the 2‐year interval in prospective follow‐up cognitive changes, introduced by our examination of the most recent CSF draw. To address this limitation, ongoing efforts are underway to expand the SAA analysis by incorporating additional time points, aiming for a more comprehensive understanding of longitudinal downstream effects resulting from SAA‐positivity. Similarly, the small sample size of the genetics cohort, not adequately powered, may have contributed to the absence of significant associations with PRS and TREM2 and GBA1 variants. Finally, tau‐PET imaging was available for a limited number of participants in the study cohort, therefore, CSF p‐tau181 was used as a surrogate biomarker for tau‐pathology. However, within the ADNI dementia participants with available tau‐PET imaging data, the inverse association between SAA+ prevalence and tau burden was replicated predominantly for temporal meta‐ROI and temporoparietal ROI, despite the small sample size (N = 55; Figure S6).
While the current SAA protocol exhibits high sensitivity for detecting LB‐pathology, caution is warranted, especially considering the decreased sensitivity in identifying limbic and amygdala‐predominant LB‐pathology. It's crucial to approach the interpretation of results with care, recognizing that SAA‐positivity may not invariably translate to symptomatic disease, as incidental LBs are commonly identified during autopsy brain investigations. 36 Furthermore, the binary outcome from the qualitative nature of the current SAA protocol poses a limitation. Developing a quantitative assay could offer valuable insights, especially in assessing the “load” of α‐syn seeds in CSF, potentially correlating with the topological spread of LB‐pathology.
5. CONCLUSION
In summary, this study expands our understanding of the utility of α‐syn SAA for the in vivo molecular assessment of comorbid LB‐pathology in the context of dementia. Understanding the prevalence and significance of LB‐pathology in the context of dementia holds promise for elucidating the complex interplay among different pathologies in the etiology and progression of neurodegenerative disorders, ultimately guiding more precise diagnostic and therapeutic approaches. Moving forward, it is imperative to broaden the detection of LB‐pathology in diverse cohorts to enhance our understanding of the causes and triggers of AD and LB co‐pathologies.
CONFLICT OF INTEREST STATEMENT
D.T., Z.H., E.B.L., R.J.P, A.J.S., L.M.S., and P.T. receive funding from NIH/NIA. M.A.N. carries out as part of contract work for NIH consultancy. C.B. have nothing to disclose. H.I. is employee of Data Tecnica International and receives NIH support for open science research. J.L., K.M., S.ME., and R.L. are full‐time employee of Amprion Inc and hold stock or stock options of Aprion Inc. A.B.S. receives funding from Intramural Research Program of the National Institutes of Health; received honoraria from Movement Disorders Journal and npjParkinson's Disease; received travel support from Chan Zuckerberg Initiative, Michael J Fox Foundation, and Weill Cornell. M.W.W. serves on Editorial Boards for Alzheimer's & Dementia, and the Journal for Prevention of Alzheimer's disease. He has served on Advisory Boards for Acumen Pharmaceutical, Alzheon, Inc., Cerecin, Merck Sharp & Dohme Corp., and NC Registry for Brain Health. He also serves on the USC ACTC grant which receives funding from Eisai for the AHEAD study. He has provided consulting to BioClinica, Boxer Capital, LLC, Cerecin, Inc., Clario, Dementia Society of Japan, Eisai, Guidepoint, Health and Wellness Partners, Indiana University, LCN Consulting, Merck Sharp & Dohme Corp., NC Registry for Brain Health, Prova Education, T3D Therapeutics, University of Southern California (USC), and WebMD. He has acted as a speaker/lecturer for China Association for Alzheimer's Disease (CAAD) and Taipei Medical University, as well as a speaker/lecturer with academic travel funding provided by: AD/PD Congress, Cleveland Clinic, CTAD Congress, Foundation of Learning; Health Society (Japan), INSPIRE Project; U. Toulouse, Japan Society for Dementia Research, and Korean Dementia Society, Merck Sharp & Dohme Corp., National Center for Geriatrics and Gerontology (NCGG; Japan), University of Southern California (USC). He holds stock options with Alzeca, Alzheon, Inc., ALZPath, Inc., and Anven. Dr Weiner received support for his research from the following funding sources: National Institutes of Health (NIH)/NINDS/National Institute on Aging (NIA), Department of Defense (DOD), California Department of Public Health (CDPH), University of Michigan, Siemens, Biogen, Hillblom Foundation, Alzheimer's Association, Johnson & Johnson, Kevin and Connie Shanahan, GE, VUmc, Australian Catholic University (HBI‐BHR), The Stroke Foundation, and the Veterans Administration. Author disclosures are available in the supporting information.
