

Abstract
The National Institute on Aging and the Alzheimer's Association convened three separate work groups in 2011 and single work groups in 2012 and 2018 to create recommendations for the diagnosis and characterization of Alzheimer's disease (AD). The present document updates the 2018 research framework in response to several recent developments. Defining diseases biologically, rather than based on syndromic presentation, has long been standard in many areas of medicine (e.g., oncology), and is becoming a unifying concept common to all neurodegenerative diseases, not just AD. The present document is consistent with this principle. Our intent is to present objective criteria for diagnosis and staging AD, incorporating recent advances in biomarkers, to serve as a bridge between research and clinical care. These criteria are not intended to provide step‐by‐step clinical practice guidelines for clinical workflow or specific treatment protocols, but rather serve as general principles to inform diagnosis and staging of AD that reflect current science.
Highlights
We define Alzheimer's disease (AD) to be a biological process that begins with the appearance of AD neuropathologic change (ADNPC) while people are asymptomatic. Progression of the neuropathologic burden leads to the later appearance and progression of clinical symptoms.
Early‐changing Core 1 biomarkers (amyloid positron emission tomography [PET], approved cerebrospinal fluid biomarkers, and accurate plasma biomarkers [especially phosphorylated tau 217]) map onto either the amyloid beta or AD tauopathy pathway; however, these reflect the presence of ADNPC more generally (i.e., both neuritic plaques and tangles).
An abnormal Core 1 biomarker result is sufficient to establish a diagnosis of AD and to inform clinical decision making throughout the disease continuum.
Later‐changing Core 2 biomarkers (biofluid and tau PET) can provide prognostic information, and when abnormal, will increase confidence that AD is contributing to symptoms.
An integrated biological and clinical staging scheme is described that accommodates the fact that common copathologies, cognitive reserve, and resistance may modify relationships between clinical and biological AD stages.
Keywords: Alzheimer's disease diagnosis, Alzheimer's disease imaging, Alzheimer's disease staging, amyloid positron emission tomography, biofluid biomarkers Alzheimer's disease, biomarkers Alzheimer's disease, preclinical Alzheimer's disease, tau positron emission tomography
1. BACKGROUND
In 2011, the National Institute on Aging and the Alzheimer's Association (NIA‐AA) convened three workgroups that published separate recommendations for the diagnosis and evaluation of Alzheimer's disease (AD) in its preclinical, mild cognitive impairment (MCI), and dementia phases. 1 , 2 , 3 , 4 In 2012, an NIA‐AA workgroup published a consensus document on the neuropathologic diagnosis of AD. 5 , 6 Several years later, the NIA‐AA convened a single workgroup to update 2011 recommendations for diagnosis and evaluation. The product of that workgroup, published in 2018, was labeled a research framework. 7 The 2018 publication stated that the framework should be updated in the future as needed in response to scientific advances.
The convening organization for these revised criteria is the AA. The AA identified a four‐person core leadership group for this effort (i.e., a steering committee) as well as a larger full workgroup. Members of the full workgroup were selected to provide a range of relevant scientific and clinical expertise; to achieve a representative sample of professional stakeholders; and to achieve a balance of academic and industry representation, sex/ethnicity, and geographic location. The steering committee also engaged expert advisors to provide reviews of the project.
Although the purpose of this document is to update the 2018 document, a set of fundamental principles emerged from prior NIA‐AA workgroups. These principles, outlined in Box 1, are carried forward and serve as the foundation or starting point for the current revised criteria.
BOX 1: Fundamental principles
It is necessary to separate syndrome (clinically identified impairment) from biology (etiology).
Alzheimer's disease (AD) is defined by its biology with the following implications.
AD is defined by its unique neuropathologic findings; therefore, detection of AD neuropathologic change by biomarkers is equivalent to diagnosing the disease.
AD exists on a continuum. The disease is first evident in vivo with the appearance of disease‐specific Core biomarkers while people are asymptomatic. Pathophysiologic mechanisms involved with processing and clearance of protein fragments may be involved very early in the disease process, but these are not yet well understood.
Symptoms are a result of the disease process and are not necessary to diagnose AD.
Unimpaired individuals with abnormal biomarker test results are at risk for symptoms due to AD. They are not at risk for a disease they already have.
Clinical syndromes commonly seen with AD may also be caused by disorders other than AD, and therefore clinical presentation alone is not diagnostic of AD.
The same AD biology may result in different phenotypic presentations.
Three major developments prompted this update. First, treatments that target core disease pathology have, for the first time, received regulatory approval. The prospect of these therapies entering clinical practice highlights the importance of conceptual alignment among clinicians, industry, and academia around diagnosis and staging of AD.
Second, the most significant advance in AD diagnostics in recent years has been the development of blood‐based markers (BBM) with some (not all) assays exhibiting accurate diagnostic performance. This now makes the biological diagnosis of AD (which previously required positron emission tomography [PET] or cerebrospinal fluid [CSF] assays) more generally accessible and is projected to revolutionize clinical care and research. The field is now in a transition phase during which BBM are being integrated with traditional CSF and PET biomarkers.
Finally, an important product of recent research is the recognition that imaging, CSF, and BBM within a pathobiological AT(N) (amyloid/tau/neurodegeneration) category are interchangeable for some, but not all, intended uses. The present document is updated to reflect this.
This is a forward‐looking document based on current scientific evidence that provides a common framework for AD diagnostic and staging criteria to inform both research and clinical care. We do not provide detailed guidance on clinical workflow or treatment protocols; formal clinical practice guidelines will appear in a subsequent document. The criteria we describe are presently operationalizable at some but by no means all centers even among major medical institutions in high‐income countries. We therefore view these criteria as a bridge between research and clinical care.
2. BIOMARKER CATEGORIZATION
Categorization of biomarkers refers to grouping biomarkers into categories that reflect a common proteinopathy pathway or pathogenic process. Categorization of biomarkers in the 2018 NIA‐AA framework assumed equivalence of CSF and imaging biomarkers within each AT(N) category. 8 Ample evidence has accumulated that this is not always the case; therefore, in these revised criteria we break from the assumption of equivalence between imaging and biofluid biomarkers within a given biomarker category.
We group biomarkers into three broad categories: core biomarkers of AD neuropathologic change (ADNPC), 5 non‐specific biomarkers that are important in AD pathogenesis but are also involved in other brain diseases, and biomarkers of common non‐AD copathologies (Table 1). Within each of these three broad categories we subcategorize biomarkers by the specific proteinopathy pathway or pathogenic process that each measures; for example, “A” biomarkers denote the amyloid beta (Aβ) proteinopathy pathway.
TABLE 1.
Categorization of fluid analyte and imaging biomarkers.
| Biomarker category | CSF or plasma analytes | Imaging |
| Core Biomarkers | ||
| Core 1 | ||
| A (Aβ proteinopathy) | Aβ 42 | Amyloid PET |
| T1 : (phosphorylated and secreted AD tau) | p‐tau217, p‐tau181, p‐tau231 | |
| Core 2 | ||
| T2 (AD tau proteinopathy) | MTBR‐tau243, other phosphorylated tau forms (e.g., p‐tau205), non‐phosphorylated mid‐region tau fragments a | Tau PET |
| Biomarkers of non‐specific processes involved in AD pathophysiology | ||
| N (injury, dysfunction, or degeneration of neuropil) | NfL | Anatomic MRI, FDG PET |
| I (inflammation) Astrocytic activation | GFAP | |
| Biomarkers of non‐AD copathology | ||
| V vascular brain injury | Infarction on MRI or CT, WMH | |
| S α‐synuclein | αSyn‐SAA a | |
Notes: P‐tau231, p‐tau205, MTBR‐tau243, and non‐phosphorylated tau fragments are included in this table because they are discussed in the text; however, these analytes have not undergone the same level of validation testing as other Core biomarkers. Biomarkers are categorized based on four criteria. First, three broad mechanistic groupings have been identified. Second, biomarkers are subclassified based on the proteinopathy or pathophysiologic pathway that each measures (e.g., A, T1, T2, N etc.). Third, within the Core category we distinguish between Core 1 and Core 2 biomarkers. Fourth, imaging and fluid analyte biomarkers are listed separately within each category.
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; αSyn‐SAA, alpha‐synuclein seed amplification assay; CSF, cerebrospinal fluid; CT, computed tomography; FDG, fluorodeoxyglucose; GFAP, glial fibrillary acidic protein; MRI, magnetic resonance imaging; MTBR, microtubule‐binding region; NfL, neurofilament light chain; PET, positron emission tomography; WMH, white matter hyperintensity.
A fluid analyte that is presently informative only when measured in CSF. No notation used if the fluid analyte is informative with plasma or CSF.
Throughout the document we distinguish between imaging and fluid biomarkers. Imaging biomarkers measure cumulative effects; capture topographic information; map onto established neuropathologic constructs; and, in the case of amyloid and tau PET, represent insoluble aggregates. 9 , 10 , 11 , 12 , 13 , 14 , 15 Fluid biomarkers reflect the net of rates of production/clearance of analytes at a given point in time.
The 2018 framework recognized the need to modify the AT(N) biomarker classification scheme to incorporate newly developed biomarkers within an existing AT(N) category; and, we have included recently developed BBM of A, T, and (N) in this update. The 2018 framework also called for incorporating new biomarker categories beyond AT(N) as appropriate. This was denoted in the 2018 document as ATX(N), where X indicated a new biomarker category beyond A, T, or (N). 7 Accordingly, Tables 1 and 2 include three new biomarker categories: inflammatory/immune mechanisms (I), vascular brain injury (V), and alpha‐synucleinopathy (S). V and S biomarkers are relevant to this document on AD diagnosis and staging because AD most often occurs with copathologies in older adults.
TABLE 2.
Intended uses for imaging, CSF, and plasma biomarker assays.
| Intended use | CSF | Plasma | Imaging |
| Diagnosis | |||
| A: (Aβ proteinopathy) | — | — | Amyloid PET |
| T1 : (phosphorylated and secreted AD tau) | — | p‐tau217 | — |
| Hybrid ratios | p‐tau181/Aβ42, t‐tau/Aβ42, Aβ42/40 | %p‐tau217 | — |
| Staging, prognosis, as an indicator of biological treatment effect | |||
| A: (Aβ proteinopathy) | — | — | Amyloid PET |
| T1 : (phosphorylated and secreted AD tau) | — | p‐tau217 | — |
| Hybrid ratios | p‐tau181/Aβ42, t‐tau/Aβ42, Aβ42/40 | %p‐tau217 | — |
| T2 : (AD tau proteinopathy) | MTBR‐tau243, other p‐tau forms (e.g., p‐tau205), non‐phosphorylated mid‐region tau fragments | MTBR‐tau243, other p‐tau forms (e.g., p‐tau205) | Tau PET |
| N (injury, dysfunction, or degeneration of neuropil) | NfL | NfL | Anatomic MRI, FDG PET |
| I (inflammation) Astrocytic activation | GFAP | GFAP | — |
| Identification of copathology | |||
| N (injury, dysfunction, or degeneration of neuropil) | NfL | NfL | Anatomic MRI, FDG PET |
| V vascular brain injury | — | — | Infarction on MRI or CT, WMH |
| S α‐synuclein | αSyn‐SAA | — | |
Notes: Table 2 lists assays while Table 1 lists analytes; therefore, plasma and CSF are listed separately in Table 2 but listed together in Table 1. The focus in this table is on plasma p‐tau217 and not p‐tau231, p‐tau181, or Aβ42/40 because p‐tau217 typically outperforms these other plasma assays in head‐to‐head comparisons. %p‐tau is the ratio p‐tau217/non‐phosphorylated‐tau217. Combinations of Core 1 biomarkers may also be used for diagnosis. P‐tau205, MTBR‐tau243, and non‐phosphorylated tau fragments have not undergone the same level of validation testing as has tau PET; however, they are included to support a “conceptual” staging scheme outlined in Table 5.
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; αSyn‐SAA, alpha‐synuclein seed amplification assay; CSF, cerebrospinal fluid; CT, computed tomography; FDG, fluorodeoxyglucose; GFAP, glial fibrillary acidic protein; MRI, magnetic resonance imaging; MTBR, microtubule‐binding region; NfL, neurofilament light chain; PET, positron emission tomography; p‐tau, phosphorylated tau; WMH, white matter hyperintensity.
Table 1 illustrates biomarker categories by pathogenic mechanism or proteinopathy pathway. CSF and plasma are listed together as fluid analytes because the same analyte is measured in CSF or plasma. Table 2 lists intended uses for biomarkers, which fall into several categories: diagnosis; staging, prognosis, as an indicator of biological treatment effect; and identification of copathologies. While Table 1 lists fluid analytes, Table 2 lists assays and, accordingly, CSF and plasma are broken into separate columns in Table 2 because assay implementation may differ between CSF and plasma. Table 2 also includes hybrid ratios, which are assays rather than individual analytes. Assays in Table 2 may be in vitro diagnostics, laboratory‐developed tests, or research‐use‐only tests. The committee used the following criteria for inclusion in Table 2: the imaging, CSF, or BBM has either received regulatory approval or has played a prominent role in recent clinical research, and, in the opinion of the committee, enough evidence exists to support its clinical value and the assumption that it may receive regulatory approval in the future.
Tables 1 and 2 categorize core and non‐core biomarkers. In the remainder of Section 2, we focus on core biomarkers of ADNPC to create a logical progression to the subsequent topics of diagnosis and staging, which use only core biomarkers. Non‐core biomarkers (i.e., N, I, V, and S) are discussed in Section 7.
Core AD biomarkers are those in the A (Aβ) and T (tau) categories (Table 1). The A category denotes biomarkers of the Aβ proteinopathy pathway. Soluble aggregation‐prone Aβ peptides are the molecular building blocks of insoluble fibrillar Aβ aggregates in plaques. Hence, fluid and imaging A biomarkers “represent different biochemical pools of the same proteinopathy pathway.” 16 Moreover, although fluid Aβ42‐based assays may become abnormal slightly before amyloid PET, 17 the two are usually highly concordant. 18 , 19 , 20
Timing relationships are different across the spectrum of T biomarkers. Phosphorylated mid‐region fragments (phosphorylated tau [p‐tau] 181, 217, and 231) become abnormal around the same time as amyloid PET and before tau PET. 21 , 22 , 23 , 24 This has led to the suggestion that secretion of tau fragments phosphorylated at specific residues (181, 217, and 231) may represent a physiologic reaction to Aβ plaques 25 and may link Aβ proteinopathy to early tau proteinopathy. 26 , 27 In contrast, other tau fragment analytes (e.g., microtubule‐binding region [MTBR‐tau243] and non‐phosphorylated mid‐region tau fragments) become abnormal later and correlate better with tau PET than with amyloid PET. 28 , 29 These observations led us to split the T biomarker category into two subcategories: T1 (biofluid analytes of soluble tau fragments that may reflect a reaction to amyloid plaques or to soluble Aβ species in plaque penumbra) and T2 (tau PET imaging or biofluid analytes that signal the presence of AD tau aggregates). Because of their time of onset, plasma p‐tau 217, 181, and 231 have been proposed as biomarkers of Aβ plaques, but this is difficult to accept conceptually because these are tau fragments, not measures of the Aβ proteinopathy pathway. Furthermore, these tau analytes do correlate with tau proteinopathy in addition to Aβ proteinopathy. 30 The T1 and T2 categories address these conceptual issues.
We introduce the concept of Core 1 and Core 2 AD biomarkers, which are differentiated by the timing of abnormality onset and intended use (Box 2). Core 1 biomarkers become abnormal around the same time as amyloid PET, and are those in the A, T1, or hybrid ratio categories (Tables 1 and 2). Because most individuals with abnormal amyloid PET have intermediate‐to‐high ADNPC (discussed in Section 3.2), the Core 1 category represents ADNPC more generally (i.e., both neuritic plaques and tangles). Core 1 biomarkers define the initial stage of AD that is detectable in vivo and can identify the presence of AD in both symptomatic and asymptomatic individuals.
BOX 2: The diagnosis of Alzheimer's disease, Core 1 and Core 2 Alzheimer's disease biomarkers
Phosphorylated mid‐region tau variants (p‐tau 217, 181, and 231) become abnormal around the same time as amyloid positron emission tomography (PET) and well before tau PET. In contrast, other tau fragments (e.g., microtubule‐binding region [MTBR]‐tau243) become abnormal later, closer to onset of tau PET. Our solution is to split the T category: T1 are early changing phosphorylated mid‐region tau fragments (p‐tau 217, 181, and 231). T2 are later‐changing biofluid tau fragments (e.g., MTBR‐tau243) along with tau PET. We then group core biomarkers into Core 1, which are A, T1, and hybrid combinations, versus Core 2, which are tau PET and T2 biofluids.
