

Abstract
INTRODUCTION
Despite a two‐fold risk, individuals of African ancestry have been underrepresented in Alzheimer's disease (AD) genomics efforts.
METHODS
Genome‐wide association studies (GWAS) of 2,903 AD cases and 6,265 controls of African ancestry. Within‐dataset results were meta‐analyzed, followed by functional genomics analyses.
RESULTS
A novel AD‐risk locus was identified in MPDZ on chromosome (chr) 9p23 (rs141610415, MAF = 0.002, p = 3.68×10−9). Two additional novel common and nine rare loci were identified with suggestive associations (P < 9×10−7). Comparison of association and linkage disequilibrium (LD) patterns between datasets with higher and lower degrees of African ancestry showed differential association patterns at chr12q23.2 (ASCL1), suggesting that this association is modulated by regional origin of local African ancestry.
DISCUSSION
These analyses identified novel AD‐associated loci in individuals of African ancestry and suggest that degree of African ancestry modulates some associations. Increased sample sets covering as much African genetic diversity as possible will be critical to identify additional loci and deconvolute local genetic ancestry effects.
Highlights
Genetic ancestry significantly impacts risk of Alzheimer's Disease (AD). Although individuals of African ancestry are twice as likely to develop AD, they are vastly underrepresented in AD genomics studies.
The Alzheimer's Disease Genetics Consortium has previously identified 16 common and rare genetic loci associated with AD in African American individuals. The current analyses significantly expand this effort by increasing the sample size and extending ancestral diversity by including populations from continental Africa.
Single variant meta‐analysis identified a novel genome‐wide significant AD‐risk locus in individuals of African ancestry at the MPDZ gene, and 11 additional novel loci with suggestive genome‐wide significance at p < 9×10−7.
Comparison of African American datasets with samples of higher degree of African ancestry demonstrated differing patterns of association and linkage disequilibrium at one of these loci, suggesting that degree and/or geographic origin of African ancestry modulates the effect at this locus.
These findings illustrate the importance of increasing number and ancestral diversity of African ancestry samples in AD genomics studies to fully disentangle the genetic architecture underlying AD, and yield more effective ancestry‐informed genetic screening tools and therapeutic interventions.
Keywords: African Americans, African genome Panel, Alzheimer's disease, genome‐wide association study
1. BACKGROUND
Based on estimations by the World Health Organization, approximately 55 million people globally suffer from dementia, and 40 million of these cases are thought to be due to Alzheimer's disease (AD). As the proportion of older individuals increases in nearly every country, the number of AD cases is expected to rise exponentially to ∼78 million in 2030 and ∼139 million by 2050 (https://www.who.int/news‐room/fact‐sheets/detail/dementia), making identification of underlying factors one of the most urgent global public health concerns.
Recent large‐scale genome‐wide association studies (GWAS) by our group and others identified over 75 loci associated with risk of AD and related dementias. 1 , 2 , 3 , 4 , 5 While these findings have significantly advanced the field by providing invaluable insights into underlying mechanistic pathways, AD genomic studies were almost exclusively conducted in individuals of European ancestry. This limits our ability to identify ancestry‐specific causative genetic variants, loci, and mechanistic pathways underlying the disease, and substantially hampers our progress toward identification of effective druggable targets to address this devastating disease in underrepresented populations.
With a two‐fold increased disease risk compared to non‐Hispanic White 6 (NHW) individuals, a ∼64% higher rate of progression to AD and related dementias (ADRD) compared to NHW, 7 , 8 a higher degree of dementia risk factors, and greater cognitive impairment and neuropsychiatric symptom severity, 9 individuals of African ancestry are at a particularly high risk of AD and its sub‐phenotypes. To identify genetic risk factors associated with AD in African American individuals, we previously conducted GWAS in 8,006 subjects of African American ancestry and identified 16 common and rare loci associated with AD, most of which appear ancestry‐specific. 10 , 11 To identify additional risk variants, genes, and mechanistic pathways, we reanalyzed these data with a 14.5% increase in sample size. To start to deconvolute regional African ancestry effects we added West African individuals with a high degree of African ancestry. Observed loci of interest were followed up through colocalization, local ancestry analysis, and analysis of Hi‐C, RNAseq, whole‐genome sequencing, and proteomic data. These analyses identified 12 novel susceptibility loci potentially associated with AD in individuals of African ancestry that have not been identified in other ancestry groups.
RESEARCH IN CONTEXT
Systematic review: Relevant literature and related efforts were screened by reviewing PubMed and dbGaP for efforts on Alzheimer's disease (AD) including studies targeting individuals of African descent.
Interpretation: Individuals of African ancestry have been largely excluded from AD genomics efforts, resulting in an extensive lack of understanding of the effects of genetic ancestry. In the largest AD genome‐wide association studies (GWAS) in individuals of African ancestry – and the first to include a Nigerian population from Yoruba with a high degree of African ancestry – we identified 12 novel AD loci and novel AD‐related mechanistic pathways and demonstrated that degree and/or origin of African genetic ancestry impacts genetic risk.
Future directions: Studies with increased sample sizes that cover as much of the African genetic diversity as possible will be critical to identify more ancestry‐specific AD‐associated loci, disentangle local genetic ancestry effects, and develop ancestry‐specific polygenic risk scores and druggable targets.
2. METHODS
2.1. Samples
To identify additional risk loci associated with AD in individuals of African ancestry, we both increased the sample size by 14.5% in this updated analysis and included, in addition to African American individuals, 705 individuals from West Africa (sampled from Ibadan, Nigeria), yielding in total 9,168 subjects of African ancestry in the analysis (2,903 cases, 6,265 controls). Summary demographics of all datasets are shown in Supplementary Table 1 and additional detailed information about each dataset is provided in the Description of Cohorts in the Supplementary Materials. Written informed consent was obtained from all participants, or, for those with cognitive impairment, from a caregiver, legal guardian, or other proxy. Study protocols for all cohorts were reviewed and approved by the appropriate institutional review boards.
2.2. Diagnosis of AD and age of onset
Participants were diagnosed for AD according to the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria. 12 , 13 Age at onset for AD patients and age at examination or death for healthy controls was available for most datasets. When not available, other information was used instead, such as age at diagnosis (Chicago Health and Aging Project [CHAP], Minority Aging Research Study/Clinical Minority Core [MARS/CORE]) or age at ascertainment (Indiana University). To restrict the analyses to cases with late‐onset AD, individuals with age <60 years at symptom onset, last examination, or death were excluded.
2.3. Genotyping
The platforms used for genome‐wide genotyping in the individual datasets are shown in Supplementary Table 2. For all datasets, samples were randomly plated to minimize potential batch effects.
2.4. Apolipoprotein E genotyping
For the Alzheimer Disease Centers, Adult Changes in Thought, National Institute in Aging–LOAD/National Cell Repository for Alzheimer Disease (NIA‐FBS/NCRAD), UM/VU, CHAP, Columbia University, Mayo Clinic, and REAAADI cohorts, apolipoprotein E (APOE) genotypes were based on haplotypes derived from single‐nucleotide polymorphisms SNPs rs7412 and rs429358. Whole‐genome sequencing genotypes of these two SNPs were used for the Ibadan cohort. For the MIRAGE and GenerAAtions cohorts, APOE genotypes were determined using the Roche Diagnostics LightCycler 480 instrument (Roche Diagnostics) and LightMix Kit ApoE C112R R158 (TIB MOLBIOL); for the University of Pittsburgh, Washington Heights Columbia Aging Project, and Indianapolis cohorts, they were determined by pyrosequencing or analysis of restriction fragment length polymorphisms; for the Religious Orders Study/Rush Memory and Aging Project (ROS/MAP) and MARS/CORE they were determined by high‐throughput sequencing of codons 112 and 158 in APOE by Agencourt Bioscience Corporation; for the Washington University samples they were determined using a taqman‐based assay from Applied Biosystems.
2.5. Genotype quality control
Standard quality control was performed separately on each dataset's genotype and sample‐level data. SNPs with call rates less than 98% or not in Hardy‐Weinberg equilibrium at p < 10−6 in controls were excluded. In addition, subjects with non‐African American ancestry according to principal components (PCs) analysis of ancestry informative markers, and study participants whose reported sex did not equal the sex designation determined by analysis of the X‐chromosome SNPs were removed. We identified latent relatedness among participants using the estimated proportion of alleles (π^) shared identical by descent (IBD) and included from each duplicate pair (π^ > 0.95) or relative pair (0.4 ≤ π^ < 0.95) one participant prioritizing first samples with non‐missing disease status followed by samples with higher SNP call rate. Relationships among individuals in family‐based cohorts (MIRAGE) were validated employing pairwise genome‐wide estimates of IBD allele sharing.
