

Abstract
Aim: The indandione nucleus, is one of the most amazing nuclei in medicinal chemistry, is used to design new derivatives.
Methods & materials: Novel indandione derivatives are prepared with different electrophilic and nucleophilic reagents to yield 3, 4, 8, 11, 14, 16, 19, 20, 21, 22 and 23. Compounds 8, 11, 16, 20 and 23 are investigated against OVCAR-3 and HeLa, using LLC-MK2 and cis-Pt as references. in silico and spectral studies were analyzed for the selected compounds.
Results: Compounds 20 and 23 at 100 ns were the most potent compounds, so molecular dynamics studies were performed.
Conclusion: Compound 23 was the most active toward the HeLa cervical cell line, and compound 20 was the most active toward the Ovcar-3 cell line.
Keywords: : anticancer, indenopyrane, indenopyrazole, in silico studies, mercaptopyrazole
Graphical Abstract

Plain language summary
Article highlights.
The research study was carried out in our laboratory as a continuous work for heterocyclization reactions.
The article represented newly synthesized compounds of indan-1,3-dione derivatives.
The compounds underwent investigation against different anticancer cell lines, molecular docking, density functional theory, molecular dynamics and pharmacokinetic studies.
Spectral data were carried out to illustrate the structure of the new series.
1. Background
The indandione nucleus is one of the intelligent nuclei in organic synthetic chemistry and organic pharmaceutical chemistry because of its variety of applications in medical sciences [1]. Indandione can be observed in many biological and pharmacological properties [2], such as anticancer and antibiotic properties [3]. It possesses activities of aldose reductase inhibition [4], antibacterial activity against Gram-positive bacteria [5], antimicrobial, antifungal [6] and anion receptors [7]. Also, indandione derivatives were used in colorimetric anion sensors using electron pulling chromophores [8], reference azodye in polycarbonate [9], preparation of good optical quality films using spin coating procedure [10], new pH indicators [11] and also was used as polymers possess good thermal stability [12]. Activated methylene derivatives are emerging as electron-poor β-dicarbonyl compounds, so they are Michael donors with activated alkenes.
On the other hand, 1,3-dicarbonyl compounds display higher reactivity and efficiency toward activated isothiocyanate [13,14]. Also, β-dicarbonyl compounds constitute a group of versatile intermediates with highly toxic and lachrymatory available reagents to prepare a variety of bioactive heterocycles [15]. Moreover, another attempt was made to investigate indane derivatives’ anticancer activities with experimental and in silico studies [16]. In this work, we aim to prepare new heterocyclic derivatives of indandione by reacting them with electrophilic and nucleophilic reagents. In addition, anticancer studies were applied to certain compounds of the newly prepared series. In silico studies effectively investigated and verified the prepared drugs’ activity; Petra/Osiris/Molinspiration (POM) theory, density functional theory (DFT) and molecular docking are the most common techniques to prove and validate the results obtained by experimental applications. In addition, molecular dynamics simulations at 100ns are an effective tool for studying the molecular properties of the compound, which helps in medicinal chemistry research [17–20]. Clinical studies of indandione derivatives were studied and mentioned in the literature [21]. This study used the in silico studies to choose the best compounds that will undergo biological studies, and it is also used to verify the results of the biological data obtained as anticancer drugs.
Structure–activity relationship (SAR) studies are applied to compounds similar to indandione derivatives. The similarity of compounds which represent the SAR of compounds 8, 20 and 23 are mentioned in Supplementary Figure S1A–C.
More clinical studies of the newly synthesized series will be continued with clinical medicinal chemistry researchers.
2. Methodology & experimental
2.1. Chemistry
All melting points were uncorrected and measured using a thermal IA 9100 apparatus. IR spectra (KBr discs) were recorded on a Shimadzu FTIR-400 spectrophotometer (Kyoto, Japan). 1H NMR spectra were determined with a Varian-300 MHz spectrometer(CA, USA). The chemical shifts are expressed on the δ (ppm) scale using TMS as the standard reference. Elemental analysis was determined on a Perkin Elmer 240 (microanalysis; MA, USA) at the microanalytical center, Cairo University, Cairo, Egypt and KASUT central laboratory.
Indan-1,3-dione (1) was prepared according to a reported procedure, in addition to a purchase from Sigma Aldrich (MI, USA), and hence no more purification of the products because chemicals are of analytical grade.
2.1.1. 1,3-Dioxo-2,3-dihydro-1H-indene-2-carbothioamide (2)
A mixture of Indian-1,3-dione 1 (1.46 g, 10 mmol) and ammonium thiocyanate (10 mmol) in Glacial AcOH (20 ml) was heated under reflux for 3 h. The precipitate formed after cooling was filtered off, dried and recrystallized from ethanol to form compound 2. Yield (61%), m. p. 204–206°C; IR (KBr cm-1) νmax: 3454 (OH), 3361 (NH2), 1709, 1679 (C=O), 1613 (C=C); 1H NMR (600 MHz, DMSO-d6) δ 8.32 (s, 2H), 8.12 (m, J = 6.2, 3.6 Hz, 2H), 7.88 (m, J = 6.2, 3.6 Hz, 2H), 5.00 (s, 1H); 13C NMR (150 MHz, Chloroform-d) δ 197.78, 193.34, 139.48, 135.38, 123.84, 66.06. Anal. Calcd for C10H7NO2S (205.23): C, 58.52; H, 3.44; N, 6.82; Found: C, 58.48; H, 3.45; N, 6.90.
2.1.2. 1-Hydroxy-3-(phenylamino)-1H-indene-2-carbothioamide (3)
A mixture of compound 2 (10 mmol) and hydrazine hydrate (2 ml) in dimethyl formamide (20 ml) was heated under reflux for 4 h, cooled and then poured onto cold water. The solid formed was filtered off, dried and recrystallized from EtOH and a few drops of glacial AcOH to give compound 3. Yield (43%), m p 224–226°C; IR (KBr cm-1) νmax: 3244 (NH2), 1706 (C=O), 1576 (C=S). 1H NMR (600 MHz, DMSO-d6) δ 9.66 (s, 1H), 8.87 (s, 2H), 7.84–7.79 (m, 2H), 7.43–7.38 (m, 2H), 7.38–7.32 (m, 1H), 7.35–7.26 (m, 2H), 6.96–6.89 (m, 1H), 6.77 (m, J = 6.9, 1.1 Hz, 1H), 5.52 (d, J = 4.9 Hz, 1H), 4.76 (d, J = 4.8 Hz, 1H).; 13C NMR (150 MHz, Chloroform-d) δ 192.49, 143.02, 141.38, 141.21, 134.66, 129.10, 127.73, 124.44, 123.72, 123.66, 121.97, 120.30, 106.87, 71.55; Anal. Calcd for C10H9N3O (187.07): C, 64.16; H, 4.85; N, 22.45; Found: C, 64.17; H, 4.84; N, 22.46.
2.1.3. 3-Amino-3a,4-dihydroindeno[1,2-c]pyrazol-4-ol (4)
A mixture of compound 2 (10 mmol) and aniline (10 mmol) in absolute ethanol (30 ml) was heated under reflux for 6 h. The formed precipitate, after cooling, was filtered off, dried and recrystallized from ethanol to yield compound 4. Yield (48%), m p 170–172°C; IR (KBr cm-1) νmax: 3411 (NH2), 1705 (C=O), 1600 (C=C), 1577 (C=S); 1H NMR (600 MHz, DMSO-d6) δ 7.91–7.86 (m, 1H), 7.75–7.71 (m, 1H), 7.45 (m, J = 8.0, 1.5 Hz, 1H), 7.38 (m, J = 8.3, 1.7 Hz, 1H), 7.02 (s, 2H), 4.99–4.92 (m, 2H), 4.86 (d, J = 4.6 Hz, 1H), 13C NMR (150 MHz, Chloroform-d) δ 166.66, 142.17, 135.74, 133.29, 128.98, 128.52, 124.53, 124.20, 71.81, 55.67.; Anal. Calcd for C16H12N2OS (280.35): C, 68.55; H, 4.31; N, 9.99; Found: C, 68.21; H, 4.47; N, 9.79.
