

Abstract
Recent advances in gene therapy and gene-editing techniques offer the very real potential for successful treatment of neurological diseases. However, drug delivery constraints continue to impede viable therapeutic interventions targeting the brain due to its anatomical complexity and highly restrictive microvasculature that is impervious to many molecules. Realizing the therapeutic potential of gene-based therapies requires robust encapsulation and safe and efficient delivery to the target cells. Although viral vectors have been widely used for targeted delivery of gene-based therapies, drawbacks such as host genome integration, prolonged expression, undesired off-target mutations, and immunogenicity have led to the development of alternative strategies. Engineered virus-like particles (eVLPs) are an emerging, promising platform that can be engineered to achieve neurotropism through pseudotyping. This review outlines strategies to improve eVLP neurotropism for therapeutic brain delivery of gene-editing agents.
Keywords: MT: Delivery Strategies, neurological diseases, blood-brain barrier, gene editing, delivery systems, engineered virus-like particles, virus pseudotyping
Graphical abstract
Hewitt and colleagues highlight the engineered virus-like particles as a promising drug delivery platform alternative to viral vectors utilized for in vivo gene editing. Several studies have examined pseudotyping delivery vectors to modulate their tissue tropism, and, in this review, the authors discuss the strategies to advance eVLP neurotropism.
Introduction
Neurological diseases are an extremely heterogeneous group of disorders affecting the CNS. They encompass almost half of all rare diseases and about 80% have a genetic basis.1 In recent years, progress in both fundamental and translational research has helped to elucidate details on the molecular signaling and genetic regulation of neurological disorders.2 Such advances have broadened the knowledge about the genetic basis of human diseases and revealed that far more disorders are genetic in origin than was previously anticipated.3 Recent technological advances such as whole-genome sequencing and whole-exome sequencing have enabled widespread identification of (CNS) disorders with a genetic etiology, with new causative mutations described for Parkinson’s disease, Alzheimer’s disease (AD), Huntington’s disease,4,5,6 and neuronal ceroid lipofuscinoses (NCLs or Batten disease).7,8 The socioeconomic burden related to these neurological diseases is escalating and is expected to continue globally. Therefore, efficient measures to counter this global health challenge are urgently needed.9 Given the structural complexity of the brain and the blood-brain barrier (BBB), the development of therapeutics to target the brain is challenging and hence many investigational drug developments have failed to translate into clinical application. Therefore, to attain the pharmacological success of gene-editing therapeutics, it is important to consider a selection of appropriate drug delivery systems for targeting and penetration of the BBB, drug administration route, and therapeutic index of the drug.10
Gene-editing technologies, such as base editing11,12 and prime editing,13 have increased in prominence as some of them have already shown success in clinical trials. As the new gene-editor variants are being developed,14,15,16,17 many other pre-clinical and clinical trials in treating certain diseases, such as for blood disorders,18 cancers,19 liver disorders,20 amyotrophic lateral sclerosis,21 Duchenne muscular dystrophy,22 spinal muscular atrophy,23,24 and neurological disorders such as Huntington’s Disease,25 genetic epilepsies,26 Parkinson’s disease,27 and Alzheimer’s28 are underway.29 To attain their full therapeutic potential, gene-editing agents are required to be encapsulated, protected from degradation, and to bind and traverse the target cell membrane to efficiently deliver the genetic cargo to the nucleus.30,31,32,33 They can be packaged as plasmid DNA or as purified proteins or ribonucleoproteins (RNPs),33 and the appropriate selection of the delivery system is crucial for the successful therapeutic outcome. Currently, delivery of gene-editing therapeutics is primarily based on viral vectors, lipid nanoparticles (LNPs), and extracellular vesicles. Currently, delivery of gene-editing therapeutics is primarily based on viral vectors, LNPs, and extracellular vesicles.33,34,35,36
In this review, we will first examine the characteristics of viral and virus-like delivery systems and then discuss the strategies to attain neurotropism in engineered virus-like particles.
Viral Vectors
Due to their infectious nature and ability to introduce genes into the host cells, viral vectors are by far the most broadly used method to deliver therapeutic gene therapeutics. A viral vector consists of a protein capsid and/or envelope, transgene, and the regulatory cassette. To package the gene-editing macromolecules, the genome sequences necessary for viral replication and capsid production are removed and replaced. Expression of the transgenes is controlled by the constituent or regulated promoters and other regulatory elements encoded in the viral genome. However, some viruses are limited in their ability to efficiently deliver gene-editing agents due to constraints in their packaging capacity. In the current gene-editing landscape, adeno-associated viruses (AAVs) and lentiviruses (LVs) are at the forefront of pre-clinical and clinical applications.37
AAV Delivery
AAV is a 20- to 25-nm, non-enveloped virus with a cargo capacity of ∼4.7 kb.36,38 Due to the relatively low immunological properties, efficient transduction, and transgene expression in a wide variety of tissues, AAVs have been broadly used for several gene therapies and have shown success in both pre-clinical and clinical settings. Approval of AAV-based gene therapies Luxturna (voretigene neparvovec-rzyl) to treat Leber congenital amaurosis and Zolgensma (onasemnogene abeparvovec) to treat spinal muscular atrophy has marked a milestone in the field of gene therapy.39,40,41,42,43
AAV serotypes identified from humans and non-human primates (NHPs) have been harnessed to deliver CRISPR-Cas9 complexes into different tissues.44,45 These AAV serotypes differ in their tropism based on the changes in the hypervariable regions of the capsid proteins. For example, AAV8 has been shown to efficiently transduce the liver tissue,20 while AAV9 transduces a variety of tissues, including muscle,22,23,24 retina,30 heart,46 and lung47 in mice. Such variations in tissue tropism are primarily attributed to the surface topology of receptor-binding proteins.44,48 Wild-type (WT) AAV serotypes have a low therapeutic index and therefore potentially heighten the risk of immunogenicity and toxicity. Moreover, their performance is likely to be impacted by the pre-existing AAV immunity within the host.49 To address these challenges, several modifications to the gene regulatory elements and vector capsid modifications have been thoroughly researched.
The WT AAV genome consists of three open reading frames (ORFs): rep, cap, and X genes, of which the rep gene encodes four proteins that mediate regulation, replication, and assembly and the cap encodes three overlapping capsid proteins: VP1 (90 kDa), VP2 (72 kDa), and VP3 (60 kDa). The icosahedral AAV capsid is a combination of approximately 50 copies of VP3, five copies of VP2, and five copies of VP1. The viral capsid sequence is a key determinant of the tropism and therefore the tropism modifications are often achieved by the chemical and genetic modifications to the capsid. These capsid-modification technologies are discussed in detail elsewhere.49,50
Modifications to the AAV capsid
To better pursue their therapeutic potential, several improvements have been made to the native AAV capsids in such a way as to interact with a repertoire of cellular receptors to mediate infections. As such, AAV capsids have been chemically or genetically modified to produce hybrid capsids to incorporate a variety of tropism properties. These techniques include, but are not limited to, capsid pseudotyping, directed evolution, rational mutagenesis, or peptide insertions to promote novel receptor-binding activity.51
Genetic modification of the capsid
Insertion of peptides into the common VP3 region is one such way to genetically modify the virus capsid. For example, I-453, I-587, I-588, I-584, and I-585 sites located within the common VP3 region are susceptible to insertions of targeting ligands. As such, the I-587 site of AAV2 capsid is known to accept peptide insertions up to a size of 34 amino acids and thus characterize AAV2 with different tissue tropism,49 whereas genetic modification of VP1 and VP2 introduces complementary split-intein domains to the N terminus of the AAV2 VP2 capsid protein, as well as to the targeting ligands. The modified capsid becomes accessible to form covalent bonds with ligands.52
High-throughput selection screening to identify targeting ligands is yet another efficient approach to identifying the modifiable capsid positions and presence of peptides or protein motifs. As such, random oligonucleotide sequence insertions into the cap ORF at sites corresponding to the top of VR-VIII or -IV generate a pool of diverse mutants.49,53 Capsid engineering is also achieved through site-directed mutagenesis of surface-exposed tyrosine (Y), serine (S), threonine (T), and lysine (K) residues.
Mutagenized Y residues of Y444F, Y500F, and Y730F to phenylalanine (F) residues promote the transduction efficacy of AAV2 in the cultured human cells more than that of their WT counterpart.54 Enhanced tropisms across various tissue types were further observed with the S residue mutants (S458V, S492V, S662V), T residue mutants (T455V, T491V, T550V, T659V),55 and K residues (K490E, K544E, and K549E).56
Chemical modification of the capsid
Chemical modification of the capsids is an alternative technique to genetic manipulation. It involves modifications to the amino acid composition of the capsid and conjugation of functionalized peptides.57 Some examples include re-directed tissue tropisms of glycated AAV2 capsids58 and 4-azidophenyl glyoxal (APGO) functionalized rAAV6 capsids.59
Mével et al. investigated chemical conjugation of a ligand by the formation of a thiourea functionality between the amino group of the capsid proteins and the reactive isothiocyanate motif incorporated into the ligand. When combined with a hepatocyte targeting ligands such as GalNAc, modified vectors resulted in a higher transduction efficiency in cultured hepatocytes.57 Overall, capsid-modification techniques are of great interest and significantly broaden the scope of gene-based therapies.
Neurotropism of AAV vectors
Engineered AAV capsids
Over the past few decades, significant research has been conducted on capsid engineering of AAV to achieve neurotropism.60 As such, new clades of primate AAVs have sparked greater interest due to their distinct transduction patterns within the nervous system. For example, rAAV9 is one such serotype that transduces neuronal and glial cell types in the CNS of murine neonates following intravenous injections, which is important for CNS diseases that require intervention during the earlier ages.61 The intravenous administration of AAV9 to the neonates and adult mice of amyotrophic lateral sclerosis (ALS) had shown an efficient transgene expression in the neonate brain, dorsal root ganglia and spinal cord motor neurons, and astrocytes,62 whereas rAAVrh.8 has been shown to transduce glial cells and neurons located within the cortex, caudate-putamen, hippocampus, corpus callosum and substantia nigra.61 Different recombinant AAVs such as rAAVrh.10, rAAV1, rAAV6, and rAAV6.2 have varying transduction profiles across different CNS cell populations. After traversing the BBB, rAAV1 primarily transduces granule cells in the cerebellum, rAAV6, and rAAV6.2 transduce Purkinje cells, while rAAVrh.10, rAAV9, rAAV7, and rAAVrh.39 efficiently transduce motor neurons in mouse neonates.63
Other modified capsids of rAAV2-retro and AAV2g9 have been shown to acquire retrograde functionality in projection neurons and also achieve neurotropism derived from both parental counterparts, AAV2 and AAV9.64,65 Moreover, point mutations introduced into the heparin-binding domain of AAV-DJ/8 capsid have been shown to successfully transduce the mouse brain.66,67 Other CNS variants of AAV-PHP.B and AAV-PHP.eB have been shown to cross the BBB and transduce astrocytes, neurons, oligodendrocytes, cerebellar Purkinje cells, and several interneuron populations more effectively than the AAV9.60,68,69 AAV-PHP.B vectors gain cellular entry by interacting with the LY6A receptors on BBB and therefore their transduction is restricted to species that express the LY6A receptor. The LY6A protein does not have a known homolog in primates and therefore it is unlikely to translate into NHPs.70,71,72,73
BI-hTFR1 is another recent example of virus capsid engineering achieved through screening an AAV9-based NNK (NNK refers to a codon sequence where "K" represents either guanine (G) or thymine (T)) capsid library for selective binding to human TfR1 receptors. This AAV9-based NNK capsid library consists of variants with random 7-mer insertions between 588 and 589 VP1 residues. The modified BI-hTFR1 delivered GBA1 robustly and increased glucocerebrosidase activity across multiple regions of humanized TFRC knockin mouse brains. Interestingly, viral transduction within the brain regions implicated in Parkinson’s disease pathology suggests a promising treatment option.74
LV-Mediated Delivery
LV is an enveloped retrovirus with two sense-RNA strands that are bound by nucleocapsid proteins. The LV genome consists of nine genes, including the gag and env. The gag gene encodes the capsid and matrix proteins, while the env gene encodes transmembrane and envelope glycoproteins.75 To render the lentiviral vectors non-pathogenic and replication deficient, all virulence genes are removed.76 These replication-defective LVs have been utilized in several clinical applications,77 such as ex vivo genetic modification of beta-thalassemia, X-linked adrenoleukodystrophy (ALD), and metachromatic leukodystrophy.78,79 Moreover, hematopoietic stem cell (HSC) transplantation of LV-transduced CD34+ cells in two X-linked ALD patients showed reverse cerebral demyelination, alleviation of neurological symptoms, and prolonged therapeutic gene expression in treated patients.78 Lovotibeglogene autotemcel is a one-time gene therapy that has been evaluated to treat sickle cell disease (SCD). It involves transplantation of autologous CD34+ stem cells transduced ex vivo with the LentiGlobin BB305 lentiviral vector encoding a modified beta-globin gene (βA-T87Q-globin) (ClinicalTrials.gov: NCT02140554).
The higher packaging capacity of LVs has been utilized to package bulk and multiplexed genome-editing tools such as CRISPR/Cas9, reporter genes, and single guide RNAs (sgRNAs).77 These all-in-one lentiviral CRISPR/Cas9 systems have been shown to transduce and sustain efficient genetic manipulations in a broad range of cell lines including the progenitor and primary cells, which are challenging to transfect.77 The tropism of LVs can be modulated by pseudotyping the virions with glycoproteins derived from different enveloped viruses with known tropism.80
Neurotropism of lentiviral vectors
The interactions between the envelope glycoproteins and the cell surface receptor on the target cells allow viral entry. Lentiviral vectors could be targeted to specific brain regions or cell sub-populations by pseudotyping the virus with different envelope glycoproteins81,82 (see Table 1).
Table 1.