2.6. Genotype imputation
Each dataset was independently phased and imputed to the African Genome Resource (AGR) reference panel utilizing the Sanger genotype imputation and phasing service (https://imputation.sanger.ac.uk/), which employs EAGLE2 for phasing and PBWT (Positional Burrows‐Wheeler Transform) for genotype imputation. The AGR reference panel provides information on 93,421,145 autosomal bi‐allelic markers and is based on 4,956 samples that includes – in addition to all of the African and non‐African populations from the 1000 Genomes Phase 3 reference panel – also ∼2000 samples from Uganda (Baganda, Banyarwanda, Barundi and others) and ∼100 samples each from Ethiopia (Gumuz, Wolayta, Amhara, Oromo, Somali), Egypt, Namibia (Nama/Khoesan), and South Africa (Zulu). The AGR reference panel showed the highest concordance rate with whole‐genome sequence data in sub‐Saharan African participants compared to other reference panels, including TOPMed. 14 Data were filtered to exclude common variants (MAF ≥ 0.01) with imputation quality score < 0.4, rare variants (MAF < 0.01) with imputation quality < 0.7, and variants present in less than 30% of AD cases and 30% of controls. The final SNP set for analysis included 33,089,606 genotyped and imputed variants.
2.7. Association analysis
Genome‐wide single‐variant association analyses of common and rare variants were performed individually on each dataset using SNPTEST. 15 , 16 , 17 Age, sex, and population stratification (as determined by the first three principal components (PCs) calculated individually on each dataset) were entered as covariates in Model 1; APOEe4 allele dosage (coded as 0,1,2) was entered as an additional covariate in Model 2. Logistic regression was used for case‐control datasets and generalized estimating equations (GEE) as implemented in GWAF 18 were used for family‐based datasets (i.e., MIRAGE). Associations with extreme beta coefficients (|β| > 5) were filtered out. Within‐study results were subsequently meta‐analyzed with METAL 19 employing an inverse‐variance based model with genomic control. Variants showing significant heterogeneity between studies (I2 > 75%) were removed. The GenABEL package 20 was used to estimate genomic inflation (λ). A p‐value threshold of 5 × 10−8 was employed to determine disease‐associated genetic variants with genome‐wide significance; a threshold of p < 10−6 was applied to determine variants with suggestive significance.
2.8. Gene‐based analysis
Genome‐wide gene‐based analyses were performed employing MAGMA implemented in the FUMA software. 21 , 22 Setting a 35 kb window upstream and a 10 kb window downstream of the genes and including only variants with MAF > 0.001 and present in >30% of cases and controls, these analyses were first adjusted for PCs, age, and sex and subsequently in addition for APOEe4 allele dosage. Genome‐wide significance was determined using Bonferroni correction (p = 0.05/19277 genes tested = 2.59×10−6).
2.9. Pathway analysis
Pathway analyses were performed with MAGMA, 21 which performs SNP‐wise gene analysis of summary statistics with correction for LD between variants and genes to test whether sets of genes are jointly associated with a phenotype (i.e., AD), compared to other genes across the genome. 9,988 gene‐sets from gene ontology (GO) 23 pathways were used in the analyses. These analyses were performed using the same parameters as described above in the gene‐based analysis section.
2.10. Local ancestry
To estimate local ancestry within African American datasets, we initially merged each array dataset with the Human Genome Diversity Project (HGDP) reference panel individually, utilizing PLINK v2 software. 24 , 25 This integration involved 98 African and 109 European individuals from the HGDP reference populations. Subsequently, the merged datasets were phased employing the SHAPEIT tool version 2 with default configurations using the 1000 Genomes Phase 3 reference panel. 26 , 27 Finally, we inferred the local ancestries using the discriminative modeling approach implemented in RFMix with the PopPhased option and a minimum node size of 5. 28
2.11. Colocalization analyses
To identify potentially causative genes at identified top loci, we performed Bayesian colocalization analyses employing the coloc.abf function in the coloc R package (version 4.0.3) 29 for a wide range of cis‐eQTL datasets (Supplementary Table 3) setting default priors to P1 = 1×10−4, P2 = 1×10−4, and P12 = 1×10−5. A posterior probability PP.H4 > 0.75 was used to declare strong evidence of the eQTL‐GWAS pair influencing both the expression and GWAS trait at a locus.
2.12. Hi‐C analysis
To identify potentially causative genes and illustrate the functional mechanisms at identified top loci, we performed Hi‐C analysis using brain autopsy samples as described before. 30 Autopsy material was obtained from the Alzheimer's Disease Research Centers (ADRC) at Emory University, Northwestern University, and the John P. Hussman Institute for Human Genomics (HIHG). All samples were acquired with informed consent for research use and approved by the institutional review board of each center. Hi‐C libraries from eight donors (four African American and four NHW individuals) were pooled for the analysis. In situ Hi‐C library was prepared using a protocol adapted from Rao et al with a four‐cutter enzyme. 31 For each library, 340∼860 million of paired‐end reads at 150 bp length were obtained. Chromatin loops were called using HiCorr 32 to correct bias and LoopEnhance 33 to remove noise. In addition, layered H3K27ac ChIP‐seq data from seven cell lines from the ENCODE project, 34 H3K27ac and H3K4me3 ChIP‐seq data from adult brains with NCBI GEO accession number GSE116825 32 were used to annotate functional DNA elements near the chromatin loops.
2.13. Bulk RNA sequence analysis in human brain
Brain RNAseq data from over 2,100 samples from post‐mortem brains of more than 1,100 individuals from the ROSMAP study, the Mount Sinai Brain Bank (MSBB), and the Mayo Clinic were scrutinized using the AGORA AD knowledge Portal (https://agora.adknowledgeportal.org/) to identify genes differentially expressed between AD cases and controls.
2.14. Bulk RNA sequence analysis in zebrafish
Amyloid toxicity was induced as previously described 35 , 36 in the adult telencephalon zebrafish brain. At 3 days after cerebroventricular injection, the brains were dissected and deep sequencing for bulk RNA was performed. 35 Data can be accessed at GSE74326 at GEO (https://www.ncbi.nlm.nih.gov/geo).
2.15. Analysis of human cerebrospinal fluid proteomic data
Cerebrospinal fluid (CSF) was collected from 500 NHW, African American, and Caribbean Hispanic individuals from the Dominican Republic and New York City. CSF biomarkers of AD including P‐tau181, Aβ40, Aβ42, total‐tau, neurofilament light chain (NfL), and glial fibrillary acidic protein (GFAP) were measured. CSF was depleted of abundant proteins followed by precipitation, cysteine reduction/alkylation, and proteolytic cleavage by trypsin. Peptides were measured using a Q Exactive HF mass spectrometer (Thermo Scientific). Association of individual and co‐abundant modules of proteins were tested with the clinical diagnosis of AD, as well as biologically defined AD pathological process based on CSF P‐tau181 and other biomarker levels.
3. RESULTS
3.1. Single‐variant meta‐analysis
The results of the single‐variant meta‐analyses are summarized in Table 1 and Figure 1. Single marker analyses identified one novel genome‐wide significant disease‐associated locus, and 11 novel loci with suggestive associations that are approaching genome‐wide significance at p < 9×10−7. The novel genome‐wide significant locus, associated with a rare variant, is located within MPDZ on chromosome (chr) 9p23 (rs141610415, MAF = 0.002, p = 3.68×10−9) and has strong regional support by variants in LD (Supplementary Figure 1). The 11 novel loci approaching genome‐wide significance include two common loci centered at 2p25.3 at LINC01250/TSSC1 and 3p25 at SRGAP3, and nine rare loci centered at 2p25 (KIDINS220), 2q22 (TEX41), 4q22 (UNC5C), 6q21 (IYD/PLEKHG1), 7p22 (SDK1), 8q21 (MMP16), 12q23 (ASCL1), 16q23 (CNTNAP4), and 17q23 (TANC2) (see Table 1). Eight of these 11 loci have strong regional support by variants in LD (see Supplementary Figure 1), and all have consistent directions of effect across most individual datasets (see Supplementary Figure 2). There was no genomic inflation in either model (Model 1: λ = 0.95; Model 2: λ = 0.96; see Supplementary Figure 3 for QQ‐plots).
TABLE 1.