The diagnosis of Alzheimer's disease (AD) can be established by abnormality on specific Core 1 biomarkers (see Table 2); however, not all available Core 1 biomarker tests have sufficient accuracy to be suitable for diagnosis. Currently, we regard the following to be diagnostic of AD: amyloid PET, cerebrospinal fluid (CSF) Aβ 42/40, CSF p‐tau 181/Aβ 42, CSF t‐tau/Aβ 42, “accurate” plasma assays (defined below), or combinations of these. In most situations different Core 1 biomarkers should be interchangeable for the detection of AD neuropathologic change (ADNPC) and, hence, for the diagnosis of AD. Because nearly all symptomatic individuals and the vast majority of asymptomatic individuals with abnormal amyloid PET will have intermediate/high AD neuropathologic change, the Core 1 category represents ADNPC more generally (i.e., both plaques and tangles). Core 1 biomarkers define the initial stage of AD that is detectable in vivo and can be used diagnostically for (1) early detection of AD in people without symptoms and (2) confirmation that AD is an underlying pathology in someone with symptoms.
Core 2 biomarkers do not detect the initial presence of disease and thus may not rule out AD pathology; however, because amyloidosis is nearly always a prerequisite for neocortical AD tauopathy, Core 2 biomarkers are highly associated with Aβ pathology and, therefore, may be sufficient to confirm (rule in) AD pathology (although rare exceptions exist). When combined with Core 1 biomarkers, Core 2 biomarkers can be used to stage biological disease severity and (1) provide information on the likelihood that symptoms are associated with AD, (2) inform on the likely rate of progression in symptomatic individuals, and (3) inform on the risk of short‐term progression in people without symptoms.
Only biomarkers that have been proven to be accurate with respect to an accepted reference standard should be used for clinical diagnostic purposes, and the same criteria apply to PET, CSF, or blood‐based biomarkers. We recommend, as a minimum requirement, an accuracy of 90% for the identification of moderate/frequent neuritic plaques at autopsy (or an approved surrogate, which, at this point, would be amyloid PET or CSF) in the intended‐use population. For blood‐based biomarker assays, this translates to an accuracy equivalent to that of approved CSF assays. We focus on accuracy (true positive + true negative)/(true positive + true negative + false positive + false negative) as a concise metric because it is equally important that a test used clinically is correct when the test result is positive and is correct when it is negative. The specification of accurate “in the intended‐use population” addresses positive and negative predictive values, which depend on the prior probability of AD in the population of interest.
Core 2 biomarkers are those in the T2 category (Tables 1 and 2) and include tau PET and certain soluble tau fragments associated with tau proteinopathy (e.g., MTBR‐tau243), but also pT205 and non‐phosphorylated mid‐region tau fragments. 21 , 29 Core 2 biomarkers become abnormal later in the evolution of AD and are more closely linked with the onset of symptoms than are Core 1 biomarkers. Core 2 biomarkers, when combined with Core 1, may be used to stage biological disease severity.
CSF assays/platforms and PET ligands that have received regulatory approval for clinical use are listed in Table S1 in supporting information. Readers are referred to recent reviews for details describing specific fluid biomarker assays and PET ligands. 31 , 32 , 33
3. DIAGNOSIS
In this update we propose that abnormality on specific Core 1 biomarkers is sufficient to diagnose AD (Table 2). Specifically, we propose that the following can be diagnostic of AD: amyloid PET; CSF Aβ 42/40, CSF p‐tau 181/Aβ 42, CSF t‐tau/Aβ 42; or “accurate” plasma assays where “accurate” can be defined as accuracy that is equivalent to approved CSF assays in detecting abnormal amyloid PET in the intended‐use population (Box 2). This definition is consistent with recent recommendations on minimum acceptable performance criteria for BBM. 34 Combinations of Core 1 biomarkers may also be used as diagnostic tests.
Core 2 biomarkers have many uses (Table 2, Box 2) but most often would not be used as standalone diagnostic tests for AD. Caution should be used in interpreting an abnormal Core 2 result in the presence of a normal Core 1 result, because, as discussed in Section 4.3, abnormal amyloid PET (or a biofluid surrogate) is nearly always a prerequisite for neocortical AD tauopathy. 35 The A−T2+ biomarker profile 36 is not consistent with a diagnosis of AD. 37 First, this combination is rare. 38 , 39 , 40 Second, when it does occur, it is often due to quantitative values close to cutpoints that may fall on one side versus the other owing to measurement variation. 41 As discussed later, biomarkers have sensitivity limits and a normal A biomarker result does not mean that the brain is devoid of plaques, but rather that if plaques are present, the burden does not rise to the detection threshold. Third, from a neuropathologic perspective, A−T2+ corresponds to primary age‐related tauopathy, which is not considered to represent AD. 5 , 42
3.1. Rationale for diagnosis of AD by Core 1 biomarkers
Natural history studies show that biomarkers in the Core 1 category become abnormal well before symptoms arise (Figure 1). 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 Our rationale for diagnosing AD by the presence of specific Core 1 biomarkers (Table 2) is that biomarkers that coincide with the onset of abnormal amyloid PET define the initially detectable stage of AD. An analogy can be drawn with slowly progressive cancer that can be detected by biomarker testing before symptoms arise. Although symptoms may not appear for years, the disease nonetheless exists at this initially detectable stage and will eventually produce symptoms if the individual lives long enough. Although many individuals die with ADNPC without experiencing symptoms, this can be attributed to the exponential rise in all‐cause mortality rates with older age. 52 , 53 Mortality from unrelated diseases prior to symptom onset from AD should not be interpreted to indicate that ADNPC is benign. Individuals with Down syndrome (DS; trisomy 21) overexpress the amyloid precursor protein and Aβ. Virtually all individuals with DS have sufficient ADNPC to meet neuropathological criteria for a diagnosis of AD by their mid‐40s. 54 Age at onset and mortality in DS are compatible with full genetic penetrance as in autosomal dominant AD (ADAD). 55 One study estimated lifetime risk of dementia to be 95%, 56 with the average age of onset of clinical symptoms in the mid‐50s, 54 when mortality rates from unrelated disorders are far lower than in older age. Remaining life expectancy is an important consideration in clinical management, but mortality from unrelated causes should not be a criterion used to define what is and what is not a disease. The biological definition of AD is consistent with the distinction between a disease and an illness. A disease is a pathogenic condition, while the term illness denotes signs and symptoms that result from the disease. Importantly, defining a disease by its biology rather than by a syndromic description has been status quo for years in other areas of medicine (e.g., oncology) and is becoming a unifying concept common to all neurodegenerative diseases, as exemplified by recent efforts in Parkinson's disease, 57 , 58 Huntington's disease, 59 and amyotrophic lateral sclerosis. 60
FIGURE 1.
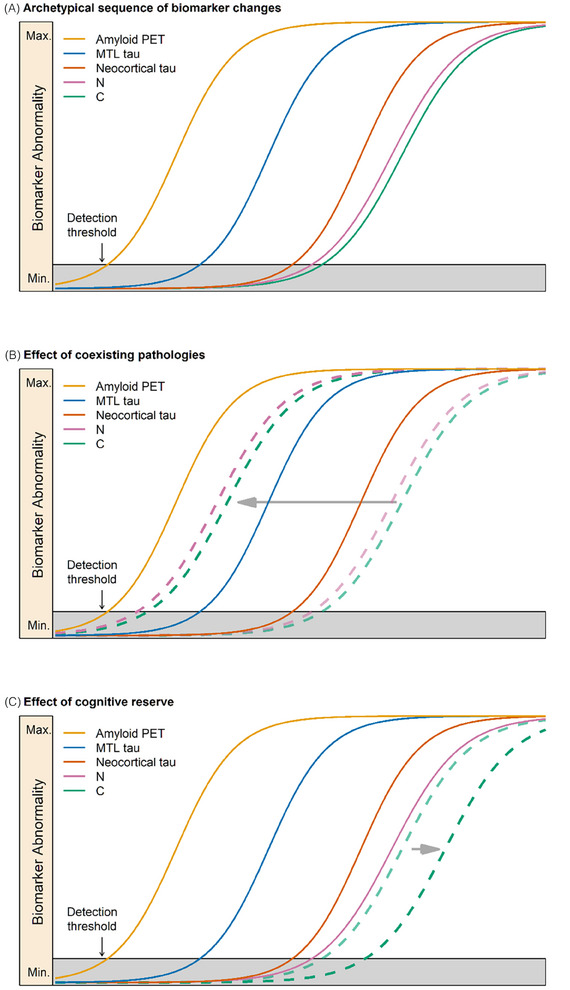
Staging illustrated with imaging biomarkers along with modifying effects of copathology and cognitive reserve. A, Prototypical temporal evolution in Alzheimer's disease (AD): sequential evolution of amyloid and tau positron emission tomography (PET), followed by neurodegeneration and clinical symptoms. Time is on the x axis and magnitude of biomarker or clinical abnormality is on the y axis; (A) also illustrates an idealized evolution of AD imaging biomarkers in an individual with only AD neuropathologic change (MTL refers to medial temporal lobe uptake on tau PET). B, The effect of neurodegenerative copathology in a person with biological AD stage A (i.e., A+T2−) but severe neurodegeneration and clinical symptoms that are out of proportion to the degree of tauopathy. This is denoted by a leftward shift (horizontal gray arrow) of neurodegeneration (N) and clinical symptoms (C) relative to the pure AD temporal sequence. C, The effect of exceptional cognitive reserve. This is denoted by a rightward shift, later in time (horizontal gray arrow), in the appearance of cognitive symptoms (C, dashed green line) relative to the average temporal biomarker sequence.
In the 2018 research framework, an A+T+ biomarker profile was required for a designation of AD based on the AT(N) biomarker classification scheme. However, with evolved understanding, the T category has been split into T1 and T2 in these revised criteria, and we now define AD as an abnormality on Core 1 biomarkers (Table 2). This definition effectively anchors the onset of AD in vivo to the onset of abnormal amyloid PET (Figure 1). However, an often overlooked but important fact is that amyloid PET is not sensitive to low levels of ADNPC. The US Food and Drug Administration (FDA)–approved amyloid PET tracers cannot reliably detect sparse neuritic plaques but can detect Consortium to Establish a Registry for Alzheimer's Disease (CERAD) moderate/frequent neuritic plaque density with high reliability. 9 , 10 , 11 , 61 , 62 In the experience of most neuropathologists, the vast majority of cases with moderate/frequent neuritic plaque burden will also have Braak III to VI neurofibrillary tangle (NFT) scores and thus will meet criteria for intermediate/high ADNPC. 5 Extending this logic to PET leads to the conclusion that an abnormal amyloid PET study should nearly always indicate intermediate/high ADNPC. However, to assess this supposition more rigorously, we examined the December 2023 data freeze of the National Alzheimer's Coordinating Center (NACC) autopsy database for participants who had CERAD moderate/frequent neuritic plaque density scores and had been assigned both a Braak NFT stage and Clinical Dementia Rating (CDR). Four thousand eight hundred eighty‐nine individuals met these criteria. Of these, 4637 were symptomatic (i.e., CDR > 0) and 4390/4637 (95%) were Braak NFT stage III to VI and therefore had a neuropathologic diagnosis of intermediate/high ADNPC. Thus, among symptomatic individuals, knowing only that someone has moderate/frequent plaques (i.e., an abnormal amyloid PET study) means that that individual will nearly always (95% of the time) qualify for a pathologic diagnosis of intermediate‐to‐high ADNPC. Diagnosing AD by abnormal amyloid PET (or biofluid Core 1 biomarkers validated against amyloid PET) in symptomatic individuals is therefore not a deviation from the accepted criteria for AD 5 because this will nearly always indicate intermediate/high ADNPC. Abnormal Core 1 biomarkers should be regarded as indicators of ADNPC generally (both plaques and tangles), even though the assays themselves may be specific measures of Aβ (A) or tau (T1) pathophysiology.
To address diagnosing AD by biomarkers in cognitively unimpaired individuals, we examined the Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program 63 database and found 123 individuals who were cognitively unimpaired at death and had moderate/frequent CERAD neuritic plaque scores. One hundred seven (87%) of these were also Braak III to VI and thus met criteria for intermediate/high ADNPC. The December 2023 NACC data freeze mentioned above contained 252 asymptomatic (CDR 0) individuals with moderate/frequent neuritic plaques and who also had been assigned a Braak NFT stage; 186/252 (74%) were Braak III to VI, while 226/252 (91%) were Braak II to VI. Thus, we can conclude that while most (74%−87%) asymptomatic individuals with an abnormal amyloid PET scan (i.e., moderate/frequent neuritic plaques) will meet criteria for intermediate/high ADNPC at that time, a proportion will not. But nearly all (91%) will either meet criteria at that time or show evidence of progression of NFT pathology beyond the entorhinal cortex (i.e., Braak II–VI).
The issue of future progression in turn raises the question, among cognitively unimpaired persons who do meet current neuritic plaque criteria (i.e., abnormal amyloid PET) but may not meet Braak NFT stage criteria for intermediate/high ADNPC, is it likely that they would have had they lived longer? To address this, we queried the Mayo Clinic Study on Aging 64 and Alzheimer's Disease Research Center database for individuals who: (1) had a positive ante mortem amyloid PET scan (defined as Centiloid ≥ 25) while cognitively unimpaired, (2) later progressed to dementia or MCI, (3) had an autopsy. Of the 34 individuals who did meet these restrictive criteria, 32 (94%) received a neuropathological diagnosis of intermediate/high likelihood ADNPC. Based on this evidence, we believe the following statement is accurate: Most cognitively unimpaired persons with an abnormal amyloid PET scan should presently meet neuropathologic pathologic criteria for intermediate/high ADNPC, and those that do not will very likely do so in the future if they live long enough.
3.2. Anchoring Core 1 biomarkers for AD diagnosis to reference standards
The amyloid PET visual reading method on which regulatory approval of florbetapir was based is highly accurate (sensitivity 96%, specificity 100%) at discriminating CERAD none/sparse versus moderate/frequent neuritic plaques in individuals who had an autopsy within 1 year of the PET scan. 11 Visual reads of other approved PET tracers demonstrated similar sensitivity/specificity with respect to a neuropathologic reference standard. 61 , 62 Quantification of amyloid PET is also accurate at distinguishing intermediate/high ADNPC from none/low ADNPC. 10 Regulatory approval of CSF assays (Table S1) was anchored to positive/negative visual reads of amyloid PET: sensitivity/specificity (or positive percent agreement/negative percent agreement) of approved CSF assays were 88%/93% and 85%/94% against this reference standard for a single cutpoint (this becomes more complicated for two cutpoints). 65 , 66 , 67 Although autopsy was not the standard used for regulatory approval, CSF assays do distinguish between intermediate/high and none/low ADNPC with high accuracy. 68
Currently, no BBMs have received regulatory approval for any intended use, although this is expected to change soon. Diagnostic accuracy varies substantially among various plasma p‐tau and Aβ42/40 assays. 69 , 70 , 71 Accuracy estimates with respect to an amyloid PET or CSF reference standard using a single preselected cutpoint or area under the receiver operating curve (i.e., accuracy over all cutpoints) range from 0.6 (60%) to > 0.9 (90%). 69 , 72 , 73 , 74 , 75 Thus, some plasma assays, particularly some p‐tau217 assays alone or in combination, have accuracy that is equivalent to that of approved CSF assays. 69 , 74 , 76 , 77 Accuracy must be defined in the intended‐use population. Thus, our definition of Core 1 plasma tests that suffice as standalone diagnostic tests for AD is those with a minimum accuracy of 90% to detect abnormal amyloid PET in the intended‐use population, or, more simply, plasma tests whose diagnostic performance is equivalent to that of approved CSF assays (Box 2). Our position is that BBM that achieve this diagnostic performance benchmark should be considered on equal footing with established PET and CSF biomarkers for diagnosis of AD.
To date, clinical symptoms have not been used as the reference standard for regulatory approval of AD biomarkers. Our position is that AD is defined by its biology and therefore a biomarker that can accurately detect ADNPC or a validated surrogate is sufficient to establish the diagnosis of the disease.
3.3. Blood‐based versus CSF biomarkers
CSF p‐tau is typically not used as a standalone diagnostic test; rather, diagnostic CSF assays are hybrid ratios (Table 2): p‐tau181/Aβ42, total tau/Aβ42, or Aβ42/40. 65 , 66 , 67 In contrast, some plasma p‐tau assays have demonstrated very good clinical performance in clinical trials and observational studies as a standalone biomarker. 75 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85
The fold difference between individuals with and without Aβ pathologic change is ≈ 50% for CSF Aβ42/40 but only 10% to 15% for plasma Aβ42/40. 70 , 72 , 86 , 87 , 88 This limited diagnostic range accounts for the generally worse clinical robustness of plasma Aβ42/40 assays compared to CSF assays or plasma p‐tau217 assays. 89 , 90 , 91
3.4. Biofluid assay development transparency
Specific regulations are established by national and international laboratory medicine associations, and regulations for the use of laboratory tests include the International Medical Device Regulations, FDA regulations, European Commission In Vitro Diagnostics regulations, and Clinical Laboratory Improvement Amendments (CLIA) of 1988. The common principle is that for clinical use of biomarker tests, documentation and proof need to be provided at the following levels: (1) scientific validity, which includes details of the reference standard (i.e., autopsy, approved CSF assays, or amyloid PET); (2) analytical validation, including criteria for test precision, bias, and linearity, which are addressed by the Clinical and Laboratory Standards Institute guidelines; and (3) clinical validation in the intended‐use population, showing clinical accuracy, positive and negative predictive value at the medical decision limit (i.e., predetermined cutpoints) in each intended‐use population, and safety (which includes the effect of incorrect test diagnosis). The information provided should also include details of the population(s) tested, such as demographic data (e.g., sex, age, race) and pertinent clinical data (e.g., degree of cognitive impairment).