2.1.4. General procedure for the reaction of compound 2 with α,β-unsaturated systems
To a stirred cold sodium ethoxide solution (prepared from sodium metal [0.23 g] in absolute ethanol [30 ml]), a mixture of compound 2 (10 mmol), 1,3-diphenylprop-2-en-1-one and (phenylmethylidene)propanedinitrile (10 mmol) in of absolute ethanol (20 ml) was added dropwise. The reaction mixture was heated under reflux for 8 h. The solution was concentrated under reduced pressure, poured into crushed ice and neutralized with dilute hydrochloric acid. The solid formed was filtered off, washed with water, dried and recrystallized from the proper solvent.
2.1.5. 1-Amino-4-benzoyl-3-phenylindeno[2,1-c]thiopyran-9(3H)-one (8)
Black powder (solvent of crystallization: ethanol/drops of acetic acid); Yield 2.73 g (69%), m p 266–268°C; IR (KBr cm-1) νmax: 3416 (NH2), 1708, 1681 (2C=O), 1612 (C=N), 1598 (C=C); 1H NMR (600 MHz, DMSO-d6) δ 8.05 (dd, J = 8.1, 1.5 Hz, 1H), 8.02 (s, 2H), 7.91 (dd, J = 8.1, 1.6 Hz, 1H), 7.76–7.71 (m, 2H), 7.66 (m, J = 7.9, 1.5 Hz, 1H), 7.59–7.48 (m, 2H), 7.45–7.39 (m, 4H), 7.39–7.30 (m, 3H), 5.34 (s, 1H), 13C NMR (150 MHz, Chloroform-d) δ 192.52, 185.19, 164.46, 149.92, 138.25, 137.92, 136.68, 136.24, 132.45, 130.94, 130.25, 128.81, 128.36, 128.18, 128.00, 127.63, 125.52, 124.32, 124.08, 105.33, 45.53.Anal. Calcd for C25H17NO2S (395.48): C, 75.93; H, 4.33; N, 3.54; Found: C, 75.97; H, 4.31; N, 3.31.
2.1.6. 1-Imino-9-oxo-3-phenyl-1,9-dihydroindeno[2,1-c]thiopyran-4-carbonitrile (11)
Brown powder (butanol/drops of acetic acid); Yield (67%), mp 240–242°C; IR (KBr cm-1) νmax: 3405 (NH), 2189 (CN), 1706 (C=O), 1616 (C=N), 1581 (C=C); 1H NMR (600 MHz, DMSO-d6) δ 10.77 (s, 1H), 8.45 (dd, J = 8.0, 1.9 Hz, 1H), 7.81 (td, J = 8.0, 1.5 Hz, 1H), 7.75 (m, J = 7.6, 1.7 Hz, 3H), 7.46 (m, J = 7.9, 1.5 Hz, 1H), 7.43–7.37 (m, 2H), 7.33 (m, J = 8.4, 6.8, 1.5 Hz, 1H). 13C NMR (150 MHz, Chloroform-d) δ 189.15, 156.02, 151.26, 140.58, 138.97, 137.68, 131.09, 130.90, 129.92, 129.35, 129.14, 125.35, 124.59, 124.14, 119.49, 118.44, 101.09. Anal. Calcd for C19H10N2OS (314.36): C, 72.59; H, 3.21; N, 8.91; Found: C, 72.57; H, 3.29; N, 8.79.
2.1.7. 1-Amino-3-methyl-8H-indeno[1,2-c]thiophen-8-one (14)
A mixture of compound 2 (10 mmol) and ethyl prop-2-enoate (10 mmol) was taken in ethanolic sodium hydroxide (30 ml). The reaction mixture was heated under reflux for 8 h. The solution was concentrated under reduced pressure, poured into crushed ice and neutralized with dilute hydrochloric acid. The formed solid was filtered off, washed with water, dried and recrystallized from absolute ethanol to form 14; Yield (38%), m p 270–272°C; IR (KBr cm-1) νmax: 3418 (NH2), 1707 (C=O), 1610 (C=C); 1H NMR (600 MHz, DMSO-d6) δ 8.01–7.94 (m, 2H), 7.67 (m, J = 7.6, 1.5 Hz, 1H), 7.58 (m, J = 7.6, 1.3 Hz, 1H), 6.64 (s, 2H), 2.23 (s, 3H). 13C NMR (150 MHz, Chloroform-d) δ 186.10, 149.88, 143.81, 138.17, 132.27, 131.84, 128.83, 125.33, 125.03, 123.29, 117.46, 17.91. Anal. Calcd for C12H9NOS (215.27): C, 66.95; H, 4.21; N, 6.51; Found: C, 66.91; H, 4.39; N, 6.90.
2.1.8. 5-Imino-5,10c-dihydroindeno[1’,2’:4,5]thiopyrano[2,3-c]furan-1,3,6(3aH)-trione (16)
A solution of compound 2 (10 mmol) and maleic anhydride (10 mmol) in pyridine (20 ml) was heated under reflux for 6 h, cooled and then poured onto ice-cold water containing a few drops of HCl. The solid formed was filtered off, washed with water, then dried and recrystallized from n-butanol to form 16; Yield (42%); m p 240–242°C; IR (KBr cm-1) νmax: 3364 (NH), 1717 (C=O); 1H NMR (600 MHz, DMSO-d6) δ 10.46 (s, 1H), 7.82 (dd, J = 8.3, 1.5 Hz, 1H), 7.71–7.64 (m, 2H), 7.47 (td, J = 7.9, 1.5 Hz, 1H), 5.27 (d, J = 6.8 Hz, 1H), 4.99 (d, J = 6.8 Hz, 1H), 13C NMR (150 MHz, Chloroform-d) δ 189.96, 170.23, 167.26, 167.23, 157.51, 139.75, 134.57, 130.70, 129.73, 128.66, 124.20, 123.89, 47.92, 47.41; Anal. Calcd for C14H7NO4S (285.27): C, 58.94; H, 2.47; N, 4.91; Found: C, 59.0; H, 2.53; N, 4.90.
2.1.9. 2-Amino-4-thioxoindeno[1,2-b]pyran-5(4H)-one (19)
A mixture of compound 2 (10 mmol) and ethyl cyanoacetate (10 mmol) in absolute ethanol (30 ml) with a few drops of TEA was heated under reflux for 8 h. The reaction mixture was cooled, diluted with water and neutralized with acetic acid. The solid formed was filtered off, washed with water, dried and recrystallized from methanol and a few drops of acetic acid to form 19. Yield (83%); m p 212–214°C; IR (KBr cm-1) νmax: 3400 (NH2), 1708 (C=O), 1578 (C=S); 1H NMR (600 MHz, DMSO-d6) δ 8.05 (dd, J = 7.9, 1.7 Hz, 1H), 7.77 (m, J = 8.1, 1.7 Hz, 1H), 7.51 (m, J = 7.9, 1.5 Hz, 1H), 7.39 (m, J = 7.9, 1.7 Hz, 1H), 7.37 (s, 2H), 6.54 (s, 1H), 13C NMR (150 MHz, Chloroform-d) δ 203.75, 190.00, 160.93, 159.77, 136.89, 135.47, 131.01, 130.07, 124.50, 124.15, 122.06, 103.29.; Anal. Calcd for C12H7NO2S (229.25): C, 62.87; H, 3.08; N, 6.11; Found: C, 63.00; H, 3.16; N, 6.09.
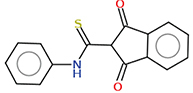
2.1.10. 1,3-Dioxo-N-phenyl indane-2-carbothioamide (20)
A mixture of Indian-1,3-dione 1 (10 mmol) and phenyl isothiocyanate (1.35 g, 10 mmol) in absolute ethanol (30 ml) with a few drops of TEA was heated under reflux for 2 h. The formed precipitate, after cooling was filtered off, dried and recrystallized from ethanol to yield 0.70 g (25%) of 20, mp 186–188°C; IR (KBr cm-1) νmax: 3228 (NH), 1657, 1630 (C=O), 1597 (C=C); 1H NMR (600 MHz, DMSO-d6) δ 11.73 (s, 1H), 8.12 (dd, J = 6.1, 3.6 Hz, 2H), 7.88 (dd, J = 6.2, 3.6 Hz, 2H), 7.58–7.53 (m, 2H), 7.35–7.29 (m, 2H), 7.11 (tt, J = 6.9, 1.2 Hz, 1H), 5.23 (s, 1H). 13C NMR (150 MHz, Chloroform-d) δ 197.87, 195.37, 139.12, 138.92, 135.38, 129.21, 123.98, 123.77, 122.30, 60.96., Anal. Calcd for C16H11NO2S (281.33): C, 68.31; H, 3.94; N, 4.98. Found: C, 68.20; H, 4.00; N, 4.81.
2.1.11. 3-Hydroxy-1-oxo-1H-indene-2-carbodithioic acid (21)
A mixture of Indian-1,3-dione 1 (10 mmol), carbon disulfide (10 mmol) and KOH (0.5 g) in absolute ethanol (50 ml) was heated under reflux for 4 h. After cooling and neutralization, the formed precipitate was filtered off, dried and recrystallized from butanol to form compound 21. Yield (57%); m p 220–222°C; IR (KBr cm-1) νmax: 3418 (OH), 1714, 1664 (2C=O); 1H NMR (600 MHz, DMSO-d6) δ 8.20 (dd, J = 8.1, 1.5 Hz, 1H), 7.74 (dd, J = 8.1, 1.7 Hz, 1H), 7.51 (td, J = 7.9, 1.7 Hz, 1H), 7.45 (td, J = 7.9, 1.5 Hz, 1H), 5.65 (s, 1H). 13C NMR (150 MHz, Chloroform-d) δ 211.03, 187.56, 181.92, 137.45, 136.58, 131.01, 130.01, 124.70, 124.26, 122.19.; Anal. Calcd for C10H6O2S2 (222.28): C, 54.03; H, 2.72; Found: C, 54.30; H, 2.70.
2.1.12. 3-(Pyridin-2-ylamino)-1H-inden-1-one (22)
A mixture of Indian-1,3-dione 1 (10 mmol) and 2-aminopyridine (10 mmol) in absolute ethanol (30 ml) was heated under reflux for 6 h. The reaction mixture was cooled, diluted with water and neutralized by acetic acid. The solid formed was filtered off, washed with water, dried and recrystallized from benzene/pet. Ether (1:2) to form compound 22. Yield (61%); m p 270–272°C; IR (KBr cm-1) νmax: 3423 (NH), 1672 (C=O), 1617 (C=C); 1H NMR (600 MHz, DMSO-d6) δ 10.10 (d, J = 0.6 Hz, 1H), 8.06–8.02 (m, 1H), 7.85 (dd, J = 7.9, 1.3 Hz, 1H), 7.45 (DTD, J = 12.1, 7.4, 1.8 Hz, 2H), 7.39 (m, J = 8.2, 2.0, 0.9 Hz, 1H), 7.33 (m, J = 8.3, 7.0, 1.3 Hz, 1H), 6.56 (dd, J = 7.2, 3.9 Hz, 2H), 6.30 (t, J = 5.6 Hz, 1H), 4.68 (dd, J = 5.5, 0.8 Hz, 2H). 13C NMR (150 MHz, Chloroform-d) δ 191.90, 158.46, 148.17, 138.02, 137.59, 134.92, 130.45, 129.97, 129.29, 127.97, 113.17, 108.15, 44.81.; Anal. Calcd for C14H10N2O (222.25): C, 75.66; H, 4.54; N, 12.60. Found: C, 75.60; H, 4.56; N, 12.48.
