

Abstract
INTRODUCTION
More robust non‐human primate models of Alzheimer's disease (AD) will provide new opportunities to better understand the pathogenesis and progression of AD.
METHODS
We designed a CRISPR/Cas9 system to achieve precise genomic deletion of exon 9 in cynomolgus monkeys using two guide RNAs targeting the 3′ and 5′ intron sequences of PSEN1 exon 9. We performed biochemical, transcriptome, proteome, and biomarker analyses to characterize the cellular and molecular dysregulations of this non‐human primate model.
RESULTS
We observed early changes of AD‐related pathological proteins (cerebrospinal fluid Aβ42 and phosphorylated tau) in PSEN1 mutant (ie, PSEN1‐ΔE9) monkeys. Blood transcriptome and proteome profiling revealed early changes in inflammatory and immune molecules in juvenile PSEN1‐ΔE9 cynomolgus monkeys.
DISCUSSION
PSEN1 mutant cynomolgus monkeys recapitulate AD‐related pathological protein changes, and reveal early alterations in blood immune signaling. Thus, this model might mimic AD‐associated pathogenesis and has potential utility for developing early diagnostic and therapeutic interventions.
Highlights
A dual‐guide CRISPR/Cas9 system successfully mimics AD PSEN1‐ΔE9 mutation by genomic excision of exon 9.
PSEN1 mutant cynomolgus monkey‐derived fibroblasts exhibit disrupted PSEN1 endoproteolysis and increased Aβ secretion.
Blood transcriptome and proteome profiling implicate early inflammatory and immune molecular dysregulation in juvenile PSEN1 mutant cynomolgus monkeys.
Cerebrospinal fluid from juvenile PSEN1 mutant monkeys recapitulates early changes of AD‐related pathological proteins (increased Aβ42 and phosphorylated tau).
Keywords: Alzheimer's disease, amyloid β, cynomolgus monkey, inflammation and immune response, phosphorylated tau, plasma proteome, PSEN1
1. BACKGROUND
Alzheimer's disease (AD) is the most prevalent chronic neurodegenerative disorder among the elderly. 1 By 2050, an estimated 152.8 million people will suffer from dementia, 2 with AD accounting for approximately 50 to 70% of cases. 3 The most prominent pathological features in the brains of patients with AD are the deposition of insoluble amyloid beta (Aβ) peptides in senile plaques, the formation of neurofibrillary tangles composed of hyperphosphorylated tau protein, and neuroinflammation driven by activated microglia. 4 , 5 While sporadic AD is the most common form, some AD cases are inherited in an autosomal‐dominant manner, which are termed “familial AD.” Symptoms of familial AD typically manifest at an earlier age than sporadic AD. 6 Genetic studies have identified pathogenic mutations in the genes that encode amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2), which result in the overproduction of toxic Aβ peptides and, hence, familial AD. This forms the foundation of the “amyloid hypothesis,” which posits that the accumulation of Aβ triggers AD pathogenesis. 7 , 8
Animal models are crucial tools for studying human diseases and developing treatments. To understand the pathogenesis of AD, transgenic mice that express gene mutations in proteins associated with AD pathogenesis, such as APP, PSEN1, PSEN2, and tau, are widely used as animal models of AD. 9 , 10 Meanwhile, novel, humanized, knock‐in AD mouse models (eg, humanized APP and apolipoprotein E) are also being developed to better mimic the molecular pathways and pathophysiological changes associated with AD. 11 However, because of the substantial differences in the genetic background, brain structure, cognitive behavior, and immune system between humans and mice, the development of treatment methods based on mouse models remains unsatisfactory.
Compared to rodents, non‐human primates are more evolutionarily similar to humans, possessing similar brain structures and higher cognitive functions. 12 Meanwhile, the use of gene‐editing technologies in non‐human primates can recapitulate human brain disease mechanisms at the gene level, making them ideal animal models for studying neurological diseases such as autism. 13 , 14 , 15 Regarding AD, the development of non‐human primate models may provide new opportunities to better recapitulate the full spectrum of the disease, which would help characterize the dynamics of the molecular networks that underlie AD pathogenesis and develop potential early diagnostic and intervention strategies.
Current AD monkey models were established by injecting Aβ or tau protein into the brain or deleting PSEN1 exon 9 in marmosets using TALEN technology. 16 , 17 , 18 Previous gene‐edited marmoset models considered homozygous deletion of PSEN1 exon 9 to be lethal. 18 Marmosets generally exhibit Aβ deposits in the brain around 7 years of age and have several benefits, including small size, multiple births, and ease of feeding and operation. 19 However, marmosets do not have an immune response to phosphorylated tau, and only approximately 6% of animals survive to old age (ie, >7 years). 19 Meanwhile, Old World monkeys, such as cynomolgus monkeys (Macaca fascicularis), exhibit both Aβ deposition and tau accumulation and have a longer lifespan, making them more useful for observing the progress of AD during aging. 19
In this study, we generated a cynomolgus monkey model with familial AD mutation using CRISPR/Cas9 (ie, Streptococcus pyogenes Cas9 [SpCas9]) technology to delete PSEN1 exon 9 and confirmed the model's viability. Skin fibroblasts derived from PSEN1 mutant monkeys exhibited signatures of familial AD, including disrupted PSEN1 endoproteolysis as well as increased Aβ42 secretion and Aβ42/40 ratio. Blood transcriptome and proteome profiling suggests early alterations of key inflammatory and immune molecules in juvenile PSEN1 mutant monkeys. Meanwhile, early changes in AD‐related pathological proteins (increased cerebrospinal fluid [CSF] Aβ42 and p‐tau217) can be observed in juvenile PSEN1 mutant cynomolgus monkeys. Thus, our model might be a robust supplement to existing AD models, with broad potential applications for studying the mechanisms of AD as well as developing preclinical diagnostic and intervention strategies.
2. METHODS
2.1. Animals
This study utilized cynomolgus monkeys as donors of ovulation‐oocyst mother cells. The selected monkeys were 4 to 12 years old and weighed 3.2 to 5.8 kg. We performed embryo transplantation using genetic engineering techniques. 13 To ensure fertility, we chose three healthy male monkeys aged 5 to 11 years and weighing 6.7 to 9.3 kg as sperm donors. We conducted all animal experiments in strict accordance with institutional animal care and use guidelines, which were approved by the Institutional Animal Care and Use Committee (IACUC) of the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences (approved case ID: SIAT‐IACUC‐230111‐NS‐LZH‐A1094). We housed the monkeys in a temperature‐controlled facility maintained at 22 ± 1°C and 50 ± 5% relative humidity. A 12‐h light‐dark cycle was set with light illumination from 0700 h. We provided the monkeys with commercial monkey grains (Beijing Keao Xieli) twice daily and water ad libitum. We provided one treat of seasonal fruit daily. Experienced veterinarians carefully monitored the monkeys twice daily to evaluate and maintain their health status. The primate facility used in this study is accredited by AAALAC International.
RESEARCH IN CONTEXT
Systematic review: The authors evaluated the use of a non‐human primate model for studying familial Alzheimer's disease (AD). While there are reports of successful non‐human primate models of AD involving injection of pathological proteins into the brain, deletion of PSEN1 exon 9 in marmosets, and transgenic expression of mutant APP in monkeys, there is a need for a more accurate Old World monkey model that mimics disease progression in AD.
Interpretation: Our results suggest the successful generation of cynomolgus monkeys with familial AD mutation. Juvenile PSEN1 mutant cynomolgus monkeys recapitulate early changes of AD‐related pathological proteins (increased cerebrospinal fluid Aβ42 and phosphorylated tau). Blood transcriptome and proteome profiling revealed early changes of inflammatory and immune molecules in PSEN1 mutant monkeys.
Future directions: Future research should focus on expanding the cohort of PSEN1 mutant monkeys and longitudinally monitoring dynamic changes in key molecules (eg, inflammatory and immune molecules, Aβ and tau species), pathologies (eg, amyloid plaques and tau tangles), brain structures, and behaviors at different developmental stages. Such efforts will provide critical insights into the molecular mechanisms that underlie AD pathogenesis and progression, thereby facilitating the development of potential early diagnostic and therapeutic strategies.
2.2. Cas9 mRNA production, zygote injection, and embryo transplantation
To prepare CRISPR mRNAs for embryonic injection, we performed in vitro transcription as previously described. 13 Briefly, we linearized and utilized the SpCas9 plasmid as a template for in vitro transcription using an mMESSAGE mMACHINE T7 ULTRA kit (Life Technologies). We subsequently purified the resultant SpCas9 mRNA using a MEGA Clear kit (Life Technologies) and eluted it in RNase‐free water.
For gene editing, we performed laparoscopic procedures to obtain oocytes approximately 32 to 36 h after human chorionic gonadotropin administration. We sourced the oocytes from follicles 2 to 8 mm in diameter and cultured them in prematuration medium. We selected metaphase II oocytes for intracytoplasmic sperm injection and confirmed successful fertilization by the presence of two pronuclei. Seven to eight hours after sperm injection, we exposed the embryos to a mixture of mRNAs comprising 50 ng/µL SpCas9, 25 ng/µL guide RNA 1, and 25 ng/µL guide RNA 2. We cultured the resultant embryos in a cleavage medium. On day 3, we transferred them to the oviducts of stage‐matched recipient female monkeys. We employed ultrasonography 4 weeks after the transfer to assess the implantation success and pregnancy of the recipient.
2.3. Fibroblast culture
We obtained skin samples from the outer auricles using skin punches. We immediately placed the tissues in prechilled fibroblast medium (DMEM containing 15% FBS, 1% GlutaMAX, and 1% penicillin/streptomycin) and quickly transferred them to a tissue culture hood. We then sterilized the tissues by immersion in 75% ethanol for approximately 3 min, cut them into small pieces, and seeded them onto Matrigel‐coated plates. We subsequently covered the skin pieces with sterile coverslips and slowly added fibroblast medium. Spindle‐shaped fibroblasts typically appeared after incubation for approximately 4 to 7 days in a CO2 incubator at 37°C. When the primary cells reached approximately 90% confluence (typically after 10 to 15 days), we passaged the cells at a ratio of 1:3 to 1:6 using 0.125% trypsin/EDTA (marked as Passage 1). We further expanded and cryopreserved these fibroblast cells for subsequent experiments.
2.4. Genotyping of embryos and monkeys
To evaluate the editing efficiency of our gene‐editing strategy, we collected 17 embryos 3 days after injection. We performed whole‐genome amplification for embryos using a Discover‐sc Single Cell WGA Kit (Vazyme) according to the manufacturer's instructions. We subsequently performed polymerase chain reaction (PCR) to amplify fragments of approximately 800 bp from genomic DNA using primers targeting the sequences up‐ and downstream of PSEN1 exon 9. We then analyzed the fragments on a 1.5% agarose gel. The forward and reverse primer sequences for PCR were 5′‐TTGAATTATAATCACCATCTGAGGC‐3′ and 5′‐AGGTTCTTACTTTTACATGTTTGGA‐GAGAG −3′, respectively.