Surface-engineered lentiviral vectors for brain-targeted drug delivery
| Brain region | VSV-G | MuLV | Mokola | LCMV | CHIKVG | VEEV | RRV-G | RV-G | RabiesFuG/B2 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Forebrain | ||||||||||
| Striatum | HIV-1/EIAV | HIV-1 | HIV-1/EIAV | EIAV/FIV | HIV-1 | HIV-1 | – | EIAV/HIV-1 | – | Mazarakis et al.,81 Won et al.,82 Desmaris et al.,83 Eleftheriadou et al.,84 Kato et al.,85 Watso et al.,86 Miletic et al.,87 Stein et al.,88 Trabalza et al.89 |
| Thalamus | HIV-1 | – | HIV-1 | HIV-1 | – | HIV-1 | – | EIAV/HIV-1 | – | Mazarakis et al.,81 Kato et al.,85 Watson et al.,86 Trabalza et al.89 |
| Hypothalamus | – | – | – | – | – | – | – | EIAV | – | Mazarakis et al.81 |
| Subthalamic nucleus | – | – | – | – | – | – | – | EIAV | – | Mazarakis et al.81 |
| Amygdala | – | – | – | – | – | – | – | EIAV | – | Mazarakis et al.81 |
| External capsule | HIV-1 | HIV-1 | HIV-1 | – | – | – | – | – | – | Desmaris et al.,83 Watson et al.86 |
| Olfactive neuron bodies | HIV-1 | – | HIV-1 | FIV | – | – | – | – | – | Desmaris et al.,83 Stein et al.88 |
| Mitral cells in the olfactory bulb | – | – | – | – | – | HIV-1 | – | – | – | Trabalza et al.89 |
| Olfactory tubercles | – | – | HIV-1 | – | – | – | – | – | – | Cannon et al.90 |
| Globus pallidus | EIAV | – | – | – | – | – | – | EIAV | – | Wong et al.82 |
| Subventricular zone (SVZ) | – | – | – | FIV | – | – | – | – | – | Stein et al.88 |
| Midbrain | ||||||||||
| Ventral midbrain | – | – | – | – | – | – | – | HIV-1 | – | Kato et al.85 |
| Hippocampal dentate gyrus | – | HIV-1 | – | – | HIV-1 | – | – | – | – | Eleftheriadou et al.,84 Watson et al.86 |
| Substantia nigra | HIV | – | – | – | – | – | – | – | – | Cannon et al.90 |
| Substantia nigra pars compacta | – | – | – | – | – | HIV-1 | – | EIAV | – | Mazarakis et al.,81 Trabalza et al.89 |
| Substantia nigra pars reticulata | EIAV | – | – | – | – | – | – | – | – | Wong et al.82 |
| Hippocampus | – | – | HIV-1 | – | HIV-1 | – | – | – | – | Desmaris et al.83 |
| Nigral-dopaminergic neurons | HIV-1 | – | EIAV | – | – | – | – | – | – | Desmaris et al.,83 Eleftheriadou et al.,84 Colin et al.91 |
| Hindbrain | ||||||||||
| Cerebellum | – | – | HIV-1 | – | – | – | – | – | – | Colin et al.91 |
| Cell sub-populations | ||||||||||
| Astrocytes | HIV-1 | HIV-1 | HIV-1 | FIV | HIV-1 | – | FIV | – | – | Cannon et al.,90 Eleftheriadou et al.,84 Miletic et al.,87 Stein et al.,88 Colin et al.,91 Kang et al.,92 Pertusa et al.93 |
| Neurons | FIV/SIVmac | – | HIV-1 | – | – | – | – | – | – | Desmaris et al.83; Kang et al.92; Lieh et al.,94 |
| Glial cells | HIV1/EIAV/SIVmac | SIVmac | Bergmann glial cells | – | – | – | SIVmac | – | HIV-1 | Mazarakis et al.,81 Desmaris et al.,83 Miletic et al.,87 Colin et al.,91 Lieh et al.,94 Kato et al.,95 Hirano et al.96 |
| Brain parenchyma | SIVmac | – | – | – | – | – | – | – | – | Lieh et al.94 |
| Cortex | EIAV | – | – | – | – | – | – | EIAV/HIV-1 | – | Mazarakis et al.,81 Wong et al.,82 Kato et al.85 |
| White matter | HIV-1 | – | HIV-1 | HIV-1 | – | – | – | – | Watson et al.86 | |
| Oligodendrocytes | HIV-1 | – | HIV-1 | – | – | – | FIV | – | – | Watson et al.,86 Kang et al.92 |
Lentiviral vectors pseudotyped with various envelope glycoproteins confer an enormous potential for gene therapy of human neurological disease. Primate and non-primate LVs derived from the HIV, feline immunodeficiency virus (FIV), simian immunodeficiency virus, and EIAV are pseudotyped with envelope glycoproteins originated from VSV, rabies virus glycoprotein (RV-G), LCMV, MV, MuLV, Ross River virus glycoprotein (RRV-G), Chikungunya virus glycoprotein (CHIKV-G), Venezuelan equine encephalitis virus glycoprotein (VEEV-G), and fusion glycoprotein B type (FuG-B) or a variant of FuG-B (FuG-B2). Recent pre-clinical studies utilizing these pseudotyped vectors have demonstrated modified neurotropism within different hosts of rats, mice, and monkeys.
In a previous study, Desmaris and co-workers assessed the neurotropism of lentivrial vectors pseudotyped with lyssavirus envelope glycoproteins and injected into the striatum region of the mouse. After 4 weeks from the initial administration, it was observed that Mokola-G pseudotyped lentiviral vectors achieved a robust β-glucuronidase expression compared to the vesicular stomatitis virus G protein (VSV-G) pseudotyped lentiviral vectors.83 In another study, astrocyte-specific LV-viral vectors were developed to target gene expression in both neurons and neuronal astrocytes. To further improve the broad distribution of lentiviral vectors they combined retrograde transport properties to target the brain.6 The astrocytic gene expression of the lentiviral vectors was achieved by pseudotyping with VSV-G envelope glycoproteins and as well as using promoters such as H1 polymerase III and G1B3 polymerase II, which are known to be highly active in astrocytes. VSV-G envelope glycoprotein interacts with low-density lipoprotein (LDL) receptors located on neurons and astrocytes to mediate endocytosis of the virus.97
Biodistribution of lentiviral vectors can be altered using a chimeric fusion glycoprotein variant B2 (FuG/B2) envelope glycoprotein that is characterized by retrograde transport properties. Injecting into the striatum of mice leads to strong transduction in the cortex, amygdala, thalamus, and substantia nigra, indicating potential for enhancing brain therapeutic interventions in the future.6 Another similar study showed that lentiviral vectors pseudotyped with rabies virus glycoprotein (RV-G) can promote retrograde axonal gene transfer and efficient neuronal transductions in the CNS. Interestingly, it was observed that RV-G-pseudotyped equine infectious anemia virus (EIAV) results in higher transduction within dopaminergic neurons of the substantia nigra pars compacta. Overall, pseudotyping lentiviral vectors with RV-G protein has been shown to enhance the transduction at the site of injection as well as the distal neurons.6
Improved neurotropism of these viral vectors plays a vital role in the treatment of neurological diseases. In mouse brain afflicted with Parkinson’s disease, there was an improved neurotropism of lentiviral vectors that were pseudotyped with vesicular stomatitis virus (VSV), Mokola virus (MV), lymphocytic choriomeningitis virus (LCMV), or Moloney murine leukemia virus (MMLV) envelope glycoproteins. When the VSV-lentiviral vectors and MV-lentiviral vectors were delivered into the substantia nigra of rats, a robust expression in midbrain neurons was observed while LCMV- and murine leukemia virus (MuLV)-pseudotyped lentiviral vector expressions were exclusive to astrocytes.90 Extended tropism studies using lentiviral vectors pseudotyped with chikungunya virus glycoprotein reported a selective astrocytic and cell subpopulation transduction pattern within the CNS.84 Therefore, it is apparent that envelope modifications of viral vectors have been useful in targeting specific tissues including the brain.
Drawbacks of Using Viral Vectors
Despite the therapeutic potential, the effective clinical application of gene-editing technologies remains challenging due to the potential for undesired off-target editing. Several strategies have attempted to reduce or eliminate off-target cleavage, including truncated guide RNA with modified scaffolds containing chemical modifications,98 and modified CRISPR effector proteins (Cas9 orthologs, high-fidelity variants).99 The efficiency of gene-editing technologies is greatly influenced by both the overall expression level and duration of the gene-editing agents within the cells.36 Prolonged expression of AAV-mediated gene editing has the propensity to cause off-target mutations over time. More precise spatiotemporal control of transgene expression can be achieved by incorporating tissue-specific promoters into the AAV gene cassette.100
Although AAVs are an excellent in vivo delivery platform due to the size limits of the AAV, it is not possible to efficiently package larger gene-editing agents, guide RNA, promoters, and other cis-regulatory elements within a single AAV particle. To overcome packaging restrictions, split intein to package the gene editor into two individual AAV vectors has been pursued in recent studies. To be reconstituted to form the full-length gene editor and thereby achieve genome editing, both AAV vectors should be delivered into the same cell, hence the editing efficiency may be decreased.100 Therefore, to attain the full therapeutic potential of dual AAVs, a higher dose of AAVs is recommended. However, much higher titers of AAV have been suggested to cause dorsal root ganglion toxicity in NHPs and piglets60,101,102 and life-threatening events in patients that received AAV-based gene therapy.60,103
Immunogenicity is another hurdle impeding viral vector efficacy, which can also lead to safety concerns. For example, AAV-based gene therapies have been found to elicit capsid-specific T cell responses in some people, which could persist for years, affecting the duration of transgene expression.104 Moreover, pre-existing immunity to WT AAV has been shown to induce both humoral and T cell-mediated immunity while reducing successful gene transfer.105
Supporting the notion that AAV vectors can be genotoxic, another study reported the incidence of hepatocellular carcinomas and angiosarcomas after AAV gene transfer in a mouse model of mucopolysaccharidosis type VII (MPS VII).106 Another 10-year post-gene-therapy study revealed oncogenic genome integration of AAV8/AAV9 in six of nine experimental canine candidates and hence raised the safe clinical application.107,108 Therefore, transient expression of genome-editing agents is crucial, and this is typically accomplished by using non-viral delivery systems.
Moreover, selecting the appropriate model is important for achieving clinical success, yet it can be challenging. As such, pre-clinical outcomes experimented in murine models may not necessarily be translatable to higher-order species. For example, systemic AAV-PHP.B vectors have been challenging to translate to NHPs as the BBB permeability of the vector did not extend to NHPs.71,72 In their study, Hordeaux et al. showed that the neurotropic properties of AAV-PHP.B are limited to C57BL/6J mouse strain and underperforms within the BALB/cJ mouse strain, as well as in non-human primates.73
Lentiviral vector is yet another important delivery method reported for its functions in in vivo gene therapy applications and stable cell transduction. Examples of how lentiviral vectors can be pseudotyped with different envelope proteins from those native to the neurotropic viruses are discussed above. However, lentiviral vectors have been shown to integrate into the host genome causing adverse effects such as insertional mutagenesis. One such gene therapy study reported that HSCs transduced with the BB305 lentiviral vector for SCD adversely developed acute myeloid leukemia (AML).109 Another study utilizing a gamma (γ) retrovirus vector expressing interleukin-2 receptor γ-chain (γc) resulted in vector-induced leukemia through enhancer-mediated mutagenesis in four patients with X-linked severe combined immunodeficiency.110
Therefore, to mitigate the potential for insertional mutagenesis, integrase-deficient LVs (IDLVs) were developed by mutating the integrase protein and thereby minimizing the proviral integration.111,112,113 These IDLVs can be used where short-term expression is required and are currently under evaluation for gene-based therapies and vaccines.114
Although the modified virus serotypes are useful to target distinct cell populations, safety drawbacks have prevented certain viral vector-based approaches from being utilized for clinical applications, and, therefore, the search for alternative delivery strategies has been intensified.35
Virus-like Particles
Gene-editing agents can be delivered in different forms such as DNA or mRNA; however, ribonucleoproteins (RNPs) offer critical advantages. Virus-like particles (VLPs) are non-infectious assemblies of viral proteins that have been utilized for vaccine development115 and have recently been engineered to deliver gene-editing agents.35,116 As they are derived from existing viral scaffolds, they have combined advantages of both viral and non-viral delivery systems. VLPs can be made from different types of viruses, such as avian sarcoma leukosis virus,117 but most reported VLPs are based on retroviruses.118 The modular nature of the retroviruses has enabled them to further improve cellular tropism. A recent study showed that MLV VLP-Cas9-sgRNA RNPs mediated efficient genome editing in cell lines, primary cells, and in mice. Moreover, in vivo injection of VLPs using retro-orbital injections targeting 4-hydroxyphenylpyruvate dioxygenase (Hpd) resulted in successful editing in mouse hepatocytes of tyrosinemic mice.118
Engineered VLPs
Recently, Banskota and co-workers developed engineered VLPs (eVLPs) using an MMLV retrovirus scaffold to encapsulate Cas9 nuclease or base editor ribonucleoproteins (RNPs) and examined the gene-editing potential.33,35 They fused the base editor to the C terminus of the Friend murine leukemia virus (FMLV) gag polyprotein via a linker peptide. When cultured cells were transduced at a higher dose, V1-VLPs mediated a higher editing efficiency. However, single intravenous injections into mice resulted in a low editing of 2% in hepatocytes.33,35
To harness the eVLP platform as a delivery system, several structural arrangements were made. As such, improvements were made with a strong focus on cargo packaging, cargo release, nuclear localization, and component stoichiometry. Subsequently, to improve the cargo release upon eVLP maturation, protease-cleavable peptide linkers (TSTLLMENSS) were added to the second-generation engineered V2.4 eVLPs.35
To further improve the potential of eVLPs, nuclear export signals (NESs) and nuclear localization signals (NLSs) were added to the opposite sites of the cleavable linkers. This optimal design to the V3.4 eVLPs resulted in improved cytoplasmic cargo localization in eVLP producer cells while retaining nuclear cargo localization in the transduced target cells.35
Additional optimizations to balance the V4 eVLPs structural proteins and cargo component stoichiometry were shown to further increase the protein delivery potency into mammalian cells both in vitro and in vivo (see Figure 1). Collectively, these improvements resulted in 5- to 26-fold editing efficiencies in contrast to first-generation V1 VLPs. As such, V4 eVLPs mediated editing in various cell cultures and different tissues in mice. This is an improvement over V1 VLPs that was only explicitly determined in the liver (a 26-fold increase). Overall, V4 eVLPs offer a safe therapeutic delivery platform for both base editing35 and prime editing agents116 and has the opportunity to modulate cell tropism to improve the targeted delivery of therapeutics.33,35
Figure 1.

Systemic structural improvement strategies of eVLPs
Systemic structural improvement strategies of eVLPs to improve cargo release, cargo packaging, and localization and component stoichiometry. Four generations of BE-eVLPs were produced by optimizing a cleavable linker sequence to improve efficient cargo (denine base editor [ABE]) release upon particle maturation (v2 BE-eVLPs), then nuclear export signals (NESs) were added to enhance cytoplasmic cargo localization to produce v3 BE-eVLPs and component stoichiometry of gag-BE: gag-pro-pol was performed to generate V4 BE-eVLPs. Redesigned from Banskota et al.35 Created with BioRender.com.
Neurotropism of eVLPs
It has been suggested that eVLPs can be functionally pseudotyped with different viral envelope glycoproteins to alter their tropism. It has been suggested that these eVLPs can be pseudotyped with different viral envelope glycoproteins to alter their tropism. They pseudotyped V4 BE-eVLPs with neurotrophic FuG-B2 envelope glycoprotein, which had previously been used to pseudotype lentiviral vectors. This glycoprotein contains the extracellular and transmembrane domains of the RV-G envelope glycoprotein and the cytoplasmic domain of VSV-G. When the cultured mouse neuro-2a cells were transduced with FuG-B2-V4 BE-eVLPs, it resulted in efficient transduction and editing, thereby confirming altered tropism.35
Overall, these observations establish the potential to advance the neurotropism of eVLPs for brain drug delivery.