3.5. Conservative treatment of values near a cutpoint; the indeterminant zone
The definition of an abnormal test value requires creating a cutpoint in the continuous range of values for a biomarker. Cutpoints denoting normal versus abnormal values may be selected by various means 92 and will vary with the fluid assay; for PET, it will depend on the specific ligand and details of the analytic pipeline for quantitative analyses. Furthermore, criteria for cutpoint selection depend on the intended use. Sensitivity and specificity are obviously inversely related, and optimizing one versus the other will depend on the intended use as well as the prior probability of AD in the relevant population.
Regardless of the biofluid assay or imaging modality, a level of diagnostic uncertainty exists for values at or near a cutpoint. When using a CSF, blood‐based, or PET biomarker quantitatively for diagnosis, a useful approach would be to report results with two cutpoints 93 that divide the continuous range of values into confidently normal, confidently abnormal, and indeterminant. Values that fall within the indeterminant zone might prompt additional testing. 94 The width of the indeterminant zone would depend on biomarker precision and intended use. 95 Higher biomarker precision would allow a narrower indeterminant zone and vice versa. The upper cutpoint can be more conservative to increase positive predictive value. Regulatory approval for assays is usually based on a single validated cutpoint and does not require designation of an indeterminant zone; however, the package insert for one approved CSF assay does include a range described as “likely consistent with a positive amyloid PET scan result,” which conveys the notion of an indeterminant zone. 65
For imaging, visual reads usually provide a normal/abnormal determination, but the approach of labeling some studies as indeterminant is common in clinical radiology and serves the same function as the indeterminant zone in quantitative analyses. While regulatory approval of amyloid and tau PET ligands was based on visual reads, the field is moving toward greater use of quantitative methods. 96
3.6. Clinical judgment
Clinical judgement must always be used in the practice of medicine, and the application of diagnostic tests for AD is no exception. Limitations of currently available biomarkers (Box 3) underscore the importance of clinical judgment when used clinically.
BOX 3: Limitations of biomarkers
Lack of certified biofluid reference methods and materials (except for cerebrospinal fluid [CSF] amyloid beta [Aβ]42, where these are available).
Biomarkers (positron emission tomography [PET], CSF, and blood) are less sensitive than neuropathologic examination for detection of early/mild Alzheimer's disease neuropathologic change (ADNPC). Disease staging by PET (or fluid biomarkers) is not equivalent to neuropathological staging, for example, tau PET ligand uptake in different Braak areas is not equivalent to Braak neuropathological staging. While the sensitivity limits of biomarkers could be viewed as a weakness, they could also be viewed as a strength because abnormal Core 1 biomarkers indicate that ADNPC more generally rather than just neuritic plaques alone is very likely present.
Thoroughly studied biomarkers are not available for all relevant diseases; therefore, it cannot be known with certainty in vivo what diseases in addition to AD are present in any individual, or what the proportional disease‐specific burden is among various pathologic entities. This leads to No. 4.
The proportion of the cognitive deficit observed in any individual that is attributable to AD versus other neuropathologic entities cannot be known with certainty. Probabilistic estimates can be made based on combinations of biomarker results and clinical judgment.
Because copathology is common, clinical judgment is always required to address this question: Is AD a cause of—or a dominant component of—a patient's symptoms (Box 4)? The nature of the syndromic presentation may indicate the likelihood that AD is or is not a dominant contributor to symptoms. For example, in someone with Parkinsonism and visual hallucinations who also has positive Core 1 biomarkers, the clinician's judgment is needed to assess the degree to which cognitive symptoms are likely attributable to AD versus neuronal synuclein disease (NSD). 57 In such a situation, additional testing may be clinically indicated. An abnormal Core 2 biomarker would suggest that AD is s a dominant contributor to symptoms, while a normal Core 2 biomarker would suggest that AD may be less so.
BOX 4: Implications for clinicians
The biologically based diagnosis of Alzheimer's disease (AD) is meant to assist rather than supplant the clinical evaluation of individuals with cognitive impairment. The revised criteria in this document are, in large part, a response to rapid advances in fluid‐based biomarkers, especially blood, and the approval of drugs specifically targeting amyloid beta (Aβ) pathology for individuals with early symptomatic AD—specifically mild cognitive impairment and mild dementia. Both advances are of high relevance to clinical use in the immediate and near future.
First, the clinical use of AD biomarkers is presently intended for the evaluation of symptomatic individuals, not cognitively unimpaired individuals. We highlight the distinction between can and should. AD can be diagnosed in asymptomatic individuals, but we do not believe this should be done for clinical purposes at this time.
Second, although the presence of abnormal Core 1 biomarkers is sufficient for confirming AD pathology in a symptomatic individual, it does not preclude the search for other contributors to the clinical symptoms, particularly other common copathologies. That said, higher biological stages, reflecting tau tangle pathology, increase the likelihood that the cognitive symptoms of an individual are due to AD pathology, and further inform on the prognosis. The integrated biological and clinical staging approach (see Table 7) may aid clinical judgment in assessing the contribution of AD to the clinical syndrome.
Third, the work group asserts that AD biomarkers are fundamental to making an accurate diagnosis and determining likely contributions to the patient's symptoms. Although it is expected that clinicians will use AD biomarkers to determine potential eligibility for recently approved Aβ‐specific therapies, clinical applications also include counseling and tailoring medications for symptomatic treatment.
Fourth, the development of Core biomarker categories wherein amyloid, tau fluid, and/or positron emission tomography biomarkers can be used for diagnosing or staging is intended to provide greater flexibility to clinicians regarding access to specific biomarkers and their judgment as to which biomarker(s) is most appropriate.
Fifth, because many of the currently available or soon to be available biomarker measures highlighted in this document have been used in clinical trials, they may provide greater opportunities for clinicians to make decisions about treatments for AD (therapeutic response and/or duration of treatments).
Clinical judgment is also essential when Core 1 biomarkers are discordant with the clinical impression: for example, a negative test result in a patient in whom the clinical presentation suggests a high probability of AD. In such a situation, additional testing is logical.
Clinical judgment is also required to assess the potential effects of confounding conditions on biomarker results. For example, head trauma or cardiorespiratory arrest may transiently increase p‐tau values. 97 Elevated p‐tau181 has been reported in autopsy‐verified cases of amyotrophic lateral sclerosis with little to no AD copathology. 98 Certain medications, 99 CSF dynamics disorders, 100 and impaired renal function can alter some plasma biomarker values. 101 , 102 , 103 These and other 104 potentially confounding situations should be apparent to the clinician.
Because of the essential role of clinical judgment, we recommend that biomarker testing should be performed only under the supervision of a clinician. Furthermore, clinicians should not be restricted by payers if additional testing seems clinically indicated. This is particularly pertinent for BBM given their much wider projected accessibility.
3.7. Intended uses
The major intended use for the biological diagnosis of AD in clinical trials is as an inclusion criterion. While a purely symptomatic therapy may not require documentation of AD biology, therapy directed toward a biological target requires confirmation of that biology.
Establishing an etiologic diagnosis is an essential aspect of good medical practice. Intended uses for a biological diagnosis of AD in clinical care include determining eligibility for treatments targeting core disease pathology based on drug registration criteria; additionally, they include counseling and tailoring medications for symptomatic (i.e., non–disease‐modifying) treatment. 105 , 106
At the present time disease‐targeted therapies have not been approved for cognitively unimpaired individuals with AD. For this reason, we currently recommend against diagnostic testing in cognitively unimpaired individuals outside the context of observational or therapeutic research studies (Box 4). This recommendation would change in the future if disease‐targeted therapies, currently being evaluated in trials, demonstrate benefit in preventing cognitive decline and are approved for clinical use in individuals with preclinical AD.
4. BIOLOGICAL DISEASE STAGING
We distinguish staging the severity of AD biology with biomarkers from staging the severity of clinical symptoms. This section addresses the former. Staging of AD applies only to individuals in whom the disease has been diagnosed by means of Core 1 biomarkers and does not apply to individuals who are not in the AD pathway. We structured this document to reflect this (i.e., diagnosis is the first step and only then does staging of AD become relevant).
4.1. Approaches to biological staging
In the 2018 framework, the “plus/minus” combinations of AT(N) were used as an informal staging scheme; individuals in the AD continuum were expected to progress from A+T−N− to A+T+N− to A+T+N+. 7 However, in 2018 we used the term biomarker profile rather than staging to avoid confusion with clinical staging. In the current update, however, we recommend an explicit scheme for staging the biological severity of AD that is distinct from staging the severity of clinical impairment, and then integrate these two staging axes.
Two general approaches may be taken for biological disease staging. Staging may be based on the order of biomarker events in the natural history of the disease, in which each event is categorized as present/abnormal (+) or absent/normal (−). This approach assumes that an archetypical order of biomarker events can be established through natural history studies; this sequence of biomarker events is then the de facto staging scheme. Alternatively, biological staging may be based on the magnitude of a continuous biomarker denoting progressively more severe disease. This latter approach is widely used for some diseases (e.g., estimated glomerular filtration rate [eGFR] for chronic kidney disease) but poses complexity for AD wherein two defining proteinopathies exist rather than a single physiologic readout.
4.2. Biological staging scheme overview
We recommend a biological staging scheme that uses only core biomarkers. N biomarkers certainly add prognostic information; 107 , 108 , 109 , 110 however, the temporal relationships between core AD biomarkers and N biomarkers are inconsistent across individuals. Biological staging implies that a person should progress from initial to advanced stages in sequence, and N biomarkers do not always follow a stereotypical A+ to T2+ to N+ sequence. People with abnormal Core 1 biomarker(s), who, by our definition, have AD, may develop significant neurodegeneration owing primarily to copathologies (Figures 1 and 2). The same reasoning applies to I biomarkers.
FIGURE 2.
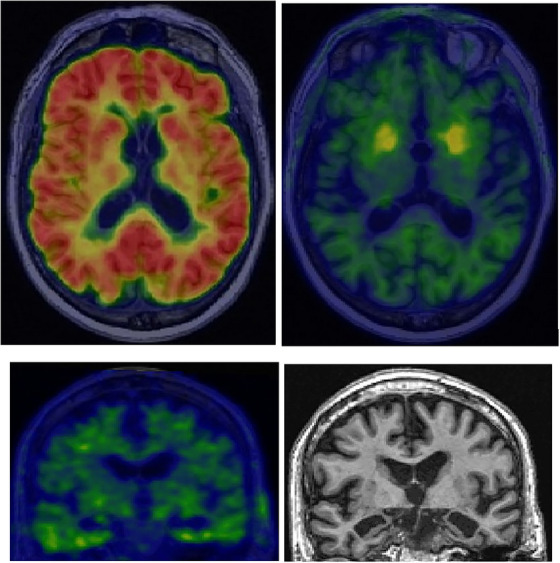
Copathology and T2N mismatch. An 89‐year‐old man with slowly progressive amnestic dementia. He carried a clinical diagnosis of probable Alzheimer's disease (AD) for several years and was receiving symptomatic treatment. AT2N imaging, however, revealed an abnormal amyloid positron emission tomographic (PET) scan (top left) but an unremarkable tau PET scan (top right and bottom left) that was insufficiently abnormal to explain the degree of atrophy or cognitive impairment (tau PET color scale reference is provided visually by the off‐target uptake in the basal ganglia; top right). The magnetic resonance imaging scan (bottom right) showed marked bilateral hippocampal atrophy that was consistent with the cognitive impairment but inconsistent with the level of tauopathy (i.e., T2N mismatch). The A+T2−N+ biomarker profile, along with the atrophy pattern on magnetic resonance imaging, suggested that the patient likely had comorbid AD and limbic‐predominant age‐related TAR DNA‐binding protein 43 encephalopathy (LATE).
We propose a four‐stage scheme based on the sequence of events observed in natural history studies: stage A, initial changing biomarkers; stage B, early changing; stage C, intermediate changing; stage D, advanced changing (Figure 1). Staging by amyloid and tau PET or with a combination of T1 fluid markers and tau PET is clinically viable at the present time and is our focus for biological staging (Tables 3 and 4). We also describe a conceptual staging scheme based on fluid biomarkers alone (Table 5). We do not attempt to link PET and fluid biomarker stages but do use the same naming convention within each modality.
TABLE 3.
Biological staging.
| Initial‐stage biomarkers | Early‐stage biomarkers | Intermediate‐stage biomarkers | Advanced‐stage biomarkers | |
| (A) | (B) | (C) | (D) | |
| PET | Amyloid PET | Tau PET medial temporal region | Tau PET moderate neocortical uptake | Tau PET high neocortical uptake |
| A+T2− | A+T2MTL+ | A+T2MOD+ | A+T2HIGH+ | |
| Core 1 fluid | CSF Aβ42/40, p‐tau181/Aβ42, t‐tau/Aβ42, and accurate a Core 1 plasma assays can establish that an individual is in biological stage A or higher, but cannot discriminate between PET stages A–D at present. | |||
Notes: Staging may be accomplished by (1) a combination of amyloid PET and tau PET or (2) a combination of Core 1 fluid biomarkers (which would establish biological stage A or higher) plus tau PET (which would be used to discriminate between stages). The approach to determining A+ versus A− with amyloid PET may need special consideration in autosomal dominant Alzheimer's disease (ADAD) and Down syndrome AD (DSAD).
Abbreviations: Aβ, amyloid beta; CSF, cerebrospinal fluid; PET, positron emission tomography; p‐tau, phosphorylated tau.
TABLE 4.
Operationalization of biological staging by positron emission tomography (PET).
| Stage | Amyloid PET | Tau PET medial temporal | Tau PET moderate neocortical uptake | Tau PET high neocortical uptake | AT2 notation |
|---|---|---|---|---|---|
| A | + | – | – | – | A+T2− |
| B | + | + | – | – | A+T2MTL+ |
| C | + | + | + | – | A+T2MOD+ |
| D | + | + | + | + | A+T2HIGH+ |
TABLE 5.
Conceptual biological staging with fluid biomarkers.
| Initial‐stage biomarkers | Early‐stage biomarkers | Intermediate‐stage biomarkers | Advanced‐stage biomarkers | |
|---|---|---|---|---|
| (A) | (B) | (C) | (D) | |
| Fluid staging | CSF Aβ42/40, p‐tau181/Aβ42, t‐tau/Aβ42, and accurate b plasma assays | Other p‐tau forms (e.g., p‐tau205 a ) | MTBR‐tau243 a | Non‐phosphorylated tau fragments a |
Note: PET and fluid measures are not equivalent; hence, stages A–D with PET should not be treated as equivalent to stages A–D for these fluid biomarkers.
Abbreviations: Aβ, amyloid beta; CSF, cerebrospinal fluid; MTBR, microtubule‐binding region; PET, positron emission tomography; p‐tau, phosphorylated tau.
Validation of p‐tau205, MTBR‐tau243, and non‐phosphorylated tau fragments as early‐, intermediate‐, and advanced‐stage fluid markers, respectively, is conceptual for now, awaiting further studies.
4.3. Biological staging with amyloid PET and tau PET
Unlike fluid biomarkers, imaging captures both topographic and magnitude information. Separate staging schemes for amyloid and tau PET have been proposed using either topographic distribution 111 , 112 , 113 , 114 , 115 , 116 , 117 , 118 or cutpoints in the continuous distribution of values from a defined region of interest (ROI). 92 , 118 , 119 , 120 However, PET staging that integrates both amyloid and tau PET has not been described, and a comprehensive disease staging scheme for AD should include both biomarker categories.
Highly reproducible results from observational studies show that abnormal amyloid PET often exists as an isolated finding in older individuals who are cognitively unimpaired and who do not have neocortical tau PET uptake or obvious neurodegeneration. 35 , 40 , 43 , 44 , 45 , 49 , 51 , 121 , 122 In contrast, high levels of neocortical tau PET are rarely seen in the absence of abnormal amyloid PET and are invariably accompanied by neurodegeneration and clinical symptoms. 35 , 40 In individuals with abnormal tau PET, clinical symptoms and neurodegeneration are closely related, both temporally and functionally, with location and magnitude of uptake on tau PET but not amyloid PET. 35 , 121 , 122 , 123 , 124 , 125 , 126 , 127 This set of findings is consistent with a stereotypical sequence of unidirectional imaging biomarker events that can be summarized as follows: abnormal amyloid PET (A) precedes tau PET (T2), which, in turn, leads to neurodegeneration (N) and clinical symptoms (C), A to T2 to N to C. 46 , 49 , 121 , 122 , 128 Amyloidosis appears to facilitate topographic spread of tauopathy, with the latter most commonly, but not always, beginning in medial temporal areas. 111 , 115 , 129 , 130
Therefore, we propose the following biological staging scheme for amyloid and tau PET (Tables 3 and 4). Stage A (initial)—abnormal amyloid PET with no uptake on tau PET (A+T2−); stage B (early)—abnormal amyloid PET plus tau PET uptake that is restricted to medial temporal areas (A+T2MTL+); stage C (intermediate)—abnormal amyloid PET plus tau PET uptake in the moderate standardized uptake value ratio (SUVR) range on a neocortical ROI (A+T2MOD+); and stage D (advanced)—abnormal amyloid PET plus tau PET uptake in the high SUVR range in the same neocortical ROI (A+T2HIGH+).