Results of single‐variant meta‐analysis.
| Model 1 | Model 2 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nearest gene(s) | dbSNP | Chromosome | Position | Minor/major allele | MAF | β | SE | p‐Value | Direction | I2 p‐value | β | SE | p‐Value | Direction | I2 p‐value |
| Common novel loci | |||||||||||||||
| LINC01250/TSSC1 | rs78857220 | 2 | 3070309 | A/G | 0.018 | 0.69 | 0.14 | 8.70×10−7 * | +–++++–+++?+?++ | 0.27 | 0.61 | 0.15 | 4.68×10−5 | +–++++–+++?+?++ | 0.35 |
| SRGAP3 | rs396323 | 3 | 9215296 | T/C | 0.101 | 0.32 | 0.06 | 3.74×10−7 * | ++‐++++‐++++++‐++ | 0.55 | 0.29 | 0.07 | 8.87×10−6 | ++–++++++++++‐++ | 0.33 |
| Rare novel loci | |||||||||||||||
| KIDINS220 | rs183088158 | 2 | 8806098 | T/C | 0.008 | 1.17 | 0.27 | 1.08×10−5 | ???++‐+++?+???‐++ | 0.45 | 1.45 | 0.28 | 2.50×10−7 * | ???++++++?+???‐++ | 0.67 |
| TEX41 | rs191256330 | 2 | 145902343 | C/T | 0.005 | 1.39 | 0.37 | 1.83×10−4 | ???+++?+‐+++???++ | 0.73 | 1.59 | 0.32 | 7.22×10−7 * | ???+++++‐+++???++ | 0.74 |
| UNC5C | rs969240869 | 4 | 96471106 | A/G | 0.001 | 4.50 | 0.91 | 7.69×10−7 * | ??????+???+?????? | 0.89 | |||||
| IYD/PLEKHG1 | rs117762284 | 6 | 150819452 | A/G | 0.008 | 2.29 | 0.45 | 4.21×10−7 * | +?++???+?++?????? | 0.80 | 1.50 | 0.34 | 1.10×10−5 | +?‐+??++?++?????? | 0.26 |
| SDK1 | rs181767458 | 7 | 4268872 | G/C | 0.006 | 1.30 | 0.30 | 1.63×10−5 | ‐+?++?++?+++???++ | 0.11 | 1.61 | 0.33 | 8.74×10−7 * | ‐+??+?++?+++???++ | 0.04 |
| MMP16 | rs138744190 | 8 | 90157165 | G/A | 0.004 | 1.71 | 0.41 | 2.51×10−5 | +????+++?+?+????‐ | 0.28 | 2.10 | 0.42 | 6.05×10−7 * | +????+++?+?+????‐ | 0.11 |
| MPDZ | rs141610415 | 9 | 13220518 | C/T | 0.002 | 3.50 | 0.68 | 2.40×10−7 * | ????+?+???+?????? | 0.63 | 4.37 | 0.74 | 3.68×10−9 * | ????+?+???+?????? | 0.88 |
| ASCL1 | rs556001137 | 12 | 103570373 | T/G | 0.003 | 2.21 | 0.45 | 8.12×10−7 * | ??????++?++???+++ | 0.65 | 2.21 | 0.46 | 1.43×10−6 | ??????++?++???+++ | 0.48 |
| rs138206541 | 12 | 103716273 | T/G | 0.003 | 2.03 | 0.45 | 7.58×10−6 | ?‐??+?++?++????++ | 0.22 | 2.41 | 0.49 | 7.39×10−7 * | ?‐??+?++?++????++ | 0.23 | |
| CNTNAP4 | rs1010752317 | 16 | 76535070 | C/A | 0.003 | 1.87 | 0.42 | 7.31×10−6 | ++?++?++?+++???+? | 0.99 | 2.26 | 0.46 | 7.71×10−7 * | ++?++?++?+++???+? | 0.96 |
| TANC2 | rs56285182 | 17 | 61288858 | T/C | 0.006 | 1.26 | 0.31 | 4.96×10−5 | ++?+++++??+‐???++ | 0.08 | 1.73 | 0.35 | 5.60×10−7 * | ++?+++++??+????++ | 0.81 |
| Loci previously reported in Kunkle et al. (2021) | |||||||||||||||
| EDEM1 | rs168193 | 3 | 5302077 | G/A | 0.241 | −0.21 | 0.05 | 2.65×10−6 | +——‐+—–+‐? | 0.46 | −0.18 | 0.05 | 2.59×10−4 | +——‐+–+–+‐? | 0.43 |
| ALCAM | rs2633682 | 3 | 104409208 | A/C | 0.333 | −0.21 | 0.05 | 3.11×10−6 | ——?———+ | 0.39 | −0.14 | 0.04 | 4.29×10−4 | —————‐+ | 0.49 |
| GPC6 | rs9516245 | 13 | 94159800 | C/T | 0.039 | 0.46 | 0.10 | 3.30×10−6 | +++++++++++++++++ | 0.35 | 0.52 | 0.10 | 5.50×10−7 * | +++++++++++++++++ | 0.27 |
| VRK3 | rs3745495 | 19 | 50524332 | G/A | 0.104 | 0.25 | 0.06 | 4.32×10−6 | +‐+‐++++‐+++‐+‐+‐ | 0.16 | 0.30 | 0.06 | 3.18×10−6 | +‐+‐++++‐+++++‐+‐ | 0.07 |
| SIPA1L2 | rs115684722 | 1 | 232376163 | T/A | 0.007 | 1.45 | 0.89 | 0.10 | ‐????+?+????????‐ | 0.03 | 0.14 | 0.56 | 0.80 | ‐????++?????????‐ | 0.76 |
| WDR70 | rs184179037 | 5 | 37483940 | T/C | 0.006 | 1.34 | 0.30 | 9.37×10−6 | –+?+?+??+++????? | 0.38 | 1.30 | 0.33 | 7.69×10−5 | –+?+?+??+++????? | 0.10 |
| API5 | rs569584007 | 11 | 43166842 | G/T | 0.005 | 0.75 | 0.41 | 0.07 | ??????+??+++?+?+‐ | 0.46 | 1.81 | 0.41 | 7.50×10−6 | ??????+??‐++?+?+‐ | 0.003 |
| ACER3 | rs115816806 | 11 | 76541840 | G/A | 0.006 | 1.66 | 0.62 | 0.007 | ?‐??+??+????????+ | 0.12 | 1.54 | 0.63 | 0.01 | ?‐??+??+????????+ | 0.15 |
| PIK3C2G | rs75739461 | 12 | 18471546 | A/G | 0.011 | 0.73 | 0.20 | 3.39×10−4 | ‐?+++?++‐++????+‐ | 0.56 | 0.92 | 0.22 | 1.99×10−5 | +?‐++?++‐++????+‐ | 0.46 |
| ARRDC4/IGF1R | rs570487962 | 15 | 97992685 | C/A | 0.012 | −1.76 | 0.36 | 1.33×10−6 | ???????+?????‐??? | 0.05 | −2.35 | 0.42 | 1.65×10−8 * | ???????+?????‐??? | 0.02 |
| RBFOX1 | rs79537509 | 16 | 8288401 | T/G | 0.008 | 1.32 | 0.27 | 1.38×10−6 | ‐+‐?++++?++????+? | 0.49 | 1.43 | 0.30 | 1.34×10−6 | ‐+‐?++++?++????+? | 0.37 |
| Loci previously reported in Reitz et al. (2013) | |||||||||||||||
| ABCA7 | rs115550680 | 19 | 1050420 | G/A | 0.061 | 0.33 | 0.08 | 4.67×10−5 | ‐++++‐++‐++++++‐? | 0.11 | 0.45 | 0.09 | 4.30×10−7 * | ‐++++‐++‐+++++++? | 0.11 |
| HMHA1 | rs115553053 | 19 | 1082844 | T/C | 0.047 | 0.40 | 0.09 | 2.51×10−5 | ‐+++–++‐++++++++ | 0.26 | 0.50 | 0.10 | 6.97×10−7 * | ++++–++‐++++++++ | 0.15 |
| GRIN3B | rs115882880 | 19 | 1001777 | A/C | 0.098 | 0.29 | 0.07 | 1.18×10−5 | ‐++++‐+++‐++++++? | 0.08 | 0.35 | 0.07 | 8.58×10−7 * | ‐++++‐++++++++++? | 0.09 |
| NSG2/MSX2 | rs145848414 | 5 | 174014114 | A/G | 0.035 | 0.45 | 0.11 | 3.74×10−5 | ‐+++++++++++?+++‐ | 0.57 | 0.47 | 0.12 | 7.31×10−5 | ‐+++++++++++?+++‐ | 0.42 |
| APOE | rs157591 | 19 | 45423934 | A/G | 0.121 | 0.69 | 0.06 | 3.57×10−29 * | ++++++++‐+++++++? | 0.16 | −0.05 | 0.08 | 0.49 | ‐+++‐++—–++++? | 0.50 |
Abbreviations: dbSNP, Single Nucleotide Polymorphism Database; MAF, minor allele frequency.