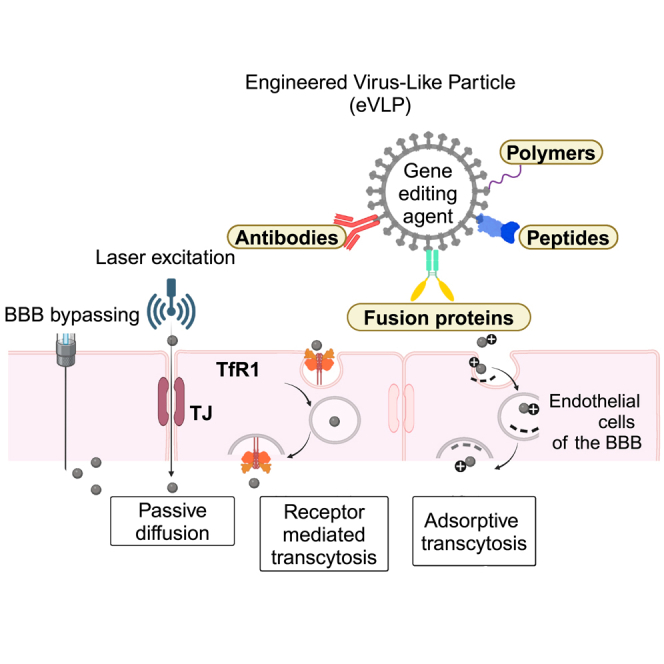
Advancing the Neurotropism of eVLPs to Target and Cross the BBB
While the modification of capsids/envelopes of AAVs and LVs can be adapted to modulate the neurotropism of eVLPs, this section focuses on alternative approaches that could be considered to develop eVLPs with enhanced neurotropism. The BBB acts as an anatomical and physiological barrier that maintains homeostasis in the CNS. The BBB strictly regulates the passage of nutrients and metabolites, thereby protecting the CNS from harmful toxins, pathogens, injury, and disease. It is composed of brain endothelial cells (BECs), a capillary basement membrane (BM), pericytes, and astrocytic foot processes. Tight junctions located between the endothelial cells inhibit the paracellular diffusion of polar molecules, macromolecules, and cells.119
When developing eVLPs to target brain drug delivery, it is important to consider the following parameters120:
-
(1)
Physiochemical modifications of the therapeutics to target transport across the BBB
-
(2)
Transient biophysical disruption to the BBB to allow transport across the BBB
-
(3)
Alternative routes to administer the drug
Physiochemical modifications of the therapeutics to target transport across the BBB.
There are two routes by which small molecules may cross the BBB:
-
(1)
Paracellular pathway
-
(2)
Transcellular pathway (transcytosis)
The paracellular pathway involves translocating the molecules between BECs and it appears to be exclusively a passive process that is regulated by the tight junctions between endothelial cells. Transcytosis translocates molecules actively or passively through the luminal side of the BEC membrane, traversing the BEC cytoplasm, and then passing across the basolateral side of the BEC into the interstitium of the brain. Passive diffusion of molecules across BECs is dependent on factors such as lipophilicity, electrical charge, and molecular weight. Macromolecules with high molecular weight and hydrophilicity, such as VLPs, are inclined to utilize two main active mechanisms121:
-
(1)
Receptor-mediated transcytosis (RMT) (see Table 2)
-
(2)
Adsorptive-mediated transcytosis (AMT)
Table 2.
Receptor-mediated delivery of therapeutics across the BBB
| BBB resident receptors | Receptor targeting strategy |
|---|---|
| IR | anti-IR antibodies: murine mAb, 83–14 mAbs to the HIR; Boado et al.122 |
| TfR | anti-TfR antibodies: murine mAb, OX26 to the rat TfR; Lee et al.123 rat mAbs, 8D3 and RI7-217, to the mouse TfR; Lee et al.123 murine mAb, 128.1 to the human TfR; Walus et al.124 fusion proteins: anti-rat TfR IgG3-Av to deliver biotinylated molecules across the BBB; Penichet et al.125 |
| LDLR | fusion protein: targeting the LDLR by fusion of 38 amino acids from the ApoB protein to a therapeutic protein; Masliah et al.126 fusogen: G protein of VSV-G; Strebinger et al.127 |
| LRP1 | angiopep-2: a 19-amino-acid-long oligopeptide that binds to the LRP1; Demeule et al.128 |
| Sodium-dependent glutathione transporters | glutathione: enable active transport through sodium-dependent glutathione transporters Reginald-Opara et al.129 |
| Nicotinic Ach nAChR receptor | fusion protein: targeting nAChR by fusion of 29 amino acids from the RV-G; Kumar et al.130 peptides: a synthetic peptide, KC2S derived from toxin b of the king cobra; Zhan et al.131 |
| Endothelial cell growth factor receptor-2 peptides | cell-penetrating peptide: CAYGRKKRRQRRR peptide sequence of TAT-B can induce internalization of molecules with high molecular weights in HBMECs; Cooper et al.132 |
| Luminal alpha(2,3)-sialoglycoprotein receptor | sdAbs FC5: llama single-domain antibodies; Muruganandam et al.133 |
| NT receptors NTSR1 and NTSR2 | 46.1 antibody: interact with NT receptors expressed on brain cells (NTSR1 and NTSR2); Georgieva et al.134 |
| Other | MiniAp-4: shorter peptidomimetic analog of apamin, renowned for higher BBB permeability via the receptor-mediated transcytosis mechanism; Oller-Salvia et al.135 |
Resident receptors on the BBB have been the target of numerous studies aiming to deliver drugs to the brain. This table provides a summary of strategies used to target these receptors, including the IR, TfR, LDLR, LRP1, sodium-dependent glutathione transporters, nicotinic Ach nAChR receptor, endothelial cell growth factor receptor-2 peptides, luminal alpha (2,3)-sialoglycoprotein receptor, and NT receptors NTSR1 and NTSR2.
In RMT, the delivery of macromolecules is based on the interactions between the ligands and specific receptors on BECs. The receptor and the ligand complex are then packaged into a vesicle through endocytosis and subsequently transported across the BEC cytoplasm. Once they reach the basolateral membrane vesicles, they are bound to it and eventually exocytosed, whereas AMT involves interactions between the cationic molecules and cell membrane binding sites, which leads to endocytosis and subsequent vesicular transcytosis.120
The following section focuses on receptor-mediated strategies that can potentially be utilized to advance the neurotropism of the eVLPs. To facilitate direct comparisons with eVLPs, we have also considered examples from VLPs, LNPs, and liposome modifications and how they have resulted in internalization via transferrin receptor (TfR) RMT.
RMT
Several receptors, including the insulin receptor (IR), TfR, melanotransferrin receptor (MTfR), LDL receptor-related protein 1 (LRP1), and the folate receptor (FR), are known to undergo RMT at the BBB. These endogenous receptors located on the luminal membrane of the brain capillary endothelium are crucial for the uptake of larger molecules such as insulin, leptin, and iron transferrin (Tf).136 Of these BBB receptors, TfR1 has been one of the primary targets investigated for RMT.
TfR
Transcytosis of Tf via TfR 1
TfR 1 (TfR1) is highly expressed in the BBB endothelium and has been extensively researched for its potential for brain drug delivery.136 However, to utilize the TfR1 for brain drug delivery, it is important to understand the process of TfR-mediated iron uptake at the BBB. In physiological conditions, plasma iron is bound to Tf (holo-Tf), which has shown a high binding affinity to the TfR. Both the ligand and the receptor complex are internalized via clathrin-mediated endocytosis. Acidification of the endosome (a pH drops to ∼5.5) results in conformational changes in Tf directing to iron release. Particularly holo-Tf dissociates and becomes apo-Tf and eventually the endocytic vesicle is recycled back to the plasma membrane in search of more free iron.137 Antibodies directed against the TfR are found to promote therapeutic delivery across the BBB and into the brain parenchyma.138 Further examples of developing therapeutics in the context of antibodies against TfR1, fusion proteins, and fusogens are discussed below.
Anti-TfR antibodies
Several strategies have been investigated to utilize antibodies as a carrier for the delivery of drugs across the BBB. Such an anti-TfR antibody system takes advantage of the high abundance of TfR1 on BBB and their ability to shuttle molecules across the BBB. Examples include species-selective rat (OX26)139,140 and mouse (Ri7, 8D3)141,142 antibodies binding to the TfR and transporting the therapeutics into the brain parenchyma. Bioengineering has enabled the design of distinct antibody types; immunoglobulin G (IgG)-like and non-IgG-like antibodies, whose binding properties, size, and half-life can be further modulated.143
Anti-human TfR antibody
TfR1-binding antibody therapeutics include Pabinafusp alfa, a TfR monoclonal antibody (mAb)-iduronate-2-sulfatase fused to an anti-human TfR antibody (JR 141) for neuropathic mucopolysaccharidosis II. Clinical studies involving JR 141 have been shown to ameliorate heparan sulfate (HS) levels in cerebrospinal fluid (CSF) neurodegeneration in patients with neuropathic mucopolysaccharidosis II (ClinicalTrials.gov: NCT04573023).144
Another study reported the re-engineering of erythropoietin (EPO) as a potential brain drug delivery target via the TfRs. Although the native form of EPO does not cross the BBB, research has indicated that the fusion protein of cTfRmAb-EPO can mediate its penetration into the brain. As such, fusion of a 166-amino-acid EPO to the carboxyl terminus of the heavy chain of a chimeric mAb against the mouse TfR resulted in elevated levels of brain uptake and pharmacologic increases in exogenous EPO in the mouse brain following the systemic injection.145 Moreover, an enhanced uptake of rsCD4 across the rodent and primate BBB has been shown after conjugation to anti-TfR antibodies 128.1 mAb.124
TfR targeting engineered antibody fragments
In recent years, smaller recombinant antibody fragments such as monovalent antibody fragments Fab and scFv have emerged as alternative targeting domains.146 For example, in 2020, Denali Therapeutics reported a BBB transport vehicle (TV) technology, which utilizes an engineered Fc polypeptide that binds to hTfR.147 The Denali team has shown significant therapeutic effects with the TV-based technology, successfully targeting lysosomal enzyme iduronate 2-sulfatase (IDS) DNL310 (ClinicalTrials.gov:NCT05371613) to treat Hunter syndrome (or MPS II)148 and N-sulfoglucosamine sulfohydrolase (SGSH) DNL126 to treat neuropathic and systemic forms of the Sanfilippo syndrome A (ClinicalTrials.gov:NCT06181136).
Another TV-based enzyme replacement therapy was developed to treat frontotemporal dementia (GRN-FTD) caused by the GRN mutations. In a murine model of GRN-FTD, Progranulin-conjugated protein transport vehicles (PGRN-PTV) restored the deficit levels of the lysosomal protein PGRN and was shown to rescue various Grn−/− pathologies, including microgliosis, lipofuscinosis, and neuronal damage.149
Brainshuttle (BS) by Roche is another antibody therapeutic developed for AD, which is currently being investigated in the phase I/II trial (ClinicalTrials.gov: 04639050). The BS-mAb module involves the delivery of an anti-amyloid beta (Aβ) antibody fused to a single-chain IgG antigen-binding fragment (fab).150
IR
IR targeting antibodies and engineered antibody fragments
Similar to the TfR targeting, the IR expressed at the BBB could be utilized as a target for RMT-based brain drug delivery. Due to the low serum half-life and reduced survival capacity in the blood, insulin is unlikely to complement brain drug delivery. Antibodies recognizing the IR have been developed, such as murine 83-14 mAb to the human IR (HIR), which is a promising brain drug targeting vector that could be used in humans.151
Another example includes modifications to the lysosomal enzyme palmitoyl protein thioesterase (PPT1) as IgG enzyme-fusion protein (AGT-194). The IgG domain of this fusion protein is an mAb targeting a BBB transporter receptor transporter such as the IR. The clinical application of AGT-194 has been reported to penetrate the BBB and deliver into the parenchyma, alleviating neuropathologies.152
Boado et al. reported on an enzyme replacement therapy (ERT) for mucopolysaccharidosis type I (MPS-I, Hurler’s syndrome). HIR mAb-IDU was developed by fusing human α-L-iduronidase (IDUA) to the carboxyl terminus of the heavy chain of a chimeric mAb to the human IR. A side-by-side comparison was performed to evaluate the BBB penetration of IDUA alone and the HIRmAb-IDUA fusion protein in vivo. The higher uptake of the fusion protein suggested HIRmAb-IDU as a potential ERT therapy.122
Functionalizing gene editor-eVLPs with antibodies designed against the endogenous BBB receptors could potentially open new prospects to deliver gene-based therapies into the brain. However, it is essential to understand the mechanisms and limitations involved in designing antibodies. As such, binding affinity and valency of the anti-TfR antibodies appear to be a key determinant of their BBB traversing characteristics.
Fundamental aspects relevant to eVLPs-antibodies
Once bound to the TfR from the blood, vectors are internalized through endocytosis, and the resulting vesicles are forwarded to one of three different pathways, such as being transcytosed into the CNS, trafficked into the lysosome, or recycled back to the apical cell surface.153 Therefore, to ensure the targeted delivery of therapeutics, it is important to recognize and correlate where improvements would be necessary. As such, evaluating the affinity of antibodies, their TfR binding mode, and antibody density will be useful to determine their transcytosis capacity.