CONSENT STATEMENT
ADNI study was approved by each ADNI study site's respective institutional review board and informed written consents were obtained from all participants.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This work was supported in part by the Intramural Research Program of the National Institute on Aging (NIA), and the Center for Alzheimer's and Related Dementias (CARD), within the Intramural Research Program of the National Institute on Aging and theNational Institute of Neurological Disorders and Stroke(AG000546). Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Data analysis for this study was partially funded by NIH/NIA U19AG024904.
Tosun D, Hausle Z, Iwaki H, et al. A cross‐sectional study of α‐synuclein seed amplification assay in Alzheimer's disease neuroimaging initiative: Prevalence and associations with Alzheimer's disease biomarkers and cognitive function. Alzheimer's Dement. 2024;20:5114–5131. 10.1002/alz.13858
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report.
Contributor Information
Duygu Tosun, Email: duygu.tosun@ucsf.edu.
Cornelis Blauwendraat, Email: cornelis.blauwendraat@nih.gov.
DATA AVAILABILITY STATEMENT
The ADNI data used in this study were obtained from the ADNI database (https://adni.loni.usc.edu). All ADNI data are shared without embargo through the LONI Image and Data Archive (IDA).
REFERENCES
- 1. Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person‐specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018;83:74‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kovacs GG, Milenkovic I, Wöhrer A, et al. Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‐based autopsy series. Acta Neuropathol. 2013;126:365‐384. PMID: 23900711. [DOI] [PubMed] [Google Scholar]
- 3. Concha‐Marambio L, Pritzkow S, Shahnawaz M, Farris CM, Soto C. Seed amplification assay for the detection of pathologic alpha‐synuclein aggregates in cerebrospinal fluid. Nat Protoc. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Siderowf A, Concha‐Marambio L, Lafontant DE, et al. Assessment of heterogeneity among participants in the Parkinson's progression markers initiative cohort using α‐synuclein seed amplification: a cross‐sectional study. Lancet Neurol. 2023;22:407‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arnold MR, Coughlin DG, Brumbach BH, et al. α‐synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological α‐synuclein in the context of co‐pathology and non‐LBD diagnoses. Ann Neurol. 2022;92:650‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pilotto A, Bongianni M, Tirloni C, Galli A, Padovani A, Zanusso G. CSF alpha‐synuclein aggregates by seed amplification and clinical presentation of AD. Alzheimer's Dement: J Alzheimer's Assoc. 2023;19:3754‐3759. [DOI] [PubMed] [Google Scholar]
- 7. Palmqvist S, Rossi M, Hall S, et al. Cognitive effects of Lewy body pathology in clinically unimpaired individuals. Nat Med. 2023;29:1971‐1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quadalti C, Palmqvist S, Hall S, et al. Clinical effects of Lewy body pathology in cognitively impaired individuals. Nat Med. 2023;29:1964‐1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89:88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimer's Dement: J Alzheimer's Assoc. 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jagust WJ, Landau SM, Koeppe RA, et al. The Alzheimer's disease neuroimaging initiative 2 PET Core: 2015. Alzheimer's Dement: J Alzheimer's Assoc. 2015;11:757‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Joshi AD, Pontecorvo MJ, Clark CM, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with alzheimer's disease and cognitively normal subjects. J Nucl Med. 2012;53:378‐384. [DOI] [PubMed] [Google Scholar]
