

Abstract
Botryosphaeriaceae species are the major causal agents of walnut dieback worldwide, along with Diaporthe species. Botryosphaeria dothidea and Neofusicoccum parvum are the only two Botryosphaeriaceae species associated with this recently emergent disease in France, and little is known about their diversity, structure, origin and dispersion in French walnut orchards. A total of 381 isolates of both species were genetically typed using a sequence-based microsatellite genotyping (SSR-seq) method. This analysis revealed a low genetic diversity and a high clonality of these populations, in agreement with their clonal mode of reproduction. The genetic similarity among populations, regardless of the tissue type and the presence of symptoms, supports the hypothesis that these pathogens can move between fruits and twigs and display latent pathogen lifestyles. Contrasting genetic patterns between N. parvum populations from Californian and Spanish walnut orchards and the French ones suggested no conclusive evidence for pathogen transmission from infected materials. The high genetic similarity with French vineyards populations suggested instead putative transmission between these hosts, which was also observed with B. dothidea populations. Overall, this study provides critical insight into the epidemiology of two important pathogens involved in the emerging dieback of French walnut orchards, including their distribution, potential to mate, putative origin and disease pathways.
Keywords: Botryosphaeriaceae, Juglans regia, Microsatellites, Genetic diversity, Emerging disease
Subject terms: Genetic markers, Haplotypes, Population genetics, Microbial ecology, Molecular ecology, Population dynamics, Microbial communities, Environmental microbiology, Fungi, Pathogens
Introduction
Botryosphaeriaceae is a large family of ascomycetous fungi that are either saprophytes, endophytes or pathogens and are mainly associated with woody hosts1. Presently, based on their morphological and genetic characteristics, 22 genera and more than 100 species belong to the Botryosphaeriaceae family2–4. Within this family, most genera (notably Botryosphaeria, Diplodia, Dothiorella, Lasiodiplodia, and Neofusicoccum) can develop within plants as latent pathogens, infecting healthy tissues and remaining dormant before being able to switch to a pathogenic lifestyle. This behavioral shift can be explained by biotic and abiotic stress conditions such as environmental changes, wounds caused by humans or insects and disease development caused by other microorganisms1,5,6.
Species belonging to the Botryosphaeriaceae family show a worldwide distribution as well as low host specificity1,7 and are responsible for diseases in both forest and agricultural environments, particularly in woody plants, due to their pathogenic arsenal8. Some species are known to be highly ubiquitous, such as B. dothidea, which is associated with 500 different host species, D. sapinea (128) and D. seriata (158), Dot. sarmentorum (114), L. pseudotheobromae (161) and L. theobromae (788), Macrophomina phaseolina (551), N. australe (110), N. parvum (294) and N. ribis (273)9. Zlatković et al. (2018) further identified 10 Botryosphaeriaceae species associated with 47 species of ornamental and forest trees belonging to Gymnosperms and Angiosperms in the Western Balkans10. In agroecosystems, Botryosphaeriaceae affect numerous economically important fruit and nut trees, such as citrus11, mango12, litchi12, apple13, cocoa14, grapevine15, pistachio16, almond17, olive18 and walnut trees19,20.
English walnut (Juglans regia L.) is the most widely cultivated species in the world for nut production21. It was historically cultivated in Central Asia, but the crop is now broadly cultivated on all continents, mainly in Asia, America, and Europe22. The top three largest producers in the world are China, followed by the United-States and Iran. With a production of 37,740 tons associated with approximately 27,000 hectares in 2021, France rank among the top three European countries23. French walnut orchards are historically affected by walnut blight, anthracnose and brown apical necrosis diseases caused by Xanthomonas campestris pv. juglandis, Ophiognomonia leptostyla and a fungal complex of Alternaria and Fusarium spp. respectively24–26. Nevertheless, since 2015, walnut dieback symptoms have been extensively reported in France20,26,27 such as twig necrosis, defoliation and cankers as well as fruit necrosis and blight to complete host dieback, associated with significant yield losses. This emergence is most likely related to the twofold increase in dedicated areas since the 2000s and to climate change, reflected by the unusual succession of hot summers in France23,28–30. Such symptoms have already been described in Mediterranean climate areas, such as California, Spain and Italy31,33, and similarly to these nut-growing regions, fungal isolations from French symptomatic walnut fruits and twigs led to the identification of a fungal complex. This complex is mainly represented in France by Diaporthe eres, only two Botryosphaeriaceae species, B. dothidea and N. parvum, but also included Colletotrichum fioriniae and C. godetiae, and Fusarium juglandicola20.
These emergent symptoms and causal agents, although widespread in tree nut orchards abroad as well as in other French crops such as vineyards, encouraged us to explore the source and pathways of such biological invasion, which is key to understanding the pathogen origin34. Human-mediated introduction of pathogen agents in new environments, as well as host jumps, are one of the main causes for disease emergence35–39, which is even more likely for Botryosphaeriaceae species considering their wide host range and low host specificity39,40. To identify origin and pathways of biological invasion, population genetics appears to be a useful tool to provide relevant insights into the diversity and origin of Botryosphaeriaceae populations newly isolated from French walnut trees.
Several recent molecular markers, including inter-simple sequence repeats (ISSR)41,42 and simple sequence repeats (SSR)40,43–47, have been used to study the genetic diversity and structure of B. dothidea and N. parvum populations from many hosts. Microsatellite markers, or SSRs, are powerful genetic markers widely used in fungal population genetic studies. These ubiquitous DNA sequences of eukaryotic genomes are highly polymorphic, codominant and their sequencing is highly reproducible48,49. These studies consistently revealed low genetic diversity of B. dothidea and N. parvum populations in cultivated hosts. In contrast, fairly high levels of genetic diversity could be found in native hosts. Populations from native hosts tend to have greater genetic diversity, as well as populations that have been well developed for a long time on cultivated hosts43,45, than emergent and introduced populations40,44,46,47.
In this study, microsatellite markers were developed and genotyped using microsatellite sequencing (SSR-seq50) for both B. dothidea and N. parvum species. Our objectives were to (i) assess the diversity of the main Botryosphaeriaceae populations newly isolated from French walnut orchards, (ii) compare these walnut populations with populations originating from countries and crops characterized by a moderate (Spanish walnut orchards) to those with a long history of disease occurrence, including Californian walnut and almond orchards as well as French vineyards, and (iii) study the genetic differentiation of populations according to geographic location, host and disease development stage.
Results
Distribution of B. dothidea and N. parvum isolates in walnut orchards from the two main French production areas
A total of 182 B. dothidea and 323 N. parvum isolates were collected from 326 walnut husks and 315 twigs from the two French production regions during three sampling campaigns, including some isolates that originated from the same tissue. The prevalence of N. parvum was significantly higher in the South-West than in the South-East (p < 0.001), contrary to that of B. dothidea, which was significantly more prevalent in the South-East than in the South-West, regardless of the campaigns (p < 0.001; Table 1). Similarly, N. parvum was significantly more prevalent than B. dothidea in the South-West (p < 0.001) and, conversely, in the South-East (p < 0.001; Table 1). Considering tissues, both species were significantly more prevalent in walnut husks than in twigs, regardless of campaigns and production area (28.5% vs 11.1%, p = 0.02, for B. dothidea, and 26.1% vs 21.9%, p = 0.04, for N. parvum; Table 1). Moreover, both species were similarly prevalent in symptomatic and asymptomatic tissues, regardless of the tissue type (Table 1).
Table 1.
Prevalence of B. dothidea and N. parvum species in the two geographical areas and the two tissue types in 2020 and 2021.
| Criterion | Condition (total number of samples) | Prevalence of B. dothidea in % (total number of samples) | Prevalence of N. parvum in % (total number of samples) |
|---|---|---|---|
| Geographical origin | South-East (281) | 31.7aA (89) | 18.1bB (51) |
| South-West (360) | 10.8bB (39) | 28.6aA (103) | |
| Total | 641 | 20.0 (128) | 24.0 (154) |
| Tissue type | Husks (326) | 28.5aA (93) | 26.1aA (85) |
| Twigs (315) | 11.1bB (35) | 21.9bA (69) | |
| Total | 641 | 20.0 (128) | 24.0 (154) |
| Presence of symptoms on husks (2021) | Asymptomatic husks (54) | 22.2aA (12) | 7.4aA (4) |
| Symptomatic husks (212) | 28.3aA (60) | 25.9aA (55) | |
| Total | 266 | 27.1 (72) | 20.3 (59) |
| Presence of symptoms on twigs (2021) | Asymptomatic twigs (63) | 4.8aA (3) | 11.1aA (7) |
| Symptomatic twigs (252) | 12.7aA (32) | 24.6aA (62) | |
| Total | 259 | 13.5 (35) | 26.6 (69) |
Prevalence in symptomatic and asymptomatic tissues was only evaluated in 2021. Different lowercase letters indicate significant differences between conditions of the same criterion for each species at the α = 0.05 threshold based on the chi-squared test. Different uppercase letters indicate significant differences between the two species for each condition at the α = 0.05 threshold based on the chi-squared test.
Genetic diversity and genotypic variations of French populations of B. dothidea and N. parvum
A total of 11 and 27 loci, involving 138 and 223 Botryosphaeriaceae strains were retained for B. dothidea and N. parvum population analysis, respectively (Table 2). Botryosphaeria dothidea strains were divided into a priori populations based on French production areas, leading to two distinct populations, namely Bd_SW for the southwestern production area (N = 39 individuals) and Bd_SE for the southeastern production area (N = 94 individuals). Similarly, the N. parvum population was divided into Np_SW (N = 107 individuals) and Np_SE (N = 49 individuals). The descriptive statistics and diversity indices are summarized in Table 2.