To analyze the exon 9 genotype of the PSEN1‐edited monkeys, we extracted genomic DNA separately from each monkey's umbilical cord, blood, and skin‐derived fibroblasts by using a QIAamp DNA Mini Kit (QIAGEN, 51306), Rapid Blood Genomic DNA Isolation Kit (Sangon Biotech, B518223), and QuickExtract DNA extraction solution (Epicenter, QE09050), respectively, according to the manufacturers’ instructions. Each genomic PCR (50 µL) contained 25 µL 2X KAPA HiFi HotStart ReadyMix (Roche, KK2602), a 200‐ to 500‐ng DNA template, and 20 pmol of each primer. The forward and reverse primer sequences were 5′‐ATTGCCTGCCTGGTTTCTGTTA‐3′ and 5′‐ACTCTGGCCATGTTAGACAAGAAGT‐3′, respectively. The annealing temperature was 62°C. After agarose gel electrophoresis, we photographed the PCR products using a Gel Dox XR+ (Bio‐Rad) and then submitted them to Sanger sequencing.
2.5. Gene expression analysis
We extracted total RNA from the fibroblasts using TRIzol. We reversed‐transcribed 500 to 1000 ng total RNA using Hifair III 1st Strand cDNA Synthesis SuperMix (YEASEN, 11141) according to the manufacturer's instructions. To genotype exon 9 in PSEN1 mRNA by RT‐PCR, we amplified amplicons spanning exons 8 to 10 using DreamTaq Green PCR Master Mix (Thermo Fisher Scientific, K1082). Each 25‐µL PCR contained 12.5 µL DreamTaq Green PCR Master Mix, 10 pmol forward and reverse primer each, and 0.2 µL cDNA template. The forward and reverse primer sequences were 5′‐GTGGCTGTTTTGTGTCCGAA‐3′ and 5′‐CAGCAAGGATGCTGCTGGAA‐3′, respectively. The annealing temperature was 58°C. We photographed the PCR products using a Gel Dox XR+ (Bio‐Rad) and validated them by Sanger sequencing.
To quantify the total PSEN1 mRNA in fibroblast using qRT‐PCR, we amplified amplicons spanning exons 4 and 5 of PSEN1 mRNA using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, 4367659) on a QuantStudio 7 Flex System. To quantify blood mRNA from 5‐month‐old control and mutant monkeys, we extracted total RNA using the PAXgene Blood miRNA Kit (Qiagen, 763134), and the reverse transcription and qRT‐PCR procedures were the same as for fibroblasts. We used ACTB as an internal control. The qRT‐PCR primers are shown in Table S1.
2.6. Off‐target analysis
To evaluate off‐target effects, we tested the genomes of the five mutant monkeys and their respective parents. The off‐target sites of the two single‐guide RNAs were predicted using CRISPOR (http://crispor.tefor.net/), and we designed corresponding primers for the top five potential sites (Table S2). We used DreamTaq Green PCR Master Mix for the first nine PCRs and Q5 DNA Polymerase (NEB, M0491) with GC Enhancer for the last one because of the high GC content in the target region. We then submitted the PCR products of the predicted sites to Sanger sequencing.
2.7. Biomarker measurement
When the fibroblasts reached ∼90% confluence, we replaced the media with fresh culture medium. After 48 h, we harvested the media and stored them at −80°C. We also collected protein lysates for the normalization of Aβ. We subsequently measured the Aβ concentrations in the media using a V‐PLEX Aβ Peptide Panel 1 (6E10) Kit (Mesoscale, K15200E‐2). CSF samples were collected from PSEN1 mutant and control monkeys at around 1.5 years of age. Briefly, CSF samples were withdrawn from the cistern magna under general anesthesia between 9:00 and 11:00 a.m. CSF samples were then centrifuged, and the supernatants were aliquoted and stored at −80°C until further analysis. We collected blood samples in EDTA tubes (BD Vacutainer) between 9:00 and 10:00 a.m. from 1.5‐year‐old control and mutant monkeys and centrifuged them at 1500 × g for 10 min at 4°C. We then collected, aliquoted, and stored the plasma at −80°C until further analysis. The levels of Aβ, p‐tau81, p‐tau217, total tau, NfL, and glial fibrillary acidic protein (GFAP) were measured by Mesoscale V‐PLEX Aβ Peptide Panel 1 (6E10) Kit (K15200E‐2), S‐PLEX NHP tau (pT181) Kit (K156AGMS‐1), S‐PLEX NHP tau (pT217) Kit (K156APFS‐1), and S‐PLEX Neurology Panel 1 (NHP) Kit (K15640S‐1), respectively, according to the instructions. It should be noted that these kits use human standards for standard curve plotting, which may have some effect on the calculated absolute values when we detect the corresponding monkey proteins.
2.8. Western blotting
We lysed fibroblasts with 2% SDS sample buffer on ice and transferred them to a 1.5‐mL Eppendorf tube for brief sonication as previously described. 20 We separated the protein samples by 8% SDS‐PAGE and transferred them to 0.22‐µm nitrocellulose membranes (at 0.2 A for 90 min). We blocked the membranes with 5% non‐fat dry milk in Tris‐buffered saline with 0.05% Tween20 (TBST). We then incubated the membranes with the following primary antibodies on a shaker overnight at 4°C: presenilin 1 (D39D1) antibody (CST, 5643S, 1:1000); anti‐presenilin 1 antibody (loop amino acids 275‐367), CT (MilliporeSigma, MAB5232, 1:500); anti‐presenilin 1 antibody (amino acids 21‐80), NT (MilliporeSigma, MAB1563, 1:500); purified anti‐presenilin 1 (N terminus) antibody (BioLegend, 823401, 1:500); N‐cadherin monoclonal antibody (Thermo Fisher Scientific, 33‐3900, 1:1000); and α‐tubulin antibody (Sigma, T6199, 1:1000). After washing three times in TBST for 10 min, we incubated the membranes for 1 h with anti‐mouse/rabbit/rat IgG, HRP‐linked antibody (1:2000) at room temperature, rinsed them three times with TBST for 10 min, and developed them using an ECL reagent (SuperSignal West Pico Plus Chemiluminescent Substrate, Thermo Fisher Scientific).
2.9. Whole‐genome sequencing
For whole‐genome sequencing, we collected the ear skin tissues of five mutant monkeys and their parents using skin punches. Novogene performed genomic DNA extraction, DNA library preparation (NEBNext Ultra DNA Library Prep Kit for Illumina, NEB), and sequencing (Illumina NovaSeq platform with the PE150 strategy). We obtained approximately 60× coverage for whole‐genome sequencing of raw data and filtered the data by using an in‐house script to remove paired reads that met the following criteria: (a) either read contains >10 nucleotide adapter sequences; (b) either read contains >15 N bases; and (c) either read contains >75 low‐quality bases (Phred quality < 5). Next, we mapped the high‐quality reads to the reference genome of Macaca_fascicularis_5.0 (GCF_000364345.1) with Burrows‐Wheeler Aligner (BWA) software 21 and removed the dislodged duplication with SAMtools 22 and Picard software (https://github.com/broadinstitute/picard). Finally, we performed single‐nucleotide polymorphism and insertion and deletion calling with SAMtools software (parameters: ‐C 50 ‐ mpileup ‐m2 ‐F 0.002 ‐d 1000), retained the variations with mapping quality >20 and depth >4×, and performed functional annotation using ANNOVAR software. 23
2.10. RNA sequencing
We collected blood samples (1 to 2 mL) from 5‐month‐old control and mutant monkeys into PAXgene RNA tubes. Novogene performed total RNA extraction, removal of globin mRNA (GLOBINclear‐Human Kit, Invitrogen), cDNA library preparation (NEBNext Ultra RNA Library Prep Kit for Illumina, NEB), and sequencing (Illumina NovaSeq platform with the PE150 strategy). Similar to the whole‐genome sequencing data filtration, we removed the paired reads containing adapter, poly‐N, and low‐quality reads from the raw data. We then mapped the high‐quality reads to the reference genome of Macaca fascicularis with Hisat2 software, predicted the novel transcripts with StringTie software, and quantified the gene expression level with featureCount software. Finally, we normalized gene expression among all samples using the edgeR package in R, 24 performed differential expression analysis with the DESeq2 package in R, and detected the enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) categories for the differentially expressed genes with the clusterProfiler package in R. 25
2.11. Plasma protein measurement
We collected blood samples (approximately 1 mL) from 4‐month‐old control and mutant monkeys in EDTA tubes (BD Vacutainer) and centrifuged them at 1500 × g for 10 min at 4°C. We then collected, aliquoted, and stored the plasma at −80°C. The levels of 1162 plasma proteins were quantified by Olink Proteomics using proximity extension assay. The following 13 Olink target 96 panels were measured: Neurology (91501), Neuro exploratory (91502), Inflammation (91301), Oncology II (91402), Oncology III (91403), Cardiovascular II (91202), Cardiovascular III (91203), Immune response (91701), Cardiometabolic (91802), Development (91703), Metabolism (91801), Cell regulation (91702), and Organ damage (91901). Detected plasma protein levels were presented in normalized protein expression units and were further analyzed as previously described. 26 For validation of plasma proteins, we used the enzyme‐linked immunosorbent assay (ELISA) to detect CD160 (CUSABIO, CSB‐EL004881HU) and C1QA (CUSABIO, CSB‐EL003637HU) in the plasma of 1.5‐year‐old monkeys.
2.12. Statistical analysis
We used GraphPad Prism 6 for statistical analysis and data visualization. We analyzed the data from qRT‐PCR, Western blot, and Aβ measurement experiments using unpaired Student's t tests for comparisons between two groups.
We used t tests to detect the differentially expressed genes between groups, adjusted the multiple test results with the false discovery rate method, and plotted heatmaps of the differentially expressed genes with the pheatmap package in R. We calculated the fold changes of the gene expression among the groups, retained the genes with p < .05, and plotted the volcano plot with the ggplot2 package in R (|log2(fold change)| > 0.585 and p adj < .05). To present the KEGG enrichment results, we used the ggplot2 package in R. 27 We used STRING software to predict the corresponding proteins for the differentially expressed genes, calculated the interactions among the proteins, and assembled the protein network. 28
3. RESULTS
3.1. CRISPR/Cas9‐mediated genome editing in cynomolgus monkey embryos
We utilized CRISPR/Cas9, a highly precise and efficient tool for targeted genome manipulation, to generate a novel monkey model harboring PSEN1 exon 9 deletion (PSEN1‐ΔE9). To achieve this, we designed a dual‐guide CRISPR/Cas9 system to flank the target exon with distances of 270 and 211 bp (Figure 1A). We detected PCR products lacking exon 9 at approximately 250 bp, while amplicons with exon 9 appeared at approximately 800 bp. Remarkably, our analysis revealed the occurrence of nine homozygous and four heterozygous mutants out of the 17 tested embryos (Figure S1A), highlighting the superb efficiency of the dual‐guide CRISPR/Cas9 system for mediating targeted gene editing in monkey embryos.
