

Abstract
Background:
This prospective study investigates the pattern of pareses in 5q-associated spinal muscular atrophy (SMA) to identify disease-specific characteristics and potential differences from amyotrophic lateral sclerosis (ALS) and spinobulbar muscular atrophy (SBMA). Detailed knowledge about pareses patterns in SMA facilitates differential diagnosis and supports therapeutic monitoring.
Methods:
Between January 2021, and June 2021, 66 SMA patients (59.1% male, aged 33.6 ± 15.2 years) were included in the study. Most patients had SMA type II (n = 28) or SMA type III (n = 28), seven patients had SMA type I, and three patients had SMA type IV. We analyzed the pattern of pareses using the UK Medical Research Council (MRC) scoring system.
Results:
In both, upper and lower limbs muscle weakness was less pronounced in distal (upper limbs: MRC median 3.0 (interquartile range 1.5–3.5); lower limbs: 1.5 (0.5–3.0)) compared to proximal muscle groups (upper limbs: 2.0 (1.5–2.6); p < 0.001; lower limbs: 0.5 (0.5–1.5); p < 0.001). Thenar muscles were stronger than other small hand muscles (3.0 (2.0–3.5) vs 3.0 (1.5–3.5); p = 0.004). Muscles had more strength in upper (2.3 (1.5–3.1)) compared to lower limbs (1.1 (0.5–2.3); p < 0.001) and in flexors compared to extensors.
Conclusion:
We identified a specific pattern of muscle paresis in SMA which is different from the pattern of paresis in ALS and SBMA. As a rule of thumb, the pattern of pareses is similar, but not identical to ALS in distal, but different in proximal muscle groups.
Keywords: amyotrophic lateral sclerosis, motor neuron disease, paresis pattern, spinal muscular atrophy
Introduction
5q-associated spinal muscular atrophy (SMA) is an autosomal-recessive motor neuron disorder, clinically characterized by muscle weakness and atrophy caused by degeneration of alpha motor neurons due to a homozygous deletion or mutation in the survival motor neuron 1 (SMN1) gene. SMN1 is located on chromosome 5q13 and is one of two genes encoding the SMN protein.1,2 The incidence of SMA in Europe is about 1/10,000 livebirths with a carrier frequency of 1/50. 3 Based on the onset of clinical symptoms, the achievement of motor milestones, and life expectancy, SMA is divided into four subtypes (SMA type 0–4) according to the International SMA Consortium. Within the three main types (SMA type 1–3), SMA type 1 (Werdnig–Hoffmann disease) represents the infantile and thus most severe form, while SMA types 2 and 3 (Kugelberg–Welander disease) are defined as late-onset forms and are characterized by intermediate (SMA type 2) or mild (SMA type 3) types of progression. In SMA type 4, muscle weakness begins in the second or third decade of life. 4
The severity of SMA phenotypes is determined primarily by the number of survival motor neuron 2 (SMN2) gene copies. 5
Nusinersen, the first approved drug treatment option for SMA is an antisense oligonucleotide that increases SMN protein expression via modification of the splicing process of the pre-mRNA of SMN2.6–8 Risdiplam is a small molecule and oral medication acting in a similar way.9,10 Onasemnogene abeparvovec-xioi is a gene replacement therapy which comprises an adeno-associated viral vector with the human SMN1 gene.11–14
The application of these disease-modifying treatment (DMT) leads to considerable improvements of muscle strength and motor function in SMA patients as demonstrated by clinical trials and real-world data.14–20 These effects are particularly evident in SMA patients treated early or presymptomatically,20,21 which has led to the implementation of SMA into newborn screening in many countries. 22 The emergence of new SMA phenotypes under these therapeutic options is to be expected. 23
The knowledge of a specific pattern of pareses is essential to better classify and monitor therapy-associated changes in motor function. It facilitates the differential diagnosis of SMA versus amyotrophic lateral sclerosis (ALS) and other motor neuron diseases in adults, for example, spinobulbar muscular atrophy (SBMA). Therefore, it was the goal of this study to determine the pattern of paresis in SMA and compare the results with ALS and SBMA.