This PET staging scheme incorporates five elements. Both amyloid PET and tau PET are included to capture the two defining proteinopathies. Within tau PET, the scheme incorporates both topography (by distinguishing between medial temporal lobe and neocortical uptake) and uptake magnitude in the neocortical meta‐ROI. In addition, the neocortical meta‐ROI will capture staging for both typical and atypical/hippocampal sparing AD presentations. 131 , 132 We also point out that continuous measures of uptake in the neocortical tau PET ROI, while not a staging method, can provide a standardized anatomic target for quantification. Finally, this PET scheme can serve as a reference standard for validation of fluid‐based staging schemes, as outlined in Section 4.5.
Amyloid PET, like tau PET, exists on a continuous scale, and higher amyloid PET SUVR or Centiloid values are associated with more advanced disease and worse outcomes. 53 , 133 However, rather than incorporating a separate continuous amyloid PET scale into the PET staging scheme, amyloid PET is denoted in a binary manner, with the recognition that increasing amyloid PET uptake will be captured by progressively worse tau PET stages. 130 , 133 , 134
4.4. Biological staging with Core 1 fluid biomarkers and tau PET
Currently approved treatments targeting Aβ require documentation of Aβ pathology for treatment eligibility. It is anticipated that many patients will undergo testing with fluid Core 1 biomarkers to assess eligibility. Individuals in whom ADNPC has been established by fluid Core 1 biomarkers (Table 2) could then undergo tau PET; a single fluid test plus a single tau PET study could then be used for biological staging rather than amyloid PET and tau PET. Core 1 fluid biomarkers (Table 2) can establish that an individual is in stage A or higher, but may not accurately discriminate between stages A through D as defined by tau PET. 118 , 135
4.5. Biological staging with fluids
The onset of abnormal fluid Core 1 biomarkers occurs around the time of amyloid PET abnormalities and earlier than neocortical tau PET abnormalities. 21 , 22 , 23 , 136 In contrast, more recently developed tau assays (e.g., MTBR‐tau243 and non‐phosphorylated mid‐domain tau fragments) are more closely linked with the onset of abnormal tau PET and correlate better with tau PET than with amyloid PET; pT205 also correlates more strongly with tau PET than with amyloid PET. 136 , 137 , 138 , 139 , 140 From these data, a sequence of events has been proposed, with pathologic tau species appearing in the following order: p‐tau181, 217, or 231; then pT205; then MTBR‐tau243; then non‐phosphorylated mid‐domain tau fragments. 29 , 136 , 138 , 139 Based on these data, a fluid‐only staging scheme (Table 5) can be envisioned that mirrors the A through D scheme described earlier. P‐tau‐T205 136 and MTBR‐tau243 141 can be measured in both CSF and plasma. This fluid‐only staging scheme is regarded as conceptual at present, and specifics of this conceptual scheme are likely to change given the rapidly changing nature of the fluid biomarker field.
4.6. Caveats
We do not specify specific proprietary fluid assays, PET ligands, or numeric cutpoints for staging purposes in this document; these would fall under the purview of clinical practice guidelines.
Several caveats are specific to tau PET. First, care must be taken to identify off‐target tau ligand binding, which is not relevant to AD staging. Second, we recognize that PET‐detectable medial temporal tauopathy does not always precede neocortical tauopathy, particularly in atypical presentations. 132 However, medial temporal to neocortical spread is by far the most common pattern. 115 Third, we use topographic location of ligand uptake as one element of staging (medial temporal vs. neocortical), but we do not specify an inflexible set of anatomic ROIs to define the medial temporal or the neocortical meta‐ROIs for tau quantitation. Neocortical areas that reflect intermediate and advanced stages by virtue of association with diagnostic utility and prediction of cognitive decline include inferior and lateral temporal and inferior and medial parietal lobes; sampling of at least some of these areas should be included in a neocortical tau PET meta‐ROI. 107 , 110 , 116 , 127 , 129 , 142 Similarly, the medial temporal ROI could include the hippocampus (for some ligands), entorhinal cortex, and amygdala. Efforts are underway to standardize quantification of tau PET for all tracers (for example, the CenTauR scale 143 ) in the same way that the Centiloid scale 144 is the standardized method for quantifying amyloid PET. Fourth, the distinction between moderate (stage C) and high (stage D) neocortical tau uptake could be operationalized in different ways. Methods for quantification of tau PET is an area of active research, and selecting the best cutpoint to distinguish moderate versus high uptake will be informed by upcoming research findings.
PET is less sensitive than neuropathologic examination (Box 3), and PET‐based staging is not equivalent to autopsy‐based staging. However, PET‐based staging clearly has prognostic value. 145 For example, a large multi‐cohort meta‐analysis 40 using a PET‐based staging scheme very similar to what we describe, found a 40‐fold range in hazard ratios (HRs) based on PET stages. Specifically, relative to the A−T2− reference group, the HR for progression from cognitively unimpaired to dementia was 1.5 for A+T2− individuals (stage A in our scheme), HR 5.6 for A+ individuals with tau PET uptake confined to the medial temporal lobe (stage B in our scheme), and HR 39.9 for A+ individuals with neocortical tau PET uptake (stages C and D in our scheme).
The Centiloid scale is the accepted method for quantifying amyloid PET; however, this method is based on the anatomic distribution of ligand uptake in sporadic AD. 144 Florid striatal amyloid PET uptake often occurs early in individuals with ADAD and DS AD (DSAD), which is not the case in sporadic AD. 146 , 147 Therefore, the approach to determining A+ versus A− may need special consideration in ADAD and DSAD.
4.7. Intended uses
As with other diseases, more advanced biological AD stage predicts a worse clinical prognosis (Figure 1). 40 , 53 , 107 , 108 , 109 , 110 , 148 In addition, more advanced biological stage provides greater confidence that AD is meaningfully contributing to symptoms.
Biological staging in clinical trials would sharpen inclusion or stratification criteria by identifying individuals who should respond to treatment in a similar fashion, thus decreasing biological heterogeneity. Inclusion in the TRAILBLAZER‐ALZ and TRAILBLAZER‐ALZ2 studies was based on an abnormal amyloid PET result and on tau PET stage. 149 , 150 In the A4 and AHEAD studies, while inclusion was based on an abnormal amyloid PET result, study assignment within the trial was based on amyloid PET severity/stage. 151 , 152
5. CLINICAL STAGING
5.1. Numeric clinical staging
In the 2018 research framework, 7 we described a six‐stage numeric clinical staging scheme that is brought forward largely unchanged into this revision, and readers are referred to the earlier document for additional details. Clinical staging of AD applies only to individuals who are in the AD pathophysiologic continuum. The six clinically defined stages are as follows (Table 6): (1) biomarker evidence of AD in asymptomatic individuals; (2) transitional decline, which denotes the earliest detectable clinical symptoms that might be due to AD in individuals who are cognitively unimpaired; (3) objective cognitive impairment of insufficient severity to result in significant functional loss (i.e., inefficient activities of daily living but still independent); 4 to 6, loss of independence with progressively worse functional loss.
TABLE 6.
Clinical staging for individuals on the Alzheimer's disease continuum.
| Stage 0 Asymptomatic, deterministic gene a |
|---|
| No evidence of clinical change. Biomarkers in normal range. |
| Stage 1 Asymptomatic, biomarker evidence only |
|---|
| Performance within expected range on objective cognitive tests. |
| No evidence of recent cognitive decline or new symptoms. |
| Stage 2 Transitional decline: mild detectable change, but minimal impact on daily function |
|---|
| Normal performance within expected range on objective cognitive tests. |
| Decline from previous level of cognitive or neurobehavioral function that represents a change from individual baseline within the past 1 to 3 years, and has been persistent for at least 6 months. |
| May be documented by evidence of subtle decline on longitudinal cognitive testing, which may involve memory or other cognitive domains but performance still within normal range. |
| May be documented through subjective report of cognitive decline. |
| May be documented with recent‐onset change in mood, anxiety, motivation not explained by life events. |
| Remains fully independent with no or minimal functional impact on activities of daily living (ADLs) |
| Stage 3 Cognitive impairment with early functional impact |
|---|
| Performance in the impaired/abnormal range on objective cognitive tests. |
| Evidence of decline from baseline, documented by the individual's report or by an observer's (e.g., study partner) report or by change on longitudinal cognitive testing or neurobehavioral assessments. |
| Performs daily life activities independently but cognitive difficulty may result in detectable functional impact on complex ADLs (i.e., may take more time or be less efficient but still can complete—either self‐reported or corroborated by an observer). |
| Stage 4 Dementia with mild functional impairment |
|---|
| Progressive cognitive and mild functional impairment on instrumental ADLs, with independence in basic ADLs. |
| Stage 5 Dementia with moderate functional impairment |
|---|
| Progressive cognitive and moderate functional impairment on basic ADLs requiring assistance. |
| Stage 6 Dementia with severe functional impairment |
|---|
| Progressive cognitive and functional impairment, and complete dependence for basic ADLs. |
Individuals with Down syndrome may not be fully independent even in stage 0 because of underlying intellectual disability. In these individuals, decline in functional independence from baseline may be a more appropriate indicator of stage.
Numeric clinical stages 1 through 6 (Table 6) bear a close resemblance to the Global Deterioration Scale, 153 with the important distinction that the latter was created before the development of disease‐specific AD biomarkers. The six‐stage numeric scheme also closely resembles staging in the FDA guidance for conduct of clinical trials in early AD. 154
Stage 2 is called out as a distinct transitional stage between asymptomatic (stage 1) and mildly impaired (stage 3) and resembles “stage 3 preclinical AD” in the 2011 NIA‐AA guidelines. 1 This stage is defined by one or more of three components: objective cognitive decline, subjective cognitive decline, or subtle neurobehavioral difficulties. All three of these components can be attributable to AD, but they also can be attributable to factors other than AD, particularly neurobehavioral symptoms (e.g., depression, anxiety, apathy), 155 , 156 , 157 which are often not associated with neurodegenerative disease. An individual may be assigned to stage 2 based on neurobehavioral symptoms alone (i.e., without objective or subjective cognitive decline), but individuals must have cognitive impairment to be placed into stages 3 through 6. Advances in unsupervised digital cognitive testing may improve the ability to reliably detect the subtle cognitive alterations characteristic of stage 2 through repeated testing, but this remains to be determined.
The nature of cognitive decline or impairment in stages 2 through 6 may involve any cognitive domain(s), not only memory. Clinical staging is based on the severity of cognitive/functional impairment rather than on phenotype, but different phenotypic presentations of AD are well known. Five characteristic AD phenotypes are recognized: amnestic or “typical,” language variant, visuospatial variant, behavioral variant, and dysexecutive variant. 158 , 159 Different phenotypes often overlap within an individual, and the severity of impairment within each domain can be variable.
Although we describe clinical AD stages, it is important to bear two principles in mind. First, cognitive performance and cognitive decline are continuous processes. Dividing this continuous process into stages has value in clinical trials and clinical practice, but staging represents a categorical construct that is superimposed on a continuous process. Second, the severity of clinical impairment is the product of all neuropathological insults experienced by an individual, not only AD. The presence and severity of symptoms in an individual with abnormal AD biomarkers often cannot be ascribed solely to AD with confidence, particularly in older persons because of the likely presence of comorbid pathologic change (Box 3).
5.2. Stage 0
The change we propose in clinical staging from 2018 is the addition of stage 0. Stage 0 represents part of the AD continuum and is defined as genetically determined AD (which includes ADAD or DSAD) 46 , 160 in an individual who is biomarker negative and clinically asymptomatic (Table 6). The rationale is that these individuals have the disease from birth, prior to onset of brain pathologic change or symptoms. A person with DSAD or ADAD would move from stage 0 into stage 1 when a diagnostic Core 1 biomarker(s) became positive. The idea of stage 0 as genetically determined disease that has not yet manifested clinically or with biomarkers is conceptually consistent with recent staging proposals for Huntington's and NSD. 57 , 58 , 59
5.3. Risk alleles
We have not included AD risk alleles in the staging scheme because the presence of risk alleles does not indicate with certainty the presence or severity of ADNPC in an individual at a given point in time. This contrasts with Core biomarkers, which do. We therefore regard risk alleles as a risk factor for AD, not diagnostic of or a stage of AD.
Knowledge of apolipoprotein E (APOE) genotype has, however, assumed heightened clinical importance in the context of anti‐Aβ immunotherapy. The risk of amyloid‐related imaging abnormalities (ARIAs) is substantially greater in APOE ε4 homozygotes than in heterozygotes and non‐carriers. 161 Consequently, screening for APOE is recommended in the FDA label for lecanemab, and counseling around risk is recommended for homozygotes. 162
5.4. Syndromic staging
Often, the first step that patients undergo will be a clinical evaluation prior to any knowledge of biomarker results that would support a diagnosis of AD. During this initial clinical evaluation, clinicians may characterize the level of impairment based on a syndromic assessment (e.g., MCI or dementia). Approximate mapping of typical syndromes onto numeric AD clinical staging is as follows. Numeric clinical stages 1 and 2 correspond to cognitively unimpaired, with the latter corresponding somewhat to subjective cognitive decline; numeric stage 3 roughly corresponds to MCI; 163 , 164 , 165 and numeric stages 4, 5, and 6 correspond to mild, moderate, and severe dementia, respectively. Unlike numeric clinical staging, syndromic classification is not conditioned on a biological AD diagnosis and is applicable to individuals who may or may not have AD.
For individuals with biologically confirmed AD, we believe that numeric staging provides a clarifying framework for categorizing the clinical continuum of AD. The term prodromal AD has been used to denote individuals with abnormal AD biomarkers who have clinically evident impairment that falls short of dementia. Our position is that such an individual has the disease, not a prodrome of the disease, and our terminology in this instance would be “AD clinical stage 3” (Table 6). The same logic applies to unimpaired individuals with abnormal biomarker test results; we would categorize these individuals as AD clinical stage 1 or 2, not as “at risk for AD” because they already have the disease.
6. INTEGRATED BIOLOGICAL AND CLINICAL STAGING
As in the 2018 NIA‐AA framework, clinical staging and biological disease staging in our revised criteria are regarded as quasi‐independent variables. The symptomatic consequence of biological AD is modified by interindividual differences in copathologies, resistance, and reserve (i.e., education and other social determinants of health). 166 , 167 , 168 Consequently, the degree of cognitive/functional impairment does not follow in lockstep with biological AD severity (i.e., a range of possible relationships between biological AD stage and clinical stage will be found across the population; Figure 1). While clinical staging and biological staging must be performed independently, these two types of staging information can be integrated while still preserving independence of content.
As shown in Table 7, we propose an integrated biological and clinical staging scheme in which clinical stages are denoted in the columns using the numeric six‐stage scheme plus stage 0. Biological stages are denoted in the rows. Integrated stages appear in the cells. This format is intended to convey the concept that biological AD stage and clinical severity are related, but do not move in lockstep in all individuals.
TABLE 7.
Integrated biological and clinical staging.
| Stage 0 | Clinical Stage 1 | Clinical Stage 2 | Clinical Stage 3 | Clinical Stages 4–6 | |
|---|---|---|---|---|---|
| Initial biological stage (A) | X | 1A | 2A | 3A | 4‐6A |
| Early biological stage (B) | X | 1B | 2B | 3B | 4‐6B |
| Intermediate biological stage (C) | X | 1C | 2C | 3C | 4‐6C |
| Advanced biological stage (D) | X | 1D | 2D | 3D | 4‐6D |
Note: The typical expected progression trajectory is along the diagonal shaded cells, from 1A to 4–6D. However, considerable individual variability exists in the population. Individuals who lie above the diagonal (i.e., worse clinical stage than expected for biological stage) often have greater than average comorbid pathology. Individuals who lie below the diagonal (i.e., better clinical stage than expected for biological stage) may have exceptional cognitive reserve or resilience.
To avoid confusion when integrating numeric clinical staging with biological staging, we use numbers for clinical staging and letters for biological staging (Table 7). For example, clinical stage 2 and biological stage A are integrated stage 2A.
7. NEURODEGENERATION, INFLAMMATION, VASCULAR, AND α‐SYNUCLEIN (NIVS) BIOMARKER CATEGORIES
Tables 1 and 2 categorize core and non‐core biomarkers. We describe the latter here.