FIGURE 1.
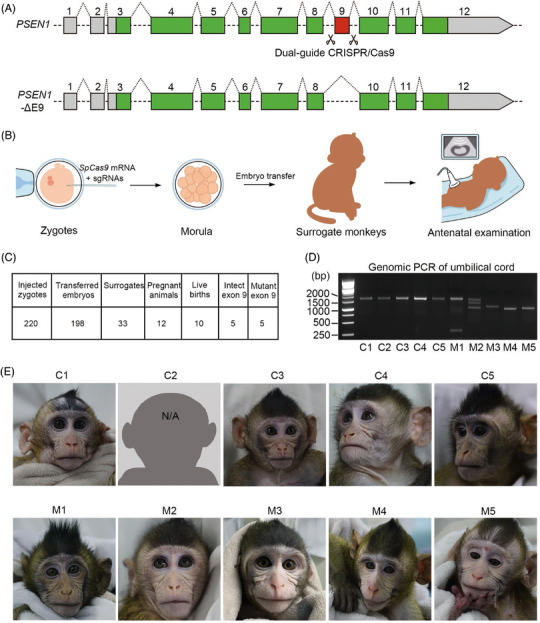
Generation of cynomolgus monkeys with PSEN1 exon 9 deletion. (A) Schematic representation of dual‐guide CRISPR/Cas9 system used to generate cynomolgus monkeys (Macaca fascicularis) with PSEN1 exon 9 deletion (PSEN1‐ΔE9). The exonic coding sequences in PSEN1 mRNA are indicated in green and red. Excision of exon 9 using two guide RNAs (ie, sgRNA1 and sgRNA2, with scissor icons indicating cutting sites) located in the flanking introns results in exon 9 being deleted from the genome. (B) Schematic representation showing generation of PSEN1‐ΔE9 cynomolgus monkeys. (C) Summary of animal generation. (D) Genomic PCR of PSEN1 exon 9 from cynomolgus monkey umbilical cord. (E) Photo of founder PSEN1‐ΔE9 monkeys. Monkey C2 died at 5.5 months of age.
3.2. Generation of cynomolgus monkeys with PSEN1‐ΔE9 mutation
To generate PSEN1‐ΔE9 cynomolgus monkeys, we mixed SpCas9 mRNA with the two single‐guide RNAs and injected them into metaphase II zygotes (Figure 1B). Following zygote injection, we successfully transferred 198 out of 220 injected zygotes to 33 surrogate females. Twelve pregnancies were established, for an overall success rate of 36.36% (Figure 1C). Four of these pregnancies were lost after embryo transfer. Ultimately, eight pregnancies were successfully carried to full term (∼155 days), and 10 live births were delivered via natural birth or cesarean section (Figure 1C). Genotyping of multiple tissues identified five PSEN1‐ΔE9 mutant monkeys (Figures 1D,E, S1B), which tended to gain weight faster than those without the mutation (Figure S1C–S1E).
3.3. Off‐target analysis of PSEN1 mutant cynomolgus monkeys
We amplified the top five potential off‐target sites of each guide RNA from the genomic DNA of the umbilical cord of five PSEN1‐ΔE9 monkeys, and no off‐target effects were observed at these predicted sites (Table S3). We also performed whole‐genome sequencing on these five PSEN1‐ΔE9 monkeys and their respective parents to systematically assess genome‐wide off‐target effects. Compared to the Macaca_fascicularis_5.0 reference genome, the total variants of PSEN1‐ΔE9 monkeys and their parents ranged from 16,307,815 to 16,529,540 (Figure S2A–S2E). More specifically, 312,199 to 342,881 de novo variants were specific to PSEN1‐ΔE9 monkeys compared to their corresponding parents (Figure S2A–S2E), which is comparable to that in a recently reported PSEN1 mutant marmoset model. 18 Venn diagram analysis revealed that only 78 PSEN1‐ΔE9 monkey‐specific variants were shared by all mutant monkeys, whereas no variants were located in coding regions (ie, exons) (Figure S2F, S2G). These results indicate that our dual‐guide CRISPR/Cas9 system for PSEN1 exon 9 skipping is highly specific and has minimal off‐target effects in PSEN1 mutant cynomolgus monkeys.
3.4. Precise exon 9 deletion in PSEN1‐ΔE9 cynomolgus monkey‐derived fibroblasts
To evaluate the functional outcome of PSEN1‐ΔE9 mutation in cynomolgus monkeys after genome editing, we cultured primary fibroblast lines from the ear lobes of control and PSEN1‐ΔE9 cynomolgus monkeys (Figure 2A). We first validated the precise exclusion of exon 9 from PSEN1 mRNA in cynomolgus monkey fibroblasts using RT‐PCR. The exon 9 genotypes at the mRNA level suggested that two heterozygous and three homozygous PSEN1‐ΔE9 cynomolgus monkeys were generated, indicating that the exon 9 genotypes at the mRNA level were consistent with the genomic genotype (Figures 2B, S3A, S3B; Table 1). Of note, one of the three homozygous PSEN1‐ΔE9 monkeys, M5, carried two exon 10 copies at one of the two PSEN1‐ΔE9 alleles (Figure 2B); further confirmation is required to determine the functional effects of this alteration. Quantification of PSEN1 mRNA by qRT‐PCR suggested no significant effect of exon 9 deletion on total PSEN1 mRNA expression in cynomolgus monkey fibroblasts (Figure 2C).
FIGURE 2.
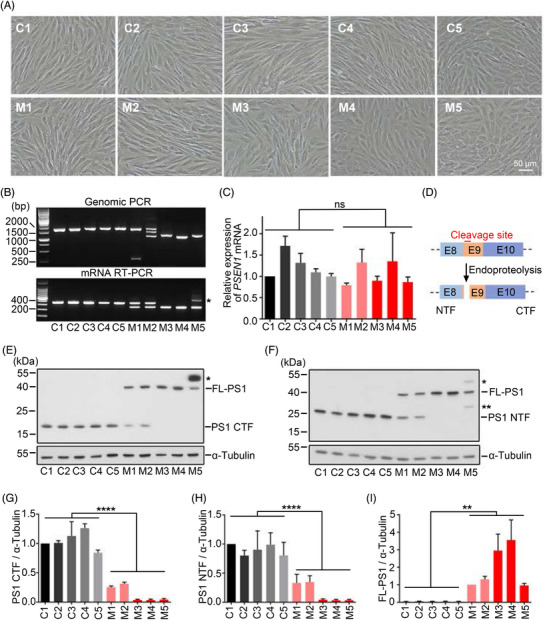
Precise deletion of exon 9 disrupts PSEN1 endoproteolysis in cynomolgus monkey‐derived fibroblasts. (A) Representative images of fibroblast lines cultured from cynomolgus monkey ear skin tissue. Scale bar: 50 µm. (B) PCR genotyping of PSEN1 exon 9 from cynomolgus monkey fibroblasts at genome and RNA levels. Upper: DNA gel of PSEN1 genomic PCR products from cynomolgus monkey‐derived fibroblasts. Lower: DNA gel of PSEN1 mRNA RT‐PCR products from cynomolgus monkey fibroblasts. Monkey M5 carries two copies of exon 10 at one of the two PSEN1‐ΔE9 alleles; its gel electrophoresis band is larger than the other lanes (asterisk). (C) Quantification of total PSEN1 mRNA levels by qRT‐PCR in cynomolgus monkey‐derived fibroblasts. Data are normalized to those of fibroblasts from monkey C1 (n = 4 independent experiments). (D) Schematic diagram of PSEN1 endoproteolysis site encoded by exon 9. (E) Western blot analysis of PSEN1 protein from fibroblast lysates detected by PSEN1 C‐terminal antibody (CST, 5643S). FL‐PS1, uncleaved full‐length PSEN1 with exon 9 deletion; PS1 CTF, C‐terminal fragment by endoproteolysis of PSEN1. Asterisk indicates uncleaved full‐length PSEN1 with exon 9 deletion and exon 10 duplication. (F) Western blot analysis of PSEN1 protein from fibroblasts lysate detected by PSEN1 N‐terminal antibody (Millipore, MAB1563). PS1 NTF, N‐terminal fragment by endoproteolysis of PSEN1. Single asterisk indicates uncleaved full‐length PSEN1 with exon 9 deletion and exon 10 duplication. Double asterisk indicates an uncharacterized protein band. (G) Quantification of PS1 CTF with reference to tubulin normalized to levels of C1 fibroblasts in (E) (n = 3 independent experiments). (H) Quantification of PS1 NTF with reference to tubulin and normalized to levels of C1 fibroblasts in (F) (n = 3 independent experiments). (I) Quantification of FL‐PS1 with reference to tubulin and normalized to levels of M1 fibroblasts in (F) (n = 3 independent experiments). Error bars indicate SEM (**p < .01, ****p < .0001 vs. control; n.s., not significant; unpaired t test).
TABLE 1.
Summary of PSEN1 exon9‐edited cynomolgus monkeys in this study.
| Animal code | Date of birth (y/m/d) | Gender | Exon 9 genotype | ΔExon 9 in mRNA |
|---|---|---|---|---|
| C1 | 2022/07/18 | M | Wild type | – |
| C2 a | 2022/07/25 | F | Wild type | – |
| C3 | 2022/07/25 | F | Wild type | – |
| C4 | 2022/08/17 | M | Wild type | – |
| C5 | 2022/08/17 | F | Wild type | – |
| M1 | 2022/08/15 | M | Mutant | 45.2 ± 1.3% |
| M2 | 2022/08/31 | F | Mutant | 51.6 ± 3.2% |
| M3 | 2022/10/13 | M | Mutant | ∼ 100% |
| M4 | 2022/08/17 | F | Mutant | ∼ 100% |
| M5 b | 2022/08/19 | M | Mutant | ∼ 100% |
Note: The percentage of ΔExon 9 in mRNA was calculated as the ratio of the normalized band intensity between RT‐PCR amplicons without and with exon 9 in cynomolgus monkey‐derived fibroblasts. Data were presented as means ± SD from three independent experiments.
Abbreviations: F, female.; M, male; y/m/d, year/month/day.
C2 and C3: same biological parents and same surrogate monkey.
C4 and C5: same biological parents and same surrogate monkey.
M4 and M5: same biological parents and different surrogate monkeys.
Died of natural cause.
Exon 10 duplication in one of the two ΔExon 9 alleles.