Methods
Patients
Sixty-six patients were prospectively examined between January 2021 and June 2021 at the Department of Neurology of Ulm, University Hospital (Germany).
All patients had a genetically confirmed 5q-associated SMA with a homozygous deletion of exons 7, 8, or both, or with compound heterozygous mutations. The study was approved by the Ethics Committee of Ulm University (No. 19/12). Informed written consent was provided by all patients or their legal representative.
Measurement of muscle weakness
Muscle strength was measured by the Medical Research Council (MRC) scale.24,25 The MRC is a standard scale for the quantification of muscle strength and has been used as a validated tool in a wide variety of clinical studies. Muscle strength is evaluated on a scale between 0/5 (no contraction), 1/5 (flicker or trace of contraction), 2/5 (active movement with gravity eliminated), 3/5 (active movement against gravity), 4/5 (active movement against moderate resistance), and 5/5 (full strength).
Muscle strength was evaluated in eight predefined muscle groups of upper and lower limbs, including elbow flexors and extensors, hand flexors and extensors, knee extensors and flexors, and foot elevators and flexors. Due to the high incidence of deformities and contractures preventing proper examination, muscle groups of shoulder and pelvic girdle were not tested.
Statistics
For descriptive statistics, median and interquartile range (IQR) are given. For comparison of muscle groups, a two-step procedure was applied: First, differences between corresponding muscle groups on the right and left side of each limb were analyzed using the two-sided sign test. As this first step showed no significant differences between the right and left sides for all muscle groups, MRC measurements from both sides were pooled and analyzed collectively. Subsequently, the two-sided sign test was used to analyze differences between muscle groups. To investigate the differences between SMA types I, II, and III across various muscle groups, the Mann–Whitney U test was used. A result was considered as significant if the p-value was <0.05 (two-sided). Statistical analyses were performed using SPSS, version 26 (IBM Corp. Released 2019. IBM SPSS Statistics for Windows, Version 26.0. Armonk, NY: IBM Corp).
Results
Sixty-six patients (59.1% male, aged 33.6 ± 15.2 years) were included. A total of 10.6% of patients were classified as SMA type 1, 42.4% type 2, 42.4% type 3, and 4.5% type 4. 6.4% had two copies of the SMN2 gene, 63.8% had three copies, 27.7% had four copies, and 2.1% had only one copy. Fifty-four patients (81.8%) were treated with nusinersen and 12 patients (18.2%) with risdiplam. For a summary of patient characteristics see Table 1.
Table 1.
Patient characteristics.
| Total | SMA 1 | SMA 2 | SMA 3 | SMA 4 | |
|---|---|---|---|---|---|
| N | 66 | 7 | 28 | 28 | 3 |
| Sex (% male) | 59.1 | 42.9 | 57.1 | 64.3 | 66.7 |
| Age (years, mean ± SD) | 33.6 (±15.2) | 17.9 (±4.5) | 26.4 (±11.3) | 43.0 (±13.6) | 49.0 (±9.6) |
| Copies of SMN2 | 2–3 | 2–4 | 2–5 | 4 | |
| Nusinersen (N, %) | 54 (81.8%) | 5 (7.6%) | 20 (30.3%) | 26 (39.4) | 3 (4.6%) |
| Risdiplam (N, %) | 12 (18.2%) | 2 (3.0%) | 8 (12.1%) | 2 (3.0%) | 0 |
| HFMSE (median, range) | 2 (0–19) | 13 (2–66) | 58 (58–62) | ||
| CHOP INTEND (median, range) | 11 (3–25) | 29 (1–39) | |||
| 6 min walking test in meters (median, range) | 510 (353–690) | 565 (556–648) |
CHOP INTEND, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; HFMSE, Hammersmith Functional Rating Scale Expanded; SMA, spinal muscular atrophy; SMN, survival motor neuron.