Model 1 is adjusted for age, sex, and population stratification (PCs).
Model 2 is adjusted for age, sex, PCs, and APOE.
rs969240869 was filtered out in Model 1 due to extreme beta coefficients (|β| > 5).
Significant at p < 9×10−7.
FIGURE 1.
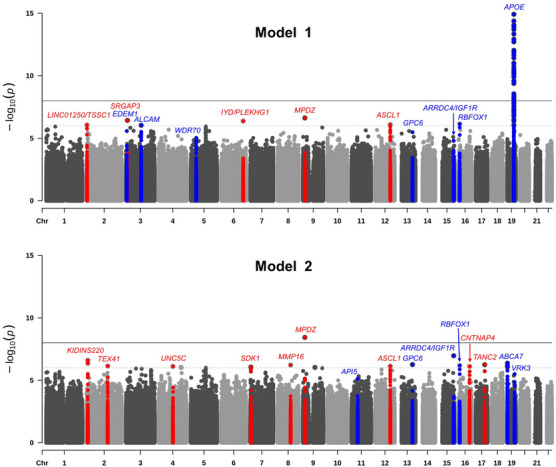
Manhattan plots showing negative log10‐transformed p‐values from the single‐variant meta‐analysis adjusted for age, sex, and population stratification (Model 1) and age, sex, population stratification, and APOE (Model 2). The solid black horizontal line represents a genome‐wide significance threshold of p = 5 × 10−8, and the dotted line represents a suggestive threshold of p = 5 × 10−6. All novel loci significant at p < 9 × 10−7 are shown in red, and all previously reported loci significant at p < 9 × 10−6 are shown in blue. The y‐axis has been truncated, the lowest p value on chromosome 19 is 3.11 × 10−65.
In addition to US‐based datasets of African American individuals, the present analyses also included samples of Yoruba from Ibadan with a high degree of African ancestry (see Supplementary Figure 4 for principal component plots of all datasets). Comparison of effect sizes of identified loci between the datasets with high (i.e., Ibadan dataset) and lower degree of African ancestry (all other US‐based African American datasets), showed comparable effects between datasets with higher and lower degree of African ancestry at all loci that were present in both groups of datasets except for chr2:3070309, chr2:145902343, and chr12:103570373, which showed differential association. These three loci showed a higher effect size in the Yoruba samples (see Supplementary Figure 2), indicating that degree of African ancestry at these loci may modify the effect. Closer examination of the local association and LD patterns in these three regions showed comparable LD patterns between African and African American datasets at the two chr2 loci, but differential association and LD patterns at the chr12 locus (see Supplementary Figure 5), suggesting that this locus may show differential genetic ancestry. The top variants at chr4:96471106 (rs969240869), chr6:150819452 (rs117762284), chr9:13220518 (rs141610415), and chr16:76535070 (rs1010752317) observed in the metanalysis were absent in the Ibadan dataset, which is consistent with the absence of these alleles in Yoruba according to reference data (https://gnomad.broadinstitute.org/).
While in the African American datasets the strongest association at the chromosome 12 locus is observed at 103,538 kb, the high African ancestry Ibadan dataset shows genome‐wide significant association at p = 3.8 × 10−8 (rs547590324) 180 kb upstream within the ASCL1 gene. Analysis of local ancestry at the two meta‐analysis top locus markers from both models (Model 1: rs556001137; Model 2: rs138206541; see Table 1) showed that both markers appear on African and Amerindian but not European local ancestry backgrounds (Supplementary Table 4). Comparison of the allele frequencies of the three top markers only observed with significant association in the African Ibadan dataset (rs547590324, rs144730555, rs148789350; see Supplementary Figure 5C) across the African American and Ibadan datasets, as well as gnomAD, 1000 Genomes, and the Human Genome Diversity Project (HGDP), suggest that these markers are present in West African individuals but less frequent in Hispanic individuals and virtually absent in other African regions (ie. Kenya, South Africa), and those of European and Asian ancestry. (https://gnomad.broadinstitute.org/variant/12‐102960732‐G‐A?dataset = gnomad_r3; https://gnomad.broadinstitute.org/variant/12‐102957658‐C‐G?dataset = gnomad_r3, https://gnomad.broadinstitute.org/variant/12‐102956366‐C‐A?dataset = gnomad_r3).
Of the 16 loci identified in our previous analyses, 10 , 11 all but 2 (SIPA1L2 and ACER3, both associated with rare variants) replicated at a p‐value of < 9 × 10−5. Of the variants previously implicated in either AD, ADRD, or AD by proxy in African American individuals by other studies, 37 , 38 , 39 , 40 , 41 , 42 rs112404845 in COBL (p = 6.13×10−5), rs2234258 in TREM2 (p = 1.74×10−5), and rs73505251 in ABCA7 (p = 2.13×10−5) showed suggestive significance, and variants in SLC4A1AP (rs17006206), POLN (rs1923775), RP11‐785H20.1 (rs956225), RP11‐116D17.2 (rs10850408), ENOX1 (rs17460623), AKAP9 (rs144662445; rs149979685), TREM2 (rs2234256; rs73427293), BZRAP1‐AS1 (rs263251), AC010967.2/SCARNA16 (rs58443395), RP11‐157D6.1/CD2AP (rs7738720), and RP11‐192P9.1/TRPS1 (rs76427927) were replicated with nominal significance (see Supplementary Table 5). Of the GWAS loci implicated in NHW individuals 1 , 3 besides APOE and ABCA7, only the variants in BIN1, IDUA, UNC5CL, UMAD1, USP6NL, FERMT2, and SCIMP showed nominal association (see Supplementary Table 6).
3.2. Gene‐based and pathway analyses
Only APOE surpassed the Bonferroni corrected p‐value threshold for gene‐based analyses. However, we also identified two novel loci (C9orf139 and SNX31) that are suggestively associated with AD in individuals of African ancestry at a suggestive significance threshold of p < 9×10−5 (see Table 2 and Supplementary Figures 6 and 7), in addition to four regions (LARP1B, TREM2, SERPINB13, and ABCA7) that we previously reported. 10 , 11 Gene‐based results for all previously reported genes in NHW 1 , 3 and African ancestry 37 , 38 , 39 , 40 , 41 , 42 are reported in Supplementary Tables 7 and 8. Gene‐set analyses identified 16 pathways at p < 5 × 10−4 (see Table 3). Notably, in addition to lipid metabolism, immune response, transcription/DNA repair, and intracellular trafficking that are also associated with AD in NHW, 1 , 3 sodium transport emerged as a novel prominent pathway that has not been reported before in analyses restricted to individuals of African ancestry or in analyses in other ethnic groups.
TABLE 2.
Top regions identified in gene‐based analysis.
| Model 1 | Model 2 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Chromosome | Start BP (hg37) | Stop BP (hg37) | No. of SNPs | No. of parameters | N | Z Statistic | p‐Value | No. of SNPs | No. of parameters | N | Z Statistic | p‐Value |
| Novel regions | |||||||||||||
| C9orf139 | 9 | 139886916 | 139941234 | 358 | 74 | 9095 | 3.28 | 5.27×10−4 | 356 | 68 | 8917 | 3.85 | 5.92×10−5 * |
| SNX31 | 8 | 101575116 | 101710643 | 1163 | 151 | 9095 | 3.01 | 1.31×10−3 | 1156 | 146 | 8917 | 3.81 | 6.83×10−5 * |
| Previously reported regions | |||||||||||||
| APOE | 19 | 45374011 | 45422650 | 342 | 85 | 9095 | 9.18 | 2.23×10−20 ** | 343 | 85 | 8917 | 1.77 | 0.04 |
| LARP1B | 4 | 128947423 | 129154086 | 1053 | 81 | 9095 | 3.81 | 6.88×10−5 * | 1211 | 93 | 8917 | 2.32 | 0.01 |
| TREM2 | 6 | 41116244 | 41165924 | 380 | 62 | 9095 | 3.55 | 1.93×10−4 | 379 | 72 | 8917 | 4.14 | 1.76×10−5 * |
| SERPINB13 | 18 | 61219223 | 61281873 | 560 | 76 | 9095 | 3.43 | 2.97×10−4 | 555 | 78 | 8917 | 4.02 | 2.94×10−5 * |
| ABCA7 | 19 | 1005102 | 1075571 | 765 | 116 | 9095 | 3.14 | 8.46×10−4 | 765 | 116 | 8917 | 3.78 | 7.76×10−5 * |
Abbreviations: BP, base pair; SNP, single nucleotide polymorphism.