Exon 9 encodes the main endoproteolysis site of PSEN1 protein, which can be cleaved into C‐ and N‐terminal fragments by an undefined mechanism(s) (Figure 2D). PSEN1‐ΔE9 mutations are reported to abolish PSEN1 protein cleavage. 29 , 30 Western blot analysis of cynomolgus monkey fibroblasts with C‐ and N‐terminal antibodies against PSEN1 showed that full‐length PSEN1 underwent proteolytic cleavage while the absence of exon 9 abrogated this biological event, resulting in fewer C‐ and N‐terminal fragments and increased full‐length PSEN1 production with dose‐dependent effects (Figure 2E–I). These effects were corroborated by other C‐ and N‐terminal antibodies against PSEN1 (Figure S3C, S3D). Moreover, these PSEN1‐ΔE9‐induced changes in PSEN1 protein expression are consistent with previous studies involving induced pluripotent stem cells and marmoset models. 18 , 31 Thus, our analysis of cynomolgus monkey‐derived fibroblasts indicates that our dual‐guide CRISPR/Cas9‐mediated genomic excision of exon 9 successfully mimics AD‐related PSEN1‐ΔE9 mutations.
3.5. PSEN1‐ΔE9 cynomolgus monkey‐derived fibroblasts exhibit an elevated secreted Aβ42/40 ratio
To further investigate the downstream biological effects of PSEN1‐ΔE9 mutation on the key pathological features of AD, we examined the protein substrates of the γ‐secretase complex, for which PSEN1 serves as a core catalytic subunit. 32 , 33 , 34 APP undergoes sequential cleavage by β‐secretase and γ‐secretase to produce Aβ. 4 Compared to culture media from control fibroblasts, we observed increased Aβ42 secretion and Aβ42/40 ratio in PSEN1‐ΔE9 fibroblast culture media; meanwhile, Aβ40 levels were not significantly affected (Figure 3A–C). In addition, the Aβ42/40 ratio was strongly correlated with the degree of exon 9 deletion, further suggesting the presence of exon 9 genotype‐dependent core AD phenotypic changes (Figure 3D). These results are also similar to observations in PSEN1‐ΔE9 marmosets and isogenic human stem cells. 18 , 31 Taken together, these results indicate the successful generation of pathogenic PSEN1‐ΔE9 mutant cynomolgus monkeys that recapitulate AD‐associated amyloid pathology.
FIGURE 3.
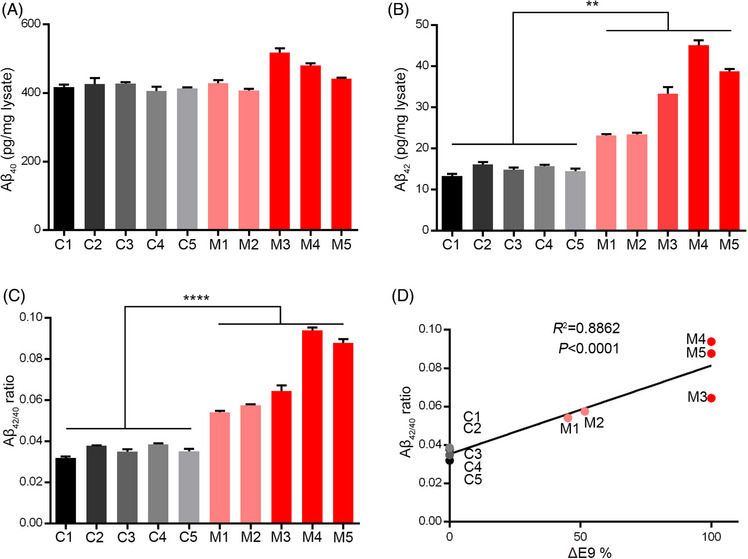
Increased Aβ42/40 ratio in PSEN1‐ΔE9 cynomolgus monkey‐derived fibroblasts. (A–C) Aβ40 level (A), Aβ42 level (B), and Aβ42/40 ratio (C) in culture media from cynomolgus monkey‐derived fibroblasts (n = 3 wells per condition; **p < .01, ****p < .0001 vs. control; unpaired t test). Error bars show SEM. (D) Correlations between Aβ42/40 ratio in fibroblast culture media and percentages of exon 9 deletion in PSEN1 mRNA.
3.6. Blood transcriptome analysis of PSEN1‐ΔE9 cynomolgus monkeys
Given the widespread protein expression of PSEN1 and substrate diversity (∼150 reported membrane protein substrates 34 ) of γ‐secretase – of which PSEN1 is a key subunit – early molecular changes may have occurred in young PSEN1‐ΔE9 monkeys. Therefore, we performed blood transcriptome analysis on 5‐month‐old control and PSEN1‐ΔE9 monkeys to evaluate the potential effects of PSEN1‐ΔE9 on young monkeys (Figure 4A; Table S4). Compared to the control monkeys, there were 943 and 325 down‐ and upregulated genes, respectively, in homozygous PSEN1‐ΔE9 monkeys (p < .05; Figure 4A,B). In addition, the downregulated genes are enriched in KEGG pathways related to primary immunodeficiency and intestinal immune network pathway, whereas the upregulated genes are enriched in pathways associated with immune response, type 1 diabetes, cell adhesion molecules, and endocytosis (Figures 4C,D, S4A, S4B). These results suggest that familial AD mutations may lead to widespread immune signal dysregulation at a very early stage. Of note, diabetes mellitus is strongly associated with the risk of AD, such that AD has been referred to as “type 3 diabetes.” 35 , 36 , 37 The enrichment of KEGG pathways related to type 1 diabetes due to PSEN1‐ΔE9 in this monkey model implicates a potential shared mechanism between diabetes and AD. Furthermore, STRING network analysis of the down‐ and upregulated genes depicts a potential core function of Proto‐oncogene tyrosine‐protein kinase Src and chemokine ligands and receptors (eg, CCL2, CCL3, CCL4, and CCR3) in PSEN1‐ΔE9‐mediated cellular dysregulation (Figure 4E,F). Notably, STRING network analyses also indicated that the Aβ functional receptor FPR2 38 , 39 may be an important node protein that interacts with the core chemokine ligands and receptors (Figure 4F). The increased expression of blood FPR2, chemokine ligands, and receptors in PSEN1‐ΔE9 monkeys was validated by qRT‐PCR (Figure S4C) and may be an indicator of the enhanced blood toxic effects of amyloidogenic peptides.
FIGURE 4.
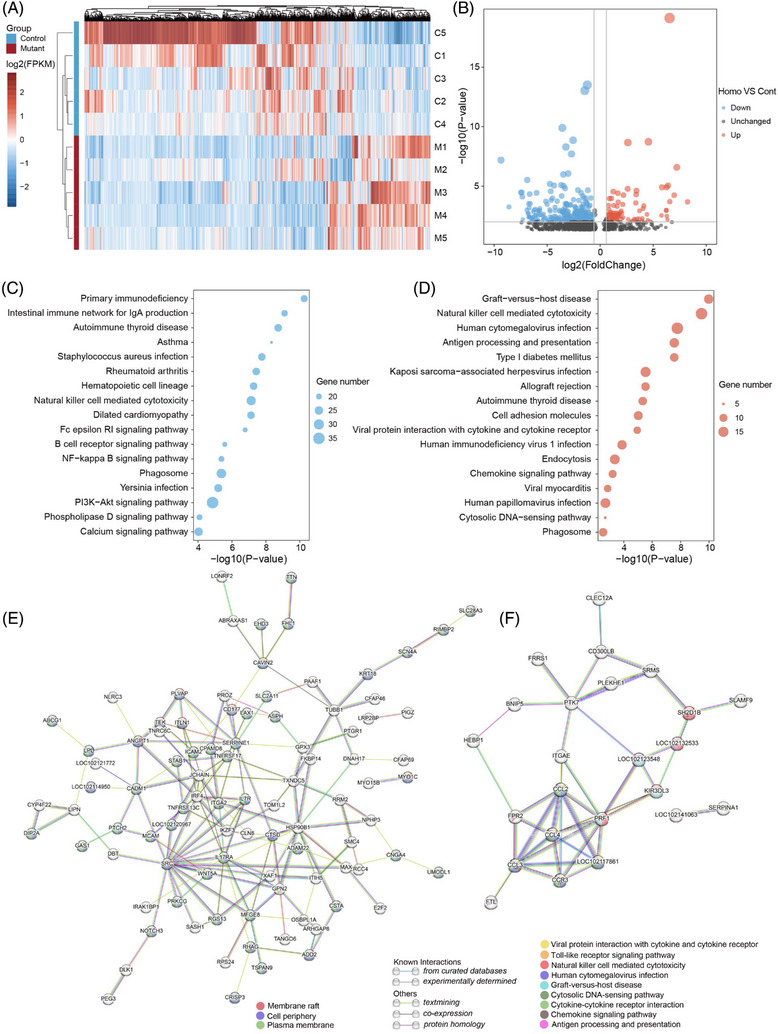
Dysregulated immune pathways in blood transcriptome of PSEN1‐ΔE9 cynomolgus monkeys. (A) Heatmap of differentially expressed genes (DEGs) (p < .05) from blood RNA sequencing of monkeys with PSEN1 exon 9 deletion (PSEN1‐ΔE9, M1 to M5) and controls (C1 to C5). (B) Volcano plot showing DEGs between homozygous PSEN1‐ΔE9 mutant monkeys (M3 to M5) and controls (C1 to C5). Horizontal gray line represents −log10(0.01); vertical gray lines represent log2(0.67) and log2(1.5), respectively. (C, D) KEGG pathway analysis of downregulated (C) and upregulated (D) genes between homozygous PSEN1‐ΔE9 mutant monkeys (M3 to M5) and controls (C1 to C5). (E, F) STRING assembly for functional network analysis of downregulated (E) and upregulated (F) DEGs between homozygous PSEN1‐ΔE9 mutant monkeys (M3 to M5) and controls (C1 to C5). In the networks, downregulated DEGs (p < .01) and upregulated DEGS (p < .01, fold change > 2) are shown. Nodes and edges represent predicted proteins and interactions, respectively.