7.1. Biomarkers that are non‐specific but important in AD pathogenesis
In this revision we identify two categories of biomarkers that are not specific to AD but are important in the AD pathogenic pathway. These are N and I biomarkers.
In the 2018 research framework, we placed (N) in parentheses to emphasize that, in contrast to A and T, (N) biomarkers were not specific for AD. 7 We no longer use this parentheses notation because it should be clear that N biomarkers do not belong in the same category as core biomarkers. While neurodegeneration and neuronal injury are obviously important steps in AD pathogenesis, abnormalities in N biomarkers occur in many other conditions, including non‐AD neurodegenerative diseases, traumatic brain injury, ischemic injury, and others (Figure 1).
Fluid N biomarkers denote active neuronal injury or more subtle neuronal dysfunction. For example, NfL (neurofilament light chain) is a marker of large‐caliber axonal injury that can be measured in CSF or plasma and becomes abnormal in various disorders including multiple sclerosis, amyotrophic lateral sclerosis, and traumatic brain injury. 31 , 32 , 97 , 169 The absence of total tau from the fluid biomarker N category (Tables 1 and 2) is a departure from the 2018 NIA‐AA research framework. 7 CSF and plasma total tau begin to increase early in the disease course in ADAD 21 and closely correlate with fluid p‐tau in ADAD and sporadic AD. 97 This could be taken as evidence that total tau should be considered a T biomarker. However, CSF and plasma total tau also increase dramatically in Creutzfeldt–Jacob disease, head trauma, anoxia, cerebral infarction, as well as peripheral neuropathies, which has been taken as evidence that total tau belongs in the N category. 8 , 97 , 170 When all evidence is considered it is unclear how best to categorize this measure in the current document.
Imaging N biomarkers represent the net result of cumulative insults to the neuropil. Neurodegenerative loss of neurons and synapses results in volume loss (or decreased cortical thickness) on magnetic resonance imaging (MRI) 171 , 172 , 173 and fluorodeoxyglucose hypometabolism. Like their fluid counterparts, imaging N biomarkers are not specific to AD and may result from a variety of prior or ongoing brain insults. 174 , 175
Synaptic loss and dysfunction are an important feature of neurodegenerative diseases. Various synaptic CSF markers have been used for research purposes. 31 , 32 , 175 PET imaging of synapses has also entered the research arena based on ligands that bind to the synaptic vesicle glycoprotein 2A, a presynaptic component that may be lost with neurodegeneration. 176 , 177 A future direction for the field could be to identify more specific roles that various synaptic biomarkers could play and possibly to break out synaptic biomarkers from the broader N category. Electroencephalography may be one of the synaptic measures because it provides insight into synaptic connectivity and is widely available. 178
Biomarkers of inflammatory/immune processes (I) are divided into two subcategories: reactivity of astrocytes and microglia. A substantial body of evidence indicates that immune/inflammatory mechanisms are important in AD pathogenesis. 179 , 180 , 181 In addition, a growing list of interventional strategies targets immune/inflammatory pathways. 182 Despite the importance of these mechanisms, there is a dearth of available I biomarkers. An I marker that may gain clinical use is glial fibrillary acidic protein (GFAP), which is a marker of astrocytic reactivity. GFAP can be measured in plasma or CSF but seems to perform better in plasma for reasons that are not well understood. Although not specific to AD, it is associated with early amyloid PET, higher risk of incident dementia, and faster rates of cognitive decline. 31 , 32 , 175 , 183 , 184 , 185 , 186 , 187 Another I biomarker that has received recent attention is soluble TREM2, which reflects microglial reactivity and can be measured in CSF. 188 , 189 , 190
7.2. Biomarkers of common non‐AD copathologies
We list biomarkers of α‐synuclein (S) and vascular brain injury (V) in Tables 1 and 2 under the heading of biomarkers of non‐AD copathology. Αlpha‐synuclein seed amplification assays (αSyn‐SAA) in CSF have gained attention in Parkinson's disease and dementia with Lewy bodies, 191 , 192 which have recently been relabeled as NSD. 57 αSyn‐SAA are sensitive and specific for ante mortem identification of cortical α‐synuclein pathologic change as a primary pathology or as a copathology. 193 , 194 Cortical α‐synuclein pathologic change is associated with cognitive decline independent of amyloid and tau pathology in both cognitively unimpaired and cognitively impaired individuals. 195 , 196 These assays are less sensitive to α‐synuclein inclusions in multisystem atrophy in which the cellular location and conformation of inclusions differ from those in NSD. 197 , 198 Development of PET ligands for α‐synuclein is an active area of research but, at present, no ligands are available for the detection of α‐synuclein copathology in patients with AD. 199 , 200 Dopamine transporter (DAT) single‐photon emission computed tomography (SPECT) or PET are DAT imaging methods used clinically to assess loss of striatal dopaminergic neurons in the evaluation of patients with suspected NSD. 57 , 201 , 202 DAT scanning plays a prominent role in recent staging criteria for NSD. 57
Cerebrovascular disease (V) is an umbrella term that encompasses different forms of vascular pathology or vascular brain injury. Various diagnostic modalities or imaging findings exist; however, at this point a single summary measure composed of different imaging findings has not been widely accepted. Macroscopic cerebral infarctions, including both large cortical and subcortical infarctions and lacunes, on anatomic MRI or computed tomography are the most definitive biomarker of ischemic vascular brain injury and are widely used for this purpose in clinical care (Tables 1 and 2). State‐of‐the‐art methods in neuroimaging of small‐vessel disease 203 are reviewed in the recent STRIVE‐2 guidelines. 204 While diffusion‐weighted imaging is used routinely in clinical practice to identify acute cerebral infarction, quantitative diffusion MRI has gained traction as a method to detect loss of microscopic tissue integrity due to small vessel disease in research. 205 , 206 , 207 However, diffusion MRI (a broad field that encompasses many approaches) findings are also abnormal in neurodegenerative diseases, traumatic brain injury, and so forth. The same reasoning applies to perfusion MRI (arterial spin labeling or variants). Thus, these modalities are not disease specific. White matter hyperintensities on MRI have long been interpreted to indicate microvascular ischemic injury. 208 However, white matter hyperintensities may also be attributed to distal axonopathy, autoimmune demyelination, and loss of blood–brain barrier integrity from cerebral amyloid angiopathy (CAA). 209
The vascular markers described earlier are linked with traditional systemic vascular risk factors and cerebral ischemia. CAA merits special mention because while the disorder is one of cerebral vessels, the etiology is disordered processing of Aβ rather than traditional systemic vascular risk factors, and CAA is commonly observed in association with Aβ plaques in AD. CAA represents the aggregation of Aβ in cerebral vessel walls leading to vessel fragility. 210 This, in turn, can lead to spontaneous leakage or exudate of intravascular contents, including heme products, into the sulcal space or brain parenchyma. The result is seen on MRI as superficial siderosis or cerebral microbleeds, typically in a lobar distribution. 211 A serious potential complication is lobar hemorrhage. 212 MRI evidence of CAA (microbleeds or siderosis) increases the risk of ARIA in patients undergoing some forms of anti‐Aβ immunotherapy, and, thus, detection plays an important role in clinical care. 213
8. MULTIMODAL BIOMARKER PROFILES AND IDENTIFICATION OF COMORBID PATHOLOGIC CHANGE
We distinguish multimodal biomarker “profiles” from AD biological staging. Biomarker profiles may involve the use of core and non‐core biomarkers to characterize the general neuropathophysiological state of an individual beyond or in addition to the presence of AD. Biological staging of AD applies only to individuals in whom AD has been detected by core biomarkers; in contrast, biomarker profiles are applicable to all individuals in the population.
Using markers outlined in Tables 1 and 2, a full multimodal biomarker profile would appear as AT1T2NISV, with results indicated (± dichotomized or a continuous quantitative scale) as appropriate to each category. Full profiles require extensive biomarker phenotyping; however, partial profiles may be useful conceptually and in clinical practice to characterize individuals.
One potential use of multimodal biomarker profiles is to provide simple conceptual organization and practical shorthand notation to characterize persons with comorbid pathologies. With advancing age, copathologies are the rule and isolated AD is the exception. Thus, AD must be considered in the context of copathologies. Common age‐related brain pathologies that underlie cognitive impairment or dementia other than AD in older persons are cerebrovascular disease, NSD, and limbic‐associated TAR DNA‐binding protein (TDP‐43) encephalopathy (LATE). 214 , 215 , 216 , 217 , 218 , 219 , 220 , 221 , 222 , 223 LATE merits special mention because while it is a common and clinically important contributor to late‐life cognitive impairment, no confirmed disease‐specific biomarkers exist currently. 214 CSF dynamics disorders may contribute to impairment and can be detected by MRI. 224 Examples of direct indicators of copathology include a positive αSyn‐SAA test result (A+T2+S+) or multiple infarctions (A+T2+V+) in someone who also had biomarker evidence of AD. However, there are also useful indirect indicators that one or more non‐AD copathologies likely is present, as described below.
To this point we have not emphasized N biomarkers, but a useful indirect indicator of copathology is a “T2N” mismatch in an AT2N profile. 225 , 226 , 227 , 228 Neurodegeneration in AD is closely related in time and topography to tau deposition. The T2−N+ biomarker profile (i.e., T2N mismatch) therefore indicates the presence of neurodegeneration or neuronal injury due to a disease(s) other than AD. An archetypical example of this is an older person with a progressive amnestic clinical course and an A+T2−N+ biomarker profile, in which N+ is represented by severe medial temporal lobe atrophy on MRI or hypometabolism on PET (Figures 1 and 2). Such a person has AD biological stage A (denoted by A+T2−), but likely also has LATE (denoted by T2−N+). 214
8.1. Intended uses
Indicators of copathology may be useful clinically when considering the etiology of symptoms, prognosis, and treatment decisions. For example, a cognitively impaired individual with an A+T2−N+ biomarker profile may not respond to anti‐Aβ immunotherapy in the same manner as someone who has an A+T2+N− or A+T2+N+ biomarker profile.
In clinical trials, indicators of copathology could be used as exclusionary criteria in phase 2 trials in which a biologically homogeneous cohort with purer AD is desirable to maximize statistical power. Individuals with indicators of copathology could be included in phase 3 AD trials, with preplanned subset analyses, to establish efficacy in a broader population.
9. TREATMENT EFFECTS
The focus of this document is on criteria for diagnosis and staging of AD; detailed discussion of the roles of biomarkers as outcome measures or indicators of target engagement in clinical trials is beyond the scope of this work. Nonetheless, the recent regulatory approval of treatments targeting core AD pathology promises to be transformative. Anti‐Aβ immunotherapy can dramatically reduce the load of amyloid plaque in a time‐ and dose‐dependent manner; change downstream biomarkers (including CSF and plasma p‐tau and total tau 150 , 161 , 229 , 230 and plasma GFAP 161 , 230 ) in the direction of normalization; and slow accumulation of tau PET. 161 , 229 Most importantly, recent trials have demonstrated that anti‐Aβ immunotherapy that substantially reduces fibrillar Aβ levels measured on PET can slow the rate of cognitive decline in early symptomatic AD. 149 , 150 , 161 , 229 Consistent results across both successful and failed immunotherapy trials show that the amount of amyloid PET reduction is associated with the degree of clinical benefit. These findings linking biology to clinical manifestations, which have been replicated across independent therapeutic programs, 149 , 150 , 161 , 229 provide solid empiric support for a biological definition of AD.
While Aβ may be reduced to subdetection levels on PET, this does not mean that the disease pathophysiology has been eradicated. Individuals followed after cessation of Aβ immunotherapy have shown decreasing plasma Aβ42/40, small recurrent accumulation of amyloid on PET, and clinical progression similar to that in patients receiving placebo. 231 The underlying AD pathophysiologic process is therefore still active in an individual who has had fibrillar amyloid removed to below PET detection levels. The biological diagnosis and staging schemes outlined earlier are based on observations of the natural history of the disease. Successful disease‐modifying therapies alter the relationships between biomarkers that are present in the natural evolution of the disease. For example, an individual who has been treated with an anti‐Aβ monoclonal antibody may change from A+ at baseline to A− after treatment, but the pathogenic process is still present. This state has recently been labeled “Treatment Related Amyloid Clearance (TRAC)” by an AA–convened workgroup. Therefore, the staging schemes outlined earlier should be regarded as tools for diagnosis, staging/prognosis, and treatment assignment but not as indicators of the stage of the natural history of the disease posttreatment.
Anti‐Aβ immunotherapies that reduce plaque load often result in higher rates of whole‐brain volume loss or ventricular enlargement in treated versus placebo individuals. 149 , 229 , 232 , 233 Interestingly, the hippocampus seems to be spared from this effect. 150 , 161 Explanations for the volume reduction effect include therapy‐induced fluid shifts or a reduction in volume of amyloid plaque and peri‐plaque inflammation. 232 It has become apparent that slowing of the rate of whole‐brain volume loss by successful amyloid plaque removal, which was anticipated based on natural history studies, has not been seen in the relatively short duration of most clinical trials. Slowing whole‐brain atrophy rates may occur over much longer time scales with successful therapy, but this remains to be shown. Serial whole‐brain volume measures may only be considered a measure of the rate of neurodegeneration in the absence of abrupt changes in plaque volume or brain edema. MRI does have an important role in anti‐amyloid therapy in trials and in clinical use as a means of identifying ARIAs for safety purposes. 213 , 234
10. DIVERSITY AND NEED FOR MORE REPRESENTATIVE COHORTS
The need for more representative cohorts for observational studies and clinical trials has been pointed out repeatedly, and the committee endorses this position. 85 , 235 , 236 , 237 Much of the published AD biomarker data have been derived from highly educated, non‐Hispanic White cohorts and these biomarkers have not yet been tested extensively in broadly representative populations. Relationships among biomarkers, genetic variants like APOE ε4, and clinical outcomes may differ by race/ethnicity. 238 , 239 , 240 , 241 , 242 The prevalence of APOE ε4, 243 prevalence of biomarker abnormalities, and mixed pathology may also differ by race/ethnicity; however, it is often unclear to what degree this may be influenced by enrollment biases in existing cohort studies. 102 , 244 , 245 , 246 A relatively consistent theme across much of the current literature is that groupwise differences in the prevalence of AD biomarker abnormalities do not explain the higher prevalence of dementia in Black and Hispanic groups compared to non‐Hispanic White groups. 247 , 248 It is unclear to what extent this may be attributable to a higher prevalence of non‐AD pathologies in underrepresented groups (e.g., cerebrovascular disease) or to additional factors. 244
Representativeness encompasses many factors, including race and ethnicity, but also social determinants of health such as socioeconomic status, education, geographic location, and lifestyle. Definitive observational studies with more representative cohorts are needed to assess natural history relationships among biomarkers, genetics, comorbidities, and clinical outcomes. Furthermore, randomization rates and eligibility rates for AD clinical trials vary disproportionately by race/ethnicity, education, and socioeconomic status. Therefore, more representative cohorts are also needed in trials to assess whether treatments are effective across race/ethnicity and the range of social determinants of health. 102 , 246 , 249
11. FUTURE DIRECTIONS
In recent years the field has moved from diagnosing and characterizing AD based on clinical presentation alone to diagnosing the disease biologically. Biologically based diagnosis and staging is now transitioning from priorities dominated by research alone to the priorities required for both research and clinical care. Future directions could include the following.
Observational studies and clinical trials should be conducted with more representative cohorts. Furthermore, nearly all observational studies in underrepresented groups that have included biomarkers have been in convenience samples with inevitable selection biases. True epidemiolocal and real‐world data studies of biomarker properties in representative groups are needed to ascertain relationships that are valid at the population level.
Standardization of biofluid assays, tau PET quantification methods, and cutpoints is needed. As in other diseases, the exact thresholds for abnormality may evolve over time as additional data inform prognostic value.
Improved understanding of various posttranslational modifications of tau may enhance fluid‐based biological staging.
With improved understanding of the role of immune/inflammatory processes, microglia, and astrocyte biology in AD pathogenesis, we envision a more prominent role for I biomarkers in biological characterization and prognosis, especially if brain‐specific changes can be detected in blood.
As clinical trials targeting mechanisms other than anti‐Aβ immunotherapy are performed, the effects of these interventions on biomarkers and clinical outcomes should be included in future criteria.
We envision creating a comprehensive system to stratify risk of onset and progression by incorporating all biomarkers (core AD, non‐core, and biomarkers of non‐AD copathology) along with demographics and genetics. Practical operationalization of a complete AT1T2NISV diagnosis and staging system (with the future addition of TDP‐43) is aspirational at this point but could be conceivable with the development of multiplex BBM assays.
The committee recognizes that the feasibility of implementing these criteria for biologically based diagnosis and staging of AD in clinical practice varies across regions, even within World Bank–designated high‐income countries. We anticipate that the increasing availability of BBM will make these criteria more widely deployable within World Bank–designated low‐ to middle‐income countries where PET‐ and CSF‐based biomarkers may not be readily available.
