

Abstract
Stereocilia protrude up to 100 μm from the apical surface of vertebrate inner ear hair cells and are packed with cross-linked filamentous actin (F-actin). They function as mechanical switches to convert sound vibration into electrochemical neuronal signals transmitted to the brain. Several genes encode molecular components of stereocilia including actin monomers, actin regulatory and bundling proteins, motor proteins and the proteins of the mechanotransduction complex. A stereocilium F-actin core is a dynamic system, which is continuously being remodeled while maintaining an outwardly stable architecture under the regulation of F-actin barbed-end cappers, severing proteins and crosslinkers. The F-actin cores of stereocilia also provide a pathway for motor proteins to transport cargos including components of tip-link densities, scaffolding proteins and actin regulatory proteins. Deficiencies and mutations of stereocilia components that disturb this “dynamic equilibrium” in stereocilia can induce morphological changes and disrupt mechanotransduction causing sensorineural hearing loss, best studied in mouse and zebrafish models. Currently, at least 23 genes, associated with human syndromic and nonsyndromic hearing loss, encode proteins involved in the development and maintenance of stereocilia F-actin cores. However, it is challenging to predict how variants associated with sensorineural hearing loss segregating in families affect protein function. Here, we review the functions of several molecular components of stereocilia F-actin cores and provide new data from our experimental approach to directly evaluate the pathogenicity and functional impact of reported and novel variants of DIAPH1 in autosomal-dominant DFNA1 hearing loss using single-molecule fluorescence microscopy.
Introduction
Hearing loss is a common neurosensory deficit of 1.33 per 1000 newborn babies (Fortnum et al. 2001; Korver et al. 2017; Watkin and Baldwin 2012). More than 50% of congenital hearing loss is caused by a hereditable deficiency, most often a single gene defect. Prenatal infections, especially cytomegalovirus, and other factors including premature birth, injuries at birth and/or drugs are also contributors to newborn hearing loss (Chien et al. 2016; Korver et al. 2017; Marazita et al. 1993). Management of congenital sensorineural hearing loss requires long-term interventions using hearing aids, cochlear implants or brain-stem implants in some cases to develop sound-based speech (Alderson-Day and Fernyhough 2015; Nikolopoulos and Vlastarakos 2010; Wong et al. 2019). Sign languages are available when these devices are ineffective or not chosen. However, it can be difficult to achieve normal speech recognition and language development even with the most advanced hearing aids and cochlear implants (Ruben 2018; Tomblin et al. 2014). More than 30% of congenital deafness as well as noise induced hearing loss are related to damage of stereocilia, mechanosensory organelles that protrude from the apical surface of inner ear sensory hair cells. Regeneration or repair of damaged auditory hair cells, associated supporting cells and the entire mosaic architecture of the sensory epithelium is a major goal of hearing researchers in management of sensorineural hearing loss (Chien et al. 2016; Duan et al. 2004).
This review focuses on the actin cytoskeleton of cochlear hair cell stereocilia that function as mechanical switches to convert sound vibrations into electrochemical signals conveyed to the brain (Fig. 1). In all vertebrates, on the apical surface of each hair cell, stereocilia are organized in rows of increasing height, positioned close to each other and interconnected by extracellular filamentous links. In human hair cells, tightly packed F-actin bundles of each stereocilium within the longest row extend from the apical surface to a length of 4–7 μm for inner hair cells and 3–6 μm for outer hair cells (Wright 1984). Each tip of a stereocilium in the shorter row is connected to the side of an adjacent longer row stereocilium by a filamentous tip-link. Deflection of stereocilia toward the longest row applies tension to tip-links and opens mechanotransduction channels located at the tips of the shorter-row stereocilia (Gillespie and Muller 2009). Stereocilia also emanate from the apical surface of vestibular hair cells, and their ranks are less prominent than for auditory hair bundles. The lengths of vestibular hair cell stereocilia bundles can be much longer allowing them to detect linear and rotational accelerations of the head.
Fig. 1.
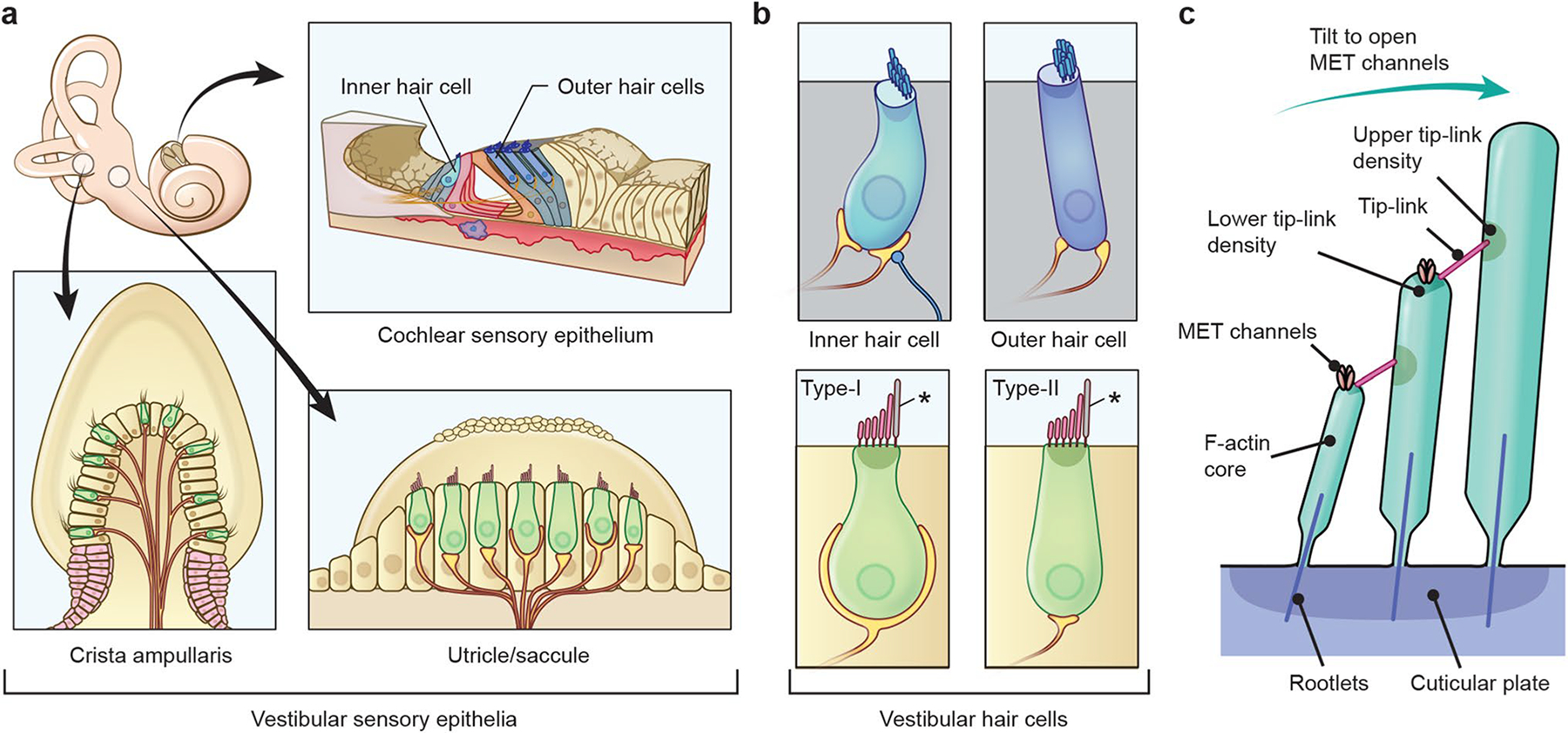
Sensory epithelia of the inner ear, hair cells and their stereocilia in mammals. a In the inner ear, there are three distinct types of sensory epithelia. The cochlear sensory epithelium detects and attenuates sound vibration using one row of inner hair cells and three rows of outer hair cells, which are a part of the organ of Corti. Vestibular sensory epithelia of utricle and saccule detect horizontal and vertical accelerations. The three cristae ampullaris detect angular accelerations of the head. b Alignment of stereocilia on the apical surface of hair cells. Each outer hair cell has three rows of V-shaped stereocilia while stereocilia of inner hair cell are organized in a straighter line. Vestibular hair cell stereocilia (type I and type II) are long and also align in a staircase manner although the alignment is not as organized as inner and outer hair cells. Asterisks indicate kinocilia, which are composed of microtubules. c Architecture of stereocilia F-actin cores and tip-links. Stereocilia shafts consist of F-actin, which are bundled more tightly near their bases to form rootlets and anchor stereocilia in the cuticular plate. Tip-links are tethered to F-actin cores via protein complexes in upper and lower tip-link densities. Tension caused by unidirectional deflection of all stereocilia within a bundle opens the mechanotransduction channels (MET channels) near the tip-links
The actin cytoskeleton of stereocilia is both a durable and a dynamic structure, being continuously remodeled while maintaining an overall architecture. For the lifespan of mammals, the F-actin architecture of stereocilia remains largely unchanged, while actin monomers are constantly turning over, mainly at the distal ends (Narayanan et al. 2015). However, the F-actin crosslinkers, espin and plastin, are refreshed along the entire length of stereocilia (Miyoshi et al. 2021; Roy and Perrin 2018). We posit that a comprehensive understanding of the mechanisms governing lifelong homeostasis of these stereocilia macromolecules will not only elucidate the pathophysiology of human syndromic and non-syndromic sensorineural hearing loss but eventually will provide the underpinnings of necessary knowledge for formulating therapeutic strategies to protect and, perhaps someday, restore hearing.
Currently, mutations in more than 130 genes have been identified as causes of human syndromic and non-syndromic sensorineural hearing loss (https://hereditaryhearingloss.org/). At least 23 of these genes encode proteins associated with the development and maintenance of stereocilia F-actin cores including actin monomers, actin regulatory proteins, bundling proteins, motor proteins and the mechanotransduction protein complex (Table 1). However, it is challenging to predict how each protein molecule contributes to homeostasis in stereocilia F-actin cores and how mutations identified in families segregating sensorineural hearing loss affect protein function. We speculate that in vivo functional analyses of protein molecules will be crucial given the recent exhaustive search capabilities available with whole exome and genome sequencing. These sequencing technologies can provide a long list of candidate causal variants some of which will no doubt prove to be just rare benign polymorphisms. An efficient pipeline to screen the functions of variant protein molecules is required to conclude that a variant is associated with hearing loss. Here, we review the genes encoding protein molecules that establish and maintain the F-actin cores of stereocilia. We then highlight how single-molecule fluorescence microscopy-based functional analysis can be employed to evaluate novel candidate variants of DIAPH1, a gene known to be associated with DFNA1 human deafness (Lynch et al. 1997).
Table 1.
Genes encoding actin and actin-associated proteins in stereocilia necessary for human hearing
| Genes | Locus | Encoded protein | Known function(s) in stereocilia |
|---|---|---|---|
|
| |||
| DIAPH1 | DFNA1 | Protein diaphanous homolog 1 | Mediates processive actin elongation activity (constitutively active DIAPH1 is pathogenic) |
| MYO7A | DFNA11, DFNB2, USH1B | Unconventional myosin 7A | Transports and assembles upper tip-link complex, tip-link tensing |
| ACTG1 | DFNA20/26 * | Actin, cytoplasmic 2 (γ-cytoplasmic actin) | Assembles F-actin cores, repairs gaps in filaments |
| MYO6 | DFNA22, DFNB37 | Unconventional myosin 6 | Transports PTPRQ and associated proteins to stereocilia bases (toward pointed ends of F-actin) |
| PTPRQ | DFNA73, DFNB84 | Phosphatidylinositol phosphatase (PTPRQ) | Forms ankle-links |
| PLS1 | DFNA ** | Plastin-1 | Crosslinks F-actin in stereocilia cores |
| MYO15A | DFNB3 | Unconventional myosin 15A | Transports the components of elongation complex to stereocilia tips, such as WHRN and EPS8 |
| CDH23 | DFNB12, USH1D | Cadherin 23 | Composes tip-links with Protocadherin-15 |
| USH1C | DFNB18, USH1C | Harmonin | Scaffolds upper tip-link complex |
| PCDH15 | DFNB23, USH1F | Protocadherin-15 | Composes tip-links with Cadherin 23 |
| RDX | DFNB24 | Radixin | Links between plasma membrane and F-actin cores |
| GRXCR1 | DFNB25 | Glutaredoxin domain-containing cysteine-rich protein 1 (GRXCR1) | Crosslinks F-actin in stereocilia cores |
| TRIOBP | DFNB28 | TRIO and F-actin-binding protein (TRIOBP) | Croslinks F-actin and forms rootlets (isoforms 4 and 5) |
| MYO3A | DFNB30, DFNA ** | Unconventional myosin 3A | Recruits ESPN isoform 1 (involved in arrangement of stereocilia) |
| WHRN | DFNB31, USH2D | Whirlin | Forms an elongation complex at the tips with EPS8, involved in forming the staircase of stereocilia |
| ESPN | DFNB36 | Espin | Crosslinks F-actin in stereocilia cores |
| PDZD7 | DFNB57 | PDZD7 | Scaffolds ankle-link complex |
| TPRN | DFNB79 | Taperin | Protein at the stereocilia taper |
| GRXCR2 | DFNB101 | Glutaredoxin domain-containing cysteine-rich protein 2 (GRXCR2) | Crosslinks F-actin in stereocilia cores |
| EPS8 | DFNB102 | Epidermal growth factor receptor kinase substrate 8 (EPS8) | Barbed-end capper, involved in forming the staircase of stereocilia |
| EPS8L2 | DFNB106 | Epidermal growth factor receptor kinase substrate 8-like protein 2 (EPS8L2) | Barbed-end capper, regulating streocilia staircase architecture |
| USH1G | USH1G | Usher syndrome type-1G protein (SANS) | Scaffolds upper tip-link complex |
| ACTB | N/A* | Actin, cytoplasmic 1 (β-cytoplasmic actin) | Assembles F-actin cores |
Gain-of-function is associated with Baraitser-Winter cerebrofrontofacial (BWCFF) syndrome accompanying sensorineural hearing loss in 30–80% of patients (Riviere et al. 2012; Verloes et al. 2015; Yates et al. 2017)
Locus not assigned
Structure and dynamics of actin in living cells
Actin genes and products
Six paralogous actin genes are present in a mammalian genome: three α-actin genes (α-skeletal muscle, α-cardiac muscle and α-smooth muscle), one β-actin gene (β-cytoplasmic) and two γ-actin genes (γ-cytoplasmic and γ-smooth muscle) (Drummond et al. 2012; Vedula et al. 2017). In human, these six genes are named ACTA1, ACTC1, ACTA2, ACTB, ACTG1 and ACTG2, respectively (Vedula and Kashina 2018). Actin is a 42-kDa globular protein that can form a polymer (F-actin) measuring approximately 7 nm in diameter with a double-helix repeat every 37 nm (13 actin monomers) (Fig. 2, a and b). An actin filament has a “barbed end” and a “pointed end”. Actin monomers are more likely to polymerize to barbed ends. Actin is one of the most highly conserved proteins in eukaryotic cells, whose amino acid sequence differs by no more than 20% even between algae and human (Hanukoglu et al. 1983). The amino acid sequence is also highly similar between paralogs, and about 90% of the sequence is identical between the six mammalian actin genes (Khaitlina 2001). Different actin genes can replace the function of another in some organs, such as in the heart (Kumar et al. 1997). However, the unique phenotypes of knockout mice of the actin paralogs clearly indicate that each actin gene has a distinct biological function. Lack of Actb in mice causes early embryonic lethality (Tondeleir et al. 2014), while lack of Acta1 or Actc1 results in perinatal lethality accompanied by muscle weakness and myofibril disorganization, respectively (Crawford et al. 2002; Kumar et al. 1997). Mice lacking Acta2 or Actg1 are viable, but Acta2-knockout mice show defects in vascular contractility and blood pressure regulation (Schildmeyer et al. 2000), while Actg1-knockout mice suffer from growth abnormalities and progressive hearing loss (Belyantseva et al. 2009). Mutations of ACTG1 also cause autosomal-dominant, nonsyndromic hearing loss, DFNA20/26 (Morell et al. 2000; van Wijk et al. 2003; Zhu et al. 2003).