2.1.13. Bis [3-(phenyl amino)-1H-inden-1-one-2-yl]-1,2-ethandione (23)
A mixture of Indian-1,3-dione 1 (2.92 g, 20 mmol), aniline (1.86 g, 20 mmol) and diethyl oxalate (1.46 g, 10 mmol) was taken in 20 ml of absolute ethanol. To this, sodium ethoxide solution prepared from sodium metal (0.46 g) in absolute ethanol (30 ml) was added and the reaction mixture was heated under reflux for 6 h. The solution was concentrated under reduced pressure, poured into crushed ice and neutralized with dilute hydrochloric acid. The formed solid was filtered off, washed with water, dried and recrystallized from benzene/pet. ether (2:1) to yield compound 23; Yield (35%); mp 184–186°C; IR (KBr cm-1) νmax: 1706, 1686, 1669 (3CO); 1H NMR (600 MHz, DMSO-d6) δ 8.09–8.04 (m, 1H), 7.73–7.67 (m, 2H), 7.67–7.61 (m, 1H), 7.38–7.32 (m, 2H), 7.29–7.24 (m, 2H), 7.10 (tt, J = 6.8, 1.5 Hz, 1H), 5.45 (s, 1H). 13C NMR (150 MHz, Chloroform-d) δ 199.58, 189.30, 161.27, 151.98, 136.95, 136.36, 133.17, 132.69, 128.73, 124.84, 124.23, 123.98, 121.73, 64.38; Anal. Calcd for C32H20N2O4 (496.52): C, 77.41; H, 4.06; N, 5.64; Found: C, 77.15; H, 4.34; N, 6.00.
2.2. Biology
2.2.1. Cell culture, treatment & MTT assay
The cell lines were provided and purchased from VACERA (Cairo, Egypt). The cells were maintained in RPMI-1640 (Sigma-Aldrich, MO, USA). Both types of media were supplemented with 2mM L-glutamine (Lonza, Verviers, Belgium), 10% FBS (Sigma, MO, USA) and 1% penicillin/streptomycin (Lonza) [22]. The routine tissue culture work was described according to the Freshney group (1); all cells were incubated at 37°C in a 5% CO2 atmosphere (NuAire, Caerphilly, UK) and all cancer cell lines were cultured. The cells were plated at a density of 5000 in triplicates in a 96-well plate. The next day, the cells were treated with the indicated compound(s) at the indicated concentrations of 0.5 mg/mol in a final volume of 100 μl media. Cell viability was assessed after 72 h using an MTT solution (Promega, WI, USA) [23]. Then, 20 μl of the reagent was added to each well, the plate was incubated for 3 h, fluorescence was subsequently measured (570 nm) using a plate reader and the IC50 values were calculated using Prism 10.
2.2.2. Cell death investigation flow cytometric analysis of annexin V/PI staining & cell cycle
The cells were seeded into six-well culture plates (3–5 × 105 cells/well) and incubated overnight at 37°C, under 5% CO2. The cells were then treated with indicated compounds for 48 h. Next, media supernatants and the cells were collected and washed with ice-cold PBS. Next, the cells were stained with propidium iodide (PI; 10 mg/ ml) for 30 min and analyzed for DNA contents using flow cytometry analysis using an FL2 (λex/em 535/617 nm) signal detector (ACEA Novocyte™ flow cytometer, ACEA Biosciences, Inc, CA, USA). For each sample, 12,000 events are acquired. Cell cycle distribution is calculated using a CytoFLex machine and analyzed using CytExpert software. Flow cytometric methodologies (annexin V/PI staining with cell cycle analyses) are carried out as previously described [24].
2.3. In silico-studies
2.3.1. Molecular docking
Molecular operation environment software (MOE2019) was utilized to dock the complexes toward the human serum albumin (HSA; Protein Data Bank [PDB] ID: 1H9Z), HeLa caspase3 (PDB ID: 5IAE) and ovarian cancer targets (PDB ID: 3W2S) [17,25]. We used the docking protocol described in our previous work [17,26]. After the crystal structure was downloaded from the PDB (www.rcsb.org). The water molecules, co-ligand and metal ions were removed. The final form was obtained after 3D protonation and the correction process. The MOE site finder generated the active binding sites to create the dummy sites as the binding pocket. The default docking parameters were triangle matcher for replacing the molecule and London dG for rescoring the docking scores. The DFT-optimized structures of the compounds were used to generate the best five binding poses with flexible molecule rotation. The hydrogen bonds formed between the elastase and the investigated compound were used to rank the binding affinity and were presented as the free binding energy (S, kcal/mol). The higher negative values of the docking scores were presented along with 2D and 3D structures.
2.3.2. Pharmacokinetics
POM theory is a method to calculate prepared compounds’ bio-calculations, toxicities and drug scores. Synthesized compounds were also selected for the in silico POM study to calculate general properties and predict the drug score for various bioactivities. Data were analyzed and compared with standard antibacterial and antitumor drugs. Osiris and Molinspiration are cheminformatics-based software tools that help predict possible side effects, calculate molecular properties and forecast the compounds’ bioactivity scores [16].
2.3.3. Density functional theory
The Spartan ‘20 program was used to perform quantum chemistry calculations using the DFT method. In addition, spartan ‘20 was used to display all the data files. The DFT at 6–311G++(d,p) basis set/B3LYP approach was utilized to optimize the organic chemical structure of the compounds under investigation. Chemoffice 19.0 software was used to create the original chemical structure [27].
2.3.4. Molecular dynamics
The molecular dynamics (MD) simulations studies were conducted on the synthesized compounds 20 and 23 to comprehend the binding interaction and affinity between them and macromolecule protein, human serum albumin complexed with myristic acid and warfarin R-(+) enantiomer (PDB: 1H9Z) for a duration of 100 ns. The ligands and protein were initially refined through the Schrodinger software's LigPrep and Protein preparation wizard module (Schrödinger Release 2023–4: LigPrep, Schrödinger, LLC, NY, USA).
The MD simulation was utilized to comprehend the conformational adjustments and interactions between ligands and proteins over time. The study was conducted on the Desmond module of the Schrodinger software [28]. First, the protein–ligand complex was added to the project table for the MD simulation study. Then, the analysis was conducted in three steps. (1) System builder: In the system builder panel, the protein–ligand complex was placed within an orthorhombic boundary box surrounded by salt and water [29]. (2) Energy minimization: The next step involved energy minimization, during which the protein–ligand complex was solvated with SPC (single point charge) water and force field, and OPLS was used. (3) Molecular dynamics: Finally, MD simulation was carried out for 100 ns using the NPT ensemble (isothermal–isobaric ensemble with constant temperature, pressure and the number of particles) at 1.01 bars atmospheric pressure and 300K temperature, with the other settings remaining default [30].
3. Results & discussion
3.1. Chemistry
Indane-1,3-dione (1) was reacted with ammonium thiocyanate in glacial AcOH to give 3-Hydroxy-1-oxo-1H-indene-2-carbothioamide (2) in good yield (Figure 1 A).
Figure 1.