Table 2.
Descriptive statistics and indices of genetic diversity of B. dothidea and N. parvum populations collected from the two main French walnut-producing regions (South-West _SW and South-East _SE).
| Species | Population | Nb of individuals | Nb of loci | AR | uh (s.d.) | P(%) | MLG | eMLG | H | λc | E5 | Uncorrected data set | Clone-corrected data set | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IA | rbarD | IA | rbarD | ||||||||||||
| B. dothidea | Bd_SW | 39 | 11 | 2.08 | 0.142 (0.060) | 63.64 | 10 | 10 | 1.54 | 0.71 | 0.61 | 1.08* | 0.20* | 0.44 | 0.07 |
| Bd_SE | 94 | 1.62 | 0.077 (0.028) | 72.73 | 12 | 7.35 | 0.92 | 0.39 | 0.42 | 1.34* | 0.26* | − 0.15 | − 0.03 | ||
| N. parvum | Np_SW | 107 | 27 | 2.59 | 0.148 (0.034) | 85.19 | 33 | 20.58 | 2.68 | 0.92 | 0.73 | 1.14* | 0.09* | 1.35* | 0.10* |
| Np_SE | 49 | 1.64 | 0.040 (0.012) | 37.04 | 7 | 7 | 0.73 | 0.30 | 0.39 | 2.96* | 0.35* | 1.35* | 0.15* | ||
s.d. standard deviation, AR allelic richness, uh unbiased haploid genetic diversity, P(%) percentage of polymorphic loci, MLG number of observed multilocus genotypes, eMLG number of multilocus genotypes expected at the smallest sample size, H Shannon–Wiener index, λc corrected Simpson’s index, E5 evenness, IA index of association, rbarD standardized index of association.
*Indicates a significant p-value for the IA and rbarD indices at α = 0.05.
Among the 11 SSR loci used for B. dothidea population analysis, the number of alleles ranged from one to four, with an average of 2.272 alleles per locus (regardless of production area), with three monomorphic loci in the Bd_SE population, and four in the Bd_SW one. Overall, both populations showed a high level of polymorphic loci and similar allelic richness values. Moreover, they showed low levels of genetic diversity, unevenly distributed among MLGs, although the distribution was more homogeneous in the SW population than in the SE population (E5 = 0.61 and 0.42, respectively; Table 2). The clonality rate (percentage of strains that share a MLG) within the two populations ranged from more than 75% (Bd_SW) to 86% (Bd_SE), further illustrating relatively moderate genotypic diversity and equitability, although it was still slightly greater in the former than in the latter population (H = 1.54 and 0.92, and λc = 0.69 and 0.38, respectively; Table 2). Indeed, more than 75% of strains belonged to only one MLG in the Bd_SE population versus two MLGs in the Bd_SW population. Interestingly, no MLGs were found to be shared by strains of the two populations, indicating that the 10 and 12 MLGs associated with the Bd_SW and Bd_SE populations, respectively, were uniquely associated with the production area.
Neofusicoccum parvum strains were genotyped based on 27 SSR loci with allele counts ranging from one to five (mean number of alleles per locus of 2.815, regardless of production area). The Np_SW population represented a greater percentage of polymorphic loci (23 loci vs 10 loci, respectively) and a higher allelic richness (AR = 2.59 vs 1.54; Table 2). Overall, the number of strains belonging to MLGs was much more evenly distributed in the Np_SW population than in the Np_SE population (E5 = 0.73 and 0.39, respectively), which was further confirmed by the considerably higher unbiased haploid genetic diversity and λc values in the Np_SW population (uh = 0.148 and 0.040; and λc = 0.91 and 0.29, respectively; Table 2). Indeed, the same MLG was shared by 41 out of 49 strains of the Np_SE population, whereas 86 out of 107 strains from the Np_SW population belonged to 12 different MLGs (from 2 to 19 strains per MLG). The Np_SE population was represented by more than 85% clonal strains, highlighted by relatively low diversity indices (H = 0.73). The second population was much more diverse (H = 2.68) and thus was associated with a lower rate of clonal strains (69%; Table 2). Interestingly, two MLGs were shared by the two production regions but were only shared by one isolate from the other population.
Botryosphaeria dothidea population analysis revealed partial sexual reproduction, although clonality was high for both species
Linkage disequilibrium was determined to assess the prevalence of clonal versus sexual reproduction within both populations. In addition to the uncorrected dataset, we also considered the clone-corrected dataset to mitigate the effect of clonality caused by a high rate of clonal strains for each MLG. As shown by the high number of strains sharing the same MLG for most of the populations, clonality was hypothesized to be the main reproduction mode for both B. dothidea and N. parvum.
In the case of B. dothidea, the uncorrected dataset showed significant p-values for both the IA and rbarD indices in both SW and SE populations. However, the clone-corrected dataset failed to reject the null hypothesis of random mating. This may indicate potential sexual reproduction within the two populations (Table 2). Note that negative values were obtained for the Bd_SE population with the clone-corrected dataset. These values indicated that the observed variance in the distance between strains was lower than the expected variance under the assumption of panmixia.
Conversely, all N. parvum populations showed clonal reproduction as the main mode of reproduction regardless of the dataset, as highlighted by relatively high rbarD values and significant p-values at a threshold of 0.05 (Table 3).
Table 3.
Number of isolates of B. dothidea and N. parvum collected from French walnut orchards, genotyped using SSR-seq and used in the population study after manual clone-correction of datasets as described in SSR quality filtering.
| Sampling (nb of samples) | B. dothidea isolates | N. parvum isolates | ||||
|---|---|---|---|---|---|---|
| Collected | Genotyped | Population study | Collected | Genotyped | Population study | |
| South-East | 124 | 124 | 94 | 96 | 69 | 49 |
| 2020 (from symptomatic husks only; 30) | 18 | 18 | 15 | 14 | 10 | 7 |
| 2021 | 106 | 106 | 79 | 82 | 59 | 42 |
| Husk | 79 | 79 | 61 | 50 | 40 | 30 |
| Asymptomatic (15) | 16 | 16 | 12 | 4 | 4 | 3 |
| Symptomatic (68) | 63 | 63 | 49 | 46 | 36 | 27 |
| Twig | 27 | 27 | 18 | 32 | 19 | 12 |
| Asymptomatic (15) | 5 | 5 | 3 | 2 | 1 | 1 |
| Symptomatic (72) | 22 | 22 | 15 | 30 | 18 | 11 |
| South-West | 58 | 57 | 39 | 227 | 131 | 107 |
| 2020 (from symptomatic husks only; 30) | 9 | 8 | 6 | 34 | 21 | 19 |
| 2021 | 49 | 49 | 33 | 193 | 110 | 88 |
| Husk | 20 | 20 | 16 | 54 | 35 | 27 |
| Asymptomatic (15) | 1 | 1 | 1 | 0 | 0 | 0 |
| Symptomatic (60) | 19 | 19 | 15 | 54 | 35 | 27 |
| Twig | 29 | 29 | 17 | 139 | 75 | 61 |
| Asymptomatic (15) | 0 | 0 | 0 | 5 | 5 | 5 |
| Symptomatic (72) | 29 | 29 | 17 | 134 | 70 | 56 |
| Total isolates | 182 | 181 | 133 | 323 | 200 | 156 |
Geographical origin impacted the structure of B. dothidea populations but not that of N. parvum populations
The AMOVAs performed on the uncorrected and clone-corrected datasets for B. dothidea populations led to opposite results. The uncorrected dataset showed that genetic differentiation was more structured according to regions (64.66%) than according to strains (35.34%), while the opposite was found for the clone-corrected dataset (79.26% of the genetic diversity was associated with strains vs 20.74% with regions; Supplementary Table S1). Nonetheless, the impact of geographical origin was further confirmed with DAPC (Fig. 1) and the Mantel test, which revealed a significant association between the genetic and geographic distances of B. dothidea isolates (R2 = 0.42 and p = 0.01). This was consistent with the absence of shared MLGs between the two populations, as described previously.
Figure 1.

Discriminant Analysis of Principal Components of the Botryosphaeria dothidea (A) and Neofusicoccum parvum (B) populations. Populations were distinguished according to their geographical origin (SW South-West, SE South-East), tissue (H Husk, T Twig) and symptoms (A Asymptomatic, S Symptomatic). The SW_HA and SE_TA populations of B. dothidea and the SE_HA and SE_TA populations of N. parvum were not represented because they were represented by only one individual.
The AMOVA performed on N. parvum populations based on geographical origin indicated that the genetic diversity was mainly explained by strains (77.69% of the variation in the uncorrected dataset and 93.39% of the variation in the clone-corrected dataset; Supplementary Table S1). For this species, no significant relationship between genetic and geographic distances was detected, which was confirmed by both the Mantel test (R2 = 0.0004 and p = 0.250) and the DAPC, which revealed relatively high genetic proximity between MLGs from the two production areas (Fig. 1).
Population structure was not impacted by the tissue type, the presence of symptoms or over time
Further analyses were conducted to compare MLGs associated with strains from walnut husks and twigs in the two production areas for both B. dothidea and N. parvum species and from asymptomatic and symptomatic tissues.