Fig. 2.
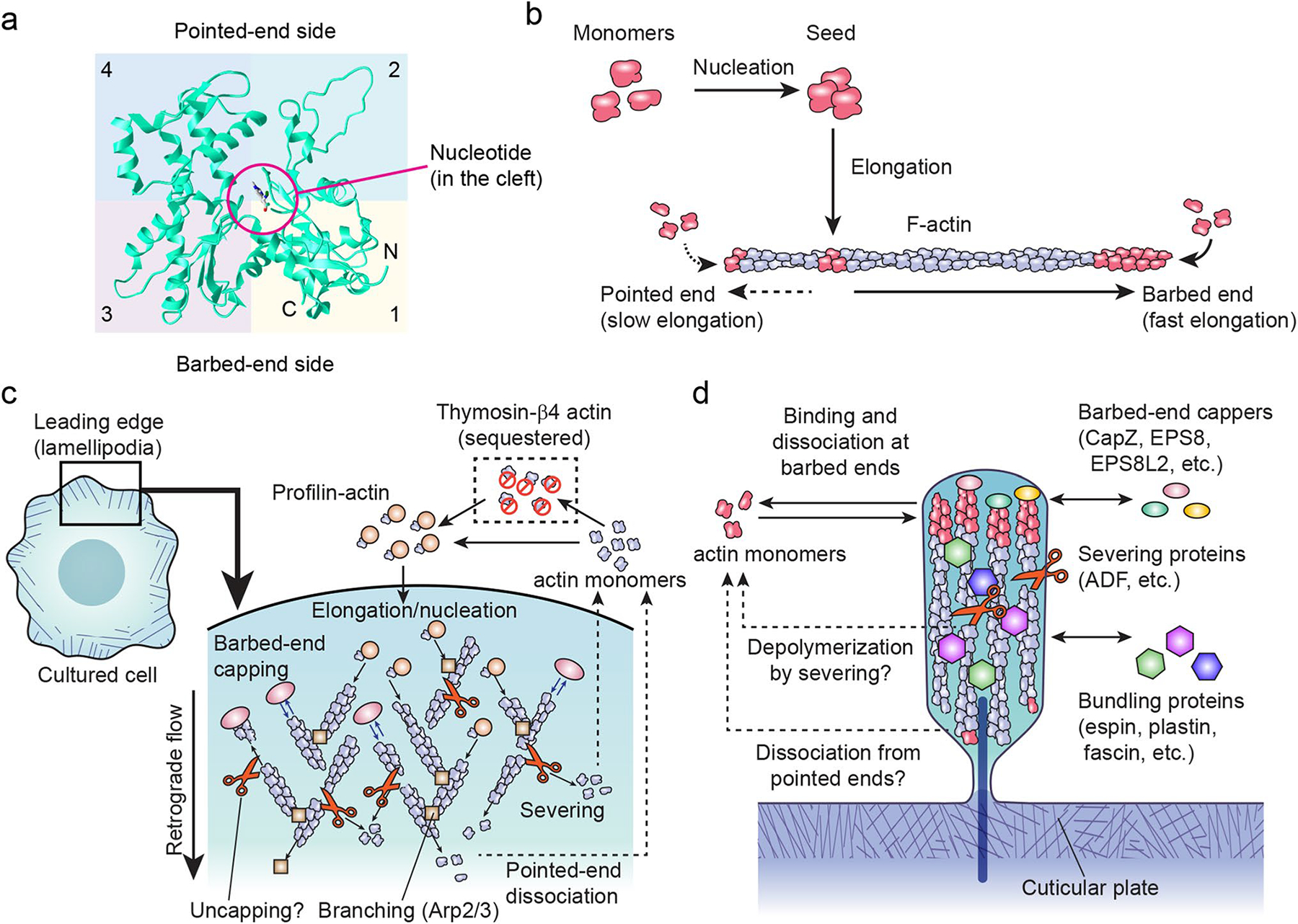
Sructure and dynamics of actin monomers and filaments (F-actin). a Tertiary structure of an ACTB actin monomer (PDB number: 3j82) (Hanc et al. 2015). A large cleft divides subdomains 1 and 2 from subdomains 3 and 4. ATP or ADP and a cation are held in a pocket deep in the cleft (ADP shown). b Assembly of F-actin from actin monomers in vitro. Formation of F-actin seeds from monomers is a slow nucleation step. F-actin has two ends, a barbed end that can rapidly elongate and a pointed end that can elongate but more slowly. c Actin dynamics in the lamellipodia, a thin veil-like structure at the leading edge of cells. Quasi-two-dimensional actin mesh is dynamically remodeled and tracked toward the center of cells by the retrograde flow. Lamellipodia can be used to study the dynamics of actin monomers, barbed-end cappers, Arp2/3 complex and severing proteins, many of which play crucial roles in stereocilia. Exchange of ATP and ADP in actin monomers is not shown to simplify the diagram. d Dynamics of stereocilia components. Proteins, such as actin, barbed-end cappers and bundling proteins, turn over through processes involving binding and dissociation that are hypothesized to be mechanisms to maintain the entire architecture of stereocilia but are not well understood, especially those that balance polymerization and depolymerization of the F-actin and replace damaged components in stereocilia F-actin cores
An actin monomer has four subdomains, 1–4, with a large cleft dividing subdomains 1 and 2 from subdomains 3 and 4 (Fig. 2a). A nucleotide, either ATP or ADP, coupled with a divalent cation, typically M g2+, is held in the cleft by hydrogen and ion bonds to the surrounding amino acid residues (Kabsch et al. 1990; Schutt et al. 1993; Sheterline et al. 1995). Nucleotides control de novo nucleation and elongation of existing F-actin in addition to stabilizing the tertiary structures of actin monomers (Asakura 1961; Dominguez 2009). For example, ATP-actin is more likely to polymerize than ADP-actin both at the barbed and pointed ends. The critical concentration at which actin monomers are in equilibrium with F-actin is approximately 20-fold lower for ATP-actin, 0.1 μM, compared with that of ADP-actin at 1.8 μM (Pollard 1986). The nucleotide also controls the depolymerization of F-actin. After polymerization, ATP-actin rapidly hydrolyzes their ATP into ADP and phosphate with a half-life of approximately 2 seconds (s) and then slowly releases the phosphate with a half-life of approximately 350 s to become ADP-actin, which is more likely to dissociate from both ends of F-actin than ATP-actin (Pollard 2016; Pollard et al. 2008). Binding of other divalent cations, such as C a2+, also alters the speeds of actin polymerization and ATP hydrolysis and affects the critical concentration (Scipion et al. 2018; Strzelecka-Golaszewska et al. 1978). The ratio of Ca2+-actin to M g2+-actin can be higher in a Ca2+-rich condition such as in stereocilia, and the polymerization properties may be different in such environment since ATP hydrolysis of Ca2+-actin is six times slower than with Mg2+-actin (Bergeron et al. 2010; Blanchoin and Pollard 2002). Among the amino acid residues surrounding the nucleotide and the divalent cation, at least two missense mutations in ACTG1, p.K213R and p.M305T, are reported to cause DFNA20/26 due to loss-of-function (Merino et al. 2018; Miyajima et al. 2020; Park et al. 2013; Yuan et al. 2016).
Actin dynamics at the leading edge of living cells
Lamellipodia of motile cells provide useful models to understand the “dynamic equilibrium” of an actin cytoskeleton (Fig. 2c). The veil-like lamellipodium continuously protrudes and retracts resembling waves coming to the seashore. Additionally, F-actin bundle-containing filopodia protrude from the edges of lamellipodia. The quasi-two-dimensional actin mesh in lamellipodia is continuously remodeled to drive the “waves” while retracting toward the center of cells (retrograde flow). Cytoplasm in lamellipodia contains as high as 50–200 μM of ATP-actin. The critical concentration of ATP-actin at which ATP-actin monomers are in equilibrium with F-actin is only 0.1 μM indicating that lamellipodia without regulatory mechanisms would be packed with F-actin and eventually the cycle of polymerization and depolymerization would stall (Lodish et al. 2000; Pollard 1986).
Polymerization and depolymerization of actin monomers in a live cell are controlled by many actin regulatory proteins. Elongation of existing actin filaments is suppressed by barbed-end cappers, such as CapZ (an α/β heterodimer formed by CAPZA1 or CAPZA2, and CAPZB) (Pollard and Cooper 1984). Depolymerization of existing filaments is enhanced by severing proteins, such as cofilin and ADF (Bamburg et al. 1980; Poukkula et al. 2011). AIP1 preferentially promotes disassembly of ADF/cofilin-decorated actin filaments (Okada et al. 2006). In addition, polymerization activities of actin monomers are modulated mainly by two ubiquitous proteins of contrasting functions, thymosin-β4 and profilin. Thymosin-β4, a 43 amino acid peptide in human, can sequester a large pool of actin monomers and suppress nucleation and binding to existing filaments (Pollard 2016; Safer et al. 1991). Profilin, a 16 kDa protein, also suppresses nucleation although binding to the barbed ends of existing actin filaments is not inhibited (Pollard and Cooper 1984; Vinson et al. 1998). Furthermore, profilin competes with thymosin-β4 and accelerates barbed-end elongation by catalyzing the exchange of actin-bound ADP to ATP (Mockrin and Korn 1980; Pantaloni and Carlier 1993). Inhibition of nucleation by these two proteins may be crucial to allow nucleation in a controlled manner, such as from the Arp2/3 complex in lamellipodia. Typical cytoplasm contains a high concentration of profilin–ATP-actin (50–100 μM in most eukaryotic cells), which substantially exceeds the critical concentration of ATP-actin and suggests that barbed-end cappers and severing proteins play crucial roles to balance polymerization and depolymerization in living cells (Pollard 2016; Pollard et al. 2000).
Single-molecule fluorescence microscopy has helped to elucidate actin cytoskeletal dynamics in lamellipodia, distinguishing and resolving single fluorescent molecules from the background signal and revealing actin and regulatory protein behavior in live cells. Thin lamellipodia of Xenopus XTC cells are useful to determine dissociation rates and trajectories of actin and actin regulatory proteins (Watanabe and Mitchison 2002). We reported that CapZ dissociates from barbed ends of lamellipodial actin with a half-life of 1–2 s, while for in vitro biochemistry experiments CapZ remains on barbed ends for more than a few hours (Miyoshi et al. 2006; Pollard and Cooper 1984). Fast dissociation was also observed for other proteins that can bind to barbed ends, such as EPS8, gelsolin (GSN) and VASP (Miyoshi et al. 2006). These results indicate that a common “uncapping” mechanism removes these proteins from barbed ends. Severing proteins may depolymerize barbed ends more aggressively than other parts of F-actin to cause uncapping because the dissociation rate of CapZ decreased significantly after removal of cytoplasm by permeabilization, stabilization of F-actin using Jasplakinolide and inhibition of cofilin by LIM-kinase overexpression (Miyoshi et al. 2006). These findings are consistent with the localization of severing proteins at the tips of stereocilia in mice, where barbed ends accumulate (McGrath et al. 2020).
Dynamics of stereocilia components
Microvilli are transient structures on the cell apical surface and usually appear and disappear within a few minutes (Gorelik et al. 2003; Ida et al. 2017; Meenderink et al. 2019). Compared with actin protrusions of microvilli, stereocilia have a unique characteristic that their overall architecture is largely unchanged for the lifetime of a mammal (Fig. 2d) (Jia et al. 2009). Actin dynamics is also different between microvilli and stereocilia. F-actin in microvilli treadmills from the barbed ends at the tips toward the pointed ends near the apical surface (Meenderink et al. 2019), while we now know that stereocilia replenish actin monomers mainly at the barbed ends near the tips without treadmilling the F-actin core (Drummond et al. 2015; Narayanan et al. 2015; Perrin and Ervasti 2010; Rzadzinska et al. 2004; Zhang et al. 2012). One unresolved question is the timing and molecular events responsible for cessation of F-actin treadmilling in a subset of microvilli on the apical surface of nascent hair cells during their transition to stereocilia. Espin, a major F-actin bundling protein in stereocilia, likely contributes to this transition since expression of espin in cultured cells can suddenly stop the treadmilling in filopodia (Loomis et al. 2003; Zheng et al. 2015).
Several of the actin regulatory proteins that function in lamellipodia also participate in stereocilia actin dynamics. CapZ is localized at the tips of shorter-row stereocilia and required for the regulation of their length (Avenarius et al. 2017). The severing proteins, cofilin-1 (CFL1) and ADF (DSTN), also localize at stereocilia tips suggesting an aggressive depolymerization dynamics near their barbed ends, as reported for the fast turnover of barbed-end capper CapZ in lamellipodia (McGrath et al. 2020; Miyoshi et al. 2006). One difference from the lamellipodial actin meshwork is that F-actin in stereocilia is tightly packed by the crosslinkers, such as plastin-1, espin and fascin-2 (Krey et al. 2016; Sekerkova et al. 2011). Unexpectedly, these crosslinkers can move freely along the entire length of stereocilia perhaps to allow for the paracrystal-like packing of F-actin (Miyoshi et al. 2021; Roy and Perrin 2018). The molecular mechanisms that maintain the entire architecture of stereocilia are not fully understood, especially those phenomena that balance polymerization and depolymerization of the F-actin cores and replace damaged actin monomers in the F-actin stereocilia cores (Belyantseva et al, 2009). Elucidating the dynamics of protein molecules that govern the homeostasis of stereocilia macromolecules will provide a basis to formulate therapeutic strategies for protecting hearing and perhaps reversing sensorineural hearing loss.
Hereditary hearing loss due to variants of actin and actin-associated proteins in stereocilia
ACTB and ACTG1
The ACTB and ACTG1 genes encode the almost identical β-cytoplasmic actin and γ-cytoplasmic actin. Both actins are found in stereocilia (Hofer et al. 1997) and differ by only four residues among the first nine amino acids at their N-termini, MDDDAALVV (ACTB, NP_001092.1) and MEEEAALVI (ACTG1, NP_001092.1), in mammals and birds (Perrin and Ervasti 2010). At least during development of the inner ear, these two actin genes can substitute functionally for one another as indicated by normal appearing morphology of early postnatal Actb-null or Actg1-null stereocilia (Perrin et al. 2010). Gain-of-function variants of ACTB and ACTG1 are associated with Baraitser–Winter cerebrofrontofacial (BWCFF) syndrome (OMIM #243,310) characterized by craniofacial features (widely spaced eyes, bulbous nose with broad nasal tip and prominent nasal bridge, congenital non-myopathic ptosis, prominent metopic ridge and highly arched eyebrows) and intellectual disability (Baraitser and Winter 1988). The prevalence of sensorineural hearing loss in cases of BWCFF syndrome ranges from 30 to 80% (Riviere et al. 2012; Verloes et al. 2015; Yates et al. 2017). The function gained by ACTB and ACTG1 variants in the BWCFF syndrome appears to be upregulated polymerization of F-actin. Lymphoblasts expressing either of two ACTB variants discovered in BWCFF patients, p.S155F and p.R196H, accumulated F-actin in cytoplasm and increased the number of filopodia-like protrusions (Riviere et al. 2012). Another BWCFF variant of ACTB, p.E117K, assembles into unusually stable F-actin by a combination of increased polymerization speed and a resistance to depolymerization (Johnston et al. 2013). However, gain-of-function ACTB variants do not always result in BWCFF syndrome. Two studies report dystonia-deafness syndrome caused by the gain-of-function variant p.R183W (Procaccio et al. 2006; Skogseid et al. 2018).