CONFLICT OF INTEREST STATEMENT
Clifford R. Jack Jr. is employed by Mayo Clinic. Within the past 36 months, he served on a Data Safety Monitoring Board for Roche pro bono; no payments to the individual or institution were involved. In addition, he holds index funds. J. Scott Andrews is employed by Takeda Pharmaceuticals and is a minor shareholder for the company. He has a leadership or fiduciary role in the Clinical Trials on Alzheimer's Disease (CTAD) Task Force—Clinical Meaningfulness and Optimizing Therapies, AD PACE Executive Steering Committee, and UsAgainstAlzheimer's—Clinical Meaningfulness Forum. He is a former employee of Eli Lilly and Company and is a minor shareholder for the company. Thomas G. Beach is employed by Banner Health. He has received consulting fees from Aprinoia Therapeutics and Acadia Pharmaceuticals. He consulted for Biogen. He has received payment or honoraria from the World PD Coalition, Mayo Clinic Florida, Stanford University, and the IOS Press‐Journal of Parkinson's Disease and support for attending meetings from the Alzheimer's Association, AD/PD/Kenes Group, Mayo Clinic Florida, Universitätsklinikum Hamburg‐Eppendorf, and the International Movement Disorders Association. In addition he has recieved funds from the National Institutes of Health for reviewing grant proposals. He also has a leadership/fiduciary role and stock options with Vivid Genomics. Teresa Burracchio is employed by the US Food and Drug Administration and has no financial conflicts to disclose. Billy Dunn is a consultant and has received consulting fees for his role as an advisor for Arch Venture Partners, Cerveau Technologies, Epilepsy Foundation, F‐PRIME Capital, Loulou Foundation, and Michael J. Fox Foundation. He has held a past position of leadership or fiduciary role in the Virginia Neurological Society (past president) and Prothena Inc. (Director). He holds stock options with Prothena Inc. Ana Graf is employed by Novartis Pharma AG and has stock options in the company. Oskar Hansson is employed by Lund University. He has grants or contracts from ADx, AVID Radiopharmaceuticals, Biogen, Eli Lilly, Eisai, Fujirebio, GE Healthcare, Pfizer, and Roche. He also has been paid consulting fees by AC Immune, Amylyx, Alzpath, BioArctic, Biogen, Cerveau, Eisai, Eli Lilly, Fujirebio, Genentech, Merck, Novartis, Novo Nordisk, Roche, Sanofi, and Siemens. In the past 2 years, he has received consultancy/speaker fees from AC Immune, Alzpath, BioArctic, Biogen, Bristol Meyer Squibb, Cerveau, Eisai, Eli Lilly, Fujirebio, Merck, Novartis, Novo Nordisk, Roche, Sanofi, and Siemens. Carole Ho is employed by Denali Therapeutics. She has stock options with Denali Therapeutics. She is also a Board Director for Beam Therapeutics and NGM Therapeutics and has stock options with both companies. William Jagust is employed by University of California, Berkley. He has received consulting fees from Biogen, Clario, Eisai, Lilly, and Prothena and has stock with Optoceutics and Molecular Medicine. Eric McDade is employed by Washington University, St Louis. He has received royalties for work on “Methods of diagnosing AD with phosphorylation changes,” which is licensed to C2N with royalties to himself and Washington University. He has received consulting fees from Alzamend (SAB member), Sanofi, AstraZeneca, Roche, Grifols, Merck, and Sage. He has also received payments from Neurology Live; Kaplan‐Projects in Knowledge; and support for travel from Foundation Alzheimer, Alzheimer's Association, and Eisai. He has a patent planned, issued, or pending on “Methods of diagnosing AD with phosphorylation changes” and has participated on a Data Safety Monitoring Board or Advisory Board for Alector and Eli Lilly, both roles for which he was paid. He has a leadership/fiduciary role in Alzamend (paid). Jose Luis Molinuevo is a full‐time employee of H. Lundbeck A/S. Ozioma Okonkwo is employed by University of Wisconsin School of Medicine & Public Health. He has received consulting fees from Mayo Clinic Rochester and IUPUI, and has held a leadership or fiduciary role in the International Neuropsychological Society (Board Member) and previously held an advisory role for Society for Black Neuropsychology. Luca Pani is employed by the University of Miami and the University of Modena and Reggio Emilia and has no financial conflicts to disclose. In unrelated areas, he holds options/stocks from Relmada Therapeutics (US) and NetraMark (Canada). Michael Rafii is employed by University of Southern California and the Alzheimer's Therapeutics Research Institute (ATRI). He has received consulting fees from AC Immune and Ionis. He has participated on a Data Safety Monitoring Board or Advisory Board for Alzheon, Aptah Bio, Biohaven, Embic, Keystone Bio, Prescient Imaging, and Positrigo. Philip Scheltens is employed by Life Science Partners and is Professor Emeritus Amsterdam University Medical Center. He has an unpaid leadership position as Chair of World Dementia Council. He holds stock options in EQT AB. Eric Siemers is employed by Acumen Pharmaceuticals, Inc. He has received consulting fees from Cogstate Ltd., Cortexyme Inc., Partner Therapeutics Inc., Vaccinex Inc., Gates Ventures LLC, and Hoffman La Roche Ltd and payments were made to Siemers Integration LLC. He has participated on a Data Safety Monitoring Board for Hoffman La Roche Ltd. and has had a leadership or fiduciary role with the Alzheimer's Association and BrightFocus Foundation, both unpaid. He holds stock options and is a shareholder for Acumen Pharmaceuticals, and is a shareholder for Eli Lilly and Company. Heather Snyder is a full‐time employee of the Alzheimer's Association, Chicago, IL, and has a spouse who is employed by Abbott Laboratories in an unrelated area. She has no financial conflicts to disclose. Reisa Sperling is employed by Brigham and Women's Hospital. She has received consulting fees from Abbvie, AC Immune, Acumen, Alector, Alnylam, Biohaven, Bristol‐Myers Squibb, Cytox, Genentech, Ionis, Janssen, Merck, NervGen, Neuraly, Neurocentria, Oligomerix, Prothena, Roche, Shionogi, and Vaxxinity. Charlotte E. Teunissen is employed by Amsterdam UMC. She is recipient of ABOARD, which is a public–private partnership receiving funding from ZonMW (#73305095007) and Health∼Holland, Topsector Life Sciences & Health (PPP‐allowance; #LSHM20106). She is also a contract researcher for ADx Neurosciences, AC‐Immune, Aribio, Axon Neurosciences, Beckman‐Coulter, BioConnect, Bioorchestra, Brainstorm Therapeutics, Celgene, Cognition Therapeutics, EIP Pharma, Eisai, Eli Lilly, Fujirebio, Grifols, Instant Nano Biosensors, Merck, Novo Nordisk, Olink, PeopleBio, Quanterix, Roche, Siemens, Toyama, Vivoryon, and the European Commission. She has received payment or honoraria from Eli Lilly, Grifols, Novo Nordisk, Olink, and Roche, where all payments were made to her institution. She also serves on editorial boards of Medidact Neurologie/Springer; and on Neurology: Neuroimmunology & Neuroinflammation. She is editor of Alzheimer Research and Therapy. Maria Carrillo is a full‐time employee of the Alzheimer's Association and has a daughter in the neuroscience program at USC. She has no financial conflicts to disclose. Eliezer Masliah and Laurie Ryan served as advisory members to the workgroup. They are employed by National Institute on Aging at the National Institutes of Health and have no financial conflicts to disclose. Author disclosures are available in the supporting information.
Supporting information
Supporting Information
ICMJE Disclosure Form
ACKNOWLEDGMENTS
Clifford R. Jack Jr. received grant funding from the National Institutes of Health, the Alexander family professorship, and the GHR Foundation. He has also received funding from the Alzheimer's Association for travel. Thomas G. Beach has received grant funding from the National Institutes of Health, Veterans Administration, State of Arizona, Life Molecular Imaging, Michael J. Fox Foundation for Parkinson's Research, Avid Radiopharmaceuticals/Eli Lilly, and Gates Foundation. Oskar Hansson has acquired research support (for the institution) from AVID Radiopharmaceuticals, Biogen, C2N Diagnostics, Eli Lilly, Eisai, Fujirebio, GE Healthcare, and Roche. William Jagust received grants or contracts paid to his institution from the National Institute on Aging, Roche/Genentech, Alzheimer's Association, and BrightFocus Foundation. Eric McDade received grant funding from NIA, Eli Lilly, Hoffmann‐La Roche, Alzheimer's Association, and GHR Foundation all directly to his institution. Ozioma Okonkwo received support from the following grants, paid to his institution: R01AG062167, R01AG077507, U19AG024904, U19AG078109, U19AG073153, R01AG066203, R01AG070028, R01AG027161, and RF1AG052324. Michael Rafii received grants or contracts from Eisai (AHEAD Study) and Eli Lilly (A4 Study), which were paid to his institution. Philip Scheltens received grants or contracts from Novo Nordisk, Toyama, UCB, AC Immune, and Alzheon, all paid to his institution. Reisa Sperling received grants or contracts from the National Institute on Aging, Eli Lilly (public–private partnership trial funding), Eisai (public–private partnership trial funding), Alzheimer's Association, and GHR Foundation. Charlotte E. Teunissen received grants or contracts for Research of the European Commission (Marie Curie International Training Network, grant agreement No 860197; MIRIADE), Innovative Medicines Initiatives 3TR (Horizon 2020, grant no 831434) EPND (IMI 2 Joint Undertaking (JU), grant No. 101034344) and JPND (bPRIDE), National MS Society (Progressive MS alliance), Alzheimer Drug Discovery Foundation, Alzheimer Association, Health Holland, the Dutch Research Council (ZonMW), including TAP‐dementia, a ZonMw funded project (#10510032120003) in the context of the Dutch National Dementia Strategy, Alzheimer Drug Discovery Foundation, The Selfridges Group Foundation, and Alzheimer Netherlands.
Jack CR, Andrews JS, Beach TG, et al. Revised criteria for diagnosis and staging of Alzheimer's disease: Alzheimer's Association Workgroup. Alzheimer's Dement. 2024;20:5143–5169. 10.1002/alz.13859
Contributing committee members: Eliezer Masliah and Laurie Ryan.
Clifford R. Jack Jr, Billy Dunn, Heather M. Snyder, Eliezer Masliah, and Maria C. Carrillo are the members of the steering committee.
REFERENCES
- 1. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Assocation workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging and Alzheimer's Association Workgroup. Alzheimers Dement. 2011;7:270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging and the Alzheimer's Assocation Workgroup. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jack CR Jr., Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jack CR Jr., Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jack CR Jr., Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87:539‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lowe VJ, Lundt ES, Albertson SM, et al. Neuroimaging correlates with neuropathologic schemes in neurodegenerative disease. Alzheimers Dement. 2019;15:927‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. La Joie R, Ayakta N, Seeley WW, et al. Multisite study of the relationships between antemortem [(11)C]PIB‐PET Centiloid values and postmortem measures of Alzheimer's disease neuropathology. Alzheimers Dement. 2019;15:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid‐beta plaques: a prospective cohort study. Lancet Neurol. 2012;11:669‐678. [DOI] [PubMed] [Google Scholar]
- 12. Murray ME, Lowe VJ, Graff‐Radford NR, et al. Clinicopathologic and 11C‐Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain. 2015;138:1370‐1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thal DR, Beach TG, Zanette M, et al. [(18)F]flutemetamol amyloid positron emission tomography in preclinical and symptomatic Alzheimer's disease: specific detection of advanced phases of amyloid‐beta pathology. Alzheimers Dement. 2015;11:975‐985. [DOI] [PubMed] [Google Scholar]
- 14. Lowe VJ, Lundt ES, Albertson SM, et al. Tau‐positron emission tomography correlates with neuropathology findings. Alzheimers Dement. 2020;16:561‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fleisher AS, Pontecorvo MJ, Devous MD Sr., et al. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 2020;77:829‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Villemagne VL, Lopresti BJ, Dore V, et al. What is T+? A gordian knot of tracers, thresholds, and topographies. J Nucl Med. 2021;62:614‐619. [DOI] [PubMed] [Google Scholar]
- 17. Palmqvist S, Mattsson N, Hansson O. Cerebrospinal fluid analysis detects cerebral amyloid‐beta accumulation earlier than positron emission tomography. Brain. 2016;139:1226‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512‐519. [DOI] [PubMed] [Google Scholar]
- 19. Schindler SE, Gray JD, Gordon BA, et al. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement. 2018;14:1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Landau SM, Lu M, Joshi AD, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta‐amyloid. Ann Neurol. 2013;74:826‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barthelemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26:398‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Janelidze S, Berron D, Smith R, et al. Associations of Plasma Phospho‐Tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA Neurol. 2021;78:149‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mattsson‐Carlgren N, Andersson E, Janelidze S, et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer's disease. Sci Adv. 2020;6:eaaz2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Therriault J, Vermeiren M, Servaes S, et al. Association of phosphorylated tau biomarkers with amyloid positron emission tomography vs tau positron emission tomography. JAMA Neurol. 2023;80:188‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sato C, Barthelemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;98:861‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mattsson‐Carlgren N, Janelidze S, Bateman RJ, et al. Soluble P‐tau217 reflects amyloid and tau pathology and mediates the association of amyloid with tau. EMBO Mol Med. 2021;13:e14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pichet Binette A, Franzmeier N, Spotorno N, et al. Amyloid‐associated increases in soluble tau relate to tau aggregation rates and cognitive decline in early Alzheimer's disease. Nat Commun. 2022;13:6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Horie K, Barthelemy NR, Spina S, et al. CSF tau microtubule‐binding region identifies pathological changes in primary tauopathies. Nat Med. 2022;28:2547‐2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salvado G, Horie K, Barthelemy NR, et al. Novel CSF tau biomarkers can be used for disease staging of sporadic Alzheimer's disease. medRxiv. 2023. Online Ahead of Print. [Google Scholar]
- 30. Salvado G, Ossenkoppele R, Ashton NJ, et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads. EMBO Mol Med. 2023;15:e17123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Teunissen CE, Verberk IMW, Thijssen EH, et al. Blood‐based biomarkers for Alzheimer's disease: towards clinical implementation. Lancet Neurol. 2022;21:66‐77. [DOI] [PubMed] [Google Scholar]
- 32. Hansson O. Biomarkers for neurodegenerative diseases. Nat Med. 2021;27:954‐963. [DOI] [PubMed] [Google Scholar]
- 33. Hansson O, Blennow K, Zetterberg H, Dage J. Blood biomarkers for Alzheimer's disease in clinical practice and trials. Nat Aging. 2023;3:506‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schindler S, Galasko D, Pereira A, et al. I use in symptomatic patients: recommendations from the Global CEO Initiative on Alzheimer's Disease. Nature Rev Neurol. 2024. [DOI] [PubMed] [Google Scholar]
- 35. Jack CR Jr., Wiste HJ, Botha H, et al. The bivariate distribution of amyloid‐beta and tau: relationship with established neurocognitive clinical syndromes. Brain. 2019;142:3230‐3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Costoya‐Sanchez A, Moscoso A, Silva‐Rodriguez J, et al. Increased medial temporal tau positron emission tomography uptake in the absence of amyloid‐beta positivity. JAMA Neurol. 2023;80:1051‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jack CR Jr., Knopman DS, Chetelat G, et al. Suspected non‐Alzheimer disease pathophysiology – concept and controversy. Nat Rev Neurol. 2016;12:117‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Erickson P, Simren J, Brum WS, et al. Prevalence and clinical implications of a β‐amyloid‐negative, tau‐positive cerebrospinal fluid biomarker profile in Alzheimer disease. JAMA Neurol. 2023;80:969‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jack CR Jr., Wiste HJ, Weigand SD, et al. Age‐specific population frequencies of cerebral beta‐amyloidosis and neurodegeneration among people with normal cognitive function aged 50‐89 years: a cross‐sectional study. The Lancet Neurology. 2014;13:997‐1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ossenkoppele R, Pichet Binette A, Groot C, et al. Amyloid and tau PET‐positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat Med. 2022;28:2381‐2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Landau SM, Mormino EC. Tau pathology without Aβ‐A limited PART of clinical progression. JAMA Neurol. 2023;80:1025‐1027. [DOI] [PubMed] [Google Scholar]
- 42. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128:755‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446‐452. [DOI] [PubMed] [Google Scholar]
- 44. Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jack CR Jr., Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. New Engl J Med. 2012;367:795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benzinger TL, Blazey T, Jack CR Jr., et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci U S A. 2013;110:E4502‐4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y, Yen D, Hendrix RD, et al. Timing of biomarker changes in sporadic Alzheimer's disease in estimated years from symptom onset. Ann Neurol. 2024;95:951‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guo T, Korman D, Baker SL, Landau SM, Jagust WJ, Alzheimer's Disease Neuroimaging I . Longitudinal cognitive and biomarker measurements support a unidirectional pathway in Alzheimer's disease pathophysiology. Biol Psychiatry. 2021;89:786‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jia J, Ning Y, Chen M, et al. Biomarker changes during 20 years preceding Alzheimer's disease. N Engl J Med. 2024;390:712‐722. [DOI] [PubMed] [Google Scholar]
- 51. Jack CR Jr., Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355‐1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brookmeyer R, Evans DA, Hebert L, et al. National estimates of the prevalence of Alzheimer's disease in the United States. Alzheimers Dement. 2011;7:61‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jack CR Jr., Therneau TM, Lundt ES, et al. Long‐term associations between amyloid positron emission tomography, sex, apolipoprotein E and incident dementia and mortality among individuals without dementia: hazard ratios and absolute risk. Brain Commun. 2022;4:fcac017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lott IT, Head E. Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol. 2019;15:135‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Iulita MF, Garzon Chavez D, Klitgaard Christensen M, et al. Association of Alzheimer disease with life expectancy in people with down syndrome. JAMA Netw Open. 2022;5:e2212910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McCarron M, McCallion P, Reilly E, Dunne P, Carroll R, Mulryan N. A prospective 20‐year longitudinal follow‐up of dementia in persons with Down syndrome. J Intellect Disabil Res. 2017;61:843‐852. [DOI] [PubMed] [Google Scholar]
- 57. Simuni T, Chahine LM, Poston KL, et al. Biological definition of neuronal alpha‐synuclein disease: towards an integrated staging system for research. Lancet Neurol. 2023;23:178‐190. [DOI] [PubMed] [Google Scholar]
- 58. Hoglinger GU, Adler CH, Berg D, et al. A biological classification of Parkinson's disease: the SynNeurGe research diagnostic criteria. Lancet Neurol. 2024;23:191‐204. [DOI] [PubMed] [Google Scholar]
- 59. Tabrizi SJ, Schobel S, Gantman EC, et al. A biological classification of Huntington's disease: the Integrated Staging System. Lancet Neurol. 2022;21:632‐644. [DOI] [PubMed] [Google Scholar]
- 60. Benatar M, Wuu J, McHutchison C, et al. Preventing amyotrophic lateral sclerosis: insights from pre‐symptomatic neurodegenerative diseases. Brain. 2022;145:27‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Curtis C, Gamez JE, Singh U, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015;72:287‐294. [DOI] [PubMed] [Google Scholar]
- 62. Sabri O, Sabbagh MN, Seibyl J, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer's disease: phase 3 study. Alzheimers Dement. 2015;11:964‐974. [DOI] [PubMed] [Google Scholar]
- 63. Beach TG, Adler CH, Sue LI, et al. Arizona Study of aging and neurodegenerative disorders and brain and body donation program. Neuropathology. 2015;35:354‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30:58‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Administration UFaD . Evaluation of automatic class III designation for Lumipulse G β‐amyloid ratio (1‐42/1‐40) Decision Summary. In: FDA, ed. 2023.