Synthesis of the new indane derivatives part 1. (A) Synthesis of compound series 2–8. (B) The new synthesized series 11 and 14.
The IR spectrum of 2 showed strong absorption bands at 1709 and 1679 cm-1 corresponding to two C=O groups and 3361 cm-1 for the NH2 group and an absorption band at 3454 cm-1 attributed to OH. 1H NMR spectrum of 2 showed signals at δ 11.50 and δ 9.10 due to the OH and NH2 (exchangeable with D2O). Compound 2 was a good starting material for directly synthesizing azoles and azines. Thus, the reaction of 2 with hydrazine hydrate in DMF afforded indenopyrazole 3. The mechanism for constructing 3 involves the initial formation of a non-isolated hydrazone intermediate and subsequent intramolecular cyclo-condensation by loss of SH2 molecule. The structure of indenopyrazole 3 is well characterized by IR and 1H NMR spectra. 1H NMR spectrum of 3 showed two singlet signals for NH2 and OH groups at δ 6.39 ppm and δ 5.17 ppm, respectively, 2 methinyl protons at δ 4.17 and 5.9 in addition to the aromatic protons around δ 7.38–8.81 ppm. The IR spectrum of 3 displays characteristic bands for OH and NH at 3400 and 3244 cm-1. The reaction of indan-1,3-dione derivative 2 with aniline in DMF yielded 2-carbothioamide derivative 4. The IR spectrum 4 showed different bands at 3411, 3400 and 1705 cm-1 for NH2, OH and C=O groups, respectively.
1,3-Dipolar cycloaddition reaction of aliphatic thioamidic compounds to α,β-unsaturated systems was reported [20]. They have been widely used inefficiently and conveniently, synthesizing thiopyrane and thiazine derivatives. So, the reaction of indane derivative 2 with benzal acetophenone produced thiopyrane derivative 8, not-indene derivatives 6 or 7. The transformation of compound 2 into the thiopyrane derivative was preceded by intermediate 5, followed by cyclo condensation by loss of the H2O molecule.1H NMR spectrum of compound 8 exhibited a signal at δ 6.89 ppm assigned for NH2 (D2O exchangeable). The IR spectrum of 8 revealed the presence of two ketonic groups at 1681 and 1708 cm-1 and the NH2 group at 3360 cm-1.
Treating the indane derivative 2 with a molar amount of benzylidene malononitrile leads to the formation of the condensed thiopyrane 11 [21] through intermediate non-isolable Michael type adduct 9, which undergo intermolecular cyclodehydration and subsequent loss of HCN. The IR spectrum showed 11 display absorption bands at 3405, 2189 and 1706 cm-1 assigned to the imine proton, cyano function and carbonyl group. The structure of indenothiopyrane derivative 11 was confirmed by 1H NMR spectrum, which showed NH group proton at δ 7.93 ppm, cyclization of ketonic compound 2 with ethyl acrylate afforded indenothiophene derivative 14. The appearance of a broad peak at 3418 cm-1 due to NH2 in the IR spectrum and the stretching band for the carbonyl group at 1707 cm-1 confirmed the construction of compound 14 Figure 1B. 1H NMR spectrum of compound 14 exhibited signals at δ 9 and 1.9 ppm assigned for NH2 (D2O exchangeable) and CH3, respectively.
The reaction of compound 2 with maleic anhydride in pyridine undergoing intramolecular cyclization gives a good yield of the fused thiophene derivative 16. Figure 2A 1H NMR spectrum 16 showed the thiophene NH group singlet signal at δ 9.48 ppm (D2O exchangeable). The mechanism of the reaction proceeds via the addition of thiolate anion to the α,β-unsaturated ketone to give the intermediate 15. The intramolecular cyclo condensation of 15 with loss of H2O molecule has proceeded to afford the thiophene derivative 16.
Figure 2.
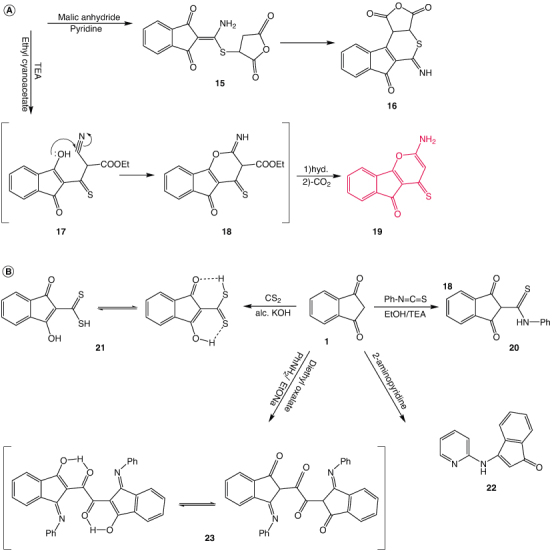
Synthesis of new series of indane derivatives part 2. (A) The new synthesized series 16 and 19. (B) The new synthesized series 20, 21, 22 and 23.
Treatment of enaminodiketones 2 with ethyl cyanoacetate using triethylamine gave the indenopyrane derivative 19. The reaction is expected to proceed via initial condensation of active methylene compound with enone 2 to afford the ketonic intermediate 17. Intermediate 17 undergoes in situ cycloaddition of enolic OH to cyano function, followed by a 1,3-hydrogen shift to yield intermediate 18. The intermediate 18 is then hydrolyzed and decarboxylated to afford the final product 19.
The structure of the latter compound 19 was deduced from its spectra and elemental analyses. The IR spectrum showed bands at 3400, 1708 and 1578 cm-1 attributed to NH2, C=O and C=S groups, respectively. 1H NMR spectrum of 19 indicates a singlet at δ 6.99 attributed to NH2. Treatment of the bifunctional compound 1 with phenyl isothiocyanate yielded the thioanilide derivative 20. The IR spectrum 20 contains bands at 3228 cm-1 for NH, and 1657 cm-1 is attributed to C=O groups. The 1H NMR spectrum of the adduct 20 showed the presence of the thioamide NH proton signal (D2O exchangeable) at δ 11.01 ppm, in addition to the methinyl proton at δ 4.49 ppm. Figure 3B represents the reaction of the potassium salt of indandione formed from the reaction of compound 1 with alcoholic KOH. This salt was reacted with carbon disulfide in ethanol to give indandione dithiocarboxylic acid 21, which was easily isomerized into enol form 21b Figure 2B 1H NMR of compound 21 showed the presence of OH and SH signals at δ 13.68 1.5 ppm, respectively.
Figure 3.
3D and 2D snapshots for the interactions of the investigated compounds. (A) 3D site view and 2D snapshots show the interaction between HSA (Protein Data Bank ID: 1H9Z) with compounds 20 and 23. (B) 3D site view and 2D snapshots show the interaction between capsase 3 (Protein Data Bank ID: 5IEA) with compounds 20 and 23. (C) 3D site view and 2D snapshots show the interaction between 3W2S with compounds 20 and 23.