Overall, more MLGs were detected in the husk-associated populations than in the twig-associated ones (Supplementary Table S2). Minimum spanning networks showed that most of the MLGs associated with husks were also identified in twigs for the two species in both regions (Fig. 2). This was consistent with the low percentage of variation observed between tissues via AMOVA (Supplementary Table S3) and the overlap of ellipses between the twig and husk populations, as shown by the DAPC (Fig. 1). The number of MLGs was greater in twigs than in husks in the Np_SW population (Fig. 2C and Supplementary Table S2). All the MLGs that were shared between husks and twigs-based populations were predominant for both species in the two areas (Fig. 2). Unlike in the South-East area, a few MLGs from the South-West area were uniquely isolated from twigs (3 out of 10 MLGs for B. dothidea and 16 out of 33 MLGs for N. parvum). Unbiased genetic diversity values (uh) were relatively low, indicating that all tissue-based populations displayed relatively uneven and heterogeneous MLG repartition and high levels of clonality (Supplementary Table S2).
Figure 2.

Minimum spanning networks of MLGs of B. dothidea (A,B) and N. parvum (C,D) populations. The networks were based on Bruvo’s distance and were generated for the southwestern (SW) and southeastern (SE) production areas, as well as for walnut husks (H) and twigs (T). (A) Populations of B. dothidea in the South-West; (B) Populations of B. dothidea in the South-East; (C) Populations of N. parvum in the South-West; (D) Populations of N. parvum in the South-East. MLGs are represented by nodes (circles) and edges (lines) representing genetic distance between individuals. The size of the nodes corresponds to the number of individuals sharing the same MLG.
Concerning the distribution of populations associated with symptomatic and asymptomatic husks and twigs, strains from both species sharing the same MLG could be isolated from both types of tissues. In addition, MLGs from symptomatic and asymptomatic tissues were genetically closely-related (Fig. 1). Interestingly, for both species, the four SE populations (based on tissues and symptoms) shared the same predominant MLG; this was also the case for the Bd_SW population, while Np_SW population was more genotypically differentiated between tissues and their disease status. Notably, one highly represented MLG in the Np_SW population collected from symptomatic husks and twigs was absent in asymptomatic samples. Conversely, two MLGs, represented by only one individual each, were found only in asymptomatic twigs of the Np_SW population.
Moreover, comparisons between strains collected from walnut husks in 2020 and 2021 revealed that these populations were relatively similar. More than 60% of the B. dothidea strains from walnut husks in 2020 and 2021 in the southwestern area shared the same two MLGs, versus more than 75% in the southeastern area. These similarities between populations were confirmed by DAPC (Supplementary Fig. S1). Similarly, more than 40% of the N. parvum strains from walnut husks in 2020 and 2021 in the southwestern area shared the same two MLGs, versus more than 80% in the southeastern area. This greater similarity between strains from the southeastern French production region over time was also confirmed by DAPC (Supplementary Fig. S1).
Comparison of B. dothidea and N. parvum populations from French walnut orchards with populations from other countries and/or crops
Populations of B. dothidea and N. parvum from French walnut orchards (SW and SE) were compared with other populations from different geographical and host origin: B. dothidea and N. parvum populations isolated from grapevines in France (Bd_GRA, N = 5 individuals; and Np_GRA, N = 43 individuals, respectively), N. parvum populations isolated from walnut trees in California (Np_CAL_W, N = 5 individuals) and Spain (Np_SPA, N = 5 individuals), and from almond trees in California (Np_CAL_A, N = 14 individuals). Geographical and plant origins of these populations are listed in Supplementary Table S4.
Surprisingly, a certain level of genetic proximity was found between the Bd_SW and Bd_GRA populations, as illustrated in the DAPC where the SW and GRA populations were clustered together on the opposite side of the SE one (Fig. 3). Despite this higher genetic similarity between Bd_SW and Bd_GRA, only one MLG was shared between these two populations while one MLG was also shared between Bd_SE and Bd_GRA (Supplementary Fig. S2).
Figure 3.

Discriminant Analysis of Principal Components (DAPC) for B. dothidea (Bd) populations. Populations were distinguished according to their origin: French walnut-producing regions (SW South-West, SE South-East) and French vineyards (GRA).
The UPGMA dendrogram based on Nei’s genetic distance highlighted an even stronger genetic proximity between the Np_SW, Np_SE and Np_GRA populations than that reported above with B. dothidea (Fig. 4). Indeed, the Np_SE and Np_SW populations shared three and five MLGs, respectively, with the Np_GRA population (Supplementary Fig. S2). Taken together, these observations further supported the lack of differentiation according to geographical origin of the N. parvum populations. Interestingly, among the individuals collected from grapevines in France (see Supplementary Table S4 for geographical origin), those that corresponded to the MLGs shared between the Np_SW/Np_SE and Np_GRA populations were all collected in the South-West area (Nouvelle-Aquitaine and Occitanie regions). In contrast, grapevine strains from the North-East area (Grand Est region) did not share any MLGs with the French walnut populations. Furthermore, the population collected from walnut trees in Spain displayed a low genetic proximity to the Np_SW, Np_SE and Np_GRA populations according to the UPGMA dendrogram, and no MLGs among the five from this Spanish population were common to the previous populations (Fig. 4 and Supplementary Fig. S2). The same was true for the populations collected from the Californian walnut (CAL_W) and almond (CAL_A) orchards. This was confirmed by UPGMA, which highlighted a higher proximity between the Californian and Spanish populations than between the other three populations (Fig. 4).
Figure 4.
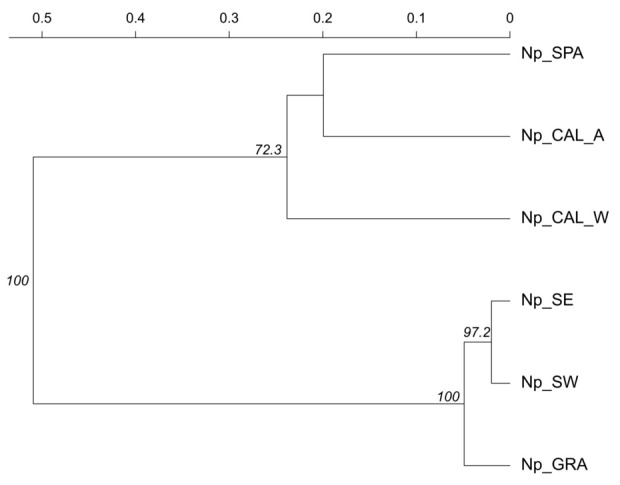
Unweighted Paired Group Mean Arithmetic (UPGMA) dendrogram between four N. parvum (Np) populations. The dendrogram was based on Nei’s distance and was generated for populations from southwestern and southeastern French regions (SW and SE, respectively), French vineyards (GRA), Californian walnut (CAL_W) and almond (CAL_A) trees and Spanish walnut trees (SPA). Bootstrap values above 50 are displayed at nodes.
Discussion
The current study documented the genetic diversity and distribution of B. dothidea and N. parvum populations associated with walnut dieback in the two main French walnut production areas. Interestingly, walnut dieback symptoms were associated with the presence of ten Botryosphaeriaceae species in addition to two species of Diaporthe in California at the beginning of the 2010s19, while six different species have been associated with dieback symptoms in France20,51,52, among which B. dothidea and N. parvum were the only representatives of the Botryosphaeriaceae family. The emergence of this disease in France led us to investigate the genetic and genotypic diversity and population structure of B. dothidea and N. parvum from tissues showing walnut dieback symptoms as well as asymptomatic ones.
Analysis of both B. dothidea and N. parvum populations from South-West and South-East of France revealed relatively low genetic diversity, which is consistent with the emerging nature of the disease in France. The same conclusions were also obtained from previous studies using different methods such as RAPD to study B. dothidea populations from blackberry, eucalyptus, pecan, pistachio, walnut and willow in California53 or UP-PCR to study N. parvum populations from vineyards in New-Zealand54. However, the latter markers are much less resolutive than SSRs55. Interestingly, the genetic diversity of both species isolated from different hosts and countries has also been reported to range from moderate40,47 to relatively high43,45,46 based on microsatellite marker genotyping.
The reproductive mode is known to contribute to the level of genetic diversity, with populations undergoing sexual reproduction being more likely to exhibit greater genetic diversity56. In this study, B. dothidea and N. parvum populations were associated with a dominant clonal reproduction pattern. Indeed, Botryosphaeriaceae species tend to exhibit relatively low levels of sexual reproduction and primarily rely on asexual reproduction under field conditions1,33. Similarly, both B. dothidea and N. parvum were reported to be represented by a high number of individuals sharing the same MLG and to have low genetic diversity40,45,47,53, suggesting an essential role for clonal reproduction56. Both sexual and asexual reproduction modes, which are associated with high and low genetic diversity, respectively, were detected based on linkage disequilibrium by Sakalidis et al., who studied the diversity of N. parvum populations collected in different countries, highlighting the ability of this species to reproduce in both ways46. Although evidence for linkage equilibrium was not found in N. parvum populations, in the B. dothidea clone-corrected dataset, significant linkage equilibrium was observed, suggesting the occurrence of recombination or sexual reproduction57. This finding is consistent with previous observations of B. dothidea populations from grapevines in China43 as well as the presence of homothallic mating types in these two species, as revealed by their genome analysis6,58,59. The predominance of clonal strains is also characteristic of the emergence of diseases, as shown by Ma et al. in strains associated with recent panicle and shoot blight disease in pistachios in California53. Moreover, Botryosphaeriaceae species appear to exhibit higher genetic diversity in native hosts and environments, such as N. parvum isolated from Syzygium cordatum in South-Africa45. Further investigations on the presence of reproductive structures on the surface of walnut branches could provide more information about the reproductive mode of these species in walnut orchards in France. Taken together, our observations are consistent with the recent emergence of the disease in France20.