Model 1 is adjusted for age, sex, and population stratification (PCs).
Model 2 is adjusted for age, sex, PCs, and APOE.
Suggestively associated at p < 9×10−5.
Significant at the Bonferroni corrected threshold of 2.59×10−6.
TABLE 3.
Results of pathway analysis.
| GO number | Pathway | Model | No. of genes | β | SE | p‐Value |
|---|---|---|---|---|---|---|
| GO:0010915 | regulation of very‐low‐density lipoprotein particle clearance | 1 | 4 | 3.25 | 0.59 | 2.07×10−8 |
| GO:0051103 | DNA ligation involved in DNA repair | 1,2 * | 9 | 1.54 | 0.36 | 1.11×10−5 |
| GO:0015347 | sodium‐independent organic anion transmembrane transporter activity | 1 | 16 | 0.84 | 0.20 | 2.11×10−5 |
| GO:0043252 | sodium‐independent organic anion transport | 1 | 15 | 0.81 | 0.21 | 7.82×10−5 |
| GO:0034596 | phosphatidylinositol phosphate 4‐phosphatase activity | 2 | 8 | 1.14 | 0.30 | 9.25×10−5 |
| GO:0010669 | epithelial structure maintenance | 2 | 28 | 0.55 | 0.15 | 1.53×10−4 |
| GO:0050890 | cognition | 2 | 281 | 0.17 | 0.05 | 1.70×10−4 |
| GO:0000405 | bubble DNA binding | 2 | 8 | 1.00 | 0.29 | 2.81×10−4 |
| GO:0019199 | transmembrane receptor protein kinase activity | 1 | 80 | 0.32 | 0.09 | 3.64×10−4 |
| GO:0097350 | neutrophil clearance | 1,2 * | 5 | 1.18 | 0.35 | 3.95×10−4 |
| GO:0070327 | thyroid hormone transport | 1 | 5 | 1.09 | 0.33 | 4.14×10−4 |
| GO:0042180 | cellular ketone metabolic process | 1 | 188 | 0.19 | 0.06 | 4.15×10−4 |
| GO:0045898 | regulation of RNA polymerase II transcriptional preinitiation complex assembly | 1 | 14 | 0.70 | 0.21 | 4.26×10−4 |
| GO:0009649 | entrainment of circadian clock | 1 | 26 | 0.55 | 0.16 | 4.36×10−4 |
| GO:0060260 | regulation of transcription initiation from RNA polymerase II promoter | 1 | 26 | 0.52 | 0.16 | 4.39×10−4 |
| GO:0043194 | axon initial segment | 2 | 20 | 0.59 | 0.18 | 4.96×10−4 |
Model 1 is adjusted for age, sex, and population stratification (PCs).
Model 2 is adjusted for age, sex, PCs, and APOE.
Results shown for Model 1.
3.3. Colocalization analyses
Colocalization analyses at the identified loci using the coloc package showed colocalization of eQTL and GWAS signals at the chr 3 locus near SRGAP3 associated with a common variant (PP.H4 = 0.75; lead eQTL variant: rs17744749).
3.4. Hi‐C analysis
Hi‐C analysis revealed that the top SNP rs396323 in SRGAP3 strongly interacts with the proximal promoter of SRGAP3 and with RAD18 at a weaker strength (Supplementary Figure 8). The rs396323 co‐localizes with a H3K27AC peak in both ENCODE cell lines and adult brains, suggesting that chromatin loops involving rs396323 represent promoter‐enhancer interactions. In addition, the second strongest association signals at SRGAP3 came from three less common variants within chr3: 9150527‐9155067 (MAF < 0.05, p = 9.01 × 10−5 ∼1.54 × 10−4). This block of DNA co‐localizes with a brain specific H3K27AC peak and interacts with both SRGAP3 and RAD18 promoters in a similar fashion as rs396323 (Supplementary Figure 8). The variants in this block (chr3: 9150527‐9155067) are not in LD with the top variant (rs396323) at this locus (R 2 < 0.1) and represent an independent signal at the locus. Notably, both rs396323 and the variants at chr3: 9150527‐9155067 interact with the lead e‐QTL variant (rs17744749) identified by the colocalization analyses near SRGAP3 described above.
3.5. Human and zebrafish brain tissue RNAseq and CSF proteomic data analyses
In brain RNAseq data from over 2,100 samples from post mortem brains of more than 1,100 individuals from the ROSMAP study, the Mount Sinai Brain Bank (MSBB) and the Mayo Clinic (https://agora.adknowledgeportal.org/), all nearest genes at all novel loci identified in single marker association or gene‐based analyses are differentially expressed between AD cases and controls (see Supplementary Figure 9). In brain RNAseq analyses in zebrafish, orthologs for MPDZ, SRGAP3, SDK1, TANC2, MMP16, and UNC5C were downregulated after inducing amyloid toxicity, while the ortholog of ASCL1 was upregulated (see Supplementary Figure 10). In CSF proteomic data, levels of UNC5C were associated with amount of GFAP, a measure of astrogliosis (coef for association = −0.012; p = .004).
3.6. Analysis of whole‐genome sequencing data
To provide additional validation of the genome‐wide significant variant in MPDZ (rs141610415; chr9:13220518), we performed chi‐square analyses on the whole‐genome sequence data of individuals of African ancestry from the latest Alzheimer's Disease Sequencing Project (ADSP) R4 36K release (https://www.niagads.org/news/alzheimer%E2%80%99s‐disease‐sequencing‐project‐update‐round‐2‐harmonized‐phenotypes‐and‐r4‐36k). Also in this dataset, the minor allele from this top GWAS variant occurred more frequently in cases than controls (allele count in cases = 16; total alleles in cases = 3330; allele count in controls = 6; total alleles in controls = 6362; χ2 = 12.74; p = 3.58×10−4). In addition, this variant was found to be in high LD with another variant (rs181009479; chr9:13240984; R 2 = 0.67) with a high CADD score of 16.26 and a minor allele that is also significantly more frequent in cases than controls (allele count in cases = 22; total alleles in cases = 3346; allele count in controls = 20; total alleles in controls = 6378; χ2 = 5.26; p = 0.02).
4. DISCUSSION
This GWAS on AD in individuals of African ancestry, which is the first to include individuals from continental Africa, nominated 1 novel locus at P = 3.68×10−9 and 11 novel suggestive loci at p ≤ 9×10−7. At least one locus appears influenced by regional origin of African ancestry. Gene‐based analyses nominated two additional suggestive loci. Of over 80 known loci implicated in NHW individuals, 1 , 3 only 8 were associated at nominal significance level or stronger in addition to APOE.
Most of the novel loci identified in this study cluster in known AD pathways. The novel top locus, associated with a rare variant (rs141610415) with strong regional support, is located within MPDZ on chromosome 9p23. MPDZ is highly expressed in brain and encodes a modular scaffold protein 43 , 44 that is localized near the junctions of neuronal synapses. 45 It is involved in regulation of synaptic transmission 46 and is a vital component of the NMDAR signaling complex in excitatory synapses of hippocampal neurons critical for learning and memory. 45 According to GTEx data from brain tissue, 47 MPDZ contains six splice site variants, three of which are in a haplotype block with the top variant identified in our analyses (see Supplementary Figure 11). As described above, the minor allele from our top variant is also more frequent in cases than controls in ADSP whole‐genome sequence data from individuals of African ancestry and is in high LD with another variant (also more frequent in cases than controls) with a CADD score of 16. According to Agora, MPDZ is highly expressed in brain; is differentially expressed between AD cases and controls in the anterior cingulate cortex, cerebellum, inferior frontal gyrus, parahippocampal gyrus, and superior temporal gyrus; and received a very high AD‐risk score of 3.91 out of 5 based on GWAS, eQTL, transcriptomic, proteomic, and other multi‐omic data (https://agora.adknowledgeportal.org/). 48
The loci at 2p25 and 3p25 are common and robustly present in at least 15 of the 17 contributing datasets. The top variant at 2p25 (rs78857220) is located within a long intergenic non‐coding RNA (lincRNA). lincRNAs play an essential role in RNA transcription, translation, regulation of gene expression and chromatin remodeling, impacting cell proliferation, survival, and differentiation. 49 Human and animal studies link lincRNAs to various neurodegenerative diseases including AD. 50 , 51 Notably, LINC01250 identified in our study also emerged as a top hit in a recent GWAS of brain amyloid deposition. 52
The top variant at 3p25 (rs396323) is located within SRGAP3, which is involved in nervous system development 53 , 54 and has been implicated in cognitive functioning 55 and intellectual disability. 56 , 57 Colocalization and Hi‐C data analyses demonstrating colocalization and interaction of the GWAS locus with functional eQTLs and regulatory elements in SRGAP3 provide strong functional support for this signal. SRGAP3 is highly expressed in brain and is downregulated in the parahippocampal gyrus and temporal cortex in AD cases compared to cognitively healthy individuals. 58 In line with this finding, in our study SRGAP3 expression was downregulated in zebrafish brain after inducing amyloid toxicity.