-
(1)
Bi-specific antibody format
Re-engineering of the therapeutic antibody requires the engineering of a bi-specific antibody (BSA). One arm of the antibody is required to be bound to the receptor (i.e.,; TfR) while the other arm is utilized to attach to the therapeutic target. Compared to the unmodified, bi-specific antibodies have been shown to have a higher brain penetrant ability and to be more uniformly distributed within the brain.154,155
-
(2)
Affinity of antibodies
Antibodies’ affinity for the receptor has an important influence on their ability to dissociate from the receptors and be released into the brain. In 2011, Yu and co-workers reported that anti-TfR antibodies with high affinity to TfR remained associated with the BBB and prevented the antibody-drug conjugates (ADCs) from releasing into the BBB endothelium. Conversely, the reduction of affinity enhanced the RMT of the anti-TfR antibody across the BBB and mediated a broad parenchymal distribution 24 h after dosing. Therefore, to deliver an amount deemed therapeutically useful, it is important to consider that the antibodies are at their low-affinity mode to induce transcytosis and also to disassociate from TfR once it reaches the brain side.156
Using Tf-conjugated nanoparticles, Wiley et al. demonstrated a similar impact on nanoparticle delivery at the BBB. It was evident that nanoparticles (∼80 nm in diameter) with a higher avidity were restricted by BBB endothelium, while the high systemic dosing of low-avidity nanoparticles (∼80 nm in diameter) was shown achieve a higher brain uptake (avidity 0.89 nM optimal dissociation constant (KD) to TfR) and subsequently into the brain parenchyma.157
-
(3)
TfR binding mode
The TfR-binding mode of an antibody fragment to the TfR is another important aspect of the transcytosis capacity. To determine the monovalent versus bivalent engagement of the TfR, the ADCs were prepared to contain either one or two anti-TfR Fabs conjugated to an anti-β-amyloid mAb and tested in an in vitro BBB model. It was shown that the bivalent binding promoted their trafficking into the lysosomes and thus prevented the transcytosis while monovalents successfully entered the CNS. Therefore, the binding mode to the TfR is crucial for successful transport of antibodies across the BBB.158
Findings from a recent study involving gold nanoparticles (AuNPs) also indicated the importance of antibody valency contributing to the higher parenchymal accumulation of AuNPs of >70 nm.159
-
(4)
Antibody density
Johnsen et al. rationalized that the ligand density contributes to the efficient brain uptake of TfR-targeted AuNPs and chemotherapeutic drug-loaded liposomes. To make comparisons, both AuNPs and liposomes were functionalized with the same density of rat anti-mouse TfR antibody, RI7217 per surface area of the particle and potential for further increase in brain uptake was evaluated using ligand densities ranging from 0.15, 0.3, and 0.6 × 103 antibodies/μm2. Interestingly, AuNPs functionalized with the highest density were shown to achieve an enhanced accumulation in brain capillaries and were transported into the murine brain parenchyma.160
In contrast, achieving high affinity to RMT receptors may be optimal for certain carrier antibodies, such as those targeting IRs, to enhance therapeutic delivery mechanisms. Therapeutic delivery using anti-IR has been investigated for delivering enzymes and therapeutic antibodies.161 HIR mAb-IDUA fusion (valanafusp alpha) is one such therapeutic protein for Hurler syndrome caused by a deficiency of the enzyme IDUA. It combines an HIR mAb with IDUA. The HIR antibody maintains high-affinity binding to IR, resulting in increased IDUA enzymatic activity.162 Another pre-clinical in vivo study on developing mAbs targeting the Abeta amyloid peptide in AD showed that high-affinity HIR binding of HIR mAb-anti-amyloid resulted in a 10-fold increase in brain uptake compared to the anti-amyloid antibody controls.163
Antibodies or Engineered Antibody Fragments to Target Other BBB Receptors
LDL receptors
The LDL receptor (LDLR) expressed in brain capillary endothelial cells mediates the transport of lipoproteins and a diverse array of other ligands across the BBB via RMT. LDLR-targeted drug delivery can be achieved by conjugating a protein(s) with high affinity to the LDLR. One such example is the application of the apolipoprotein B (ApoB)-LDLR binding domain approach for the development of CNS-penetrating peptides for AD. This involved targeting the LDLR by the fusion of 38 amino acids from the ApoB protein to a therapeutic protein. When combined with the LDLR binding domain, a significant brain penetration at the BBB was observed.126
Therefore, to engineer cell trafficking properties, eVLPs can be potentially modified with cell-penetrating ligands such as N-methyl-D-aspartate (NMDA) receptor peptides, nicotinic acetylcholine receptor (nAChR) peptides, and trans-acting activation transduction (TAT) peptide.
nAChR-targeting peptides
A 29-amino-acid peptide derived from the RV-G fused to a sequence of nine arginine residues is suggested to drive an absorptive-mediated transport across the BBB. This peptide effectively targets the nicotinic acetylcholine receptor (nAChR) of which the expression is restricted to astrocytes and neurons.130
KC2S is a synthetic peptide derived from toxin b of the king cobra (Ophiophagus hannah) that has a high binding affinity with nAChRs. In cultured brain capillary endothelial cells, KC2S-linked micelles were taken up and delivered systemically, while the dye-loaded KC2S-micelles were observed to accumulate slowly in the brain, and drug-loaded KC2S-micelles provided a modest survival benefit in an orthotopic glioma model. In their study, Zhan et al. further showed that loop 2 of O. hannah toxin b binds with neuronal nAChRs and enhances intracranial drug delivery in mice.131
Growth factor receptor-2-targeting peptides
HIV-derived trans-activating regulatory protein (TAT) is a key activator of HIV-1 transcription. This 86- to 101-amino-acid protein has been shown to penetrate BECs in both in vitro and in vivo models. Cooper and co-workers showed that the CAYGRKKRRQRRR peptide sequence of TAT-B can induce internalization of molecules with high molecular weights in human brain microvascular endothelial cells (HBMECs).132 TAT is known to bind to several cell surface receptors, including vascular endothelial cell growth factor receptor-2 on BECs.164 In vivo studies resulted in BBB penetration of intravenously administered TAT peptide in the hippocampus, occipital cortex, and hypothalamus of the mouse brain.85 Therefore, peptide-mediated BBB penetration has important implications for future therapeutic developments.164
Luminal alpha (2,3)-sialoglycoprotein receptor-targeting antibody FC5
Single-domain antibodies (sdAbs) have been investigated for creating bi-functional proteins or bi-specific antibodies. Such characteristics offer several benefits compared to the current peptide and antibody-based methods. One such novel single-domain llama antibody, FC5, transmigrates across the BBB and is initiated by interacting with cell surface α (2,3)-sialoglycoprotein and then internalized via clathrin-coated endocytic vesicles. Both in vitro and in vivo studies have shown the ability of FC5 to cross the intact BBB and, hence, its potential for FC5-drug conjugates brain drug delivery strategies.133
NTSR1 and NTSR2 receptor targeting antibody 46.1
Georgieva et al. recently identified the antibody 46.1, which was generated following screening of a human single-chain Fv (ScFv) phage library. Following the intravenous administration, antibody 46.1 was observed to accumulate in the post-vascular brain. To further evaluate the brain delivery properties, the scFv-Fc form of the antibody was fused with a 13-amino-acid peptide neurotensin (NT) payload. Intravenously administered 46.1-scFv-Fc-LL-NTScFv 46.1 mediated the transport of NT across the BBB and accumulated in the median preoptic nucleus and striatum. Overall, the 46.1 antibody is capable of transporting drug cargo into the CNS.134
BBB traversing peptide shuttles and analogs
Peptides are another successful strategy to traverse the BBB and have been shown to have advantages over mAbs. Anand et al. developed a bi-functional nanocontainer Tat (FAM)P22-MVIIA using cell-penetrating HIV-Tat modified Salmonella typhimurium bacteriophage P22 capsids. When tested in vitro, these modified VLPs were efficiently taken up by the RBMVEC-BBB model via an endocytic pathway.165
To accommodate the brain delivery, Pang et al. designed a virus-like particle/RNAi nanocomplex modified with an apolipoprotein E (ApoE) peptide. The ApoE has an inherent affinity for the LDLR on the BBB. When tested in vivo, the modified virus-like particle/RNAi nano complexes (VLP/RNAi) (diameter of 30 nm) were observed to penetrate the BBB and ameliorate malignant brain tumors in a mouse model of glioblastomas (GBMs).166
Moreover, peptides derived from venoms have demonstrated the potential as BBB penetrating peptide shuttles. However, given the cytotoxic nature, venoms in their crude stage cannot be used for therapeutic applications. Such development of minimized versions of chlorotoxin (CTX) (MinCTX-3) and apamin (MiniAp-4) peptide toxins has shown the potential for brain-targeted drug delivery.167
MiniAp-4
Apamin is a bicyclic 18-mer peptide derived from Apis mellifera bee venom and has a higher BBB permeability. However, the therapeutic use of apamin is restricted due to higher toxicity, immunogenicity, and structural complexity. MiniAp-4 is a shorter peptidomimetic analog of apamin, renowned for higher BBB permeability via the receptor-mediated transcytosis mechanism. MiniAp-4 has been shown to carry covalently bound macromolecules and nanoparticles. As such, 12-nm AuNPs cargoes tethered with MiniAp-4 have been shown to actively cross the BBB and deliver cargo into the brain parenchyma of mice.135
MiniCTX-3
CTX is a 36-amino-acid peptide neurotoxin isolated from the venom of the giant yellow Israeli scorpion Leiurus quinquestriatus and a synthetic chlorotoxin (TM-601) is currently utilized to deliver anti-cancer therapeutics for glioma.168 MiniCTX-3 is a synthetic peptide based on CTX peptide that is protease resistant. It has been shown to transport both small molecules as well as AuNPs of 12-nm diameter in a human BBB cellular model.169
Given their higher BBB permeability and stability and BBB-traversing ability, these peptide shuttles could potentially be utilized to improve the neurotropism of eVLPs.
Fusogens
Tropism of viruses, VLPs, and engineered virus-like particle delivery systems can be modulated via pseudotyping strategies. This involves the fusion of a viral protein (or fusogen) on the surface of the vector particle. Pseudotyping allows the viral membrane to fuse with the target host cell membrane receptors, thereby releasing the cargo into the cytoplasm of the target cell. G protein of vesicular stomatitis virus (VSV-G) is one such popular glycoprotein that interacts with LDLRs and possibly other LDLR family members and hence the broad range of cell tropism. Several other viral envelope proteins or their analogs have been investigated for their different tropism characteristics and it is important to engineer them to optimize their performance.127
Recently, Strebinger et al. described a technique known as delivery to intended recipient cells through envelope design (DIRECTED) to program viral vector tropism via receptor-antibody interactions. It involves cell fusion components of both natural and engineered origin and includes various cell-targeting strategies. Once the cells are transduced, interactions between the glycoprotein and cognate cell receptor promote cell targeting and entry. Certain fusogen membrane fusions are pH dependent and independent of the engagement of a cellular receptor. Once they have gained entry, the endosomal process results in successful payload delivery. To redirect the vector tropism, they developed strategies such as using a chimeric antibody-binding protein or an SNAP-mediated covalent linkage (SNAP-tag) to recruit or immobilize antibodies on the viral envelope. Overall abundance of cell surface receptors, endocytosis of target receptor, endosomal formation, and pH-dependent conformational changes are important. Due to the programmable and compatibility nature of DIRECTED particles, it can be applied for enveloped delivery systems such as lentiviral particles, eVLPs, and Cre recombinase VLPs (CreVLPs) and has been anticipated to enhance the applicability across various cells including the brain.127
As has been reported for VLPs,118 we reason that the use of multiple glycoproteins to pseudotype eVLPs could overcome receptor saturation and therefore realize the combined benefits of different glycoprotein tropisms.
Surface Engineering Using Polymers
VLP surface can be engineered by introducing surface-accessible amino acids to provide conjugation sites for the synthesis of non-metallic nanomaterials via covalent bonding. Examples include but are not limited to VLP derivatives based on tobacco mosaic virus (TMV) carboxylic acid moiety of aspartate and glutamate reactions with amines, which have been functionalized with biotin, chromophores, and crown ethers while Y can conjugate with poly(ethylene glycol) (PEG).170 We envisage modifications such as polymer coating of eVLPs may prove valuable to improve their brain-targeted delivery.
PEG coating
The capacity to achieve brain penetration with larger vectors is important for the clinical translation of therapeutics. Given the success achieved so far with nanoparticles of similar diameter (100–150 nm), there is potential for improving eVLPs through PEGylation to enhance brain delivery. Compared to the non-polymer-coated nanoparticles, the polymer-coated nanoparticles show an improvement in transport across the BBB and accumulation in the brain. Nance et al. demonstrated that nanoparticles as large as 114 nm in diameter can diffuse through the human brain cortex tissue and in the rat brain but only when they are densely coated with PEG. The proof of concept was pre-clinically evaluated using paclitaxel-polymer-coated nanoparticles. This study also highlighted the importance of the surface density of PEG on a nanoparticle. When quantified using nuclear magnetic resonance (NMR)-based methods, it was estimated that a 100-nm nanoparticles should be PEGlyated with about nine PEG molecules (molecular weight = 5 kDa) per 100 nm2 of particle surface.171
Additionally, cationic bovine serum albumin (CBSA)-conjugated PEG-poly(d,l-lactide-co-glycolide) (PLGA) nanoparticles (CBSA-NPs) (diameter of ∼100 nm) indicated a higher biodistribution in the brain tissue. A dosage of 60 mg/kg CBSA-NP in mice caudal vein demonstrated a higher accumulation of CBSA-NP in the lateral ventricle, third ventricle, and periventricular region compared to the control.172 Moreover, PEGylated nanoparticles (Maleimide-PEG3500-PLA40000 and methoxyPEG2600-PLA40000) conjugated with OX26 mAbs also resulted in a robust gene expression throughout the CNS, including neurons, choroid plexus epithelium, and the brain microvasculature of the monkey brain.173
Magnetic targeting for BBB delivery
Magnetic targeting of nanoparticles is an alternative approach that enables them to reach deep parenchymal targets. This involves the application of an external magnetic field (EMF), thereby guiding the pathway to the nanoparticles across the BBB. The EMF can be exerted either by the magnets implanted intracranially or placed outside the skull of mice. Systemic administration of polystyrene nanospheres with trapped magnetic nanoparticles (MNPs) of ∼100 nm were shown to accumulate in the brain parenchyma. Although this technique could open new prospects for brain-targeted drug delivery, damage to the brain cells induced by the magnetic field is yet to be evaluated.174
Overall, current techniques on brain-targeted delivery provide insights into the further development of eVLPs. We have considered examples of VLPs, LNPs, and liposome modifications that can hamper the applications of the strategies into clinically useful eVLPs.
Transient biophysical disruption to the BBB to allow transport across the BBB
To improve drug delivery through the paracellular pathway, efforts have been made to alter the properties of the BBB. As such, Li and co-workers demonstrated a reversible modulation of the BBB by laser stimulation.175 Transcranial picosecond laser stimulation of AuNPs enhanced the BBB permeability and AuNPs were diffused through the tight junction complementing the paracellular pathway. AuNPs were modified by antibody BV11 to specifically target junctional adherence molecules (JAMs),176 which are highly expressed in brain vascular endothelial cells. They reported that the BBB permeability changes are completely reversible and do not lead to significant disruption in the spontaneous vasomotion or the structure of the neurovascular unit. Subcutaneous injections of AuNPs are functionalized by anti-JAM-A antibody BV11 on the scalp and then a surgical procedure is performed to peel the scalp to expose the skull to apply laser through the intact skull. It allowed the entry of immunoglobulins, viral vectors, and liposomes and suggested opening new avenues for brain drug delivery applications.175 Hence, we anticipate that a similar strategy could be considered to improve the brain target delivery of eVLPs.