- 13. Royse SK, Minhas DS, Lopresti BJ, et al. Validation of amyloid PET positivity thresholds in centiloids: a multisite PET study approach. Alzheimer's research & therapy. 2021;13:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol. 2019;18:1091‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nalls MA, Duran R, Lopez G, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013;70:727‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crane PK, Choi S‐E, Gibbons LE, et al. Cognitive assessments in ADNI: lessons learned from the ADNI psychometrics project. Alzheimer's & Dementia. 2021;17:e056474. [Google Scholar]
- 20. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2022. [DOI] [PubMed] [Google Scholar]
- 21. Brockmann K, Quadalti C, Lerche S, et al. Association between CSF alpha‐synuclein seeding activity and genetic status in Parkinson's disease and dementia with Lewy bodies. Acta Neuropathol Commun. 2021;9:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Markesbery WR, Jicha GA, Liu H, Schmitt FA. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol. 2009;68:816‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spina S, La Joie R, Petersen C, et al. Comorbid neuropathological diagnoses in early versus late‐onset Alzheimer's disease. Brain. 2021;144:2186‐2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi‐center longitudinal observational studies: the Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology. 2015;35:390‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lippa CF, Fujiwara H, Mann DM, et al. Lewy bodies contain altered alpha‐synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ. Antibodies to alpha‐synuclein detect Lewy bodies in many Down's syndrome brains with Alzheimer's disease. Ann Neurol. 1999;45:353‐357. [DOI] [PubMed] [Google Scholar]
- 27. Majbour NK, Chiasserini D, Vaikath NN, et al. Increased levels of CSF total but not oligomeric or phosphorylated forms of alpha‐synuclein in patients diagnosed with probable Alzheimer's disease. Sci Rep. 2017;7:40263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arezoumandan S, Cousins KAQ, Ohm DT, et al. Tau maturation in the clinicopathological spectrum of Lewy body and Alzheimer's disease. Annals of clinical and translational neurology. 2024. [DOI] [PMC free article] [PubMed]
- 29. Landau SM, Lee J, Murphy A, et al. Individuals with Alzheimer's disease and low tau burden: Characteristics and implications. Alzheimer's & dementia: the journal of the Alzheimer's Association. 2024. [DOI] [PMC free article] [PubMed]
- 30. Wilson RS, Yang J, Yu L, et al. Postmortem neurodegenerative markers and trajectories of decline in cognitive systems. Neurology. 2019;92:e831‐e840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boyle PA, Yang J, Yu L, et al. Varied effects of age‐related neuropathologies on the trajectory of late life cognitive decline. Brain. 2017;140:804‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson JL, Corrada MM, Kovacs GG, et al. Non‐Alzheimer's contributions to dementia and cognitive resilience in The 90+ Study. Acta Neuropathol. 2018;136:377‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsigelny IF, Crews L, Desplats P, et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases. PLoS One. 2008;3:e3135. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Dasari AKR, Kayed R, Wi S, Lim KH. Tau interacts with the C‐terminal region of α‐synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry. 2019;58:2814‐2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brenowitz WD, Hubbard RA, Keene CD, et al. Mixed neuropathologies and associations with domain‐specific cognitive decline. Neurology. 2017;89:1773‐1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nichols E, Merrick R, Hay SI, et al. The prevalence, correlation, and co‐occurrence of neuropathology in old age: harmonisation of 12 measures across six community‐based autopsy studies of dementia. Lancet Healthy Longev. 2023;4:e115‐e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
The ADNI data used in this study were obtained from the ADNI database (https://adni.loni.usc.edu). All ADNI data are shared without embargo through the LONI Image and Data Archive (IDA).