The additional identified loci, all associated with rare variants with suggestive significance, also cluster near/in genes involved in biologically plausible pathways. KIDINS220 encodes a scaffold protein involved in neuronal survival, neurite outgrowth, and synaptic plasticity. 59 , 60 In rodents, knockdown of Kidins220 leads to memory deficits. 61 TEX41 encodes a lincRNA that, as described above, can lead to disease by impacting RNA transcription, translation, regulation of gene expression, or chromatin remodeling. 49 UNC5C encodes a netrin receptor protein directing axon extension and cell migration during neural development. 62 , 63 , 64 Notably, variants in UNC5C have been previously identified in multiplex families with AD, an association that has been replicated in independent case‐control studies, 65 and the gene encoding UNC5C‐like (UNC5CL) protein is an implicated risk locus in NHW 1 that has also been shown in a methylation study to be associated with key AD neuropathologic changes. 66 The present study is the first to report association at this locus in individuals of African ancestry. PLEKHG1 encodes a protein involved in Rho GTPase signaling that has been associated with rate of cognitive decline, 67 white matter hyperintensities, 68 and high blood pressure in African American individuals. 69 SDK1 encodes an immunoglobulin superfamily cell adhesion protein required for synaptic connectivity between neurons. 70 MMP16 encodes a matrix metalloproteinase family protein. Matrix metalloproteinases (MMPs) have been implicated in several neuropathological processes including inflammation, blood‐brain barrier (BBB) damage, and neuronal cell death. 71 ASCL1 encodes a protein that regulates neurogenesis and neuronal differentiation. 72 While the Ibadan dataset has a limited sample size and the possibility of false‐positive findings cannot be excluded, comparison of allele frequencies, association, and LD patterns of identified loci between datasets with high and lower degree of African ancestry indicate differential association patterns at this locus suggesting an influence by origin of local African ancestry. CNTNAP4 encodes a neurexin superfamily protein involved in neural development and synaptic transmission, 73 , 74 and variants in this gene have been associated with AD and cognitive impairment in NHW individuals. 75 TANC2 encodes a scaffolding protein inhibiting mTOR signaling controlling long‐term synaptic efficacy and memory storage. 76 The two loci identified in gene‐based analyses encode a member of the SNX‐FERM family involved in intracellular trafficking (SNX31) 77 and a lincRNA (C9orf139/LINC02908). In brain RNAseq data from over 1,100 individuals from the ROSMAP study, the Mount Sinai Brain Bank (MSBB), and the Mayo Clinic all these genes are differentially expressed between AD cases and controls (https://agora.adknowledgeportal.org/). In line with this notion, RNAseq analyses showed that zebrafish orthologs for MPDZ, SRGAP3, SDK1, TANC2, MMP16, and UNC5C are downregulated and the ortholog of ASCL1 is upregulated after inducing amyloid toxicity. In addition, CSF proteomic data showed that levels of UNC5C are associated with amount of GFAP, a marker of astroglial activation and gliosis during neurodegeneration, further supporting the involvement of these genes in AD etiology. Imputation quality for all novel variants was excellent, and there was no evidence of inflation, minimizing the possibility of spurious associations.
Our pathway analyses also suggest that at least a subset of the causative molecular pathways (immunity, lipid processing, intracellular trafficking, DNA repair, and transcription) overlap with those in NHW individuals, although largely with different disease‐associated genes within these pathways. However, a novel AD pathway emerging from our analysis is sodium‐independent organic anion transmembrane transporter (OAT) activity. OATs are transporter proteins delivering a range of hydrophobic organic anions including neurotransmitter and amyloid beta metabolites across the BBB. There is significant evidence that BBB dysfunction and dysregulation of BBB transporters are involved in AD etiology. 78
This study has limitations. First, given the paucity of available African American samples for genomic research on AD and the need to maximize statistical power, we combined all samples into one discovery set and relied on a range of multi‐omic data for functional validation. Additional validation will likely need to be derived from experimental studies. Second, while this is the largest GWAS on directly assessed AD in African ancestry individuals to date, our sample size was underpowered to detect associations with very rare variants or rare variants exerting very small effects. It should also be noted that, while our findings at several loci are supported by additional functional data, further fine mapping analyses of all loci are needed to determine the specific causative variants under these signals.
In summary, while our findings support the notion that the principal molecular pathways implicated in AD etiology in individuals of African ancestry largely overlap with those in NHW, they suggest that the specific disease‐associated loci within these pathways differ. They further suggest that, even within individuals of African ancestry, genetic association with AD differs and is influenced by local genetic ancestry. These observations have critical implications for our quest to fully disentangle the genetic influences on the biology of AD and to develop effective, population‐specific druggable targets. First, our findings provide significant support for the importance of lipid metabolism, native immune response, intracellular trafficking, nervous system development, and synaptic plasticity in AD etiology and suggest that these pathways are critical in disease etiology across ethnic groups. At the same time, they suggest that there are also pathways whose contributions differ in individuals of African ancestry compared to other populations. OAT activity was identified as a novel disease mechanism in these individuals of African ancestry, while amyloid and tau pathology are not represented among the top pathways. It is important to note, however, that associations with amyloid and tau pathways may be revealed as larger studies of AD in African ancestry populations are conducted, as was the case in NHW populations. 3 The observation of differential association patterns modulated by regional origin of African ancestry underscores the importance of comprehensive analysis of local ancestry at disease‐associated genetic loci, and the urgent need to assemble larger sample sets from Africa that cover as much of the African genetic diversity as possible. 79 It is likely that both an overall increase of the total sample size as well as higher and broader representation of individuals of high African ancestry, as is currently underway through ADSP efforts, will facilitate the identification of additional disease‐associated loci and significantly help to disentangle local genetic ancestry effects.
CONFLICTS OF INTERESTS STATEMENT
C.K. is an advisor to Neuron D GmbH, Germany. No other authors have any competing interests to disclose. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects provided informed consent.