In some patients, certain muscle groups retained full strength (MRC grade 5). Elbow flexors, hand flexors, and hand extensors (in at least one arm) showed full strength in 12.1% of the patients, followed by foot flexors (5.3%), foot elevators (3.8%), knee extensors (2.3%), elbow extensors (1.5%), and knee flexors (0.8%).
Median and range of MRC scores obtained in different muscle groups are shown in Figure 1.
Figure 1.

MRC scores for different muscle groups (median, minimum to maximum) and comparisons between muscle groups.
***p < 0.01.
MRC, Medical Research Council.
Elbow flexors (3.0 (2.0–3.0)) were stronger than elbow extensors (1.0 (0.0–2.0); p < 0.001), the same was true for hand flexors (3.0 (2.0–3.8)) if compared with hand extensors (3.0 (1.0–3.0); p = 0.004). Overall, hand muscles (3.0 (1.5–3.5)) were stronger than elbow muscles (2.0 (1.5–2.7); p < 0.001).
Finger flexors (3.0 (2.0–4.0)) were stronger than finger extensors (3.0 (1.0–3.0); p < 0.001) and thumb flexors (3.0 (3.0–4.0)) were stronger than thumb extensors (3.0 (1.0–3.0); p < 0.001). Overall, thumb muscles were stronger than muscles other fingers (3 (2.0–3.5) vs 3 (1.5–3.5); p = 0.004).
Knee flexors (1.0 (1.0–2.0)) were stronger than knee extensors (0.0 (0.0–1.0)); p < 0.001), and foot flexors (2.0 (2.0–3.0)) were stronger than foot elevators (1.0 (0.0–3.0); p = 0.001). Foot muscles (1.5 (0.5–3.0)) were stronger than knee muscles (0.5 (0.5–1.5); p < 0.001). On average, upper limbs (2.3 (1.5–3.1)) were stronger than lower limbs (1.1 (0.5–2.3); p < 0.001). In summary, compared to ALS, the pattern of pareses was similar in distal muscle groups, but opposite in proximal muscle groups (Figure 2). In SMA, lower extremities, proximal muscle groups, and extensors are predominantly affected (Figure 3).
Figure 2.
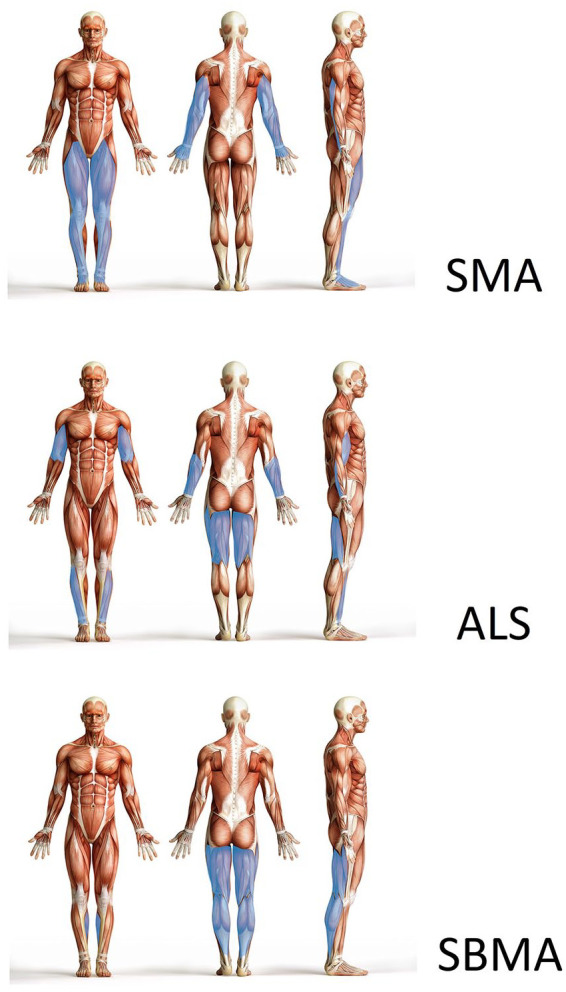
Pattern of pareses in SMA versus ALS versus SBMA. More affected muscle groups are marked with blue color. Compared to ALS, elbow and knee extensors are more severely affected in SMA, while elbow and knee flexors are less severely affected. In distal muscle groups, the pattern of paresis is similar. In SBMA, thigh and calf flexors are more severely affected than extensors. In arm muscle groups, all muscles are affected quite equally.