Comparisons between strains associated with walnut husks and twigs revealed a high percentage of identity between MLGs of the two types of tissues and a relatively low genetic distance. These connections between walnut husks and twigs support the hypothesis raised by Michailides et al., that walnut dieback disease often originates in nuts, with pathogen agents transferring from them to twigs through the peduncle60. A second hypothesis would be that infection could also be directly initiated in twigs through wounds and scars caused by the fall of fruits and leaves from walnut trees or even pruning wounds60,61. Pruning wounds are identified as the primary entry point for infection of twigs and branches by Botryosphaeriaceae species in other hosts, such as vines62, and in walnut trees, wounds have been shown to remain susceptible to Botryosphaeriaceae infection for at least 4 months63. These different wounds could either constitute a breach for exogenous infections of Botryosphaeriaceae or generate abiotic and biotic stresses that could lead to the behavioral shift of Botryosphaeriaceae species from endophytes (that is, present in the plant before wounds occurred) to pathogens.
Thus, we also considered isolates from asymptomatic tissues to evaluate the lifestyle of these latent pathogens. The results showed that for both species, the populations isolated from symptomatic and asymptomatic husks and twigs were genetically similar, indicating that the strains exhibiting latent pathogen lifestyle in asymptomatic tissues were the same as those exhibiting a pathogenic lifestyle in symptomatic ones. It was evidenced that Botryosphaeriaceae species are capable of causing latent infections on young walnut trees in nurseries33, further supporting the hypothesis that these species can remain latent for extended periods and switch to a pathogenic lifestyle under abiotic or biotic stresses1,5,64. For example, the presence of the walnut scale (Quadraspidiotus juglansreginae) exacerbates the symptoms caused by Botryosphaeriaceae species in walnut orchards in California61. In France, the first observation of dieback symptoms occurred in August 2003, concurrently with the occurrence of a severe heatwave before dieback symptoms, reported again from 2015 onwards, corresponding to an unusual succession of hot summers20,27,29,30. This temporal link between the occurrence of dieback symptoms and summer heatwaves could be explained by the following two non-mutually exclusive hypotheses: (i) environmental stresses have an impact on the activation of latent infections by Botryosphaeriaceae species, and (ii) higher temperatures increase the sporulation and growth of Botryosphaeriaceae that have a high optimal growth temperature33.
In addition, we found that most strains from symptomatic walnut husks sampled in 2020 were detected in 2021 for both species, suggesting a certain stability, at least over a year of experimentation. Further investigations on individuals collected over multiple years could provide additional information about the spatiotemporal structure of the populations of the two species. It could also provide more information on the stability of populations over time and potential changes in these populations, as shown in B. dothidea populations from pistachios in California65.
The results also showed that the structure of N. parvum populations is independent of geographical origin, tissue type or the presence of symptoms, indicating a lack of genetic differentiation based on these factors. This observation would imply a possible dispersion of individuals between the two regions through wind or insects33, allowing for longer distance dispersal than spore dispersal by rain splashes. In addition, our results clearly showed a significantly greater level of N. parvum genotypic diversity in the South-West area. This fact, together with a significantly higher prevalence of the species in the same area, also suggest that the origin of contamination may be older and/or that environmental conditions as well as agronomic practices may favor the implantation of N. parvum compared to the South-East area. Past observations in France may support the hypothesis that walnut trees in the southwestern area are more subject to abiotic stress and thus more prone to fungal diseases. In fact, the first observations of fungal walnut dieback caused by Phytophthora species were reported during the 1990s only in the South-West production region66, even though the “intensive” cultivation of walnut trees has been going on for longer in the southeastern area (i.e. at the beginning of the 1900s67) than in the South-West one (around the 1950s68). Moreover, a reduction in production due to unhealthy, unproductive and weak walnut trees was reported in the South-West in the early 1980s, while at the same time, the South-East was seen as the best area for growing walnut trees69. Conversely, the geographical origin of B. dothidea populations significantly impacted their genetic characteristics based on the uncorrected dataset, and no MLGs were shared between the two populations. Therefore, the emergence of B. dothidea on walnut trees may result from separate inoculum sources between the South-West and the South-East areas of France.
Interestingly, B. dothidea and N. parvum populations collected from French walnut trees showed differences in behavior and structure that could be the result of several parameters, such as host specificity, the mechanisms of spore dispersal, and the date of introduction or pathogenicity criteria, such as aggressiveness. In their study, Marsberg et al. (2017) highlighted that on the one hand, the low host specificity displayed by Botryosphaeriaceae contributes to a lack of spatial differentiation, as observed for N. parvum populations in France, while on the other hand, the dispersal of Botryosphaeriaceae spores by rain and water at relatively short distances may lead to geographic genetic structure, such as that observed for B. dothidea populations6. Additionally, considering that reproductive mode plays a crucial role in population structure45, the highlighted traces of sexual reproduction displayed by B. dothidea populations could also explain their specific structure. Furthermore, a recently-introduced population is more likely to have no particular structure47, and some studies have hypothesized that the introduction of B. dothidea may be older, originating from Europe6, which could be a second partial explanation. The difference in aggressiveness between the two species is also likely to explain the differences observed. Indeed, it has been shown that N. parvum is more aggressive than B. dothidea19,31,70,71, and greater aggressiveness involves longer and more numerous lesions as well as a greater level of reproduction72. A higher aggressiveness may also reflect a greater ability to colonize the host. This could also partially explain the greater number of collected N. parvum individuals compared to B. dothidea individuals. This higher occurrence of N. parvum in walnut orchards displaying dieback symptoms has also been demonstrated in surveys carried out in California19, Croatia73 and Turkey74. It has been shown that some cryptic species, such as N. parvum and N. ribis, are better invaders due to their better ability to colonize non-native and human-disturbed environments45.
Furthermore, the analyses revealed that N. parvum populations exhibited genetic similarity between the two walnut production regions and showed proximity to strains collected from French vineyards in the southwestern regions. In contrast, the N. parvum French populations clearly differed from the Californian and Spanish populations. Interestingly, some individuals from French walnut trees and vineyards even shared identical MLGs. Shared MLGs were also detected between the B. dothidea population from the South-West (Bd_SW) and French southwestern vineyards. Thus, B. dothidea and N. parvum populations infecting walnut trees could share common origin with populations collected from vineyards or originate from vineyards, the two species are indeed commonly associated with grapevine trunk diseases in France9. Both hypotheses are supported by the fact that Botryosphaeriaceae can colonize numerous wild woody plants1, and that viticulture is historically older than walnut orchards cultivation in the French regions studied67,68. As described previously, climate change could play a significant role in causing abiotic stresses on plants, leading to disease onset and facilitating the geographical and host expansion of pathogenic agents, as predicted by Bastista et al.75. Human-mediated transmission is a second possible pathway, specifically by introducing infected material. In the history of walnut cultivation, the English walnut (Juglans regia L.), which originated in Central Asia, was exported to North-America primarily as rootstock to enhance the characteristics and cultivation of walnuts and to improve species crossbreeding76. Initially, the majority of Californian walnut trees were grafted onto Juglans nigra rootstocks, an endemic species77. Currently, scions grafted onto Juglans nigra rootstock are still produced in Californian nurseries and exported to different regions, including France78. Thus, some French walnut producers cultivate J. regia x J. nigra hybrids. However, the lack of genetic proximity between strains of French and Californian walnut trees does not confirm this origin of infection; and indicates that these strains may have had separate origins or undergone distinct evolutionary processes, although additional sampling would be needed in walnut-producing countries to confirm this result. Neofusicoccum parvum strains from Spain were also genetically distant from French ones. Nevertheless, the implantation and evolutionary pressure of Botryosphaeriaceae populations are likely to be quite similar between those two regions, considering that walnut dieback, as in France, was more recently observed in Spain than in California32. Further isolations and investigations of B. dothidea and N. parvum populations from other hosts and countries should help to shed light into the emergence of walnut dieback in France.
The present study aimed to explore the genetic diversity and population structure of B. dothidea and N. parvum strains collected from walnut trees using SSR-sequencing. Our results showed relatively low genetic diversity and a main clonal reproduction pattern, although French populations of B. dothidea displayed traces of sexual reproduction. Unlike those from N. parvum, B. dothidea populations from the two production areas did not share any MLGs and showed geographic genetic structure. Some strains collected from symptomatic tissues could also be isolated from asymptomatic ones, further confirming the ability of both species to switch from endophytic to pathogenic lifestyles. The genetic similarity of the N. parvum and B. dothidea walnut populations from the South-West of France to grapevine populations from the same country indicates a possible host jump from grapevines to walnuts. In addition, the observed genetic distance between French and Spanish N. parvum populations and with Californian populations suggested that introduced contaminated materials are less likely to be an important source of inoculum. This study provides critical insight into the epidemiology of two essential pathogens involved in emergent dieback disease in French walnut orchards, including their distribution, potential to mate, putative origin and disease pathways.
Materials and methods
Collection of fungal isolates
The plant and fungal samples were collected in accordance with the Nagoya Protocol to the Convention on Biological Diversity. Symptomatic and asymptomatic walnut tissues were collected from different trees in 12 French orchards (P1–P12) during September 2020 and August 2021 for husks, and during May 2021 for twigs. A total of six orchards were surveyed in the South-East area (SE; P1–P6, mainly the Auvergne-Rhône-Alpes region), and six orchards were surveyed in the South-West area (SW; P7–P12, mainly the Nouvelle-Aquitaine region; Fig. 5). Two orchards were cultivated with the cultivar Serr (P7 and P9) while the remaining orchards were cultivated with the variety Franquette. The collected walnut husks were characterized by blights and necrosis, while symptomatic twigs showed necrosis, defoliation and cankers. In each orchard, five symptomatic walnut husks were sampled during September 2020, while 24 symptomatic and six asymptomatic walnut husks and the same number of twigs were sampled during the two other campaigns. All the samples were stored at 4 °C until analysis.