The synthesis of acyclic enaminone 22 was achieved by condensation of 1,3-indandione 1 with 2-aminopyridine [22]. The IR spectrum of compound 22 showed absorption bands at 3423 cm-1 for (NH), 1672 cm-1 for (C=O) and 1617 cm-1 for (C=C). 1H NMR spectrum revealed signals at δ 8.98 ppm for NH and δ 6.50 ppm for CH ethylenic. The polyketones derivative 23 was obtained by alkaline condensation of two equivalents of 3-(phenylamino)-1H-inden-1-one produced in one pot reaction of 1 with diethyl oxalate. Compound 23 gives two types of enols (endo- and exo-cyclic) [23,24]. Spectral data revealed the structure of compound 23. The IR spectrum showed peaks at 1706, 1686 and 1669 cm-1 for tautomeric carbonyl groups. 1H NMR spectrum showed signals for the different tautomeric forms at δ 10.66 ppm of OH enolic and at δ 4.04 ppm of 2 methinyl protons.
3.2. Biology
All the synthesized compounds are tested against two cell lines, Ovcar-3 for ovarian cancer and HeLa for cervical cancer. In addition, all the results obtained were compared and validated to normal cell-line LCC-MK2 (Table 1) [26]. Graphical statistical analysis is represented in Supplementary Figure S2. Compounds 8, 20 and 23 showed the most potent and effective ones for the HeLa and the Ovarian cell lines [31]; the IC50 values with standard deviations are illustrated in Table 1 & Supplementary Table S1. In addition, the IC50 of LCC-MK2 showed that these compounds are moderately safe, as most IC50 values are less than 200 µg/ml. The high potency compound for the ovarian cell line is 20, while the HeLa cervical cell line is 23.
Table 1.
Summarization of the docking score energies and the biological data obtained.
| Compound | D.S.-HAS | D.S.-3W2S | D.S.-5IEA | IC50-Ovcar-3 | IC50-HeLa |
|---|---|---|---|---|---|
| 8 | -6.9443 | -7.5797 | -7.0834 | 16.007 ± 3.09 | 7.49 ± 0.78 |
| 11 | -5.8002 | -5.6802 | -5.7586 | 38.374 ± 0.511 | 10.74 ± 0.521 |
| 16 | -6.1812 | -5.9625 | -5.0964 | 23.1 ± 0.151 | 17.11 ± 0.17 |
| 19 | -6.8284 | -5.9033 | -5.4098 | 18.19 ± 0.914 | 14.56 ± 0.29 |
| 20 | -6.3258 | -6.7516 | -6.2030 | 6.858 ± 0.032 | 7.72 ± 0.47 |
| 23 | -8.5147 | -7.8940 | -8.1286 | 12.39 ± 0 | 6.74 ± 1.87 |
3.3. In silico-studies
3.3.1. Molecular docking
Molecular docking is a computational software routinely used to understand protein–receptor interaction with complexes. The docking process was carried out by simulating the interaction of the prepared compounds with three types of protein–receptor: HSA (PDB ID: 1H9Z; transport protein), Ovcar-3 Ovarian cancer (PDB ID: 3W2S; transferase/transferase inhibitor), and HeLa Caspase 3 V266F (PDB ID: 5IAE; hydolyase/hydrolayse inhibitor) [26], which were selected according to the literature and previous studies [32]. The docking score energy obtained by dock HSA on compounds 8, 11, 16, 19, 20 and 23 were -6.9, -5.8, -6.18, -6.83, -6.32, -8.51 kcal/mol respectively, this indicated that compounds 8, 19, 20 and 23 have the highest docking score. The docking score energies for HSA are represented in Supplementary Table S2 (Supplementary materials). The docking interactions are described in Supplementary Table S3 (Supplementary materials). 2D and 3D snapshots of the interaction between HSA and the selected compounds are mentioned in Supplementary Figure S3. While The 3D and 2D interactions of compounds 20 and 23 are illustrated in Figure 3A. Accordingly, the preferred compounds docked on HeLa Caspase 3 V266F, and the docking score energies showed that compounds 8, 20 and 23 are of highest values of -7.08, -6.203 and -8.13 kcal/mol, respectively. The docking score energies for and the docking interactions of HeLa caspase are represented in Supplementary Tables S4 & S5 (Supplementary materials), while the 3D and 2D interactions are illustrated in Supplementary Figure S4 (Supplementary materials). The 3D and 2D interactions of compounds 20 and 23 are illustrated in Figure 3B. Ovarian cancer docked with the selected compounds, and the highest docking score energies were for compounds 8, 20 and 23 with values -7.57, -6.75 and -7.84 kcal/mol, respectively. The docking score energies for and the docking interactions Ovarian cancer are illustrated in Supplementary Tables S6 & S7 (Supplementary materials), while the 3D and 2D interactions are illustrated in Supplementary Figure S4. Docking score energies and docking interactions of compounds are represented in Table 1, the 3D and 2D interactions of compounds 20 and 23 are illustrated in Figure 3C. From the results obtained, one can notice that compound 23 is the most effective compound.
The results obtained from the molecular docking studies are very similar and compatible with the biological assessment results. The summary collective results of both docking scores and biological studies are represented in Table 1
3.3.2. POM & pharmacokinetics studies
3.3.2.1. Evaluation of physicochemical characteristics & drug likeness
Selected compounds 8, 11, 16, 19 and 23 had their physicochemical characteristics and drug-likeness methodically evaluated using the POM platform. The goal of this thorough investigation was to find substances that have the potential to be developed into new drugs [33–39].
3.3.2.2. Optimum structure & lipinski properties
The computed findings of this investigation, including the ideal structure and Lipinski characteristics, are shown in Supplementary Table S8 (Supplementary materials). Remarkably, compounds 8 and 23 seem to have excellent qualities for developing new drugs. Compound 8 exhibits acceptable limits for both cLogP (lipophilicity) at 4.8 and clog (solubility) at 5.7 while having a molecular weight (Mw) of 395. Given that it has a drug score (DS) of 0.3 and a drug-likeness (DL) of 1.9, it may have therapeutic candidate potential. Compound 20, which has a Mw of 281.5, exhibits a potential drug-likeness with a DL score of 0.9 and a DS of 0.7. Its cLogP and cLogS values are 2.5 and 4.8, respectively [33–36].
3.3.2.3. Toxicity hazard assessment
Included toxicity hazards, physicochemical attributes and drug-likeness. Notably, the potential for reproductive impacts, irritant effect, tumorigenicity and mutagenicity were considered. According to Supplementary Table S9 (Supplementary materials) results from the Osiris program, most of the produced compounds showed neither mutagenicity nor irritative qualities. Interestingly, most of these compounds showed a high potential for adverse effects on reproduction, with compound 8 having the lowest potential for reproductive toxicity [36–38].
3.3.2.4. Hydrophilicity behavior & cLogP
The partition coefficient, or cLogP, was computed to assess the hydrophilicity behavior. It is generally advised to have a cLogP value of fewer than five, as this is frequently linked to sound absorption and penetration [34,35]. Surprisingly, all the data computed for the compounds matches this criterion, indicating positive hydrophilicity characteristics.
3.3.2.5. Compliance with Lipinski rules
This investigation also considered compliance with the Lipinski norms [35–37], a set of regulations essential to drug development. The majority of the synthesized compounds satisfy the Lipinski requirements and fall within an acceptable range of these guidelines, as the data clearly show. This suggests that the compounds have great potential as therapeutic possibilities. The systematic evaluation of physicochemical properties, drug-likeness and toxicity hazards is essential when selecting products for biological activities. The assessment data indicate compounds chosen to demonstrate favorable characteristics in these crucial areas, positioning them as promising candidates for further research and potential development of drugs.
Finally, The most potential compounds noticed are 20 and 23, and their bio-calculation and assessment of toxicities and drug scores are represented in Table 2A & B.
Table 2.
Different physico properties of compounds 20 and 23.
| A: Optimum structure and bio-calculation of prepared compounds 20, 23 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Optimum structure | Lipinski rules prediction | Drug-likeness prediction | |||||||
20 |
miLogP IPSA natoms MW nON nOHNH nviolations nrotb volume |
2.15 46.17 20 281.34 3 1 0 3 239.67 |
GPCR ligand Ion channel modulator Kinase inhibitor Nuclear receptor ligand Protease inhibitor Enzyme inhibitor |
-0.35 -0.39 -0.54 -0.23 -0.57 -0.03 |
|||||