Different phenotypes associated with loss-of-function variants of the genes encoding ACTB or ACTG1 proteins indicate that these two actin paralogues cannot completely substitute for a loss of the other despite their nearly identical amino acid sequences. Two elegant studies suggest that both the amino acid sequences and the genomic context including non-coding regions are important for the functions of these two genes. In these studies, genetically-engineered mice using CRISPR/Cas9 and TALEN technologies express ACTG1 and ACTB from the endogenous Actb and Actg1 loci, respectively. Mice expressing ACTG1 protein under the control of the endogenous Actb promotor and mice expressing actin almost identical to ACTB under the control of the Actg1 promotor showed a normal phenotype in overall morphology and appearance of all major organs examined (Vedula and Kashina 2018). Expanding on these observations, a contemporaneous study using mice expressing ACTG1 protein from the endogenous Actb locus, revealed a progressive elevation of the hearing threshold beginning in the high frequencies and degeneration of outer hair cell stereocilia by six months of age in homozygous mice (Patrinostro et al. 2018). Taken together, the interchangeability of the Actb and Actg1 protein coding sequences in the limited number of organs examined indicates that the nucleotide sequence of noncoding regions, such as the promoters and other regulatory elements, the untranslated regions (UTRs) and also the mRNAs translation dynamic differences may be important determinants of unique ACTB and ACTG1 functions (Patrinostro et al. 2018; Vedula and Kashina 2018). It is also apparent that in some cell types, the few amino acid sequence difference between ACTB and ACTG1 is likely responsible for their distinct functions, since these two actins are not able to substitute for one another in the long-term maintenance of stereocilia (Patrinostro et al. 2018), as concluded also from the analysis of the progressive hearing loss phenotype in Actg1-knockout mice (Belyantseva et al. 2009).
A microdeletion of 7p22.1 including the ACTB gene and a missense mutation of ACTB result in a pleiotropic phenotype including malformations of the heart and the renal tract, a distinctive facial feature of interrupted wavy eyebrows, dense eyelashes, a wide nose and mouth and a prominent chin as well as intellectual disability and a developmental delay (Cuvertino et al. 2017; Shimojima et al. 2016). In contrast, dominant missense mutations damaging ACTG1 function predominantly cause DFNA20/26 sensorineural hearing loss (Miyajima et al. 2020; Morell et al. 2000; van Wijk et al. 2003; Zhu et al. 2003). The onset of sensorineural hearing loss in cases of DFNA20/26 ranges widely from first decade to sixth decade (Yuan et al. 2016). Mouse models reveal that ACTG1 likely contributes to repair processes within damaged stereocilia. Mice subjected to intense sound incorporate ACTG1 into gaps within stereocilia F-actin cores (Belyantseva et al., 2009). Mice engineered to express reduced levels of ACTG1 show elevation of ABR thresholds although less so than the severe consequences to hearing loss of Actg1-null mice (Perrin et al. 2010).
Barbed-end cappers and severing proteins associated with human deafness
Currently, mutations in two genes encoding barbed-end cappers, EPS8 and EPS8L2, underlie human deafness genetically mapped to the DFNB102 and DFNB106 loci, respectively (Behlouli et al. 2014; Furness et al. 2013). Epidermal growth factor receptor pathway substrate 8, EPS8, was initially identified as a substrate of EGFR and is involved in the transduction of signals between a small GTPase (Ras) regulating cell proliferation and another small GTPase (Rac) to cause actin remodeling and membrane ruffling in fibroblasts (Fazioli et al. 1993; Scita et al. 1999). EPS8 also binds to the barbed ends of F-actin and can directly inhibit actin elongation (Croce et al. 2004). EPS8L2 (EPS8-like protein 2) is one of three EPS8-related proteins and is predicted to remodel actin cytoskeleton in response to EGF signaling (Tocchetti et al. 2003). Although it is unclear how EPS8 and EPS8L2 contribute to the actin dynamics in stereocilia, experiments using mice suggest that these two proteins bind to the barbed ends of F-actin near the tips of stereocilia and are involved in maintaining the staircase arrangement of stereocilia. EPS8 mainly localizes at the tips of tallest row stereocilia while Eps8L2 distributes to the tips of the mechanosensitive shorter-row stereocilia (Furness et al. 2013; Manor et al. 2011). Consistent with this finding, mutant proteins discovered from patients, EPS8 (p.N30*) and EPS8L2 (p.S339Afs*15), likely disrupt the F-actin-binding domain at the C-termini (Behlouli et al. 2014; Furness et al. 2013). Different distributions of these proteins are likely to be a result of different interacting partners. EPS8 is reported to be a cargo of MYO15 (Hadi et al. 2020; Manor et al. 2011). It is uncertain if EPS8L2 is also a cargo of a myosin or passively diffuses to the tips of stereocilia (Manor et al. 2011).
Knockouts of other F-actin barbed-end cappers and severing proteins are also associated with sensorineural hearing loss in mouse. CapZ is an abundant barbed-end capper in stereocilia. A conditional knockout of CapZB results in shortened or lost stereocilia in mice (Avenarius et al. 2017). Stereocilia contain the severing proteins, cofilin-1 (CFL1) and ADF (DSTN), and also AIP1 (WDR1), an F-actin-binding protein enhancing severing (McGrath et al. 2020; Narayanan et al. 2015). Loss-of-function in ADF and AIP1 causes hearing loss and shortened stereocilia in rows 2 and 3 of the staircase in mice (Narayanan et al. 2015). Stereocilia also contain twinfilin-2 (TWF2), a protein consisting of two ADF homology domains (Paavilainen et al. 2007). However, the exact function of twinfilins in actin dynamics is still unresolved as to whether it binds to ADP–actin, is involved in filament severing, is a barbed-end capper or is an uncapper of barbed ends (Hakala et al. 2021; Poukkula et al. 2011). In addition to other barbed-end cappers and severing proteins, it is likely that TWF2 is involved in length regulation of stereocilia after being transported to tips by MYO7A (Peng et al. 2009; Rzadzinska et al. 2009). Interestingly, knockout of CapZ, ADF and AIP1 results in shortened stereocilia and yet these actin regulatory proteins are thought to suppress elongation or promote depolymerization of existing F-actin.
Actin crosslinkers
Actin filaments in a stereocilium are bundled together by the crosslinkers espin (ESPN), espin-like protein (ESPNL), plastin-1 (PLS1), plastin-3 (PLS3), fascin-1 (FSCN1), fascin-2 (FSCN2), GRXCR1, GRXCR2, Xin-related protein 2 (XIRP2) and TRIOBP (Kitajiri et al. 2010; Krey et al. 2016; Odeh et al. 2010; Scheffer et al. 2015; Shin et al. 2010; Zheng et al. 2000). Among the genes encoding these actin crosslinkers, pathogenic recessive variants of GRXCR1, TRI-OBP, ESPN and GRXCR2 are associated with hearing loss in human at the DFNB25, DFNB28, DFNB36 and DFNB101 loci, respectively (Imtiaz et al. 2014; Kitajiri et al. 2010; Naz et al. 2004; Schraders et al. 2010). One case study reported that three missense mutations in PLS1 cause autosomal-dominant hearing loss (Morgan et al. 2019). Experimental demonstration using a functional assay for pathogenicity of these substitutions would add strength to this conclusion.
Most of the actin crosslinkers, such as espin (ESPN), plastin1 (PLS1) and fascin-2 (FSCN2), are found along the entire length of stereocilia F-actin cores and contribute to the paracrystal-like packing of actin filaments (Krey et al. 2016; Sekerkova et al. 2011). The Jerker mouse has a mutation in the Espn gene and develops thin, short stereocilia compared to the wild-type (Sekerkova et al. 2011). Two studies reported that stereocilia lacking PLS1 show hexagonal packing of F-actin, which is different from heterogenous F-actin packing in wild-type stereocilia (Krey et al. 2016; Metlagel et al. 2019). However, these two reports describe discrepant phenotypes of Pls1-null stereocilia perhaps due to different methodologies. Krey and co-authors reported a decrease in the diameters of Pls1-null stereocilia. In this study, utricular stereocilia were embedded in agarose melted at 42 °C, detached from cell bodies after cooling and then fixed in paraformaldehyde. Metlagel et al. performed CryoET using cryofixed vestibular stereocilia and reported an increase in the diameters of Pls1-null stereocilia. Therefore, morphological change of Pls1-null stereocilia is an unresolved question although Pls1-null and wild-type stereocilia may well behave differently during the treatments used in these studies. Mice expressing mutant FSCN2 (p.R102H) develop stereocilia with heterogeneous packing, but the distance between filaments is larger than those in wild-type stereocilia (Krey et al. 2016). The F-actin packing is disorganized when XIRP2 is lacking (Scheffer et al. 2015). Absence of functional GRXCR1 in pirouette mice results in thin and slightly shortened stereocilia. GRXCR1 localizes along the entire length of auditory and vestibular stereocilia (Odeh et al. 2010) and based on data from zebrafish, may regulate the assembly of harmonin-sans Usher protein complex (Blanco-Sanchez et al., 2018).
In contrast to other crosslinkers, TRIOBP is unique in its localization to stereocilia rootlets and actin-based cytoskeletal structures in supporting cells of the organ of Corti (Kitajiri et al. 2010). The TRIOBP/Triobp genes in human and mouse encode three major isoform classes, TRIOBP-1, TRIOBP-4 and TRIOBP-5. Mice lacking both the TRIOBP-4 and TRIOBP-5 are congenitally profoundly deaf although initially stereocilia appear morphologically normal. Subsequently, stereocilia rootlets fail to develop and hair cells in these mice degenerate by P16. In contrast, stereocilia rootlets in mice lacking only TRIOBP-5 are thin and dysmorphic. Data from a recent study indicate that TRIOBP-5 is critical to form thick, stiff rootlets (Katsuno et al. 2019). Mice lacking TRIOBP-5 but expressing TRIOBP-4 develop less rigid stereocilia vulnerable to external force. The reinforcement of rootlets is likely provided by the ability of TRIOBP-5 to homopolymerize by its C-terminal coiled-coil domains since lack of these domains leads to frequent rootlet breakage and malformations. Loss of TRIOBP-5 also causes an expansion of TRIOBP-4 bundling within the actin core, which extends up to stereocilia tips, and these mice show progressive hearing loss (Katsuno et al. 2019) as do humans with pathogenic variants of TRIOBP that are limited to exons unique to TRIOBP-5 mRNA (Pollak et al. 2017; Wesdorp et al. 2017).
Variants of myosins and cadherins associated with human deafness
Myosins move on F-actin using the energy of ATP hydrolysis. Myosins are classified into “conventional” myosins (also known as myosin II) originally found in muscles and “unconventional” myosins that were discovered in many other tissues (Cheney and Mooseker 1992). Two genes encoding conventional, non-muscle myosins, MYH9 and MYH14, are associated with autosomal-dominant hearing loss DFNA17 and DFNA4A, respectively, although their exact functions in the inner ear are still debated (Donaudy et al. 2004; Lalwani et al. 2000). Among unconventional myosins, variants of MYO3A, MYO6, MYO7A and MYO15A are linked to autosomal recessive hearing loss, DFNB30, DFNB37, DFNB2 and DFNB3, respectively (Ahmed et al. 2003a; Liu et al. 1997; Walsh et al. 2002; Wang et al. 1998; Weil et al. 1997). Variants of MYO6 and MYO7A are also associated with autosomal-dominant hearing loss, DFNA22 and DFNA11, respectively (Liu et al. 1997; Melchionda et al. 2001; Weil et al. 1997). Furthermore, mutations in MYO7A cause the most severe Type 1 form of Usher syndrome characterized by congenital, bilateral, profound sensorineural hearing loss, vestibular areflexia and adolescent-onset retinitis pigmentosa (Koenekoop et al. 1993). A few studies report that missense mutations in the kinase domain or motor domain of MYO3A can cause autosomal-dominant hearing loss (Dantas et al. 2018; Doll et al. 2020; Grati et al. 2016). Cargo transport in stereocilia is a major function of unconventional myosins (Friedman et al. 2020).
The amino acid sequence of the motor domains of unconventional and conventional myosins is well conserved, although their kinetic properties are adapted for unique functions (Bird et al. 2014). In contrast, the tail domains of myosins are divergent and have evolved several functions including auto-regulating the activity of its motor domain and delivering a variety of cargo including proteins and lipids (Li and Zhang 2020; Li et al. 2008; Yao et al. 2015). Among the unconventional myosins currently associated with hearing loss, MYO6, MYO7A and MYO15A have well-folded tail domains, while the tail domain of MYO3A is predicted to be almost unfolded (Li and Zhang 2020). The tail domain of MYO6 is characterized by two consecutively arranged regions for cargo binding, an N-terminal helical cargo-binding domain (HCBD) and a C-terminal cargo-binding domain (CBD), which is likely to interact with a protein tyrosine phosphatase receptor type Q (PTPRQ), a membrane protein crucial to tether the stereocilia membrane to F-actin (Goodyear et al. 2003; Sakaguchi et al. 2008). MYO6 is the only unconventional myosin that moves toward the pointed end of F-actin at the base of stereocilia and is thought to transport PTPRQ toward the base of stereocilia on the apical surface of hair cells (Avraham et al. 1997). The loss of MYO6 in the deaf Snell’s waltzer mouse (sv) results in fused stereocilia and degeneration of the organ of Corti (Avraham et al. 1995).
Cadherin 23 (CDH23) and protocadherin 15 (PCDH15) are two proteins comprising stereocilia tip-links that gate the mechanotransduction channel and are encoded by the CDH23 and PCDH15 genes, respectively. Recessive variants of CDH23 and PCDH15 are associated either with type 1 Usher syndrome 1D and 1F or non-syndromic deafness DFNB12 or DFNB23, respectively, (Ahmed et al. 2003b; Bork et al. 2001). MYO7A is concentrated at the upper tip-link density (Grati and Kachar 2011) near the intracellular domain of cadherin 23 by binding to the CEN region of USH1G (SANS) using the N-terminal MyTH4-FERM (NMF) and to the PDZ3 domain of USH1C (Harmonin) using the C-terminal MyTH4-FERM domain (CMF) (Li et al. 2016; Wu et al. 2011). The tail of MYO15A also has tandem MyTH4-FERM domains structurally homologous to that of MYO7A although there is a ~ 370-residue insertion within the F1 domain of NMF (Liang et al. 1999). The short isoform of MYO15A that lacks the long N-terminal domain elongates stereocilia to a predetermined length (Belyantseva et al., 2003) and forms an elongation complex at the tips of stereocilia by transporting whirlin, a PDZ-domain-containing scaffold protein, using the PDZ ligand of MYO15A (Belyantseva et al. 2005). The short isoform of MYO15A is also reported to transport the barbed-end capper EPS8 to stereocilia tips and controls the lengths and probably the diameters of stereocilia (Hadi et al. 2020; Manor et al. 2011). The long isoform of MYO15A is localized at the lower tip-link density near PCDH15 and is essential for maintaining the length of the mechanotransducing stereocilia (Fang et al., 2015).