Figure 5.
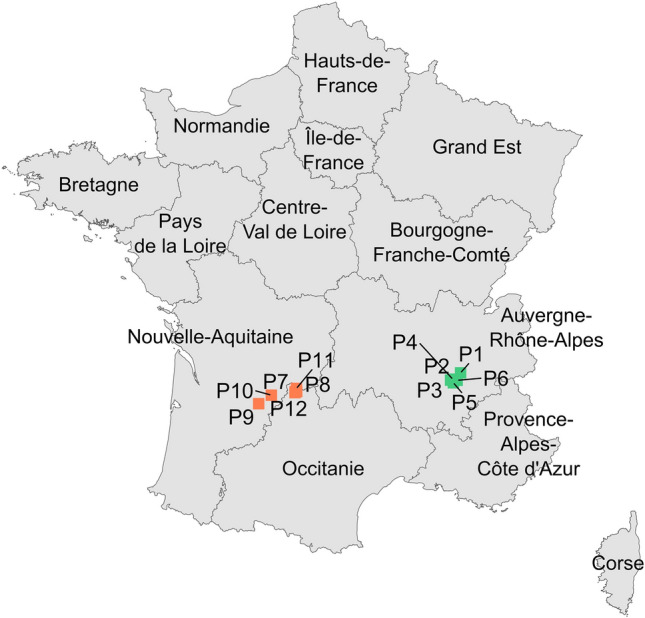
Sampling locations of Botryosphaeria dothidea and Neofusicoccum parvum isolates from French walnut orchards. Green and orange dots represent orchards sampled in the South-East and South-West walnut-producing areas, respectively.
Walnut and twig samples were surface-sterilized and cut as described in Belair et al., and five pieces of each sample were placed onto potato dextrose agar (PDA) supplemented with 10 mL/L of a penicillin–streptomycin solution (10,000 units penicillin and 10 mg streptomycin/mL, Sigma Aldrich, Saint-Louis, Missouri, USA)51. Fungal isolates were collected on PDA media and grown for a 4-day incubation period at 25 °C in the dark and a 3-day incubation period at room temperature (23 ± 2 °C) under natural light.
Representatives Botryosphaeriaceae isolates were molecularly characterized by amplifying and sequencing the internal transcribed spacer (ITS; ITS5 and ITS4 primers79), the translation elongation factor 1-alpha (EF1-α; EF1-728F and EF1-986R primers80), and the β-tubulin gene (β-tub; Bt2a and Bt2b primers81). DNA extractions were performed using the KingFisher Duo Prime System (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and the Mag-BindⓇ Universal Pathogen DNA extraction kit (Omega Bio-Tek Inc., VWR International, Radnor, Pennsylvania, USA) following the manufacturer’s instructions. DNA solutions were quantified with a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). Sanger sequencing was performed by Eurofins Genomics platform (Cologne, Germany). Contig assembly and taxonomic assignment were performed with Geneious Prime 2021.1.1.
A chi-squared test was performed to compare the prevalence (defined as the percentage of husks or twigs contaminated by B. dothidea and N. parvum) of the two species between the two tissue types, the two sampled areas or between asymptomatic and symptomatic tissues. Statistical analyses were conducted using the chisq.test() R function to compare the proportions82.
Fungal isolates from French vineyards from UMR SAVE83 (Santé et Agroécologie du Vignoble) as well as from Californian walnut and almond trees and Spanish walnut trees from the Kearney Agricultural Research and Extension Center (University of California) and Agronomy Department of the University of Córdoba (Spain) were added to the analysis. A part of representatives isolates from UMR SAVE were already molecularly characterized84. The other part, as well as the isolates from California and Spain were identified at the species level by amplifying and sequencing three loci as described above.
Phylogenetic analyses
Sequences of the three loci (ITS, EF1-α and β-tub) were aligned independently using MAFFT v7 online service85,86 with the Auto algorithm. The resulting alignments were edited using Gblocks 0.91b87,88 with all the options enabled to allow less stringent selection. As no phylogenetic incongruence between the different loci was detected (p = 0.02) by performing an Incongruence Length Difference (ILD) test on 1,000 replicates using PAUP v4.0a16689, the three individual alignments were concatenated to build a whole dataset.
Phylogenetic analyses were based on Maximum Likelihood (ML) and Bayesian Inference (BI). For ML analysis, the determination of the best models and the phylogenetic reconstruction were conducted using MEGA-X90. The robustness of the analysis was evaluated by 1,000 bootstrap replications. For BI analysis, the best models were estimated using the bModelTest add-on in Bayesian Evolutionary Analysis Utility (BEAUti2)91 simultaneously with the BI analysis using Bayesian Evolutionary Analysis Sampling Trees (BEAST)2 v2.6.1 package92. The analysis was performed using the Markov Chain Monte Carlo (MCMC) method by performing 3 independent repetitions of 1 × 107 generations each, with sampling a tree at every 1000 generations. Convergence of the independent BI analyses was checked using Tracer v1.7.193 and a consensus tree was obtained by a frequency of resampling of 10,000 and a burn-in fixed at 10% using LogCombiner and Treeannotator. For both analyses, Neoscytalidium dimidiatum CBS 499.66 was selected as an outgroup. The two phylogenetic trees obtained with ML and BI analyses were compared using FigTree v1.4.494. A tree based on the topology of ML tree and annotated with bootstrap and posterior probability node supports was built (Supplementary Fig. S3), with nodes with bootstrap support < 60% collapsed using TreeCollapserCL 495.
Fungal isolate purification and DNA extraction
All the isolates were single-thalli purified from 14-day-old cultures. Mycelial suspensions were produced by scraping surface mycelia with sterile rakes and 5 mL of a solution of sterile distilled water and TweenⓇ 80 (2 drops of TweenⓇ 80 for 1 L of solution; Sigma Aldrich). Serial dilutions in sterile distilled water and TweenⓇ 80 down to 10–3 were performed before surface plating (100 µL of 10–2 and 10–3 mycelial solutions) onto PDA media. Petri dishes were incubated at 25 °C in the dark and single-thallus growth was observed using a binocular loupe after 24 h of incubation. Purified isolates were then collected and isolated on new PDA media and incubated at 25 °C in the dark for seven days. Of our collection of 323 N. parvum isolates, 200 were further selected for SSR-sequencing by covering the range of origin and using the same number of isolates for each sampling condition. Additionally, if an tissue was infected with a high number of isolates, two to four randomly selected isolates were genotyped (Table 3).
Sequence-based microsatellite genotyping
Microsatellite marker genotyping, from whole-genome microsatellite detection to primer design, as well as microsatellite sequencing and bioinformatic analyses, were performed at the Genome Transcriptome Facility of Bordeaux (PGTB, Bordeaux, France). A global description of the workflow is available in the publication of Lepais et al.50. Briefly, the QDD v 3.1 pipeline96 was used to detect microsatellites and to design associated primer pairs based on genome sequences retrieved from the National Center for Biotechnology Information (NCBI) (accession numbers: GCA_011503125.2 for B. dothidea and GCA_020912385.1 for N. parvum; https://www.ncbi.nlm.nih.gov/; accessed on 08 April 2022). A total of 60 microsatellite primer pairs were tested for amplification via simplex PCR before amplification of the successful locus via multiplex PCR. Amplicons were indexed and sequenced in a portion of a P1 flow cell of an Illumina NextSeq 2000 sequencer in a paired-end 2 × 150 bp configuration. Then, data analyses were performed using a bioinformatic pipeline50 integrating the FDSTools analysis toolkit97, leading to complete genotypic profiles for each strain integrating allelic polymorphisms of 35 loci and 34 loci for N. parvum and B. dothidea, respectively. Moreover, the sequences of 95 strains of each species were amplified and sequenced twice to ensure reproducibility of the sequenced SSRs, estimate the allelic error rate and compute the overall missing data rate for each locus.
SSR quality filtering
First, loci with more than 20% missing data and/or more than 3% allelic error were discarded. In addition, since both species are haploid, loci with more than 20% heterozygosity, suggesting a sequencing error, were also discarded. The genotypes of repeated individuals were compared, and a representative was conserved for further analyses, provided that both genotypes were identical. The homozygous genotype was retained if the genotypes were not identical (due to heterozygous loci). Individuals with more than 20% heterozygosity or missing data were also discarded. Finally, isolates from French walnut orchards were clone-corrected by discarding individuals isolated from the same tissue with the same multilocus genotype (MLG) to retain only one representative per sample (hereafter uncorrected data sets; Table 3). Clones and MLGs were delineated using the poppr v2.9.4 R package98 by assessing the allelic distance between pairs of individuals sequenced twice (Supplementary Fig. S4). Based on these results, MLGs were considered different if they differed by at least one locus.