23 |
miLogP IPSA natoms MW nON nOHNH nviolations nrotb volume |
4.52 93.01 38 496.52 6 0 0 5 432.70 |
GPCR ligand Ion channel modulator Kinase inhibitor Nuclear receptor ligand Protease inhibitor Enzyme inhibitor |
-0.11 -0.38 -0.25 -0.06 -0.25 -0.05 |
|||||
| B: Assessment of toxicities and drug scores of prepared compounds 20, 23 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | Toxicity risks | Drug Score predictions | |||||||
| Mw | Mut | Tum | Irr | Rep | cLogP | cLogS | DL | DS | |
| 20 | 281 |

|

|

|

|
2.5 | 4.8 | 0.9 | 0.7 |
| 23 | 496 |

|

|

|

|
3.9 | 7.2 | -3.3 | 0.2 |
High toxic ( ); Slight ( ); Slight ( ), No Toxic ( ), No Toxic ( ). Mut: Mutagenic; Tum: Tumorigenic; Tumorigenic: Reproductive Effective; Irri: Irritant; DS: Drug Score ). Mut: Mutagenic; Tum: Tumorigenic; Tumorigenic: Reproductive Effective; Irri: Irritant; DS: Drug Score
| |||||||||
| C: The HOMO and LUMO energies of compounds 8–23 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | Highest HOMO energy (ev) | Lowest LUMO energy (ev) | |||||||
| 20 | -8.78 | -1.28 | |||||||
| 23 | -9.27 | -0.97 | |||||||
HOMO: Highest occupied molecular orbital; LUMO: Lowest occupied molecular orbital.
3.3.3. Density functional theory studies
The DFT/B3LYP on Spartan 20 approach was used in the current work to perform quantum chemical computations to optimize the selected structures [40,41]. The DFT (B3LYP) method with 6–311G++(d,p) using Spartan 20 basis set was applied in this test. The electrostatic potential map, the optimized structure and its highest occupied molecular orbital (HOMO) and lowest occupied molecular orbital (LUMO) values are represented in Supplementary Figure S5 (Supplementary material). The HOMO energy expresses the ability of the compound to give electrons as an electron donor. It is localized mainly on compounds 8, 11, 16, 20 and 23.
On the other hand, the LUMO energy displayed by a site can act as an electron attractive, i.e., electron acceptor. The electrostatic potential maps of the compounds showed areas with electron localization throughout the molecules, with red and blue colors representing electron-rich (negative) and deficient (positive) localization, respectively Supplementary Figure S6 (Supplementary material). The highest HOMO energy and the least LUMO energy for the tested compounds are represented in Supplementary Table S10 (Supplementary material). Finally, Supplementary Table S11 (Supplementary material) describes the DFT calculations that revealed favorable energetic parameters for the selected compounds 8, 11, 16, 20 and 23. The atomic charge distributions of the compounds 8, 11, 16, 20 and 23 are depicted in Supplementary Figure S7 (Supplementary material). The data retrieved for most relevant compounds 20 and 23 are shown in Table 2C for the HOMO and LUMO energies, and Figure 4A (HOMO and LUMO molecular orbitals for compounds 20, 23), Figure 4B (electrostatic potential map of compounds 20, 23) and Figure 4C 9the charge distribution of the selected compounds).
Figure 4.

Density functional theory studies of compounds 20 and 23. (A) HOMO and LUMO molecular orbitals for compounds. (B) Electrostatic potential map of compounds. (C) The charge distribution of the selected compounds.
HOMO: Highest occupied molecular orbital; LUMO: Lowest occupied molecular orbital.
3.3.4. Molecular dynamics
To analyze the stability and fluctuations of the protein–ligand complexes, we subjected the compounds 20 and 23 in complex with human serum albumin complexed with myristic acid and warfarin R-(+) enantiomer (PDB: 1H9Z) to MD simulation for 100 ns.
We first analyzed the Root Mean-Square Deviation (RMSD) graphs of the protein–ligand complexes with compound 20 (Figure 5A). The protein fluctuated slightly around 30ns throughout the simulation and remained stable in the 2.0–7 Å range. On the other hand, the ligand initially showed fluctuations around 10ns and a drift of around 40ns, but after that, it attained stability throughout the fluctuation. We also used Root Mean-Square Fluctuation (RMSF) to find the perturbations in the protein residues during the 100ns simulation. The protein RMSF analysis revealed fluctuations in the backbone residues between amino acids 150–200 (3.2 Å) and 300–400 residues (3.2 Å), as shown in Figure 5B. The protein–ligand contacts diagram (Figure 5C) showed that Lys195 (54%) formed a π–cation interaction with the aromatic ring of the ligand and Arg218 (68%) showed an H-bond with the carbonyl group of the ligand. In comparison, His242 (70%) and Thr239 (72%) were involved in the hydrogen bond interaction with the hydroxy group. The residual amino acid interaction with functional pharmacophores of 20 is illustrated in Figure 5D.
Figure 5.