3.7. Characteristics of plasma proteome in PSEN1‐ΔE9 cynomolgus monkeys
To reveal potential early blood proteomic changes in PSEN1‐ΔE9 monkeys, we performed plasma proteome profiling on the 4‐month‐old control and PSEN1‐ΔE9 monkeys by high‐throughput proximity extension assay. Among the 1162 plasma proteins examined, 144 were differentially expressed in PSEN1‐ΔE9 monkeys compared with the control group (p < .05) – the expressions of 127 and 17 of which were up‐ and downregulated, respectively (Figure 5A). Moreover, the blood RNA levels of eight of the 127 upregulated plasma proteins were also upregulated (Figure S5A). Cell‐type enrichment analysis showed that approximately 86.8% and 68.06% of the 144 plasma proteins were expressed in peripheral blood cells and endothelial cells, respectively (Figure S5B). Biological function classification unveiled the involvement of plasma proteins in the Inflammation, Oncology, Cardiovascular diseases, and Neurology panels (Figure S5C). To gain further insights into the functional significance of these plasma proteins, we employed Gene Ontology (GO) annotation, which revealed prominent enrichment of upregulated proteins in functional categories associated with inflammatory and immune responses as well as adhesion molecule‐related biological processes (Figure 5B). Indeed, inflammatory and immune response‐associated molecules, such as CD160, CXCL10, MCP‐2, CXCL9, CLEC4G, and CLEC10A, were among the top 20 differentially expressed plasma proteins in PSEN1‐ΔE9 cynomolgus monkeys (Figure 5C; Tables 2, S5). Of note, elevated blood levels of CXCL9 and CXCL10 are strongly associated with AD. 40 In addition, activation of CXCL9/CXCL10–CXCR3 signaling promotes both plaque formation and behavioral deficits in AD mouse models 41 and modulates CD8+ T‐cell infiltration and neurodegeneration in an AD neuroimmune axis model. 42 Meanwhile, several other differentially expressed plasma proteins associated with inflammation and immune response, including IL12RB1, C1QA, IL‐17A, and IL3RA, are also reported to be closely associated with AD pathogenesis (Table S5), suggesting that this non‐human primate model may develop the early molecular changes of inflammatory and immune signaling similar to that in AD pathogenesis. Meanwhile, cell adhesion molecules, such as NINJ1, CYR61, PTPRM, and PECAM1, are another functional feature of the top 40 differentially expressed plasma proteins in PSEN1‐ΔE9 cynomolgus monkeys (Figure 5D; Tables 2, S5). It is also noteworthy that cell adhesion molecules may be intimately associated with inflammatory and immune responses. 43 For example, the plasma adhesion molecules NINJ1 and PECAM1 are reported to promote the trans‐endothelial migration of immune cells under inflammatory conditions. 44 , 45 We also observed perturbed blood expression of the γ‐secretase and APP downstream molecules, CNTNAP2 and WASF1, 46 , 47 in PSEN1‐ΔE9 monkeys (Figure 5E); to some extent, this may reflect the altered activity of γ‐secretase containing mutant PSEN1. Selected immune‐associated plasma proteins (CD160 and C1QA) were further validated by ELISA in PSEN1 mutant monkeys (Figure S5D, S5E). Taken together, profiling of the plasma proteome of suggests that juvenile PSEN1‐ΔE9 mutant monkeys may begin to develop abnormal peripheral inflammatory and immune signaling.
FIGURE 5.
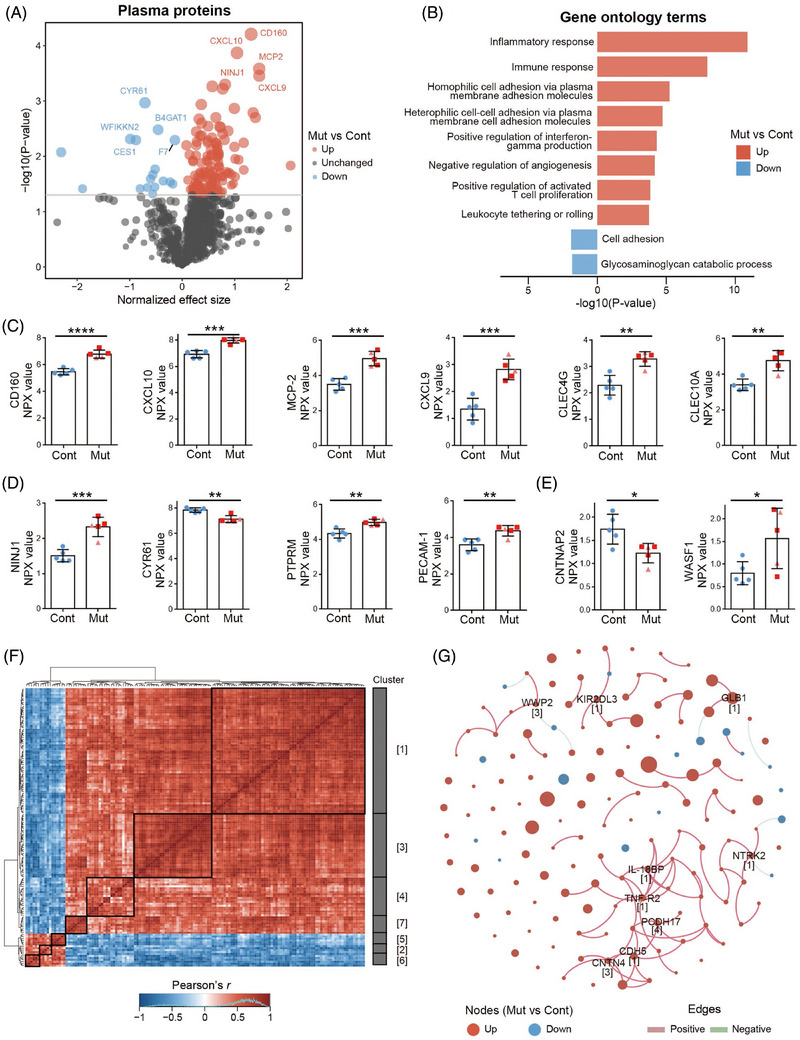
Early plasma inflammatory and immune molecular dysregulation in PSEN1‐ΔE9 cynomolgus monkeys. (A) Volcano plot of plasma proteome in mutant monkeys with PSEN1 exon 9 deletion (PSEN1‐ΔE9, M1 to M5) and controls (C1 to C5). The top five down‐ and upregulated plasma proteins are labeled. (B) Gene Ontology (GO) terms of differentially expressed plasma proteins between mutant and control monkeys. (C–E) Representative inflammatory and immune‐related (C), cell adhesion‐related (D), and γ‐secretase‐ and APP‐related (E) differentially expressed plasma proteins in PSEN1‐ΔE9 cynomolgus monkeys. Blue dots, light red triangles, and red squares indicate control monkeys, heterozygous PSEN1‐ΔE9 mutant monkeys, and homozygous PSEN1‐ΔE9 mutant monkeys, respectively. Error bars indicate SD (*p < .05, **p < .01, ***p < .001, ****p < .0001 vs. control; unpaired t test). (F) Correlation heatmap of differentially expressed plasma proteins. Each row and column represent a differentially expressed plasma protein in the mutant monkeys. Red and blue indicate positive and negative correlations between protein pairs, respectively. (G) Correlation network of the differentially expressed plasma proteins. Each node represents a protein, and node size is proportional to the p value (log10 scale). Pink and green edges indicate positive and negative relationships between proteins, respectively. Hub proteins and their corresponding clusters are labeled.
TABLE 2.
Top 10 significant differentially expressed plasma proteins between PSEN1‐ΔE9 and control cynomolgus monkeys.
| Protein | Description | Effect size | p |
|---|---|---|---|
| CD160 | Cell surface receptors on NK and T cells | 1.31335 | 0.00006 |
| CXCL10 | C‐X‐C motif chemokine ligand | 1.04437 | 0.00013 |
| MCP‐2 | C‐C motif chemokine ligand | 1.46795 | 0.00026 |
| CXCL9 | C‐X‐C motif chemokine ligand | 1.46890 | 0.00035 |
| NINJ1 | Homophilic transmembrane adhesion molecule | 0.81718 | 0.00050 |
| GLB1 | Beta‐galactosidase catalyzing degradation of galactosylceramide | 0.57322 | 0.00054 |
| FURIN | Endoprotease involved in cerebral amyloid angiopathy | 0.77216 | 0.00060 |
| CYR61 | Secreted protein that promotes adhesion of endothelial cells | −0.70882 | 0.00108 |
| LEPR | Leptin receptor involved in fat metabolism | 0.36351 | 0.00116 |
| CLEC4G | C‐type lectin involved in immune response | 0.99321 | 0.00136 |
Note: NPX values of plasma proteins of PSEN1‐ΔE9 and control monkeys were compared by t tests and differentially expressed plasma proteins were ranked by p value.
Subsequent investigation of the intercorrelations among the PSEN1‐ΔE9 monkey‐associated proteins yielded seven co‐regulated clusters (Figure 5F). Within the co‐regulation network, we further identified nine hub proteins, including TNF‐R2, WWP2, and NTRK2, which play critical roles in maintaining network stability (Figure 5G). Notably, six, two, and one of these hub proteins belonged to clusters 1, 3, and 4, respectively, suggesting their significance in representing alterations of the plasma proteome in the PSEN1‐ΔE9 monkeys (Figure 5G). Furthermore, we explored the plasma proteins that exhibited similar regulatory patterns in both PSEN1‐ΔE9 monkeys and human patients with AD. 26 We identified six proteins (C1QA, CSF‐1, DPEP1, FGF‐5, SPINK4, and VCAM1) that were upregulated in both PSEN1‐ΔE9 monkeys and patients with AD (Figure S5F), which were associated with the functional categories of aging (p = .022) and inflammatory response (p = .064) (Figure S5F). Thus, the characteristics of plasma proteins in the PSEN1‐ΔE9 monkey model may indicate the early features of plasma protein changes in patients with AD, highlighting the value and potential of this non‐human primate model for disease modeling and translational research on AD.
3.8. Characteristics of AD‐related pathological proteins in PSEN1‐ΔE9 cynomolgus monkeys
Fluid biomarkers (eg, AD biomarkers Aβ and p‐tau) have the potential to diagnose or predict the likelihood of developing AD. Cross‐sectional analyses of autosomal‐dominant AD mutation carriers suggest a higher CSF Aβ42 level in the very early disease process (estimated to be approximately 20 to 30 years prior to the onset of symptoms) than non‐carriers and then, as the disease progresses, significantly lower than non‐carriers, about 10 to 20 years before symptom onset. 48 , 49 , 50 To confirm whether PSEN1 mutant monkeys recapitulated key early features associated with AD, we examined AD‐ and neurodegeneration‐related biomarkers in the CSF of PSEN1‐ΔE9 monkeys. We found that Aβ42 was significantly elevated in homozygous PSEN1‐ΔE9 monkeys, whereas the Aβ42/40 ratio showed a ΔE9 genotype‐dependent increase in PSEN1 mutant monkeys (Figure 6A–C). For tau biomarkers, it has been estimated that in human studies, increases in p‐tau217 and p‐tau181 (p‐tau/total tau ratio) precede insoluble tau aggregates (tau positron emission tomography [PET] positivity) by several years, even before the threshold for amyloid PET positivity. 51 , 52 , 53 We found that CSF from homozygous PSEN1‐ΔE9 monkeys exhibited a significantly elevated p‐tau217/total tau ratio (p = .044), while p‐tau181/total tau (p = .054) also tended to be elevated (Figure 6D–I). It appears that p‐tau/total tau trends in PSEN1 mutant monkeys match with the changes of CSF Aβ42/40 ratios (Figure 6C,F,I), supporting the notion that the CSF p‐tau217 and p‐tau181 may begin to increase almost simultaneously with the initial Aβ changes. 51 , 52 Meanwhile, total tau, neurofilament light (NfL), and GFAP, biomarkers associated with neurodegeneration and inflammation, were less affected in CSF at this age (Figure 6J–L).