The MYO3A tail domain is mostly disordered but has a conserved THDI (tail homology domain I), which interacts with the espin isoform 1 (Espin1) and boosts elongation of stereocilia (Salles et al. 2009). Through the ARB1 and ARB2 motifs in the THDI domain, MYO3A interacts with two Espin1 molecules via the Ankyrin (ANK) repeats (Liu et al. 2016). Double-knockout mice of MYO3A and MYO3B result in an abnormal arrangement of stereocilia bundles with a deficient staircase architecture (Lelli et al. 2016).
Scaffolding proteins
Scaffolding proteins provide linkages between F-actin of the cell cortex and membrane proteins (Bretscher et al. 2002). Among the genes encoding scaffold proteins in stereocilia, harmonin (USH1C), radixin (RDX), whirlin (WHRN) and PDZD7 are associated with autosomal recessive hearing loss DFNB18, DFNB24, DFNB31 and DFNB57, respectively (Ahmed et al. 2002; Booth et al. 2015; Khan et al. 2007; Mburu et al. 2003; Ouyang et al. 2002). More debilitating variants of USH1C and USH1G encoding harmonin (USH1C) and SANS (USH1G) also cause Usher syndrome type 1 (Bitner-Glindzicz et al. 2000; Weil et al. 2003), while variants of whirlin are associated with Usher syndrome type 2D (Ebermann et al. 2007). RDX is a member of the ERM family of three proteins (ezrin, radixin and moesin) characterized by an N-terminal FERM (Four-point one, ezrin, radixin, moesin) domain and a C-terminal F-actin-binding domain (Bretscher et al. 2002). The FERM domain of RDX is reported to interact with several membrane proteins including CD43, CD44 and ICAM-1/2/3, but the partners of RDX in hair cells are yet to be determined (Shaw 2001; Tsukita et al. 1994; Yonemura et al. 1998). Rdx-null mice show degeneration of cochlear stereocilia at approximately P14 and subsequent hearing loss although there is no obvious malformation of stereocilia at P1 (Kitajiri et al. 2004). In Rdx-null mice, in vestibular hair cells but not cochlear hair cells, a radixin deficiency appears to be partially substituted for by ezrin, another member of the ERM family (Kitajiri et al. 2004). Whirlin (WHRN) is a PDZ-domain-containing scaffold protein, which is transported via an interaction with the PDZ ligand of MYO15A (Belyantseva et al. 2005). Whrn mutant mice (whirler) are deaf and have short stereocilia arrested in an immature state (Mogensen et al. 2007). PDZD7 has three PDZ domains, a similar domain composition to harmonin and whirlin, and is involved in scaffolding the ankle-link complex (Du et al. 2020; Grati et al. 2012). Monoallelic deleterious mutations of PDZD7 and GPR98 were reported as a digenic case of Usher syndrome type 2 (Ebermann et al. 2010).
Harmonin (USH1C) and SANS (USH1G), encoded by the Usher syndrome genes USH1C and USH1G, are transported to stereocilia upper tip-link densities by binding to the MyTH4-FREM domain of MYO7A (Grati and Kachar 2011). These proteins are necessary for tensioning tip-links that gate the mechanotransduction channels near the tips of stereocilia (Kros et al. 2002; Li et al. 2016; Wu et al. 2011). Among the three splice isoforms of harmonin (harmonin-a, -b and -c), cochlear hair cells express the longest isoform, harmonin-b, which has three PDZ domains (Verpy et al. 2000). Harmonin-b interacts with CDH23 both in retina and in the upper tip-link density using one or two PDZ domains (PDZ1 and PDZ2 in retina; PDZ2 in stereocilia) (Siemens et al. 2002). The PDZ3 domain interacts with the MyTH4-FERM domain of MYO7A (Yu et al. 2017). Mice expressing harmonin-b with a missense mutation in the PDZ2 domain are deaf with mislocalized CDH23 and disrupted morphology of stereocilia (Grillet et al. 2009). Harmonin-b also binds directly to F-actin using sequence between its PDZ2 and PDZ3 domains (Michalski et al. 2009). SANS is another critical component of the upper tip-link complex with the C-terminal SAM-PBM domains interacting with the PDZ1 domain of harmonin-b (Yu et al. 2017). Ush1g−/− mice show hearing loss due to the lack of mechanoelectrical transduction currents (Caberlotto et al. 2011).
DIAPH1 and DFNA1 deafness
Dominant variants of DIAPH1 are associated with DFNA1 hearing loss (Lynch et al. 1997). The pathology associated with variants of DFNA1 is unique from other F-actin regulatory proteins. DIAPH1 is a formin homology protein with a processive F-actin elongation activity (Fig. 3a). The average speed of movement can reach 2.0 μm/s, which corresponds to polymerization of ~ 700 actin subunit per second (Higashida et al. 2004). With such aggressive actin elongation activity, ectopic expression of DIAPH1 forms a large amount of F-actin in cultured cells (Watanabe et al. 1997). The actin elongation activity of FH1 and FH2 domains is suppressed by an autoinhibitory interaction between DID and DAD domains of DIAPH1, which is released by the binding of Rho, a small GTPase, to the DIAPH1 GBD domain (Watanabe et al. 1997). The function of DIAPH1 appears to be unnecessary to the development of stereocilia although abnormal activation of DIAPH1 is pathogenic (Ueyama et al. 2016). Loss of DIAPH1 function is associated with syndromic microcephaly, blindness and early-onset seizures in human but not with hearing loss (Al-Maawali et al. 2016; Ercan-Sencicek et al. 2015). Neither hearing loss nor abnormal morphology of inner ear was observed in mice lacking a functional Diaph1 gene (Ueyama et al. 2016). However, a constitutively active mutation of DIAPH1 that has a truncated DAD domain (p.R1213X) does cause hearing loss both in human and mouse as a result of stereocilia degeneration (Ueyama et al. 2016). Variants of DIAPH1, such as p.A265S, p.A1210Sfs*31, p.A1210GfsX3130 and p.E1192_Q1220del30, located in the DID and DAD domains have been identified in families segregating hearing loss, suggesting a relationship between a gain-of-function of DIAPH1 and hearing loss (Kim et al. 2019; Neuhaus et al. 2017; Westbury et al. 2018). In mice, ectopically expressed GFP-DIAPH1 (p.R1213X), a constituitively active form, is observed at stereocilia tips and in the apical surface of hair cells as well as in Deiters’ cells and pillar cells (Ninoyu et al. 2020). Although the DFNA1 inner ear pathology associated with variants of DIAPH1 is still not entirely understood, we hypothesize that abnormally formed F-actin, monomer depletion or both due to upregulated actin polymerization may disturb the normal, routine maintenance of stereocilia F-actin cores.
Fig. 3.
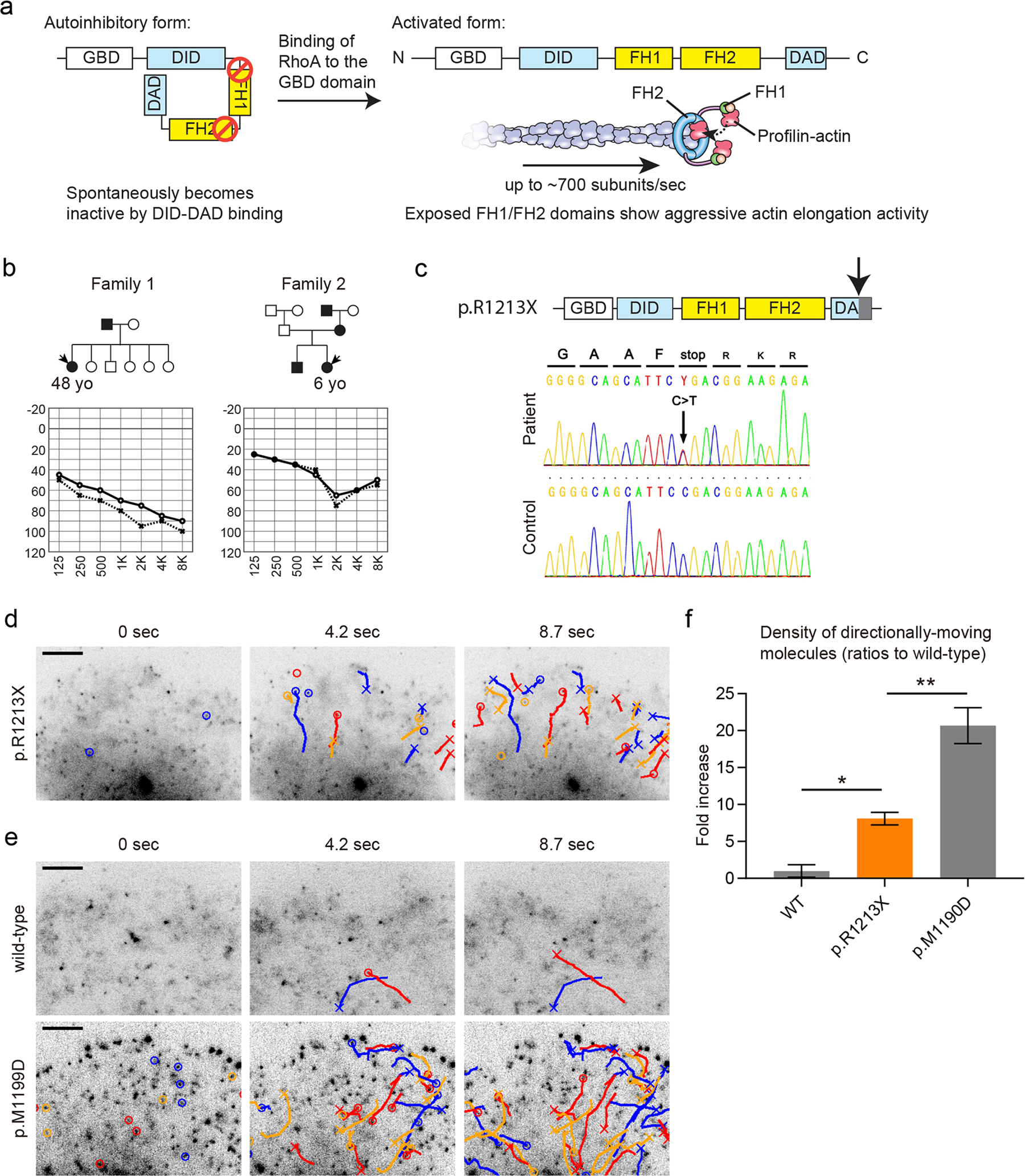
Functional analysis of the DIAPH1 (p.R1213X) variant using single-molecule fluorescence microscopy. a Autoinhibitory mechanism regulating DIAPH1 actin elongation activity. The DID and DAD domains spontaneously interact with each other. Binding of RhoA to the GBD domain of DIAPH1 releases inhibition and allows for aggressive actin elongation activity by the FH1-FH2 domains of DIAPH1. The FH1 domain recruits profilin-actin while the FH2 domain holds onto the barbed end of F-actin to cause processive and high-speed actin elongation, which reaches an average speed of ~ 700 actin subunits/sec. b Two Japanese families segregating hearing loss analyzed to identify the DIAPH1 (p.R1213X) variant. Audiograms of probands are also shown. c Diagrams and Sanger sequencing results of DIAPH1 (p.R1213X) variant found in families shown in (b). The p.R1213X variant truncates the C-terminus of the DAD domain (downward pointing arrow). The DNA sequence is from the proband in Family 1. d Detection of abnormal, constitutive activation of DIAPH1 (p.R1213X) using single-molecule fluorescence microscopy. Time-lapse images of Xenopus XTC cells expressing GFP-DIAPH1 (p.R1213X) are shown. GFP-DIAPH1 (p.R1213X) molecules show frequent directional movements driven by actin elongation activity of FH1-FH2 domains, which were abnormally exposed by the disrupted DID-DAD interaction. Molecules moving directionally for more than two frames are indicated by circles and trajectories. Crosses indicate disappearances. Bar, 5 μm. e Time-lapse images of negative and positive controls, wild-type GFP-DIAPH1 and GFP-DIAPH1 (p.M1199D). Only a few molecules show directional movements in XTC cells expressing wild-type GFP-DIAPH1. Moving molecules were frequently observed in XTC cells expressing GFP-DIAPH1 (p.M1199D), which is a variant lacking the DID-DAD autoinhibitory interaction (Lammers et al. 2005). Bar, 5 μm. f Semi-quantitative comparison of directionally moving molecules showing moderate activation of DIAPH1 (p.R1213X). The densities of moving molecules were determined and normalized to the expression levels of GFP-DIAPH1 as described in our previous study (Ueyama et al. 2016). Directional movements of DIAPH1 (p.R1213X) were significantly more frequent than wild-type DIAPH1 and less frequent than DIAPH1 (p.M1199D). One-way ANOVA showed p < 0.0001. Post hoc Tukey’s multiple comparison is indicated by asterisk (p < 0.05) and double asterisks (p < 0.01). Number of replicates: n = 5 for wild-type, n = 6 for p.R1213X and p.M1199D. Error bars, SEMs
Single-molecule fluorescence microscopy for functional analysis of DIAPH1 variants
Here, we introduce our experimental approach to use single-molecule fluorescence microscopy in cultured cells for functional analyses of novel DIAPH1 variants (Figs. 3 and 4). In Xenopus XTC cells expressing GFP-DIAPH1, single-molecule fluorescence microscopy can visualize processive actin elongation activities of DIAPH1 molecules as directional movements of fluorescent puncta (Higashida et al. 2004). Using this technique, we evaluated the actin elongation activity of a novel p.R1213X variant found in two Japanese families segregating hearing loss (Fig. 3b and c). This variant was predicted to show abnormal, constitutively active actin elongation activity due to the truncation at the C-terminus of the autoinhibitory DAD domain. We prepared XTC cells expressing GFP-DIAPH1 (p.R1213X) and evaluated the frequency of directionally moving molecules (Fig. 3d, Movie S1, indicated by circles and trajectories in time-lapse images) (Ueyama et al. 2016). We also prepared XTC cells expressing wild-type GFP-DIAPH1 for a negative control and GFP-DIAPH1 (p.M1199D) for a positive control, which is a mutant with no DID-DAD interaction (Fig. 3e, Movie S1, indicated by circles and trajectories in time-lapse images) (Lammers et al. 2005). Directionally moving molecules were frequently observed in cells expressing GFP-DIAPH1 (p.R1213X) and GFP-DIAPH1 (p.M1199D) compared with cells expressing wild-type GFP-DIAPH1. These data indicate the disrupted DID–DAD autoinhibitory interaction of these two variants. We also performed semi-quantitative comparison using the density of moving molecules normalized to the expression levels of GFP-DIAPH1 (Ueyama et al. 2016). DIAPH1 (p.R1213X) was considered to be moderately active since the density of activated DIAPH1 (p.R1213X) molecules was significantly higher than wild-type DIAPH1 and also lower than DIAPH1 (p.M1199D) (Fig. 3f). From the phenotype of transgenic mice expressing DIAPH1 (p.R1213X), which show degeneration of stereocilia after approximately 10 weeks of age, we concluded that abnormal actin elongation activity of DIAPH1 is associated with DFNA1 deafness (Ueyama et al. 2016).