Molecular dynamic studies of compound 20. (A) The interaction between a protein–ligand (PL) in the form of the Root-Mean Square Deviation (RMSD) for compound 20. (B) The protein's Root Mean Square Fluctuation (RMSF). (C) The PL contacts of compound 20. (D) The residual interaction (2D) between the protein amino acids and compound 20 functional pharmacophores.
Similarly, the RMSD analysis of the human serum albumin complexed with compound 23 (PDB: 1H9Z) revealed that the protein exhibited some drift around 40ns and then became stable within the range of 1.2–4.4 Å. Similarly, the RMSD of ligand revealed that it remained stable during the 100ns simulation and within the protein's active site, as shown in Figure 6A. The RMSF analysis of the protein revealed that fluctuations occurred around 100–200 (4 Å) and 350 residues (3.2 Å), as depicted in Figure 6B. The protein–ligand contact diagram Figure 6C showed that Arg222 (99%) formed hydrogen bond interaction with the carbonyl group of the ligand, Arg218 (173%) formed hydrogen bond interaction with the hydroxy as well as with the carbonyl group, and Arg160 (33%) formed π–cation interaction with the aromatic ring of ligand. Additionally, other amino acid residues such as Lys195 (43%) and Lys194 (95%) formed hydrogen bond interaction with the hydroxy of ligand and Phe211 (33%) formed π–π stacking hydrophobic interaction. The residual amino acid interaction with functional pharmacophores of 23 is illustrated in Figure 6D.
Figure 6.