Alternative Routes to Bypass the BBB
Barrier bypass delivery methods
Different techniques, such as intranasal (IN) delivery, intrathecal (IT) delivery, and interstitial delivery, are currently being used to bypass the BBB, and more information on these strategies has been reviewed in detail elsewhere.120
IN delivery
The IN cavity is a highly vascular absorptive surface located at a closer proximity to the brain. Intranasally administered drugs may pass through the olfactory epithelium and/or nasal epithelium and subsequently enter the systemic vasculature to be absorbed by lymphatics or continue to pass through a paracellular route into the neurons. The IN route has been widely used in veterinary clinical neurology applications, such as to deliver therapies for seizures.120
In addition, endocytosis of the drug by olfactory sensory neurons or trigeminal nerve endings within the nasal mucosa has also been shown to mediate axonal transport of the drug through the olfactory of trigeminal nerve pathways. Repeated transsynaptic processes within these neurons promote robust distribution of the drug into different brain regions. Moreover, intranasally administered drugs avoid the first-pass hepatic effect and hence sustain a higher bioavailability of the drug in the CNS. Furthermore, IN delivery offers practical advantages such as a non-invasive nature, not requiring any expertise to administer the drug, and reduced adverse effects.120
IT delivery
IT administration is a well-established drug delivery technique that has been widely used to treat both humans and animals.177 It involves direct administration of therapeutics into the CSF that flows through the thecal sac.178 Examples include the administration of analgesia or therapeutics to treat spasticity (via baclofen pump)179 and CNS neoplasms.178 IT delivery can also be achieved through intracisternal magna (ICM) or intracerebroventricular (ICV) injection, by which the CSF can be accessed. The ICV administration of drugs using an implanted device such as the Ommaya reservoir has been widely used to treat pediatric and adult patients to treat CNS disorders.178 For example, cerliponase alfa, or recombinant human tripeptidyl peptidase 1 (TPP1), is a US Food and Drug Administration (FDA)-approved ERT infused through an Ommaya reservoir to treat children with CLN2 Batten disease.180,181 Advantages of IT injections include consistent surgical procedures, reduced dosage, and smaller drug volumes. However, risks associated with the invasive technical approaches raise safety concerns. As with all surgical manipulations, IT injections are also associated with disadvantages such as the potential for backflow, damage to the brain tissue, and variable distribution beyond the injection site. Distribution of the therapeutics is majorly dependent on the diffusion rate of the drugs into the brain cells from the injection site and hence the drug concentration is decreased logarithmically.10 It can also be technically challenging in different species, given their differences in size, age, and anatomical limitations.177,178
Interstitial delivery
Although the IN and IT routes bypass the BBB, direct administration of small molecules and macromolecular compounds into the interstitium is considered the most direct delivery route. This technique often achieves higher therapeutic drug concentrations in the brain and poses a reduced systemic drug exposure.120
Direct interstitial delivery can be performed by a parenchymal bolus injection, implantation of biocompatible and biodegradable materials, and convection-enhanced delivery (CED). CED is another strategy known for homogeneous and efficient brain drug delivery. Classically, a small hydrostatic pressure gradient built from the syringe pump drives the distribution of the drug to bypass the BBB. CED has been shown to improve the biodistribution of AAVs in the brains of large animals and humans.182
For example, the AAV2-aromatic l-amino acid decarboxylase (AADC) enzyme, which catalyzes the conversion of levodopa (primary drug) to dopamine, was delivered via CED to the bilateral putamen to safely increase the AADC levels in Parkinson's disease patients (ClinicalTrials.gov: NCT03065192). More recently, Stahl and co-workers observed robust editing in the mouse striatum mediated by Cas9 RNPs and AAVs delivered via CED. They showed that bilateral CED injections of engineered cell-penetrant 4x-Cas9-2x NLS fusion protein improved the efficient delivery of Cas9 RNPs within the mouse brain. However, adverse effects such as fluid backflow, white matter edema, and formation of air bubbles have been reported relating to CED, highlighting the importance of improved brain drug delivery systems.183,184
However, similar to other invasive techniques, interstitial delivery has its drawbacks. As such, its need for repeated administrations, and specifically the bolus injections, may cause uncontrolled spatial distribution of drugs in the interstitium that could potentially cause reflux along the injection tract or expose non-target areas of the brain.120,185
Safety considerations with VLPs
The clinical success of the eVLPs entails their ability to bind and deliver the gene-editing agent to the target cells. Access to the target cells and endosomal escape within the cells is achieved by surface modifications or specific targeting moieties derived from different viral envelopes. However, higher concentrations of viral envelope proteins are cytotoxic and therefore using non-viral proteins to promote VLP cell entry and endosomal escape is encouraged.186 It has also been implicated that vectors pseudotyped with certain virus envelope glycoproteins could potentially be susceptible to inactivation by the human serum complement. For example, VSV-G-pseudotyped lentiviral vectors generated in human cell lines have been found to be inactivated either by restricted vector membrane incorporation or by functional blockage by human cell membrane complement control proteins such as CD-55 or CD-59. This observation highlights the importance of developing vectors with amphotropic envelopes for in vivo applications.187
Improved particle survival and circulation could be achieved by re-engineering the eVLP surface. Previously, Milani and co-workers produced lentiviral vector particles lacking polymorphic class-I major histocompatibility complexes that inhibited human primary T cell activation. Lentiviral vector surface modification was achieved by inactivating beta-2 microglobulin (B2M) genes in producer cells.188 Pre-existing immune responses to viral envelope proteins such as VSV-G may contribute to eliciting an immunogenic response within the host.189
Summary
There is an imperative need for therapeutic intervention to improve brain-targeted drug delivery, but efficient drug penetration is limited by the tightly regulated BBB. There have been extensive efforts to overcome brain drug delivery challenges and recent advances in gene-editing technology are useful in treatments and translational neuroscience.3,190 Due to the structural complexity of the brain and the BBB, gene therapy vectors are being injected directly into regions of disease pathology through IT, intraventricular, or intravascular routes.191 To circumvent the BBB, direct injection into brain parenchyma and intraventricular infusion has been utilized in most of the clinical trials. For example, intraventricular infusion of cerliponase alfa in children with CLN2 disease has proved a clinical success (ClinicalTrials.gov: NCT01907087 and NCT02485899).192 However, these invasive delivery techniques have an inherent risk of surgery-related side effects, and, therefore, alternative, less invasive delivery strategies are required.
Alternative delivery platforms such as VLPs have been long studied as a potential drug delivery modality. These VLP particles lack viral genetic material and are therefore considered safer than viral-mediated gene delivery. Recently developed engineered VLPs can be utilized to package and efficiently deliver therapeutic gene-editing proteins and has proved to have minimal off-target effects. They strategically developed the “bottlenecks” of conventional VLPs related to cargo packaging, localization, and cargo release. This fourth-generation eVLP has shown a significant 16 times more cargo packaging capacity as well as a 26-fold increased gene-editing activity within cells and animal models.35 eVLPs have a combination of key characteristics of both viral and non-viral delivery and they have been shown to efficiently deliver therapeutic RNPs, avoiding the risk of host genome integration and prolonged expression of therapeutic agents.33,35 In contrast to the recurring challenges associated with the viral and non-viral delivery platforms, eVLPs are likely to have an increased delivery potency in cultured cells as well as efficient delivery in animals.
However, in the process of engineering eVLPs for brain delivery, it is important to consider several factors, such as avoiding relying on naturally existing ligands, evaluating the antibody fusion format, affinity and effector function of the antibody, therapeutic efficacy, biodistribution, and the brain uptake efficiency. RMT of macromolecules is achieved through modifications to the vectors targeting specific transporter or transporters, such as the TfR, LDL receptor-related proteins (LRP-1 and LRP-2), or IR.193 Pseudotyping with viral glycoproteins is one such modification that has been successful for re-targeting retroviral Cas9-VLPs to different cell targets in humans or other non-rodent species.194 In eVLPs, the scaffold is derived from the MMLV, which is known to be pseudotyped with different envelope glycoproteins.35,195 Similarly, eVLPs can also be pseudotyped with different glycoproteins,35,116 and interest continues in exploring the use of other targeting moieties, such as antibodies or receptor ligands, conjugated to the surface of the eVLPs. The choice of cognate RMT system is an important aspect when designing a BBB delivery vector.193 The modifiable nature of the VLP surfaces facilitates the binding of peptides and proteins via chemical conjugation, genetic engineering, or a combination of both techniques.196 Typical constituents of the VLP capsid surfaces, such as K, cysteine, glutamic acid, and methionine, can be directed for surface modification with various biomolecules, including proteins, antigens, and small molecules.197
Moreover, it is also important to acknowledge the different species-specific RMT receptor expression profiles,70 and therefore pre-clinical translation of modified eVLPs should involve comprehensive evaluations across different animal models such as rodents and NHPs. Furthermore, the discovery of novel BBB-restricted RMT targets continues to be imperative. In this regard, recent advancements in RNA sequencing (RNA-seq) and proteomics techniques have proved instrumental in identifying novel CNS-specific RMT targets.161
In summary, eVLP-based gene-editing delivery systems offer a safe and promising alternative to existing techniques like viral and non-viral vectors. Moreover, the combined advantages of both viral and non-viral delivery systems and the programmable nature of the eVLP structure make eVLPs adaptable to improve the delivery of genetic cargo. As more CNS target drug delivery strategies are developed, we will gain new insights on how to further improve the precision of eVLPs for gene therapy delivery to the brain.
Acknowledgments
A.W.H. is supported by the Australian National Health and Medical Research Council Fellowship. We are grateful for funding from Retina Australia and the Batten Disease Support and Research Association Australia.
Author contributions
All authors contributed equally.
Declaration of interests
The authors declare no competing interests.
References
- 1.Qi M., Fan S., Wang Z., Yang X., Xie Z., Chen K., Zhang L., Lin T., Liu W., Lin X., et al. Identifying Common Genes, Cell Types and Brain Regions Between Diseases of the Nervous System. Front. Genet. 2019;10:1202. doi: 10.3389/fgene.2019.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayrignac X., Carra-Dallière C., Marelli C., Taïeb G., Labauge P. Adult-Onset Genetic Central Nervous System Disorders Masquerading as Acquired Neuroinflammatory Disorders: A Review. JAMA Neurol. 2022;79:1069–1078. doi: 10.1001/jamaneurol.2022.2141. [DOI] [PubMed] [Google Scholar]
- 3.FitzPatrick L., Bird A. Genetic therapies for neurological disorders. Hum. Genet. 2022;141:1085–1091. doi: 10.1007/s00439-021-02399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foo J.-N., Liu J.-J., Tan E.-K. Whole-genome and whole-exome sequencing in neurological diseases. Nat. Rev. Neurol. 2012;8:508–517. doi: 10.1038/nrneurol.2012.148. [DOI] [PubMed] [Google Scholar]
- 5.Roberts J.S., Patterson A.K., Uhlmann W.R. Genetic testing for neurodegenerative diseases: Ethical and health communication challenges. Neurobiol. Dis. 2020;141 doi: 10.1016/j.nbd.2020.104871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humbel M., Ramosaj M., Zimmer V., Regio S., Aeby L., Moser S., Boizot A., Sipion M., Rey M., Déglon N. Maximizing lentiviral vector gene transfer in the CNS. Gene Ther. 2021;28:75–88. doi: 10.1038/s41434-020-0172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cotman S.L., Karaa A., Staropoli J.F., Sims K.B. Neuronal ceroid lipofuscinosis: impact of recent genetic advances and expansion of the clinicopathologic spectrum. Curr. Neurol. Neurosci. Rep. 2013;13:366. doi: 10.1007/s11910-013-0366-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mole S., Williams R., Goebel H. OUP Oxford; 2011. The Neuronal Ceroid Lipofuscinoses (Batten Disease) [Google Scholar]
- 9.Feigin V.L., Vos T., Nichols E., Owolabi M.O., Carroll W.M., Dichgans M., Deuschl G., Parmar P., Brainin M., Murray C. The global burden of neurological disorders: translating evidence into policy. Lancet Neurol. 2020;19:255–265. doi: 10.1016/S1474-4422(19)30411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardridge W.M. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan J., Oyler-Castrillo P., Ravisankar P., Ward C.C., Levesque S., Jing Y., Simpson D., Zhao A., Li H., Yan W., et al. Improving prime editing with an endogenous small RNA-binding protein. Nature. 2024;628:639–647. doi: 10.1038/s41586-024-07259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doman J.L., Pandey S., Neugebauer M.E., An M., Davis J.R., Randolph P.B., McElroy A., Gao X.D., Raguram A., Richter M.F., et al. Phage-assisted evolution and protein engineering yield compact, efficient prime editors. Cell. 2023;186:3983–4002.e26. doi: 10.1016/j.cell.2023.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L., Zhang S., Xue N., Hong M., Zhang X., Zhang D., Yang J., Bai S., Huang Y., Meng H., et al. Engineering a precise adenine base editor with minimal bystander editing. Nat. Chem. Biol. 2023;19:101–110. doi: 10.1038/s41589-022-01163-8. [DOI] [PubMed] [Google Scholar]