Fig. 4.
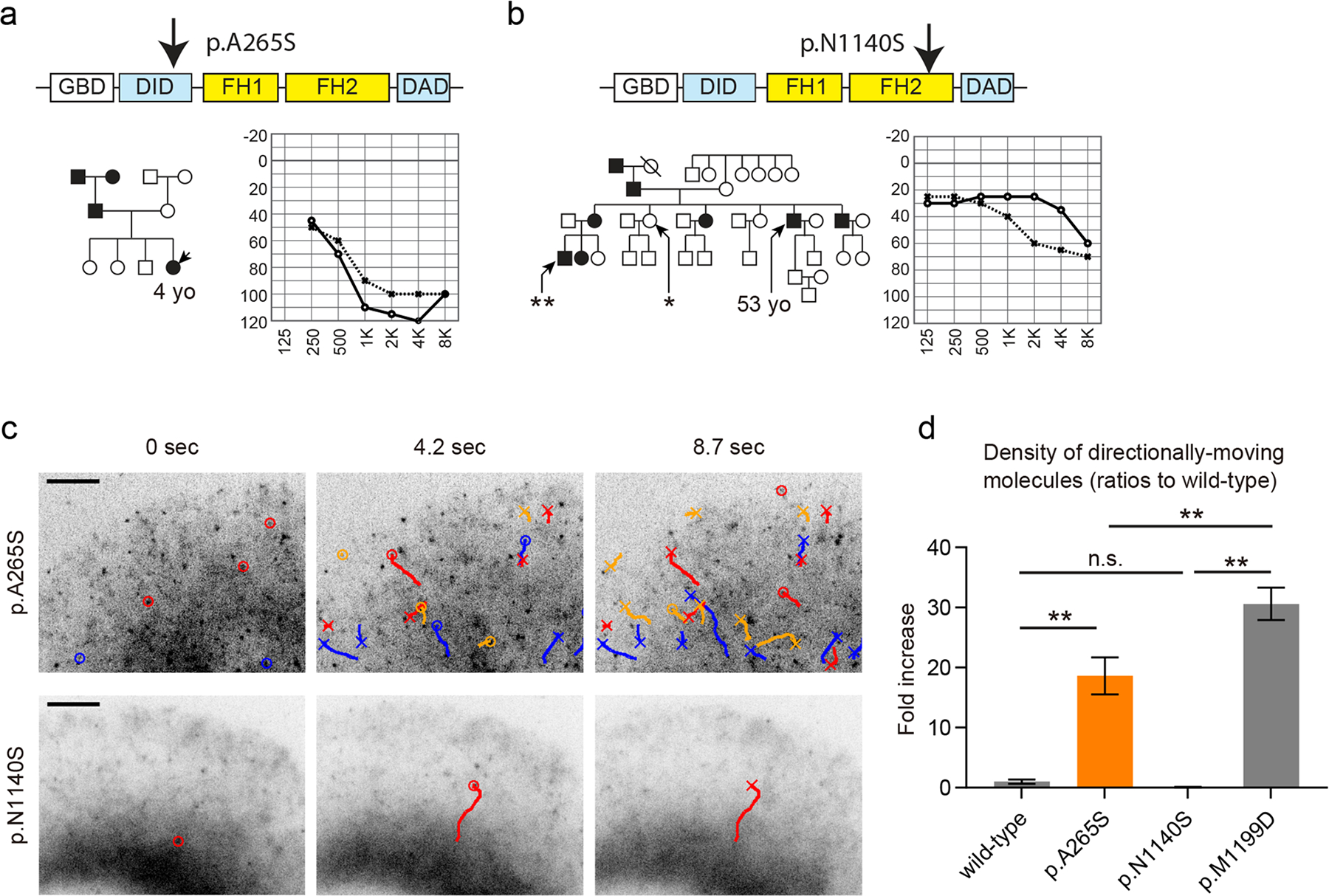
Pathogenicity screening of DIAPH1 variants using single-molecule fluorescence microscopy. a A Korean family segregating hearing loss. The p.A265S variant was found in this family and considered to be pathogenic using single-molecule fluorescence microscopy analyses in (c). An audiogram of the proband is shown. b A Japanese family segregating hearing loss. The p.N1140S variant was found in the proband of this family and predicted to be a benign polymorphism based upon our single-molecule fluorescence microscopy assay in (c). The p.N1140S variant was subsequently found in a female with normal hearing (asterisk), and another male with hearing loss in this family did not have the p.N1140S variant (double asterisks). An audiogram of the proband is shown. c Functional analyses of the p.A265S and p.N1140S variants using single-molecule fluorescence microscopy and Xenopus XTC cells expressing GFP-fused variants. GFP-DIAPH1 (p.A265S) showed frequent directional movements similar to p.R1213X indicating the pathogenicity of p.A265S variant. In contrast, the p.N1140S variant is likely a benign polymorphism since GFP-DIAPH1 (p.N1140S) did now show frequent directional movements. The movement distances were also similar to wild-type DIAPH1 despite the amino-acid substitution in the FH2 domain. Indications of markers are similar to Fig. 3d. Bar, 5 μm. d Semi-quantitative comparison of directionally moving molecules showing the constitutive activation of DIAPH1 (p.A265S) and the intact autoinhibition of DIAPH1 (p.N1140S). The densities of moving molecules were normalized by the expression levels as described in our previous study (Ueyama et al. 2016). Directional movements of DIAPH1 (p.A265S) were significantly more frequent than wild-type DIAPH1 and less frequent than DIAPH1 (p.M1199D) while DIAPH1 (p.N1140S) did not show an increase of moving molecules. New XTC cells were prepared to obtain data for the wild-type and p.M1199D in this graph. One-way ANOVA showed p < 0.0001. Post-hoc Tukey’s multiple comparison is indicated by double asterisks (p < 0.01). Number of replicates: n = 5 for p.N1140S, n = 6 for wild-type, p.A265S and p.M1199D. Error bars, SEMs
Single-molecule fluorescence microscopy was also used as a tool to screen for the pathogenicity of novel variants of DIAPH1 (Fig. 4). Two missense mutations, p.A265S and p.N1140S, were identified in a Korean family and a Japanese family, respectively (Fig. 4, a and b). The p.A265S variant was predicted to be pathogenic since GFP-DIAPH1 (p.A265S) expressed in XTC cells showed frequent directional movements (Fig. 4c, Movie S2, indicated by circles and trajectories in time-lapse images) similar to p.R1213X (Fig. 3d) (Kim et al. 2019). These data suggest that the p.A265S variant in the DID domain disrupts the autoinhibitory DID–DAD interaction. In contrast, the p.N1140S variant appears to be a benign polymorphism since GFP-DIAPH1 (p.N1140S) expressed in XTC cells showed only a few directional movements (Fig. 4c, Movie S2, indicated by circles and trajectories in time-lapse images), which is a phenotype indistinguishable from XTC cells expressing wild-type GFP-DIAPH1 (Fig. 3e). The p.N1140S substitution in the FH2 domain was unlikely to disrupt processive actin elongation activity of DIAPH1 since GFP-DIAPH1 (p.N1140S) could travel long distances similarly to wild-type GFP-DIAPH1. Subsequently, the p.N1140S variant was found in a healthy control in the same pedigree (Fig. 4b, a female with asterisk), and another patient in this pedigree with hearing loss did not have this variant (Fig. 4b, a male double asterisks) supporting the assumption that this variant of DIAPH1 is a benign polymorphism. Semi-quantitative comparison also suggests that p.A265S is moderately active while the autoinhibition of p.N1140S is intact (Fig. 4d). Compared with conventional, macroscopic biochemistry experiments, our single-molecule microscopy-based approach can directly visualize the function of molecules, such as the autoinhibitory DID–DAD interaction of DIAPH1 and the actin elongation activity of FH1–FH2 domains. As we demonstrated in this section, direct analysis of a protein function could represent an efficient methodology to screen variants found in patients with hearing loss.
Conclusion
In the analyses of multiplex pedigrees segregating hearing loss, recent advances in whole exome or genome sequencing permit exhaustive searches for candidate variants. However, it remains even more challenging in affected isolated cases of deafness to determine if a protein variant or a noncoding variant is truly pathogenic or a rare benign polymorphism. Thus, individuals with rare inherited disorders may be left without fully solved molecular genetic diagnoses. Methodologies that can directly evaluate the function of protein variants, such as single-molecule fluorescence microscopy, may be another valuable tool in the armamentarium to experimentally evaluate putative pathogenic variants of hearing loss.
Actin and its regulatory proteins form a dynamic system that is continuously remodeled in living cells. Stereocilia are also dynamic structures that, while maintaining the overall shape due to a relative stability of the F-actin core, possess an ongoing replenishment of many of their constituents, such as actin monomers and crosslinkers, through their constant binding and dissociation. Variants in human and mouse actin and partner proteins can disrupt this “dynamic equilibrium” in stereocilia resulting in abnormal morphological changes, damaged mechanotransduction and often sensorineural hearing loss caused by the death or dysfunction of hair cells. Single-molecule fluorescence microscopy helps to elucidate the molecular mechanisms of hearing loss as we detected abnormal actin elongation activity of DIAPH1 variants. Insight from a molecular diagnosis and understanding the underlying wild-type and defective functions are important for making headway towards therapeutics to protect and restore hearing.
Supplementary Material
Acknowledgements
We thank Drs. Mhamed Grati and Dennis Winkler for valuable comments and Ms. Erina He for her beautiful diagrams. Informed written consent was obtained from all subjects. This study was approved by the Ethics Committee of Shinshu University School of Medicine (No. 576: 2 May 2017), the Institutional Review Board of Seoul National University Bundang Hospital (IRB-B-1007-105-402) and the ethics committees of all other participating institutions listed in a previous report (Nishio and Usami 2015).
Funding
TM, IAB and TBF were supported (in part) by NIDCD intramural research funds DC000039. HS was supported by NIBIB intramural research funds. This review was also supported by JSPS Overseas Research Fellowships to TM and a research fund from Chungnam National University to BJK. SU was supported by the Health and Labor Sciences Research Grant for Research on Rare and Intractable Diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labor and Welfare of Japan (20FC1048) and Grants-in-Aid from the Japan Agency for Medical Research and Development (AMED) (20ek0109363h0003). BYC was funded by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHDI), the ministry of Health & Welfare, Republic of Korea (2018R1A2B2001054) and SNUBH Intramural research fund (16-2019-006).
Abbreviations
- ATP
Adenosine triphosphate
- ADP
Adenosine diphosphate
- DFNA
Locus for dominantly inherited nonsyndromic deafness
- DFNB
Locus for recessively inherited nonsyndromic deafness
- GFP
Green fluorescent protein
- USH
Usher syndrome
Footnotes
Declarations
Conflict of interests The authors declare no conflict of interest associated with this manuscript.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00439-021-02304-0.
References
- Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, Menon PS, Deshmukh D, Griffith AJ, Riazuddin S, Friedman TB, Wilcox ER (2002) Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet 110:527–531. 10.1007/s00439-002-0732-4 [DOI] [PubMed] [Google Scholar]
- Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, Mohiddin SA, Fananapazir L, Caruso RC, Husnain T, Khan SN, Riazuddin S, Griffith AJ, Friedman TB, Wilcox ER (2003a) Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet 72:1315–1322. 10.1086/375122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Riazuddin S, Griffith AJ, Friedman TB, Belyantseva IA, Wilcox ER (2003b) PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 12:3215–3223. 10.1093/hmg/ddg358 [DOI] [PubMed] [Google Scholar]
- Alderson-Day B, Fernyhough C (2015) Inner speech: development, cognitive functions, phenomenology, and neurobiology. Psychol Bull 141:931–965. 10.1037/bul0000021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Maawali A, Barry BJ, Rajab A, El-Quessny M, Seman A, Coury SN, Barkovich AJ, Yang E, Walsh CA, Mochida GH, Stoler JM (2016) Novel loss-of-function variants in DIAPH1 associated with syndromic microcephaly, blindness, and early onset seizures. Am J Med Genet A 170A:435–440. 10.1002/ajmg.a.37422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura S (1961) The interaction between G-actin and ATP. Arch Biochem Biophys 92:140–149. 10.1016/0003-9861(61)90228-4 [DOI] [PubMed] [Google Scholar]
- Avenarius MR, Krey JF, Dumont RA, Morgan CP, Benson CB, Vijayakumar S, Cunningham CL, Scheffer DI, Corey DP, Muller U, Jones SM, Barr-Gillespie PG (2017) Heterodimeric capping protein is required for stereocilia length and width regulation. J Cell Biol 216:3861–3881. 10.1083/jcb.201704171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA (1995) The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat Genet 11:369–375. 10.1038/ng1295-369 [DOI] [PubMed] [Google Scholar]
- Avraham KB, Hasson T, Sobe T, Balsara B, Testa JR, Skvorak AB, Morton CC, Copeland NG, Jenkins NA (1997) Characterization of unconventional MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice. Hum Mol Genet 6:1225–1231. 10.1093/hmg/6.8.1225 [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Harris HE, Weeds AG (1980) Partial purification and characterization of an actin depolymerizing factor from brain. FEBS Lett 121:178–182. 10.1016/0014-5793(80)81292-0 [DOI] [PubMed] [Google Scholar]
- Baraitser M, Winter RM (1988) Iris coloboma, ptosis, hypertelorism, and mental retardation: a new syndrome. J Med Genet 25:41–43. 10.1136/jmg.25.1.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behlouli A, Bonnet C, Abdi S, Bouaita A, Lelli A, Hardelin JP, Schietroma C, Rous Y, Louha M, Cheknane A, Lebdi H, Boudjelida K, Makrelouf M, Zenati A, Petit C (2014) EPS8, encoding an actin-binding protein of cochlear hair cell stereocilia, is a new causal gene for autosomal recessive profound deafness. Orphanet J Rare Dis 9:55. 10.1186/1750-1172-9-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, Griffith AJ, Friedman TB (2005) Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 7:148–156. 10.1038/ncb1219 [DOI] [PubMed] [Google Scholar]
- Belyantseva IA, Perrin BJ, Sonnemann KJ, Zhu M, Stepanyan R, McGee J, Frolenkov GI, Walsh EJ, Friderici KH, Friedman TB, Ervasti JM (2009) Gamma-actin is required for cytoskeletal maintenance but not development. Proc Natl Acad Sci U S A 106:9703–9708. 10.1073/pnas.0900221106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron SE, Zhu M, Thiem SM, Friderici KH, Rubenstein PA (2010) Ion-dependent polymerization differences between mammalian beta- and gamma-nonmuscle actin isoforms. J Biol Chem 285:16087–16095. 10.1074/jbc.M110.110130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird JE, Takagi Y, Billington N, Strub MP, Sellers JR, Friedman TB (2014) Chaperone-enhanced purification of unconventional myosin 15, a molecular motor specialized for stereocilia protein trafficking. Proc Natl Acad Sci U S A 111:12390–12395. 10.1073/pnas.1409459111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, Barnes PD, O’Brien RE, Farndon PA, Sowden J, Liu XZ, Scanlan MJ, Malcolm S, Dunne MJ, Aynsley-Green A, Glaser B (2000) A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat Genet 26:56–60. 10.1038/79178 [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Pollard TD (2002) Hydrolysis of ATP by polymerized actin depends on the bound divalent cation but not profilin. Biochemistry 41:597–602. 10.1021/bi011214b [DOI] [PubMed] [Google Scholar]
- Booth KT, Azaiez H, Kahrizi K, Simpson AC, Tollefson WT, Sloan CM, Meyer NC, Babanejad M, Ardalani F, Arzhangi S, Schnieders MJ, Najmabadi H, Smith RJ (2015) PDZD7 and hearing loss: More than just a modifier. Am J Med Genet A 167A:2957–2965. 10.1002/ajmg.a.37274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Riazuddin S, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ (2001) Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 68:26–37. 10.1086/316954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A, Edwards K, Fehon RG (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 3:586–599. 10.1038/nrm882 [DOI] [PubMed] [Google Scholar]
- Caberlotto E, Michel V, Foucher I, Bahloul A, Goodyear RJ, Pepermans E, Michalski N, Perfettini I, Alegria-Prevot O, Chardenoux S, Do Cruzeiro M, Hardelin JP, Richardson GP, Avan P, Weil D, Petit C (2011) Usher type 1G protein sans is a critical component of the tip-link complex, a structure controlling actin polymerization in stereocilia. Proc Natl Acad Sci U S A 108:5825–5830. 10.1073/pnas.1017114108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheney RE, Mooseker MS (1992) Unconventional myosins. Curr Opin Cell Biol 4:27–35. 10.1016/0955-0674(92)90055-h [DOI] [PubMed] [Google Scholar]