Supporting information
Supporting Information
ICMJE Disclosure Form
ACKNOWLEDGMENTS
The National Institutes of Health, National Institute on Aging (NIH‐NIA) supported this work through the following grants: ADGC, U01 AG032984, RC2 AG036528; Samples from the National Cell Repository for Alzheimer's Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24‐AG041689); GCAD, U54 AG052427; NACC, U01 AG016976; NIA FBS (Columbia University), U24 AG026395, U24 AG026390, R01AG041797; Banner Sun Health Research Institute P30 AG019610; Boston University, P30 AG013846, U01 AG10483, R01 CA129769, R01 MH080295, R01 AG017173, R01 AG025259, R01 AG048927, R01AG33193, R01 AG009029; Columbia University, P50 AG008702, R37 AG015473, R01 AG037212, R01 AG028786; Duke University, P30 AG028377, AG05128; Emory University, AG025688; Group Health Research Institute, UO1 AG006781, UO1 HG004610, UO1 HG006375, U01 HG008657; Indiana University, P30 AG10133, R01 AG009956, RC2 AG036650; Johns Hopkins University, P50 AG005146, R01 AG020688; Massachusetts General Hospital, P50 AG005134; Mayo Clinic, P50 AG016574, R01 AG032990, KL2 RR024151; Mount Sinai School of Medicine, P50 AG005138, P01 AG002219; New York University, P30 AG08051, UL1 RR029893, 5R01AG012101, 5R01AG022374, 5R01AG013616, 1RC2AG036502, 1R01AG035137; North Carolina A&T University, P20 MD000546, R01 AG28786‐01A1; Northwestern University, P30 AG013854; Oregon Health & Science University, P30 AG008017, R01 AG026916; Rush University, P30 AG010161, R01 AG019085, R01 AG15819, R01 AG17917, R01 AG030146, R01 AG01101, RC2 AG036650, R01 AG22018; TGen, R01 NS059873; REAADI study is supported by NIA grant AG052410; University of Alabama at Birmingham, P50 AG016582; University of Arizona, R01 AG031581; University of California, Davis, P30 AG010129; University of California, Irvine, P50 AG016573; University of California, Los Angeles, P50 AG016570; University of California, San Diego, P50 AG005131; University of California, San Francisco, P50 AG023501, P01 AG019724; University of Kentucky, P30 AG028383, AG05144; University of Michigan, P50 AG008671; University of Pennsylvania, P30 AG010124; University of Pittsburgh, P50 AG005133, AG030653, AG041718, AG07562, AG02365; University of Southern California, P50 AG005142; University of Texas Southwestern, P30 AG012300; University of Miami, R01 AG027944, AG010491, AG027944, AG021547, AG019757; University of Washington, P50 AG005136, R01 AG042437; University of Wisconsin, P50 AG033514; Vanderbilt University, R01 AG019085; and Washington University, P50 AG005681, P01 AG03991, P01 AG026276. The Kathleen Price Bryan Brain Bank at Duke University Medical Center is funded by NINDS grant # NS39764, NIMH MH60451 and by Glaxo Smith Kline. Support was also from the Alzheimer's Association (LAF, IIRG‐08‐89720; MP‐V, IIRG‐05‐14147), the US Department of Veterans Affairs Administration, Office of Research and Development, Biomedical Laboratory Research Program, and BrightFocus Foundation (MP‐V, A2111048). P.S.G.‐H. is supported by Wellcome Trust, Howard Hughes Medical Institute, and the Canadian Institute of Health Research. Genotyping of the TGEN2 cohort was supported by Kronos Science. The TGen series was also funded by NIA grant AG041232 to AJM and MJH, The Banner Alzheimer's Foundation, The Johnnie B. Byrd Sr. Alzheimer's Institute, the Medical Research Council, and the state of Arizona and also includes samples from the following sites: Newcastle Brain Tissue Resource (funding via the Medical Research Council, local NHS trusts and Newcastle University), MRC London Brain Bank for Neurodegenerative Diseases (funding via the Medical Research Council),South West Dementia Brain Bank (funding via numerous sources including the Higher Education Funding Council for England (HEFCE), Alzheimer's Research Trust (ART), BRACE as well as North Bristol NHS Trust Research and Innovation Department and DeNDRoN), The Netherlands Brain Bank (funding via numerous sources including Stichting MS Research, Brain Net Europe, Hersenstichting Nederland Breinbrekend Werk, International Parkinson Fonds, Internationale Stiching Alzheimer Onderzoek), Institut de Neuropatologia, Servei Anatomia Patologica, Universitat de Barcelona. ADNI data collection and sharing was funded by the National Institutes of Health Grant U01 AG024904 and Department of Defense award number W81XWH‐12‐2‐0012. ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The results published here are in whole or in part based on data obtained from Agora, a platform initially developed by the NIA‐funded AMP‐AD consortium that shares evidence in support of AD target discovery. Agora is available at: https://agora.adknowledgeportal.org/. The RNAseq results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org). Study data were provided by the Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161 (ROS), R01AG15819 (ROSMAP; genomics and RNAseq), R01AG17917 (MAP), R01AG30146, R01AG36042 (5hC methylation, ATACseq), RC2AG036547 (H3K9Ac), R01AG36836 (RNAseq), R01AG48015 (monocyte RNAseq) RF1AG57473 (single nucleus RNAseq), U01AG32984 (genomic and whole exome sequencing), U01AG46152 (ROSMAP AMP‐AD, targeted proteomics), U01AG46161(TMT proteomics), U01AG61356 (whole genome sequencing, targeted proteomics, ROSMAP AMP‐AD), the Illinois Department of Public Health (ROSMAP), and the Translational Genomics Research Institute (genomic). Additional phenotypic data can be requested at www.radc.rush.edu. The data from Mount Sinai Brain Bank (MSBB) were generated from postmortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by Dr. Eric Schadt from Mount Sinai School of Medicine. This study was supported by NIH grants: U19AG074865 (MPV, CR, JLH, WB, BK, GB, AO, RA), R01AG072547 (MPV, CR, GB, JLH, RA), U01AG058654 (JLH, MPV, WSB, ERM, LAF, FR, MC, JMV), R01AG048927 (LAF), R01AG064614 (CR), U24AG056270 (RM, CR), P30AG066462 (CR), AG057659 (MPV), AG062943 (MPV), U01AG072579 (JMV), RF1AG059018 (JMV).
Ray NR, Kunkle BW, Hamilton‐Nelson K, et al. Extended genome‐wide association study employing the African genome resources panel identifies novel susceptibility loci for Alzheimer's disease in individuals of African ancestry. Alzheimer's Dement. 2024;20:5247–5261. 10.1002/alz.13880
Nicholas R. Ray and Brian W. Kunkle authors contributed equally.
DATA AVAILABILITY STATEMENT
This study was approved by all appropriate university institutional review boards and was performed in accordance with the ethical standards described in the 1964 Declaration of Helsinki and its later amendments. This study specifically addresses the topic of diversity, equity, and inclusion by targeting individuals of African descent, who are vastly underrepresented in AD research.
REFERENCES
- 1. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wightman DP, Jansen IE, Savage JE, et al. A genome‐wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat Genet. 2021;53:1276‐1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tang M‐X, Cross P, Andrews H, et al. Incidence of AD in African‐Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology. 2001;56:49‐56. [DOI] [PubMed] [Google Scholar]
- 7. Mehta KM, Yeo GW. Systematic review of dementia prevalence and incidence in United States race/ethnic populations. Alzheimers Dement. 2017;13:72‐83. [DOI] [PubMed] [Google Scholar]
- 8. Steenland K, Goldstein FC, Levey A, Wharton W. A meta‐analysis of Alzheimer's Disease incidence and prevalence comparing African‐Americans and Caucasians. J Alzheimers Dis. 2016;50:71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lennon JC, Aita SL, Bene VAD, et al. Black and White individuals differ in dementia prevalence, risk factors, and symptomatic presentation. Alzheimers Dement. 2022;18:1461‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kunkle BW, Schmidt M, Klein H‐U, et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta‐analysis. JAMA Neurol. 2021;78:102‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reitz C, Jun G, Naj A, et al. Variants in the ATP‐binding cassette transporter (ABCA7), apolipoprotein E ϵ4, and the risk of late‐onset Alzheimer disease in African Americans. JAMA. 2013;309:1483‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NiNCdS‐AdrdA Work Group under the auspices of department of health and human Services Task Force on Alzheimer's disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 13. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sengupta D, Botha G, Meintjes A, et al. Performance and accuracy evaluation of reference panels for genotype imputation in sub‐Saharan African populations. Cell Genom. 2023;3:100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Consortium TWTCC . Genome‐wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marchini J, Howie B. Genotype imputation for genome‐wide association studies. Nat Rev Genet. 2010;11:499‐511. [DOI] [PubMed] [Google Scholar]
- 17. Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome‐wide association studies by imputation of genotypes. Nat Genet. 2007;39:906‐913. [DOI] [PubMed] [Google Scholar]
- 18. Chen M‐H, Yang Q. GWAF: an R package for genome‐wide association analyses with family data. Bioinformatics. 2010;26:580‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aulchenko YS, Ripke S, Isaacs A, Van Duijn CM. GenABEL: an R library for genome‐wide association analysis. Bioinformatics. 2007;23:1294‐1296. [DOI] [PubMed] [Google Scholar]
- 21. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene‐set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe K, Taskesen E, Van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commum. 2017;8:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Consortium TGO. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325‐D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cann HM, de Toma C, Cazes L, et al. A human genome diversity cell line panel. Science. 2002;296:261‐262. [DOI] [PubMed] [Google Scholar]