Population genetic diversity
GenAlEx v6.50399 was used to determine the characteristics of each data set and population, e.g. unbiased haploid genetic diversity (uh) and percentage of polymorphic loci (P(%)), as well as the Mantel test100. Moreover, the allelic richness (AR) at the smallest population size across combinations of populations and loci was assessed using the PopGenReport v3.0.7 R package101. The poppr v2.9.4 R package was also used to assess complementary estimators such as the number of multilocus genotypes (MLG), the number of expected MLGs (eMLG) at the smallest sample size, Shannon (H)102 and corrected Simpson’s indices (λc)103, and the evenness of MLGs (E.5, which corresponds to MLGs distribution among populations104). Simpson’s index was corrected by multiplying by N/(N-1), where N represents the total number of individuals of the given population. Shannon index and evenness were estimated with a bootstrap value of 1000 and rarefaction at the smallest population size, as indicated in Grünwald et al.105. Clone-correction was applied for each population using poppr to retain only one representative strain for each MLG in each population (hereafter clone-corrected datasets). Linkage disequilibrium was estimated using the index of association and its standardized form (Ia106 and rbarD107, respectively) for both uncorrected and clone-corrected datasets, with 999 permutations. Genotype accumulation curves were generated to ensure that the loci used in the analysis were sufficiently numerous and discriminative to assess the genotypic diversity of the B. dothidea and N. parvum populations with a resampling without replacement value of 1000.
Population structure and differentiation
Analyses of MOlecular VAriance (AMOVA) were performed using the ade4 R package108 implemented in poppr on both uncorrected and clone-corrected datasets. Minimum spanning networks were constructed based on Bruvo’s distance109 using the two R packages poppr and magrittr v2.0.3110. Discriminant Analyses of Principal Components (DAPC) were conducted with the adegenet v2.1.10 R package in poppr111,112. The optimal number of principal components (PCs) was assessed using cross-validation with the xvalDapc() function. A dendrogram of genetic distance among populations of N. parvum was constructed using the Unweighted Pair Group Method with Arithmetic mean (UPGMA) method and Nei’s distance113,114 with the poppr and adegenet R packages. The number of bootstraps was set to 1000 and missing data were treated using the “mean” method.
Supplementary Information
Acknowledgements
We would like to thank the growers for giving us access to their orchards in this project. We are also grateful to the Genome Transcriptome Facility of Bordeaux (10.15454/1.5572396583599417E12) for performing sequence-based microsatellite genotyping, with the help of Emilie Chancerel, Zoé Delaporte and Erwan Guichoux. The Botryosphaeriaceae collection in French vineyards was financially supported by the Bordeaux Wine Trade Council (CIVB) and FranceAgrimer (found for agricultural and rural development) in the National vine dieback plan (PNDV Mycovir Project).
Author contributions
G.L.F., F.P. and A.P. acquired funding and supervised the scientific management of the PhD work. M-F.C.-C. and G.C. collected and provided isolates from French vineyards. T.J.M., J.M., and V.M.G.M. collected and provided isolates from California and Spain. C.M., M.-N.H., A.M., and Y.L. sampled the walnut tissues. S.T. provided crucial technical support. M.B., F.P., and O.L. performed the analyses and generated all the tables and figures. M.B. wrote the manuscript in collaboration with F.P. and A.P. All authors reviewed and approved the final manuscript.
Funding
This work, which was conducted in the framework of the CARIBOU project, was funded by the French Ministry of Agriculture and Food under CASDAR funds (AAP RT 2020 CARIBOU project coordinated by the CTIFL). This project was also funded by the French Brittany region (ARED Grant #1797 SHERWOOD project) and the French Ministry of Higher Education, Research and Innovation (2020 CDE EGAAL Doctoral School).
Data availability
The datasets generated and analyzed during the current study are available in the Recherche Data Gouv repository, 10.57745/J8ERW1.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-67613-6.
References
- 1.Slippers, B. & Wingfield, M. J. Botryosphaeriaceae as endophytes and latent pathogens of woody plants: Diversity, ecology and impact. Fungal Biol. Rev.21, 90–106 (2007). 10.1016/j.fbr.2007.06.002 [DOI] [Google Scholar]
- 2.Dissanayake, A. J., Philips, A. J. L., Li, X. H. & Hyde, K. D. Botryosphaeriaceae: Current status of genera and species. Mycosphere7, 1001–1073 (2016). 10.5943/mycosphere/si/1b/13 [DOI] [Google Scholar]
- 3.Slippers, B., Crous, P. W., Jami, F., Groenewald, J. Z. & Wingfield, M. J. Diversity in the Botryosphaeriales: Looking back, looking forward. Fungal Biol.121, 307–321 (2017). 10.1016/j.funbio.2017.02.002 [DOI] [PubMed] [Google Scholar]
- 4.Pem, D. Mycosphere notes 275–324: A morpho-taxonomic revision and typification of obscure Dothideomycetes genera (incertae sedis). Mycosphere10, 1115–1246 (2019). 10.5943/mycosphere/10/1/22 [DOI] [Google Scholar]
- 5.Manawasinghe, I. S. et al. Mycosphere Essays 14: Assessing the aggressiveness of plant pathogenic Botryosphaeriaceae. Mycosphere7, 883–892 (2016). 10.5943/mycosphere/si/1b/7 [DOI] [Google Scholar]
- 6.Marsberg, A. et al.Botryosphaeria dothidea: A latent pathogen of global importance to woody plant health. Mol. Plant Pathol.18, 477–488 (2017). 10.1111/mpp.12495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batista, E., Lopes, A. & Alves, A. What do we know about Botryosphaeriaceae? An overview of a worldwide cured dataset. Forests12, 313 (2021). 10.3390/f12030313 [DOI] [Google Scholar]
- 8.Belair, M. et al. Pathogenicity factors of Botryosphaeriaceae associated with grapevine trunk diseases: New developments on their action on grapevine defense responses. Pathogens11, 951 (2022). 10.3390/pathogens11080951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Batista, E., Lopes, A. & Alves, A. MDRBOT. https://mdr-bot-cesam-ua.shinyapps.io/bot_database/ (2022).
- 10.Zlatković, M., Wingfield, M. J., Jami, F. & Slippers, B. Host specificity of co-infecting Botryosphaeriaceae on ornamental and forest trees in the Western Balkans. For. Path.48, e12410 (2018). 10.1111/efp.12410 [DOI] [Google Scholar]
- 11.Bezerra, J. D. P. et al. Genetic diversity and pathogenicity of Botryosphaeriaceae species associated with symptomatic citrus plants in Europe. Plants10, 492 (2021). 10.3390/plants10030492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aiello, D. et al. Woody canker and shoot blight caused by Botryosphaeriaceae and Diaporthaceae on mango and litchi in Italy. Horticulturae8, 330 (2022). 10.3390/horticulturae8040330 [DOI] [Google Scholar]
- 13.Díaz, G. A. et al. Characterization and pathogenicity of Diplodia, Lasiodiplodia, and Neofusicoccum species causing Botryosphaeria canker and dieback of apple trees in central Chile. Plant Dis.106, 925–937 (2022). 10.1094/PDIS-06-21-1291-RE [DOI] [PubMed] [Google Scholar]
- 14.Huda-Shakirah, A. R., Mohamed Nor, N. M. I., Zakaria, L., Leong, Y.-H. & Mohd, M. H. Lasiodiplodia theobromae as a causal pathogen of leaf blight, stem canker, and pod rot of Theobroma cacao in Malaysia. Sci. Rep.12, 8966 (2022). 10.1038/s41598-022-13057-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Úrbez-Torres, J. R. The status of Botryosphaeriaceae species infecting grapevines. Phytopathol. Mediterr.50, S5–S45 (2011). [Google Scholar]
- 16.Gusella, G. et al. Etiology of Botryosphaeria panicle and shoot blight of pistachio (Pistacia vera) caused by Botryosphaeriaceae in Italy. Plant Dis.106, 1192–1202 (2022). 10.1094/PDIS-08-21-1672-RE [DOI] [PubMed] [Google Scholar]
- 17.Inderbitzin, P., Bostock, R. M., Trouillas, F. P. & Michailides, T. J. A six locus phylogeny reveals high species diversity in Botryosphaeriaceae from California almond. Mycologia102, 1350–1368 (2010). 10.3852/10-006 [DOI] [PubMed] [Google Scholar]
- 18.Moral, J., Muñoz-Díez, C., González, N., Trapero, A. & Michailides, T. J. Characterization and pathogenicity of Botryosphaeriaceae species collected from olive and other hosts in Spain and California. Phytopathology100, 1340–1351 (2010). 10.1094/PHYTO-12-09-0343 [DOI] [PubMed] [Google Scholar]
- 19.Chen, S. F., Morgan, D. P., Hasey, J. K., Anderson, K. & Michailides, T. J. Phylogeny, morphology, distribution, and pathogenicity of Botryosphaeriaceae and Diaporthaceae from English walnut in California. Plant Dis.98, 636–652 (2014). 10.1094/PDIS-07-13-0706-RE [DOI] [PubMed] [Google Scholar]
- 20.Laloum, Y. et al. Projet CARIBOU : Élancer la recherche pour cerner le dépérissement du noyer. INFOS CTIFL386, 48–58 (2022). [Google Scholar]
- 21.Martínez-García, P. J. et al. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J.87, 507–532 (2016). 10.1111/tpj.13207 [DOI] [PubMed] [Google Scholar]
- 22.McGranahan, G. & Leslie, C. Walnut. In Fruit Breeding vol. 8 827–846 (Springer, Boston, MA, 2012).
- 23.FAOSTAT. Statistical database - World and France total production. Walnut with shell. License: CC BY-NC-SA 3.0 IGO. https://www.fao.org/faostat/en/#data/QCL (2021).
- 24.Belisario, A., Maccaroni, M., Corazza, L., Balmas, V. & Valier, A. Occurrence and etiology of brown apical necrosis on Persian (English) walnut fruit. Plant Dis.86, 599–602 (2002). 10.1094/PDIS.2002.86.6.599 [DOI] [PubMed] [Google Scholar]
- 25.Giraud, M., Prunet, J.-P., Chevalier, A. & Ramain, S. L. bactériose du noyer: Évolution des populations de Xanthomonas en verger. INFOS CTIFL230, 38–42 (2007). [Google Scholar]
- 26.Giraud, M. & Verhaeghe, A. Fiche bio-agresseur: Anthracnose à Colletotrichum sp. en verger de noyers. Arboric. Fruitière 2 (2015).