- Chien WW, Isgrig K, Roy S, Belyantseva IA, Drummond MC, May LA, Fitzgerald TS, Friedman TB, Cunningham LL (2016) Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Mol Ther 24:17–25. 10.1038/mt.2015.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien WW, Isgrig K, Roy S, Belyantseva IA, Drummond MC, May LA, Fitzgerald TS, Friedman TB, Cunningham LL (2016) Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Mol Ther 24(1):17–25. 10.1038/mt.2015.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K, Flick R, Close L, Shelly D, Paul R, Bove K, Kumar A, Lessard J (2002) Mice lacking skeletal muscle actin show reduced muscle strength and growth deficits and die during the neonatal period. Mol Cell Biol 22:5887–5896. 10.1128/mcb.22.16.5887-5896.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce A, Cassata G, Disanza A, Gagliani MC, Tacchetti C, Malabarba MG, Carlier MF, Scita G, Baumeister R, Di Fiore PP (2004) A novel actin barbed-end-capping activity in EPS-8 regulates apical morphogenesis in intestinal cells of Caenorhabditis elegans. Nat Cell Biol 6:1173–1179. 10.1038/ncb1198 [DOI] [PubMed] [Google Scholar]
- Cuvertino S, Stuart HM, Chandler KE, Roberts NA, Armstrong R, Bernardini L, Bhaskar S, Callewaert B, Clayton-Smith J, Davalillo CH, Deshpande C, Devriendt K, Digilio MC, Dixit A, Edwards M, Friedman JM, Gonzalez-Meneses A, Joss S, Kerr B, Lampe AK, Langlois S, Lennon R, Loget P, Ma DYT, McGowan R, Des Medt M, O’Sullivan J, Odent S, Parker MJ, Pebrel-Richard C, Petit F, Stark Z, Stockler-Ipsiroglu S, Tinschert S, Vasudevan P, Villa O, White SM, Zahir FR, Study DDD, Woolf AS, Banka S (2017) ACTB Loss-of-Function Mutations Result in a Pleiotropic Developmental Disorder. Am J Hum Genet 101:1021–1033. 10.1016/j.ajhg.2017.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas VGL, Raval MH, Ballesteros A, Cui R, Gunther LK, Yamamoto GL, Alves LU, Bueno AS, Lezirovitz K, Pirana S, Mendes BCA, Yengo CM, Kachar B, Mingroni-Netto RC (2018) Characterization of a novel MYO3A missense mutation associated with a dominant form of late onset hearing loss. Sci Rep 8:8706. 10.1038/s41598-018-26818-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll J, Hofrichter MAH, Bahena P, Heihoff A, Segebarth D, Muller T, Dittrich M, Haaf T, Vona B (2020) A novel missense variant in MYO3A is associated with autosomal dominant high-frequency hearing loss in a German family. Mol Genet Genomic Med 8:e1343. 10.1002/mgg3.1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez R (2009) Actin filament nucleation and elongation factors–structure-function relationships. Crit Rev Biochem Mol Biol 44:351–366. 10.3109/10409230903277340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaudy F, Snoeckx R, Pfister M, Zenner HP, Blin N, Di Stazio M, Ferrara A, Lanzara C, Ficarella R, Declau F, Pusch CM, Nurnberg P, Melchionda S, Zelante L, Ballana E, Estivill X, Van Camp G, Gasparini P, Savoia A (2004) Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am J Hum Genet 74:770–776. 10.1086/383285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond MC, Belyantseva IA, Friderici KH, Friedman TB (2012) Actin in hair cells and hearing loss. Hear Res 288:89–99. 10.1016/j.heares.2011.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond MC, Barzik M, Bird JE, Zhang DS, Lechene CP, Corey DP, Cunningham LL, Friedman TB (2015) Live-cell imaging of actin dynamics reveals mechanisms of stereocilia length regulation in the inner ear. Nat Commun 6:6873. 10.1038/ncomms7873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Zou L, Ren R, Li N, Li J, Wang Y, Sun J, Yang J, Xiong W, Xu Z (2020) Lack of PDZD7 long isoform disrupts ankle-link complex and causes hearing loss in mice. FASEB J 34:1136–1149. 10.1096/fj.201901657RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan M, Venail F, Spencer N, Mezzina M (2004) Treatment of peripheral sensorineural hearing loss: gene therapy. Gene Ther 11(Suppl 1):S51–S56. 10.1038/sj.gt.3302369 [DOI] [PubMed] [Google Scholar]
- Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, Millan JM, Aller E, Mitter D, Bolz H (2007) A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet 121:203–211. 10.1007/s00439-006-0304-0 [DOI] [PubMed] [Google Scholar]
- Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, Lopez I, Schafer E, Roux AF, Dafinger C, Bernd A, Zrenner E, Claustres M, Blanco B, Nurnberg G, Nurnberg P, Ruland R, Westerfield M, Benzing T, Bolz HJ (2010) PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest 120:1812–1823. 10.1172/JCI39715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan-Sencicek AG, Jambi S, Franjic D, Nishimura S, Li M, El-Fishawy P, Morgan TM, Sanders SJ, Bilguvar K, Suri M, Johnson MH, Gupta AR, Yuksel Z, Mane S, Grigorenko E, Picciotto M, Alberts AS, Gunel M, Sestan N, State MW (2015) Homozygous loss of DIAPH1 is a novel cause of microcephaly in humans. Eur J Hum Genet 23:165–172. 10.1038/ejhg.2014.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazioli F, Minichiello L, Matoska V, Castagnino P, Miki T, Wong WT, Di Fiore PP (1993) Eps8, a substrate for the epidermal growth factor receptor kinase, enhances EGF-dependent mitogenic signals. EMBO J 12:3799–3808. 10.1002/j.1460-2075.1993.tb06058.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortnum HM, Summerfield AQ, Marshall DH, Davis AC, Bamford JM (2001) Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ 323:536–540. 10.1136/bmj.323.7312.536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman TB, Belyantseva IA, Frolnkov GI (2020) Myosins and hearing. In: Coluccio L (ed) Myosins: a superfamily of molecular motors, 2nd edn, vol 1239. Springer International Publishing, pp 317–330 [Google Scholar]
- Furness DN, Johnson SL, Manor U, Ruttiger L, Tocchetti A, Offenhauser N, Olt J, Goodyear RJ, Vijayakumar S, Dai Y, Hackney CM, Franz C, Di Fiore PP, Masetto S, Jones SM, Knipper M, Holley MC, Richardson GP, Kachar B, Marcotti W (2013) Progressive hearing loss and gradual deterioration of sensory hair bundles in the ears of mice lacking the actin-binding protein Eps8L2. Proc Natl Acad Sci U S A 110:13898–13903. 10.1073/pnas.1304644110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PG, Muller U (2009) Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell 139:33–44. 10.1016/j.cell.2009.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear RJ, Legan PK, Wright MB, Marcotti W, Oganesian A, Coats SA, Booth CJ, Kros CJ, Seifert RA, Bowen-Pope DF, Richardson GP (2003) A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J Neurosci 23:9208–9219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik J, Shevchuk AI, Frolenkov GI, Diakonov IA, Lab MJ, Kros CJ, Richardson GP, Vodyanoy I, Edwards CR, Klenerman D, Korchev YE (2003) Dynamic assembly of surface structures in living cells. Proc Natl Acad Sci U S A 100:5819–5822. 10.1073/pnas.1030502100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati M, Kachar B (2011) Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc Natl Acad Sci U S A 108:11476–11481. 10.1073/pnas.1104161108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati M, Shin JB, Weston MD, Green J, Bhat MA, Gillespie PG, Kachar B (2012) Localization of PDZD7 to the stereocilia ankle-link associates this scaffolding protein with the Usher syndrome protein network. J Neurosci 32:14288–14293. 10.1523/JNEUROSCI.3071-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati M, Yan D, Raval MH, Walsh T, Ma Q, Chakchouk I, Kannan-Sundhari A, Mittal R, Masmoudi S, Blanton SH, Tekin M, King MC, Yengo CM, Liu XZ (2016) MYO3A Causes Human Dominant Deafness and Interacts with Protocadherin 15-CD2 Isoform. Hum Mutat 37:481–487. 10.1002/humu.22961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T, Lillo C, Dumont RA, Hintermann E, Sczaniecka A, Schwander M, Williams D, Kachar B, Gillespie PG, Muller U (2009) Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron 62:375–387. 10.1016/j.neuron.2009.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadi S, Alexander AJ, Velez-Ortega AC, Frolenkov GI (2020) Myosin-XVa Controls Both Staircase Architecture and Diameter Gradation of Stereocilia Rows in the Auditory Hair Cell Bundles. J Assoc Res Otolaryngol 21:121–135. 10.1007/s10162-020-00745-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakala M, Wioland H, Tolonen M, Kotila T, Jegou A, Romet-Lemonne G, Lappalainen P (2021) Twinfilin uncaps filament barbed ends to promote turnover of lamellipodial actin networks. Nat Cell Biol 23:147–159. 10.1038/s41556-020-00629-y [DOI] [PubMed] [Google Scholar]
- Hanc P, Fujii T, Iborra S, Yamada Y, Huotari J, Schulz O, Ahrens S, Kjaer S, Way M, Sancho D, Namba K, Reis e Sousa C, (2015) Structure of the Complex of F-Actin and DNGR-1, a C-Type Lectin Receptor Involved in Dendritic Cell Cross-Presentation of Dead Cell-Associated Antigens. Immunity 42:839–849. 10.1016/j.immuni.2015.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanukoglu I, Tanese N, Fuchs E (1983) Complementary DNA sequence of a human cytoplasmic actin. Interspecies divergence of 3’ non-coding regions. J Mol Biol 163:673–678. 10.1016/0022-2836(83)90117-1 [DOI] [PubMed] [Google Scholar]
- Higashida C, Miyoshi T, Fujita A, Oceguera-Yanez F, Monypenny J, Andou Y, Narumiya S, Watanabe N (2004) Actin polymerization-driven molecular movement of mDia1 in living cells. Science 303:2007–2010. 10.1126/science.1093923 [DOI] [PubMed] [Google Scholar]
- Hofer D, Ness W, Drenckhahn D (1997) Sorting of actin isoforms in chicken auditory hair cells. J Cell Sci 110(Pt 6):765–770 [DOI] [PubMed] [Google Scholar]
- Ida H, Takahashi Y, Kumatani A, Shiku H, Matsue T (2017) High speed scanning ion conductance microscopy for quantitative analysis of nanoscale dynamics of microvilli. Anal Chem 89:6015–6020. 10.1021/acs.analchem.7b00584 [DOI] [PubMed] [Google Scholar]
- Imtiaz A, Kohrman DC, Naz S (2014) A frameshift mutation in GRXCR2 causes recessively inherited hearing loss. Hum Mutat 35:618–624. 10.1002/humu.22545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Yang S, Guo W, He DZ (2009) Fate of mammalian cochlear hair cells and stereocilia after loss of the stereocilia. J Neurosci 29:15277–15285. 10.1523/JNEUROSCI.3231-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Wen KK, Keppler-Noreuil K, McKane M, Maiers JL, Greiner A, Sapp JC, Center NIHIS, Demali KA, Rubenstein PA, Biesecker LG (2013) Functional analysis of a de novo ACTB mutation in a patient with atypical Baraitser-Winter syndrome. Hum Mutat 34:1242–1249. 10.1002/humu.22350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC (1990) Atomic structure of the actin:DNase I complex. Nature 347:37–44. 10.1038/347037a0 [DOI] [PubMed] [Google Scholar]
- Katsuno T, Belyantseva IA, Cartagena-Rivera AX, Ohta K, Crump SM, Petralia RS, Ono K, Tona R, Imtiaz A, Rehman A, Kiyonari H, Kaneko M, Wang YX, Abe T, Ikeya M, Fenollar-Ferrer C, Riordan GP, Wilson EA, Fitzgerald TS, Segawa K, Omori K, Ito J, Frolenkov GI, Friedman TB, Kitajiri SI (2019) TRIOBP-5 sculpts stereocilia rootlets and stiffens supporting cells enabling hearing. JCI Insight. 10.1172/jci.insight.128561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitlina SY (2001) Functional specificity of actin isoforms. Int Rev Cytol 202:35–98. 10.1016/s0074-7696(01)02003-4 [DOI] [PubMed] [Google Scholar]
- Khan SY, Ahmed ZM, Shabbir MI, Kitajiri S, Kalsoom S, Tasneem S, Shayiq S, Ramesh A, Srisailpathy S, Khan SN, Smith RJ, Riazuddin S, Friedman TB, Riazuddin S (2007) Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum Mutat 28:417–423. 10.1002/humu.20469 [DOI] [PubMed] [Google Scholar]
- Kim BJ, Ueyama T, Miyoshi T, Lee S, Han JH, Park HR, Kim AR, Oh J, Kim MY, Kang YS, Oh DY, Yun J, Hwang SM, Kim NKD, Park WY, Kitajiri SI, Choi BY (2019) Differential disruption of autoinhibition and defect in assembly of cytoskeleton during cell division decide the fate of human DIAPH1-related cytoskeletopathy. J Med Genet 56:818–827. 10.1136/jmedgenet-2019-106282 [DOI] [PubMed] [Google Scholar]
- Kitajiri S, Fukumoto K, Hata M, Sasaki H, Katsuno T, Nakagawa T, Ito J, Tsukita S, Tsukita S (2004) Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. J Cell Biol 166:559–570. 10.1083/jcb.200402007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajiri S, Sakamoto T, Belyantseva IA, Goodyear RJ, Stepanyan R, Fujiwara I, Bird JE, Riazuddin S, Riazuddin S, Ahmed ZM, Hinshaw JE, Sellers J, Bartles JR, Hammer JA 3rd, Richardson GP, Griffith AJ, Frolenkov GI, Friedman TB (2010) Actin-bundling protein TRIOBP forms resilient rootlets of hair cell stereocilia essential for hearing. Cell 141:786–798. 10.1016/j.cell.2010.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenekoop R, Arriaga M, Trzupek K, Lentz J (1993) Usher Syndrome Type I. University of Washington, Seattle, GeneReviews [PubMed] [Google Scholar]
- Korver AM, Smith RJ, Van Camp G, Schleiss MR, Bitner-Glindzicz MA, Lustig LR, Usami SI, Boudewyns AN (2017) Congenital hearing loss. Nat Rev Dis Primers 3:16094. 10.1038/nrdp.2016.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, Krystofiak ES, Dumont RA, Vijayakumar S, Choi D, Rivero F, Kachar B, Jones SM, Barr-Gillespie PG (2016) Plastin 1 widens stereocilia by transforming actin filament packing from hexagonal to liquid. J Cell Biol. 10.1083/jcb.201606036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros CJ, Marcotti W, van Netten SM, Self TJ, Libby RT, Brown SD, Richardson GP, Steel KP (2002) Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat Neurosci 5:41–47. 10.1038/nn784 [DOI] [PubMed] [Google Scholar]