FIGURE 6.
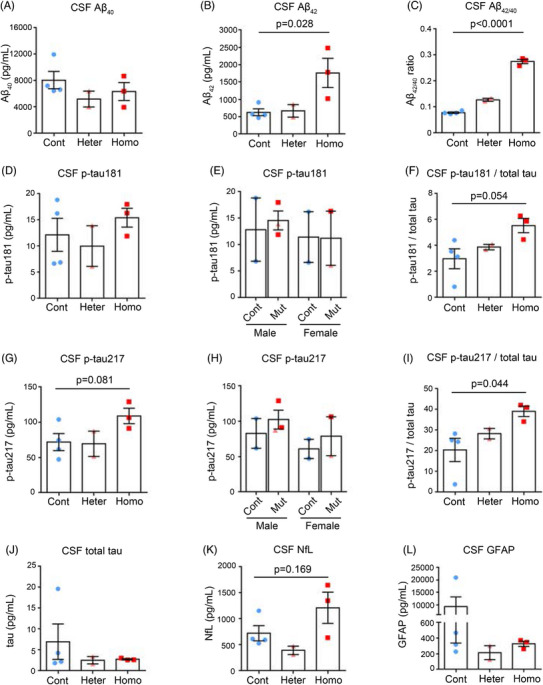
Characteristics of AD‐related pathological proteins in cerebrospinal fluid of PSEN1‐ΔE9 cynomolgus monkeys. (A–C) CSF Aβ40 level (A), Aβ42 level (B), and Aβ42/40 ratio (C) from 1.5‐year‐old cynomolgus monkeys. (D–F) CSF p‐tau181 level (D), CSF p‐tau181 level plotted by sex (E), and CSF p‐tau181/total tau (F) from cynomolgus monkeys. (G–I) CSF p‐tau217 level (G), CSF p‐tau217 level plotted by sex (H), and CSF p‐tau217/total tau (I) from cynomolgus monkeys. (J–L) CSF total tau level (J), CSF NfL level (K), and CSF GFAP level (L) from cynomolgus monkeys. Cont, control monkeys; Heter, heterozygous PSEN1‐ΔE9 monkeys; Homo, homozygous PSEN1‐ΔE9 monkeys. Unpaired t test. Error bars show SEM.
We also assessed plasma samples from PSEN1‐ΔE9 monkeys for tau species, NfL, and GFAP biomarkers. In PSEN1‐ΔE9 monkeys, the overall pattern of plasma AD‐related pathological protein changes was similar to that in CSF, but to a lesser extent, and females appeared to be relatively more affected, which needs to be further confirmed due to the small sample size (Figure S6A–S6I). Taken together, early changes in AD‐related pathological proteins (increased CSF Aβ42 and p‐tau217/total tau ratio) were observed in the CSF of juvenile PSEN1 mutant monkeys, highlighting the potential of using this cynomolgus monkey model for AD modeling and translational research.
4. DISCUSSION
In this study, we generated five PSEN1 mutant cynomolgus monkeys using a dual‐guide, RNA‐mediated, CRISPR/Cas9 genome‐editing approach. Our approach demonstrated a remarkable editing efficiency of 76.5% in zygotes and 50% in live newborns. This high efficiency can be attributed to the use of two guide RNAs that target the 3′ and 5′ intronic sequences of PSEN1 exon 9. 54 In addition, this dual‐guide approach resulted in the permanent removal of the entire PSEN1 exon 9 sequence from the genome rather than destroying the exon 9 splicing site by the single‐cut TALEN approach. 18 In contrast to the random insertions or deletions induced by the single‐guide RNA approach, another advantage of this dual‐guide approach is that it can generate monkey models with defined in‐frame deletions for exon skipping 55 or defined frameshifting mutations for targeted gene knockout. 56 Alternatively, base editing – a genome‐editing technology that enables the direct conversion of one base to another without double‐stranded DNA cleavage 57 – has been used to generate monkey models of Hutchinson–Gilford progeria syndrome and STXBP1 encephalopathy. 58 , 59 However, in our case, such base editing would likely induce bystander editing on the genomic sequence surrounding the PSEN1 exon 9 splicing sites, which may result in unintended effects. 60 , 61
PSEN1 mutations are the most common cause of familial AD. 62 PSEN1‐ΔE9 mutations are well characterized in multiple families carrying genomic deletion of exon 9 or point mutations of the splicing sites. 63 , 64 Therefore, these mutations are suitable for precise and efficient introduction by genome‐editing‐mediated exon deletion or disruption of splicing sites. 56 In the human and cynomolgus monkey genomes, PSEN1‐ΔE9 mutations lead to the in‐frame deletion of the exon 9‐coding auto‐endoproteolysis site in addition to an amino acid change (ie, S290C, TCA to TGC) at the junction between exons 9 and 10. However, in the mouse genome, PSEN1‐ΔE9 mutations lead to a premature stop codon mutation (ie, S290X, TCA to TGA) and PSEN1 knockout, which are expected to result in embryonic lethality in not only mice but marmosets as well. 18 , 65 , 66 Notably, we generated one male and one female monkey carrying nearly pure homozygous PSEN1‐ΔE9 mutations, demonstrating the tolerance of the homozygous PSEN1‐ΔE9 mutation during normal development in monkeys. These findings highlight the species differences between primates and rodents and underscore the advantages of using monkey models to recapitulate AD genetics and pathogenesis more accurately. More importantly, by doubling the chance of generating offspring carriers, we anticipate that these homozygous monkeys will greatly accelerate the generation of a mutant monkey cohort for translational research.
The mutant monkeys in this study exhibited disrupted PSEN1 endoproteolysis. This led to the presence of full‐length PSEN1 protein in fibroblasts derived from the mutant monkeys, which resembles the characteristics of human neuronal models with PSEN1 mutations. 31 Meanwhile, these fibroblasts exhibited signatures of familial AD pathogenesis, including increased Aβ42 secretion and Aβ42/40 ratio. These findings align with the increased presence amyloid plaques in the brain and are inversely correlated with Aβ42 levels in the plasma and CSF of human patients with AD. Furthermore, blood transcriptome profiling of mutant monkeys revealed molecular dysregulation associated with AD. The dysregulated pathways involve immune genes, diabetes mellitus, endocytosis, and phagocytosis, implicating their roles in the pathogenesis of AD. Notably, protein–protein interaction network analysis further identified chemokine ligands and receptors (eg, CCL2, CCL3, CCL4, and CCR3) as potential hub genes among the differentially expressed genes. Interestingly, CCL2 (also known as MCP‐1) level is associated with faster cognitive decline during the early stages of AD, and the rs1799864 allele is associated with a higher CCL2 level in patients with AD or mild cognitive impairment. 67 , 68 STRING network analysis also suggests that FPR2 may be an important node protein that interacts with these hub chemokine ligands and receptors. Given that FPR2 is an important receptor of immune cells that mediates the proinflammatory and neurotoxic activities of Aβ42, it may be a promising anti‐inflammation therapeutic target for AD. 38 , 39 Whether blockade of FPR2 can delay the onset of AD symptoms in non‐human primate models awaits confirmation.
The natural occurrence of typical Aβ plaques and immunoreactive deposits of PHF‐tau (AT8) has been observed in cynomolgus monkeys over 20 years of age. 69 , 70 Human carriers of the PSEN1‐ΔE9 mutation typically develop symptoms between the ages of 45 and 57 years. 63 , 64 Based on the estimated timing of neuroimaging change in familial AD studies, 51 , 71 it is likely that Aβ deposition (amyloid PET) in heterozygous PSEN1‐ΔE9 monkeys may begin to be obvious around the equivalent ages of 7 to 11 years, with AD‐related symptom onset in mid‐life (around the age of 15 years). Given the current young age of the PSEN1 mutant monkeys, we focused on characterizing the early molecular changes in the blood and CSF in this study.
Our investigation of the plasma proteome and CSF proteins in a monkey model with familial AD mutation revealed the early changes of immune proteins and AD‐related pathological proteins in juvenile monkeys. Some of these inflammatory‐ and immune‐related molecules, such as C1QA, IL12RB1, CXCL9, and CXCL10, have been reported to play key roles in AD pathogenesis. 41 , 42 , 72 C1QA is a subunit of serum complement subcomponent C1q. The brain region‐specific upregulation of C1q precedes pre‐plaque synaptic loss, and depletion of C1QA reduces microglia‐mediated early synaptic loss in AD mouse models. 72 Meanwhile, the chemokines CXCL9, CXCL10, and CXCL11 share the same receptor: CXCR3. 41 Blood levels of CXCL9 and CXCL10 are strongly associated with AD, 40 and activation of CXCL9/CXCL10–CXCR3 signaling promotes plaque formation and behavioral deficits in AD mouse models. 41 A recent study also demonstrates the important role of the CXCL10–CXCR3 signaling in modulating CD8+ T‐cell infiltration and promoting neurodegeneration in an AD neuroimmune axis model. 42 , 43 Elevated CSF Aβ42/40 and p‐tau217/total tau ratios in juvenile PSEN1‐ΔE9 cynomolgus monkeys may indicate a similar pathological protein change to the early stages of the AD process. 48 , 51 , 52 , 73 Taken together, our findings suggest that, although we examined our mutant monkeys at a much younger age than when disease symptoms appear, the mutant monkeys may already exhibit AD‐associated peripheral immune molecular dysregulation as well as elevation in pathological proteins in the CSF during the developmental stage. 74
Transgenic cynomolgus monkeys and rhesus monkeys that overexpress mutant APP have been developed as non‐human primate models to study AD. 75 , 76 These models display elevated levels of Aβ and accelerated development of Aβ plaques, respectively. In the transgenic cynomolgus monkeys, the ratio of plasma Aβ42 to Aβ40 was approximately 20‐fold higher than that in control monkeys, whereas in our PSEN1‐ΔE9 monkeys, the CSF Aβ42/40 ratio was increased about three‐fold compared to controls. CSF total tau levels were significantly higher in the mutant APP transgenic rhesus monkey than in control monkeys at 2 years of age, whereas CSF total tau levels appeared to be unchanged in our 1.5‐year‐old PSEN1‐ΔE9 monkeys. The PSEN1‐ΔE9 gene‐edited monkey model reported in this study represent a single mutation knock‐in model, where the mutant PSEN1 gene is expressed and regulated in the endogenous context. In light of previous studies comparing APP transgenic mice with knock‐in mice, 77 gene‐edited cynomolgus monkeys offer a valuable and complementary approach to enhance the modeling of AD pathogenesis.