ALS, amyotrophic lateral sclerosis; SMA, spinal muscular atrophy; SBMA, spinobulbar muscular atrophy.
Figure 3.
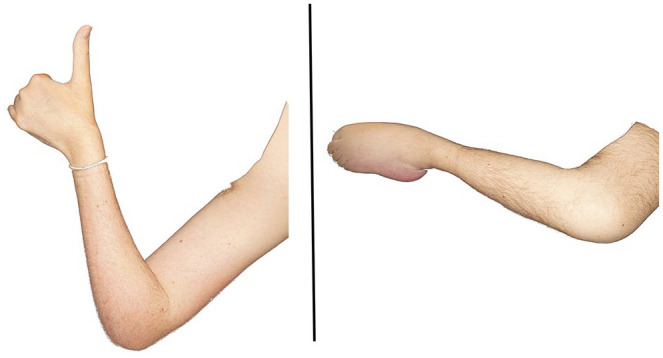
Pattern of muscle atrophy in SMA. Patient with SMA type 2, showing the typical pattern of pareses and atrophy of upper (left) and lower (right) extremities in SMA, predominantly affecting extensors, lower limb muscles, and proximal muscle groups.
SMA, spinal muscular atrophy.
The descriptive analysis of muscle group data for types I, II, and III of SMA showed that all SMA types shared a specific pattern of muscle weakness. Strength levels were lowest in SMA I, followed by SMA II, and were highest in SMA III (SMA I, II, and III; see Table 2). Significant differences in muscle strength were found in multiple areas. For the elbow, knee, hand, and foot muscles, strength was generally lower in SMA I compared to SMA II and III, and also lower in SMA II compared to SMA III, with statistical significance in most comparisons (p-values ranging from <0.001 to <0.02).
Table 2.
Descriptive analysis of muscle groups data by types 1–4.
| Muscle group | SMA type 1 | SMA type 2 | SMA type 3 | SMA type 4 | ||||
|---|---|---|---|---|---|---|---|---|
| N | 7 | 28 | 28 | 3 | ||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Elbow extensors | 0.5 | 0.1 | 0.7 | 0.2 | 2.1 | 1.4 | 3.0 | 0.0 |
| Elbow flexors | 1.4 | 1.2 | 2.4 | 1.0 | 3.3 | 1.0 | 5.0 | 0.0 |
| Hand extensors | 1.2 | 1.1 | 1.5 | 1.2 | 3.6 | 1.0 | 4.3 | 0.8 |
| Hand flexors | 1.3 | 1.1 | 2.3 | 1.2 | 3.4 | 1.0 | 4.5 | 0.5 |
| Finger extensors | 1.2 | 1.1 | 1.6 | 1.0 | 3.3 | 0.8 | 3.8 | 0.8 |
| Finger flexors | 1.7 | 1.1 | 2.4 | 1.1 | 3.6 | 1.0 | 4.2 | 0.8 |
| Thumb extensors | 1.0 | 0.5 | 2.1 | 1.1 | 3.2 | 0.8 | 3.5 | 0.5 |
| Thumb flexors | 1.7 | 0.9 | 2.6 | 0.9 | 3.8 | 0.8 | 4.5 | 0.8 |
| Knee extensors | 0.1 | 0 | 0.3 | 0.2 | 1.1 | 1.0 | 2.7 | 0.5 |
| Knee flexors | 1.0 | 0.6 | 1.1 | 0.6 | 2.0 | 1.1 | 3.7 | 0.5 |
| Foot elevators | 0.2 | 0.1 | 1.2 | 1.1 | 2.6 | 1.6 | 4.0 | 0.6 |
| Foot depressors | 0.9 | 0.4 | 1.5 | 1.0 | 2.6 | 1.2 | 3.8 | 0.8 |
SMA, spinal muscular atrophy.