- 17.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E., et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohammadian Gol T., Ureña-Bailén G., Hou Y., Sinn R., Antony J.S., Handgretinger R., Mezger M. CRISPR medicine for blood disorders: Progress and challenges in delivery. Front. Genome Ed. 2022;4 doi: 10.3389/fgeed.2022.1037290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breier D., Peer D. Genome editing in cancer: Challenges and potential opportunities. Bioact. Mater. 2023;21:394–402. doi: 10.1016/j.bioactmat.2022.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trevisan M., Masi G., Palù G. Genome editing technologies to treat rare liver diseases. Transl. Gastroenterol. Hepatol. 2020;5:23. doi: 10.21037/tgh.2019.10.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang T., Je G., Pacut P., Keyhanian K., Gao J., Ghasemi M. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells. 2022;11 doi: 10.3390/cells11132066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erkut E., Yokota T. CRISPR Therapeutics for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms23031832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day J.W., Finkel R.S., Chiriboga C.A., Connolly A.M., Crawford T.O., Darras B.T., Iannaccone S.T., Kuntz N.L., Peña L.D.M., Shieh P.B., et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:284–293. doi: 10.1016/S1474-4422(21)00001-6. [DOI] [PubMed] [Google Scholar]
- 24.Day J.W., Mendell J.R., Mercuri E., Finkel R.S., Strauss K.A., Kleyn A., Tauscher-Wisniewski S., Tukov F.F., Reyna S.P., Chand D.H. Clinical Trial and Postmarketing Safety of Onasemnogene Abeparvovec Therapy. Drug Saf. 2021;44:1109–1119. doi: 10.1007/s40264-021-01107-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alkanli S.S., Alkanli N., Ay A., Albeniz I. CRISPR/Cas9 Mediated Therapeutic Approach in Huntington’s Disease. Mol. Neurobiol. 2023;60:1486–1498. doi: 10.1007/s12035-022-03150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpenter J.C., Lignani G. Gene Editing and Modulation: the Holy Grail for the Genetic Epilepsies? Neurotherapeutics. 2021;18:1515–1523. doi: 10.1007/s13311-021-01081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahman M.U., Bilal M., Shah J.A., Kaushik A., Teissedre P.-L., Kujawska M. CRISPR-Cas9-Based Technology and Its Relevance to Gene Editing in Parkinson’s Disease. Pharmaceutics. 2022;14 doi: 10.3390/pharmaceutics14061252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhardwaj S., Kesari K.K., Rachamalla M., Mani S., Ashraf G.M., Jha S.K., Kumar P., Ambasta R.K., Dureja H., Devkota H.P., et al. CRISPR/Cas9 gene editing: New hope for Alzheimer’s disease therapeutics. J. Adv. Res. 2022;40:207–221. doi: 10.1016/j.jare.2021.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharaf-Eldin W. Technologies of gene editing and related clinical trials for the treatment of genetic and acquired diseases: a systematic review. Egypt. J. Med. Hum. Genet. 2024;25:30. [Google Scholar]
- 30.Levy J.M., Yeh W.-H., Pendse N., Davis J.R., Hennessey E., Butcher R., Koblan L.W., Comander J., Liu Q., Liu D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Musunuru K., Chadwick A.C., Mizoguchi T., Garcia S.P., DeNizio J.E., Reiss C.W., Wang K., Iyer S., Dutta C., Clendaniel V., et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434. doi: 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- 32.Rothgangl T., Dennis M.K., Lin P.J.C., Oka R., Witzigmann D., Villiger L., Qi W., Hruzova M., Kissling L., Lenggenhager D., et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021;39:949–957. doi: 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raguram A., Banskota S., Liu D.R. Therapeutic in vivo delivery of gene editing agents. Cell. 2022;185:2806–2827. doi: 10.1016/j.cell.2022.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Haasteren J., Li J., Scheideler O.J., Murthy N., Schaffer D.V. The delivery challenge: fulfilling the promise of therapeutic genome editing. Nat. Biotechnol. 2020;38:845–855. doi: 10.1038/s41587-020-0565-5. [DOI] [PubMed] [Google Scholar]
- 35.Banskota S., Raguram A., Suh S., Du S.W., Davis J.R., Choi E.H., Wang X., Nielsen S.C., Newby G.A., Randolph P.B., et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell. 2022;185:250–265.e16. doi: 10.1016/j.cell.2021.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taha E.A., Lee J., Hotta A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J. Contr. Release. 2022;342:345–361. doi: 10.1016/j.jconrel.2022.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Bulcha J.T., Wang Y., Ma H., Tai P.W.L., Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Targeted Ther. 2021;6:53. doi: 10.1038/s41392-021-00487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drouin L.M., Agbandje-McKenna M. Adeno-associated virus structural biology as a tool in vector development. Future Virol. 2013;8:1183–1199. doi: 10.2217/fvl.13.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maguire A.M., Simonelli F., Pierce E.A., Pugh E.N., Jr., Mingozzi F., Bennicelli J., Banfi S., Marshall K.A., Testa F., Surace E.M., et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naso M.F., Tomkowicz B., Perry W.L., 3rd, Strohl W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs. 2017;31:317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duan W., Guo M., Yi L., Liu Y., Li Z., Ma Y., Zhang G., Liu Y., Bu H., Song X., Li C. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2020;27:157–169. doi: 10.1038/s41434-019-0116-1. [DOI] [PubMed] [Google Scholar]
- 42.Wang L., Yang Y., Breton C., Bell P., Li M., Zhang J., Che Y., Saveliev A., He Z., White J., et al. A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aax5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis J.R., Wang X., Witte I.P., Huang T.P., Levy J.M., Raguram A., Banskota S., Seidah N.G., Musunuru K., Liu D.R. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nat. Biomed. Eng. 2022;6:1272–1283. doi: 10.1038/s41551-022-00911-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Z., Asokan A., Samulski R.J. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol. Ther. 2006;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Bartel M., Schaffer D., Büning H. Enhancing the Clinical Potential of AAV Vectors by Capsid Engineering to Evade Pre-Existing Immunity. Front. Microbiol. 2011;2:204. doi: 10.3389/fmicb.2011.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chai A.C., Cui M., Chemello F., Li H., Chen K., Tan W., Atmanli A., McAnally J.R., Zhang Y., Xu L., et al. Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice. Nat. Med. 2023;29:401–411. doi: 10.1038/s41591-022-02176-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M., et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lau C.-H., Suh Y. In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease. F1000Res. 2017;6:2153. doi: 10.12688/f1000research.11243.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Büning H., Srivastava A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019;12:248–265. doi: 10.1016/j.omtm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam A.K., Frabutt D., Li L., Xiao W. Chemical Modifications of the Capsid for Redirecting and Improving the Efficacy of Adeno-Associated Virus Vectors. Hum. Gene Ther. 2021;32:1433–1438. doi: 10.1089/hum.2021.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goswami R., Subramanian G., Silayeva L., Newkirk I., Doctor D., Chawla K., Chattopadhyay S., Chandra D., Chilukuri N., Betapudi V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019;9:297. doi: 10.3389/fonc.2019.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muik A., Reul J., Friedel T., Muth A., Hartmann K.P., Schneider I.C., Münch R.C., Buchholz C.J. Covalent coupling of high-affinity ligands to the surface of viral vector particles by protein trans-splicing mediates cell type-specific gene transfer. Biomaterials. 2017;144:84–94. doi: 10.1016/j.biomaterials.2017.07.032. [DOI] [PubMed] [Google Scholar]
- 53.Adachi K., Nakai H. A New Recombinant Adeno-Associated Virus (Aav)-Based Random Peptide Display Library System: Infection-Defective Aav1.9-3 As A Novel Detargeted Platform For Vector Evolution. Gene Ther. Regul. 2010;5:31–55. doi: 10.1142/S1568558610000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Markusic D.M., Herzog R.W., Aslanidi G.V., Hoffman B.E., Li B., Li M., Jayandharan G.R., Ling C., Zolotukhin I., Ma W., et al. High-efficiency transduction and correction of murine hemophilia B using AAV2 vectors devoid of multiple surface-exposed tyrosines. Mol. Ther. 2010;18:2048–2056. doi: 10.1038/mt.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aslanidi G.V., Rivers A.E., Ortiz L., Song L., Ling C., Govindasamy L., Van Vliet K., Tan M., Agbandje-McKenna M., Srivastava A. Optimization of the capsid of recombinant adeno-associated virus 2 (AAV2) vectors: the final threshold? PLoS One. 2013;8 doi: 10.1371/journal.pone.0059142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B., Ma W., Ling C., Van Vliet K., Huang L.-Y., Agbandje-McKenna M., Srivastava A., Aslanidi G.V. Site-Directed Mutagenesis of Surface-Exposed Lysine Residues Leads to Improved Transduction by AAV2, But Not AAV8, Vectors in Murine Hepatocytes In Vivo. Hum. Gene Ther. Methods. 2015;26:211–220. doi: 10.1089/hgtb.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mével M., Bouzelha M., Leray A., Pacouret S., Guilbaud M., Penaud-Budloo M., Alvarez-Dorta D., Dubreil L., Gouin S.G., Combal J.P., et al. Chemical modification of the adeno-associated virus capsid to improve gene delivery. Chem. Sci. 2019;11:1122–1131. doi: 10.1039/c9sc04189c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horowitz E.D., Weinberg M.S., Asokan A. Glycated AAV vectors: chemical redirection of viral tissue tropism. Bioconjugate Chem. 2011;22:529–532. doi: 10.1021/bc100477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pearce H.A., Qian H., Connell T.U., Huang D., Gottstein C., Donnelly P.S., Peter K., Gregorevic P., Hagemeyer C.E. Site-Specific Glycation and Chemo-enzymatic Antibody Sortagging for the Retargeting of rAAV6 to Inflamed Endothelium. Mol. Ther. Methods Clin. Dev. 2019;14:261–269. doi: 10.1016/j.omtm.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Challis R.C., Ravindra Kumar S., Chen X., Goertsen D., Coughlin G.M., Hori A.M., Chuapoco M.R., Otis T.S., Miles T.F., Gradinaru V. Adeno-Associated Virus Toolkit to Target Diverse Brain Cells. Annu. Rev. Neurosci. 2022;45:447–469. doi: 10.1146/annurev-neuro-111020-100834. [DOI] [PubMed] [Google Scholar]
- 61.Yang B., Li S., Wang H., Guo Y., Gessler D.J., Cao C., Su Q., Kramer J., Zhong L., Ahmed S.S., et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. 2014;22:1299–1309. doi: 10.1038/mt.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H., Yang B., Mu X., Ahmed S.S., Su Q., He R., Wang H., Mueller C., Sena-Esteves M., Brown R., et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol. Ther. 2011;19:1440–1448. doi: 10.1038/mt.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murlidharan G., Sakamoto K., Rao L., Corriher T., Wang D., Gao G., Sullivan P., Asokan A. CNS-restricted Transduction and CRISPR/Cas9-mediated Gene Deletion with an Engineered AAV Vector. Mol. Ther. Nucleic Acids. 2016;5:e338. doi: 10.1038/mtna.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tervo D.G.R., Hwang B.-Y., Viswanathan S., Gaj T., Lavzin M., Ritola K.D., Lindo S., Michael S., Kuleshova E., Ojala D., et al. A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron. 2016;92:372–382. doi: 10.1016/j.neuron.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grimm D., Lee J.S., Wang L., Desai T., Akache B., Storm T.A., Kay M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Solis C.A., Ho A., Holehonnur R., Ploski J.E. The Development of a Viral Mediated CRISPR/Cas9 System with Doxycycline Dependent gRNA Expression for Inducible In vitro and In vivo Genome Editing. Front. Mol. Neurosci. 2016;9:70. doi: 10.3389/fnmol.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.-L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.-L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang Q., Chan K.Y., Tobey I.G., Chan Y.A., Poterba T., Boutros C.L., Balazs A.B., Daneman R., Bloom J.M., Seed C., Deverman B.E. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS One. 2019;14 doi: 10.1371/journal.pone.0225206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matsuzaki Y., Konno A., Mochizuki R., Shinohara Y., Nitta K., Okada Y., Hirai H. Intravenous administration of the adeno-associated virus-PHP.B capsid fails to upregulate transduction efficiency in the marmoset brain. Neurosci. Lett. 2018;665:182–188. doi: 10.1016/j.neulet.2017.11.049. [DOI] [PubMed] [Google Scholar]
- 72.Liguore W.A., Domire J.S., Button D., Wang Y., Dufour B.D., Srinivasan S., McBride J.L. AAV-PHP.B Administration Results in a Differential Pattern of CNS Biodistribution in Non-human Primates Compared with Mice. Mol. Ther. 2019;27:2018–2037. doi: 10.1016/j.ymthe.2019.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hordeaux J., Wang Q., Katz N., Buza E.L., Bell P., Wilson J.M. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. 2018;26:664–668. doi: 10.1016/j.ymthe.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang Q., Chan K.Y., Lou S., Keyes C., Wu J., Botticello-Romero N.R., Zheng Q., Johnston J., Mills A., Brauer P.P., et al. An AAV capsid reprogrammed to bind human Transferrin Receptor mediates brain-wide gene delivery. bioRxiv. 2023 doi: 10.1101/2023.12.20.572615. Preprint at. [DOI] [PubMed] [Google Scholar]
- 75.Beasley B.E., Hu W.-S. cis-Acting elements important for retroviral RNA packaging specificity. J. Virol. 2002;76:4950–4960. doi: 10.1128/JVI.76.10.4950-4960.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blessing D., Déglon N. Adeno-associated virus and lentivirus vectors: a refined toolkit for the central nervous system. Curr. Opin. Virol. 2016;21:61–66. doi: 10.1016/j.coviro.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 77.Kabadi A.M., Ousterout D.G., Hilton I.B., Gersbach C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L., et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 79.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Bougnères P., Schmidt M., Kalle C.V., Fischer A., Cavazzana-Calvo M., Aubourg P. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012;507:187–198. doi: 10.1016/B978-0-12-386509-0.00010-7. [DOI] [PubMed] [Google Scholar]
- 80.Cronin J., Zhang X.-Y., Reiser J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 2005;5:387–398. doi: 10.2174/1566523054546224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mazarakis N.D., Azzouz M., Rohll J.B., Ellard F.M., Wilkes F.J., Olsen A.L., Carter E.E., Barber R.D., Baban D.F., Kingsman S.M., et al. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum. Mol. Genet. 2001;10:2109–2121. doi: 10.1093/hmg/10.19.2109. [DOI] [PubMed] [Google Scholar]