- 66. Administration UFaD . FDA Permits Marketing for New Test to Improve Diagnosis of Alzheimer's Disease. In: Administration USFaD, ed. 2022.
- 67. Administration UFaD . Center for Devices and Radiological Health. Elecsys β‐Amyloid (1‐42) CSF II, Elecsys Phospho‐Tau (181P) CSF: 510(k) substantial equivalence determination decision summary. In: FDA, ed. 2023.
- 68. Mattsson‐Carlgren N, Grinberg LT, Boxer A, et al. Cerebrospinal fluid biomarkers in autopsy‐confirmed Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2022;98:e1137‐e1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Janelidze S, Bali D, Ashton NJ, et al. Head‐to‐head comparison of 10 plasma phospho‐tau assays in prodromal Alzheimer's disease. Brain. 2023;146:1592‐1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Janelidze S, Teunissen CE, Zetterberg H, et al. Head‐to‐head comparison of 8 plasma amyloid‐beta 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78:1375‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zicha S, Bateman RJ, Shaw LM, et al. Comparative analytical performance of multiple plasma Aβ42 and Aβ40 assays and their ability to predict positron emission tomography amyloid positivity. Alzheimers Dement. 2022;19:956‐966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐β biomarkers for Alzheimer's disease. Nature. 2018;554:249‐254. [DOI] [PubMed] [Google Scholar]
- 73. Thijssen EH, La Joie R, Strom A, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer's disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet. 2021;20:739‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Therriault J, Servaes S, Tissot C, et al. Equivalence of plasma p‐tau217 with cerebrospinal fluid in the diagnosis of Alzheimer's disease. Alzheimers Dement. 2023;19:4967‐4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020;26:379‐386. [DOI] [PubMed] [Google Scholar]
- 76. Ashton NJ, Brum WS, Di Molfetta G, et al. Diagnostic accuracy of a plasma phosphorylated tau 217 immunoassay for Alzheimer disease pathology. JAMA Neurol. 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Groot C, Cicognola C, Bali D, et al. Diagnostic and prognostic performance to detect Alzheimer's disease and clinical progression of a novel assay for plasma p‐tau217. Alzheimers Res Ther. 2022;14:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mielke MM, Hagen CE, Xu J, et al. Plasma phospho‐tau181 increases with Alzheimer's disease clinical severity and is associated with tau‐ and amyloid‐positron emission tomography. Alzheimers Dement. 2018;14:989‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19:422‐433. [DOI] [PubMed] [Google Scholar]
- 80. Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of plasma phospho‐tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA Neurol. 2020;324:772‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Palmqvist S, Tideman P, Cullen N, et al. Prediction of future Alzheimer's disease dementia using plasma phospho‐tau combined with other accessible measures. Nat Med. 2021;27:1034‐1042. [DOI] [PubMed] [Google Scholar]
- 83. Therriault J, Benedet AL, Pascoal TA, et al. Association of plasma P‐tau181 with memory decline in non‐demented adults. Brain Commun. 2021;3:fcab136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Moscoso A, Grothe MJ, Ashton NJ, et al. Longitudinal associations of blood phosphorylated tau181 and neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 2021;78:396‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Brickman AM, Manly JJ, Honig LS, et al. Plasma p‐tau181, p‐tau217, and other blood‐based Alzheimer's disease biomarkers in a multi‐ethnic, community study. Alzheimers Dement. 2021;17:1353‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐beta PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13:841‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schindler SE, Bollinger JG, Ovod V, et al. High‐precision plasma beta‐amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647‐e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Brand AL, Lawler PE, Bollinger JG, et al. The performance of plasma amyloid beta measurements in identifying amyloid plaques in Alzheimer's disease: a literature review. Alzheimers Res Ther. 2022;14:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rabe C, Bittner T, Jethwa A, et al. Clinical performance and robustness evaluation of plasma amyloid‐beta(42/40) prescreening. Alzheimers Dement. 2023;19:1393‐1402. [DOI] [PubMed] [Google Scholar]
- 91. Cullen NC, Janelidze S, Mattsson‐Carlgren N, et al. Test‐retest variability of plasma biomarkers in Alzheimer's disease and its effects on clinical prediction models. Alzheimers Dement. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jack CR Jr., Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Klunk WE, Cohen A, Bi W, et al. Why We Need Two Cutoffs for Amyloid‐Imaging: Early Versus Alzheimer's‐Like Amyloid‐Positivity. Alzheimer's Association International Conference; 2012; Alzheimer's Associaton: P453‐P454. [Google Scholar]
- 94. Brum WS, Cullen NC, Janelidze S, et al. A two‐step workflow based on plasma p‐tau217 to screen for amyloid beta positivity with further confirmatory testing only in uncertain cases. Nat Aging. 2023;3:1079‐1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Brum WS, Ashton NJ, Simren J, et al. Biological variation estimates of Alzheimer's disease plasma biomarkers in healthy individuals. Alzheimers Dement. 2023;20:1284‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Landau SM, Horng A, Jagust WJ, Alzheimer's Disease Neuroimaging I. Memory decline accompanies subthreshold amyloid accumulation. Neurology. 2018;90:e1452‐e1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zetterberg H, Blennow K. Moving fluid biomarkers for Alzheimer's disease from research tools to routine clinical diagnostics. Mol Neurodegener. 2021;16:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cousins KAQ, Shaw LM, Shellikeri S, et al. Elevated Plasma Phosphorylated Tau 181 in Amyotrophic Lateral Sclerosis. Ann Neurol. 2022;92:807‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Brum WS, Docherty KF, Ashton NJ, et al. Effect of neprilysin inhibition on Alzheimer disease plasma biomarkers: a secondary analysis of a randomized clinical trial. JAMA Neurol. 2023;81:197‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Graff‐Radford J, Jones DT, Wiste HJ, et al. Cerebrospinal fluid dynamics and discordant amyloid biomarkers. Neurobiol Aging. 2022;110:27‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mielke MM, Dage JL, Frank RD, et al. Performance of plasma phosphorylated tau 181 and 217 in the community. Nat Med. 2022;28:1398‐1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. O'Bryant SE, Petersen M, Hall J, Johnson LA, Team H‐HS. Medical comorbidities and ethnicity impact plasma Alzheimer's disease biomarkers: important considerations for clinical trials and practice. Alzheimers Dement. 2023;19:36‐43. [DOI] [PubMed] [Google Scholar]
- 103. Pichet Binette A, Janelidze S, Cullen N, et al. Confounding factors of Alzheimer's disease plasma biomarkers and their impact on clinical performance. Alzheimers Dement. 2023;19:1403‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Huber H, Ashton NJ, Schieren A, et al. Levels of Alzheimer's disease blood biomarkers are altered after food intake‐A pilot intervention study in healthy adults. Alzheimers Dement. 2023;19:5531‐5540. [DOI] [PubMed] [Google Scholar]
- 105. Rabinovici GD, Gatsonis C, Apgar C, et al. Association of amyloid positron emission tomography with subsequent change in clinical management among medicare beneficiaries with mild cognitive impairment or dementia. JAMA. 2019;321:1286‐1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Monane M, Johnson KG, Snider BJ, et al. A blood biomarker test for brain amyloid impacts the clinical evaluation of cognitive impairment. Ann Clin Transl Neurol. 2023;10:1738‐1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Strikwerda‐Brown C, Hobbs DA, Gonneaud J, et al. Association of elevated amyloid and tau positron emission tomography signal with near‐term development of Alzheimer disease symptoms in older adults without cognitive impairment. JAMA Neurol. 2022;79:975‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Salvado G, Larsson V, Cody KA, et al. Optimal combinations of CSF biomarkers for predicting cognitive decline and clinical conversion in cognitively unimpaired participants and mild cognitive impairment patients: a multi‐cohort study. Alzheimers Dement. 2023;19:2943‐2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ebenau JL, Timmers T, Wesselman LMP, et al. ATN classification and clinical progression in subjective cognitive decline: the SCIENCe project. Neurology. 2020;95:e46‐e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jack CR Jr., Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321:2316‐2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Therriault J, Pascoal TA, Lussier FZ, et al. Biomarker modeling of Alzheimer's disease using PET‐based Braak staging. Nat Aging. 2022;2:526‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Grothe MJ, Barthel H, Sepulcre J, et al. In vivo staging of regional amyloid deposition. Neurology. 2017;89:2031‐2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Mattsson N, Palmqvist S, Stomrud E, Vogel J, Hansson O. Staging beta‐amyloid pathology with amyloid positron emission tomography. JAMA Neurol. 2019;76:1319‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Collij LE, Heeman F, Salvado G, et al. Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology. 2020;95:e1538‐e1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cho H, Choi JY, Hwang MS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol. 2016;80:247‐258. [DOI] [PubMed] [Google Scholar]
- 116. Scholl M, Lockhart SN, Schonhaut DR, et al. PET Imaging of tau deposition in the aging human brain. Neuron. 2016;89:971‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Schwarz AJ, Shcherbinin S, Slieker LJ, et al. Topographic staging of tau positron emission tomography images. Alzheimers Dement (Amst). 2018;10:221‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Jack CR Jr., Wiste HJ, Algeciras‐Schimnich A, et al. Predicting amyloid PET and tau PET stages with plasma biomarkers. Brain. 2023;146:2029‐2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wang L, Benzinger TL, Su Y, et al. Evaluation of tau imaging in staging alzheimer disease and revealing interactions between beta‐amyloid and tauopathy. JAMA Neurol. 2016;73:1070‐1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Villemagne VL, Dore V, Bourgeat P, et al. The tau MeTeR scale for the generation of continuous and categorical measures of tau deposits in the brain: results from 18F‐AV1451 and 18F‐THK5351 tau imaging studies. Alzheimer's dement. 2016;12:244.26218444 [Google Scholar]
- 121. Jack CR Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Jack CR Jr., Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Smith R, Cullen NC, Pichet Binette A, et al. Tau‐PET is superior to phospho‐tau when predicting cognitive decline in symptomatic AD patients. Alzheimers Dement. 2023;19:2497‐2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ossenkoppele R, Schonhaut DR, Scholl M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016;139:1551‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Brier MR, Gordon B, Friedrichsen K, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med. 2016;8:338ra366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gordon BA, Blazey TM, Christensen J, et al. Tau PET in autosomal dominant Alzheimer's disease: relationship with cognition, dementia and other biomarkers. Brain. 2019;142:1063‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ossenkoppele R, Smith R, Mattsson‐Carlgren N, et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head‐to‐head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 2021;78:961‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76:915‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Berron D, Vogel JW, Insel PS, et al. Early stages of tau pathology and its associations with functional connectivity, atrophy and memory. Brain. 2021;144:2771‐2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Leuzy A, Smith R, Cullen NC, et al. Biomarker‐based prediction of longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2022;79:149‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tetzloff KA, Graff‐Radford J, Martin PR, et al. Regional distribution, asymmetry, and clinical correlates of tau uptake on [18F]AV‐1451 PET in atypical Alzheimer's disease. J Alzheimers Dis. 2018;62:1713‐1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Vogel JW, Young AL, Oxtoby NP, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27:871‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Knopman DS, Lundt ES, Therneau TM, et al. Association of initial beta‐amyloid levels with subsequent flortaucipir positron emission tomography changes in persons without cognitive impairment. JAMA Neurol. 2021;78:217‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sanchez JS, Becker JA, Jacobs HIL, et al. The cortical origin and initial spread of medial temporal tauopathy in Alzheimer's disease assessed with positron emission tomography. Sci Transl Med. 2021;13:eabc0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Mattsson‐Carlgren N, Collij LE, Stomrud E, et al. Plasma biomarker strategy for selecting patients with Alzheimer disease for antiamyloid immunotherapies. JAMA Neurol. 2024;81:69‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Montoliu‐Gaya L, Benedet AL, Tissot C, et al. Mass spectrometric simultaneous quantification of tau species in plasma shows differential associations with amyloid and tau pathologies. Nat Aging. 2023;3:661‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Horie K, Barthelemy NR, Sato C, Bateman RJ. CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer's disease. Brain. 2021;144:515‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Barthelemy NR, Saef B, Li Y, et al. CSF tau phosphorylation occupancies at T217 and T205 represent improved biomarkers of amyloid and tau pathology in Alzheimer's disease. Nat Aging. 2023;3:391‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Horie K, Salvado G, Barthelemy NR, et al. CSF MTBR‐tau243 is a specific biomarker of tau tangle pathology in Alzheimer's disease. Nat Med. 2023;29:1954‐1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lantero‐Rodriguez J, Montoliu‐Gaya L, Benedet AL, et al. CSF p‐tau205: a biomarker of tau pathology in Alzheimer's disease. Acta Neuropathol. 2024;147:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Horie K, Koppisetti RK, Janelidzde S, et al. Plasma MTBR‐tau243 is a specific biomarker of tau tangle pathology in Alzheimer's disease. Clinical Trials on Alzheimer's Disease 2023. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Jack CR Jr., Wiste HJ, Schwarz CG, et al. Longitudinal tau PET in ageing and Alzheimer's disease. Brain. 2018;141:1517‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Villemagne V, Leuzy A, Bohorquez SMS, et al. CenTauR: towards a universal scale and masks for standardizing tau imaging studies. Alzheimer's and Dementia. 2023;15:e12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimer's Dement. 2015;11:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Therriault J, Schindler SE, Salvado G, et al. Biomarker‐based staging of Alzheimer disease: rationale and clinical applications. Nat Rev Neurol. 2024. [DOI] [PubMed] [Google Scholar]
- 146. Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin‐1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27:6174‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Cohen AD, McDade E, Christian B, et al. Early striatal amyloid deposition distinguishes Down syndrome and autosomal dominant Alzheimer's disease from late‐onset amyloid deposition. Alzheimers Dement. 2018;14:743‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Sperling RA, Mormino EC, Schultz AP, et al. The impact of amyloid‐beta and tau on prospective cognitive decline in older individuals. Ann Neurol. 2019;85:181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384:1691‐1704. [DOI] [PubMed] [Google Scholar]
- 150. Sims JR, Zimmer JA, Evans CD, et al. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER‐ALZ 2 randomized clinical trial. JAMA. 2023;330:512‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6:228fs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Rafii MS, Sperling RA, Donohue MC, et al. The AHEAD 3‐45 study: design of a prevention trial for Alzheimer's disease. Alzheimers Dement. 2023;19:1227‐1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Reisberg B, Ferris SH, de Leon MJ, Crook T. The Global Deterioration Scale for assessment of primary degenerative dementia. The American journal of psychiatry. 1982;139:1136‐1139. [DOI] [PubMed] [Google Scholar]
- 154. Administration UFaD . Early Alzheimer's Disease: Developing Drugs for Treatment In: Services USDoHaH, Administration FaD, eds. Office of Communications, Division of Drug Information: US Food and Drug Administration, 2024.