However, no significant differences were found in muscle strength between SMA types I and II for the elbow extensors, hand extensors, knee flexors, and knee extensors (p > 0.05).
Discussion
Our findings support the assumption of a specific pattern of pareses in SMA. This pattern is characterized by a pronounced muscle weakness of lower limbs and preferentially proximal muscle groups.
The pattern is distinctly different from ALS, the most frequent motor neuron disease in adults. In ALS, pronounced weakness in thumb muscles, hand/finger extensors, elbow flexors, knee flexors, and foot extensors is a specific diagnostic feature of the disease. 26 Also typical is the paresis pattern of the intrinsic hand muscles which involve pronounced weakness and atrophy of the abductor pollicis brevis (APB) and first dorsal interosseous (FDI) muscles with relative sparing of the abductor digiti minimi (ADM) (“split-hand” syndrome). 27 Cortical influences, in particular a pathogenetic role of monosynaptic corticomotoneuronal (CM) input, have been suggested to explain this pattern.27–30 Accordingly, it is reasonable to assume that in ALS muscle groups receiving the strongest direct CM innervation by phylogenetically young monosynaptic inputs are most severely affected.26,31–35
In SBMA, proximal legs are more affected than arms, and the tongue shows a pattern distinguishable from ALS with a higher fat fraction in muscle magnetic resonance imaging (MRI). 36 From a clinical point of view, severe tongue atrophy in SBMA goes along with a striking lack of dysarthria. Also, the lower limbs in SBMA show a higher amount of fatty infiltration compared to ALS. 36 In the thigh, a relative sparing of the medial muscle group (adductor magnus, sartorius, soleus) with early involvement of the posterior thigh muscles and quadriceps are observed. In the lower leg, the posterior deep and superficial calf compartment (gastrocnemius, soleus, and tibialis posterior) is predominantly affected with a relative sparing of tibialis anterior. These MRI results correlate well with the functional rating scales (lower limb part of ALS Functional Rating Scale – Revised and SBMA Functional Rating Scale) in Klickovic et al. 36 Regarding the paresis pattern in the arms, involvement is more diverse: According to literature, finger extensors and elbow flexors are most commonly affected, 37 but distal and proximal arm muscles (extensors and flexors) are almost equally affected. In contrast to ALS, SBMA involves not only motor neuron degeneration but also considerable muscle involvement which may partly explain the distinct muscle vulnerability and paresis pattern. 38
In contrast to ALS, very few studies examined the specific pattern of pareses in SMA. In line with our study, Günther et al. 39 showed that adult SMA patients suffer from a “reversed split-hand” phenomenon, which discriminates SMA from ALS and controls with a high sensitivity and specificity. In their electrophysiological study using Motor Unit Number Index (MUNIX), 39 the most affected muscle in SMA was the FDI, followed by the ADM, whereas the APB was relatively well preserved. There are reports of patients with other pure lower motor neuron diseases, who have also been described as having a “split-hand.”40,41 In patients with spinal and bulbar muscular atrophy (SBMA, Kennedy’s disease), a “split-hand” was described in 57% of patients. 42 We assume that the “reversed split-hand” phenomenon might represent a specific pattern of paresis in SMA and can therefore help to clinically differentiate SMA from ALS and other motor neuron diseases in adults.
The pattern of paresis in proximal and distal muscle groups of upper and lower extremities can also be utilized to distinguish adult-onset SMA from ALS and SBMA. As a rule of thumb, the pattern of paresis is similar in distal muscle groups (apart from the hand patterns), but differs in proximal muscle groups. In both SMA and ALS, the typical pattern of paresis in distal muscle groups involves a pronounced affection of finger/hand extensors and foot elevators. On the other hand, elbow and knee extensors are more severely affected in SMA, whereas elbow and knee flexors are more severely affected in ALS. In SBMA, the posterior leg flexors in the thigh and lower leg are more severely affected, while in the less affected arms, a specific pattern for flexors or extensors could not be determined in proximal and distal muscle groups.