- 82.Wong L.-F., Azzouz M., Walmsley L.E., Askham Z., Wilkes F.J., Mitrophanous K.A., Kingsman S.M., Mazarakis N.D. Transduction patterns of pseudotyped lentiviral vectors in the nervous system. Mol. Ther. 2004;9:101–111. doi: 10.1016/j.ymthe.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 83.Desmaris N., Bosch A., Salaün C., Petit C., Prévost M.C., Tordo N., Perrin P., Schwartz O., de Rocquigny H., Heard J.M. Production and neurotropism of lentivirus vectors pseudotyped with lyssavirus envelope glycoproteins. Mol. Ther. 2001;4:149–156. doi: 10.1006/mthe.2001.0431. [DOI] [PubMed] [Google Scholar]
- 84.Eleftheriadou I., Dieringer M., Poh X.Y., Sanchez-Garrido J., Gao Y., Sgourou A., Simmons L.E., Mazarakis N.D. Selective transduction of astrocytic and neuronal CNS subpopulations by lentiviral vectors pseudotyped with Chikungunya virus envelope. Biomaterials. 2017;123:1–14. doi: 10.1016/j.biomaterials.2017.01.023. [DOI] [PubMed] [Google Scholar]
- 85.Kato S., Inoue K.-I., Kobayashi K., Yasoshima Y., Miyachi S., Inoue S., Hanawa H., Shimada T., Takada M., Kobayashi K. Efficient gene transfer via retrograde transport in rodent and primate brains using a human immunodeficiency virus type 1-based vector pseudotyped with rabies virus glycoprotein. Hum. Gene Ther. 2007;18:1141–1151. doi: 10.1089/hum.2007.082. [DOI] [PubMed] [Google Scholar]
- 86.Watson D.J., Kobinger G.P., Passini M.A., Wilson J.M., Wolfe J.H. Targeted transduction patterns in the mouse brain by lentivirus vectors pseudotyped with VSV, Ebola, Mokola, LCMV, or MuLV envelope proteins. Mol. Ther. 2002;5:528–537. doi: 10.1006/mthe.2002.0584. [DOI] [PubMed] [Google Scholar]
- 87.Miletic H., Fischer Y.H., Neumann H., Hans V., Stenzel W., Giroglou T., Hermann M., Deckert M., Von Laer D. Selective transduction of malignant glioma by lentiviral vectors pseudotyped with lymphocytic choriomeningitis virus glycoproteins. Hum. Gene Ther. 2004;15:1091–1100. doi: 10.1089/hum.2004.15.1091. [DOI] [PubMed] [Google Scholar]
- 88.Stein C.S., Martins I., Davidson B.L. The lymphocytic choriomeningitis virus envelope glycoprotein targets lentiviral gene transfer vector to neural progenitors in the murine brain. Mol. Ther. 2005;11:382–389. doi: 10.1016/j.ymthe.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 89.Trabalza A., Georgiadis C., Eleftheriadou I., Hislop J.N., Ellison S.M., Karavassilis M.E., Mazarakis N.D. Venezuelan equine encephalitis virus glycoprotein pseudotyping confers neurotropism to lentiviral vectors. Gene Ther. 2013;20:723–732. doi: 10.1038/gt.2012.85. [DOI] [PubMed] [Google Scholar]
- 90.Cannon J.R., Sew T., Montero L., Burton E.A., Greenamyre J.T. Pseudotype-dependent lentiviral transduction of astrocytes or neurons in the rat substantia nigra. Exp. Neurol. 2011;228:41–52. doi: 10.1016/j.expneurol.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Colin A., Faideau M., Dufour N., Auregan G., Hassig R., Andrieu T., Brouillet E., Hantraye P., Bonvento G., Déglon N. Engineered lentiviral vector targeting astrocytes in vivo. Glia. 2009;57:667–679. doi: 10.1002/glia.20795. [DOI] [PubMed] [Google Scholar]
- 92.Kang Y., Stein C.S., Heth J.A., Sinn P.L., Penisten A.K., Staber P.D., Ratliff K.L., Shen H., Barker C.K., Martins I., et al. In vivo gene transfer using a nonprimate lentiviral vector pseudotyped with Ross River Virus glycoproteins. J. Virol. 2002;76:9378–9388. doi: 10.1128/JVI.76.18.9378-9388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pertusa M., García-Matas S., Mammeri H., Adell A., Rodrigo T., Mallet J., Cristòfol R., Sarkis C., Sanfeliu C. Expression of GDNF transgene in astrocytes improves cognitive deficits in aged rats. Neurobiol. Aging. 2008;29:1366–1379. doi: 10.1016/j.neurobiolaging.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 94.Liehl B., Hlavaty J., Moldzio R., Tonar Z., Unger H., Salmons B., Günzburg W.H., Renner M. Simian immunodeficiency virus vector pseudotypes differ in transduction efficiency and target cell specificity in brain. Gene Ther. 2007;14:1330–1343. doi: 10.1038/sj.gt.3302988. [DOI] [PubMed] [Google Scholar]
- 95.Kato R., Wolfe D., Coyle C.H., Wechuck J.B., Tyagi P., Tsukamoto T., Nelson J.B., Glorioso J.C., Chancellor M.B., Yoshimura N. Herpes simplex virus vector-mediated delivery of neurturin rescues erectile dysfunction of cavernous nerve injury. Gene Ther. 2009;16:26–33. doi: 10.1038/gt.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hirano M., Kato S., Kobayashi K., Okada T., Yaginuma H., Kobayashi K. Highly efficient retrograde gene transfer into motor neurons by a lentiviral vector pseudotyped with fusion glycoprotein. PLoS One. 2013;8 doi: 10.1371/journal.pone.0075896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Finkelshtein D., Werman A., Novick D., Barak S., Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ryan D.E., Taussig D., Steinfeld I., Phadnis S.M., Lunstad B.D., Singh M., Vuong X., Okochi K.D., McCaffrey R., Olesiak M., et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2018;46:792–803. doi: 10.1093/nar/gkx1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guo C., Ma X., Gao F., Guo Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023;11 doi: 10.3389/fbioe.2023.1143157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang D., Zhang F., Gao G. CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell. 2020;181:136–150. doi: 10.1016/j.cell.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hordeaux J., Buza E.L., Dyer C., Goode T., Mitchell T.W., Richman L., Denton N., Hinderer C., Katz N., Schmid R., et al. Adeno-Associated Virus-Induced Dorsal Root Ganglion Pathology. Hum. Gene Ther. 2020;31:808–818. doi: 10.1089/hum.2020.167. [DOI] [PubMed] [Google Scholar]
- 103.Agarwal S. High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020;38:910. doi: 10.1038/s41587-020-0642-9. [DOI] [PubMed] [Google Scholar]
- 104.Li H., Lasaro M.O., Jia B., Lin S.W., Haut L.H., High K.A., Ertl H.C.J. Capsid-specific T-cell responses to natural infections with adeno-associated viruses in humans differ from those of nonhuman primates. Mol. Ther. 2011;19:2021–2030. doi: 10.1038/mt.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Verdera H.C., Kuranda K., Mingozzi F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020;28:723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Donsante A., Vogler C., Muzyczka N., Crawford J.M., Barker J., Flotte T., Campbell-Thompson M., Daly T., Sands M.S. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors [Review of Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors] Gene Ther. 2001;8:1343–1346. doi: 10.1038/sj.gt.3301541. [DOI] [PubMed] [Google Scholar]
- 107.Nakai H., Yant S.R., Storm T.A., Fuess S., Meuse L., Kay M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.-A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Philippidis A. After Analysis, Bluebird Bio Says Vector “Very Unlikely” Cause of Acute Myeloid Leukemia. Hum. Gene Ther. 2021;32:332–334. doi: 10.1089/hum.2021.29159.bfs. [DOI] [PubMed] [Google Scholar]
- 110.Hacein-Bey-Abina S., Pai S.-Y., Gaspar H.B., Armant M., Berry C.C., Blanche S., Bleesing J., Blondeau J., de Boer H., Buckland K.F., et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2014;371:1407–1417. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Banasik M.B., McCray P.B., Jr. Integrase-defective lentiviral vectors: progress and applications. Gene Ther. 2010;17:150–157. doi: 10.1038/gt.2009.135. [DOI] [PubMed] [Google Scholar]
- 112.Yew C.-H.T., Gurumoorthy N., Nordin F., Tye G.J., Wan Kamarul Zaman W.S., Tan J.J., Ng M.H. Integrase deficient lentiviral vector: prospects for safe clinical applications. PeerJ. 2022;10 doi: 10.7717/peerj.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kymäläinen H., Appelt J.U., Giordano F.A., Davies A.F., Ogilvie C.M., Ahmed S.G., Laufs S., Schmidt M., Bode J., Yáñez-Muñoz R.J., Dickson G. Long-term episomal transgene expression from mitotically stable integration-deficient lentiviral vectors. Hum. Gene Ther. 2014;25:428–442. doi: 10.1089/hum.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luis A. The Old and the New: Prospects for Non-Integrating Lentiviral Vector Technology. Viruses. 2020;12 doi: 10.3390/v12101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nooraei S., Bahrulolum H., Hoseini Z.S., Katalani C., Hajizade A., Easton A.J., Ahmadian G. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 2021;19:59. doi: 10.1186/s12951-021-00806-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.An M., Raguram A., Du S.W., Banskota S., Davis J.R., Newby G.A., Chen P.Z., Palczewski K., Liu D.R. Engineered virus-like particles for transient delivery of prime editor ribonucleoprotein complexes in vivo. Nat. Biotechnol. 2024 doi: 10.1038/s41587-023-02078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaczmarczyk S.J., Sitaraman K., Young H.A., Hughes S.H., Chatterjee D.K. Protein delivery using engineered virus-like particles. Proc. Natl. Acad. Sci. USA. 2011;108:16998–17003. doi: 10.1073/pnas.1101874108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mangeot P.E., Risson V., Fusil F., Marnef A., Laurent E., Blin J., Mournetas V., Massouridès E., Sohier T.J.M., Corbin A., et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019;10:45. doi: 10.1038/s41467-018-07845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fu H., McCarty D.M. Crossing the blood-brain-barrier with viral vectors. Curr. Opin. Virol. 2016;21:87–92. doi: 10.1016/j.coviro.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 120.Partridge B., Eardley A., Morales B.E., Campelo S.N., Lorenzo M.F., Mehta J.N., Kani Y., Mora J.K.G., Campbell E.-O.Y., Arena C.B., et al. Advancements in drug delivery methods for the treatment of brain disease. Front. Vet. Sci. 2022;9 doi: 10.3389/fvets.2022.1039745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen Y.-L., Bao C.-J., Duan J.-L., Xie Y., Lu W.-L. Overcoming biological barriers by virus-like drug particles for drug delivery. Adv. Drug Deliv. Rev. 2023;203 doi: 10.1016/j.addr.2023.115134. [DOI] [PubMed] [Google Scholar]
- 122.Boado R.J., Zhang Y., Zhang Y., Xia C.-F., Wang Y., Pardridge W.M. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol. Bioeng. 2008;99:475–484. doi: 10.1002/bit.21602. [DOI] [PubMed] [Google Scholar]
- 123.Lee H.J., Engelhardt B., Lesley J., Bickel U., Pardridge W.M. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J. Pharmacol. Exp. Therapeut. 2000;292:1048–1052. [PubMed] [Google Scholar]
- 124.Walus L.R., Pardridge W.M., Starzyk R.M., Friden P.M. Enhanced uptake of rsCD4 across the rodent and primate blood-brain barrier after conjugation to anti-transferrin receptor antibodies. J. Pharmacol. Exp. Therapeut. 1996;277:1067–1075. [PubMed] [Google Scholar]
- 125.Penichet M.L., Kang Y.S., Pardridge W.M., Morrison S.L., Shin S.U. An antibody-avidin fusion protein specific for the transferrin receptor serves as a delivery vehicle for effective brain targeting: initial applications in anti-HIV antisense drug delivery to the brain. J. Immunol. 1999;163:4421–4426. [PubMed] [Google Scholar]
- 126.Masliah E., Spencer B. Applications of ApoB LDLR-Binding Domain Approach for the Development of CNS-Penetrating Peptides for Alzheimer’s Disease. Methods Mol. Biol. 2015;1324:331–337. doi: 10.1007/978-1-4939-2806-4_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Strebinger D., Frangieh C.J., Friedrich M.J., Faure G., Macrae R.K., Zhang F. Cell type-specific delivery by modular envelope design. Nat. Commun. 2023;14:5141. doi: 10.1038/s41467-023-40788-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Demeule M., Régina A., Ché C., Poirier J., Nguyen T., Gabathuler R., Castaigne J.-P., Béliveau R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharmacol. Exp. Therapeut. 2008;324:1064–1072. doi: 10.1124/jpet.107.131318. [DOI] [PubMed] [Google Scholar]
- 129.Reginald-Opara J.N., Tang M., Svirskis D., Chamley L., Wu Z. The role of glutathione conjugation on the transcellular transport process of PEGylated liposomes across the blood brain barrier. Int. J. Pharm. 2022;626 doi: 10.1016/j.ijpharm.2022.122152. [DOI] [PubMed] [Google Scholar]
- 130.Kumar P., Wu H., McBride J.L., Jung K.-E., Kim M.H., Davidson B.L., Lee S.K., Shankar P., Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 131.Zhan C., Yan Z., Xie C., Lu W. Loop 2 of Ophiophagus hannah toxin b binds with neuronal nicotinic acetylcholine receptors and enhances intracranial drug delivery. Mol. Pharm. 2010;7:1940–1947. doi: 10.1021/mp100238j. [DOI] [PubMed] [Google Scholar]
- 132.Cooper I., Sasson K., Teichberg V.I., Schnaider-Beeri M., Fridkin M., Shechter Y. Peptide derived from HIV-1 TAT protein destabilizes a monolayer of endothelial cells in an in vitro model of the blood-brain barrier and allows permeation of high molecular weight proteins. J. Biol. Chem. 2012;287:44676–44683. doi: 10.1074/jbc.M112.395384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Muruganandam A., Tanha J., Narang S., Stanimirovic D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB (Fed. Am. Soc. Exp. Biol.) J. 2002;16:240–242. doi: 10.1096/fj.01-0343fje. [DOI] [PubMed] [Google Scholar]
- 134.Georgieva J.V., Katt M., Ye Z., Umlauf B.J., Wenthur C.J., Shusta E.V. The 46.1 Antibody Mediates Neurotensin Uptake into the CNS and the Effects Depend on the Route of Intravenous Administration. Pharmaceutics. 2022;14 doi: 10.3390/pharmaceutics14081706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Oller-Salvia B., Sánchez-Navarro M., Ciudad S., Guiu M., Arranz-Gibert P., Garcia C., Gomis R.R., Cecchelli R., García J., Giralt E., Teixidó M. MiniAp-4: A Venom-Inspired Peptidomimetic for Brain Delivery. Angew. Chem. 2016;55:572–575. doi: 10.1002/anie.201508445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abbott N.J., Patabendige A.A.K., Dolman D.E.M., Yusof S.R., Begley D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 137.Widera A., Norouziyan F., Shen W.-C. Mechanisms of TfR-mediated transcytosis and sorting in epithelial cells and applications toward drug delivery. Adv. Drug Deliv. Rev. 2003;55:1439–1466. doi: 10.1016/j.addr.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 138.Johnsen K.B., Burkhart A., Thomsen L.B., Andresen T.L., Moos T. Targeting the transferrin receptor for brain drug delivery. Prog. Neurobiol. 2019;181 doi: 10.1016/j.pneurobio.2019.101665. [DOI] [PubMed] [Google Scholar]
- 139.Moos T., Morgan E.H. Restricted transport of anti-transferrin receptor antibody (OX26) through the blood-brain barrier in the rat. J. Neurochem. 2001;79:119–129. doi: 10.1046/j.1471-4159.2001.00541.x. [DOI] [PubMed] [Google Scholar]