- 27.Bressolier, A. Participez à l’enquête sur le dépérissement du noyer. Médiafel par l’arboriculture fruitière et culture légumièrehttps://www.arboriculture-fruitiere.com/articles/technique-fruit/participez-lenquete-sur-le-deperissement-du-noyer (2021).
- 28.European Environment Agency. Global and European temperatures. https://www.eea.europa.eu/ims/global-and-european-temperatures (2023).
- 29.Singh, B. K. et al. Climate change impacts on plant pathogens, food security and paths forward. Nat. Rev. Microbiol.21, 640–656 (2023). 10.1038/s41579-023-00900-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.World Meteorological Organization. Temperatures in Europe increase more than twice global average. https://public.wmo.int/en/media/press-release/temperatures-europe-increase-more-twice-global-average (2022).
- 31.Gusella, G., Giambra, S., Conigliaro, G., Burruano, S. & Polizzi, G. Botryosphaeriaceae species causing canker and dieback of English walnut (Juglans regia) in Italy. For. Path.51, (2021).
- 32.López-Moral, A. et al. Etiology of Branch Dieback and shoot blight of English walnut caused by Botryosphaeriaceae and Diaporthe species in southern Spain. Plant Dis.104, 533–550 (2020). 10.1094/PDIS-03-19-0545-RE [DOI] [PubMed] [Google Scholar]
- 33.Moral, J., Morgan, D., Trapero, A. & Michailides, T. J. Ecology and epidemiology of diseases of nut crops and olives caused by Botryosphaeriaceae fungi in California and Spain. Plant Dis.103, 1809–1827 (2019). 10.1094/PDIS-03-19-0622-FE [DOI] [PubMed] [Google Scholar]
- 34.Robert, S., Ravigne, V., Zapater, M.-F., Abadie, C. & Carlier, J. Contrasting introduction scenarios among continents in the worldwide invasion of the banana fungal pathogen Mycosphaerella fijiensis. Mol. Ecol.21, 1098–1114 (2012). 10.1111/j.1365-294X.2011.05432.x [DOI] [PubMed] [Google Scholar]
- 35.Billones-Baaijens, R. et al. Management of Botryosphaeriaceae species infection in grapevine propagation materials. Phytopathol. Mediterr.54, 355–367 (2015). [Google Scholar]
- 36.Dunn, A. M. & Hatcher, M. J. Parasites and biological invasions: parallels, interactions, and control. Trends Parasitol.31, 189–199 (2015). 10.1016/j.pt.2014.12.003 [DOI] [PubMed] [Google Scholar]
- 37.Frankel, S. J. et al.Phytophthora introductions in restoration areas: Responding to protect California native flora from human-assisted pathogen spread. Forests11, 1291 (2020). 10.3390/f11121291 [DOI] [Google Scholar]
- 38.Na, D.-H., Lee, J. S., Shin, H.-D., Ono, Y. & Choi, Y.-J. Emergence of a new rust disease of Virginia creeper (Parthenocissus quinquefolia) through a host range expansion of Neophysopella vitis. Mycobiology50, 166–171 (2022). 10.1080/12298093.2022.2076366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thines, M. An evolutionary framework for host shifts—jumping ships for survival. New Phytol.224, 605–617 (2019). 10.1111/nph.16092 [DOI] [PubMed] [Google Scholar]
- 40.Mehl, J. W. M., Slippers, B., Roux, J. & Wingfield, M. J. Overlap of latent pathogens in the Botryosphaeriaceae on a native and agricultural host. Fungal Biol.121, 405–419 (2017). 10.1016/j.funbio.2016.07.015 [DOI] [PubMed] [Google Scholar]
- 41.Slippers, B. et al. Development of simple sequence repeat markers for Botryosphaeria spp. with Fusicoccum anamorphs. Mol. Ecol. Notes4, 675–677 (2004).
- 42.Zhou, S., Smith, D. R. & Stanosz, G. R. Differentiation of Botryosphaeria species and related anamorphic fungi using Inter Simple or Short Sequence Repeat (ISSR) fingerprinting. Mycol. Res.105, 919–926 (2001). 10.1016/S0953-7562(08)61947-4 [DOI] [Google Scholar]
- 43.Manawasinghe, I. S. et al. Novel microsatellite markers reveal multiple origins of Botryosphaeria dothidea causing the Chinese grapevine trunk disease. Fungal Ecol.33, 134–142 (2018). 10.1016/j.funeco.2018.02.004 [DOI] [Google Scholar]
- 44.Nagel, J. H., Cruywagen, E. M., Machua, J., Wingfield, M. J. & Slippers, B. Highly transferable microsatellite markers for the genera Lasiodiplodia and Neofusicoccum. Fungal Ecol.44, 100903 (2020). 10.1016/j.funeco.2019.100903 [DOI] [Google Scholar]
- 45.Pavlic-Zupanc, D., Wingfield, M. J., Boissin, E. & Slippers, B. The distribution of genetic diversity in the Neofusicoccum parvum / N. ribis complex suggests structure correlated with level of disturbance. Fungal Ecol.13, 93–102 (2015).
- 46.Sakalidis, M. L., Slippers, B., Wingfield, B. D., Hardy, G. E. St. J. & Burgess, T. I. The challenge of understanding the origin, pathways and extent of fungal invasions: global populations of the Neofusicoccum parvum–N. ribis species complex. Divers. Distrib.19, 873–883 (2013).
- 47.Zlatković, M., Wingfield, M. J., Jami, F. & Slippers, B. Genetic uniformity characterizes the invasive spread of Neofusicoccum parvum and Diplodia sapinea in the Western Balkans. For. Path.49, e12491 (2019). 10.1111/efp.12491 [DOI] [Google Scholar]
- 48.Jarne, P. & Lagoda, P. J. L. Microsatellites, from molecules to populations and back. Trends Ecol. Evol.11, 424–429 (1996). 10.1016/0169-5347(96)10049-5 [DOI] [PubMed] [Google Scholar]
- 49.Tautz, D. & Renz, M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res.12, 4127–4138 (1984). 10.1093/nar/12.10.4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lepais, O. et al. Fast sequence-based microsatellite genotyping development workflow. PeerJ8, e9085 (2020). 10.7717/peerj.9085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belair, M., Pensec, F., Jany, J.-L., Floch, G. L. & Picot, A. Profiling walnut fungal pathobiome associated with walnut dieback using community-targeted DNA metabarcoding. Plants12, 2383 (2023). 10.3390/plants12122383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trouillas, F. P., Hébrard, M.-N., Aurelle, V., Jargeat, P. & Tranchand, E. Etiology of walnut fruit rot, spur and twig dieback in southwestern France. Acta Hortic. (2023).
- 53.Ma, Z., Boehm, E. W. A., Luo, Y. & Michailides, T. J. Population structure of Botryosphaeria dothidea from pistachio and other hosts in California. Phytopathology91, 665–672 (2001). 10.1094/PHYTO.2001.91.7.665 [DOI] [PubMed] [Google Scholar]
- 54.Baskarathevan, J., Jaspers, M. V., Jones, E. E., Cruickshank, R. H. & Ridgway, H. J. Genetic and pathogenic diversity of Neofusicoccum parvum in New Zealand vineyards. Fungal Biol.116, 276–288 (2012). 10.1016/j.funbio.2011.11.010 [DOI] [PubMed] [Google Scholar]
- 55.Sunnucks, P. Efficient genetic markers for population biology. Trends Ecol. Evol.15, 199–203 (2000). 10.1016/S0169-5347(00)01825-5 [DOI] [PubMed] [Google Scholar]
- 56.McDonald, B. A. & Linde, C. Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol.40, 349–379 (2002). 10.1146/annurev.phyto.40.120501.101443 [DOI] [PubMed] [Google Scholar]
- 57.Xu, J. Fundamentals of fungal molecular population genetic analyses. Curr. Issues Mol. Biol.8, 75–90 (2006). [PubMed] [Google Scholar]
- 58.Lopes, A., Phillips, A. J. L. & Alves, A. Mating type genes in the genus Neofusicoccum: Mating strategies and usefulness in species delimitation. Fungal Biol.121, 394–404 (2017). 10.1016/j.funbio.2016.08.011 [DOI] [PubMed] [Google Scholar]
- 59.Nagel, J. H., Wingfield, M. J. & Slippers, B. Evolution of the mating types and mating strategies in prominent genera in the Botryosphaeriaceae. Fungal Genet. Biol.114, 24–33 (2018). 10.1016/j.fgb.2018.03.003 [DOI] [PubMed] [Google Scholar]
- 60.Michailides, T. J. et al. Managing Botryosphaeria/Phomopsis cankers and anthracnose blight of walnut in California. In Walnut Research Reports 325–346 (California Walnut Board, Folsom, CA, 2013).
- 61.Moral, J., Morgan, D. & Michailides, T. J. Management of Botryosphaeria canker and blight diseases of temperate zone nut crops. Crop Prot.126, 104927 (2019). 10.1016/j.cropro.2019.104927 [DOI] [Google Scholar]
- 62.Rosace, M. C., Legler, S. E., Salotti, I. & Rossi, V. Susceptibility of pruning wounds to grapevine trunk diseases: A quantitative analysis of literature data. Front. Plant Sci.14, (2023). [DOI] [PMC free article] [PubMed]
- 63.Michailides, T. J. et al. Epidemiology and management of Botryosphaeria/Phomopsis canker and blight and anthracnose blight of walnut in California. In Walnut Research Reports 349–376 (California Walnut Board, Folsom, CA, 2016).