To comprehensively investigate the emergence and progression of AD‐related molecular changes and pathologies in this PSEN1 mutant monkey model, we will continuously monitor early changes in blood molecules and AD‐related fluid biomarkers through annual assessments of blood transcriptome, plasma proteome, and plasma and CSF biomarker levels prior to symptom onset. As the monkeys grow and age, we will initiate regular pathological, structural, and behavioral tests, such as annual amyloid PET, fluorodeoxyglucose PET, tau PET, magnetic resonance imaging, and assessments of spatial working memory and associative learning ability. 78 These assessments will be performed in conjunction with changes in biomarkers and according to the estimated timing of the corresponding pathology. Additionally, brain tissue samples will be considered for further analyses, such as staining to determine amyloid deposition and single‐cell sequencing, at the appropriate time. By longitudinally monitoring the full pre‐symptomatic lifecycle of molecular, biomarker, pathological, and structural changes in PSEN1 mutant monkeys, we aim to develop a more comprehensive understanding of the pathophysiological roadmap of AD. In conjunction with the PSEN1 mutant marmoset models, 18 , 79 this newly developed gene‐edited PSEN1‐ΔE9 cynomolgus monkey model can greatly contribute to the improvement or development of diagnostic tools for AD, enabling earlier and more accurate diagnosis and intervention.
In conclusion, our study demonstrates the high efficiency of a dual‐guide, RNA‐mediated, CRISPR/Cas9 genome‐editing approach for generating genomic deletions in monkey models. We used this approach to successfully generate cynomolgus monkeys with the PSEN1‐ΔE9 mutation, which displayed key characteristic features of familial AD pathogenesis. Notably, our findings revealed early dysregulation of blood inflammatory and immune molecules associated with AD. These results provide evidence supporting the notion that systemic immune dysregulation may play a pivotal role in the pathogenesis of AD. 80 In addition, AD‐related pathological protein changes (increased CSF Aβ42 and p‐tau217) were observed in juvenile monkeys with PSEN1 mutation. Hence, our gene‐edited PSEN1 mutant monkey model may be a valuable tool for investigating the early pathogenesis of AD and developing potential early diagnostic and therapeutic interventions.
AUTHOR CONTRIBUTIONS
Tao Ye, Yu Chen, and Zhonghua Lu conceived of and designed the study. Mengqi Li, Mingfeng Guan, Jianbang Lin, and Jiayi Zhu performed most of the experiments. Jiayi Zhu and Wanying Yi performed the Olink experiment. Ming Guo, Yefei Chen, Ying Zou, Daiqiang Wu, Junxin Xu, Yingying Fan, Shuangshuang Ma, and Yuewen Chen helped conduct the experiments. Mengqi Li, Mingfeng Guan, Jiayi Zhu, Yinhu Li, Tao Ye, Zhonghua Lu, and Yu Chen analyzed the data. Yinhu Li performed the bioinformatics analysis of the sequencing and Olink data. Mengqi Li, Mingfeng Guan, Jianbang Lin, Yijing Chen, and Tao Ye drafted the manuscript. Tao Ye, Yu Chen, Kaichuan Zhu, Jun Xu, Lixin Yang, Ji Dai, and Zhonghua Lu revised the manuscript and supervised the study. All authors contributed to and approved the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
This study was not conducted on human subjects, so consent was not required.
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank the Non‐Human Primate Research Platform (the Brain Cognition and Brain Disease Institute, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences) for assisting with the animal experiments. This study was financially supported in part by the National Key R&D Program of China (2021YFE0203000, 2018YFA0801404, and 2021YFC2500103), the National Natural Science Foundation of China (NSFC)/RGC Joint Research Scheme (32061160472), the National Natural Science Foundation of China (32100778, 81961128019, and 82071187), the Guangdong Provincial Fund for Basic and Applied Basic Research (2019B1515130004, and 2022A1515011639), the Guangdong Provincial Key S&T Program (2018B030336001), the Development and Reform Commission of Shenzhen (F‐2021‐Z99‐504979), the Technology Innovation Commission of Shenzhen (JCYJ20220818100800001, JCYJ20210324102006018, JCYJ20220531100217038, ZDSYS20200828154800001, and KQTD20221101093608028), and the Innovation and Technology Commission of HKSAR (INNOHK18SC01).
Li M, Guan M, Lin J, et al. Early blood immune molecular alterations in cynomolgus monkeys with a PSEN1 mutation causing familial Alzheimer's disease. Alzheimer's Dement. 2024;20:5492–5510. 10.1002/alz.14046
Mengqi Li, Mingfeng Guan, Jianbang Lin and Kaichuan Zhu authors contributed equally
Contributor Information
Tao Ye, Email: tao.ye@siat.ac.cn.
Zhonghua Lu, Email: zh.lu@siat.ac.cn.
Yu Chen, Email: yu.chen@siat.ac.cn.
DATA AVAILABILITY STATEMENT
The raw data for blood RNA sequencing and whole‐genome sequencing in this paper have been deposited in the Sequence Read Archive under accession numbers PRJNA977131 and PRJNA993388, respectively. All other data are available from the authors upon request.
REFERENCES
- 1. 2023 Alzheimer's disease facts and figures. Alzheimers Dement. 2023;19(4):1598‐1695. doi: 10.1002/alz.13016 [DOI] [PubMed] [Google Scholar]
- 2. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7(2):e105‐e125. doi: 10.1016/s2468-2667(21)00249-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Winblad B, Amouyel P, Andrieu S, et al. Defeating Alzheimer's disease and other dementias: a priority for European science and society. Lancet Neurol. 2016;15(5):455‐532. doi: 10.1016/s1474-4422(16)00062-4 [DOI] [PubMed] [Google Scholar]
- 4. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet (London, England). 2021;397(10284):1577‐1590. doi: 10.1016/s0140-6736(20)32205-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rauchmann BS, Brendel M, Franzmeier N, et al. Microglial activation and connectivity in Alzheimer disease and aging. Ann Neurol. 2022;92(5):768‐781. doi: 10.1002/ana.26465 [DOI] [PubMed] [Google Scholar]
- 6. Tang M, Ryman DC, McDade E, et al. Neurological manifestations of autosomal dominant familial Alzheimer's disease: a comparison of the published literature with the Dominantly Inherited Alzheimer Network observational study (DIAN‐OBS). Lancet Neurol. 2016;15(13):1317‐1325. doi: 10.1016/s1474-4422(16)30229-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353‐356. doi: 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- 8. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8(6):595‐608. doi: 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakai T, Yamada K, Mizoguchi H. Alzheimer's Disease animal models: elucidation of biomarkers and therapeutic approaches for cognitive impairment. Int J Mol Sci. 2021;22(11):5549. doi: 10.3390/ijms22115549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vitek MP, Araujo JA, Fossel M, et al. Translational animal models for Alzheimer's disease: an Alzheimer's association business consortium think tank. Alzheimers Dement. 2020;6(1):e12114. doi: 10.1002/trc2.12114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Padmanabhan P, Götz J. Clinical relevance of animal models in aging‐related dementia research. Nature aging. 2023;3(5):481‐493. doi: 10.1038/s43587-023-00402-4 [DOI] [PubMed] [Google Scholar]
- 12. Feng G, Jensen FE, Greely HT, et al. Opportunities and limitations of genetically modified nonhuman primate models for neuroscience research. Proc Nat Acad Sci USA. 2020;117(39):24022‐24031. doi: 10.1073/pnas.2006515117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou Y, Sharma J, Ke Q, et al. Atypical behaviour and connectivity in SHANK3‐mutant macaques. Nature. 2019;570(7761):326‐331. doi: 10.1038/s41586-019-1278-0 [DOI] [PubMed] [Google Scholar]
- 14. Liu Z, Li X, Zhang JT, et al. Autism‐like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature. 2016;530(7588):98‐102. doi: 10.1038/nature16533 [DOI] [PubMed] [Google Scholar]
- 15. Li B, Zhao H, Tu Z, et al. CHD8 mutations increase gliogenesis to enlarge brain size in the nonhuman primate. Cell Discov. 2023;9(1):27. doi: 10.1038/s41421-023-00525-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beckman D, Ott S, Donis‐Cox K, et al. Oligomeric Aβ in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proc Nat Acad Sci USA. 2019;116(52):26239‐26246. doi: 10.1073/pnas.1902301116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beckman D, Chakrabarty P, Ott S, et al. A novel tau‐based rhesus monkey model of Alzheimer's pathogenesis. Alzheimers Dement. 2021;17(6):933‐945. doi: 10.1002/alz.12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sato K, Sasaguri H, Kumita W, et al. A non‐human primate model of familial Alzheimer's disease. bioRxiv. 2020. doi: 10.1101/2020.08.24.264259 [DOI] [Google Scholar]
- 19. Li HW, Zhang L, Qin C. Current state of research on non‐human primate models of Alzheimer's disease. Animal Model Exp Med. 2019;2(4):227‐238. doi: 10.1002/ame2.12092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tomita T, Takikawa R, Koyama A, et al. C terminus of presenilin is required for overproduction of amyloidogenic Abeta42 through stabilization and endoproteolysis of presenilin. J Neurosci. 1999;19(24):10627‐10634. doi: 10.1523/jneurosci.19-24-10627.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25(14):1754‐1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078‐2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Novak P, Kovacech B, Katina S, et al. ADAMANT: a placebo‐controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer's disease. Nat Aging. 2021;1(6):521‐534. doi: 10.1038/s43587-021-00070-2 [DOI] [PubMed] [Google Scholar]
- 24. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139‐140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Cambridge (Mass)). 2021;2(3):100141. doi: 10.1016/j.xinn.2021.100141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang Y, Zhou X, Ip FC, et al. Large‐scale plasma proteomic profiling identifies a high‐performance biomarker panel for Alzheimer's disease screening and staging. Alzheimers Dement. 2022;18(1):88‐102. doi: 10.1002/alz.12369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wickham H. ggplot2: elegant graphics for data analysis. Springer‐Verlag; 2016. [Google Scholar]
- 28. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447‐D452. doi: 10.1093/nar/gku1003. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thinakaran G, Borchelt DR, Lee MK, et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17(1):181‐190. doi: 10.1016/s0896-6273(00)80291-3 [DOI] [PubMed] [Google Scholar]
- 30. Lee MK, Borchelt DR, Kim G, et al. Hyperaccumulation of FAD‐linked presenilin 1 variants in vivo. Nat Med. 1997;3(7):756‐760. doi: 10.1038/nm0797-756 [DOI] [PubMed] [Google Scholar]