- 155. Ismail Z, Smith EE, Geda Y, et al. Neuropsychiatric symptoms as early manifestations of emergent dementia: provisional diagnostic criteria for mild behavioral impairment. Alzheimers Dement. 2016;12:195‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Johansson M, Stomrud E, Lindberg O, et al. Apathy and anxiety are early markers of Alzheimer's disease. Neurobiol Aging. 2020;85:74‐82. [DOI] [PubMed] [Google Scholar]
- 157. Johansson M, Stomrud E, Johansson PM, et al. Development of apathy, anxiety, and depression in cognitively unimpaired older adults: effects of Alzheimer's disease pathology and cognitive decline. Biol Psychiatry. 2022;92:34‐43. [DOI] [PubMed] [Google Scholar]
- 158. Graff‐Radford J, Yong KXX, Apostolova LG, et al. New insights into atypical Alzheimer's disease in the era of biomarkers. Lancet Neurol. 2021;20:222‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Dubois B, Villain N, Frisoni GB, et al. Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. Lancet Neurol. 2021;20:484‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Fortea J, Zaman SH, Hartley S, Rafii MS, Head E, Carmona‐Iragui M. Alzheimer's disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol. 2021;20:930‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388:9‐21. [DOI] [PubMed] [Google Scholar]
- 162. Administration UFaD . Leqembi Prescribing Information. 2023.
- 163. Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58:1985‐1992. [DOI] [PubMed] [Google Scholar]
- 164. Roberts JS, Karlawish JH, Uhlmann WR, Petersen RC, Green RC. Mild cognitive impairment in clinical care: a survey of American Academy of Neurology members. Neurology. 2010;75:425‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Bertens D, Vos S, Kehoe P, et al. Use of mild cognitive impairment and prodromal AD/MCI due to AD in clinical care: a European survey. Alzheimers Res Ther. 2019;11:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Vemuri P, Lesnick TG, Przybelski SA, et al. Association of lifetime intellectual enrichment with cognitive decline in the older population. JAMA Neurol. 2014;71:1017‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Jagust WJ, Mormino EC. Lifespan brain activity, beta‐amyloid, and Alzheimer's disease. Trends Cogn Sci. 2011;15:520‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Arenaza‐Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease: clarifying terminology for preclinical studies. Neurology. 2018;90:695‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bridel C, van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol. 2019;76:1035‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kmezic I, Samuelsson K, Finn A, et al. Neurofilament light chain and total tau in the differential diagnosis and prognostic evaluation of acute and chronic inflammatory polyneuropathies. Eur J Neurol. 2022;29:2810‐2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Jack CR Jr., Petersen RC, O'Brien PC, Tangalos EG. MR‐based hippocampal volumetry in the diagnosis of Alzheimer's disease. Neurology. 1992;42:183‐188. [DOI] [PubMed] [Google Scholar]
- 172. Bobinski M, de Leon MJ, Wegiel J, et al. The histological validation of post mortem magnetic resonance imaging‐determined hippocampal volume in Alzheimer's disease. Neuroscience. 2000;95:721‐725. [DOI] [PubMed] [Google Scholar]
- 173. Zarow C, Vinters HV, Ellis WG, et al. Correlates of hippocampal neuron number in Alzheimer's disease and ischemic vascular dementia. Ann Neurol. 2005;57:896‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Jack CR Jr., Dickson DW, Parisi JE, et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002;58:750‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Thijssen EH, Verberk IMW, Kindermans J, et al. Differential diagnostic performance of a panel of plasma biomarkers for different types of dementia. Alzheimers Dement (Amst). 2022;14:e12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8:348ra396. [DOI] [PubMed] [Google Scholar]
- 177. Mecca AP, Chen MK, O'Dell RS, et al. In vivo measurement of widespread synaptic loss in Alzheimer's disease with SV2A PET. Alzheimers Dement. 2020;16:974‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Gouw AA, Alsema AM, Tijms BM, et al. EEG spectral analysis as a putative early prognostic biomarker in nondemented, amyloid positive subjects. Neurobiol Aging. 2017;57:133‐142. [DOI] [PubMed] [Google Scholar]
- 179. Haass C, Selkoe D. If amyloid drives Alzheimer disease, why have anti‐amyloid therapies not yet slowed cognitive decline? PLoS Biol. 2022;20:e3001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. De Strooper B, Karran E. The cellular phase of Alzheimer's disease. Cell. 2016;164:603‐615. [DOI] [PubMed] [Google Scholar]
- 181. Bouzid H, Belk JA, Jan M, et al. Clonal hematopoiesis is associated with protection from Alzheimer's disease. Nat Med. 2023;29:1662‐1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer's disease drug development pipeline: 2021. Alzheimers Dement (N Y). 2021;7:e12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Verberk IMW, Laarhuis MB, van den Bosch KA, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic‐based cohort study. Lancet Healthy Longev. 2021;2:e87‐e95. [DOI] [PubMed] [Google Scholar]
- 184. Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Transl Psychiatry. 2021;11:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Chatterjee P, Vermunt L, Gordon BA, et al. Plasma glial fibrillary acidic protein in autosomal dominant Alzheimer's disease: Associations with Aβ‐PET, neurodegeneration, and cognition. Alzheimers Dement. 2023;19:2790‐2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Jack CR Jr., Wiste HJ, Algeciras‐Schimnich A, et al. Comparison of plasma biomarkers and amyloid PET for predicting memory decline in cognitively unimpaired individuals. Alzheimers Dement. 2024;20:2143‐2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Cicognola C, Janelidze S, Hertze J, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimers Res Ther. 2021;13:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Suarez‐Calvet M, Araque Caballero MA, Kleinberger G, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016;8:369ra178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Morenas‐Rodriguez E, Li Y, Nuscher B, et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal‐dominant Alzheimer's disease: a longitudinal observational study. Lancet Neurol. 2022;21:329‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Pereira JB, Janelidze S, Strandberg O, et al. Microglial activation protects against accumulation of tau aggregates in nondemented individuals with underlying Alzheimer's disease pathology. Nat Aging. 2022;2:1138‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Fairfoul G, McGuire LI, Pal S, et al. Alpha‐synuclein RT‐QuIC in the CSF of patients with alpha‐synucleinopathies. Ann Clin Transl Neurol. 2016;3:812‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Shahnawaz M, Tokuda T, Waragai M, et al. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of alpha‐Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol. 2017;74:163‐172. [DOI] [PubMed] [Google Scholar]
- 193. Arnold MR, Coughlin DG, Brumbach BH, et al. α‐synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological α‐synuclein in the context of co‐pathology and non‐LBD diagnoses. Ann Neurol. 2022;92:650‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Hall S, Orru CD, Serrano GE, et al. Performance of alphaSynuclein RT‐QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol Commun. 2022;10:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Palmqvist S, Rossi M, Hall S, et al. Cognitive effects of Lewy body pathology in clinically unimpaired individuals. Nat Med. 2023;29:1971‐1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Quadalti C, Palmqvist S, Hall S, et al. Clinical effects of Lewy body pathology in cognitively impaired individuals. Nat Med. 2023;29:1964‐1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Rossi M, Candelise N, Baiardi S, et al. Ultrasensitive RT‐QuIC assay with high sensitivity and specificity for Lewy body‐associated synucleinopathies. Acta Neuropathol. 2020;140:49‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Bargar C, De Luca CMG, Devigili G, et al. Discrimination of MSA‐P and MSA‐C by RT‐QuIC analysis of olfactory mucosa: the first assessment of assay reproducibility between two specialized laboratories. Mol Neurodegener. 2021;16:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Seibyl JP. alpha‐synuclein PET and parkinson disease therapeutic trials: ever the twain shall meet? J Nucl Med. 2022;63:1463‐1466. [DOI] [PubMed] [Google Scholar]
- 200. Korat S, Bidesi NSR, Bonanno F, et al. Alpha‐synuclein PET tracer development‐an overview about current efforts. Pharmaceuticals (Basel). 2021;14:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. McKeith I, O'Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I‐FP‐CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol. 2007;6:305‐313. [DOI] [PubMed] [Google Scholar]
- 202. Vlaar AM, de Nijs T, Kessels AG, et al. Diagnostic value of 123I‐ioflupane and 123I‐iodobenzamide SPECT scans in 248 patients with parkinsonian syndromes. Eur Neurol. 2008;59:258‐266. [DOI] [PubMed] [Google Scholar]
- 203. Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Duering M, Biessels GJ, Brodtmann A, et al. Neuroimaging standards for research into small vessel disease‐advances since 2013. Lancet Neurol. 2023;22:602‐618. [DOI] [PubMed] [Google Scholar]
- 205. Baykara E, Gesierich B, Adam R, et al. A novel imaging marker for small vessel disease based on skeletonization of white matter tracts and diffusion histograms. Ann Neurol. 2016;80:581‐592. [DOI] [PubMed] [Google Scholar]
- 206. Vemuri P, Graff‐Radford J, Lesnick TG, et al. White matter abnormalities are key components of cerebrovascular disease impacting cognitive decline. Brain Commun. 2021;3:fcab076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Vemuri P, Lesnick TG, Knopman DS, et al. Amyloid, vascular, and resilience pathways associated with cognitive aging. Ann Neurol. 2019;86:866‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Jagust WJ, Zheng L, Harvey DJ, et al. Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol. 2008;63:72‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Beach TG, Sue LI, Scott S, et al. Cerebral white matter rarefaction has both neurodegenerative and vascular causes and may primarily be a distal axonopathy. J Neuropathol Exp Neurol. 2023;82:457‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Greenberg SM, Bacskai BJ, Hernandez‐Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease – one peptide, two pathways. Nat Rev Neurol. 2020;16:30‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Graff‐Radford J, Lesnick T, Rabinstein AA, et al. Cerebral microbleed incidence, relationship to amyloid burden: the Mayo Clinic Study of Aging. Neurology. 2020;94:e190‐e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Koemans EA, Chhatwal JP, van Veluw SJ, et al. Progression of cerebral amyloid angiopathy: a pathophysiological framework. Lancet Neurol. 2023;22:632‐642. [DOI] [PubMed] [Google Scholar]
- 213. Sperling R, Salloway S, Brooks DJ, et al. Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11:241‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Nelson PT, Head E, Schmitt FA, et al. Alzheimer's disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011;121:571‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Beach TG, Malek‐Ahmadi M. Alzheimer's disease neuropathological comorbidities are common in the younger‐old. J Alzheimers Dis. 2021;79:389‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Power MC, Mormino E, Soldan A, et al. Combined neuropathological pathways account for age‐related risk of dementia. Ann Neurol. 2018;84:10‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Spina S, La Joie R, Petersen C, et al. Copathologies in early‐ vs late‐onset Alzheimer's disease. Alzheimer's dement. 2021;17:e056436. [Google Scholar]
- 219. Robinson JL, Richardson H, Xie SX, et al. The development and convergence of co‐pathologies in Alzheimer's disease. Brain. 2021;144:953‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology. 2007;69:2197‐2204. [DOI] [PubMed] [Google Scholar]
- 221. White L. Brain lesions at autopsy in older Japanese‐American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu‐Asia aging study. J Alzheimers Dis. 2009;18:713‐725. [DOI] [PubMed] [Google Scholar]
- 222. Kawas CH, Kim RC, Sonnen JA, Bullain SS, Trieu T, Corrada MM. Multiple pathologies are common and related to dementia in the oldest‐old: the 90+ Study. Neurology. 2015;85:535‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke. 2011;42:722‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Graff‐Radford J, Gunter JL, Jones DT, et al. Cerebrospinal fluid dynamics disorders: relationship to Alzheimer biomarkers and cognition. Neurology. 2019;93:e2237‐e2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Duong MT, Das SR, Lyu X, et al. Dissociation of tau pathology and neuronal hypometabolism within the ATN framework of Alzheimer's disease. Nat Commun. 2022;13:1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Botha H, Mantyh WG, Graff‐Radford J, et al. Tau‐negative amnestic dementia masquerading as Alzheimer disease dementia. Neurology. 2018;90:e940‐e946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Botha H, Mantyh WG, Murray ME, et al. FDG‐PET in tau‐negative amnestic dementia resembles that of autopsy‐proven hippocampal sclerosis. Brain. 2018;141:1201‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Lyu X, Duong MT, Xie L, et al. Tau‐Neurodegeneration mismatch reveals vulnerability and resilience to comorbidities in Alzheimer's continuum. medRxiv. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. 2022;9:197‐210. [DOI] [PubMed] [Google Scholar]
- 230. Pontecorvo MJ, Lu M, Burnham SC, et al. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: a secondary analysis of the TRAILBLAZER‐ALZ randomized clinical trial. JAMA Neurol. 2022;79:1250‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. McDade E, Cummings JL, Dhadda S, et al. Lecanemab in patients with early Alzheimer's disease: detailed results on biomarker, cognitive, and clinical effects from the randomized and open‐label extension of the phase 2 proof‐of‐concept study. Alzheimers Res Ther. 2022;14:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563‐1572. [DOI] [PubMed] [Google Scholar]
- 233. Novak G, Fox N, Clegg S, et al. Changes in brain volume with bapineuzumab in mild to moderate Alzheimer's disease. J Alzheimers Dis. 2016;49:1123‐1134. [DOI] [PubMed] [Google Scholar]
- 234. Cogswell PM, Barakos JA, Barkhof F, et al. Amyloid‐related imaging abnormalities with emerging Alzheimer disease therapeutics: detection and reporting recommendations for clinical practice. AJNR Am J Neuroradiol. 2022;43:E19‐E35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Ighodaro ET, Nelson PT, Kukull WA, et al. Challenges and considerations related to studying dementia in Blacks/African Americans. J Alzheimers Dis. 2017;60:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Babulal GM, Quiroz YT, Albensi BC, et al. Perspectives on ethnic and racial disparities in Alzheimer's disease and related dementias: update and areas of immediate need. Alzheimers Dement. 2019;15:292‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Brewster P, Barnes L, Haan M, et al. Progress and future challenges in aging and diversity research in the United States. Alzheimers Dement. 2019;15:995‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Howell JC, Watts KD, Parker MW, et al. Race modifies the relationship between cognition and Alzheimer's disease cerebrospinal fluid biomarkers. Alzheimers Res Ther. 2017;9:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Deters KD, Napolioni V, Sperling RA, et al. Amyloid PET imaging in self‐identified non‐hispanic black participants of the anti‐amyloid in asymptomatic Alzheimer's Disease (A4) Study. Neurology. 2021;96:e1491‐e1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Naslavsky MS, Suemoto CK, Brito LA, et al. Global and local ancestry modulate APOE association with Alzheimer's neuropathology and cognitive outcomes in an admixed sample. Mol Psychiatry. 2022;27:4800‐4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349‐1356. [PubMed] [Google Scholar]
- 242. Evans DA, Bennett DA, Wilson RS, et al. Incidence of Alzheimer disease in a biracial urban community: relation to apolipoprotein E allele status. Arch Neurol. 2003;60:185‐189. [DOI] [PubMed] [Google Scholar]
- 243. Wilkins CH, Windon CC, Dilworth‐Anderson P, et al. Racial and ethnic differences in amyloid PET positivity in individuals with mild cognitive impairment or dementia: a secondary analysis of the Imaging Dementia‐Evidence for Amyloid Scanning (IDEAS) Cohort Study. JAMA Neurol. 2022;79:1139‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Barnes LL, Leurgans S, Aggarwal NT, et al. Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology. 2015;85:528‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Gottesman RF, Schneider AL, Zhou Y, et al. The ARIC‐PET amyloid imaging study: brain amyloid differences by age, race, sex, and APOE. Neurology. 2016;87:473‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Schindler SE, Karikari TK, Ashton NJ, et al. Effect of race on prediction of brain amyloidosis by plasma abeta42/abeta40, phosphorylated tau, and neurofilament light. Neurology. 2022;99:e245‐e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Weuve J, Barnes LL, Mendes de Leon CF, et al. Cognitive aging in black and white americans: cognition, cognitive decline, and incidence of Alzheimer disease dementia. Epidemiology. 2018;29:151‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Barnes LL, Bennett DA. Alzheimer's disease in African Americans: risk factors and challenges for the future. Health Aff (Millwood). 2014;33:580‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Ramanan VK, Graff‐Radford J, Syrjanen J, et al. Association of plasma biomarkers of Alzheimer disease with cognition and medical comorbidities in a biracial cohort. Neurology. 2023;101:e1402‐e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
ICMJE Disclosure Form