- 64.Rai, M. & Agarkar, G. Plant-fungal interactions: What triggers the fungi to switch among lifestyles?. Crit. Rev. Microbiol.42, 428–438 (2016). 10.3109/1040841X.2014.958052 [DOI] [PubMed] [Google Scholar]
- 65.Ma, Z., Luo, Y. & Michailides, T. J. Spatiotemporal changes in the population qtructure of Botryosphaeria dothidea from California pistachio orchards. Phytopathology94, 326–332 (2003). 10.1094/PHYTO.2004.94.4.326 [DOI] [PubMed] [Google Scholar]
- 66.Herman, I. et al. Etude sur les dépérissements et la mortalité des noyers - Région Sud-Ouest. 1–80 (1995).
- 67.Letonnelier, G. Les origines de la culture intensive du noyer dans le Bas-Grésivaudan. Rev. Géogr. Alp.18, 303–325 (1930). 10.3406/rga.1930.4531 [DOI] [Google Scholar]
- 68.Laugénie, C. La culture du noyer en Périgord. Revue géographique des Pyrénées et du Sud-Ouest. Sud-Ouest Européen36, 135–158 (1965).
- 69.Ramos, D. & Doyle, J. Walnut research and industry survey - France, 1984. in Walnut Research Reports 49–55 (California Walnut Board, Folsom, CA, 1984).
- 70.López-Moral, A. et al. Effects of cultivar susceptibility, branch age, and temperature on infection by Botryosphaeriaceae and Diaporthe fungi on English walnut (Juglans regia). Plant Dis.106, 2920–2926 (2022). 10.1094/PDIS-09-21-2042-RE [DOI] [PubMed] [Google Scholar]
- 71.Sohrabi, M., Mohammadi, H., León, M., Armengol, J. & Banihashemi, Z. Fungal pathogens associated with branch and trunk cankers of nut crops in Iran. Eur. J. Plant Pathol.157, 327–351 (2020). 10.1007/s10658-020-01996-w [DOI] [Google Scholar]
- 72.Dyakov, Yu. T. Chapter 0 - Overview on parasitism. in Comprehensive and Molecular Phytopathology (eds. Dyakov, Yu. T., Dzhavakhiya, V. G. & Korpela, T.) 3–17 (Elsevier, Amsterdam, 2007). 10.1016/B978-044452132-3/50003-1.
- 73.Novak, A., Ivić, D., Sever, Z., Fazinić, T. & Šimunac, K. Botryosphaeria canker of walnut in Croatia. Glasilo biljne zaštite18, 316–321 (2018). [Google Scholar]
- 74.Yildiz, A., Benlioglu, S., Benlioglu, K. & Korkom, Y. Occurrence of twig blight and branch dieback of walnut caused by Botryosphaeriaceae species in Turkey. J. Plant Dis. Protect.129, 687–693 (2022). 10.1007/s41348-022-00591-x [DOI] [Google Scholar]
- 75.Batista, E., Lopes, A., Miranda, P. & Alves, A. Can species distribution models be used for risk assessment analyses of fungal plant pathogens? A case study with three Botryosphaeriaceae species. Eur. J. Plant Pathol.165, 41–56 (2023). 10.1007/s10658-022-02587-7 [DOI] [Google Scholar]
- 76.Preece, J. E. & McGranahan, G. Luther Burbank’s contributions to walnuts. HortScience50, 201–204 (2015). 10.21273/HORTSCI.50.2.201 [DOI] [Google Scholar]
- 77.McGranahan, G. & Leslie, C. Walnuts (Juglans). Acta Hortic.290, 907–974 (1991). 10.17660/ActaHortic.1991.290.20 [DOI] [Google Scholar]
- 78.Delbos, R. et al. Virus infection of walnuts in France. Acta Hortic.130, 123–132 (1983). 10.17660/ActaHortic.1983.130.21 [DOI] [Google Scholar]
- 79.White, T. J., Bruns, T., Lee, S. & Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. in PCR protocols: a guide to methods and amplifications 315–322 (Academic Press, Cambridge, MA, USA, 1990). 10.1016/B978-0-12-372180-8.50042-1.
- 80.Carbone, I. & Kohn, L. M. A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia91, 553–556 (1999). 10.1080/00275514.1999.12061051 [DOI] [Google Scholar]
- 81.Glass, N. L. & Donaldson, G. C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol.61, 1323–1330 (1995). 10.1128/aem.61.4.1323-1330.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing (2022).
- 83.Comont, G., Mayet, V. & Corio-Costet, M. F. First report of Lasiodiplodia viticola, Spencermartinsia viticola and Diplodia intermedia associated with Vitis vinifera grapevine decline in French vineyards. Plant Dis.100, 2328–2328 (2016). 10.1094/PDIS-04-16-0576-PDN [DOI] [Google Scholar]
- 84.Comont, G. et al. Characterization of the RNA mycovirome associated with grapevine fungal pathogens: Analysis of mycovirus distribution and their genetic variability within a collection of Botryosphaeriaceae isolates. Viruses16, 392 (2024). 10.3390/v16030392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform.20, 1160–1166 (2019). 10.1093/bib/bbx108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuraku, S., Zmasek, C. M., Nishimura, O. & Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res.41, W22–W28 (2013). 10.1093/nar/gkt389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol.17, 540–552 (2000). 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- 88.Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol.56, 564–577 (2007). 10.1080/10635150701472164 [DOI] [PubMed] [Google Scholar]
- 89.Swofford, D. PAUP*. Phylogenetic analysis using parsimony and other methods. Version 4.0. (Sunderland, Massachusetts, 2003).
- 90.Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol.35, 1547–1549 (2018). 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bouckaert, R. R. & Drummond, A. J. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol.17, 42 (2017). 10.1186/s12862-017-0890-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bouckaert, R. et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLOS Comput. Biol.15, e1006650 (2019). [DOI] [PMC free article] [PubMed]
- 93.Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol.67, 901–904 (2018). [DOI] [PMC free article] [PubMed]
- 94.Rambaut, A. FigTree. http://tree.bio.ed.ac.uk/software/figtree/ (2006).
- 95.Hodcroft, E. TreeCollapserCL4. http://emmahodcroft.com/TreeCollapseCL.html.
- 96.Meglécz, E. et al. QDD version 3.1: A user-friendly computer program for microsatellite selection and primer design revisited: Experimental validation of variables determining genotyping success rate. Mol. Ecol. Resour.14, 1302–1313 (2014). [DOI] [PubMed]
- 97.Hoogenboom, J. et al. FDSTools: A software package for analysis of massively parallel sequencing data with the ability to recognise and correct STR stutter and other PCR or sequencing noise. Forensic Sci. Int. Genet.27, 27–40 (2017). [DOI] [PubMed]
- 98.Kamvar, Z. N., Brooks, J. C. & Grünwald, N. J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet.6, (2015). [DOI] [PMC free article] [PubMed]
- 99.Peakall, R. & Smouse, P. E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics28, 2537–2539 (2012). [DOI] [PMC free article] [PubMed]
- 100.Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res.27, 209–220 (1967). [PubMed] [Google Scholar]
- 101.Adamack, A. T. & Gruber, B. PopGenReport: Simplifying basic population genetic analyses in R. Methods Ecol. Evol.5, 384–387 (2014). 10.1111/2041-210X.12158 [DOI] [Google Scholar]
- 102.Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J.27, 379–423 (1948). 10.1002/j.1538-7305.1948.tb01338.x [DOI] [Google Scholar]
- 103.Simpson, E. H. Measurement of diversity. Nature163, 688–688 (1949). 10.1038/163688a0 [DOI] [Google Scholar]
- 104.Pielou, E. C. Ecological diversity (John Wiley & Sons, 1975). [Google Scholar]
- 105.Grünwald, N. J., Goodwin, S. B., Milgroom, M. G. & Fry, W. E. Analysis of genotypic diversity data for populations of microorganisms. Phytopathology93, 738–746 (2003). 10.1094/PHYTO.2003.93.6.738 [DOI] [PubMed] [Google Scholar]
- 106.Brown, A. H. D., Feldman, M. W. & Nevo, E. Multilocus structure of natural populations of Hordeum spontaneum. Genetics96, 523–536 (1980). 10.1093/genetics/96.2.523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Agapow, P.-M. & Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes1, 101–102 (2001). 10.1046/j.1471-8278.2000.00014.x [DOI] [Google Scholar]
- 108.Dray, S. & Dufour, A.-B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw.22, 1–20 (2007). 10.18637/jss.v022.i04 [DOI] [Google Scholar]
- 109.Bruvo, R., Michiels, N. K., D’souza, T. G. & Schulenburg, H. A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol. Ecol.13, 2101–2106 (2004). 10.1111/j.1365-294X.2004.02209.x [DOI] [PubMed] [Google Scholar]
- 110.Bache, S. & Wickman, H. magrittr: A forward-pipe operator for R. https://magrittr.tidyverse.org/, https://github.com/tidyverse/magrittr (2022).
- 111.Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet.11, 94 (2010). 10.1186/1471-2156-11-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jombart, T. & Ahmed, I. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics27, 3070–3071 (2011). [DOI] [PMC free article] [PubMed]
- 113.Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics89, 583–590 (1978). 10.1093/genetics/89.3.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nei, M. Genetic distance between populations. Am. Nat.106, 283–292 (1972). 10.1086/282771 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available in the Recherche Data Gouv repository, 10.57745/J8ERW1.