As this was an observational study, we were unable to analyze the effect of specific SMA treatments such as nusinersen and risdiplam, on paresis patterns. However, our findings mith impact future longitudinal studies which assess the longitudinal impact of SMA therapies. Based on our results, we suggest a nuanced approach for monitoring treatment effects, focusing on muscles with moderate function (power 3–4) rather than those with severe weakness (power 1–2). This might offer a more sensitive measure of therapeutic efficacy, as these muscles show more significant and measurable responses. Similarly, CMAP assessments in muscles with moderate pareses will likely provide more meaningful data, reflecting subtle functional improvements and serving as more robust indicators of treatment success.
The emergence of new SMA phenotypes under DMT is to be expected; the question remains whether the pattern of paresis described here might change in the future, or whether treatment will only exert quantitative effects.
Acknowledgments
We thank our patients for contributing their data.
Footnotes
ORCID iDs: Zeljko Uzelac  https://orcid.org/0000-0003-2479-2025
https://orcid.org/0000-0003-2479-2025
Johannes Dorst
https://orcid.org/0000-0003-0338-5439
Angela Rosenbohm
https://orcid.org/0000-0001-9700-0338
Contributor Information
Zeljko Uzelac, Department of Neurology, Ulm University, Ulm, Germany.
Beate Schwäble, Department of Neurology, Ulm University, Ulm, Germany.
Johannes Dorst, Department of Neurology, Ulm University, Ulm, Germany; German Center for Neurodegenerative Diseases, Research Site Ulm, Ulm, Germany.
Angela Rosenbohm, Department of Neurology, Ulm University, Ulm, Germany.
Kurt Wollinsky, Department of Anesthesiology, RKU—University and Rehabilitation Clinics, Ulm University, Ulm, Germany.
Claudia D. Wurster, Department of Neurology, Ulm University, Ulm, Germany German Center for Neurodegenerative Diseases, Research Site Ulm, Ulm, Germany; Institute of Human Genetics, Ulm University Medical Center, Ulm, Germany.
Janna S. Steinbreier, Department of Neurology, Ulm University, Ulm, Germany
Albert C. Ludolph, Department of Neurology, Ulm University, Oberer Eselsberg 45, 89091 Ulm, Germany; German Center for Neurodegenerative Diseases, Research Site Ulm, Ulm, Germany.
Declarations
Ethics approval and consent to participate: The study was executed in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the University of Ulm (Nr. 19/12). Informed written consent was provided by all patients.
Consent for publication: Not applicable.
Author contributions: Zeljko Uzelac: Conceptualization; Data curation; Formal analysis; Methodology; Software; Validation; Visualization; Writing – original draft; Writing – review & editing.
Beate Schwäble: Conceptualization; Investigation; Writing – review & editing.
Johannes Dorst: Data curation; Formal analysis; Methodology; Supervision; Validation; Visualization; Writing – review & editing.
Angela Rosenbohm: Methodology; Supervision; Writing – review & editing.
Kurt Wollinsky: Investigation; Methodology; Writing – review & editing.
Claudia D. Wurster: Conceptualization; Formal analysis; Supervision; Writing – review & editing.
Janna S. Steinbreier: Investigation; Methodology; Writing – review & editing.
Albert C. Ludolph: Conceptualization; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Writing – original draft; Writing – review & editing.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Competing interests: C.D.W. has received honoraria from Biogen as an advisory board member and for lectures and as an advisory member and consultant from Hoffmann-La Roche. She also received travel expenses from Biogen. A.C.L. received financial research support from AB Science, Biogen Idec, Cytokinetics, GSK, Orion Pharam, Novartis, TauRx Therapeutics Ltd, and TEVA Pharmaceuticals. He also has received honoraria as a consultant from Mitsubishi, Orion Pharma, Novartis, Teva, and as an advisory board member of Biogen, Treeway, and Hoffmann-La Roche. A.C.L. is Associate Editor at Therapeutic Advances in Neurological Disorders; therefore, the peer review process was managed by alternative members of the Board and the submitting Editor was not involved in the decision-making process. J.D. has received honoraria from Biogen as a consultant. A.R. has received honoraria as a consultant from Amicus Therapeutics, Sanofi, Hormosan, and Fulcrum. Z.U. has received honoraria and grants from Biogen as a consultant. K.W., B.S., and J.S.S. report no disclosures.