- 31. Woodruff G, Young JE, Martinez FJ, et al. The presenilin‐1 ΔE9 mutation results in reduced γ‐secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013;5(4):974‐985. doi: 10.1016/j.celrep.2013.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bai XC, Yan C, Yang G, et al. An atomic structure of human γ‐secretase. Nature. 2015;525(7568):212‐217. doi: 10.1038/nature14892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou R, Yang G, Guo X, Zhou Q, Lei J, Shi Y. Recognition of the amyloid precursor protein by human γ‐secretase. Science. 2019;363(6428):eaaw0930. doi: 10.1126/science.aaw0930 [DOI] [PubMed] [Google Scholar]
- 34. Güner G, Lichtenthaler SF. The substrate repertoire of γ‐secretase/presenilin. Semin Cell Dev Biol. 2020;105:27‐42. doi: 10.1016/j.semcdb.2020.05.019 [DOI] [PubMed] [Google Scholar]
- 35. Biessels GJ, Despa F. Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol. 2018;14(10):591‐604. doi: 10.1038/s41574-018-0048-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arnold SE, Arvanitakis Z, Macauley‐Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168‐181. doi: 10.1038/nrneurol.2017.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, Giau VV. Type 3 diabetes and its role implications in Alzheimer's disease. Int J Mol Sci. 2020;21(9). doi: 10.3390/ijms21093165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Le Y, Gong W, Tiffany HL, et al. Amyloid (beta)42 activates a G‐protein‐coupled chemoattractant receptor, FPR‐like‐1. J Neurosci. 2001;21(2):Rc123. doi: 10.1523/JNEUROSCI.21-02-j0003.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trojan E, Tylek K, Schröder N, et al. The N‐formyl peptide receptor 2 (FPR2) agonist MR‐39 improves ex vivo and in vivo amyloid beta (1‐42)‐induced neuroinflammation in mouse models of Alzheimer's disease. Mol Neurobiol. 2021;58(12):6203‐6221. doi: 10.1007/s12035-021-02543-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou F, Sun Y, Xie X, Zhao Y. Blood and CSF chemokines in Alzheimer's disease and mild cognitive impairment: a systematic review and meta‐analysis. Alzheimers Res Ther. 2023;15(1):107. doi: 10.1186/s13195-023-01254-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krauthausen M, Kummer MP, Zimmermann J, et al. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer's disease model. J Clin Invest. 2015;125(1):365‐378. doi: 10.1172/jci66771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jorfi M, Park J, Hall CK, et al. Infiltrating CD8(+) T cells exacerbate Alzheimer's disease pathology in a 3D human neuroimmune axis model. Nat Neurosci. 2023;26(9):1489‐1504. doi: 10.1038/s41593-023-01415-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Imhof BA, Aurrand‐Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. 2004;4(6):432‐444. doi: 10.1038/nri1375 [DOI] [PubMed] [Google Scholar]
- 44. Ifergan I, Kebir H, Terouz S, et al. Role of Ninjurin‐1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann Neurol. 2011;70(5):751‐763. doi: 10.1002/ana.22519 [DOI] [PubMed] [Google Scholar]
- 45. Muller WA, Weigl SA, Deng X, Phillips DM. PECAM‐1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178(2):449‐460. doi: 10.1084/jem.178.2.449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang J, Cai F, Lu R, et al. CNTNAP2 intracellular domain (CICD) generated by γ‐secretase cleavage improves autism‐related behaviors. Signal Transduct Target Ther. 2023;8(1):219. doi: 10.1038/s41392-023-01431-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ceglia I, Reitz C, Gresack J, et al. APP intracellular domain‐WAVE1 pathway reduces amyloid‐β production. Nat Med. 2015;21(9):1054‐1059. doi: 10.1038/nm.3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):795‐804. doi: 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schindler SE, Fagan AM. Autosomal dominant Alzheimer disease: a unique resource to study CSF biomarker changes in preclinical AD. Front Neurol. 2015;6:142. doi: 10.3389/fneur.2015.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case‐control study. Lancet Neurol . 2012;11(12):1048‐1056. doi: 10.1016/s1474-4422(12)70228-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barthélemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26(3):398‐407. doi: 10.1038/s41591-020-0781-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mattsson‐Carlgren N, Andersson E, Janelidze S, et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer's disease. Sci Adv. 2020;6(16):eaaz2387. doi: 10.1126/sciadv.aaz2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Janelidze S, Stomrud E, Smith R, et al. Cerebrospinal fluid p‐tau217 performs better than p‐tau181 as a biomarker of Alzheimer's disease. Nat Commun. 2020;11(1):1683. doi: 10.1038/s41467-020-15436-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mandal PK, Ferreira LM, Collins R, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15(5):643‐652. doi: 10.1016/j.stem.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen Y, Zheng Y, Kang Y, et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum Mol Genet. 2015;24(13):3764‐3774. doi: 10.1093/hmg/ddv120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu Z, Hui Y, Shi L, et al. Efficient CRISPR/Cas9‐mediated versatile, predictable, and donor‐free gene knockout in human pluripotent stem cells. Stem Cell Rep. 2016;7(3):496‐507. doi: 10.1016/j.stemcr.2016.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR‐Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824‐844. doi: 10.1038/s41587-020-0561-9 [DOI] [PubMed] [Google Scholar]
- 58. Wang F, Zhang W, Yang Q, et al. Generation of a Hutchinson‐Gilford progeria syndrome monkey model by base editing. Protein Cell. 2020;11(11):809‐824. doi: 10.1007/s13238-020-00740-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu Z, He S, Jiang J, et al. Base‐edited cynomolgus monkeys mimic core symptoms of STXBP1 encephalopathy. Mol Ther. 2022;30(6):2163‐2175. doi: 10.1016/j.ymthe.2022.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zuo E, Sun Y, Wei W, et al. Cytosine base editor generates substantial off‐target single‐nucleotide variants in mouse embryos. Science. 2019;364(6437):289‐292. doi: 10.1126/science.aav9973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grünewald J, Zhou R, Garcia SP, et al. Transcriptome‐wide off‐target RNA editing induced by CRISPR‐guided DNA base editors. Nature. 2019;569(7756):433‐437. doi: 10.1038/s41586-019-1161-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kelleher RJ 3rd, Shen J. Presenilin‐1 mutations and Alzheimer's disease. Proc Nat Acad Sci USA. 2017;114(4):629‐631. doi: 10.1073/pnas.1619574114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Crook R, Verkkoniemi A, Perez‐Tur J, et al. A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998;4(4):452‐455. doi: 10.1038/nm0498-452 [DOI] [PubMed] [Google Scholar]
- 64. Prihar G, Verkkoniem A, Perez‐Tur J, et al. Alzheimer disease PS‐1 exon 9 deletion defined. Nat Med. 1999;5(10):1090. doi: 10.1038/13383 [DOI] [PubMed] [Google Scholar]
- 65. Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin‐1‐deficient mice. Cell. 1997;89(4):629‐639. doi: 10.1016/s0092-8674(00)80244-5 [DOI] [PubMed] [Google Scholar]
- 66. De Strooper B, Saftig P, Craessaerts K, et al. Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387‐390. doi: 10.1038/34910 [DOI] [PubMed] [Google Scholar]
- 67. Lee WJ, Liao YC, Wang YF, Lin IF, Wang SJ, Fuh JL. Plasma MCP‐1 and cognitive decline in patients with Alzheimer's disease and mild cognitive impairment: a two‐year follow‐up study. Sci Rep. 2018;8(1):1280. doi: 10.1038/s41598-018-19807-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Westin K, Buchhave P, Nielsen H, Minthon L, Janciauskiene S, Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer's disease. PLoS One. 2012;7(1):e30525. doi: 10.1371/journal.pone.0030525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nakamura S, Nakayama H, Goto N, Ono F, Sakakibara I, Yoshikawa Y. Histopathological studies of senile plaques and cerebral amyloidosis in cynomolgus monkeys. J Med Primatol. 1998;27(5):244‐252. doi: 10.1111/j.1600-0684.1998.tb00244.x [DOI] [PubMed] [Google Scholar]
- 70. Uchihara T, Endo K, Kondo H, et al. Tau pathology in aged cynomolgus monkeys is progressive supranuclear palsy/corticobasal degeneration‐ but not Alzheimer disease‐like ‐Ultrastructural mapping of tau by EDX. Acta Neuropathol Commun. 2016;4(1):118. doi: 10.1186/s40478-016-0385-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gordon BA, Blazey TM, Su Y, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018;17(3):241‐250. doi: 10.1016/s1474-4422(18)30028-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hong S, Beja‐Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712‐716. doi: 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li Y, Yen D, Hendrix RD, et al. Timing of biomarker changes in sporadic Alzheimer's disease in estimated years from symptom onset. Ann Neurol. 2024;95(5):951‐965. doi: 10.1002/ana.26891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Arber C, Lovejoy C, Harris L, et al. Familial Alzheimer's disease mutations in PSEN1 lead to premature human stem cell neurogenesis. Cell Rep. 2021;34(2):108615. doi: 10.1016/j.celrep.2020.108615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Seita Y, Morimura T, Watanabe N, et al. Generation of transgenic cynomolgus monkeys overexpressing the gene for amyloid‐β precursor protein. J Alzheimers Dis. 2020;75(1):45‐60. doi: 10.3233/jad-191081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chan AWS, Cho IK, Li CX, et al. Cerebral Aβ deposition in an Aβ‐precursor protein‐transgenic rhesus monkey. Aging brain. 2022;2:100044. doi: 10.1016/j.nbas.2022.100044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sasaguri H, Hashimoto S, Watamura N, et al. Recent advances in the modeling of Alzheimer's disease. Front Neurosci. 2022;16:807473. doi: 10.3389/fnins.2022.807473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jiang Z, Wang J, Qin Y, et al. A nonhuman primate model with Alzheimer's disease‐like pathology induced by hippocampal overexpression of human tau. Alzheimers Res Ther. 2024;16(1):22. doi: 10.1186/s13195-024-01392-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Homanics GE, Park JE, Bailey L, et al. Early molecular events of autosomal‐dominant Alzheimer's disease in marmosets with PSEN1 mutations. Alzheimers Dement. 2024;20(5):3455‐3471. doi: 10.1002/alz.13806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bettcher BM, Tansey MG, Dorothée G, Heneka MT. Peripheral and central immune system crosstalk in Alzheimer disease—a research prospectus. Nat Rev Neurol. 2021;17(11):689‐701. doi: 10.1038/s41582-021-00549-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Data Availability Statement
The raw data for blood RNA sequencing and whole‐genome sequencing in this paper have been deposited in the Sequence Read Archive under accession numbers PRJNA977131 and PRJNA993388, respectively. All other data are available from the authors upon request.