Molecular dynamic studies of compound 23. (A) The interaction between a protein–ligand (PL) in the form of the root-mean square deviation (RMSD) for compound 23. (B) The root mean square fluctuation (RMSF) of protein. (C) The PL-contacts of compound 23. (D) The residual interaction (2D) between the amino acids of protein with compound 23 functional pharmacophores.
4. Conclusion
Different derivatives of indandione were prepared in a good yield such as indeno pyrazole 3, enaminone 4, benzoyl indenothiopyrane 8, cyanoindenothiopyrane 11, methyl indenothiophene 14, thiophene derivative 16 and indenopyrane 19, carbothioamide 20, carbodithioic acid 21 and aminoindenone 22. Polyketonic compound 23. The compounds showed good anticancer activities and high docking score energies, the newly synthesized series is a promising one in synthetic medicinal chemistry which can be further applied to clinical chemistry shortly.
Supplementary Material
Acknowledgments
The authors extend their appreciation to Taif University, Saudi Arabia, for supporting this work through project number TU-DSPP-2024-19.
Funding Statement
The research was funded by Taif University Saudi Arabia, project number TU-DSPP-2024-19.
Supplemental material
Supplementary data for this article can be accessed at https://doi.org/10.1080/17568919.2024.2351350
Author contributions
Conceptualization, MGA; methodology, WS Shehab; software, MH Abdellattif; validation MAH; formal analysis, AF El-Farargy, MS Elgendy; investigation, FA Ramadan; resources, MS Elgendy; data curation, FA Ramadan; writing—original draft preparation, WS Shehab, AF El-Farargy; writing—review and editing, MH Abdellattif; visualization, MAH; supervision, MG Assy; project administration, MG Assy; funding acquisition, MS Elgendy, MAH. All authors have read and agreed to the published version of the manuscript.
Financial disclosure
The research was funded by Taif University Saudi Arabia, project number TU-DSPP-2024-19. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Papers of special note have been highlighted as: •• of considerable interest
- 1.Pluskota R, Koba M. Indandione and its derivatives - chemical compounds with high biological potential. Mini Rev Med Chem. 2018;18(15):1321–1330. doi: 10.2174/1389557518666180330101809 [DOI] [PubMed] [Google Scholar]
- 2.Li M, Yang W-L, Wen L-R, Li F-Q. Eur J Org Chem. 2008;16:2751–2758. http://onlinelibrary.wiley.com/. doi: 10.1002/ejoc.200800035 [DOI] [Google Scholar]
- 3.Kuan H-H, Chien C-H, Chen K. Org Lett. 2013;15:2880–2883. doi: 10.1021/ol4011689 [DOI] [PubMed] [Google Scholar]
- 4.Pizzirani D, Roberti M, Grimaudo SM, et al. J. Med. Chem. 2009;52:6936–6940. doi: 10.1021/jm900907s [DOI] [PubMed] [Google Scholar]
- 5.Shehab WS, EL-Farargy AF, Abdelhamid AO, et al. Synth Commun. 2019;49:3560–3572. doi: 10.1080/00397911.2019.1679538 [DOI] [Google Scholar]; •• Previous studies conducted, contain similar docking molecular dynamics and biological studies as references for this study.
- 6.Maddila SN, Maddila S, van Zyl WE, Jonnalagadda SB. RSC Adv. 2015;5:37360–37366. doi: 10.1039/C5RA06373F [DOI] [Google Scholar]
- 7.Siddappa A Patil, Renukadevi Patil, Shivaputra A Patil. Recent developments in biological activities of indanones. European J Med Chem. 2017;138:P182–198. doi: 10.1016/j.ejmech.2017.06.032 [DOI] [PubMed] [Google Scholar]
- 8.Pavlovskaya TL, Yaremenko FG, Lipson VV, Karpenko AS, et al. Beilstein J Org Chem. 2014;10:117–126. doi: 10.3762/bjoc.10.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chate AV, Dongre RM, Khaire MK, et al. Res Chem Intermed. 2018;44:6119–6136. doi: 10.1007/s11164-018-3479-9 [DOI] [Google Scholar]
- 10.Shehab WS, Assy MG, Abdellattif MH, et al. J Iran Chem Soc. 2019;16:2451–2461. doi: 10.1007/s13738-019-01712-4 [DOI] [Google Scholar]; •• Previous studies conducted relating to the work and topics in this paper.
- 11.Abdel-Galil EA, Mohamad M. Bioorg Med Chem. 2006;14:4341–4352. doi: 10.1016/j.bmc.2006.02.045 [DOI] [PubMed] [Google Scholar]
- 12.Hamdy NA, Gamal-Eldeen AM. Eur J Med Chem. 2009;44:4547–4556. doi: 10.1016/j.ejmech.2009.06.023 [DOI] [PubMed] [Google Scholar]
- 13.El-Shwiniy WH, Shehab WS, Mohamed SF, et al. Appl Organomet Chem. 2018;32:e4503–4519. doi: 10.1002/aoc.4503 [DOI] [Google Scholar]; •• Previous studies conducted relating to the work and topics in this paper.
- 14.Reis AS, Vogt AG, Pinz M, Petl MP, et al. Chem Biol Interact. 2019;311:108790. doi: 10.1016/j.cbi.2019.108790 [DOI] [PubMed] [Google Scholar]
- 15.Yamagishi H, Shirakami S, Nakajima Y, et al. Bioorg Med Chem. 2015;23:4846–4859. doi: 10.1016/j.bmc.2015.05.028 [DOI] [PubMed] [Google Scholar]
- 16.Alfaifi GH, Farghaly TA, Magda H. Abdellattif Indenyl-thiazole and indenyl-formazan derivatives: synthesis, anticancer screening studies, molecular-docking, and pharmacokinetic/ molin-spiration properties. PLOS ONE. 2023;18(3):e0274459. doi: 10.1371/journal.pone.0274459 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper contains one of the indolyl derivatives from the same family of derivatives in this article.
- 17.Shatha Ibrahim Alaqeel, Natarajan Arumugam, Vijayan Viswanathan, et al. Synthesis, computational studies and antibacterial assessment of dispirooxindolopyrrolidine integrated indandione hybrids. J Mol Struct. 2022;1267(2022):133577. doi: 10.1016/j.molstruc.2022.133577 [DOI] [Google Scholar]
- 18.Abdellattif MH, Elkamhawy A, Hagar M, et al. Novel saccharin analogs as promising antibacterial and anticancer agents: synthesis, DFT, POM analysis, molecular docking, molecular dynamic simulations, and cell-based assay. Front Pharmacol. 2022;13:958379. doi: 10.3389/fphar.2022.958379 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper contains one of the indolyl derivatives from the same family of derivatives in this article.
- 19.Hassan SA, Aziz DM, Abdullah MNJ, et al. In vitro and in vivo evaluation of the antimicrobial, antioxidant, cytotoxic, hemolytic activities and silico POM/DFT/DNA-binding and pharmacokinetic analyses of new sulfonamide bearing thiazolidin-4-ones. J Biomol Struct Dyn. 2023;42(7):3747–3763. doi: 10.1080/07391102.2023.2226713 [DOI] [PubMed] [Google Scholar]
- 20.Howsaui HB, Basaleh AS, Abdellattif MH, et al. Synthesis, structural investigations, molecular docking, and anticancer activity of some novel schiff bases and their uranyl complexes. Biomolecules. 2021;11(8):1138. doi: 10.3390/biom11081138 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Previous studies conducted relating to the work and topics in this paper.
- 21.Shchepin VV, Stepanyan YG, Silaichev PS. Russ J Gen Chem. 2008;78:929–932. doi: 10.1134/S1070363208050162 [DOI] [Google Scholar]
- 22.Freshney RI. Culture of Animal Cells. New York: John Wiley & Sons; 2010;463–479. doi: 10.1002/9780470649367 [DOI] [Google Scholar]
- 23.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
- 24.Dawood KM, Raslan MA, Abbas AA, et al. NovelBis-thiazole derivatives: synthesis and potential cytotoxic activity through apoptosis with molecular docking approaches. Front Chem. 2021;9:694870. doi: 10.3389/fchem.2021.694870 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper contains one of the indolyl derivatives from the same family of derivatives in this article.
- 25.Maciag JJ, Mackenzie SH, Tucker MB, et al. Tunable allosteric library of caspase-3 identifies coupling between conserved water molecules and conformational selection. Biophys Computat Biol. 2016;113(41):E6080–E6088. doi: 10.1073/pnas.1603549113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Haven Brandon A, Box G, Hallsworth A, et al. Identification of ovarian high-grade serous carcinoma cell lines that show estrogen-sensitive growth as xenografts in immunocompromised mice. Sci Rep. 2020;10(1):10799. doi: 10.1038/s41598-020-67533-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma M, Thakur S, Jadhav H, et al. Identification of azelastine and carvedilol as cholinesterase inhibitors via structure-based virtual screening of FDA-approved. Drugs. 2023;8(28):e202301879. [Google Scholar]
- 28.Galina Kalibaeva, Mauro Ferrario, Giovanni Ciccotti. Constant pressure-constant temperature molecular dynamics: a correct constrained NPT ensemble using the molecular virial. Mol Phys. 2023;101(6):765–778. DOI: 10.1080/0026897021000044025 [DOI] [Google Scholar]
- 29.Sun S, Li P, Wang J, et al. Novel scaffold agonists of the α2A adrenergic receptor identified via ensemble-based strategy. Molecules. 2024;29(5):1097. doi: 10.3390/molecules29051097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maciag JJ, Mackenzie SH, Tucker MB, et al. Tunable allosteric library of caspase-3 identifies coupling between conserved water molecules and conformational selection. Proc Natl Acad Sci USA. 2016;113(41):E6080–E6088. doi: 10.1073/pnas.1603549113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Titi A, Badran I, Dahmani M, et al. Rapid microwave synthesis of tetrahedral pyrazole/Co (II) complex:[NH··· Cl] synthon XRD/HSA-interactions, DFT/TD-DFT, physiochemical, antifungal, antibacterial, and POM bio-calculations. J Mol Struct. 2023;1293:136297. doi: 10.1016/j.molstruc.2023.136297 [DOI] [Google Scholar]
- 32.Dahmani M, Titi A, Et-Touhami I, et al. Novel 3-(2-thienyl) acrylic acid bridge di-triphenyltin (IV){[(Ph) 3SnCl) 2COO]-[Et3NH]+,[(Ph) 3SnCl2)]-[Et3NH]+} complex: XRD/HSA-interactions, physicochemical, thermal and POM/antifungal evaluation. Inorgan Chim Acta. 2023;557:121695. doi: 10.1016/j.ica.2023.121695 [DOI] [Google Scholar]
- 33.Titi A, Touzani R, Moliterni AA, et al. Synthesis, structural, biocomputational modeling and antifungal activity of novel armed pyrazoles. J Mol Struct. 2022;1264:33156. doi: 10.1016/j.molstruc.2022.133156 [DOI] [Google Scholar]
- 34.Titi A, Messali M, Alqurashy BA, et al. Synthesis, characterization, X-Ray crystal study and bioctivities of pyrazole derivatives: identification of antitumor, antifungal and antibacterial pharmacophore sites. J Mol Struct. 2020;1205:127625. doi: 10.1016/j.molstruc.2019.127625 [DOI] [Google Scholar]
- 35.Titi A, Almutairi SM, Alrefaei AF, et al. Novel phenethylimidazolium based ionic liquids: design, microwave synthesis, in-silico, modeling and biological evaluation studies. J Mol Liquid. 2020;315:113778. doi: 10.1016/j.molliq.2020.113778 [DOI] [Google Scholar]
- 36.Hadda TB, Rastija V, AlMalki F, et al. Ptra/Osiris/Molinspiration and molecular docking analyses of 3-hydroxy-indolin-2-one derivatives as potential antiviral agents. Curr Computer-Aided Drug Design. 2021;17(1):123–133. doi: 10.2174/1573409916666191226110029 [DOI] [PubMed] [Google Scholar]
- 37.Titi A, Warad I, Almutairi SM, et al. One-pot liquid microwave-assisted green synthesis of neutral trans-Cl2Cu (NNOH) 2: XRD/HSA-interactions, antifungal and antibacterial evaluations. Inorg Chem Commun. 2021;122:108292. doi: 10.1016/j.inoche.2020.108292 [DOI] [Google Scholar]
- 38.Titi A, Almutairi SM, Messali M, et al. New fluorinated imidazolium ionic liquids: ecofriendly synthesis, structural, in silico analysis and identification of antitumor pharmacophore sites. Inorgan Chim Acta. 2023;121619. doi: 10.1016/j.ica.2023.121619 [DOI] [Google Scholar]
- 39.Hatice Arı, Talat Özpozan, Yiğit Kabacalı, et al. Monomeric or dimeric? A theoretical and vibrational spectroscopic approach to the structural stability of 5-(4-metoxy benzoyl)-6-(4-metoxyphenyl)-3-methyl-2-thioxo-2,3-dihydropyrimidine-4(1H)-on. J Mol Struct. 2020;1222:128848. doi: 10.1016/j.molstruc.2020.128848 [DOI] [Google Scholar]
- 40.Amany Belal, Mohamed A Elanany, Ahmed A Al-Karmalawy, et al. Design of new captopril mimics as promising ACE inhibitors: ADME, pharmacophore, molecular docking and dynamics simulation with MM-PBSA and PCA calculations. J Taibah Univer Sci. 2023;17:1. doi: 10.1080/16583655.2023.2210348 [DOI] [Google Scholar]
- 41.Elyashberg M, Tyagarajan S, Mandal M, et al. Enhancing efficiency of natural product structure revision: leveraging CASE and DFT over total synthesis. Molecules. 2023;28(9):3796. doi: 10.3390/molecules28093796 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.