Data access, responsibility, and analysis: Z.U. has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Z.U. conducted and is responsible for the data analysis.
Data sharing: Individual participant data that underlie the results reported in this article, after de-identification (text, tables, and figures), as well as the study protocol will be available. Data will be available beginning 3 months and ending 5 years following article publication. Data will be shared with researchers who provide a methodologically sound proposal. Data will be shared for analyses to achieve the aims in the approved proposal. Proposals should be directed to zeljko.uzelac@rku.de; to gain access, data requestors will need to sign a data access agreement. Data are available for 5 years at https://www.uniklinik-ulm.de/neurologie.html.
Availability of data and materials: Not applicable.
References
- 1. Brzustowicz LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q1 1.2-13.3. Nature 1990; 344: 540–541. [DOI] [PubMed] [Google Scholar]
- 2. Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80: 155–165. [DOI] [PubMed] [Google Scholar]
- 3. Ogino S, Wilson RB. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet 2002; 111: 477–500. [DOI] [PubMed] [Google Scholar]
- 4. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008; 371: 2120–2133. [DOI] [PubMed] [Google Scholar]
- 5. Lorson CL, Hahnen E, Androphy EJ, et al. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A 1999; 96: 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010; 24: 1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hua Y, Vickers TA, Okunola HL, et al. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 2008; 82: 834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Passini MA, Bu J, Richards AM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 2011; 3: 72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Naryshkin NA, Weetall M, Dakka A, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014; 345: 688–693. [DOI] [PubMed] [Google Scholar]
- 10. Ratni H, Ebeling M, Baird J, et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem 2018; 61: 6501–6517. [DOI] [PubMed] [Google Scholar]
- 11. Dominguez E, Marais T, Chatauret N, et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet 2011; 20: 681–693. [DOI] [PubMed] [Google Scholar]
- 12. Foust KD, Nurre E, Montgomery CL, et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 2009; 27: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valori CF, Ning K, Wyles M, et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med 2010; 2: 35ra42. [DOI] [PubMed] [Google Scholar]
- 14. Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 2017; 377: 1713–1722. [DOI] [PubMed] [Google Scholar]
- 15. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 2017; 377: 1723–1732. [DOI] [PubMed] [Google Scholar]
- 16. Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med 2018; 378: 625–635. [DOI] [PubMed] [Google Scholar]
- 17. Hagenacker T, Wurster CD, Günther R, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol 2020; 19: 317–325. [DOI] [PubMed] [Google Scholar]
- 18. Darras BT, Masson R, Mazurkiewicz-Bełdzińska M, et al. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N Engl J Med 2021; 385: 427–435. [DOI] [PubMed] [Google Scholar]
- 19. Mercuri E, Deconinck N, Mazzone ES, et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol 2022; 21: 42–52. [DOI] [PubMed] [Google Scholar]