- Kumar A, Crawford K, Close L, Madison M, Lorenz J, Doetschman T, Pawlowski S, Duffy J, Neumann J, Robbins J, Boivin GP, O’Toole BA, Lessard JL (1997) Rescue of cardiac alpha-actin-deficient mice by enteric smooth muscle gamma-actin. Proc Natl Acad Sci U S A 94:4406–4411. 10.1073/pnas.94.9.4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalwani AK, Goldstein JA, Kelley MJ, Luxford W, Castelein CM, Mhatre AN (2000) Human Nonsyndromic Hereditary Deafness DFNA17 Is Due to a Mutation in Nonmuscle Myosin MYH9. Am J Human Genet 67:1121–1128. 10.1016/s0002-9297(07)62942-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers M, Rose R, Scrima A, Wittinghofer A (2005) The regulation of mDia1 by autoinhibition and its release by Rho*GTP. EMBO J 24:4176–4187. 10.1038/sj.emboj.7600879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli A, Michel V, Boutet de Monvel J, Cortese M, Bosch-Grau M, Aghaie A, Perfettini I, Dupont T, Avan P, El-Amraoui A, Petit C (2016) Class III myosins shape the auditory hair bundles by limiting microvilli and stereocilia growth. J Cell Biol 212:231–244. 10.1083/jcb.201509017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang M (2020) Cargo binding by unconventional myosins. In: Coluccio L (ed) Myosins: a superfamily of molecular motors, 2nd edn, vol 1239. Springer International Publishing, pp 21–40 [DOI] [PubMed] [Google Scholar]
- Li XD, Jung HS, Wang Q, Ikebe R, Craig R, Ikebe M (2008) The globular tail domain puts on the brake to stop the ATPase cycle of myosin Va. Proc Natl Acad Sci U S A 105:1140–1145. 10.1073/pnas.0709741105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, He Y, Lu Q, Zhang M (2016) Mechanistic basis of organization of the harmonin/USH1C-Mediated brush border microvilli tiplink complex. Dev Cell 36:179–189. 10.1016/j.devcel.2015.12.020 [DOI] [PubMed] [Google Scholar]
- Liang Y, Wang A, Belyantseva IA, Anderson DW, Probst FJ, Barber TD, Miller W, Touchman JW, Jin L, Sullivan SL, Sellers JR, Camper SA, Lloyd RV, Kachar B, Friedman TB, Fridell RA (1999) Characterization of the human and mouse unconventional myosin XV genes responsible for hereditary deafness DFNB3 and shaker 2. Genomics 61:243–258. 10.1006/geno.1999.5976 [DOI] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD (1997) Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet 16:188–190. 10.1038/ng0697-188 [DOI] [PubMed] [Google Scholar]
- Liu H, Li J, Raval MH, Yao N, Deng X, Lu Q, Nie S, Feng W, Wan J, Yengo CM, Liu W, Zhang M (2016) Myosin III-mediated cross-linking and stimulation of actin bundling activity of Espin. Elife. 10.7554/eLife.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H, Berk A, Zipursky S (2000) The actin cytoskeleton. In: Freeman WH & Co; (ed) Molecular cell biology, 4th edn [Google Scholar]
- Loomis PA, Zheng L, Sekerkova G, Changyaleket B, Mugnaini E, Bartles JR (2003) Espin cross-links cause the elongation of microvillus-type parallel actin bundles in vivo. J Cell Biol 163:1045–1055. 10.1083/jcb.200309093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch ED, Lee MK, Morrow JE, Welcsh PL, Leon PE, King MC (1997) Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 278:1315–1318. 10.1126/science.278.5341.1315 [DOI] [PubMed] [Google Scholar]
- Manor U, Disanza A, Grati M, Andrade L, Lin H, Di Fiore PP, Scita G, Kachar B (2011) Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr Biol 21:167–172. 10.1016/j.cub.2010.12.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE (1993) Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet 46:486–491. 10.1002/ajmg.1320460504 [DOI] [PubMed] [Google Scholar]
- Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, Rump A, Hardisty RE, Blanchard S, Coimbra RS, Perfettini I, Parkinson N, Mallon AM, Glenister P, Rogers MJ, Paige AJ, Moir L, Clay J, Rosenthal A, Liu XZ, Blanco G, Steel KP, Petit C, Brown SD (2003) Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet 34:421–428. 10.1038/ng1208 [DOI] [PubMed] [Google Scholar]
- McGrath J, Tung CY, Liao X, Belyantseva IA, Roy P, Chakraborty O, Li J, Berbari NF, Faaborg-Andersen CC, Barzik M, Bird JE, Zhao B, Balakrishnan L, Friedman TB, Perrin BJ (2020) Actin at stereocilia tips is regulated by mechanotransduction and ADF/cofilin. Curr Biol. 10.1016/j.cub.2020.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meenderink LM, Gaeta IM, Postema MM, Cencer CS, Chinowsky CR, Krystofiak ES, Millis BA, Tyska MJ (2019) Actin dynamics drive microvillar motility and clustering during brush border assembly. Dev Cell 50(545–556):e4. 10.1016/j.devcel.2019.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionda S, Ahituv N, Bisceglia L, Sobe T, Glaser F, Rabionet R, Arbones ML, Notarangelo A, Di Iorio E, Carella M, Zelante L, Estivill X, Avraham KB, Gasparini P (2001) MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet 69:635–640. 10.1086/323156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino F, Pospich S, Funk J, Wagner T, Kullmer F, Arndt HD, Bieling P, Raunser S (2018) Structural transitions of F-actin upon ATP hydrolysis at near-atomic resolution revealed by cryo-EM. Nat Struct Mol Biol 25:528–537. 10.1038/s41594-018-0074-0 [DOI] [PubMed] [Google Scholar]
- Metlagel Z, Krey JF, Song J, Swift MF, Tivol WJ, Dumont RA, Thai J, Chang A, Seifikar H, Volkmann N, Hanein D, Barr-Gillespie PG, Auer M (2019) Electron cryo-tomography of vestibular hair-cell stereocilia. J Struct Biol 206:149–155. 10.1016/j.jsb.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski N, Michel V, Caberlotto E, Lefevre GM, van Aken AF, Tinevez JY, Bizard E, Houbron C, Weil D, Hardelin JP, Richardson GP, Kros CJ, Martin P, Petit C (2009) Harmonin-b, an actin-binding scaffold protein, is involved in the adaptation of mechanoelectrical transduction by sensory hair cells. Pflugers Arch 459:115–130. 10.1007/s00424-009-0711-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima H, Moteki H, Day T, Nishio SY, Murata T, Ikezono T, Takeda H, Abe S, Iwasaki S, Takahashi M, Naito Y, Yamazaki H, Kanda Y, Kitajiri SI, Usami SI (2020) Novel ACTG1 mutations in patients identified by massively parallel DNA sequencing cause progressive hearing loss. Sci Rep 10:7056. 10.1038/s41598-020-63690-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi T, Tsuji T, Higashida C, Hertzog M, Fujita A, Narumiya S, Scita G, Watanabe N (2006) Actin turnover-dependent fast dissociation of capping protein in the dendritic nucleation actin network: evidence of frequent filament severing. J Cell Biol 175:947–955. 10.1083/jcb.200604176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi T, Zhang Q, Miyake T, Watanabe S, Ohnishi H, Chen J, Vishwasrao HD, Chakraborty O, Belyantseva IA, Perrin BJ, Shroff H, Friedman TB, Omori K, Watanabe N (2021) Semi-automated single-molecule microscopy screening of fast-dissociating specific antibodies directly from hybridoma cultures. Cell Rep 34:108708. 10.1016/j.celrep.2021.108708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockrin SC, Korn ED (1980) Acanthamoeba profilin interacts with G-actin to increase the rate of exchange of actin-bound adenosine 5’-triphosphate. Biochemistry 19:5359–5362. 10.1021/bi00564a033 [DOI] [PubMed] [Google Scholar]
- Mogensen MM, Rzadzinska A, Steel KP (2007) The deaf mouse mutant whirler suggests a role for whirlin in actin filament dynamics and stereocilia development. Cell Motil Cytoskeleton 64:496–508. 10.1002/cm.20199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell RJ, Friderici KH, Wei S, Elfenbein JL, Friedman TB, Fisher RA (2000) A new locus for late-onset, progressive, hereditary hearing loss DFNA20 maps to 17q25. Genomics 63:1–6. 10.1006/geno.1999.6058 [DOI] [PubMed] [Google Scholar]
- Morgan A, Koboldt DC, Barrie ES, Crist ER, Garcia Garcia G, Mezzavilla M, Faletra F, Mihalic Mosher T, Wilson RK, Blanchet C, Manickam K, Roux AF, Gasparini P, Dell’Orco D, Girotto G (2019) Mutations in PLS1, encoding fimbrin, cause autosomal dominant nonsyndromic hearing loss. Hum Mutat 40:2286–2295. 10.1002/humu.23891 [DOI] [PubMed] [Google Scholar]
- Narayanan P, Chatterton P, Ikeda A, Ikeda S, Corey DP, Ervasti JM, Perrin BJ (2015) Length regulation of mechanosensitive stereocilia depends on very slow actin dynamics and filament-severing proteins. Nat Commun 6:6855. 10.1038/ncomms7855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz S, Griffith AJ, Riazuddin S, Hampton LL, Battey JF Jr, Khan SN, Riazuddin S, Wilcox ER, Friedman TB (2004) Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J Med Genet 41:591–595. 10.1136/jmg.2004.018523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus C, Lang-Roth R, Zimmermann U, Heller R, Eisenberger T, Weikert M, Markus S, Knipper M, Bolz HJ (2017) Extension of the clinical and molecular phenotype of DIAPH1-associated autosomal dominant hearing loss (DFNA1). Clin Genet 91:892–901. 10.1111/cge.12915 [DOI] [PubMed] [Google Scholar]
- Nikolopoulos TP, Vlastarakos PV (2010) Treating options for deaf children. Early Hum Dev 86:669–674. 10.1016/j.earlhumdev.2010.10.001 [DOI] [PubMed] [Google Scholar]
- Ninoyu Y, Sakaguchi H, Lin C, Suzuki T, Hirano S, Hisa Y, Saito N, Ueyama T (2020) The integrity of cochlear hair cells is established and maintained through the localization of Dia1 at apical junctional complexes and stereocilia. Cell Death Dis 11:536. 10.1038/s41419-020-02743-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio SY, Usami S (2015) Deafness gene variations in a 1120 nonsyndromic hearing loss cohort: molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann Otol Rhinol Laryngol 124(Suppl 1):49S–60S. 10.1177/0003489415575059 [DOI] [PubMed] [Google Scholar]
- Odeh H, Hunker KL, Belyantseva IA, Azaiez H, Avenarius MR, Zheng L, Peters LM, Gagnon LH, Hagiwara N, Skynner MJ, Brilliant MH, Allen ND, Riazuddin S, Johnson KR, Raphael Y, Najmabadi H, Friedman TB, Bartles JR, Smith RJ, Kohrman DC (2010) Mutations in Grxcr1 are the basis for inner ear dysfunction in the pirouette mouse. Am J Hum Genet 86:148–160. 10.1016/j.ajhg.2010.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Ravi H, Smith EM, Goode BL (2006) Aip1 and cofilin promote rapid turnover of yeast actin patches and cables: a coordinated mechanism for severing and capping filaments. Mol Biol Cell 17:2855–2868. 10.1091/mbc.e06-02-0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang XM, Xia XJ, Verpy E, Du LL, Pandya A, Petit C, Balkany T, Nance WE, Liu XZ (2002) Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum Genet 111:26–30. 10.1007/s00439-002-0736-0 [DOI] [PubMed] [Google Scholar]
- Paavilainen VO, Hellman M, Helfer E, Bovellan M, Annila A, Carlier MF, Permi P, Lappalainen P (2007) Structural basis and evolutionary origin of actin filament capping by twinfilin. Proc Natl Acad Sci U S A 104:3113–3118. 10.1073/pnas.0608725104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni D, Carlier M-F (1993) How profilin promotes actin filament assembly in the presence of thymosin β4. Cell 75:1007–1014. 10.1016/0092-8674(93)90544-z [DOI] [PubMed] [Google Scholar]
- Park G, Gim J, Kim AR, Han KH, Kim HS, Oh SH, Park T, Park WY, Choi BY (2013) Multiphasic analysis of whole exome sequencing data identifies a novel mutation of ACTG1 in a nonsyndromic hearing loss family. BMC Genomics 14:191. 10.1186/1471-2164-14-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrinostro X, Roy P, Lindsay A, Chamberlain CM, Sundby LJ, Starker CG, Voytas DF, Ervasti JM, Perrin BJ (2018) Essential nucleotide- and protein-dependent functions of Actb/beta-actin. Proc Natl Acad Sci U S A 115:7973–7978. 10.1073/pnas.1807895115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng AW, Belyantseva IA, Hsu PD, Friedman TB, Heller S (2009) Twinfilin 2 regulates actin filament lengths in cochlear stereocilia. J Neurosci 29:15083–15088. 10.1523/JNEUROSCI.2782-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin BJ, Ervasti JM (2010) The actin gene family: function follows isoform. Cytoskeleton (hoboken) 67:630–634. 10.1002/cm.20475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin BJ, Sonnemann KJ, Ervasti JM (2010) beta-actin and gamma-actin are each dispensable for auditory hair cell development but required for Stereocilia maintenance. PLoS Genet 6:e1001158. 10.1371/journal.pgen.1001158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak A, Lechowicz U, Murcia Pienkowski VA, Stawinski P, Kosinska J, Skarzynski H, Oldak M, Ploski R (2017) Whole exome sequencing identifies TRIOBP pathogenic variants as a cause of post-lingual bilateral moderate-to-severe sensorineural hearing loss. BMC Med Genet 18:142. 10.1186/s12881-017-0499-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD (1986) Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J Cell Biol 103:2747–2754. 10.1083/jcb.103.6.2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD (2016) Actin and actin-binding proteins. Cold Spring Harb Perspect Biol. 10.1101/cshperspect.a018226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA (1984) Quantitative analysis of the effect of Acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry 23:6631–6641. 10.1021/bi00321a054 [DOI] [PubMed] [Google Scholar]
- Pollard TD, Blanchoin L, Mullins RD (2000) Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct 29:545–576. 10.1146/annurev.biophys.29.1.545 [DOI] [PubMed] [Google Scholar]
- Pollard TD, Earnshaw WC, Lippincott-Schwartz J, Johnson GT (2008) Cell biology [Google Scholar]
- Poukkula M, Kremneva E, Serlachius M, Lappalainen P (2011) Actin-depolymerizing factor homology domain: a conserved fold performing diverse roles in cytoskeletal dynamics. Cytoskeleton (hoboken) 68:471–490. 10.1002/cm.20530 [DOI] [PubMed] [Google Scholar]