- 25. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526:68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delaneau O, Marchini J. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun. 2014;5:3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maples BK, Gravel S, Kenny EE, Bustamante CD. RFMix: a discriminative modeling approach for rapid and robust local‐ancestry inference. Am J Hum Genet. 2013;93:278‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Griswold AJ, Celis K, Bussies PL, et al. Increased APOE ε4 expression is associated with the difference in Alzheimer's disease risk from diverse ancestral backgrounds. Alzheimers Dement. 2021;17:1179‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu L, Liu X, Huang WK, et al. Robust Hi‐C maps of enhancer‐promoter interactions reveal the function of non‐coding genome in neural development and diseases. Mol Cell. 2020;79:521‐534.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang S, Plummer D, Lu L, et al. DeepLoop robustly maps chromatin interactions from sparse allele‐resolved or single‐cell Hi‐C data at kilobase resolution. Nat Genet. 2022;54:1013‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bhattarai P, Thomas AK, Cosacak MI, et al. IL4/STAT6 signaling activates neural stem cell proliferation and neurogenesis upon amyloid‐β42 aggregation in adult zebrafish brain. Cell Rep. 2016;17:941‐948. [DOI] [PubMed] [Google Scholar]
- 36. Lee AJ, Raghavan NS, Bhattarai P, et al. FMNL2 regulates gliovascular interactions and is associated with vascular risk factors and cerebrovascular pathology in Alzheimer's disease. Acta Neuropathol. 2022;144:59‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jin SC, Carrasquillo MM, Benitez BA, et al. TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol Neurodegener. 2015;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jun GR, Chung J, Mez J, et al. Transethnic genome‐wide scan identifies novel Alzheimer's disease loci. Alzheimers Dement. 2017;13:727‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Logue MW, Schu M, Vardarajan BN, et al. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch Neurol. 2011;68:1569‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Logue MW, Schu M, Vardarajan BN, et al. Two rare AKAP9 variants are associated with Alzheimer's disease in African Americans. Alzheimers Dement. 2014;10:609‐618.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mez J, Chung J, Jun G, et al. Two novel loci, COBL and SLC10A2, for Alzheimer's disease in African Americans. Alzheimers Dement. 2017;13:119‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sherva R, Zhang R, Sahelijo N, et al. African ancestry GWAS of dementia in a large military cohort identifies significant risk loci. Mol Psychiatry. 2023;28:1293‐1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adachi M, Hamazaki Y, Kobayashi Y, et al. Similar and distinct properties of MUPP1 and Patj, two homologous PDZ domain‐containing tight‐junction proteins. Mol Cell Biol. 2009;29:2372‐2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ullmer C, Schmuck K, Figge A, Lübbert H. Cloning and characterization of MUPP1, a novel PDZ domain protein. FEBS Lett. 1998;424:63‐68. [DOI] [PubMed] [Google Scholar]
- 45. Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP‐MUPP1‐CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor‐dependent synaptic AMPA receptor potentiation. Neuron. 2004;43:563‐574. [DOI] [PubMed] [Google Scholar]
- 46. Kennedy MB. Signal‐processing machines at the postsynaptic density. Science. 2000;290:750‐754. [DOI] [PubMed] [Google Scholar]
- 47. Consortium G. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318‐1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cary GA, Wiley JC, Gockley J, et al. Genetic and multi‐omic risk assessment of Alzheimer's disease implicates core associated biological domains. Alzheimer's & Dementia: Translational Research & Clinical Interventions. 2024;10:e12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non‐coding RNA. Nat Rev Mol Cell Biol. 2018;19:143‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cao M, Li H, Zhao J, Cui J, Hu G. Identification of age‐ and gender‐associated long noncoding RNAs in the human brain with Alzheimer's disease. Neurobiol Aging. 2019;81:116‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou X, Xu J. Identification of Alzheimer's disease‐associated long noncoding RNAs. Neurobiol Aging. 2015;36:2925‐2931. [DOI] [PubMed] [Google Scholar]
- 52. Yan Q, Nho K, Del‐Aguila JL, et al. Genome‐wide association study of brain amyloid deposition as measured by Pittsburgh Compound‐B (PiB)‐PET imaging. Mol Psychiatry. 2021;26:309‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bacon C, Endris V, Rappold G. Dynamic expression of the Slit‐Robo GTPase activating protein genes during development of the murine nervous system. J Comp Neurol. 2009;513:224‐236. [DOI] [PubMed] [Google Scholar]
- 54. Zhang QP, Zhang HY, Zhang XF, et al. srGAP3 promotes neurite outgrowth of dorsal root ganglion neurons by inactivating RAC1. Asian Pac J Trop Med. 2014;7:630‐638. [DOI] [PubMed] [Google Scholar]
- 55. Bacon C, Endris V, Rappold GA. The cellular function of srGAP3 and its role in neuronal morphogenesis. Mech Dev. 2013;130:391‐395. [DOI] [PubMed] [Google Scholar]
- 56. Ellery PM, Ellis RJ, Holder SE. Interstitial 3p25 deletion in a patient with features of 3p deletion syndrome: further evidence for the role of SRGAP3 in mental retardation. Clin Dysmorphol. 2014;23:29‐31. [DOI] [PubMed] [Google Scholar]
- 57. Endris V, Wogatzky B, Leimer U, et al. The novel Rho‐GTPase activating gene MEGAP/srGAP3 has a putative role in severe mental retardation. Proc Natl Acad Sci U S A. 2002;99:11754‐11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sieberts SK, Perumal TM, Carrasquillo MM, et al. Large eQTL meta‐analysis reveals differing patterns between cerebral cortical and cerebellar brain regions. Sci Data. 2020;7:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Neubrand VE, Cesca F, Benfenati F, Schiavo G. Kidins220/ARMS as a functional mediator of multiple receptor signalling pathways. J Cell Sci. 2012;125:1845‐1854. [DOI] [PubMed] [Google Scholar]
- 60. Scholz‐Starke J, Cesca F. Stepping out of the shade: control of neuronal activity by the scaffold protein Kidins220/ARMS. Front Cell Neurosci. 2016;10:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Duffy AM, Schaner MJ, Wu SH, et al. A selective role for ARMS/Kidins220 scaffold protein in spatial memory and trophic support of entorhinal and frontal cortical neurons. Exp Neurol. 2011;229:409‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Küry S, Garrec C, Airaud F, et al. Evaluation of the colorectal cancer risk conferred by rare UNC5C alleles. World J Gastroenterol. 2014;20:204‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li Q, Wang BL, Sun FR, Li JQ, Cao XP, Tan L. The role of UNC5C in Alzheimer's disease. Ann Transl Med. 2018;6:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mur P, Sánchez‐Cuartielles E, Aussó S, et al. Scarce evidence of the causal role of germline mutations in UNC5C in hereditary colorectal cancer and polyposis. Sci Rep. 2016;6:20697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wetzel‐Smith MK, Hunkapiller J, Bhangale TR, et al. A rare mutation in UNC5C predisposes to late‐onset Alzheimer's disease and increases neuronal cell death. Nat Med. 2014;20:1452‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lang AL, Eulalio T, Fox E, et al. Methylation differences in Alzheimer's disease neuropathologic change in the aged human brain. Acta Neuropathol Commun. 2022;10:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sherva R, Tripodis Y, Bennett DA, et al. Genome‐wide association study of the rate of cognitive decline in Alzheimer's disease. Alzheimers Dement. 2014;10:45‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Traylor M, Tozer DJ, Croall ID, et al. Genetic variation in PLEKHG1 is associated with white matter hyperintensities (n = 11,226). Neurology. 2019;92:e749‐e757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Franceschini N, Fox E, Zhang Z, et al. Genome‐wide association analysis of blood‐pressure traits in African‐ancestry individuals reveals common associated genes in African and non‐African populations. Am J Hum Genet. 2013;93:545‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Goodman KM, Yamagata M, Jin X, et al. Molecular basis of sidekick‐mediated cell‐cell adhesion and specificity. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhong C, Bu X, Xu T, et al. Serum matrix metalloproteinase‐9 and cognitive impairment after acute ischemic stroke. J Am Heart Assoc. 2018;7:e007776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Woods LM, Ali FR, Gomez R, et al. Elevated ASCL1 activity creates de novo regulatory elements associated with neuronal differentiation. BMC Genomics. 2022;23:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang W, Zhou M, Lu W, et al. CNTNAP4 deficiency in dopaminergic neurons initiates Parkinsonian phenotypes. Theranostics. 2020;10:3000‐3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Spiegel I, Salomon D, Erne B, Schaeren‐Wiemers N, Peles E. Caspr3 and caspr4, two novel members of the caspr family are expressed in the nervous system and interact with PDZ domains. Mol Cell Neurosci. 2002;20:283‐297. [DOI] [PubMed] [Google Scholar]
- 75. Iakoubov L, Mossakowska M, Szwed M, Duan Z, Sesti F, Puzianowska‐Kuznicka M. A common copy number variation (CNV) polymorphism in the CNTNAP4 gene: association with aging in females. PLoS One. 2013;8:e79790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Costa‐Mattioli M, Monteggia LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci. 2013;16:1537‐1543. [DOI] [PubMed] [Google Scholar]
- 77. Yong X, Zhao L, Hu W, et al. SNX27‐FERM‐SNX1 complex structure rationalizes divergent trafficking pathways by SNX17 and SNX27. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Montagne A, Barnes SR, Sweeney MD, et al. Blood‐brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Akinyemi RO, Yaria J, Ojagbemi A, et al. Dementia in Africa: current evidence, knowledge gaps, and future directions. Alzheimers Dement. 2022;18:790‐809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
ICMJE Disclosure Form
Data Availability Statement
This study was approved by all appropriate university institutional review boards and was performed in accordance with the ethical standards described in the 1964 Declaration of Helsinki and its later amendments. This study specifically addresses the topic of diversity, equity, and inclusion by targeting individuals of African descent, who are vastly underrepresented in AD research.