- 20. Mendell JR, Al-Zaidy SA, Lehman KJ, et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol 2021; 78: 834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord 2019; 29: 842–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vill K, Schwartz O, Blaschek A, et al. Newborn screening for spinal muscular atrophy in Germany: clinical results after 2 years. Orphanet J Rare Dis 2021; 16: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schorling DC, Pechmann A, Kirschner J. Advances in treatment of spinal muscular atrophy – new phenotypes, new challenges, new implications for care. J Neuromuscul Dis 2020; 7: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Compston A. Aids to the investigation of peripheral nerve injuries. Medical Research Council: Nerve Injuries Research Committee. His Majesty’s Stationery Office: 1942; pp. 48 (iii) and 74 figures and 7 diagrams; with aids to the examination of the peripheral nervous. Brain 2010; 133: 2838–2844. [DOI] [PubMed] [Google Scholar]
- 25. Vanhoutte EK, Faber CG, van Nes SI, et al. Modifying the Medical Research Council grading system through Rasch analyses. Brain 2012; 135: 1639–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ludolph AC, Emilian S, Dreyhaupt J, et al. Pattern of paresis in ALS is consistent with the physiology of the corticomotoneuronal projections to different muscle groups. J Neurol Neurosurg Psychiatry 2020; 91: 991–998. [DOI] [PubMed] [Google Scholar]
- 27. Weber M, Eisen A, Stewart H, et al. The split hand in ALS has a cortical basis. J Neurol Sci 2000; 180: 66–70. [DOI] [PubMed] [Google Scholar]
- 28. Eisen A, Braak H, Del Tredici K, et al. Cortical influences drive amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2017; 88: 917–924. [DOI] [PubMed] [Google Scholar]
- 29. Menon P, Kiernan MC, Vucic S. ALS pathophysiology: insights from the split-hand phenomenon. Clin Neurophysiol 2014; 125: 186–193. [DOI] [PubMed] [Google Scholar]
- 30. Eisen A, Kuwabara S. The split hand syndrome in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2012; 83: 399–403. [DOI] [PubMed] [Google Scholar]
- 31. Lemon RN. Descending pathways in motor control. Annu Rev Neurosci 2008; 31: 195–218. [DOI] [PubMed] [Google Scholar]
- 32. Phillips CG, Porter R. Corticospinal neurones. Their role in movement. Monogr Physiol Soc 1977; v–xii, 1–450. [PubMed] [Google Scholar]
- 33. Kuypers HGJM. The descending pathways to the spinal cord, their anatomy and function. Prog Brain Res 1964; 11: 178–202. [DOI] [PubMed] [Google Scholar]
- 34. Porter R, Lemon R. Corticospinal function and voluntary movement. Oxford: Oxford University Press, 1995. [Google Scholar]
- 35. Braak H, Brettschneider J, Ludolph AC, et al. Amyotrophic lateral sclerosis – a model of corticofugal axonal spread. Nat Rev Neurol 2013; 9: 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klickovic U, Zampedri L, Sinclair CDJ, et al. Skeletal muscle MRI differentiates SBMA and ALS and correlates with disease severity. Neurology 2019; 93: E895–E907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dahlqvist JR, Oestergaard ST, Poulsen NS, et al. Refining the spinobulbar muscular atrophy phenotype by quantitative MRI and clinical assessments. Neurology 2019; 92: e548–e559. [DOI] [PubMed] [Google Scholar]
- 38. Hashizume A, Fischbeck KH, Pennuto M, et al. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (SBMA). J Neurol Neurosurg Psychiatry 2020; 91: 1085–1091. [DOI] [PubMed] [Google Scholar]
- 39. Günther R, Neuwirth C, Koch JC, et al. Motor Unit Number Index (MUNIX) of hand muscles is a disease biomarker for adult spinal muscular atrophy. Clin Neurophysiol 2019; 130: 315–319. [DOI] [PubMed] [Google Scholar]
- 40. Wilbourn AJ. The “split hand syndrome”. Muscle Nerve 2000; 23: 138. [DOI] [PubMed] [Google Scholar]
- 41. Schelhaas HJ, van de Warrenburg BPC, Kremer HPH, et al. The “split hand” phenomenon: evidence of a spinal origin. Neurology 2003; 61: 1619–1620. [DOI] [PubMed] [Google Scholar]
- 42. Shibuya K, Misawa S, Uzawa A, et al. Split hand and motor axonal hyperexcitability in spinal and bulbar muscular atrophy. J Neurol Neurosurg Psychiatry 2020; 91: 1189–1194. [DOI] [PubMed] [Google Scholar]