- Procaccio V, Salazar G, Ono S, Styers ML, Gearing M, Davila A, Jimenez R, Juncos J, Gutekunst CA, Meroni G, Fontanella B, Sontag E, Sontag JM, Faundez V, Wainer BH (2006) A mutation of beta -actin that alters depolymerization dynamics is associated with autosomal dominant developmental malformations, deafness, and dystonia. Am J Hum Genet 78:947–960. 10.1086/504271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere JB, van Bon BW, Hoischen A, Kholmanskikh SS, O’Roak BJ, Gilissen C, Gijsen S, Sullivan CT, Christian SL, Abdul-Rahman OA, Atkin JF, Chassaing N, Drouin-Garraud V, Fry AE, Fryns JP, Gripp KW, Kempers M, Kleefstra T, Mancini GM, Nowaczyk MJ, van Ravenswaaij-Arts CM, Roscioli T, Marble M, Rosenfeld JA, Siu VM, de Vries BB, Shendure J, Verloes A, Veltman JA, Brunner HG, Ross ME, Pilz DT, Dobyns WB (2012) De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat Genet 44(440–4):S1–2. 10.1038/ng.1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy P, Perrin BJ (2018) The stable actin core of mechanosensory stereocilia features continuous turnover of actin cross-linkers. Mol Biol Cell 29:1856–1865. 10.1091/mbc.E18-03-0196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben RJ (2018) Language development in the pediatric cochlear implant patient. Laryngoscope Investig Otolaryngol 3:209–213. 10.1002/lio2.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzadzinska AK, Schneider ME, Davies C, Riordan GP, Kachar B (2004) An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J Cell Biol 164:887–897. 10.1083/jcb.200310055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzadzinska AK, Nevalainen EM, Prosser HM, Lappalainen P, Steel KP (2009) MyosinVIIa interacts with Twinfilin-2 at the tips of mechanosensory stereocilia in the inner ear. PLoS ONE 4:e7097. 10.1371/journal.pone.0007097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safer D, Elzinga M, Nachmias VT (1991) Thymosin β4 and Fx, an actin-sequestering peptide, are indistinguishable. J Biol Chem 266:4029–4032. 10.1016/s0021-9258(20)64278-8 [DOI] [PubMed] [Google Scholar]
- Sakaguchi H, Tokita J, Naoz M, Bowen-Pope D, Gov NS, Kachar B (2008) Dynamic compartmentalization of protein tyrosine phosphatase receptor Q at the proximal end of stereocilia: implication of myosin VI-based transport. Cell Motil Cytoskeleton 65:528–538. 10.1002/cm.20275 [DOI] [PubMed] [Google Scholar]
- Salles FT, Merritt RC Jr, Manor U, Dougherty GW, Sousa AD, Moore JE, Yengo CM, Dose AC, Kachar B (2009) Myosin IIIa boosts elongation of stereocilia by transporting espin 1 to the plus ends of actin filaments. Nat Cell Biol 11:443–450. 10.1038/ncb1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer DI, Zhang DS, Shen J, Indzhykulian A, Karavitaki KD, Xu YJ, Wang Q, Lin JJ, Chen ZY, Corey DP (2015) XIRP2, an actin-binding protein essential for inner ear hair-cell stereocilia. Cell Rep 10:1811–1818. 10.1016/j.celrep.2015.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildmeyer LA, Braun R, Taffet G, Debiasi M, Burns AE, Bradley A, Schwartz RJ (2000) Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. FASEB J 14:2213–2220. 10.1096/fj.99-0927com [DOI] [PubMed] [Google Scholar]
- Schraders M, Lee K, Oostrik J, Huygen PL, Ali G, Hoefsloot LH, Veltman JA, Cremers FP, Basit S, Ansar M, Cremers CW, Kunst HP, Ahmad W, Admiraal RJ, Leal SM, Kremer H (2010) Homozygosity mapping reveals mutations of GRXCR1 as a cause of autosomal-recessive nonsyndromic hearing impairment. Am J Hum Genet 86:138–147. 10.1016/j.ajhg.2009.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutt CE, Myslik JC, Rozycki MD, Goonesekere NC, Lindberg U (1993) The structure of crystalline profilin-beta-actin. Nature 365:810–816. 10.1038/365810a0 [DOI] [PubMed] [Google Scholar]
- Scipion CPM, Ghoshdastider U, Ferrer FJ, Yuen TY, Wongsantichon J, Robinson RC (2018) Structural evidence for the roles of divalent cations in actin polymerization and activation of ATP hydrolysis. Proc Natl Acad Sci U S A 115:10345–10350. 10.1073/pnas.1806394115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scita G, Nordstrom J, Carbone R, Tenca P, Giardina G, Gutkind S, Bjarnegard M, Betsholtz C, Di Fiore PP (1999) EPS8 and E3B1 transduce signals from Ras to Rac. Nature 401:290–293. 10.1038/45822 [DOI] [PubMed] [Google Scholar]
- Sekerkova G, Richter CP, Bartles JR (2011) Roles of the espin actin-bundling proteins in the morphogenesis and stabilization of hair cell stereocilia revealed in CBA/CaJ congenic jerker mice. PLoS Genet 7:e1002032. 10.1371/journal.pgen.1002032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AS (2001) FERMing Up the Synapse. Immunity 15:683–686. 10.1016/s1074-7613(01)00237-0 [DOI] [PubMed] [Google Scholar]
- Sheterline P, Clayton J, Sparrow J (1995) Actin. Protein Profile 2:1–103 [PubMed] [Google Scholar]
- Shimojima K, Narai S, Togawa M, Doumoto T, Sangu N, Vanakker OM, de Paepe A, Edwards M, Whitehall J, Brescianini S, Petit F, Andrieux J, Yamamoto T (2016) 7p22.1 microdeletions involving ACTB associated with developmental delay, short stature, and microcephaly. Eur J Med Genet 59:502–506. 10.1016/j.ejmg.2016.09.008 [DOI] [PubMed] [Google Scholar]
- Shin JB, Longo-Guess CM, Gagnon LH, Saylor KW, Dumont RA, Spinelli KJ, Pagana JM, Wilmarth PA, David LL, Gillespie PG, Johnson KR (2010) The R109H variant of fascin-2, a developmentally regulated actin crosslinker in hair-cell stereocilia, underlies early-onset hearing loss of DBA/2J mice. J Neurosci 30:9683–9694. 10.1523/JNEUROSCI.1541-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens J, Kazmierczak P, Reynolds A, Sticker M, Littlewood-Evans A, Muller U (2002) The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc Natl Acad Sci U S A 99:14946–14951. 10.1073/pnas.232579599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skogseid IM, Rosby O, Konglund A, Connelly JP, Nedregaard B, Jablonski GE, Kvernmo N, Stray-Pedersen A, Glover JC (2018) Dystonia-deafness syndrome caused by ACTB p.Arg183Trp heterozygosity shows striatal dopaminergic dysfunction and response to pallidal stimulation. J Neurodev Disord 10:17. 10.1186/s11689-018-9235-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strzelecka-Golaszewska H, Prochniewicz E, Drabikowski W (1978) Interaction of actin with divalent cations. Eur J Biochem 88:219–227. 10.1111/j.1432-1033.1978.tb12441.x [DOI] [PubMed] [Google Scholar]
- Tocchetti A, Confalonieri S, Scita G, Di Fiore PP, Betsholtz C (2003) In silico analysis of the EPS8 gene family: genomic organization, expression profile, and protein structure. Genomics 81:234–244. 10.1016/s0888-7543(03)00002-8 [DOI] [PubMed] [Google Scholar]
- Tomblin JB, Oleson JJ, Ambrose SE, Walker E, Moeller MP (2014) The influence of hearing aids on the speech and language development of children with hearing loss. JAMA Otolaryngol Head Neck Surg 140:403–409. 10.1001/jamaoto.2014.267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondeleir D, Noelanders R, Bakkali K, Ampe C (2014) Beta-actin is required for proper mouse neural crest ontogeny. PLoS ONE 9:e85608. 10.1371/journal.pone.0085608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Oishi K, Sato N, Sagara J, Kawai A, Tsukita S (1994) ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J Cell Biol 126:391–401. 10.1083/jcb.126.2.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama T, Ninoyu Y, Nishio SY, Miyoshi T, Torii H, Nishimura K, Sugahara K, Sakata H, Thumkeo D, Sakaguchi H, Watanabe N, Usami SI, Saito N, Kitajiri SI (2016) Constitutive activation of DIA1 (DIAPH1) via C-terminal truncation causes human sensorineural hearing loss. EMBO Mol Med 8:1310–1324. 10.15252/emmm.201606609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk E, Krieger E, Kemperman MH, De Leenheer EM, Huygen PL, Cremers CW, Cremers FP, Kremer H (2003) A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J Med Genet 40:879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedula P, Kashina A (2018) The makings of the “actin code”: regulation of actin’s biological function at the amino acid and nucleotide level. J Cell Sci. 10.1242/jcs.215509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedula P, Kurosaka S, Leu NA, Wolf YI, Shabalina SA, Wang J, Sterling S, Dong DW, Kashina A (2017) Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence. Elife. 10.7554/eLife.31661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verloes A, Di Donato N, Masliah-Planchon J, Jongmans M, Abdul-Raman OA, Albrecht B, Allanson J, Brunner H, Bertola D, Chassaing N, David A, Devriendt K, Eftekhari P, Drouin-Garraud V, Faravelli F, Faivre L, Giuliano F, Guion Almeida L, Juncos J, Kempers M, Eker HK, Lacombe D, Lin A, Mancini G, Melis D, Lourenco CM, Siu VM, Morin G, Nezarati M, Nowaczyk MJ, Ramer JC, Osimani S, Philip N, Pierpont ME, Procaccio V, Roseli ZS, Rossi M, Rusu C, Sznajer Y, Templin L, Uliana V, Klaus M, Van Bon B, Van Ravenswaaij C, Wainer B, Fry AE, Rump A, Hoischen A, Drunat S, Riviere JB, Dobyns WB, Pilz DT (2015) Baraitser-Winter cerebrofrontofacial syndrome: delineation of the spectrum in 42 cases. Eur J Hum Genet 23:292–301. 10.1038/ejhg.2014.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C (2000) A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet 26:51–55. 10.1038/79171 [DOI] [PubMed] [Google Scholar]
- Vinson VK, De La Cruz EM, Higgs HN, Pollard TD (1998) Interactions of Acanthamoeba profilin with actin and nucleotides bound to actin. Biochemistry 37:10871–10880. 10.1021/bi980093l [DOI] [PubMed] [Google Scholar]
- Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, Lee MK, Kanaan M, King MC, Avraham KB (2002) From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci U S A 99:7518–7523. 10.1073/pnas.102091699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, Morton CC, Morell RJ, Noben-Trauth K, Camper SA, Friedman TB (1998) Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science 280:1447–1451. 10.1126/science.280.5368.1447 [DOI] [PubMed] [Google Scholar]
- Watanabe N, Mitchison TJ (2002) Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science 295:1083–1086. 10.1126/science.1067470 [DOI] [PubMed] [Google Scholar]
- Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S (1997) p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J 16:3044–3056. 10.1093/emboj/16.11.3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkin P, Baldwin M (2012) The longitudinal follow up of a universal neonatal hearing screen: the implications for confirming deafness in childhood. Int J Audiol 51:519–528. 10.3109/14992027.2012.673237 [DOI] [PubMed] [Google Scholar]
- Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M, Ayadi H, Petit C (1997) The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet 16:191–193. 10.1038/ng0697-191 [DOI] [PubMed] [Google Scholar]
- Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C (2003) Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 12:463–471. 10.1093/hmg/ddg051 [DOI] [PubMed] [Google Scholar]
- Wesdorp M, van de Kamp JM, Hensen EF, Schraders M, Oostrik J, Yntema HG, Feenstra I, Admiraal RJC, Kunst HPM, Tekin M, Kanaan M, Kremer H, Pennings RJE (2017) Broadening the phenotype of DFNB28: Mutations in TRIOBP are associated with moderate, stable hereditary hearing impairment. Hear Res 347:56–62. 10.1016/j.heares.2016.12.017 [DOI] [PubMed] [Google Scholar]
- Westbury SK, Downes K, Burney C, Lozano ML, Obaji SG, Toh CH, Sevivas T, Morgan NV, Erber WN, Kempster C, Moore SF, Thys C, Papadia S, Ouwehand WH, Diseases NB-R, Laffan MA, Gomez K, Freson K, Rivera J, Mumford AD (2018) Phenotype description and response to thrombopoietin receptor agonist in DIAPH1-related disorder. Blood Adv 2:2341–2346. 10.1182/bloodadvances.2018020370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Kozin ED, Kanumuri VV, Vachicouras N, Miller J, Lacour S, Brown MC, Lee DJ (2019) Auditory Brainstem Implants: Recent Progress and Future Perspectives. Front Neurosci 13:10. 10.3389/fnins.2019.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A (1984) Dimensions of the cochlear stereocilia in man and the guinea pig. Hear Res 13:89–98. 10.1016/0378-5955(84)90099-6 [DOI] [PubMed] [Google Scholar]
- Wu L, Pan L, Wei Z, Zhang M (2011) Structure of MyTH4-FERM domains in myosin VIIa tail bound to cargo. Science 331:757–760. 10.1126/science.1198848 [DOI] [PubMed] [Google Scholar]
- Yao LL, Cao QJ, Zhang HM, Zhang J, Cao Y, Li XD (2015) Melanophilin Stimulates Myosin-5a Motor Function by Allosterically Inhibiting the Interaction between the Head and Tail of Myosin-5a. Sci Rep 5:10874. 10.1038/srep10874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates TM, Turner CL, Firth HV, Berg J, Pilz DT (2017) Baraitser-Winter cerebrofrontofacial syndrome. Clin Genet 92:3–9. 10.1111/cge.12864 [DOI] [PubMed] [Google Scholar]
- Yonemura S, Hirao M, Doi Y, Takahashi N, Kondo T, Tsukita S, Tsukita S (1998) Ezrin/radixin/moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2. J Cell Biol 140:885–895. 10.1083/jcb.140.4.885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IM, Planelles-Herrero VJ, Sourigues Y, Moussaoui D, Sirkia H, Kikuti C, Stroebel D, Titus MA, Houdusse A (2017) Myosin 7 and its adaptors link cadherins to actin. Nat Commun 8:15864. 10.1038/ncomms15864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Gao X, Huang B, Lu J, Wang G, Lin X, Qu Y, Dai P (2016) Phenotypic heterogeneity in a DFNA20/26 family segregating a novel ACTG1 mutation. BMC Genet 17:33. 10.1186/s12863-016-0333-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DS, Piazza V, Perrin BJ, Rzadzinska AK, Poczatek JC, Wang M, Prosser HM, Ervasti JM, Corey DP, Lechene CP (2012) Multi-isotope imaging mass spectrometry reveals slow protein turnover in hair-cell stereocilia. Nature 481:520–524. 10.1038/nature10745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Sekerková G, Vranich K, Tilney LG, Mugnaini E, Bartles JR (2000) The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102:377–385. 10.1016/s0092-8674(00)00042-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Beeler DM, Bartles JR (2015) Characterization and regulation of an additional actin-filament-binding site in large isoforms of the stereocilia actin-bundling protein espin. J Cell Sci 128:2208. 10.1242/jcs.174052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, Elfenbein JL, Fisher RA, Leal SM, Smith RJ, Friderici KH (2003) Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet 73:1082–1091. 10.1086/379286 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.