- 140.Friden P.M., Walus L.R., Musso G.F., Taylor M.A., Malfroy B., Starzyk R.M. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc. Natl. Acad. Sci. USA. 1991;88:4771–4775. doi: 10.1073/pnas.88.11.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Manich G., Cabezón I., del Valle J., Duran-Vilaregut J., Camins A., Pallàs M., Pelegrí C., Vilaplana J. Study of the transcytosis of an anti-transferrin receptor antibody with a Fab’ cargo across the blood-brain barrier in mice. Eur. J. Pharmaceut. Sci. 2013;49:556–564. doi: 10.1016/j.ejps.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 142.Cabezón I., Manich G., Martín-Venegas R., Camins A., Pelegrí C., Vilaplana J. Trafficking of Gold Nanoparticles Coated with the 8D3 Anti-Transferrin Receptor Antibody at the Mouse Blood-Brain Barrier. Mol. Pharm. 2015;12:4137–4145. doi: 10.1021/acs.molpharmaceut.5b00597. [DOI] [PubMed] [Google Scholar]
- 143.Ma J., Mo Y., Tang M., Shen J., Qi Y., Zhao W., Huang Y., Xu Y., Qian C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.626616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Okuyama T., Eto Y., Sakai N., Minami K., Yamamoto T., Sonoda H., Yamaoka M., Tachibana K., Hirato T., Sato Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019;27:456–464. doi: 10.1016/j.ymthe.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhou Q.-H., Boado R.J., Lu J.Z., Hui E.K.-W., Pardridge W.M. Re-engineering erythropoietin as an IgG fusion protein that penetrates the blood-brain barrier in the mouse. Mol. Pharm. 2010;7:2148–2155. doi: 10.1021/mp1001763. [DOI] [PubMed] [Google Scholar]
- 146.Holliger P., Hudson P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005;23:1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 147.Kariolis M.S., Wells R.C., Getz J.A., Kwan W., Mahon C.S., Tong R., Kim D.J., Srivastava A., Bedard C., Henne K.R., et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aay1359. [DOI] [PubMed] [Google Scholar]
- 148.Ullman J.C., Arguello A., Getz J.A., Bhalla A., Mahon C.S., Wang J., Giese T., Bedard C., Kim D.J., Blumenfeld J.R., et al. Brain delivery and activity of a lysosomal enzyme using a blood-brain barrier transport vehicle in mice. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aay1163. [DOI] [PubMed] [Google Scholar]
- 149.Logan T., Simon M.J., Rana A., Cherf G.M., Srivastava A., Davis S.S., Low R.L.Y., Chiu C.-L., Fang M., Huang F., et al. Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell. 2021;184:4651–4668.e25. doi: 10.1016/j.cell.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Weber F., Bohrmann B., Niewoehner J., Fischer J.A.A., Rueger P., Tiefenthaler G., Moelleken J., Bujotzek A., Brady K., Singer T., et al. Brain Shuttle Antibody for Alzheimer’s Disease with Attenuated Peripheral Effector Function due to an Inverted Binding Mode. Cell Rep. 2018;22:149–162. doi: 10.1016/j.celrep.2017.12.019. [DOI] [PubMed] [Google Scholar]
- 151.Coloma M.J., Lee H.J., Kurihara A., Landaw E.M., Boado R.J., Morrison S.L., Pardridge W.M. Transport across the primate blood-brain barrier of a genetically engineered chimeric monoclonal antibody to the human insulin receptor. Pharm. Res. (N. Y.) 2000;17:266–274. doi: 10.1023/a:1007592720793. [DOI] [PubMed] [Google Scholar]
- 152.Hahn A., Sato Y., Ikeda T., Sonoda H., Schmidt M., Pfrimmer C., Boado R.J., Pardridge W.M. Treatment of CLN1 disease with a blood-brain barrier penetrating lysosomal enzyme. Mol. Genet. Metab. Rep. 2022;33 doi: 10.1016/j.ymgmr.2022.100930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lajoie J.M., Shusta E.V. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annu. Rev. Pharmacol. Toxicol. 2015;55:613–631. doi: 10.1146/annurev-pharmtox-010814-124852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Faresjö R., Bonvicini G., Fang X.T., Aguilar X., Sehlin D., Syvänen S. Brain pharmacokinetics of two BBB penetrating bispecific antibodies of different size. Fluids Barriers CNS. 2021;18:26. doi: 10.1186/s12987-021-00257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Syvänen S., Hultqvist G., Gustavsson T., Gumucio A., Laudon H., Söderberg L., Ingelsson M., Lannfelt L., Sehlin D. Efficient clearance of Aβ protofibrils in AβPP-transgenic mice treated with a brain-penetrating bifunctional antibody. Alzheimer's Res. Ther. 2018;10:49. doi: 10.1186/s13195-018-0377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yu Y.J., Zhang Y., Kenrick M., Hoyte K., Luk W., Lu Y., Atwal J., Elliott J.M., Prabhu S., Watts R.J., Dennis M.S. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002230. [DOI] [PubMed] [Google Scholar]
- 157.Wiley D.T., Webster P., Gale A., Davis M.E. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc. Natl. Acad. Sci. USA. 2013;110:8662–8667. doi: 10.1073/pnas.1307152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Niewoehner J., Bohrmann B., Collin L., Urich E., Sade H., Maier P., Rueger P., Stracke J.O., Lau W., Tissot A.C., et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron. 2014;81:49–60. doi: 10.1016/j.neuron.2013.10.061. [DOI] [PubMed] [Google Scholar]
- 159.Johnsen K.B., Bak M., Kempen P.J., Melander F., Burkhart A., Thomsen M.S., Nielsen M.S., Moos T., Andresen T.L. Antibody affinity and valency impact brain uptake of transferrin receptor-targeted gold nanoparticles. Theranostics. 2018;8:3416–3436. doi: 10.7150/thno.25228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Johnsen K.B., Bak M., Melander F., Thomsen M.S., Burkhart A., Kempen P.J., Andresen T.L., Moos T. Modulating the antibody density changes the uptake and transport at the blood-brain barrier of both transferrin receptor-targeted gold nanoparticles and liposomal cargo. J. Contr. Release. 2019;295:237–249. doi: 10.1016/j.jconrel.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 161.Zhao P., Zhang N., An Z. Engineering antibody and protein therapeutics to cross the blood-brain barrier. Antib. Ther. 2022;5:311–331. doi: 10.1093/abt/tbac028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Boado R.J., Pardridge W.M. Brain and Organ Uptake in the Rhesus Monkey in Vivo of Recombinant Iduronidase Compared to an Insulin Receptor Antibody-Iduronidase Fusion Protein. Mol. Pharm. 2017;14:1271–1277. doi: 10.1021/acs.molpharmaceut.6b01166. [DOI] [PubMed] [Google Scholar]
- 163.Boado R.J., Lu J.Z., Hui E.K.-W., Pardridge W.M. IgG-single chain Fv fusion protein therapeutic for Alzheimer’s disease: Expression in CHO cells and pharmacokinetics and brain delivery in the rhesus monkey. Biotechnol. Bioeng. 2010;105:627–635. doi: 10.1002/bit.22576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Banks W.A., Robinson S.M., Nath A. Permeability of the blood-brain barrier to HIV-1 Tat. Exp. Neurol. 2005;193:218–227. doi: 10.1016/j.expneurol.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 165.Anand P., O’Neil A., Lin E., Douglas T., Holford M. Tailored delivery of analgesic ziconotide across a blood brain barrier model using viral nanocontainers. Sci. Rep. 2015;5 doi: 10.1038/srep12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pang H.-H., Huang C.-Y., Chou Y.-W., Lin C.-J., Zhou Z.-L., Shiue Y.-L., Wei K.-C., Yang H.-W. Bioengineering fluorescent virus-like particle/RNAi nanocomplexes act synergistically with temozolomide to eradicate brain tumors. Nanoscale. 2019;11:8102–8109. doi: 10.1039/c9nr01247h. [DOI] [PubMed] [Google Scholar]
- 167.Fuster C., Varese M., García J., Giralt E., Sánchez-Navarro M., Teixidó M. Expanding the MiniAp-4 BBB-shuttle family: Evaluation of proline cis-trans ratio as tool to fine-tune transport. J. Pept. Sci. 2019;25 doi: 10.1002/psc.3172. [DOI] [PubMed] [Google Scholar]
- 168.Mamelak A.N., Jacoby D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601) Expet Opin. Drug Deliv. 2007;4:175–186. doi: 10.1517/17425247.4.2.175. [DOI] [PubMed] [Google Scholar]
- 169.Díaz-Perlas C., Varese M., Guardiola S., García J., Sánchez-Navarro M., Giralt E., Teixidó M. From venoms to BBB-shuttles. MiniCTX3: a molecular vector derived from scorpion venom. Chem. Commun. 2018;54:12738–12741. doi: 10.1039/c8cc06725b. [DOI] [PubMed] [Google Scholar]
- 170.Lee K.Z., Basnayake Pussepitiyalage V., Lee Y.-H., Loesch-Fries L.S., Harris M.T., Hemmati S., Solomon K.V. Engineering Tobacco Mosaic Virus and Its Virus-Like-Particles for Synthesis of Biotemplated Nanomaterials. Biotechnol. J. 2021;16 doi: 10.1002/biot.202000311. [DOI] [PubMed] [Google Scholar]
- 171.Nance E.A., Woodworth G.F., Sailor K.A., Shih T.-Y., Xu Q., Swaminathan G., Xiang D., Eberhart C., Hanes J. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lu W., Zhang Y., Tan Y.-Z., Hu K.-L., Jiang X.-G., Fu S.-K. Cationic albumin-conjugated pegylated nanoparticles as novel drug carrier for brain delivery. J. Contr. Release. 2005;107:428–448. doi: 10.1016/j.jconrel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 173.Shi N., Pardridge W.M. Noninvasive gene targeting to the brain. Proc. Natl. Acad. Sci. USA. 2000;97:7567–7572. doi: 10.1073/pnas.130187497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kong S.D., Lee J., Ramachandran S., Eliceiri B.P., Shubayev V.I., Lal R., Jin S. Magnetic targeting of nanoparticles across the intact blood-brain barrier. J. Contr. Release. 2012;164:49–57. doi: 10.1016/j.jconrel.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Li X., Vemireddy V., Cai Q., Xiong H., Kang P., Li X., Giannotta M., Hayenga H.N., Pan E., Sirsi S.R., et al. Reversibly Modulating the Blood-Brain Barrier by Laser Stimulation of Molecular-Targeted Nanoparticles. Nano Lett. 2021;21:9805–9815. doi: 10.1021/acs.nanolett.1c02996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tachibana K., Iwashita Y., Wakayama E., Nishino I., Nishikaji T., Kondoh M. Tight Junction Modulating Bioprobes for Drug Delivery System to the Brain: A Review. Pharmaceutics. 2020;12 doi: 10.3390/pharmaceutics12121236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Butt M.T. Morphologic changes associated with intrathecal catheters for direct delivery to the central nervous system in preclinical studies. Toxicol. Pathol. 2011;39:213–219. doi: 10.1177/0192623310391679. [DOI] [PubMed] [Google Scholar]
- 178.Fowler M.J., Cotter J.D., Knight B.E., Sevick-Muraca E.M., Sandberg D.I., Sirianni R.W. Intrathecal drug delivery in the era of nanomedicine. Adv. Drug Deliv. Rev. 2020;165–166:77–95. doi: 10.1016/j.addr.2020.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pin T.W., McCartney L., Lewis J., Waugh M.-C. Use of intrathecal baclofen therapy in ambulant children and adolescents with spasticity and dystonia of cerebral origin: a systematic review. Dev. Med. Child Neurol. 2011;53:885–895. doi: 10.1111/j.1469-8749.2011.03992.x. [DOI] [PubMed] [Google Scholar]
- 180.Kim A., Grover A., Hammon K., de Hart G., Slasor P., Cherukuri A., Ajayi T., Jacoby D., Schulz A., Specchio N., et al. Clinical Pharmacokinetics and Pharmacodynamics of Cerliponase Alfa, Enzyme Replacement Therapy for CLN2 Disease by Intracerebroventricular Administration. Clin. Transl. Sci. 2021;14:635–644. doi: 10.1111/cts.12925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Schwering C., Kammler G., Wibbeler E., Christner M., Knobloch J.K.-M., Nickel M., Denecke J., Baehr M., Schulz A. Development of the “Hamburg Best Practice Guidelines for ICV-Enzyme Replacement therapy (ERT) in CLN2 Disease” Based on 6 Years Treatment Experience in 48 Patients. J. Child Neurol. 2021;36:635–641. doi: 10.1177/0883073821989154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lonser R.R., Akhter A.S., Zabek M., Elder J.B., Bankiewicz K.S. Direct convective delivery of adeno-associated virus gene therapy for treatment of neurological disorders. J. Neurosurg. 2020;134:1751–1763. doi: 10.3171/2020.4.JNS20701. [DOI] [PubMed] [Google Scholar]
- 183.Voges J., Reszka R., Gossmann A., Dittmar C., Richter R., Garlip G., Kracht L., Coenen H.H., Sturm V., Wienhard K., et al. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann. Neurol. 2003;54:479–487. doi: 10.1002/ana.10688. [DOI] [PubMed] [Google Scholar]
- 184.Casanova F., Carney P.R., Sarntinoranont M. Effect of needle insertion speed on tissue injury, stress, and backflow distribution for convection-enhanced delivery in the rat brain. PLoS One. 2014;9 doi: 10.1371/journal.pone.0094919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rossmeisl J.H. New treatment modalities for brain tumors in dogs and cats. The Veterinary Clinics of North America. Small Animal Practice. 2014;44:1013–1038. doi: 10.1016/j.cvsm.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 186.He J., Yu L., Lin X., Liu X., Zhang Y., Yang F., Deng W. Virus-like Particles as Nanocarriers for Intracellular Delivery of Biomolecules and Compounds. Viruses. 2022;14 doi: 10.3390/v14091905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.DePolo N.J., Reed J.D., Sheridan P.L., Townsend K., Sauter S.L., Jolly D.J., Dubensky T.W., Jr. VSV-G pseudotyped lentiviral vector particles produced in human cells are inactivated by human serum. Mol. Ther. 2000;2:218–222. doi: 10.1006/mthe.2000.0116. [DOI] [PubMed] [Google Scholar]
- 188.Milani M., Annoni A., Bartolaccini S., Biffi M., Russo F., Di Tomaso T., Raimondi A., Lengler J., Holmes M.C., Scheiflinger F., et al. Genome editing for scalable production of alloantigen-free lentiviral vectors for in vivo gene therapy. EMBO Mol. Med. 2017;9:1558–1573. doi: 10.15252/emmm.201708148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lyu P., Lu B. New Advances in Using Virus-like Particles and Related Technologies for Eukaryotic Genome Editing Delivery. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms23158750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Duarte F., Déglon N. Genome Editing for CNS Disorders. Front. Neurosci. 2020;14 doi: 10.3389/fnins.2020.579062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sweeney M.D., Zhao Z., Montagne A., Nelson A.R., Zlokovic B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019;99:21–78. doi: 10.1152/physrev.00050.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Schulz A., Ajayi T., Specchio N., de Los Reyes E., Gissen P., Ballon D., Dyke J.P., Cahan H., Slasor P., Jacoby D., et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018;378:1898–1907. doi: 10.1056/NEJMoa1712649. [DOI] [PubMed] [Google Scholar]
- 193.Jones A.R., Shusta E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. (N. Y.) 2007;24:1759–1771. doi: 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hamilton J.R., Tsuchida C.A., Nguyen D.N., Shy B.R., McGarrigle E.R., Sandoval Espinoza C.R., Carr D., Blaeschke F., Marson A., Doudna J.A. Targeted delivery of CRISPR-Cas9 and transgenes enables complex immune cell engineering. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Battini J.L., Heard J.M., Danos O. Receptor choice determinants in the envelope glycoproteins of amphotropic, xenotropic, and polytropic murine leukemia viruses. J. Virol. 1992;66:1468–1475. doi: 10.1128/jvi.66.3.1468-1475.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schwarz B., Uchida M., Douglas T. Biomedical and Catalytic Opportunities of Virus-Like Particles in Nanotechnology. Adv. Virus Res. 2017;97:1–60. doi: 10.1016/bs.aivir.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Martino M.L., Crooke S.N., Manchester M., Finn M.G. Single-Point Mutations in Qβ Virus-like Particles Change Binding to Cells. Biomacromolecules. 2021;22:3332–3341. doi: 10.1021/acs.biomac.1c00443. [DOI] [PMC free article] [PubMed] [Google Scholar]